
Abstract
ZFTA–RELA is the most recurrent genetic alteration seen in paediatric supratentorial ependymoma (EPN) and is sufficient to initiate tumours in mice1. Despite its oncogenic potential, ZFTA–RELA (ZR) is observed nearly exclusively in childhood EPN, with tumours located distinctly in the supratentorial brain of the central nervous system1. We proposed that specific chromatin modules accessible during brain development would render distinct cell lineage programs at direct risk of transformation by ZR. To test this hypothesis, we performed combined single-nucleus assay for transposase-accessible chromatin and RNA (snMultiome) sequencing of the developing mouse forebrain compared with ZR-driven mouse and human EPN. We demonstrated that specific developmental lineage programs present in transient progenitor cells and regulated by PLAG/L family transcription factors were at risk of neoplastic transformation. Binding of this chromatin network by ZR or other PLAG/L family motifs targeting fusion oncoproteins led to persistent chromatin accessibility at oncogenic loci and oncogene expression. Cross-species analysis of mouse and human ZR EPN revealed significant cell type heterogeneity indicating incomplete neurogenic and gliogenic differentiation, with a small percentage of cycling progenitor-like or radial glial-like cells that established a putative tumour cell hierarchy. In vivo lineage tracing studies identified neoplastic clones that aggressively dominated tumour growth and established the entire EPN cellular hierarchy. These findings identify developmental epigenomic states that are critical for fusion-oncoprotein-driven transformation and show how these states continue to shape tumour progression.
Similar content being viewed by others


Main
Paediatric cancers are characterized by silent genomes and fewer disease-driving mutations than their adult counterparts2,3, and aberrant epigenomes have predominant roles in tumour initiation and development of many childhood cancers4. Fusion oncoproteins are common drivers of paediatric cancers and often define their own disease subtypes, for instance, EWSR1–FLI1 in Ewing sarcoma5, PAX3–FOXO1 in rhabdomyosarcoma6, ETV6–NTRK3 in infantile fibrosarcoma7 and ZFTA–RELA (ZR) in ependymoma (EPN)1. Although significant research has focused on molecular characterization of these fusion oncoproteins, the mechanism by which they drive transformation in the context of unique childhood developmental programs is incompletely understood. Specifically, how some of these fusion oncoproteins uniquely intersect and rewire developmental epigenetic states to transform cells remains unclear. To elucidate these mechanisms, we focused on EPN, the third most common paediatric brain tumour, as a disease model8. EPNs are aggressive, chemo-resistant paediatric brain tumours that are characterized by ‘quiet’ genomes with few recurrent genetic alterations but exhibit profoundly aberrant epigenomes1,9. ZFTA gene fusion events (including ZFTA–RELA) are observed nearly exclusively in EPNs and occur specifically in the brain cortex, suggesting a direct relationship between ZR transformation and cortical progenitor populations1. EPNs have been reported to arise from a type of radial glial cell (RGC) or gliogenic progenitor cell present during embryonic brain development10,11,12. RGCs are multipotent progenitor cells and can give rise to neurons and glia in two waves of differentiation during embryonic development13,14,15,16,17. Although EPN cells express many RGC markers10,11,18, the underlying molecular basis that places these and similar cycling progenitor cells at risk of transformation is not understood. In this study, we sought to understand the epigenomic landscapes of developmental cell lineages that render specific cell types vulnerable to transformation by fusion oncoproteins such as ZR. This was accomplished by dual single-nucleus RNA and assay for transposase-accessible chromatin (ATAC) (herein termed snMultiome) sequencing of the developing mouse brain from embryonic day 12.5 (E12.5) to postnatal day 6 (P6), a time of rapid progenitor cell expansion, as well as integrated characterization across mouse and human ZR EPN tumours. We applied in vivo barcoding technology (TrackerSeq19) to interrogate ZR-driven mouse EPN models and determine the dynamics of malignant clonal diversity through characterization of single-nucleus transcriptional programs and labelled lineage barcodes (LBs). Our investigation of the snMultiome landscapes of EPN across development and disease provide valuable insights into the cell lineage epigenomic programs that shape EPN initiation and progression. These findings may have broader ramifications for our understanding of how fusion oncoproteins leverage distinct chromatin modules in specific cell lineage programs to drive tumour development and cellular heterogeneity.
Embryonic chromatin accessibility and cancer
ZR functions as an oncogenic transcription factor (TF), activating neoplastic gene expression programs18,20,21. This led us to propose that ZR would remodel the developing epigenome and patterns of chromatin accessibility to activate oncogene expression. To test this hypothesis, we isolated putative EPN cells of origin, RGCs1,11, from FABP7–eGFP mice and transduced cells with a lentiviral vector expressing ZR or an empty vector control (Fig. 1a and Extended Data Fig. 1a). Across independent replicates, very minimal changes in patterns of chromatin accessibility were observed in ZR-transformed RGCs profiled by ATAC using sequencing (ATAC-seq) (Fig. 1b). This was in sharp contrast to ZR-driven activation of several thousand transcriptional targets and expression of a core 93-gene signature associated with ZR oncogenic activity (Fig. 1c). These findings suggested that patterns of chromatin accessibility are already established in normal neural progenitor programs and subsequently commissioned by binding of the ZR fusion oncoprotein to induce oncogenic activation.

a, Schematic illustrating the experimental workflow for generating bulk RNA-seq and ATAC-seq data from control (CTL) and ZR-transformed RGCs. b, Genome-wide changes in chromatin accessibility between control RGCs and ZR-transformed RGCs based on ATAC-seq, shown across two technical replicates (rep. 1 and rep. 2). c, Volcano plot showing differential gene expression from RNA-seq analysis between control and ZR-transformed RGCs; known ZR signature genes are highlighted. d, Schematic illustrating the CSI method for analysis of the binding affinity of ZR for PLAG family motifs. e, CSI rank 1 sequence showing the GC-rich PLAG family binding motif. f, Schematic depicting the methodology for generating snMultiome data from E12.5 to P6 mouse forebrain. g, Uniform manifold approximation and projection (UMAP) plot illustrating the distribution of annotated cell types following integration of the snMultiome data. h, Proportions of cell types identified in each of seven normal mouse forebrain samples. i, Results of pseudotime analysis showing lineage trajectories from RGCs to excitatory and inhibitory neurons. j, Landscape of cell-type-specific motif activation. CycProg, cycling progenitor cell; EC, endothelial cell. k, PLAG family motif activation scores across cell types. Dot size represents the percentage of activated cells. l, PLAGL2 motif scores across human developing brain cell types, derived from data published in ref. 25. CGE, caudal ganglionic eminence; CT, corticothalamic; dLGE, dorsal lateral ganglionic eminence; EN, excitatory neuron; ET, extratelencephalic; IPC, intermediate progenitor cell; IT, intratelencephalic; MGE, medial ganglionic eminence; NP, near-projecting; oRG, outer radial glia; PV, parvalbumin; SNCG, synuclein-gamma; SST, somatostatin; tRG, truncated radial glia; VIP, vasoative intestinal peptide; vRG, ventral radial glia. m, Average snATAC peak signal at known ZR binding sites in RGCs and neurons across embryonic stages E12.5, E14.5 and E16.5 in mouse forebrain development. The box plot shows the median (centre line), interquartile range (box) and range (whiskers). Statistical significance between groups at each time point was assessed using a two-sided Wilcoxon rank-sum test without adjustment for multiple comparisons (****P < 2.2 × 10−16). n, Scatter plot illustrating average snATAC peak signals in RGCs and neurons at overlapping ZR binding sites, with ZR-associated gene sites highlighted in red.
Although previous CUT&RUN experiments have suggested that PLAGL1/2 binding motifs are shared by ZR, the exact binding sequence and preference of the ZR fusion oncoprotein has remained unknown18. To understand the DNA sequence binding preferences of ZR, we adapted the cognate site identification (CSI) assay22 for use with ZR-expressing HEK293T cell lysates. This involved immunoprecipitation of HA-tagged ZR-expressing cell lysates with a library of 1 trillion unique combinations of double-stranded DNA fragments across a stretch of 20 base pairs. DNA fragments bound by HA-tagged ZR were amplified and reincubated with fresh lysate for subsequent rounds of immunoprecipitation. All rounds (three in total) were then barcoded and pooled for Illumina short-read sequencing. CSI profiling of ZR showed a very strong binding preference for PLAG/L family TF motifs engaged in a dimerized fashion and enrichment of a core GGGCC consensus binding sequence (Fig. 1d,e and Extended Data Fig. 1b). We therefore investigated the accessibility of PLAG/L family TF motifs during embryonic brain development across different developmental cell lineages.
To this end, we microdissected forebrains from mouse embryos at E12.5, E14.5, E16.5, E18.5, P0, P1 and P6, which contained transient progenitor cells, including RGCs23,24. Tissues were dissociated and subjected to single-nucleus ATAC-seq (snATAC-seq) and single-nucleus RNA sequencing (snRNA-seq), using the snMultiome assay developed by 10x Genomics (Fig. 1f). Cell types were annotated using marker sets curated from numerous published studies of the developmental mouse brain (Methods); this identified RGCs, cycling progenitor cells, neurons, astrocytes, oligodendrocyte progenitor cells (OPCs), microglia and endothelial cells, along with their transcriptomic and epigenomic signatures (Fig. 1g,h, Extended Data Fig. 1c,d and Supplementary Table 2). As expected, RGCs were actively proliferating in the early stages, giving rise to postmitotic inhibitory and excitatory neurons (Fig. 1i and Extended Data Fig. 1e–g). Critically, we found that PLAG/L family TF motifs were preferentially accessible in cycling progenitor cells, including RGCs, and chromatin accessibility was reduced upon neuronal differentiation (Fig. 1j,k and Extended Data Fig. 1h–m). These findings were consistent with observations in the developing human fetal brain25, with increased PLAGL2 TF activity in RGCs and intermediate progenitor cells that decreased upon differentiation (Fig. 1l). Examination of ZR-specific DNA binding sites in the PLAG/L TF motif data revealed cell-type-specific accessibility in RGCs versus neurons, including at known target genes such as Gli2, Jag1, Rela and Notch1 (refs. 18,21; Fig. 1m,n and Extended Data Fig. 1n). Finally, to investigate the oncogenic role of PLAG/L TF proteins, we overexpressed Plag1, Plagl1 and Plagl2 separately in mouse RGCs and compared transcriptional changes relative to ZR-expressing cells. Overexpression of the PLAG/L TFs did not lead to oncogenic ZR activation, as measured by a failure to recapitulate the 93-gene signature seen in ZR EPN (Extended Data Fig. 2a,b). Similarly, CRISPR–Cas9 knockout of Plagl1 in both a natively forming and a ZR implant model did not lead to increased survival1,11,18,21 (Extended Data Fig. 2c–e). Together, these findings pinpoint specific chromatin modules at risk of transformation in specific cell lineages and temporal windows present during brain development as an underlying basis for ZR-driven initiation of EPN.
Development restrains tumorigenesis
To study the intersection of developmental and ZR-driven oncogenic programs in EPN, we leveraged a natively forming ZR EPN mouse model established by an electroporation-based gene transfer technique called in utero electroporation (IUE) and characterized tumours using snMultiome profiling26,27. This involved stable integration of DNA transgenes expressing ZR using the PiggyBac transposon system at E16.5, predominantly targeting RGCs of the lateral ventricles with a PiggyBac transposase (PBase) vector driven by a Glast promoter18. Cells surrounding the lateral ventricles also express Plagl1 and known ZR target genes including Notch1 (Fig. 2a and Extended Data Fig. 2f,g). As controls, we generated a series of YAP1–MAMLD1 (YM)-driven EPN and glioblastoma (GBM) mouse tumours for comparative analyses (Fig. 2b,c). GBMs were established by simultaneous CRISPR–Cas9-mediated knockout of Nf1;Pten;Trp53 at E16.5 (Fig. 2b,c; denoted 3xCR). All tumour cell types had unique transcriptional programs, and application of known gene signatures (Supplementary Table 3) could robustly identify tumour cells that corroborated annotations using copy number variant (CNV) analysis (Fig. 2d and Extended Data Fig. 3).

a, Workflow for mouse IUE tumour generation and preparation of snMultiome libraries and data. b, UMAP based on weighted nearest-neighbour analysis of integrated snMultiome data from mouse IUE tumours. EPN ZR (N = 3), GBM (N = 1) and EPN YM (N = 3) tumours generated by IUE. Tumour cells were identified by expression of established tumour marker genes. c, Cells from each tumour type projected on to integrated UMAP. d, Distribution of cells exhibiting tumour gene signatures specific to tumour type. e, Reconstructed UMAP based on tumour cells and annotated tumour cell types. f, Distribution of tumour cell types across individual tumour samples. g, Scatter plot illustrating tumour-type-specific motif differences relative to other tumour types. The x axis denotes the mean motif score, and the y axis represents the average log2 fold change (FC) in motif activity. Highly differential motifs are highlighted in red and labelled. h, Activity states of PLAG family motifs across all cell types, with tumour cell groups indicated. Max., maximum; min., minimum. i, PLAG family motif activity across cell types in tumour (ZR, YM, GBM) and non-tumour cell groups. OC, oligodendrocyte cell. j, Comparison of cell cycling signal score (y axis) with ZR fusion target gene expression signal score (x axis) across the various tumour cell types.
The developmental mouse brain atlas used to identify cell types in normal development (Fig. 1) was then applied to snMultiome profiles of EPN and GBM mouse tumours. We observed diverse patterns of tumour and non-tumour cell types among ZR EPN, YM EPN and 3xCr GBM. Whereas YM EPN was almost exclusively composed of cells with signatures similar to those of RGCs (denoted RGC-like cells), suggesting a complete differentiation ‘block’28, ZR EPN tumours showed a diversity of tumour cells with signatures of RGC-like, cycling progenitor-like and neuronal-like cells (Fig. 2e,f and Extended Data Fig. 4). Notably, very few cells along the oligodendrocyte lineage were associated with neoplastic transformation in ZR EPN, in contrast to mouse GBM tumours, which demonstrated cellular expansion of the OPC compartment29,30 (Fig. 2e,f).
To further investigate the potential role of OPC epigenetic states in ZR EPN initiation, we isolated progenitor cells from E14.5 mouse brains, differentiated them to OPCs, and transduced them with a ZR-expressing lentivirus (Extended Data Fig. 5a–c). Unlike in RGCs, ZR expression in OPCs could not activate the full extent of oncogenic targets and did not result in global changes in patterns of chromatin accessibility according to ATAC-seq (Extended Data Fig. 5d,e). To understand whether ZR was still able to initiate tumours in the oligodendrocyte lineage, we performed IUE using two complementary methods: one in which the oligodendrocyte lineage was transformed with ZR at the time of surgery using an MBP promoter driving PBase; and one in which all electroporated cells were transformed with an MBP promoter driving ZR. In these ZR transformation experiments using these two methods, the oligodendrocyte lineage was either unable to form tumours or formed tumours at a significantly later time point, respectively (Extended Data Fig. 5f–h). Given that human ZR EPNs very rarely express OPC markers (for instance, OLIG2 and MBP)31, these data suggest that ZR may engage accessible PLAG/L TF motifs at an earlier time point in development, preceding the emergence of the OPC lineage.
In addition to gene expression differences, tumour types showed unique patterns of TF motif activity consistent with their underlying genetic drivers, including enrichment of PLAG/L TF motifs in ZR EPN, TEAD TF motifs in YM EPN32, and IRF TF motifs in GBM33 (Fig. 2g). Application of the 93-gene signature to mouse ZR EPN revealed widespread ZR target activity across all tumour cell states detected among RGC-like, cycling progenitor-like and neuronal-like cells (Fig. 2d). PLAG/L TF motif activity was detected across all tumour cell types and was highly correlated with ZR target activity (Fig. 2d,h,i and Extended Data Fig. 6a). This was in sharp contrast to normal mouse forebrain development, in which PLAG/L and TEAD TF motif activity decreased upon differentiation (Fig. 1j,k and Extended Data Fig. 6b).
Finally, we quantified the cell cycling state across different cell types identified in ZR-driven EPN mouse tumours (Fig. 2j). This showed that only a subpopulation of cycling progenitor cells and RGCs were actively dividing, with other cell types being mostly non-proliferative. Notably, only cycling progenitors with low-to-moderate ZR activity exhibited the highest cell proliferation signal (Fig. 2j); this was in contrast to other developmental cell types (for instance, neuronal-like), for which far fewer cells were in a proliferative state despite the presence of ZR oncogenic activity. Together, these data demonstrate that the transcriptional diversity of ZR EPN mirrors normal (albeit incomplete) developmental programs and that differentiated cell types are resistant to ZR-induced proliferation, potentially because of their more restricted epigenomic states.
Convergent epigenomic programs in brain cancer
We sought to corroborate our findings in mouse ZR EPN by studying human EPN samples. Primary tumours from 21 patient samples, comprising 6 ZR-driven EPNs, 10 posterior fossa EPNs (PF-EPNs) and 5 PLAG/L fusion-driven tumours were profiled by snMultiome sequencing (Fig. 3a,b). Upon cell type annotation, the non-tumour cells were found to include endothelial cells, immune cells and oligodendrocytes (Fig. 3b). Malignant and non-malignant cells were defined on the basis of the CNVs detected per cell (Fig. 3c, Extended Data Fig. 7 and Methods). In addition, t-distributed stochastic neighbour embedding (t-SNE) profiling of snRNA-seq data segregated PF-EPNs and resolved two groups of ZR and PLAG/L fusion-driven tumours (Fig. 3d). Consistent with mouse ZR EPN, we found that ZR fusion activity per cell could be predicted across species using a common 93-gene signature that could robustly stratify ZR-driven EPN tumours (Fig. 3e). There were significant differences in the gene expression signatures of PF-EPNs and those driven by ZFTA–RELA or PLAG/PLAGL gene fusions (n = 465 genes, adjusted P < 0.001 and average log2[fold change] > 1). Notably, at least 20 TF genes showed significantly different expression among the three groups; these included PAX3 and MECOM in PF-EPN, LHX2 and RELA in ZR EPN, and PAX2 and SALL3 in PLAG/L fusion-driven tumours (Fig. 3f and Supplementary Table 4).

a, Schematic of human snMultiome data generation from 21 patients, including ZR fusion-driven EPN (N = 6), PLAG/L fusion-driven tumours (N = 5) and PF-EPN (N = 10). b, Integrated snMultiome data from 21 patients with initial cell type annotation. UMAP was generated by joint analysis of snRNA and snATAC data using the weighted nearest-neighbour method. c, Tumour cells were categorized into non-malignant and malignant types according to their degree of CNV. d, Analysis of malignant tumour cells using tumour-specific gene expression markers classified patients into three molecular groups (PF-EPN, PLAG/L fusion and ZR EPN). Specific group characteristics were further validated by DNA methylation analysis. e, The ZR fusion signal in malignant cells was assessed across samples by expression of the 93-gene signature. f, Heatmap showing genes with significant differential expression among malignant cells predicted in the three tumour groups. TF-encoding genes with significant expression differences are highlighted. g, Heatmap displaying the top 100 differential snATAC peaks identified for each group. Motif enrichment was predicted separately for each group on the basis of the top 100 peaks, with the top 10 enriched motifs highlighted alongside the associated gene names. PLAG family motifs are highlighted in red. h, PLAG family motif activity in each tumour type. i, Comparison of the proportion of cells exhibiting open chromatin at the ZR binding region between the ZR and PLAG/L groups. Orange dots indicate regions in which the proportion of cells with open ZR binding sites was more than twofold higher in the ZR group compared with the PLAG/L group. Black-highlighted dots represent ZR fusion target genes. The green dotted line represents the regression line.
We next evaluated whether ZR and PLAG/L fused tumours converged upon shared chromatin accessibility programs enriched in developmental PLAG/L TF motifs. As expected, PLAG/L family (PLAG1, PLAGL1, PLAGL2) motifs were highly enriched in the ZR group among the top 100 differential snATAC peaks (Fig. 3g and Supplementary Table 4). PLAGL1/2 motifs were also highly enriched among the top DNA motifs in PLAG/L fused tumours, compared with PF-EPN (Fig. 3h). These data suggest a convergence upon PLAG/L family TF motifs in ZR and PLAG/L fusion-driven tumours. Direct comparison of snATAC results between ZR and PLAG/L showed many shared genomic sites (Fig. 3i), suggesting convergent oncogenic mechanisms. However, highly expressed signature genes of ZR EPN, including L1CAM, CCND1 and WNT3A, were accessible specifically in ZR EPN, indicating molecular differences (Fig. 3i and Supplementary Table 4). These findings underscore the utilization and diversity of PLAG/L TF family motifs that are leveraged by paediatric fusion oncoproteins to drive brain tumour development.
Tumour cell diversity mirrors development
We next characterized the cell types present in ZR and PF-EPN and their patterns of chromatin accessibility. Malignant cell types were distinguished based on gene expression markers curated from several published single-cell RNA sequencing (scRNA-seq) studies of human brain development (Methods). Similar to the findings in mouse ZR EPN, snMultiome sequencing identified malignant cells classified as RGC-like, cycling progenitor-like, neuronal-like, fibroblast-like, ependymal-like and astrocyte-like (Fig. 4a–d and Supplementary Table 5). Notably, ependymal-like cells were observed only in human ZR tumours and were absent from mouse ZR tumours (Figs. 4a–d and 2e,f). PF-EPN was largely composed of the same cell types, although in different proportions (Fig. 4a–d, Extended Data Fig. 8a,b and Supplementary Table 5). Given the overrepresentation of PLAG/L TF family motifs in ZR EPN (Fig. 3g–i), we next investigated the enrichment of these DNA motifs within ZR EPN cell populations. We found that PLAG/L motif activity was most elevated in cycling progenitor-like and RGC-like cells and maintained in differentiated cell types such as astrocyte-like and neuronal-like cells (Fig. 4e–g). ZR signatures were also positively correlated with PLAG/L TF motif activity (Fig. 4h,i and Extended Data Fig. 8c,d). Compared with normal cortical development (Fig. 1k,l), this suggests that ZR expression maintains chromatin accessibility of PLAG/L family motifs that are normally repressed during neuronal and glial differentiation. Finally, we predicted cell cycling signatures and compared them with ZR signatures in different malignant cell types. Only cycling progenitor-like cells with low-to-moderate ZR activity exhibited the highest cell proliferation signal, highly consistent with our findings in mouse ZR EPN (Figs. 2j and 4j). Together, these data suggest that in both mouse and human EPN development, ZR abnormally sustains the activity of epigenomic programs—specifically, the PLAG/L cistrome—beyond the normal developmental window, which typically closes upon neuronal cell differentiation.

a,c, Annotated cell types among malignant tumour cells from human PF (a) and ZR (c) snMultiome data. b,d, Expression of known marker genes for each malignant tumour cell type in PF (b) and ZR (d) tumours. e, PLAG family motif activity for each cell type in ZR and PF tumours. f, PLAG1, PLAGL1 and PLAGL2 motif activity for each cell type. g, Landscape of cell-type-specific motif activation. h, Signal intensity of ZR target genes in malignant cells of ZR tumours, indicated as high (red), medium (blue) and low (grey). i, Proportion of cells per cell type with detectable ZR target gene signal. j, Comparison of cell cycling signal score (y axis) versus ZR fusion target gene expression score (x axis) across cell types.
Dominant clones establish tumour heterogeneity
To study how developmental lineage programs intersect with tumour heterogeneity, we leveraged an in vivo barcoding system called TrackerSeq to label cells and identify barcodes using scRNA-seq19. TrackerSeq was used to barcode the developing embryonic brain at the time of EPN initiation. This was achieved by simultaneous incorporation of PiggyBac plasmids expressing ZR and transcribed RNA barcodes using IUE. Following detection by magnetic resonance imaging, tumours were dissected and sorted for positive GFP expression, which labelled successfully TrackerSeq-barcoded cells (Fig. 5a, Extended Data Fig. 9a,b and Supplementary Table 6). Our ZR EPN barcoded cohort consisted of 6 independent experiments with inclusion of multiple replicates totalling N = 11 TrackerSeq datasets. Significant tumour clonal diversity was observed in the early-stage neoplastic lesion, with four- to six-fold greater numbers of LBs detected compared with end-point tumours. However, in most cases, we observed the emergence of a largely single dominant tumour cell clone detected in end-stage tumours (Fig. 5b and Extended Data Fig. 9c–m). In TrackerSeq experiments, tumour cells were identified using a 93-ZR-gene signature (Fig. 5c,d), and dominant LBs (LB-1) were evenly distributed among tumour cells (Fig. 5e). Cell type annotations combined with trajectory analysis showed that dominant tumour cell clones encompassed the entire developmental and transcriptional diversity of mouse ZR EPN (Fig. 5f and Supplementary Table 6), revealing glial and neuronal cell lineage programs emerging from cycling progenitor-like cells. In addition, the dominant lineage largely comprised neuronal-like cells (Fig. 5g). These findings identify dominant tumour clones in mouse ZR EPN that drive tumour formation and potentially establish the entire cellular diversity of the disease.

a, Schematic of PiggyBac TrackerSeq lineage barcoding system and raw single-cell RNA data processing workflow. GFP + IUE cells were isolated and sequenced, and cells with a single LB were used for downstream analysis. b, LB abundance across six IUE biological replicates. Samples IUE-ZR-3 to IUE-ZR-5 included technical replicates. Sample IUE-ZR-1, derived from a P4 mouse brain, showed high diversity in the TrackerSeq library shortly after IUE. c, Integrated UMAP of all 11 IUE TrackerSeq datasets, with tumour cell clusters defined by ZR fusion target gene expression enclosed with a dotted line. d, UMAP visualization highlighting tumour cell clusters with ZR fusion target gene signal. e, Projection of cells harbouring dominant LBs from different samples on to tumour cell clusters in the UMAP, with assessment of barcode distribution across samples. f, UMAP reconstruction performed using tumour cells harbouring dominant LBs. Lineage trajectories were inferred by Slingshot on the basis of Seurat cluster assignments. Arrows denote two distinct differentiation trajectories identified through lineage analysis. g, Bar plot illustrating the relative proportions of tumour cell types derived from the dominant LB (LB-1). h,i, Pseudotime analysis of human ZR (h) and mouse IUE ZR (i) tumour cells showing progression from early-stage progenitor-like cells to later-stage differentiated cells. j, Schematic summarizing the main findings of our study, showing how shifts in cellular differentiation and progenitor programs correspond with changes in PLAG motif activity, providing insights into molecular mechanisms driving tumorigenesis from neurodevelopmental origins.
To characterize the cell heterogeneity and potential hierarchy governed by progenitor-like cells in human ZR EPN, we divided tumour cells from ZR EPN according to cycling progenitor or RGC-like signatures and compared them against differentiated programs known to arise from these transient progenitor cells in development, such as neurons, astrocytes and ependymal cells (Extended Data Fig. 10). A gradient of transcriptional states was observed, with cycling progenitor-like and RGC-like cell populations potentially giving rise to less frequently dividing cell types along the glial and neuronal lineages. To understand malignant cell lineage trajectories and determine which cell types develop in early versus late stages of differentiation, we performed pseudotime trajectory inference34,35 analysis of human ZR EPN; this showed that RGC-like and cycling progenitor-like cells arise ‘early’, followed by the emergence of differentiated neuronal-like, astrocyte-like and ependymal-like cells at later stages (Fig. 5h). Similarly, pseudotime trajectory inference analysis of tumour cells harbouring the dominant LB (LB-1) in TrackerSeq experiments suggested a model in which cycling progenitor-like cells could give rise to astrocyte-like and neuronal-like cells, mirroring normal (albeit incomplete) differentiation programs of the developing cortical brain (Fig. 5i). Together, these data support a model in which EPN cellular heterogeneity is established by dominant progenitor-like cells that give rise to postmitotic or infrequently dividing cells along the glial or neuronal lineages (Fig. 5j).
Discussion
Paediatric brain cancers are often considered a disease of development36, characterized by neoplastic transformation of early developmental cells. In EPN, multipotent RGCs have been proposed to be the candidate cells of origin10,11. However, the underlying mechanistic basis for this connection has remained unclear. snMultiome (ATAC + RNA) sequencing characterization of the developing mouse brain revealed a specific chromatin accessibility program in transient cycling progenitor cells (including RGCs) enriched in PLAG/L family TF motifs at direct risk of cellular transformation. ZR fusions, which are observed near-exclusively in EPN1,11,18,21, cannot significantly remodel chromatin accessibility but instead engage the already accessible PLAG/L epigenetic program for tumour progression. This led us to consider whether other paediatric cancer drivers (for instance, PAX3–FOXO1 and EWS–FLI1) arise not only in specific cell lineages but engage specific epigenomic programs active during distinct developmental windows that may be primed for cellular transformation.
PLAG/L family proteins have important roles in corticogenesis, patterning, proliferation and development37,38,39 and are overexpressed or form fusions in cancer40,41,42. However, they become downregulated and their binding sites correspondingly diminish shortly after birth. Considering the role of PLAGL1 and PLAGL2 in brain development and disease, the ability of ZR to maintain the accessibility of the PLAG/L motif beyond the embryonic stage, in contrast to its regulation during normal development, may be an important mechanism in EPN initiation. As a result, proproliferation genes that promote progenitor cell expansion—such as Ccnd1, Notch1 and Ephb2—are probably kept active through terminal glial and neuronal differentiation. In addition to binding of PLAG/L motifs, the acquisition of strong transcriptional activation domains through gene fusion is likely to play a critical part. In the case of ZFTA fusion oncoproteins, acquisition of intrinsically disordered regions that drive transcriptional condensate formation seems to be necessary for oncogenic transcription and tumour initiation43. PLAG/L fusions may have oncogenic mechanisms that converge with the acquisition of intrinsically disordered regions in EWSR1 that are critical for gene activation40,41.
Our findings may broadly implicate PLAG/L family DNA motifs as direct targets in brain tumour transformation. In support of this concept, we found shared chromatin accessibility programs that converged on PLAG/L family TF motifs when comparing ZR EPN with PLAG/L fusion-driven brain tumours, which often share EPN-associated histological features40,41. Beyond ZR and PLAG/L, PLAG1 is amplified and overexpressed in subsets of central nervous system embryonal tumours and is among the top upregulated genes in oncohistone H3 K27M-driven tumours42,44,45,46. These observations led us to propose that the PLAG/L TF network and its engagement by specific oncoproteins may have broader significance in the development of a wide variety of central nervous system malignancies.
Our study evaluated the intersection of oncogenic and developmental programs from persistent ZR expression during brain development. snMultiome analysis of the ZR-driven IUE model identified diverse cell types with distinct transcriptional and chromatin accessibility programs. These findings were validated across species by snMultiome profiling of mouse and human ZR EPN. Although we anticipated a ‘block’ in development at the RGC state, we also observed continued (albeit incomplete) differentiation through the glial and neuronal lineages, with most differentiated cells in ZR EPN classified as infrequently cycling or non-cycling. Notably, although this differentiation ‘block’ has not yet been validated in human YAP1 fusion EPN tumours, it may occur at the RGC stage, consistent with the known role of YAP1 in regulation of RGC proliferation47.
Despite ZR fusion activity across malignant cell types, cell cycle signatures were seen prominently in the progenitor cell state harbouring low-to-moderate levels of ZR target gene expression and rarely in differentiated populations such as neuronal-like and astrocyte-like cells. In normal brain development, RGCs can asymmetrically differentiate to intermediate progenitor cells, which then terminally differentiate to neurons48,49,50. This could suggest a mechanism whereby a minority of transformed ZR-driven cells accumulate in a progenitor state, with most cells continuing to differentiate along the neuronal or glial cell lineages. The elevated number of neuronal-like cells seen in these EPN tumours could suggest an origin in cells already primed to differentiate into neurons, such as RGCs and cycling progenitor cells. A caveat is that the Glast promoter used to govern ZR transformation in our experiments is not exclusively active in RGCs and cycling progenitor cells; its activity may be elevated in astrocytes and oligodendrocyte cells. This limitation is to some extent addressed by the IUE surgery itself, which targets RGCs that line the lateral ventricles at E16.5 of forebrain development. A further consideration is that the IUE transformation approach decouples endogenous regulation provided by cis-regulatory elements surrounding the native oncogene. A potentially more faithful approach would be to genetically engineer the chromosomal rearrangement that leads to ZFTA–RELA gene fusion; this may be possible in the future with advances in CRISPR genome-editing technologies.
In vivo lineage tracing (TrackerSeq) experiments of mouse ZR EPN demonstrated the emergence of dominant (often single) clones and the capability of such clones to establish the entire cellular diversity of ZR-driven EPN. Although different molecular barcoding approaches have been previously described in cancer, this has been characterized in only one mouse model of adult glioma (driven by PDGFB overexpression) and is distinct from the genetic drivers used to natively model brain tumours such as paediatric EPN51,52,53. Our findings suggest that upon expression of ZR during embryonic brain development, dominant tumour cell clones establish transcriptional diversity, which reflects normal (albeit ‘incomplete’) differentiation programs. This could have important therapeutic applications predicated on induction of cellular differentiation of cycling progenitor-like tumour cells to adopt a more differentiated state that is postmitotic or slowly mitotic, resembling normal development of glial and neuronal cells. The predominance of neuronal-like cells could suggest a lineage-specific differentiation bias, and the interplay between developmental and ZR programs remains to be defined.
Finally, on the basis of mouse and human ZR EPN, we propose a model in which a small proportion of cycling progenitor cells (including RGCs) contribute to EPN initiation and give rise to differentiated cells such as neuronal-like, ependymal-like and astrocyte-like tumour cells. Although our study does not ascribe function to these cells, we have observed hybrid, incompletely differentiated cells in patients with GBM capable of regulating neuronal activity in the tumour microenvironment54. Furthermore, we have previously demonstrated in glioma that differentiated malignant cells can contribute to tumour progression by releasing paracrine factors that maintain glioma stem cell identity and regulate tumour progression55. Our findings may also explain the highly treatment-resistant nature of EPN, in which persistent and non-dividing or slowly dividing malignant cells resist conventional cytotoxic therapies that target rapidly dividing cell types.
Methods
In utero electroporation
All animal procedures in this study were performed with approval from the St. Jude Institutional Animal Care and Use Committee. IUE was performed as described previously27, and plasmids were prepared with a NucleoBond Xtra Maxi Plus EF kit (Takara Biosciences). After anaesthesia with 4.5% isoflurane, pregnant CD1 mice at E16.5 were subjected to abdominal incision to expose the uterus. A DNA plasmid cocktail (1 μg μl−1 pBCAG-HA-ZRFUS1, 1 μg μl−1 pbCAG-eGFP-Luciferase, 1.5 μg μl−1 pX330-sgTp53, 2 μg μl−1 GLAST-PBase, 1.5 μg μl−1 mPlagl1 single-guide RNA, 0.5 μg μl−1 TrackerSeq library, FastGreen dye) was injected into the lateral ventricles with a glass pipette. Electric pulses were then delivered to the embryos by gently clasping their heads with forceps-shaped electrodes. Six 33-V pulses of 55 ms were applied at 100-ms intervals. The uterus was then repositioned into the abdominal cavity, the abdominal wall was sutured and the skin was stapled. Following birth, pups were monitored for clinical signs of tumour growth (such as seizures, circling, and head doming), as well being monitored by magnetic resonance imaging every 2 weeks. At end point, mice were collected for isolation of nuclei or immunofluorescence staining. In accordance with our St. Jude Institutional Animal Care and Use Committee protocol, end point was defined on the basis of a set of neurological symptoms (gait, hunching, kyphosis, squinting), and these limits were not exceeded in any of the experiments. Mice for isolation of nuclei were perfused with 10 ml cold PBS, and tumours were frozen in isopentane and stored at −80 °C. For survival curves, pups from a minimum of 2 mothers were included. Randomization and blinding were not applicable.
RGC isolation
RGCs were previously isolated from Ink4a-knockout mice with GFP expressed from the Blbp promoter using a Worthington Papain Dissociation system (LK003150). Cells were grown in neural basal medium (Invitrogen) supplemented with sodium pyruvate, glutamine, B27, N2, bFGF (10 ng ml−1) and rhEGF (20 ng ml−1) and tested for mycoplasma monthly. Cells were grown on treated cell culture dishes coated with Matrigel (Corning). RGCs were made ZR positive using a lentivirus generated by the Viral Vector Core at St. Jude. Mouse tumour cells were seeded into 10-cm plates 24 h before infection and infected with lentivirus with 8 μg ml−1 polybrene for 24 h. Infected cells were selected with 2 μg ml−1 puromycin for 3 days. ZR expression was confirmed by western blotting. Single-guide RNAs for Plagl1 were generated by the Center for Advanced Genome Engineering with an Addgene 52961 backbone, which included RFP. ZR-positive RGCs were infected by the same method and sorted for RFP using a BD FACSAria Fusion system. Knockout of more than 90% was confirmed by targeted deep sequencing.
OPC isolation
Primary OPC cultures were performed as previously described56,57. In brief, cortical tissues from E14.5 mouse embryos were collected, and neural stem cells were cultured as neurospheres for 4 days. Neural stem cells were dissociated and plated on poly-d-lysine-coated dishes at a density of 1.5 × 104 cells cm−2 in OPC media. They were subsequently infected with either ZR-Lenti-Cherry or control virus for 14 h. After viral infection, cells were maintained in OPC medium for 4 days before being collected for ATAC-seq and RNA-seq.
RNA-seq and ATAC-seq analyses
RNA-seq and ATAC-seq analyses were performed using Genialis Expression software (https://www.genialis.com) deployed locally on St Jude HPC infrastructure. Briefly, the RNA-seq pipeline run on the Genialis platform comprised the following steps. Raw reads were filtered to remove adaptors and poor-quality reads using BBDuk (v.37.9; https://sourceforge.net/projects/bbmap/). The resulting reads were mapped to the reference genomes (Ensembl 92) using STAR (v.2.7.0; RRID SCR_015899). FeatureCounts (v.1.6.3; RRID SCR_012919) was used for quantification of gene expression levels, followed by DEseq2 (RRID SCR_000154) for differential gene expression analysis. Genes with low expression (expression count summed over all samples of less than 10) were filtered out from the input matrix to DESeq2. The paired-end reads from ATAC-seq were trimmed using BBDuk (v.37.9) and mapped to reference genome mm10 using Bowtie2 (v.2.3.4.1). MACS2 (v.2.1.1.20160309) was then used to call peaks on the aligned reads using a P value cutoff of 0.01 (parameters –shift −75 –extsize 150 –nomodel –call-summits –nolambda –keep-dup all −P = 0.01).
TrackerSeq library generation and validation
TrackerSeq library cloning was carried out generally as described in ref. 19. In brief, the pCAG-SacB plasmid was digested with BstXI, and the 8-bit barcode was cloned into it using NEBuilder HiFi master mix (six reactions in total), followed by isopropanol purification. Purified reactions were electroporated into Endura DUOs (Lucigen) using a MicroPulser with program Ec1 (Bio-Rad). Four electroporations were carried out then recovered for 1 h at 37 °C in 2 ml of recovery media. Next, cells were plated overnight at 32 °C on 245-mm plates (Corning). The following morning, plates were scraped and cells were collected in Luria Broth (Miller), and library plasmids were purified using Endofree midiprep kits (Qiagen). For validation, 10 ng of the library plasmid prep was amplified using 2xPhusion (NEB) and sequenced by the Hartwell Center for Genome Sequencing Facility at St. Jude.
CSI DNA binding assay
HEK293T cells were transiently transfected with HA-tagged ZR fusion plasmids using Lipofectamine 2000 reagent according to the protocol (Thermo Fisher 11668019). Following expression, cells were lysed with RIPA buffer (Thermo Fisher 89900) and spun down for collection of supernatant. A DNA library (Integrated DNA Technologies) containing randomized central regions of 20 bp flanked by constant sequences complimentary to primers was converted to double-stranded DNA and brought to 74 ng μl−1 before being combined with 1% w/v bovine serum albumin, 500 ng μl−1 poly dI-DC (Thermo Fisher 20148E), 1% NP-40 (Thermo Fisher 85124), and 10× PBS. ZR-positive and ZR-negative cell lysates were incubated with this mixture for 1 h at room temperature. The mixture was then added to anti-HA beads (Thermo Fisher 88836) washed in binding buffer (10× PBS, 1% bovine serum albumin, 1% NP-40) and incubated for 30 min. Solutions were washed in binding buffer and aspirated on a magnetic plate three times before being resuspended in a PCR master mix (Lucigen Econo Taq 2×30035-1 and custom primers). Library fragments attached to beads were amplified on a Bio-Rad thermal cycler and then purified using a New England Biolabs Monarch PCR & DNA clean-up kit (T1030L). Eluted DNA library fragments from each sample were diluted to a concentration of 74 ng μl−1 and checked on a gel before being incubated with cell lysates again. Following three rounds of incubation and amplification, all purified library fragments from each round for each sample were given a unique barcode and a sequencing adaptor and then sequenced with a NovaSeq short-read sequencing amplicon kit, yielding approximately 500 million reads. Sequencing results were sorted by barcode, and the 20-bp library regions selected in each sample were ranked by enrichment and normalized to fusion negative lysate samples. Primer sequences were as follows: forward 5′-CTGATCCTACCATCCGTGCT-3′, reverse 5′-CCGCTCGGTACGAAGCTG-3′.
Nuclei isolation
Tissue (10–30 mg) was cut from human tumours and input into a 10x Nuclei Isolation Kit (PN-1000494). Kit instructions were followed, but the lysis buffer incubation time was increased to 15 min to isolate quality nuclei. Mouse embryonic forebrain was isolated with the 10x Demonstrated Protocol CG000366 RevD, but lysis buffer incubation was decreased to 2 min. IUE tumours were isolated with a Dounce homogenizer and iodixanol gradient using a modified version of the protocol described in ref. 58. Nuclei were resuspended in nuclei buffer and counted with a haemocytometer, and 10,000 were loaded on to a 10x Chromium Chip. Libraries were assessed with an Agilent TapeStation and sent for sequencing at St. Jude’s Hartwell Center on a NovaSeq 6000. Gene expression libraries had 400 million reads with a 28-10-10-90 cycle configuration, and ATAC libraries had 500 million reads with a 50-8-24-49 cycle configuration.
RNAscope
End-point ZR tumour-bearing mice were cervically dislocated, and brains were removed from the skull. Brains were flash-frozen in isopentane and frozen in OCT blocks, which were kept at −80 °C. Sections were cut on a Leica 3050 cryostat at 16 μm, and slides were kept at −80 °C until ready for use. The ACDBio Multiplex Fluorescent v.2 fresh frozen RNAscope protocol (revision B UM 323100) was followed with slight modifications: protease III was used for 30 min, opal fluorophores were used at 1:750, and DAPI was used at 1:5,000 for 20 min. The probes used were a custom C1 Zfta probe targeting [TGGCGTTTGGAGTATCTCATGGATTTCAACCCAGCGAGGCACGGCATGGTGTGCATGGTCTGCGGTAGCTCTTTGGCTACCCTGAAGCTGAGCACTATCAAACGCCACATCCGTCAGAAGCACCCGTACAGCCTGCAT], Cdk1 476081-C2, Pax6 412821-C3, Eomes 429641-C3, Grin1 431611-C3, Pdgfra 480661-C3, Mog 492981-C3 and Kif6 1760891-C3. Images were taken on a Zeiss LSM 780 at ×20 using a 9-square tile scan and maximum intensity projection of a z-stack.
Immunofluorescence
IUE surgery was performed with PBCAG-GFP at 4 μg μl−1. Brains were taken from P2 pups and placed in 4% paraformaldehyde at 4 °C with shaking for 24 h. They were then transferred to 30% sucrose until they sank (approximately 48 h). They were frozen in OCT and kept at −80 °C until ready for cutting. Brains were cut at 16 μm on a Leica 3050 cryostat, and slides were kept at −20 °C until ready for use. Slides were then permeabilized for 20 min (0.3% Triton in PBS), blocked for 1 h (5% normal donkey serum, 0.3% Triton in PBS), incubated with primary antibody overnight (GFP Aves, 1:2,000), incubated in secondary antibody for 1 h at room temperature (Thermo A78948), incubated in 1:5,000 DAPI (Thermo 62248) for 20 min, and mounted with Fluoromount G (Thermo 00-4958-02). For immunofluorescence of primary OPCs, cells were washed three times in PBS, permeabilized in PBST (0.3 % Triton/PBS) for 5 min, washed again, blocked with 10% normal goat serum in PBST for 1 h at room temperature, and incubated overnight at 4 °C with primary antibodies (Mouse OLIG2, Millipore MABN50, 1:500; Rabbit HA, sc-805, 1:1,000). After washing, cells were incubated with secondary antibodies for 1 h at room temperature, washed, stained with DAPI and mounted in VectaShield Antifade. All images were acquired on a Zeiss LSM 780 confocal microscope at ×20 using maximum intensity projections of z-stacks.
snMultiome data processing
For both human and mouse datasets, the ‘cellranger-arc count’ pipeline (10x Genomics, v.2.0.1, https://www.10xgenomics.com) was used for cell barcode detection, read alignment and quality assessment, following the standard 10x Genomics protocols. Human reads were aligned to the GRCh38 reference genome (refdata-cellranger-arc-GRCh38-2020-A-2.0.0, based on GENCODE v.32) and mouse reads to the mm10 reference genome (refdata-cellranger-arc-mm10-2020-A-2.0.0, based on GENCODE v.M23). The pipeline performed initial quality control by distinguishing intact nuclei from background and removing non-nucleus-associated reads. Subsequently, ambient RNA contamination per sample was assessed using SoupX (https://github.com/constantAmateur/SoupX) on the prefiltered data. Samples that passed both the initial quality control metrics reported in the Cell Ranger ARC QC summary and the SoupX contamination thresholds were retained for downstream analysis. We assessed data quality at the individual nucleus level and retained high-quality nuclei using Seurat (v.5.1.0, https://satijalab.org/seurat) and Signac (v.1.14.0, https://github.com/timoast/signac), applying the following criteria: total ATAC fragment count (nCount_ATAC) of at least 3,000, transcription start site enrichment scores between 2 and 15, total RNA counts (nCount_RNA) of at least 2,000, detected gene counts (nFeature_RNA) ranging from 500 to 8,000, and mitochondrial gene percentages less than 10%. For exclusion of doublets, doublet probabilities were estimated using DoubletFinder (v.2.0.4, https://github.com/chris-mcginnis-ucsf/DoubletFinder), and nuclei with high doublet scores were removed. DropletQC59 was applied to further identify and filter potential empty droplets, ensuring that only high-quality nuclei were retained for downstream analysis.
snMultiome data integration
For snATAC data of snMultiome analysis, open chromatin region peaks were called on individual samples using MACS2 (v.2.2.7, https://github.com/macs3-project/MACS) with the CallPeaks function in Signac (v.1.14.0), with intervals overlapping ENCODE blacklist regions excluded. To integrate all snATAC-seq data, we first created a unified set of peaks for quantification across datasets using the GenomicRanges package, filtering out peaks shorter than 20 bp or longer than 10,000 bp. Fragments for each sample were then recalled on the basis of this unified peak set. Top features in each snATAC-seq dataset were identified using FindTopFeatures (min.cutoff = 10), followed by TF-IDF normalization with the RunTFIDF function and latent semantic indexing (LSI) dimensionality reduction using the RunSVD function in Signac. After merging the datasets, we repeated these steps on the combined data. The snATAC data were then integrated using the FindIntegrationAnchors and IntegrateEmbeddings functions. Finally, nonlinear dimensionality reduction was performed using RunUMAP on LSI components 2 to 40. For snRNA data, normalization and data scaling were performed on the merged snRNA dataset using SCTransform v.2, followed by principal component analysis (PCA) and uniform manifold approximation and projection (UMAP) dimensionality reduction using the RunPCA and RunUMAP function in Seurat (v.5.1.0). Weighted nearest-neighbour analysis was done using Seurat with 1–30 principal components from snRNA data and 2–30 integrated LSI components from snATAC data. The resulting nearest-neighbour graph was then used for UMAP embedding and clustering following the best practice described in Seurat and Signac for multiome data analysis.
Estimation of motif activity from snATAC-seq data
Motif/TF chromatin accessibility (motif activity) was computed for a set of 841 TFs (combined mouse and human TFs) from the JASPAR 2022 database using the RunChromVAR function in Signac (v.1.14.0), and differential motif activity was computed with the FindMarkers function. Motif enrichment in the differentially accessible regions was analysed with the FindMotif function.
Cell type annotation for snMultiome and scRNA
Cell type annotation of single-cell data was performed using CellTypeEstimate (v.0.3.1; https://github.com/combiozone/CellTypeEstimate), an in-house tool adapted from the published tool ScType (https://github.com/IanevskiAleksandr/sc-type), incorporating custom code modifications and an alternative reference database. Marker sets were derived from more than 40 published studies of human and mouse brain development. References for each marker set are recorded in the ‘source’ column of the marker database files (db/*.xlsx). Following automated annotation, all labels were manually reviewed and refined to ensure biological accuracy. For tumour samples, the suffix ‘-like’ was manually appended to each annotated tumour cell type.
Processing of TrackerSeq barcode reads
We processed TrackerSeq barcode reads following the methods described in ref. 19 (https://github.com/mayer-lab/Bandler-et-al_lineage). Reads from the R2 FASTQ files were pre-processed by trimming the flanking sequences on both sides of the LBs. Barcodes shorter than 37 bp were discarded. A whitelist of cell barcodes was generated using UMI-tools. These whitelisted cell barcodes, along with their corresponding unique molecular identifiers, were appended to the read names in the LB FASTQ files to produce new, modified FASTQ files. The modified files were then used for downstream processing, including matching of LBs to cells and further filtering of the LBs. To isolate high-confidence LBs, we retained only those containing the conserved nucleotide motif ‘…CTG…ACT…GAC…TGA…CTG…ACT…GAC…’ and excluded any barcodes with ambiguous bases (N). These high-confidence LBs were matched to clean cells from the Seurat object, resulting in a set of cells with confidently assigned LBs. Cells were classified as either single LB, containing one unique LB per cell, or multiple LB, containing more than one LB per cell, on the basis of these assignments. In the single-LB group, LBs were ranked by number of associated cells, with those having the highest counts designated as dominant single LBs (typically referred to as LB-1). Only cells with a single LB were retained for downstream analyses.
TrackerSeq data analysis from quality control to cell type annotation
Raw TrackerSeq data were processed using Cell Ranger (v.9.0.0, https://www.10xgenomics.com) with mm10 (10x Genomics, refdata-cellranger-mm10-2020-A-2.0.0) as the reference genome. Then, filtered_feature_bc_matrix data were used to filter pooled cells and normalized in Seurat. We selected high-quality cells on the basis of cutoffs such as more than 500 genes, more than 2,000 unique molecular identifiers (nCount_RNA), less than 10% of mitochondrial genes, and less than the customized maximum quantile for nFeature_RNA (less than quantile(probs = 0.9) and no more than 8,000) and nCount_RNA (less than quantile(probs = 0.99)). Next, we used DoubletFinder (v.2.0.4) to estimate doublets, retaining only cells classified as ‘Singlet’, and removed empty droplets on the basis of DropletQC filtering. Afterwards, data were normalized using the SCTransform v.2 method in Seurat (v.5.1.0), regressing out the percentage of mitochondrial gene expression (percent.mt) to reduce its confounding effects. Last, high-quality cells were used to perform PCA, UMAP and clustering analyses in Seurat. Cell type annotation was then carried out using CellTypeEstimate (v.0.3.1) on the basis of the identified clusters. In downstream analyses, this information was integrated with LB-matched cells to enable more in-depth analysis. For the integration analysis, we merged all high-quality cells from independent TrackerSeq datasets using Seurat (v.5.1.0), performed SCTransform v.2 normalization (regressing out mitochondrial percentage) and PCA, applied Harmony (v.1.2.3; https://github.com/immunogenomics/harmony) with the IntegrateLayers function, and used the top 30 Harmony embeddings for UMAP visualization and clustering.
Classification of malignant cells
To classify cells as malignant or non-malignant, we inferred genome-wide CNVs using InferCNV (v.1.22.0, https://github.com/broadinstitute/infercnv) with default parameters. InferCNV was run at the sample level with integrated count matrices of snRNA-seq and snATAC-seq data, separately. The reference used immune cells and microglial cells. We first defined the CNVs as loss (InferCNV value ≤ 0.8) or gain (InferCNV value ≥ 1.2) and filtered CNV regions (hidden Markov model outputs) with fewer than 20 genes to detect large-region (≥5 Mb) chromosomal CNVs. Next, we calculated the copy number alteration ratio per CNV region. Last, we estimated malignant cells as those with large CNV regions (at least 100 CNV genes in the region) or a CNV ratio greater than 0.5 per sample.
ZR fusion signal signatures
The single-cell ZR fusion signature was calculated on the basis of 93 ZR driver genes18,20,45 using the AddModuleScore function in Seurat (v.5.1.0). We considered a score on our in-house benchmark test greater than 0.2 to indicate confidence, whereas scores of 0.1–0.2 indicated uncertainty, and scores less than 0.1 indicated distrust. Scores greater than 0.1 corresponded to the ZR target gene signal, whereas scores less than 0.1 indicated non-signal.
Cell cycling and non-cycling signatures
The CellCycleScoring function in Seurat (v.5.1.0) was used to score cells according to their cell cycle phase (G1, G2M or S). To define cycling and non-cycling cells, we recalculated the cell cycle score on the basis of cell cycle markers (cc.genes.updated.2019) using the AddModuleScore function in Seurat. The recalculated score was named cc.score. On the basis of internal testing, we defined cycling cells as those with cc.score, S.score or G2M.score greater than 0.2, and non-cycling cells as those with all scores less than 0.1.
Construction of pseudotime trajectories
Pseudotime trajectories were constructed to model cellular differentiation dynamics using two R packages, Monocle3 (v.1.3.7, https://cole-trapnell-lab.github.io/monocle3) and Slingshot (v.2.14.0). For Monocle3, the Seurat object was converted into a Monocle3 cell_data_set object. Root nodes were selected on the basis of known progenitor cell types identified in the UMAP, and cells were subsequently ordered along the trajectory for assignment of pseudotime values. For Slingshot, clustering results and dimensionality reduction coordinates from the Seurat object were used as input. Lineages were inferred on the basis of Seurat clusters or cell types, and pseudotime values were assigned to cells. The pseudotime from lineage 1 was used for downstream analyses.
Progenitor and lineage score of malignant cells
The progenitor and lineage score were calculated following the method in ref. 29, using only malignant cells identified by CNV (as described in the Methods). RGC-like or cycling progenitor-like cell types were designated as progenitors to serve as the starting point at the top. Progenitor score was estimated as the expression of the progenitor shared program minus the maximal expression of the two differentiation programs, and differentiated cells were further classified on the basis of average gene expression differences to differentiate between astrocyte- or ependymal-like and neuron-like lineages.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Data generated for this study are available at the Gene Expression Omnibus (accessions GSE269937, GSE314803, GSE314599 and GSE314911). All other data are available in the manuscript or in Supplementary Figures or Tables 1–5. All data needed to evaluate the conclusions of the paper are present in the paper and Extended Data Figs. 1–10.
Code availability
The data analysis code used in the study is available at GitHub (https://github.com/combiozone/PROJ-Multiome-EPN-ZR and https://github.com/combiozone/CellTypeEstimate).
References
Parker, M. et al. C11orf95–RELA fusions drive oncogenic NF-κB signalling in ependymoma. Nature 506, 451–455 (2014).
Sweet-Cordero, E. A. & Biegel, J. A. The genomic landscape of pediatric cancers: Implications for diagnosis and treatment. Science 363, 1170–1175 (2019).
Gröbner, S. N. et al. The landscape of genomic alterations across childhood cancers. Nature 555, 321–327 (2018).
Panditharatna, E. & Filbin, M. G. The growing role of epigenetics in childhood cancers. Curr. Opin. Pediatr. 32, 67–75 (2020).
Grünewald, T. G. P. et al. Ewing sarcoma. Nat. Rev. Dis. Primers 4, 5 (2018).
Barr, F. G. et al. Rearrangement of the PAX3 paired box gene in the paediatric solid tumour alveolar rhabdomyosarcoma. Nat. Genet. 3, 113–117 (1993).
Davis, J. L. et al. Infantile NTRK-associated mesenchymal tumors. Pediatr. Dev. Pathol. 21, 68–78 (2018).
Saleh, A. H. et al. The biology of ependymomas and emerging novel therapies. Nat. Rev. Cancer 22, 208–222 (2022).
Mack, S. C. et al. Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature 506, 445–450 (2014).
Taylor, M. D. et al. Radial glia cells are candidate stem cells of ependymoma. Cancer Cell 8, 323–325 (2005).
Johnson, R. A. et al. Cross-species genomics matches driver mutations and cell compartments to model ependymoma. Nature 466, 632–636 (2010).
Vladoiu, M. C. et al. Childhood cerebellar tumours mirror conserved fetal transcriptional programs. Nature 572, 67–73 (2019).
Deneen, B. et al. The transcription factor NFIA controls the onset of gliogenesis in the developing spinal cord. Neuron 52, 953–968 (2006).
Hirabayashi, Y. et al. Polycomb limits the neurogenic competence of neural precursor cells to promote astrogenic fate transition. Neuron 63, 600–613 (2009).
Mission, J. P., Takahashi, T. & Caviness, V. S. Jr. Ontogeny of radial and other astroglial cells in murine cerebral cortex. Glia 4, 138–148 (1991).
Noctor, S. C. et al. Neurons derived from radial glial cells establish radial units in neocortex. Nature 409, 714–720 (2001).
Noctor, S. C., Martínez-Cerdeño, V. & Kriegstein, A. R. Distinct behaviors of neural stem and progenitor cells underlie cortical neurogenesis. J. Comp. Neurol. 508, 28–44 (2008).
Arabzade, A. et al. ZFTA–RELA dictates oncogenic transcriptional programs to drive aggressive supratentorial ependymoma. Cancer Discov. 11, 2200–2215 (2021).
Bandler, R. C. et al. Single-cell delineation of lineage and genetic identity in the mouse brain. Nature 601, 404–409 (2022).
Kupp, R. et al. ZFTA translocations constitute ependymoma chromatin remodeling and transcription factors. Cancer Discov. 11, 2216–2229 (2021).
Zheng, T. et al. Cross-species genomics reveals oncogenic dependencies in ZFTA/C11orf95 fusion-positive supratentorial ependymomas. Cancer Discov. 11, 2230–2247 (2021).
Warren, C. L. et al. Defining the sequence-recognition profile of DNA-binding molecules. Proc. Natl Acad. Sci. USA 103, 867–872 (2006).
Misson, J. P. et al. Identification of radial glial cells within the developing murine central nervous system: studies based upon a new immunohistochemical marker. Brain Res. Dev. Brain Res. 44, 95–108 (1988).
La Manno, G. et al. Molecular architecture of the developing mouse brain. Nature 596, 92–96 (2021).
Wang, L. et al. Molecular and cellular dynamics of the developing human neocortex. Nature 647, 169–178 (2025).
Saito, T. & Nakatsuji, N. Efficient gene transfer into the embryonic mouse brain using in vivo electroporation. Dev. Biol. 240, 237–246 (2001).
John Lin, C. C. et al. Identification of diverse astrocyte populations and their malignant analogs. Nat. Neurosci. 20, 396–405 (2017).
Jessa, S. et al. Stalled developmental programs at the root of pediatric brain tumors. Nat. Genet. 51, 1702–1713 (2019).
Filbin, M. G. et al. Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science 360, 331–335 (2018).
Nagaraja, S. et al. Transcriptional dependencies in diffuse intrinsic pontine glioma. Cancer Cell 31, 635–652 (2017).
Otero, J. J., Rowitch, D. & Vandenberg, S. OLIG2 is differentially expressed in pediatric astrocytic and in ependymal neoplasms. J. Neurooncol. 104, 423–38 (2011).
Pajtler, K. W. et al. YAP1 subgroup supratentorial ependymoma requires TEAD and nuclear factor I-mediated transcriptional programmes for tumorigenesis. Nat. Commun. 10, 3914 (2019).
Lei, J. et al. Interferon regulatory factor transcript levels correlate with clinical outcomes in human glioma. Aging 13, 12086–12098 (2021).
Cao, J. et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502 (2019).
Street, K. et al. Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genomics 19, 477 (2018).
Filbin, M. & Monje, M. Developmental origins and emerging therapeutic opportunities for childhood cancer. Nat. Med. 25, 367–376 (2019).
Adnani, L. et al. Zac1 regulates the differentiation and migration of neocortical neurons via Pac1. J. Neurosci. 35, 13430–47 (2015).
Varrault, A. et al. Zac1 regulates an imprinted gene network critically involved in the control of embryonic growth. Dev. Cell 11, 711–722 (2006).
Adnani, L. et al. Plag1 and Plagl2 have overlapping and distinct functions in telencephalic development. Biol. Open 7, bio038661 (2018).
Sievers, P. et al. Recurrent fusions in PLAGL1 define a distinct subset of pediatric-type supratentorial neuroepithelial tumors. Acta Neuropathol. 142, 827–839 (2021).
Tauziède-Espariat, A. et al. CNS tumors with PLAGL1-fusion: beyond ZFTA and YAP1 in the genetic spectrum of supratentorial ependymomas. Acta Neuropathol. Commun. 12, 55 (2024).
Keck, M. K. et al. Amplification of the PLAG-family genes—PLAGL1 and PLAGL2—is a key feature of the novel tumor type CNS embryonal tumor with PLAGL amplification. Acta Neuropathol. 145, 49–69 (2023).
Arabzade, A. et al. Synthetic ZFTA fusions pinpoint disordered protein domain acquisition as a mechanism of brain tumorigenesis. Nat. Cell Biol. 27, 1496–1509 (2025).
Larson, J. D. et al. Histone H3.3 K27M accelerates spontaneous brainstem glioma and drives restricted changes in bivalent gene expression. Cancer Cell 35, 140–155 (2019).
Zheng, H. et al. PLAGL2 regulates Wnt signaling to impede differentiation in neural stem cells and gliomas. Cancer Cell 17, 497–509 (2010).
Krug, B. et al. Pervasive H3K27 acetylation leads to ERV expression and a therapeutic vulnerability in H3K27M gliomas. Cancer Cell 36, 338–339 (2019).
Cao, X., Pfaff, S. L. & Gage, F. H. YAP regulates neural progenitor cell number via the TEA domain transcription factor. Genes Dev. 22, 3320–34 (2008).
Koo, B. et al. Setting the clock of neural progenitor cells during mammalian corticogenesis. Semin. Cell Dev. Biol. 142, 43–53 (2023).
Johnson, M. B. et al. Single-cell analysis reveals transcriptional heterogeneity of neural progenitors in human cortex. Nat. Neurosci. 18, 637–46 (2015).
Bonnefont, J. & Vanderhaeghen, P. Neuronal fate acquisition and specification: time for a change. Curr. Opin. Neurobiol. 66, 195–204 (2021).
Walens, A. et al. Adaptation and selection shape clonal evolution of tumors during residual disease and recurrence. Nat. Commun. 11, 5017 (2020).
Ho, I. L. et al. Clonal dominance defines metastatic dissemination in pancreatic cancer. Sci. Adv. 10, eadd9342 (2024).
Ceresa, D. et al. Early clonal extinction in glioblastoma progression revealed by genetic barcoding. Cancer Cell 41, 1466–1479 (2023).
Curry, R. N. et al. Integrated electrophysiological and genomic profiles of single cells reveal spiking tumor cells in human glioma. Cancer Cell 42, 1713–1728.e6 (2024).
Wang, X. et al. Reciprocal signaling between glioblastoma stem cells and differentiated tumor cells promotes malignant progression. Cell Stem Cell 22, 514–528 (2018).
Lee, H. K. et al. Daam2-PIP5K is a regulatory pathway for Wnt signaling and therapeutic target for remyelination in the CNS. Neuron 85, 1227–1243 (2015).
Wang, C. Y. et al. Daam2 phosphorylation by CK2α negatively regulates Wnt activity during white matter development and injury. Proc. Natl Acad. Sci. USA 120, e2304112120 (2023).
Corces, M. R. et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962 (2017).
Muskovic, W. & Powell, J. E. DropletQC: improved identification of empty droplets and damaged cells in single-cell RNA-seq data. Genome Biol. 22, 329 (2021).
Acknowledgements
This study was supported by a NCI Cancer Center Support Grant, P30 CA021765, St. Jude Children’s Research Hospital Research Collaborative on Transcription Regulation in Pediatric Cancer Grant, St Jude Children’s Research Hospital institutional funds, and Alex’s Lemonade Stand Foundation ‘A’ Award; and by P01CA298963-01, R01NS128184, R01CA280203, R01CA284455, U01CA281823, DOD-IDEA (CA220510) and a DOD-IMPACT (CA220247) award (to S.C.M.). S.C.M. is supported by funding from the National Brain Tumor Society and CERN Foundation. This work was also supported by US National Institutes of Health grants R35-NS132230, R01-NS124093, R01-CA223388 to B.D. and K99CA277576 to Y.X.; National Cancer Institute Cancer Target Discovery and Development grant U01-CA217842 to B.D.; and grants from the Helis Medical Research Foundation (to H.K.L.), the Ependymoma Research Foundation (to J.Q.), the NIH/NINDS (2R01NS110859 and R01NS126287 to H.K.L.), the Human Frontier Science Program (LT0018/2022-L to Y.X.), and the Wallace Endowment established by an anonymous donor and the Petrello family (to H.K.L.). Further support was provided by the NMSS postdoctoral fellowship (FG-2407-43793 to J.Z.), P01CA096832 (S.J.B.) and F31CA265285 (K.M.B.). We acknowledge J. Norrie and the Single Cell Core for training and allowing us to use their Chromium machine, and the Christian Mayer laboratory for their generous donation of TrackerSeq plasmids and for guiding us through the initial optimization experiments. We thank A. Ansari and his team for helping us with the application and analysis of the CSI technology to study ZR, and C. Schuurmans for her generous donation of Plagl-related plasmids and valuable advice. Several other St. Jude core facilities were instrumental in this work, including the Cell and Tissue Imaging Center (CTIC), the Hartwell Center for Bioinformatics & Biotechnology (funded partially by ALSAC, NCI grant P30 CA021765), Robert Throm and the St. Jude Vector Laboratory Shared Resource (funded partially by ALSAC), the Center for Advanced Genome Engineering (CAGE; funded partially by ALSAC, NCI grant P30 CA021765) and the Department of Developmental Neurobiology Flow Cytometry Lab.
Author information
Authors and Affiliations
Contributions
A.S.K., H.S., K.B., B.D. and S.C.M., were involved in the conception and design of the project. A.S.K., S.I., K.Y., H.-C.C., T.Z., R.D., J.Z., H.L., T.B., R.P., B.H. and N.L. performed experiments. A.S.K., S.I., A.B. and E.E. were involved in mouse care. H.S. performed single-cell sequencing analysis and visualization, designed and created all single-cell figures, and managed single-cell data. H.S., A.S.K., S.V., N.L., K.Y., J.Z., H.L., S.C. and S.C.M. organized figures. S.V. performed bulk sequencing analysis and visualization. N.L. performed CSI experiments and visualization. K.L. performed flow sorting for TrackerSeq experiments. S.M.P.-M., J.P.C. and Y.-D.W. expanded and validated the TrackerSeq library. K.M.B. and S.J.B. provided guidance and assistance on OPC experiments. S.C.M., A.S.K., H.S., N.L., P.C. and S.V. wrote the manuscript, and A.S.K., H.S., S.I., N.L., S.V., K.Y., H.-C.C., E.E., T.Z., R.M.D., J.P.C., Y.-D.W., J.Z., H.L., K.L., T.B., R.P., A.B., S.M., S.C., A.A., B.H., K.M.B., G.K., T.W., E.C., M.P., A.H., D.G., J.M., Y.X., S.K.M., T.E.M., J.Q., A.D.D., L.A.S., L.W., A.M.D., N.K.F., S.A., A.L. S.J.B., D.W.E., H.K.L., S.M.P.-M., K.C.B., B.D. and S.C.M. were involved in editing the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors report no competing interests.
Peer review
Peer review information
Nature thanks Michelle Monje and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Multi-layered analysis of mouse forebrain development: cell distribution, cell type marker expression, transcription factor regulatory networks, and Plagl family motif activity.
(a) Western blot gel image showing expression of HA-tagged ZFTA-RELA (ZR) in RGC + ZR cells compared to control radial glial cells (RGCs). β-Tubulin serves as the loading control. (b) Cognate Site Identification (CSI) experiment to analyze the binding affinity of ZR for PLAG/L family motifs with results ranked by binding motif enrichment. (c) Cell distribution per sample and total cell counts in the integrated UMAP. (d) Top 5 markers for each cell type and corresponding gene expression. (e) Location of cycling cells on mouse forebrain development UMAP. (f) Gene expression of Eomes, a TF that dictates intermediate progenitor cell (IPC) to excitatory neuron (EN) differentiation, in the excitatory neuron cell group. (g) Gene expression of Gsx2, a TF that dictates IPC to inhibitory neuron (IN) differentiation, in the IN cell group. (h-i) Plagl1 and Plagl2 motif activities classified as high, middle, low, and absent categories for each cell type in development. Y-axis represents the proportion of cells per cell type at each activity level. (j, l) TF–target gene interaction network in RGCs and neurons, based on snRNA-seq gene expression data, respectively. (k, m) TF–target gene interaction network in RGCs and neurons with associated gene activity from single-nucleus ATAC-seq (snATAC) data, respectively. (n) Comparison of snATAC peak signals in the Notch1 gene region between RGCs and neuron cell types.
Extended Data Fig. 2 Independent studies on gene expression, chromatin accessibility, tissue survival, and immunofluorescence.
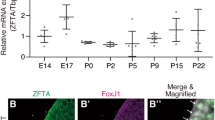
(a) Confirmation of Plag1, Plagl1, and Plagl2 overexpression in mRGCs by RNA-seq. (b) Heatmap of ZR 93 gene signature for parental, RFP, Plag1, Plagl1, Plagl2, and ZR expressing mRGCs by RNA-seq. (c) Survival curve for GLAST-PBase, pBCAG-ZR (red, n = 17) vs GLAST-PBase, pBCAG-ZR, Plagl1 KO IUE mice (blue, p = 0.8211). (d) Survival curve for mRGC ZR+ (blue, n = 5) vs mRGC ZR+ Plagl1 KO (red, n = 5) implanted mice (p = 0.7778). (e) Confirmation of Plagl1 KO for mice within survival curves. Red X denotes a mouse with no knockout which was removed from analysis. (f) Chromatin accessibility signals for Fabp7, Plagl1, and Notch1 are detected by spatial scATAC in cells located around the ventricles of mouse embryos. Figure adapted from Zhang et al. Nature 2023. (g) Immunofluorescence for GFP in P2 mice after GFP IUE. Arrows denote radial fibers typical of RGCs.
Extended Data Fig. 3 Copy number variation and correlation between ZR signal and Plagl1 motif activity in IUE tumors.
(a) Copy number variation (CNV) profiles of IUE tumors derived from snRNA-seq data across samples. (b) CNV profiles of IUE tumors, identified through UMAP-based clustering of snRNA-seq data.
Extended Data Fig. 4 Cell type–specific marker expression in IUE ZR tumors detected by RNAscope.
Confirmatory RNAscope of IUE ZR tumors for Cdk1, Zfta, and marker genes for cell types seen in ZR IUE tumors (Eomes = early excitatory neurons, Grin1 = neurons, Kif6 = ependymal cells, Mog = oligodendrocytes, Pax6 = RGCs, Pdgfra = OPCs).
Extended Data Fig. 5 Immunofluorescence staining of cell type markers and survival curves of marker-defined cell lines.
(a) Western blot gel image showing expression of HA-tagged ZR in OPC + ZR cells compared to control OPCs. GAPDH serves as the loading control. (b) Immunofluorescent staining of OPCs showing HA (green, tags ZR fusion) and Olig2 (red). (c) Quantification of HA+/Olig2+ cells in immunofluorescent staining. (d) Heatmap of ZR 93 gene expression within RGC, RGC + ZR, OPC, and OPC + ZR cell lines. (e) Global ATAC-seq peak profiles of OPC and OPC + ZR samples, based on two technical replicates. (f) Survival curve for MBP-PBase, pBCAG-ZR (blue, n = 12) GLAST-PBase, pBCAG-ZR (red, n = 47) IUE mice (p = 0.0004). (g) Survival curve for GLAST-PBase, pB-MBP-ZR (blue, n = 18) GLAST-PBase, pBCAG-ZR (red, n = 51, Chen et al. Nature 2024) IUE mice (p = 0.000118). (h) Immunofluorescent staining of GLAST-PBase, pB-MBP-ZR (top row) and GLAST-PBase, pBCAG-ZR (bottom row) tumors.
Extended Data Fig. 6 Shared mechanisms of fusion driven tumors.
(a) Correlation between Plagl1 motif score and ZR target gene signal for cycling progenitor-like, fibroblast-like, neuronal-like, and RGC-like IUE ZR tumor cells. (b) Depiction of Tead1-4 motif activity enriched in RGCs and cycling progenitor cells during embryonic brain development.
Extended Data Fig. 7 Copy number variation landscape of human ZR and PLAG/L brain tumors.
(a, b) Copy number variation (CNV) profiles of human ZR (a), PLAG/L (b), and PF (c) tumors across samples, derived from snRNA-seq data. The analysis reveals genomic alterations and distinct, sample-specific CNV patterns, particularly within each tumor cohort.
Extended Data Fig. 8 Cell type proportion and PLAGL motif–ZR target gene correlation in human ependymomas.
(a, b) Bar plots showing the proportions of cell types per sample in human ZR (a) and PF (b) tumors, highlighting cellular heterogeneity across samples. (c, d) Correlation between PLAGL1 (c) and PLAGL2 (d) motif activity and ZR target gene signal across distinct cell types in human ZR tumors, including astrocyte-like, cycling progenitor-like, ependymal-like, neuronal-like, and RGC-like cells.
Extended Data Fig. 9 Distribution of lineage barcodes and the number of cells per barcode across samples.
(a) TrackerSeq barcode diversity. (b) Proportion of cells in LB-1 across all neoplastic and non-neoplastic cell types per sample. (c-m) The bar plot displays the top 5 single lineage barcodes with the highest cell counts per sample.
Extended Data Fig. 10 Early-stage progenitor tumor cells in ZFTA-RELA tumors display cancer stem cell–like differentiation potential into diverse cell types.
Malignant tumor cell trajectories predicted by progenitor and differentiation scores demonstrate a transition from progenitor-like cells (RGC-/CycProg-like) to glial-like and neuronal-like cell types in human ZR ependymomas.
Supplementary information
Supplementary Tables (download ZIP )
Supplementary Tables 1 (dataset overview), 2 (mouse forebrain snMultiome profiling across embryonic development), 3 (mouse IUE tumour snMultiome profiling and associated datasets), 4 (human brain tumour snMultiome profiling and associated datasets), 5 (cell type annotation of human tumours and associated datasets) and 6 (TrackerSeq data analysis information).
Supplementary Data (download ZIP )
Data underlying Figs. 1–5, western blot raw images and cell type names.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kardian, A.S., Sun, H., Ippagunta, S. et al. Dominant clones leverage developmental epigenomic states to drive ependymoma. Nature (2026). https://doi.org/10.1038/s41586-026-10270-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41586-026-10270-8
