
Abstract
Optimal sleep has a vital role in promoting healthy ageing and enhancing longevity. Here we propose Sleep Chart to assess the relationship between self-reported sleep duration and 23 biological ageing clocks derived from in vivo imaging1, plasma proteomics2 and metabolomics3. First, a systemic, U-shaped pattern emerges between sleep duration and biological age gaps across nine brain and body systems and three omics technologies. The sample-specific lowest biological age gaps are achieved between 6.4 and 7.8 h of sleep duration, varying by organ and sex in the UK Biobank (aged 37–84 years). Furthermore, short (<6 h) and long (>8 h) sleep duration, compared with a normal sleep duration (6–8 h), are associated with increased risk of systemic diseases beyond the brain and all-cause mortality, with evidence from genetic correlations and time-to-incident survival predictions, such as depression and diabetes. Finally, the pathways by which long and short sleep duration are associated with late-life depression differ: ageing clocks may partially mediate the pathway for long sleep duration, while short sleep duration shows a more direct link. Although Mendelian randomization does not provide strong evidence that disease causally affects sleep, it cannot completely exclude such reverse causality. Our findings suggest a cross-organ, multi-omics U-shaped relationship between sleep duration and biological ageing clocks, highlighting the potential of sleep optimization to promote healthy ageing, lower disease risk and extend longevity.
Similar content being viewed by others


Main
Sleep is a fundamental biological process that is essential for physical restoration, cognitive functioning and overall health. Increasing evidence underscores its association with ageing4, disease susceptibility5 and longevity6. Both insufficient and excessive sleep duration have been linked to a wide range of adverse health outcomes, including cardiometabolic disease7, cognitive decline8 and psychiatric disorders, such as late-life depression (LLD)9. Importantly, sleep is probably modifiable, making it a potential target for promoting healthy ageing and reducing the burden of age-related diseases across the lifespan.
In parallel, the field of ageing research has seen rapid progress through the development of biological ageing clocks derived from imaging and multi-omics data, like magnetic resonance imaging (MRIBAG)1,10, plasma proteomics (ProtBAG)2 and metabolomics (MetBAG)3. These clocks aim to quantify the biological age of individuals across organ systems and molecular layers, enabling a more granular understanding of ageing beyond chronological or calendar age. Organ-specific biological age gaps (BAGs) derived from these clocks have been used as intuitive and personalized biomarkers to quantify biological ageing and have shown great predictive value for disease morbidity, cognition and mortality risk11. This multi-organ, multi-omics ageing clock framework offers a promising avenue to model human ageing and disease in a multisystem and personalized manner.
Previous studies have demonstrated a nonlinear, U-shaped relationship between sleep duration and several phenotype-based ageing clocks12,13,14,15, suggesting that both short (for example, <6 h) and long (for example, >8 h) sleep may accelerate neurobiological ageing. However, it remains unclear whether this relationship generalizes beyond the brain to body systems (that is, multi-organ)16,17,18,19 and omics layers (that is, multi-omics)2, and whether this relationship is similar in male and female individuals. This raises the questions of whether similar U-shaped patterns emerge in the structural, functional and molecular hallmarks of ageing in organs and tissues beyond the brain, and whether the observed U-shaped associations and empirically derived sample minimum values of the BAG–sleep relationships are consistent across different sexes and organ systems. To address these gaps, we used large-scale population biobanks, combining both individual-level and summary-level data, consolidated through the MULTI Consortium3,16,19 (Methods and Supplementary Table 1), to comprehensively map how sleep duration is associated with biological ageing across multiple systems and to elucidate the pathways through which disrupted sleep may drive age-related diseases and mortality. Our analysis focused on the UK Biobank’s (UKBB) questionnaire-derived sleep duration (field ID: 1160). Self-reported measures are less objective than actigraphy or polysomnography, capturing different yet complementary aspects of sleep biology, with only moderate correlations between modalities20. The large sample size (around 500,000) enables robust identification of nonlinear associations using generalized additive models (GAMs; Methods).
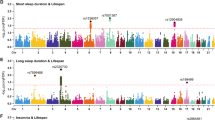
This study addresses several scientific and clinical questions. First, we examined the nonlinear, U-shaped associations, without any prior assumption of the shape of the associations, between sleep duration and 23 multi-organ BAGs derived from multi-omics data (Fig. 1). We reinforced this pattern using in vivo imaging markers (Extended Data Fig. 1) and circulating molecular phenotypes, including plasma proteomics and metabolomics (Extended Data Figs. 2 and 3). Second, we assessed whether abnormal sleep duration patterns (that is, short or long sleep duration) were adversely associated with all-cause mortality and systemic disease end points (DEs) beyond the brain (Figs. 2 and 3). Third, we examined whether short and long sleep duration are differentially associated with two distinct subtypes of LLD9 through separate mediational pathways (Fig. 4). Finally, we examine whether sleep disturbances act as modifiable risk factors for disease (or genetic predisposition), consequences of disease burden or reflect a potential bidirectional relationship (Fig. 4, Extended Data Fig. 4 and Supplementary Note 4). All results, code and summary statistics are publicly available at the SleepChart portal (https://labs-laboratory.com/sleepchart).

Sleep duration (x axis) exhibits nonlinear U-shaped relationships with 9 out of 23 BAGs across 17 organs and 3 omics types (y axis; units are normalized years based on the selected distribution): 11 ProtBAGs for plasma proteomics, 5 MetBAGs for plasma metabolomics and 7 MRIBAGs for in vivo MRI data. For each BAG, we fitted a GAM with cubic regression splines to assess the association between sleep duration and BAG, stratified by sex and sex–sleep interaction term. The solid curves depict the estimated BAG, while shaded bands represent the 95% CI. Significant signals (two-sided P < 0.05/23) are shown with asterisks. Sample-specific BAG minimum values (in hours) of sleep duration are displayed separately for male and female individuals. Sample sizes of each sleep–BAG association are presented in Supplementary Table 2. The image at the bottom right is from NIAID Visual & Medical Arts.

a, The top lead SNP, the nearest gene and cytogenetic regions associated with short (4–6 h) and long (8–10 h) sleep duration compared with a normal sleep duration (6–8 h), with significant genomic loci identified using the genome-wide significance threshold (two-sided P < 5 × 10−8). b, MAGMA gene property analysis of tissue-specific expression across 54 GTEx v.8 tissues. Statistical significance was determined using FDR-adjusted P values (<0.05). acc., accumbens; ecto., ectocervix; endo., endocervix; exp., exposed; GE, gastroesophageal; oesoph., oesophagus. c, Genetic correlation estimates based on the LDSC between the two abnormal sleep duration patterns and 527 DEs from FinnGen and PGC (two-sided P < 0.05/527). The error bars represent the s.e. of the LDSC genetic correlation (rg) estimate. An interactive webpage is available (https://labs-laboratory.com/sleepchart/sleep_gc.html) to facilitate visualization. Sample sizes of the FinnGen and PGC DEs are presented in Supplementary Table 7. CPAP, continuous positive airway pressure; T2D, type 2 diabetes. The diagrams in b and c were created using images from the Noun Project and NIAID Visual & Medical Arts.

a, Using normal sleep duration, 6–8 h as the reference, we treated sleep duration patterns as categorical variables (4–6 h for short sleep; 8–10 h for long sleep) to estimate their association with 726 comorbidity-free DEs defined by ICD-10 codes (limited to diseases with more than 50 cases). After applying Bonferroni correction for multiple comparisons (two-sided P < 0.05/726), we highlight the significant associations and annotate representative systemic diseases across various organ systems. IHD, ischaemic heart disease. b, Short and long sleep duration are associated with a higher risk of all-cause mortality. An interactive webpage is available (https://labs-laboratory.com/sleepchart/sleep_cox.html) to facilitate visualization. The error bars represent the 95% CIs of the hazard ratio (HR) estimate, which is represented by a central shape. The diagrams in a were created using images from the Noun Project and NIAID Visual & Medical Arts.

We tested whether 7 MRIBAGs mediate the effect of abnormal sleep duration on two artificial-intelligence-derived subtypes of LLD (LLD1 and LLD2)9. The participants were grouped into short (4–6 h), normal (6–8 h; as a reference) and long (8–10 h) sleep categories: sleep duration → MRIBAG → LLD1/2. For each LLD subtype and each contrast (short versus normal, long versus normal), we fit structural equation models using the lavaan package in R. The model included a direct path from the binary-coded sleep category to the LLD subtype (depicted as solid lines with warm colours) and indirect paths mediated through the MRIBAG (shown as dotted lines with cool colours). Only statistically significant pathways (two-sided P < 0.05/7) and estimated path coefficients are displayed. All models were adjusted for relevant covariates, including age, sex, body mass index, and systolic and diastolic blood pressure, among others. Standardized estimates were obtained using 1,000 nonparametric bootstrap resamples. a, Mediational models for short sleep duration → MRIBAG → LLD1. b, Mediational models for long sleep duration → MRIBAG → LLD1. c, Mediational models for short sleep duration → MRIBAG → LLD2. d, Mediational models for long sleep duration → MRIBAG → LLD2. Our primary analyses were based on the temporal ordering of data collection, with sleep duration assessed at the baseline and both MRIBAG and LLD subtypes measured at the second visit. The sample sizes of the analyses are presented in Supplementary Table 8.
Twenty-three biological ageing clocks
Using GAMs with the UKBB data, we assessed the nonlinear, U-shaped relationship between sleep duration and 23 multi-organ, multi-omics BAGs (P < 0.05/23; Fig. 1). We quantified nonlinearity in the sleep–BAG relationship using the effective degrees of freedom (e.d.f.) of the smooth term—a measure of curve complexity (Methods).
We confirmed that 9 out of the 23 BAGs exhibited statistically significant nonlinear associations (P < 0.05/23). Among the 11 ProtBAGs, the brain ProtBAG showed the strongest U-shaped association with sleep duration (e.d.f. = 3.61, P1 < 1 × 10−20). At the population mean level, female individuals exhibited significantly higher brain ProtBAG values than male individuals (P2 < 1 × 10−20). The interaction between sex and sleep duration approached nominal significance (P3 = 0.06), indicating a potential trend toward sex-specific associations. We next estimated the sample minimum of the BAG–sleep curves based on the peak of the smoothed spline curves for the brain ProtBAG, identifying 7.82 h for female and 7.70 h for male individuals. Similar U-shaped relationships were also evident for the pulmonary (e.d.f. = 3.21, P1 = 1.09 × 10−4), hepatic (e.d.f. = 3.13, P1 = 1.36 × 10−3), immune (e.d.f. = 3.47, P1 = 1.78 × 10−5) and skin (e.d.f. = 3.21, P1 = 3.12 × 10−6) ProtBAGs. Among the 5 MetBAGs, the endocrine MetBAG showed a significant U-shaped relationship (e.d.f. = 1.04, P1 = 3.97 × 10−5), and the mean population value was different between female and male individuals (P2 < 1 × 10−20, P3 = 5.72 × 10−7), with estimated sample minimum values of 6.67 h for female and 6.06 h for male individuals. Finally, among the 7 MRIBAGs, the brain MRIBAG showed the most significant U-shaped relationship (e.d.f. = 1.94, P1 = 3.85 × 10−7), and the mean population value was different between female and male individuals (P2 < 1×10−20, P3 = 0.84). The optimal sleep time was 6.48 h for female and 6.42 h for male individuals. Significant signals were also observed for the adipose (e.d.f. = 1.93, P1 = 2.84 × 10−4) and pancreas (e.d.f. = 1.95, P1 = 9.95 × 10−6) MRIBAGs. Detailed statistics, including the P value, sample size, chosen family distribution and e.d.f., are provided in Supplementary Table 2 and Supplementary Data 1.
Several sensitivity analyses were performed. Supplementary Fig. 1 displays quantile–quantile plots and residual versus fitted plots for the best model of each BAG. Supplementary Figs. 2–16 and Supplementary Table 3 present a full set of analyses to scrutinize the robustness of the U-shaped pattern, including a potential replication of the U-shaped relationship in two independent datasets21,22, although the sample sizes (n = 385 and 573) are much smaller and the populations are much older than in the UKBB. We also observed this U-shaped pattern between sleep duration and 720 organ-specific imaging-derived phenotypes (IDP; Extended Data Fig. 1), 342 organ-enriched plasma proteins (Extended Data Fig. 2 and Supplementary Fig. 17) and 107 organ-associated plasma metabolites (Methods, Extended Data Fig. 3 and Supplementary Data 2–4). A detailed discussion of these results and their implications in sleep-related hypotheses is presented in Supplementary Note 1.
Overall, across the nine significant proteomic, metabolomic and MRI-based BAGs, sample minimum values for sleep duration ranged from 6.5 to 7.8 h for female individuals and 6.4 to 7.7 h for male individuals. Given the strong evidence of the U-shaped relationship across widespread biomarkers at both the imaging and molecular levels, we classified sleep duration into short (<6 h), long (>8 h) and normal (6–8 h) categories for all downstream analyses. Although our definition is data driven by the sample minimum value range observed, it is important to note that the definition of normal sleep varies between studies and is often associated with cultural and environmental factors.
Systemic diseases and all-cause mortality
In the UKBB, our genome-wide association study (GWAS; Methods) identified 8 genetic loci (P < 5 × 10−8) associated with abnormal sleep duration patterns (Fig. 2a). A locus at 3q29 (top lead single-nucleotide polymorphism (SNP): rs34382732) was associated with short sleep duration, whereas long sleep duration was linked to multiple loci, highlighting potential genetic heterogeneity between short and long sleep patterns. We denoted the genomic loci using their top lead SNPs defined by FUMA (Supplementary Note 2), considering linkage disequilibrium; the genomic loci are presented in Supplementary Table 4. The Manhattan plot and quantile–quantile plot, as well as the linkage disequilibrium score regression (LDSC)23 intercept, are presented in Supplementary Fig. 18. We then conducted gene–property analyses24 to assess tissue-specific expression patterns of genetic signals associated with short and long sleep duration, using gene expression data from 54 human tissues provided by the Genotype-Tissue Expression Program (GTEx) v.8. Notably, only short sleep duration demonstrated significant enrichment (false discovery rate (FDR)-corrected P < 0.05) in several brain regions, including the cerebellum, caudate and hippocampus (Fig. 2b), but not long sleep duration.
Given the relatively smaller number of genomic loci in our GWASs, despite the large sample size (n > 300,000), we hypothesize that the U-shaped association between sleep duration and BAG is primarily environmentally driven and modifiable. As such, we hypothesized that this U-shaped pattern observed at the BAG level would be flattened and attenuated when examining BAG polygenic risk scores derived in our previous studies1,2,3,10. Our results supported our hypothesis (Extended Data Fig. 4). Notably, the endocrine MetBAG showed a J-shaped association, with higher genetic risk in individuals reporting shorter sleep duration, while the metabolic MetBAG demonstrated a linear relationship. Moreover, genetic correlation analyses revealed a significant positive correlation only between short sleep duration and endocrine MetBAG, and between long sleep duration and pancreas MRIBAG (P < 0.05/23) (Supplementary Table 5). These findings imply that, while most sleep–BAG relationships are not genetically mediated, certain organ systems, particularly endocrine and metabolic pathways, may share partial genetic underpinnings with habitual sleep duration.
Using summary-level data from our GWAS in the UKBB, as well as those from FinnGen and Psychiatric Genomics Consortium (PGC) for 527 DEs, genetic correlation analyses revealed 153 positive associations between abnormal sleep duration and systemic diseases across multiple organ systems (P < 0.05/527), dominated by short sleep duration (Fig. 2c). For short sleep duration, we observed robust correlations with cardiovascular diseases, including ischaemic heart disease (gc = 0.19 ± 0.04, P = 2.52 × 10−6), heart failure (gc = 0.31 ± 0.06, P = 5.80 × 10−6) and coronary atherosclerosis (gc = 0.18 ± 0.04, P = 1.51 × 10−5); metabolic disorders, such as type 2 diabetes (gc = 0.18 ± 0.04, P = 5.33 × 10−5) and insulin-requiring diabetes (gc = 0.17 ± 0.04, P = 4.45 × 10−5); and musculoskeletal conditions, like low back pain (gc = 0.40 ± 0.06, P = 1.13 × 10−12), osteoarthritis (gc = 0.19 ± 0.05, P = 7.07 × 10−5) and soft tissue disorders (gc = 0.50 ± 0.06, P = 2.04 × 10−16). Neurological and psychiatric associations included migraine (gc = 0.25 ± 0.06, P = 1.91 × 10−5), depression (gc = 0.37 ± 0.04, P = 1.57 × 10−18), anxiety (gc = 0.32 ± 0.06, P = 1.83 × 10−7), substance-use disorders (gc = 0.37 ± 0.05, P = 3.73 × 10−15) and suicidality (gc = 0.34 ± 0.06, P = 4.28 × 10−8), indicating broad involvement of the central nervous system. Pulmonary and infectious conditions, such as asthma (gc = 0.22 ± 0.05, P = 2.50 × 10−5), bronchitis (gc = 0.42 ± 0.12, P = 5.00 × 10−4) and chronic obstructive pulmonary disease (gc = 0.28 ± 0.05, P = 2.74 × 10−7), were also genetically correlated, as were gastrointestinal and hepatic diseases, including reflux (gc = 0.34 ± 0.06, P = 6.09 × 10−8), diverticulosis (gc = 0.23 ± 0.04, P = 1.14 × 10−6) and irritable bowel syndrome (gc = 0.32 ± 0.09, P = 3.00 × 10−4). By contrast, long sleep duration showed a more focused genetic correlation profile, predominantly involving brain-related phenotypes, such as major depressive disorder (MDD; gc = 0.29 ± 0.04, P = 2.57 × 10−11), schizophrenia (gc = 0.28 ± 0.03, P = 3.47 × 10−16), bipolar disorder (gc = 0.21 ± 0.03, P = 1.09 × 10−7), alcohol dependence (gc = 0.23 ± 0.05, P = 3.26 × 10−5), ADHD (gc = 0.28 ± 0.04, P = 2.24 × 10−12) and migraine (gc = 0.28 ± 0.07, P = 7.09 × 10−5), suggesting potential compensatory or indirect neuropsychiatric mechanisms (further elucidated in Fig. 4). Supplementary Data 5a presents detailed statistics for our genetic correlation analysis.
Taken together, these findings support that short and long sleep duration have distinct genetic architectures (Fig. 2a). While both show genetic correlations with various systemic diseases, short sleep duration exhibits broader systemic associations, whereas long sleep duration is more specifically linked to brain-related traits (Fig. 2c). A discussion comparing our GWAS and genetic relevance with previous studies is provided in Supplementary Note 3, Supplementary Figs. 19–21 and Supplementary Data 5b,c.
Using ICD code-based clinical diagnosis and echoing the genetic correlation results, we further assessed the relationships between abnormal sleep duration patterns and the future onset of DEs, as well as all-cause mortality, in the UKBB (Methods). We identified 153 significant associations between sleep duration patterns (both short and long) and DEs (P < 0.05/726), after correcting for multiple comparisons across end points with at least 50 cases (Fig. 3a), with a notable predominance of associations linked to short sleep duration (1.20 < hazard ratio < 6.69). These findings span multiple organ systems, supporting the systemic and direct association with disturbed sleep. Within the brain-related disorders, short sleep was significantly associated with depressive episodes (ICD code: F329), anxiety disorders (F419) and primary insomnia (G473), reinforcing the well-established link between sleep and mental health. In the metabolic domain, we observed associations with obesity (E669), type 2 diabetes (E119) and hyperlipidaemia (E780), aligning with previous evidence that short sleep disrupts metabolic homeostasis. Cardiovascular outcomes such as essential hypertension (I10), ischaemic heart disease (I209, I252) and arrhythmias (I471, I440) were also enriched, suggesting increased cardiovascular risk among individuals with short sleep. Pulmonary conditions, including chronic obstructive pulmonary disease (J449) and asthma (J459), were linked to both short and long sleep, although more strongly with short sleep. Finally, a cluster of digestive disorders, including gastritis and duodenitis (K297), gastroesophageal reflux disease (K219) and functional intestinal disorders (K590), was significantly predicted by both long and short sleep duration. These findings underscore the broad biological associations of both insufficient and prolonged sleep, highlighting distinct mechanistic pathways by which short and long sleep duration relate to systemic DEs (further elucidated in Fig. 4). We also performed a sensitivity analysis using 7–9 h as normal sleep duration (Supplementary Fig. 22), as suggested by the National Sleep Foundation25 for young/older adults.
For all-cause mortality, we found that both short (hazard ratio = 1.50, 95% confidence interval (CI) = 1.44–1.55, P < 1 × 10−20) and long sleep duration (hazard ratio = 1.40, 95% CI = 1.36–1.44, P < 1 × 10−20) patterns were positively associated with increased risk of death from any cause (Fig. 3b). Supplementary Data 6 presents detailed statistics for all survival analyses. Supplementary Note 4, Supplementary Data 7 and Extended Data Fig. 5 present results of examining the associations between two sleep disorders (insomnia and hypersomnia) and the DEs, using the TriNetX dataset, identified in Fig. 3a, as well as all-cause mortality.
From ageing clocks to LLD
Given that short sleep duration showed more significant associations with systemic DEs in our genetic correlation (Fig. 3a) and survival analysis (Fig. 3b) than long sleep duration, we hypothesize that long sleep may be linked to disease risk through more complex or indirect pathways, potentially serving as a marker of underlying health conditions or reflecting mediational, compensatory physiological responses rather than acting as a direct risk factor (Methods).
To evaluate this hypothesis in the UKBB, we performed structural equation modelling using sleep duration patterns (short or long) as exposures, the 7 MRIBAGs as mediators and two MRI-derived subtypes of LLD (LLD1 and LLD2)9 as outcomes. We selected LLD as the outcome because it is closely linked to sleep disturbances (Figs. 2 and 3) and ageing, making it a compelling model to explore potential mediating pathways. In the pathway linking short sleep to LLD1, characterized by preserved subcortical brain volume, we observed strong direct effects (c2 ranging from −0.33 to −0.24) across 6 organ systems (the c1, c2, and a1 coefficients are defined in the Methods). Among the mediators, only the adipose MRIBAG showed a significant indirect effect (a1 = 0.44 ± 0.13, c1 = −0.05 ± 0.005), suggesting a specific role for adipose ageing in mediating this relationship. By contrast, for long sleep, the associations with LLD1 were predominantly mediated through organ-specific MRIBAGs, particularly the brain (a1 = 0.54 ± 0.08, c1 = −0.11 ± 0.002) and adipose (a1 = 0.61 ± 0.09, c1 = −0.04 ± 0.005) pathways. Notably, brain MRIBAG alone accounted for 62% of the total effect through mediation, followed by the adipose and liver MRIBAGs (24%), highlighting a more indirect and organ-mediated link between long sleep and LLD. Similar patterns were observed for LLD2, which were characterized by diffuse patterns of cortical atrophy in LLD (Fig. 4). All statistics for the mediation analyses are presented in Supplementary Data 8.
Our mediation analysis treated sleep duration as a potential modifiable risk factor (not for causal inference), based on the temporal sequence of data collection: sleep data were recorded at the baseline, while brain MRI occurred during follow-up. As described in Supplementary Table 6, we conducted sensitivity analyses by alternately specifying brain MRIBAG and LLD1/2 as mediators and outcomes. However, due to the limitations of the data’s time-ordering, we cannot fully exclude the possibility that sleep disturbances result from underlying disease burden. To test this reverse causality, we conducted Mendelian randomization (MR) analyses using 5 different estimators (Supplementary Note 5 and Supplementary Data 9), from 525 DEs using FinnGen and PGC data, to the 2 binary sleep traits. These analyses did not support a widespread causal effect of disease on sleep disturbances, reinforcing the interpretation of sleep disturbances as potential risk factors. We nevertheless acknowledge the possibility of bidirectional effects. For example, a previous study26 found bidirectional causality between MDD and insomnia, but not sleep duration. Moreover, some previous studies modelled sleep duration as a continuous trait using linear MR, which does not capture potential nonlinear relationships that methods such as fractional polynomial MR can address27. To examine this further, we conducted additional sensitivity analyses focused on depression-related end points in our MR results. Specifically, depression in FinnGen (F5-DEPRESSION-DYSTHYMIA; number of cases, 48,222) was not causally linked to long sleep duration (P > 0.05). The sensitivity analyses results are presented in Extended Data Figs. 6 and 7, including the per-SNP F statistics, horizontal pleiotropy and heterogeneity test based on MR-Egger, and horizontal pleiotropy from MR-PRESSO and fractional polynomial MR.
Discussion
Here we reveal a robust and systemic U-shaped association between sleep duration and biological ageing across organ systems and omics layers. By using large-scale multimodal data from the MULTI Consortium, we demonstrate that both short (<6 h) and long (>8 h) sleep duration are linked to elevated biological ageing burden across seven organ systems and three omics types. Importantly, our findings extend previous work on phenotype-based ageing clocks by showing that this nonlinear sleep–ageing relationship generalizes across the body and is evident not only in structural and functional imaging features but also at the molecular level. This is the first study, to our knowledge, that reveals a broad agreement between sleep duration and multi-organ, multi-omics ageing clocks and links these signatures to systemic disease outcomes and mortality risk. Our results underscore the systemic biological adverse associations of disturbed sleep and provide a compelling framework for more targeted and thoughtful attention to sleep disturbance as a potential signal of emerging health issues and a partner in the quest to promote healthy ageing, reduce disease risk and extend lifespan.
The U-shaped pattern
A key contribution of this study lies in identifying consistent U-shaped associations between sleep duration and organ-specific biological ageing clocks across diverse organs, tissues and omics data types. At the molecular level, we observed that both short and long sleep duration are associated with accelerated pulmonary, brain, hepatic, immune and skin ProtBAGs, suggesting that proteomic signatures of ageing in these systems are particularly sensitive to sleep perturbations. These findings support sleep’s systemic implications on immune–inflammatory processes28, metabolic detoxification29 and neurodegenerative pathways30. Notably, endocrine MetBAG also exhibited a U-shaped pattern, highlighting the metabolic cost of sleep imbalance, possibly through dysregulation of hormonal homeostasis and glucose metabolism31. At the imaging level, U-shaped patterns in brain MRIBAG, adipose MRIBAG and pancreas MRIBAG further reinforce the idea that both central and peripheral organs experience structural and functional alterations under sleep extremes32.
In addition to the overarching U-shaped pattern, our results reveal organ- and omics-specific variability in the sample-specific BAG minimum values of the BAG-sleep relationship. While the overall sample-specific minimum values centred between 6 and 8 h, consistent with previous epidemiological recommendations6, the precise inflection point varied across tissues and omics types. For example, brain ProtBAG showed the lowest ageing burden around 7.7 h of sleep, whereas brain MRIBAG reached its inflection point closer to 6.5 h. These findings suggest that molecular ageing in the brain may require longer restorative sleep than its structural counterpart, potentially reflecting the different timescales and mechanisms by which proteomic versus imaging phenotypes capture sleep-related damage or resilience32,33. Alternatively, this can be due to a reverse causality, where longer sleep reflects brain ageing rather than causes it, pulling the right side of the U-shaped curve upward, and making the apparent sample BAG minimum values appear shorter for the brain MRIBAG (for example, 6.5 h). Moreover, UKBB’s MRI subpopulation over-represents healthier and more highly educated participants; this may attenuate or distort the right limb of the observed U-shape independent of reverse causality. By contrast, the proteomic and metabolomic BAGs are derived from broader, potentially more representative samples spanning wider health states, which could partly explain their stronger or more symmetric associations. To summarize, the differing sample-specific BAG minimum values across organ-specific clocks probably reflect the heterogeneous physiological demands and recovery processes of each organ system. Organs such as the brain may be more sensitive to sleep deprivation and circadian disruption, whereas peripheral organs (for example, liver and pancreas) may exhibit delayed or compensatory responses, resulting in distinct sleep–ageing optima.
Moreover, clear sex differences emerged in the relationship between sleep and biological ageing. Several organs displayed significant BAG differences, BAG-by-sex interactions, divergence in the population mean and sample minimum values of sleep time. For example, male individuals exhibited higher brain MRIBAG, whereas female individuals exhibited higher brain ProtBAG between sexes. This divergence probably reflects that these clocks index distinct layers of biology. The brain MRIBAG is driven by macrostructural MRI features (for example, regional volumes) that are shaped by lifelong neurodevelopmental trajectories, hormonal and sex-differential factors that have been repeatedly linked to sex differences in brain ageing and neurodegeneration. By contrast, brain ProtBAG is derived from circulating brain-enriched proteins, which may be more sensitive to systemic inflammatory/immune signalling, endocrine regulation, blood–brain barrier permeability, and glial- and vasculature-related processes that influence the release, transport or clearance of brain-linked proteins in the plasma. These differences may reflect sex-specific hormonal regulation, immune responses, activity patterns or metabolic demands, as well as socially unfavourable factors for female individuals, that probably relate to how sleep modulates ageing trajectories across organ systems—a concept supported by growing literature on sex-specific sleep physiology and ageing biology34.
Together, these results reveal that deviations from sample-specific minimum BAG values of sleep duration are not only associated with the brain but also peripheral tissues and systems critical to cardiometabolic and immune regulation, emphasizing the multi-organ biological burden imposed by disrupted sleep.
Molecular and imaging factors
Our findings provide converging evidence from imaging, proteomics and metabolomics that support the hypothesis that sleep duration exerts nonlinear effects across multiple organ systems and molecular domains. These patterns reflect the systemic nature of sleep physiology and are consistent with previous research showing that both sleep deprivation and prolonged sleep are associated with adverse outcomes across diverse biological pathways.
At the imaging level, U-shaped associations between sleep duration and organ-specific IDPs, particularly in the brain, adipose tissue and pancreas, echo previous findings that link both short and long sleep with structural brain alterations and metabolic dysregulation. For example, previous neuroimaging studies have shown that hippocampal atrophy is associated with both insufficient and excessive sleep, possibly reflecting neurotoxic or neurodegenerative processes triggered by circadian misalignment, impaired glymphatic clearance or inflammatory load32,35. Similarly, adipose tissue dysfunction and pancreatic abnormalities underlie cardiometabolic conditions such as insulin resistance and obesity, both of which have been associated with extremes of sleep36.
At the molecular level, the U-shaped relationships observed in organ-enriched plasma proteins align with several established biological hypotheses of sleep regulation. For example, immune-enriched proteins involved in neutrophil chemotaxis, complement activation and leukocyte signalling were significantly altered in individuals with suboptimal sleep. This supports the immune activation hypothesis, positing that sleep disturbances lead to, or reflect, systemic inflammation, impairing tissue repair, neurogenesis and metabolic homeostasis37. Similarly, hepatic-enriched proteins involved in coagulation and innate defence pathways reinforce the liver–sleep axis, in which disrupted sleep contributes to liver stress, oxidative damage and altered lipid metabolism38. Complementing these proteomic findings, the metabolomics results revealed alterations in metabolites involved in fatty acid metabolism, ketone body synthesis and amino acid catabolism. For example, the observed enrichment of pathways related to neurotransmitter transport and synaptic remodelling, such as tyrosine metabolism and Na+/Cl−-dependent transporter activity, supports the neurochemical plasticity hypothesis, suggesting that sleep modulates central nervous system function39 through fine-tuned molecular and ionic gradients that are essential for neural signalling.
Together, our multilayered findings reinforce the notion that sleep duration is not only a behavioural or neurological variable but a deeply embedded systemic modulator. The evidence from both structural-functional imaging and circulating biomolecules converges to support the role of sleep in maintaining organ integrity, metabolic balance and immune equilibrium. These results further reinforce sleep as an important process with far-reaching molecular and physiological implications across the body.
Evidence of adverse effects on health
Our integrated genetic and survival analyses consistently demonstrate that disturbed sleep duration patterns, both short and long, are robustly linked to a wide array of systemic DEs and increased mortality risk. Genetic correlation analyses revealed significant positive associations between abnormal sleep duration patterns and over 150 DEs across cardiovascular, metabolic, musculoskeletal, psychiatric, neurological, pulmonary and gastrointestinal systems, with short sleep duration exhibiting particularly widespread effects. These patterns were mirrored in our longitudinal survival analyses, where short and long sleep duration were both significantly associated with elevated risk for a broad spectrum of diseases and all-cause mortality. Importantly, the genetic and clinical evidence together suggest that short and long sleep duration may contribute to disease through distinct biological pathways: short sleep duration appears to exert more direct effects across multiple organ systems, potentially reflecting heightened physiological stress and systemic dysregulation, whereas long sleep duration shows a more focused association with neuropsychiatric and brain-related phenotypes, potentially reflecting compensatory mechanisms or underlying latent pathologies. These findings reinforce the role of sleep as a potentially modifiable risk factor in health and ageing management and underscore the need for mechanistically informed strategies tailored to sleep disruption, as we further illustrated in LLD.
Mechanisms in LLD
While MDD has been considered a brain-centric disorder, our findings suggest a more nuanced framework in which both short and long sleep duration link to LLD vulnerability through distinct mechanistic pathways involving multiple organ systems. Notably, short sleep duration appears to exert more direct effects on LLD. This pattern may reflect acute physiological stressors associated with sleep deprivation, including heightened sympathetic activity, immune dysregulation and hypothalamic–pituitary–adrenal axis activation, all of which can be linked to mood regulation independently of brain atrophy. These findings are consistent with previous studies linking short sleep to systemic inflammation, metabolic strain and emotional dysregulation, pointing to a fast-acting, body-to-brain pathway40.
By contrast, long sleep duration appears to be associated with LLD predominantly through indirect pathways, especially through accelerated ageing in the brain and adipose tissue. The strong mediation effects observed for brain and adipose MRIBAGs suggest that long sleep may not be a direct risk factor per se, but rather a marker of underlying physiological compensations or subclinical disease processes, potentially including neurodegeneration, energy imbalance or immune exhaustion, that gradually erode mental health resilience. The finding also parallels a cultural illustration from Moshfegh’s novel My Year of Rest and Relaxation, in which an unnamed narrator methodically increases prescription medication to achieve a year-long sleep41 to escape her grief and the world. Such patterns support emerging views of long sleep as a complex, sometimes maladaptive response to latent morbidity rather than a simple behavioural choice42. The pronounced mediational roles of the brain and other body ageing clocks are particularly striking, indicating that central and systemic ageing may jointly drive mood vulnerability in individuals with prolonged sleep. This broader multi-organ involvement suggests that depression in late life might arise from distributed physiological ageing, in which systemic decline in tissues like adipose, liver or cardiovascular systems contributes to or exacerbates neural vulnerability. This is consistent with recent multidimensional models of depression that integrate metabolic dysfunction, systemic inflammation and neuroplasticity deficits as co-occurring drivers of depressive phenotypes43. Importantly, we used MR to partially rule out the possibility of reverse causality, whereby sleep disturbances may be caused by disease burden. While a previous study44 reported a bidirectional relationship between long sleep duration and MDD, their analysis relied on a lenient P-value threshold (P < 1 × 10−5) for selecting instrumental variables, which may cause weak instrument bias. We attempted to replicate their findings using the same PGC exposure GWAS as described in ref. 45 (45,396 cases, removing UKBB samples) using a genome-wide P-value threshold (P < 5 × 10−8). After LD clumping, no independent genome-wide significant instruments remained. To further investigate this, we downloaded the most recent MDD GWAS from FinnGen (59,333 cases). Overall, although the inverse variance weighted (IVW; P = 0.003) estimator suggested a positive causal effect of genetic liability, and the significant MR-Egger intercept (P = 0.047), MR-PRESSO global pleiotropy (P = 0.002) and high heterogeneity (P = 0.0009) indicate pleiotropy-induced biases. Pleiotropy-aware methods showed attenuated effects and were sensitive to pleiotropy assumptions (balanced versus directional pleiotropy), including MR-Egger for null/negative (P = 0.24), MRMix for near-null (P = 0.41) and MR-RAPS for nominally positive (P = 0.019). Thus, while suggestive, the evidence for a causal link between MDD and long sleep duration remains inconclusive and should be interpreted cautiously (Extended Data Fig. 7).
Together, these findings highlight that, while short sleep may acutely disturb mood through direct neuroimmune and neuroendocrine perturbations, long sleep may reflect an organ-mediated pathway, linking chronic subclinical ageing processes to eventual neuropsychiatric decline. This distinction reinforces the need for tailored sleep interventions and underscores the importance of viewing LLD as a systemic, not merely cerebral, manifestation of ageing.
Limitations
Several limitations warrant consideration. First, although our findings revealed U-shaped associations between sleep duration and various phenotypes, these patterns require external validation in independent cohorts, potentially on the same scale as UKBB. Second, the reliance on self-reported, questionnaire-based sleep duration measurement may introduce recall bias or misclassification; future studies incorporating objective measures, such as polysomnography, are essential to better understand underlying mechanisms. Third, the cross-sectional design of this study limits our ability to determine causality or the direction of effect; although our current analysis positions sleep disturbance as a modifiable risk factor, longitudinal follow-up is needed to clarify whether sleep disturbance is a modifiable risk factor or a consequence of disease burden. Furthermore, proteomic and metabolomic signals fluctuate with time, illness, medication and diet, so single snapshots can misclassify biology. Longitudinal sampling may yield more reliable estimates and separate transient noise from persistent, risk-relevant biology. Fourth, our analyses were restricted to individuals of predominantly European ancestry, which limits generalizability. Studies in more ethnically diverse and under-represented populations are needed to ensure broader applicability. Fifth, despite adjusting for a wide array of covariates, residual confounding and the potential for reverse causality, particularly for long sleep as a marker of subclinical illness, cannot be fully excluded. Moreover, circadian misalignment and sleep fragmentation were not directly assessed, which may influence the observed associations between sleep duration and organ-specific biological ageing. Sixth, future research needs to extend the Sleep Chart across the lifespan to better capture dynamic patterns of sleep duration beyond adulthood.
Methods
The MULTI Consortium
The MULTI Consortium is an ongoing initiative to integrate and consolidate existing multi-organ and multi-omics data, including imaging, genetics, metabolomics and proteomics. Building on existing consortia and studies, MULTI aims to curate and harmonize the data to model human ageing and disease at scale across the lifespan. Refer to Supplementary Table 1 for comprehensive information, including the complete list of data analysed and their respective sample characteristics. The participants provided written informed consent to the corresponding studies. The MULTI Consortium is approved by the Institutional Review Board at Columbia University (AAAV6751).
UKBB
UKBB46 is a population-based research initiative comprising around 500,000 individuals from the United Kingdom between 2006 and 2010. Ethical approval for the UKBB study has been secured, and information about the ethics committee can be found online (https://www.ukbiobank.ac.uk/learn-more-about-uk-biobank/governance/ethics-advisory-committee). The main sleep data used in this study were sleep duration (field ID: 1160) based on a self-reported questionnaire collected from all 500,000 participants at the baseline. The 7 brain MRIBAGs were derived from multi-organ MRI data at the second visit, 11 ProtBAGs and 5 MetBAGs were derived using plasma proteomics and metabolomics at the baseline. Finally, we also included individual plasma proteins and metabolites in our ProWAS and MetWAS, as described below.
For the primary variable of interest in the UKBB, sleep duration (data-field: 1160) was assessed using an ACE touchscreen questionnaire asking “About how many hours sleep do you get in every 24 h? (please include naps).” The participants entered a numeric value, which underwent basic quality control: responses of less than 1 h or more than 23 h were rejected, and values below 3 h or above 12 h triggered a confirmation prompt. If participants clicked the ‘Help’ button, they were instructed that, if their sleep duration varied substantially, they should report the average number of hours slept in a 24-hour day over the past 4 weeks. For this variable, the value −1 indicates ‘Do not know’ and the value −3 indicates ‘Prefer not to answer’; these were excluded in the current work.
For the multi-organ IDPs, we used multi-organ MRI data from eight organ systems and tissues (category ID: 100003), including the brain, heart, liver, pancreas, spleen, adipose and kidney, as well as eye OCT features. The MUSE atlas-derived brain IDPs from the T1-weighted MRI10 were used for the brain MRIBAG generation. We also used neural networks to analyse the raw cardiac MRI images in our previous study and returned them to the UKBB to extract heart-specific IDPs (category ID: 157), which were used to derive the heart MRIBAG. For the other organs’ IDPs, we used the pre-derived features from the UKBB (category ID: 105). For the plasma proteomics data, we downloaded the original data (category ID: 1838), which were analysed and made available to the community by the UKBB Pharma Proteomics Project (UKB-PPP)47. The initial quality control procedures were described in the original work48; we conducted additional quality-check steps as outlined in the ‘Proteome-wide associations’ section. We also imputed missing normalized protein expression values and defined the organ-specific proteins using the HPA platform (https://www.proteinatlas.org/), as detailed in our previous work. For the plasma metabolomics data, we downloaded the original data (category ID: 220), which were analysed and made available to the community by Nightingale Health in the UKBB. Additional quality check analyses were performed as detailed in the ‘Metabolome-wide associations’ section.
FinnGen
The FinnGen49 study is a large-scale genomics initiative that has analysed over 500,000 Finnish biobank samples and correlated genetic variation with health data to understand disease mechanisms and predispositions. The project is a collaboration between research organizations and biobanks within Finland and international industry partners. For the benefit of research, FinnGen generously made its GWAS findings accessible to the wider scientific community (https://www.finngen.fi/en/access_results). This research used the publicly released GWAS summary statistics (version R9), which became available on 11 May 2022, after harmonization by the consortium. No individual data were used in the current study.
FinnGen published the R9 version of GWAS summary statistics via REGENIE software (v.2.2.4)50, covering 2,272 DEs, including 2,269 binary traits and 3 quantitative traits. The GWAS model included covariates such as age, sex, the initial 10 genetic principal components and the genotyping batch. Genotype imputation was referenced on the population-specific SISu v.4.0 panel. We included GWAS summary statistics for 521 FinnGen DEs in our analyses (Supplementary Table 7).
PGC
PGC51 is an international collaboration of researchers studying the genetic basis of psychiatric disorders. PGC aims to identify and understand the genetic factors contributing to various psychiatric disorders such as schizophrenia, bipolar disorder, MDD and others. The GWAS summary statistics were acquired from the PGC website (https://pgc.unc.edu/for-researchers/download-results/), underwent quality checks and were harmonized to ensure seamless integration into our analysis. No individual data were used from PGC. Each study detailed its specific GWAS models and methodologies, and the consortium consolidated the release of GWAS summary statistics derived from individual studies. In the current study, we included summary data for 6 brain diseases (Supplementary Table 7).
TriNetX
To evaluate real-world clinical outcomes associated with sleep traits, we used the TriNetX52 database (https://trinetx.com/)—a global federated health research platform that provides access to deidentified electronic medical records from over 70 healthcare organizations, encompassing more than 90 million patients. The TriNetX platform integrates clinical data, including diagnoses, medications, procedures and laboratory results, enabling large-scale observational analyses. We used this resource to assess associations between insomnia and hypersomnia and systemic disease outcomes across organ systems identified in the UKBB for short and long sleep duration (Fig. 3).
Baltimore longitudinal study of ageing
The main goal of the BLSA is to understand the normal ageing process. Tracking physiological and cognitive changes over time aims to identify risk factors for age-related diseases, study patterns of decline and identify predictors of healthy ageing. BLSA53,54 brain MRI, self-reported, and actigraphy-derived sleep duration (n = 385) were used to compare and replicate the U-shaped pattern observed in the main analysis for the brain MRIBAG. For self-reported sleep duration, sleep duration was assessed using a standardized questionnaire item asking “On average, in the past month, how many hours of sleep did you get each night?”. The participants select from ordered categorical response options reflecting typical nightly sleep duration: more than 7 h; more than 6 h up to 7 h; more than 5 h up to 6 h; or 5 h or fewer. This measure captures habitual sleep duration over the previous month and represents participants’ perceived average nightly sleep. We also included participants who underwent wrist actigraphy. They were instructed to wear an Actiwatch-2 wrist actigraphy (Philips-Respironics) on their non-dominant wrist for seven consecutive 24 h periods. The devices continuously recorded activity counts and ambient light levels, and the participants were asked to press the event marker each time they intended to go to sleep (lights out) and when they got up to start their day55.
The Multi-Ethnic Study of Atherosclerosis
The Multi-Ethnic Study of Atherosclerosis (MESA)21,22,56 is a medical research initiative involving over 6,000 men and women from six US communities. For this analysis, we included 573 participants with available brain MRI data, self-reported sleep duration measures, and relevant covariates such as age, sex and BMI. We leveraged the MESA cohort to attempt replication of the U-shaped association observed in the UKBB for the brain MRIBAG. Self-reported sleep duration is obtained from the Exam 5 Sleep Questionnaire, in which the participants report their habitual bedtimes and wake times separately for weekdays and weekends. The questionnaire includes items asking for bedtime and waketime in 24 h format, from which MESA derives weekday sleep duration and weekend sleep duration expressed in hours and minutes.
GAM models the relationship between sleep duration and organ ageing clocks, IDPs, proteins and metabolites
To model the association between sleep duration and the 23 multi-organ BAGs and any other phenotype, we implemented GAMs using the mgcv package in R. This approach enabled flexible modelling of complex relationships, whether linear, flat, sigmoidal or U-shaped, without requiring any prior assumptions about the underlying shape of the fitted curves. We adjusted for key demographic and physiological covariates (that is, age, sex, weight, height, waist circumference, BMI, assessment centre, diastole, systolic blood pressure, time differences for data collection (for MRIBAGs) and disease status). The participants reporting extremely short durations or those with missing sleep duration were excluded. The analysis was restricted to those reporting 4–10 h of sleep to reduce the influence of outliers. For each BAG, we fitted GAM with cubic regression splines (bs = ‘cr’) to smooth the nonlinear association between sleep duration and the BAG, considering sex and sex–sleep interaction term.
Model selection was conducted by systematically evaluating combinations of smoothing dimensions (k = 3, 5, 10, 15, 20) and distribution families (Gaussian, t, and gamma). For each candidate model, the estimated e.d.f. and smoothing parameters were optimized internally through penalized maximum-likelihood estimation. The optimal model was defined as the one yielding the lowest Akaike information criterion, indicating the best balance between model fit and complexity, while ensuring that the e.d.f. did not approach the upper limit of the specified k, thereby avoiding overfitting. We tested (1) the main effect (sleep duration; P1); (2) sex difference in population mean (P2); and (3) sex–sleep interaction terms (P3) on each BAG. The solid curves in Fig. 1 depict estimated BAG, while shaded bands represent the 95% CI. Supplementary Fig. 1 provides diagnostic plots of model fit for the optimal model. The raw BAG values were normalized to the range (0, 1) to allow for the application of different family distributions in the model.
Twenty-three multi-organ ageing clocks
In our previous work, we processed raw brain MRI data from the UKBB to extract 119 grey matter regions of interest (ROIs) from T1-weighted images, which were used to compute the brain MRIBAG. For the heart MRIBAG, we used 80 cardiac MRI-derived traits from a previous study57, which we had incorporated in an earlier study. Moreover, five abdominal MRIBAGs were derived from abdominal MRI data (category ID = 105) across multiple studies58,59,60,61,62,63, yielding a total of 7 MRIBAGs1. We also developed 11 ProtBAGs using UKBB plasma proteomics data2, along with 5 MetBAGs3 based on plasma metabolomics profiles. All 23 BAGs were developed using a nested cross-validation framework, adhering to best practices in machine learning to minimize overfitting and prevent data leakage2,64.
In our previous studies1,2,3, we described in detail the population definition and cross-validation procedures used for model training, which we summarize here. Applying a coherent machine learning framework, we assessed the performance of age-prediction models. Hyperparameter tuning was performed through nested, repeated holdout cross-validation with 50 repetitions (80% training/validation and 20% testing). Specifically, we performed a grid search for fine-tuning model-specific hyperparameters. The within-distribution, holdout test dataset was held out to unbiasedly evaluate model performance (different from the 20% test dataset from the cross-validation).
To rigorously train the age-prediction models, we first defined participants without any pathologies based on ICD code and clinical history as CN. We further split the CN into the following datasets:
-
CN within-distribution, holdout test dataset: 500 participants were randomly drawn from the CN population. Within-distribution, holdout test datasets are ideal for objectively evaluating machine learning model performance, especially in studies with large sample sizes, such as the UKBB.
-
CN training/validation dataset: 80% of the remaining CN population was used for the inner loop tenfold cross-validation for hyperparameter selection.
-
CN cross-validated test dataset: 20% of the remaining CN population was used for the outer-loop 50 repetitions.
-
Patient dataset: all patients who have at least one ICD-10-based diagnosis or clinical history.
The CN training/validation/test datasets were used for model development and were used with a nested cross-validation procedure for all machine learning models (LASSO regression and support vector regressor, elastic net and neural network), whereas the within-distribution, holdout test set provided an unbiased assessment of model performance. Model evaluation metrics included MAE and Pearson’s r. Age bias correction was applied using the approach outlined previously65.
IDP-wide associations
We assessed the association between sleep duration and 720 IDPs covering 8 organ systems and tissues using UKBB in vivo imaging data. For each IDP, we fitted the same GAM as in the ‘GAM models the relationship between sleep duration and organ ageing clocks, IDPs, proteins and metabolites’ section to capture the associations with sleep duration. These models included age, sex, body mass index, height, weight, waist circumference, assessment centre, blood pressure and disease status as covariates. Moreover, organ-specific covariates were incorporated, such as brain positioning in the scanner (lateral, transverse and longitudinal), head motion and intracranial volume for the brain IDPs. Outlier values in the IDP outcome variables, defined as ±4 s.d. from the mean, were removed to minimize the influence of extreme values. For each IDP, we extracted effect estimates and tested for the main effects of sleep, sex differences and sex-specific interactions. Predicted curves and 95% confidence intervals were generated separately for male and female individuals. When significant (P < 0.05/720), sex-specific turning points (sample minimum values of sleep duration) were also identified from the fitted curves. Descriptions of these 720 IDPs are provided in our previous study1,66 and in Supplementary Data 2.
Proteome-wide associations
Our analysis focused on the first instance of the proteomics data (“instance” = 0). We then integrated Olink files containing coding information, batch numbers, assay details and limit of detection (LOD) data (category ID: 1839) by matching them to the proteomics dataset ID. Finally, we excluded normalized protein expression values that fell below the protein-specific LOD. Descriptions of these proteins are provided in our previous study2 and in Supplementary Data 3.
We conducted ProWASs by linking sleep duration to 2,923 unique plasma proteins measured using the same GAM model. The GAM was adjusted for common covariates, including age, sex, weight, height, waist circumference, BMI, assessment centre, disease status, diastolic and systolic blood pressure, protein batch number, LOD and the first 40 genetic principal components. Multiple-testing corrections were applied using Bonferroni adjustment (P < 0.05/2,923). Given the substantial correlation structure among proteomic measures, we acknowledge this choice is conservative; we retain Bonferroni correction in the main text to minimize false positives. To identify and exclude extreme outliers, we defined an upper threshold as the mean plus 4 s.d. for each protein. We mainly focus on the 342 organ-enriched proteins defined in our previous study2.
Metabolome-wide associations
The original data (category ID: 220) were (1) calibrated absolute concentrations (or ratios) and not raw NMR spectra; and (2) before release, had already been subject to quality control procedures by Nightingale Health67. After the quality-check procedures described previously68, we performed additional quality-check steps to remove a range of unwanted technical variations, including shipping batch, 96-well plate, well position, aliquoting robot and aliquot tip. We focused our analysis on the first instance of the metabolomics data (“instance” = 0). The analysis included 327 metabolites (comprising both small molecules and lipoprotein measures), of which 107 were non-derived raw metabolites, and the remainder were composite metabolites, across 274,247 participants. Descriptions of these metabolites are provided in our previous study3 and in Supplementary Data 4.
We conducted MetWASs by linking sleep duration to 327 plasma metabolites. The GAM controlled common covariates, including age, sex, weight, height, waist circumference, BMI, assessment centre, disease status, diastolic and systolic blood pressure, and the first 40 genetic principal components. Multiple-testing corrections were applied using Bonferroni adjustment (P < 0.05/327). To identify and exclude extreme outliers, we defined an upper threshold as the mean plus 4 s.d. for each metabolite. We mainly focus on the 107 organ-associated metabolites defined in our previous study3.
Genetic analyses
We used the genotype and imputed genotype data from UKBB for all genetic analyses. Our quality-check pipeline focused on European ancestry in the UKBB (6,477,810 SNPs passing quality checks). We summarize our genetic quality check steps. First, we excluded related individuals (up to second degree) from the complete UKBB sample using the KING software for family relationship inference69. We then removed duplicated variants from all 22 autosomal chromosomes. Individuals whose genetically identified sex did not match their self-acknowledged sex were removed. Other exclusion criteria were (1) individuals with more than 3% of missing genotypes; (2) variants with minor allele frequency (MAF; dosage mode) of less than 1%; (3) variants with larger than 3% missing genotyping rate; (4) variants that failed the Hardy–Weinberg test at 1 × 10−10. To further adjust for population stratification70, we derived the first 40 genetic principal components using the FlashPCA software71. Details of the genetic quality check protocol have been described elsewhere9,10,72,73,74.
GWAS
Given the large sample sizes for both short versus normal (16,872 short and 300,420 normal) and long versus normal (25,049 long and 300,420 normal) sleep duration comparisons, we used REGENIE50 for GWAS, as it is well-suited for large-scale genetic analyses due to its computational efficiency and ability to control for population structure and relatedness, outperforming alternatives like PLINK and fastGWA in these settings. Our GWAS adjusted common covariates, including age, disease status, age-squared, sex, interactions of age with sex, BMI, waist circumference, standing height, weight, systolic/diastolic blood pressure and the first 40 genetic principal components. We applied a genome-wide significance threshold (5 × 10−8) to annotate the significant independent genomic loci.
Annotation of genomic loci
For all GWASs, genomic loci were annotated using FUMA75. For genomic loci annotation, FUMA initially identified lead SNPs (correlation r2 ≤ 0.1, distance <250 kb) and assigned them to non-overlapping genomic loci. The lead SNP with the lowest P value (that is, the top lead SNP) represented the genomic locus. Further details on the definitions of top lead SNP, lead SNP, independent significant SNP and candidate SNP can be found in Supplementary Note 2.
MAGMA tissue expression analysis
MAGMA gene-property analysis24 was conducted using gene expression data from the GTEx (v.8) to investigate tissue-specific associations with genetic variants. In contrast to differential gene expression enrichment tests focusing solely on prioritized genes, MAGMA leverages the full distribution of SNP P values across the genome, providing a more comprehensive assessment of how genetic signals relate to gene expression patterns in various tissues.
Genetic correlation
We estimated the genetic correlation (gc) using the LDSC23 software between the two abnormal sleep duration patterns and 527 DEs from the FinnGen and PGC datasets. We used precomputed LD scores from the 1000 Genomes of European ancestry, maintaining the default settings for other parameters in LDSC. Note that LDSC corrects for sample overlap, ensuring an unbiased genetic correlation estimate76. Statistical significance was determined using Bonferroni correction.
Survival analyses for risk of DEs and all-cause mortality
In the UKBB, we evaluated the predictive value of abnormal sleep duration patterns, specifically short sleep (<6 h) and long sleep (>8 h), through two sets of analyses (1) survival analysis to predict the future onset of incident single DEs defined by ICD-10 codes; and (2) survival analysis to estimate the longitudinal risk of all-cause mortality.
Survival analysis for ICD-based single DE
We used a Cox proportional hazard model while adjusting for covariates to test the associations of short/long sleep duration, compared to normal sleep duration (6–8 h), with the time to incident of ICD-based single disease entities. Notably, we excluded individuals with any disease diagnosis (except the disease of interest) to ensure the analysis was restricted to a disease-free population. The covariates age, sex, body mass index, height, weight, waist circumference and blood pressure were included as additional right-side variables in the model. To train the model, the time variable was determined by calculating the difference between the date of diagnosis of the disease for cases (or the censoring date for non-cases) and the date attending the assessment centre (field ID: 53). The participants who were diagnosed for a specific disease of interest after enrolling in the study were classified as cases; non-cases were defined by participants without any disease diagnoses.
Survival analysis for mortality risk
We used a Cox proportional hazard model while adjusting for covariates to test the associations of short/long sleep duration patterns with all-cause mortality. The covariates age, sex, body mass index, height, weight, waist circumference and blood pressure were included as additional right-side variables in the model. The hazard ratio, exp(βR), was calculated and reported as the effect size measure that indicates the influence of each biomarker on the risk of mortality. To train the model, the time variable was determined by calculating the difference between the date of death (field ID: 40000) for cases (or the censoring date for non-cases) and the date attending the assessment centre (field ID: 53). Participants who passed away after enrolling in the study were classified as cases.
SEM for mediation analysis
Using UKBB data, we used structural equation modelling (SEM)66 to examine whether organ-specific MRIBAGs, measured at the second imaging visit (2014 or later), serve as mediators in the relationship between sleep duration, assessed at the baseline visit (2007–2010), and two distinct subtypes of LLD (LLD1 and LLD2), also evaluated at the second visit. Sleep duration was categorized into binary groups reflecting short (<6 h) and long sleep (>8 h) patterns, with respect to the normal sleep duration (6–8 h).
For each MRIBAG, we specified a mediation model (sleep duration → MRIBAG → LLD) that included (1) a direct path from sleep duration to LLD subtype (c2); and (2) an indirect path from sleep duration to MRIBAG (a1), and from MRIBAG to LLD subtype (c1), with the product term (a1 × c1) representing the mediated (indirect) effect. Models were adjusted for relevant covariates, including age at assessment, sex, weight, standing height, waist circumference, BMI, diastolic blood pressure and systolic blood pressure. To assess the robustness of mediational directionality considering the time-ordering of events, we also tested reversed models (sleep duration → LLD1/2 → brain MRIBAG) to scrutinize potential inverse mediation. Significance was determined using a Bonferroni-corrected threshold (P < 0.05/7) to account for multiple comparisons across the 7 organ systems. All model estimates, including direct, indirect, total effects and proportion mediated, are reported in the Supplementary Information.
MR tests whether DEs are causally linked to short or long sleep duration relative to normal sleep duration
We conducted two-sample MR linking 525 DEs (as exposures) from FinnGen and PGC to short and long sleep duration as outcomes; however, limited statistical power prevented testing the reverse direction (sleep duration → DEs), which was partially tested by the mediation analysis (not in a strict sense of causal inference for SEM).
We used a two-sample MR approach implemented in the TwoSampleMR package77 to infer the causal relationships. We used five distinct MR methods, including the IVW method, Egger, weighted median, simple mode and weighted mode estimators. The STROBE-MR Statement78 guided our analyses to increase transparency and reproducibility, encompassing the selection of exposure and outcome variables, reporting statistics and implementing sensitivity checks to identify potential violations of underlying assumptions. First, we performed an unbiased quality check on the GWAS summary statistics. Notably, the absence of population overlapping bias79 was confirmed, given that FinnGen and UKBB participants largely represent populations of European ancestry without explicit overlap with UKBB. PGC GWAS summary data were ensured to exclude UKBB participants. Furthermore, all consortium GWAS summary statistics were based on or lifted to GRCh37. Subsequently, we selected the effective exposure variables by assessing the statistical power of the exposure GWAS summary statistics in terms of instrumental variables (IVs), ensuring that the number of IVs exceeded 7. Crucially, the function clump_data was applied to the exposure GWAS data, considering LD. The function harmonise_data was then used to harmonize the GWAS summary statistics of the exposure and outcome variables. Bonferroni correction was applied to all tested traits based on the number of effective DEs.
Finally, we conducted multiple sensitivity analyses. First, we conducted a heterogeneity test to scrutinize potential violations of the IV’s assumptions. To assess horizontal pleiotropy, which indicates the IV’s exclusivity assumption80, we used a funnel plot, single-SNP MR methods and the Egger estimator. Furthermore, we performed a leave-one-out analysis, systematically excluding one instrument (SNP/IV) at a time, to gauge the sensitivity of the results to individual SNPs. Notably, our MR analyses rely on the standard assumption of linear genetic effects on sleep duration, providing an average causal effect per unit increment in the exposure. As such, we conducted the sleep GWAS using binary traits (long sleep versus normal sleep and short sleep versus normal sleep), rather than treating sleep duration as a continuous variable. Nevertheless, the MR estimates do not directly represent the nonlinear U-shaped sleep–BAG relationships observed in our phenotypic analyses and should be interpreted as complementary to, rather than a direct mirror of, the observational sleep–BAG associations.
Ethics statement
All data used in this study were obtained from previously approved research cohorts and biobanks. The MULTI Consortium has been approved by the Institutional Review Board at Columbia University (IRB protocol: AAAV6751). Individual contributing studies received approval from their respective institutional review boards. All research was performed in accordance with relevant guidelines and regulations, and written informed consent was obtained from all participants in each study.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The GWAS summary statistics for long and short sleep duration are publicly available through the SleepChart portal (https://labs-laboratory.com/sleepchart). The GWAS summary statistics for the 23 multi-organ BAGs are publicly accessible through the MEDICINE portal (https://labs-laboratory.com/medicine/). All GWAS summary statistics are also available at Synapse (https://www.synapse.org/Synapse:syn64923248/wiki/630992 (ref. 81) and https://www.synapse.org/Synapse:syn68602065/wiki/633329 (ref. 82)). Our study used data generated by the Human Protein Atlas (https://www.proteinatlas.org) and MetaboAnalyst (https://www.metaboanalyst.ca/MetaboAnalyst/ModuleView.xhtml). GWAS summary data for the DEs were downloaded from the official websites of FinnGen (R9: https://www.finngen.fi/en/access_results) and PGC (https://pgc.unc.edu/for-researchers/download-results/). Individual data from the UKBB can be requested with proper registration online (https://www.ukbiobank.ac.uk/). The data used to link insomnia/hypersomnia with DEs are from TriNetX (https://trinetx.com/), which requires a valid registration and request. Data from the BLSA study (https://www.blsa.nih.gov/) and the MESA study (https://mesa-nhlbi.org/) can be requested with proper registration. We also shared the original PDF for the four main figures at Zenodo83 (https://zenodo.org/records/17409426). Source data are provided with this paper.
Code availability
The software and resources used in this study are all publicly available: SleepChart-R84 (https://github.com/anbai106/SleepChart), customized R code for linking sleep duration and 23 BAGs; MLNI85 (v.0.1.2; https://github.com/anbai106/mlni), BAG generation; MGCV (v.1.9.1) with R v.4.2.2 (https://cran.r-project.org/web/packages/mgcv/index.html), core package for the GAM implementation; GAMLSS (v.5.4.22) with R v.4.2.2 (https://www.gamlss.com/), replicate the nine sleep–BAG signals identified by the GAM modelling; Lifelines (v.0.27.8; https://lifelines.readthedocs.io/en/latest/), survival analysis; Lavaan (v.0.6.19; https://lavaan.ugent.be/), mediation analysis; String (v.12.0; https://string-db.org/), protein set enrichment analyses and network analysis; MetaboAnalyst (v.6.0; https://www.metaboanalyst.ca/), metabolite enrichment analysis and network analysis; REGENIE (v.4.1; https://rgcgithub.github.io/regenie/), binary sleep GWASs (v.4.1), FinnGen used v.2.4.4 for their disease end-point GWASs; LDSC (git version aa33296; https://github.com/bulik/ldsc), genetic correlation. FUMA (v.1.8.0; https://fuma.ctglab.nl/), gene mapping, genomic locus annotation; and KING (v.2.2.7; https://www.kingrelatedness.com/), kinship-based inference for GWAS.
References
The MULTI Consortium et al. MRI-based multi-organ clocks for healthy aging and disease assessment. Nat. Med. 32, 82–92 (2026).
Wen, J. Refining the generation, interpretation and application of multi-organ, multi-omics biological aging clocks. Nat. Aging 5, 1897–1913 (2025).
The MULTI Consortium et al. Multi-organ metabolome biological age implicates cardiometabolic conditions and mortality risk. Nat. Commun. 16, 4871 (2025).
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. Hallmarks of aging: an expanding universe. Cell 186, 243–278 (2023).
Diao, T. et al. Changes in sleep patterns, genetic susceptibility, and incident cardiovascular disease in China. JAMA Netw. Open 7, e247974 (2024).
Cappuccio, F. P., D’Elia, L., Strazzullo, P. & Miller, M. A. Sleep duration and all-cause mortality: a systematic review and meta-analysis of prospective studies. Sleep 33, 585–592 (2010).
Fan, M. et al. Sleep patterns, genetic susceptibility, and incident cardiovascular disease: a prospective study of 385 292 UK biobank participants. Eur. Heart J. 41, 1182–1189 (2020).
Leng, Y., McEvoy, C. T., Allen, I. E. & Yaffe, K. Association of sleep-disordered breathing with cognitive function and risk of cognitive impairment: a systematic review and meta-analysis. JAMA Neurol. 74, 1237–1245 (2017).
Wen, J. et al. Characterizing heterogeneity in neuroimaging, cognition, clinical symptoms, and genetics among patients with late-life depression. JAMA Psychiatry 79, 464–474 (2022).
Wen, J. et al. The genetic architecture of multimodal human brain age. Nat. Commun. 15, 2604 (2024).
Oh, H. S.-H. et al. Organ aging signatures in the plasma proteome track health and disease. Nature 624, 164–172 (2023).
You, Y. et al. Inverted U-shaped relationship between sleep duration and phenotypic age in US adults: a population-based study. Sci Rep. 14, 6247 (2024).
Wang, X. et al. U-shaped association between sleep duration and biological aging: evidence from the UK Biobank study. Aging Cell 23, e14159 (2024).
Tao, M.-H., Drake, C. L. & Lin, C.-H. Association of sleep duration, chronotype, social jetlag, and sleep disturbance with phenotypic age acceleration: A cross-sectional analysis. Sleep Health 10, 122–128 (2024).
Wang, M. et al. Association between sleep traits and biological aging risk: a Mendelian randomization study based on 157 227 cases and 179 332 controls. Sleep 47, zsad299 (2024).
The MULTI Consortium et al. Brain–heart–eye axis revealed by multi-organ imaging genetics and proteomics. Nat. Biomed. Eng. https://doi.org/10.1038/s41551-025-01506-5 (2025).
Wen, J. Towards a multi-organ, multi-omics medical digital twin. Nat. Biomed. Eng. 9, 1386–1389 (2025).
Boquet-Pujadas, A. et al. MUTATE: a human genetic atlas of multiorgan artificial intelligence endophenotypes using genome-wide association summary statistics. Brief. Bioinform. 26, bbaf125 (2025).
The MULTI Consortium et al. Multi-organ AI endophenotypes chart the heterogeneity of brain, eye and heart pan-disease. Nat. Mental Health 4, 203–230 (2026).
Cespedes, E. M. et al. Comparison of self-reported sleep duration with actigraphy: results from the Hispanic Community Health Study/Study of Latinos Sueño Ancillary Study. Am. J. Epidemiol. 183, 561–573 (2016).
Zhang, G.-Q. et al. The National Sleep Research Resource: towards a sleep data commons. J. Am. Med. Inform. Assoc. 25, 1351–1358 (2018).
Chen, X. et al. Racial/ethnic differences in sleep disturbances: The Multi-Ethnic Study of Atherosclerosis (MESA). Sleep 38, 877–888 (2015).
Bulik-Sullivan, B. K. et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
de Leeuw, C. A., Mooij, J. M., Heskes, T. & Posthuma, D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 11, e1004219 (2015).
Hirshkowitz, M. et al. National Sleep Foundation’s updated sleep duration recommendations: final report. Sleep Health 1, 233–243 (2015).
Pasman, J. A. et al. An encompassing Mendelian randomization study of the causes and consequences of major depressive disorder. Nat. Mental Health 3, 1002–1011 (2025).
Staley, J. R. & Burgess, S. Semiparametric methods for estimation of a nonlinear exposure-outcome relationship using instrumental variables with application to Mendelian randomization. Genet. Epidemiol. 41, 341–352 (2017).
Irwin, M. R. Sleep and inflammation: partners in sickness and in health. Nat. Rev. Immunol. 19, 702–715 (2019).
Morselli, L. L., Guyon, A. & Spiegel, K. Sleep and metabolic function. Pflugers Arch. 463, 139–160 (2012).
Anghel, L. et al. Sleep disorders associated with neurodegenerative diseases. Diagnostics 13, 2898 (2023).
Kim, T. W., Jeong, J.-H. & Hong, S.-C. The impact of sleep and circadian disturbance on hormones and metabolism. Int. J. Endocrinol. 2015, 591729 (2015).
Lo, J. C., Loh, K. K., Zheng, H., Sim, S. K. Y. & Chee, M. W. L. Sleep duration and age-related changes in brain structure and cognitive performance. Sleep 37, 1171–1178 (2014).
Mander, B. A., Winer, J. R. & Walker, M. P. Sleep and human aging. Neuron 94, 19–36 (2017).
Mong, J. A. & Cusmano, D. M. Sex differences in sleep: impact of biological sex and sex steroids. Philos. Trans. R. Soc. Lond. B 371, 20150110 (2016).
Spira, A. P. et al. Sleep duration and subsequent cortical thinning in cognitively normal older adults. Sleep 39, 1121–1128 (2016).
Itani, O., Jike, M., Watanabe, N. & Kaneita, Y. Short sleep duration and health outcomes: a systematic review, meta-analysis, and meta-regression. Sleep Med. 32, 246–256 (2017).
Irwin, M. R. & Opp, M. R. Sleep health: reciprocal regulation of sleep and innate immunity. Neuropsychopharmacology 42, 129–155 (2017).
Marjot, T., Ray, D. W., Williams, F. R., Tomlinson, J. W. & Armstrong, M. J. Sleep and liver disease: a bidirectional relationship. Lancet Gastroenterol. Hepatol. 6, 850–863 (2021).
Tononi, G. & Cirelli, C. Sleep and the price of plasticity: from synaptic and cellular homeostasis to memory consolidation and integration. Neuron 81, 12–34 (2014).
Tian, Y. E., Cole, J. H., Bullmore, E. T. & Zalesky, A. Brain, lifestyle and environmental pathways linking physical and mental health. Nat. Mental Health 2, 1250–1261 (2024).
Moshfegh, O. My Year of Rest and Relaxation (Penguin, 2018).
Dew, M. A. et al. Healthy older adults’ sleep predicts all-cause mortality at 4 to 19 years of follow-up. Psychosom. Med. 65, 63–73 (2003).
Penninx, B. W., Milaneschi, Y., Lamers, F. & Vogelzangs, N. Understanding the somatic consequences of depression: biological mechanisms and the role of depression symptom profile. BMC Med. 11, 129 (2013).
Austin-Zimmerman, I. et al. Genome-wide association studies and cross-population meta-analyses investigating short and long sleep duration. Nat. Commun. 14, 6059 (2023).
Wray, N. R. et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat. Genet. 50, 668–681 (2018).
Bycroft, C. et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018).
Li, W. UK Biobank pharma proteomics resource. Nat. Genet. 55, 1781–1781 (2023).
Sun, B. B. et al. Plasma proteomic associations with genetics and health in the UK Biobank. Nature 622, 329–338 (2023).
Kurki, M. I. et al. FinnGen provides genetic insights from a well-phenotyped isolated population. Nature 613, 508–518 (2023).
Mbatchou, J. et al. Computationally efficient whole-genome regression for quantitative and binary traits. Nat. Genet. 53, 1097–1103 (2021).
O’Donovan, M. C. What have we learned from the Psychiatric Genomics Consortium. World Psychiatry 14, 291–293 (2015).
Palchuk, M. B. et al. A global federated real-world data and analytics platform for research. JAMIA Open 6, ooad035 (2023).
Andres, G. R. et al. Normal human aging: The Baltimore Longitudinal Study of Aging. J. Gerontol. 40, 767 (1985).
Duggan, M. R. et al. Proteomic analyses reveal plasma EFEMP1 and CXCL12 as biomarkers and determinants of neurodegeneration. Alzheimers Dement. 20, 6486–6505 (2024).
He, L. et al. Age and objectively measured sleep: investigating associations and interactions by sex and race in middle-aged and older adults. Sleep Adv. 5, zpae045 (2024).
Bild, D. E. et al. Multi-ethnic study of atherosclerosis: objectives and design. Am. J. Epidemiol. 156, 871–881 (2002).
Bai, W. et al. A population-based phenome-wide association study of cardiac and aortic structure and function. Nat. Med. 26, 1654–1662 (2020).
Mojtahed, A. et al. Reference range of liver corrected T1 values in a population at low risk for fatty liver disease—a UK Biobank sub-study, with an appendix of interesting cases. Abdom. Radiol. 44, 72–84 (2019).
Whitcher, B. et al. Precision MRI phenotyping enables detection of small changes in body composition for longitudinal cohorts. Sci. Rep. 12, 3748 (2022).
Parisinos, C. A. et al. Genome-wide and Mendelian randomisation studies of liver MRI yield insights into the pathogenesis of steatohepatitis. J. Hepatol. 73, 241–251 (2020).
Karlsson, A. et al. Automatic and quantitative assessment of regional muscle volume by multi-atlas segmentation using whole-body water-fat MRI. J. Magn. Reson. Imaging 41, 1558–1569 (2015).
Borga, M. et al. Validation of a fast method for quantification of intra-abdominal and subcutaneous adipose tissue for large-scale human studies. NMR Biomed. 28, 1747–1753 (2015).
Liu, Y. et al. Genetic architecture of 11 organ traits derived from abdominal MRI using deep learning. eLife 10, e65554 (2021).
Wen, J. et al. Convolutional neural networks for classification of Alzheimer’s disease: overview and reproducible evaluation. Med. Image Anal. 63, 101694 (2020).
Beheshti, I., Nugent, S., Potvin, O. & Duchesne, S. Bias-adjustment in neuroimaging-based brain age frameworks: a robust scheme. Neuroimage Clin. 24, 102063 (2019).
Tian, Y. E. et al. Heterogeneous aging across multiple organ systems and prediction of chronic disease and mortality. Nat. Med. 29, 1221–1231 (2023).
Würtz, P. et al. Quantitative serum nuclear magnetic resonance metabolomics in large-scale epidemiology: a primer on -omic technologies. Am. J. Epidemiol. 186, 1084–1096 (2017).
Ritchie, S. C. et al. Quality control and removal of technical variation of NMR metabolic biomarker data in ~120,000 UK Biobank participants. Sci. Data 10, 64 (2023).
Manichaikul, A. et al. Robust relationship inference in genome-wide association studies. Bioinformatics 26, 2867–2873 (2010).
Price, A. L., Zaitlen, N. A., Reich, D. & Patterson, N. New approaches to population stratification in genome-wide association studies. Nat. Rev. Genet. 11, 459–463 (2010).
Abraham, G., Qiu, Y. & Inouye, M. FlashPCA2: principal component analysis of Biobank-scale genotype datasets. Bioinformatics 33, 2776–2778 (2017).
Wen, J. et al. Genomic loci influence patterns of structural covariance in the human brain. Proc. Natl Acad. Sci. USA 120, e2300842120 (2023).
Wen, J. et al. The genetic architecture of biological age in nine human organ systems. Nat. Aging 4, 1290–1307 (2024).
Wen, J. et al. Genetic and clinical correlates of two neuroanatomical AI dimensions in the Alzheimer’s disease continuum. Transl. Psychiatry 14, 420 (2024).
Watanabe, K., Taskesen, E., van Bochoven, A. & Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 8, 1826 (2017).
Bulik-Sullivan, B. et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet. 47, 1236–1241 (2015).
Hemani, G. et al. The MR-Base platform supports systematic causal inference across the human phenome. eLife 7, e34408 (2018).
Skrivankova, V. W. et al. Strengthening the reporting of observational studies in epidemiology using mendelian randomization: the STROBE-MR statement. JAMA 326, 1614–1621 (2021).
Sanderson, E. et al. Mendelian randomization. Nat. Rev. Methods Primers 2, 6 (2022).
Bowden, J. et al. A framework for the investigation of pleiotropy in two-sample summary data Mendelian randomization. Stat. Med. 36, 1783–1802 (2017).
Wen, J. MEDICINE: AI-derived GWAS summary statistics sharing portal. Synapse https://doi.org/10.7303/SYN64923248 (2025).
Wen, J. SleepChart: digitize sleep duration and biological aging clocks across organs and omics. Synapse https://doi.org/10.7303/syn68602065 (2025).
Wen, J. SleepChart source data for figures and R script. Zenodo https://doi.org/10.5281/zenodo.17409425 (2026).
Wen, J. SleepChart-R (v.0.1). GitHub https://github.com/anbai106/SleepChart (2026).
Wen, J. MLNI (v.0.1.4). GitHub https://github.com/anbai106/mlni (2025).
Besedovsky, L., Lange, T. & Haack, M. The sleep-immune crosstalk in health and disease. Physiol. Rev. 99, 1325–1380 (2019).
Yeung, C.-Y. C. et al. Circadian regulation of protein cargo in extracellular vesicles. Sci. Adv. 8, eabc9061 (2022).
Scharf, M. T., Naidoo, N., Zimmerman, J. E. & Pack, A. I. The energy hypothesis of sleep revisited. Prog. Neurobiol. 86, 264–280 (2008).
Staiger, D. & Stock, J. H. Instrumental variables regression with weak instruments. Econometrica 65, 557–586 (1997).
Acknowledgements
This research is supported through funding from the NIH-supported MULTI Consortium (J.W.; grant number: RF1AG092412). The MULTI consortium (J.W.; UKBB application number: 647044) aims to integrate multi-organ imaging and multi-omics data to advance our understanding of human ageing and disease. This study used the UKBB resource under application number 647044 (J.W.). C.D. is supported by the NIA grant: RF1 AG054409. A.Z. is supported by the Australian Research Council Future Fellowship (grant number FT220100091). M.R.D., K.A.W., E.M.S., S.M.R. and L.F. are supported by the Intramural Research Program of the NIH, NIA. We thank the members of the UKBB team for their contributions to advancing clinical research in our field (https://www.ukbiobank.ac.uk/); the participants and investigators of the FinnGen study and the PGC consortium; FinnGen (https://www.finngen.fi/en) and PGC (https://pgc.unc.edu/) for sharing the GWAS summary statistics with the scientific community; and the BLSA participants and staff for their participation and continued dedication. The BLSA protocol was approved by the Institutional Review Board of the National Institute of Environmental Health Science, National Institutes of Health (03AG0325). We acknowledge the Multi-Ethnic Study of Atherosclerosis (MESA) and the MESA SHARe project, conducted and supported by the National Heart, Lung and Blood Institute (NHLBI) in collaboration with MESA investigators, under contracts 75N92025D00022, 75N92020D00001, HHSN268201500003I, N01-HC-95159, 75N92025D00026, 75N92020D00005, N01-HC-95160, 75N92020D00002, N01-HC-95161, 75N92025D00024, 75N92020D00003, N01-HC-95162, 75N92025D00027, 75N92020D00006, N01-HC-95163, 75N92025D00025, 75N92020D00004, N01-HC-95164, 75N92025D00028, 75N92020D00007, N01-HC-95165, N01-HC-95166, N01-HC-95167, N01-HC-95168 and N01-HC-95169, with additional support from NHLBI grant R01HL127659 and NCATS grants UL1-TR-000040, UL1-TR-001079 and UL1-TR-001420. The MESA Sleep Ancillary Study was funded by NIH/NHLBI grant R01HL098433. The National Sleep Research Resource was supported by NHLBI (R24HL114473, 75N92019R002).
Author information
Authors and Affiliations
Consortia
Contributions
J.W. has full access to all the study data and is responsible for its integrity and accuracy. Study concept and design: J.W. Drafting of the manuscript: J.W. Critical revision of the manuscript for important intellectual content: C.K.O., Z.S., F.A., Z.Y., Y.E.T., M.R.D., C.Z., Y.L., Y.C., W.B., C.H.Y.F., M.S.R., P.A., G.W., P.L.D.J., J.Z., H.S.-H.O., X.Z., K.A.W., D.W.B., A.Z., E.M.S., S.M.R., L.F., C.D. and J.W. Analysis: J.W. ran all analyses for UKBB, BLSA and MESA. J.W. and Z.S. developed the SleepChart portal. F.A. ran analyses for TriNetX. C.K.O. helped with figures.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature thanks Alexandra Badea, Susan Redline, Christopher Whelan and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Sleep Chart delineates U-shaped associations between sleep duration and 720 organ-specific imaging-derived phenotypes.
a) Sleep duration was associated with 720 organ-specific imaging-derived phenotypes (IDP; x-axis) using the GAM model. Non-grey coloured IDPs indicate significant associations after Bonferroni correction for multiple comparisons across all available IDPs (two-sided P < 0.05/720). The y-axis shows the effective degrees of freedom (EDF), quantifying the degree of nonlinearity for the U-shaped patterns, with representative proteins annotated for each organ. b) The EDF brain map displays the EDF values for significant signals associated with brain IDPs derived from T1-weighted MRI. c) The EDF brain map displays the EDF values for significant signals associated with brain IDPs derived from fractional anisotropy (FA) using diffusion MRI. d-j) Representative IDPs from various organ systems and tissues illustrate U-shaped associations with sleep duration. The solid curves depict estimated IDP, while shaded bands represent the 95% confidence interval (CI).
Extended Data Fig. 2 Sleep Chart delineates U-shaped associations between sleep duration and 342 organ-enriched proteins.
a) Sleep duration was associated with 342 organ-enriched plasma proteins (x-axis) using the GAM model. These organ/tissue-enriched proteins were defined in our previous study2 via resources from the Human Protein Atlas (https://www.proteinatlas.org/) project. Non-grey coloured proteins indicate proteins with significant associations after Bonferroni correction for multiple comparisons across all available proteins (two-sided P < 0.05/2923 for all available proteins). The y-axis shows the effective degrees of freedom (EDF), quantifying the degree of nonlinearity (e.g., U-shaped associations), with representative proteins annotated for each organ. b) Representative U-shaped relationship between the FGFR2 protein and sleep duration. The solid curves depict estimated protein, while shaded bands represent the 95% confidence interval (CI). c-d) Protein-protein interaction (PPI) network was conducted using STRING v12.0 (https://string-db.org/) to examine the functional relationships among 14 immune-enriched and 29 hepatic-enriched proteins. STRING integrates evidence from experimental data, computational predictions, and literature text mining, enabling the PPI network to reveal how these proteins may functionally interact within and across biological pathways, potentially offering insights into shared mechanisms and tissue-specific processes. e) We performed protein set enrichment analysis (PSEA) using STRING for organs with more than 9 significant proteins (i.e., the hepatic and immune) to identify over-represented Gene Ontology (GO) terms across biological processes, molecular functions, and cellular components. Icon sizes in the visualization reflect the statistical significance, represented by the -log10(FDR-corrected P). The strength measurement (x-axis) is a log-scale enrichment ratio that shows the magnitude of enrichment. We group distinct GO terms along the y-axis by associating each biological pathway with potential sleep-related hypotheses informed by prior literature, such as the immune activation and inflammation hypothesis86 or the Extracellular signalling and vesicle transport hypothesis87.
Extended Data Fig. 3 Sleep Chart delineates U-shaped associations between sleep duration and 107 organ-associated metabolites.
a) Sleep duration was associated with 107 organ-associated plasma metabolites (x-axis) using the GAM model. These organ-associated metabolites were defined in our previous study3. Non-grey coloured points indicate significant associations after Bonferroni correction for multiple comparisons across all available metabolites (two-sided P1 < 0.05/327 for all available metabolites). The y-axis displays the effective degrees of freedom (EDF), quantifying the extent of nonlinearity for the U-shaped associations. We annotated representative proteins for each organ system. b-e) Representative U-shaped relationship between 4 metabolites or lipid subclasses and sleep duration. The solid curves depict estimated metabolites, while shaded bands represent the 95% confidence interval (CI). f) Metabolite set enrichment analysis (MSEA) was performed using the Over Representation Analysis (ORA) module in MetaboAnalyst (https://www.metaboanalyst.ca/), which identifies biologically meaningful patterns by assessing whether known metabolite sets (e.g., significant metabolites related to sleep duration) are overrepresented among the list of input metabolites. The analysis uses a hypergeometric test to calculate statistical significance, with multiple testing correction applied using the FDR method. Only metabolite sets with two-sided PFDR < 0.05 were considered significantly enriched. Only small-molecule metabolites, excluding lipid subclasses, that could be mapped in MetaboAnalyst were included. As the background reference, we used 3,694 metabolite and lipid pathways curated in RaMP-DB (integrating KEGG via HMDB, Reactome, and WikiPathways). We organize distinct metabolite sets along the y-axis by linking them to potential sleep-related hypotheses grounded in prior literature, including the sleep-liver axis, which is closely tied to the energy hypothesis88 of sleep.
Extended Data Fig. 4 Associations between sleep duration and the polygenic risk score (PRS) of the 23 multi-organ, multi-omics BAGs.
Unlike the BAGs, sleep duration (x-axis) did not exhibit a U-shaped relationship with the polygenic risk scores (PRS-BAG) of the 23 BAGs derived from common genetic variants (y-axis). For each PRS-BAG, we fitted a generalized additive model (GAM) with cubic regression splines to assess the association between sleep duration and BAG, stratified by sex and sex-sleep interaction term. The solid curves depict estimated PRS-BAG, while shaded bands represent the 95% confidence interval (CI). Significant signals (two-sided P-value < 0.05/23) are shown with coloured icons and the * symbol. Sample-specific BAG minimum values (in hours) of sleep duration are displayed separately for males and females.
Extended Data Fig. 5 Insomnia and hypersomnia are associated with the time to incident of the 110 disease endpoints and mortality in the TriNetX database.
Using insomnia and hypersomnia diagnoses, we replicated the significant associations shown in Fig. 3 within the UK Biobank cohort. Disease endpoints that remained significant after Bonferroni correction (two-sided P-value < 0.05/111, accounting for 110 disease endpoints and all-cause mortality) are indicated by coloured shapes, while non-significant associations are shown in grey. One additional disease category was also tested for all types of dementia. Ctrl: health control. Error bars represent the 95% confidence interval (CI) of the hazard ratio (HR) estimate, which is represented by a central shape.
Extended Data Fig. 6 Sensitivity analysis for the causal relationship from FinnGen F5-DEPRESSION-DYSTHYMIA to long sleep duration.
a) Lollipop plot of F-statistics for the 8 genome-wide significant depression instruments after preprocessing, considering linkage disequilibrium, ordered from weakest to strongest. All instruments exceed the commonly used rule-of-thumb threshold of F = 1089 (min F = 30.70; median F = 36.88; mean F = 38.62). The combined variance explained is R2-total = 0.00113, which corresponds to an approximate overall F-statistic of 38.66 (N = 274,330; k = 8), exceeding the conventional F > 10 threshold that defines adequately strong instruments in univariable Mendelian randomization. b) Scatter plot for the Mendelian randomization effect sizes of the SNP-depression association (x-axis, log OR) and the SNP-long sleep duration associations (y-axis, log OR) with standard error bars of the estimated beta values. The slopes of the 5 lines correspond to the causal effect sizes estimated by the five MR estimators, respectively. For all five methods, no statistical significance was achieved (two-sided P-value > 0.05). c) Forest plot for the single-SNP Mendelian randomization results. Each dot represents the effect (log OR), and the error bar displays the 95% CI using only one SNP; the red line shows the MR effect using all SNPs together for IVW and Egger estimators. d) Funnel plot for the relationship between the causal effect of depression on long sleep duration. Each dot represents causal effect sizes estimated (95% CI for error bars) using each SNP as a separate instrument against the inverse of the standard error of the causal estimate. e) Leave-one-SNP-out analysis of depression on long sleep duration. Each dot represents the causal effect (log OR), and the error bar displays the 95% CI by excluding that SNP from the analysis. The red line depicts the IVW estimator using all SNPs. f) MR-PRESSO confirms the absence of causality and a significant causal signal (two-sided P = 0.16). Dots summarize the three key MR-PRESSO outputs: the global test p-value, the number of outlier instruments detected, and the observed residual sum of squares (RSSobs). Using eight SNPs, the global test was not significant (two-sided P-value = 0.18), no outliers were identified, and RSSobs was 14.1, indicating no evidence of horizontal pleiotropy. MR-PRESSO was run with 1,000 simulations at α = 0.05; because no outliers were detected, no outlier-corrected estimate was produced. g) Heterogeneity (Cochran’s Q) test indicated no strong between-instrument heterogeneity: MR-Egger Q = 8.56 (df=6, two-sided P = 0.200) and IVW Q = 10.76 (df=7, two-sided P-value = 0.149) using 8 SNPs. These two-sided P-values > 0.05 suggest the causal estimates are not driven by substantial heterogeneity. h) The MR-Egger intercept was 0.0497 (SE = 0.0400; two-sided P = 0.260), indicating no evidence that directional (unbalanced) pleiotropy biases the causal estimate in this analysis. The dot represents the estimate for the intercept and the error bar for its standard error.
Extended Data Fig. 7 Sensitivity analysis for the causal relationship from the most recent MDD to long sleep duration in FinnGen.
Mendelian randomization of major depressive disorder (MDD) on long sleep duration using the FinnGen (R12) GWAS. We downloaded the R12 FinnGen MDD GWAS with an expanded patient sample (N = 59,333) to ensure adequate statistical power. a) Lollipop plot of first-stage F-statistics for the eight genome-wide significant MDD instruments after LD clumping, ordered from weakest to strongest. All instruments exceeded the conventional F > 10 threshold, suggesting sufficient instrument strength. b) Scatter plot of SNP-specific associations between MDD (x-axis, log OR) and long sleep duration (y-axis, log OR). The slopes of the five regression lines correspond to causal effect estimates from the inverse-variance weighted (IVW), weighted median, MR-Egger, simple-mode, and weighted-mode estimators. Among these, only the IVW method reached nominal significance (two-sided P = 0.003). The MR-Egger slope was negative, contrasting with the positive slopes from all other estimators. Error bars indicate the standard error of the beta coefficients. c) Funnel plot showing the relationship between individual SNP causal effects and their precision, revealing heterogeneity across variants. d) Leave-one-SNP-out analysis of the MDD → long-sleep association; each point and 95 % CI represent the IVW estimate obtained when excluding a single SNP. e) Forest plot of single-SNP causal effects with corresponding 95 % confidence intervals. f) MR-Egger intercept plot showing a significant positive intercept (0.03 ± 0.01; two-sided P = 0.047), indicating directional pleiotropy that may bias the overall causal estimate; error bar indicates the standard error of the estimated intercept. g) MR-PRESSO global test also detected horizontal pleiotropy (two-sided P = 0.002) but no outlier variants; consequently, the distortion test was not applicable. h) Cochran’s Q heterogeneity statistics for both IVW (two-sided P = 0.0009) and MR-Egger (two-sided P = 0.009) methods further confirmed substantial heterogeneity among the instruments. i) MR-RAPS models balanced (mean-zero) horizontal pleiotropy via an over-dispersion term and down-weights outliers using a robust loss, while profiling out weak-instrument bias. MR-RAPS point estimate remains positive but greatly attenuated versus IVW (two-sided P = 0.019), indicating a signal that persists under balanced-pleiotropy/weak-IV adjustments, though with increased uncertainty. Interpretation should note that MR-RAPS does not correct for directional pleiotropy; results are therefore contingent on the balanced-pleiotropy assumption. j) MRMix estimates the causal effect by modelling SNP residuals as a two-component mixture, a spike for valid IVs (no direct effect) and a normal component with variance σ² for invalid/pleiotropic IVs. By probabilistically allocating pleiotropic SNPs to the high-variance component, MRMix limits their influence on the slope and is robust to a large fraction of invalid instruments when pleiotropic effects are mean-zero on average. Here, the estimate is near-null/slightly negative (two-sided P = 0.41), with a CI spanning zero, providing no pleiotropy-robust evidence for a causal effect under the mixture model. To summarize, although IVW suggested a positive causal effect of genetic liability to MDD on long sleep (two-sided P = 0.003), multiple heterogeneity/pleiotropy diagnostics argue this is likely biased: the MR-Egger intercept is significant, the MR-PRESSO global test is positive, and Cochran’s Q indicates substantial heterogeneity. Pleiotropy-robust estimators do not corroborate the IVW signal. MR-Egger (slope) is null/negative (β = −0.36, two-sided P = 0.24), MRMix is near-null (β = −0.10, two-sided P = 0.41), and MR-RAPS, while largely attenuated and nominally positive (β = 0.22, two-sided P = 0.019), remains sensitive to pleiotropy assumptions. Taken together, the data do not provide pleiotropy-robust evidence for a causal effect; any apparent association should be viewed as suggestive at best and interpreted with caution. Error bars in panels f, j, i, and j represent the standard error of the estimated parameter, denoted by the dots in the centre.
Supplementary information
Supplementary Information (download PDF )
Supplementary Notes 1–5, Supplementary Figs. 1–22 and Supplementary Tables 1–8.
Supplementary Data (download XLSX )
Supplementary Data 1–9.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
The MULTI Consortium., O’Toole, C.K., Song, Z. et al. Sleep chart of biological ageing clocks in middle and late life. Nature (2026). https://doi.org/10.1038/s41586-026-10524-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41586-026-10524-5
