
Abstract
A polygenic score (PGS) for Alzheimer’s disease (AD) was derived recently from data on genome-wide significant loci in European ancestry populations. We applied this PGS to populations in 17 European countries and observed a consistent association with the AD risk, age at onset and cerebrospinal fluid levels of AD biomarkers, independently of apolipoprotein E locus (APOE). This PGS was also associated with the AD risk in many other populations of diverse ancestries. A cross-ancestry polygenic risk score improved the association with the AD risk in most of the multiancestry populations tested when the APOE region was included. Finally, we found that the PGS/polygenic risk score captured AD-specific information because the association weakened as the diagnosis was broadened. In conclusion, a simple PGS captures the AD-specific genetic information that is common to populations of different ancestries, although studies of more diverse populations are still needed to better characterize the genetics of AD.
Similar content being viewed by others


Main
Over the last 15 years, genome-wide association studies (GWASs) have fostered the development of powerful approaches for characterizing disease processes and the introduction of diagnostic/prognostic tools such as polygenic scores (PGSs)1,2. Given the high estimated heritability (60–80%, in twin studies) of Alzheimer’s disease (AD)3, a number of PGSs have been developed; associations with AD risk or related phenotypes have been described for almost all of the scores4,5,6,7,8,9,10. However, interstudy comparisons are complicated by marked differences in the populations analyzed, the PGS calculation methods, the summary statistics used and the variants included11. Furthermore, most PGSs have been developed from studies of European ancestry populations, and only a few studies have investigated PGSs performance in populations of different ancestries12,13,14,15.
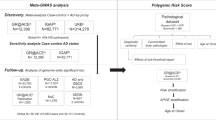
Here, we describe the generation of a PGS (PGSALZ) that includes the genome-wide significant, independent sentinel single nucleotide polymorphisms (SNPs) at the loci reported by Bellenguez et al.16, excluding the apolipoprotein E (APOE) locus (n = 83; see Supplementary Table 1 for the list of variants). We studied the associations between PGSALZ and AD risk or relevant endophenotypes in populations from 17 European countries and then extended the analysis to populations of diverse ancestries (from Asia, Africa, Latin America and North America). Finally, as already performed for other complex human diseases17,18,19,20, and with a view to improving the predictive performance of PGSALZ (refs. 2,21), we generated a cross-ancestry polygenic risk score (PRS) by integrating GWAS summary statistics from several populations.
We first evaluated the association between PGSALZ and AD risk in case–control studies of European countries (see Supplementary Table 2 for population description and adjustments used in each population and Supplementary Figs. 1–3 for PGSALZ distributions). PGSALZ was associated significantly with AD risk irrespective of APOE adjustment (Extended Data Fig. 1a and Supplementary Fig. 4). PGSALZ was similarly associated with AD risk in men and in women (Extended Data Fig. 1b and Supplementary Fig. 6). Furthermore, the score was associated with a younger age at onset (Extended Data Fig. 2). It is noteworthy that when the PGSs were adjusted for difference in PGSALZ distribution between the European populations, the association with AD remained similar (Supplementary Fig. 5).
As we did not identify any potential bias/heterogeneity when comparing PGSALZ in the European populations, we performed a combined analysis (mega-analysis) of our European datasets to assess the risk of developing AD within various PGSALZ strata: 0–2%, 2–5%, 10–20%, 20–40%, 60–80%, 80–90%, 90–95%, 95–98% and 98–100%, with the 40–60% PGSALZ stratum as the reference. We also generated a PGS that included both the sentinel AD GWAS loci and the two SNPs defining the ε2/ε3/ε4 APOE alleles. As expected, the risk of developing AD in the most extreme strata was particularly high when APOE was included (Fig. 1a). The association with PGSALZ was also significant in all strata analyzed, irrespective of APOE adjustment. In the 0–2% and 98–100% strata, PGSALZ was associated with a greater than twofold decrease in AD risk and a greater than threefold increase in AD risk, respectively, compared with the 40–60% stratum (Fig. 1a and Supplementary Table 3).

a, The risk of developing AD, by PGSALZ stratum (0–2%, 2–5%, 10–20%, 20–40%, 60–80%, 80–90%, 90–95%, 95–98% and 98–100%). The 40–60% PGSALZ stratum was used as the reference. b, Risk of developing AD, by PGSALZ stratum (0–20%, 20–40%, 60–80% and 80–100%) and by APOE genotype (by grouping together the ε2ε2/ε2ε3, ε3ε3, ε2ε4/ε3ε4 and ε4ε4 carriers). The 40–60% PGSALZ stratum was used as the reference. OR per s.d. was calculated by logistic regression adjusted for age, gender, 14 first PCs and chip center if necessary. The lines indicate the 95% CI of each OR. ε2ε2/ε2ε3 carriers (960 AD cases and 3,604 controls), ε3ε3 (15,623 AD cases and 17,782 controls), ε2ε4/ε3ε4 (8,780 AD cases and 6,242 controls) and ε4ε4 carriers (2,309 AD cases and 479 controls).
Since these results suggested that association of PGSALZ was independent of APOE, we leveraged our mega-analysis to determine how PGSALZ interacted with the APOE genotypes. We found a weak interaction between PGSALZ, the number of APOE ε4 alleles and AD risk (P = 3 × 10−4). Next, we stratified the mega-analysis into four APOE genotype groups (ε2ε2/ε2ε3, ε3ε3, ε2ε4/ε3ε4 and ε4ε4) and assessed the association between PGSALZ and AD risk per quintile (0–20%, 20–40%, 60–80% and 80–100%) for each subpopulation (reference, 40–60% stratum). PGSALZ was associated with AD risk to a similar extent in all strata, although a stronger association might be present among ε4ε4 carriers (Fig. 1b and Supplementary Table 4).
To determine whether PGSALZ is associated with AD pathophysiological processes, we analyzed GWAS data on CSF levels of Aβ42, tau and p-tau (n = 13,051 individuals), as described previously22. PGSALZ was associated with a decrement in Aβ42 levels and an increment in tau and p-tau levels, whatever the adjustment for APOE (Fig. 2a,b and Supplementary Fig. 7). We also checked for a possible association between PGSALZ and Aβ42 levels, tau and p-tau levels in quintiles (0–20%, 20–40%, 60–80% and 80–100%); again, the 40–60% stratum served as the reference. As expected, PGSALZ was associated with the lowest and highest levels of p-tau and Aβ42 in the 0–20% strata and, conversely, the highest and lowest levels of p-tau and Aβ42 in the 80–100% stratum (Fig. 2c and Supplementary Table 5).

a–c, Association of PGSALZ with the level of normalized Aβ42 (a) and p-tau (b) in cerebrospinal fluid (n = 13,004) across European ancestry populations and according to PGSALZ strata (0–20%, 20–40%, 60–80% and 80–100%) (c); the 40–60% PGSALZ stratum was used as the reference. β values were calculated by general linear model and logistic regression adjusted for APOE, age, gender, ten first PCs and chip center if necessary. The horizontal lines in the forest plots indicate the 95% CI of each β value. If heterogeneity P (HetP) < 0.05, a random effect is shown for the meta-analysis results. I2; heterogeneity.
We then extended the PGSALZ analyses to other European ancestry populations (United States, Australia), populations from India, East Asia (China, Japan and Korea), North Africa (Tunisia), sub-Saharan Africa (Central African Republic/the Congo Republic), South America (Argentina, Brazil, Chile and Colombia) and African American, Native American and Latin American ancestry populations from US studies (that is, more than 75% African American or Native American ancestry or self-reporting for Latin American populations; see Extended Data Fig. 3a and Supplementary Table 2 for a description of the population). With the exception of the analyses for Korea and Japan (where 72 and 74 SNPs, respectively, were available), most PGSs were built from 78 to 85 SNPs (including APOE variants; see Supplementary Table 1 and Supplementary Figs. 8–10 for PGSALZ distributions). The strength of the APOE ε4-AD association differed from one population to another, as observed previously23,24. The odds ratios (ORs) ranged from 1.36 in sub-Saharan Africa to 5.46 in North Africa (Extended Data Fig. 3b).
As expected, the association between PGSALZ and AD risk was strongest in European ancestry populations (United States and Australia). PGSALZ was also significantly associated with AD risk in North African, East Asian, Latin American and African American populations (Fig. 3a and Supplementary Fig. 11). Finally, PGSALZ was not associated with AD risk in the sub-Saharan African and Indian populations; this might be related to the small sample size and corresponding lack of statistical power. PGSALZ was associated with a younger age at onset in most of the populations studied, with the notable exception of the Chinese and Korean populations (Extended Data Fig. 4). Of note, the APOE ε2/ε3/ε4 alleles influenced age at onset in Chinese and Korean populations (Supplementary Fig. 12).

a, Association of PGSALZ with the risk of developing AD in multiancestry populations. The European ancestry meta-analysis includes MVP and Australia. The African American ancestry (more than 75% AA ancestry) meta-analysis includes MVP and ADSP. The East Asia meta-analysis includes China, Korea and Japan. The Latin American ancestry (self-reported) meta-analysis includes MVP and ADSP. The South America meta-analysis includes Argentina, Brazil, Chile and Colombia. b, The risk of developing AD, according to PGSALZ (logistic regression adjusted or not for APOE or included APOE variants) strata (0–20%, 20–40%, 60–80% and 80–100%) in multiancestry populations. The 40–60% PGSALZ stratum was used as the reference in each population, and results were meta-analyzed. The European ancestry meta-analysis includes MVP and Australia. The African American ancestry meta-analysis includes MVP and ADSP. The East Asia meta-analysis includes China, Korea and Japan. The Latin American ancestry meta-analysis includes MVP and ADSP. The South America meta-analysis includes Argentina, Brazil, Chile and Colombia. Ncases, number of cases; Ncontrols, number of controls. OR per s.d. was calculated by logistic regression adjusted for APOE, age, sex and specific PCs according to the study (Supplementary Table 2). The lines in the Forest plots indicate the 95% CI of each OR. If HetP < 0.05, a random effect is shown for the meta-analysis results. AA, African American; EUR, European; LA, Latin American.
To refine our analysis of these populations of diverse ancestries, we calculated the association between AD and PGSALZ quintiles (0–20%, 20–40%, 60–80% and 80–100%; reference, 40–60%) and meta-analyzed them by ancestry (Fig. 3b,c and Supplementary Tables 6 and 7). The Indian, North African and sub-Saharan African populations were excluded because of the small sample size. The strength of the association with PGSALZ decreased from the European American, East Asian and Latin American populations to the African American population, in that order (Fig. 3b and Supplementary Table 6). PGSALZ generated from a European ancestry population GWAS performed poorly in African ancestry populations.
The latter observation was strengthened by analyzing the association between PGSALZ and AD risk as a function of the African American admixture. The strength of the association decreased as the percentage of African ancestry increased, and ultimately reached a level similar to that observed in our sub-Saharan African population: the association between PGSALZ and AD risk in populations in whom more than 90% of the members were of African ancestry had an OR of 1.09 (95% confidence interval (CI) 0.98–1.21; P = 1.4 × 10−1, adjusted for APOE). Of note, a similar pattern was observed in the Native American population of the Alzheimer Disease Sequencing Project: the strength of the association decreased as the Native American ancestry percentage increased, from OR = 1.21 (95% CI, 1.12–1.32; P = 5.3 × 10−6) and OR = 1.14 (95% CI, 1.05–1.25; P = 2.6 × 10−3) to OR = 1.12 (95% CI, 1.02–1.24; P = 1.4 × 10−2 in the populations with more than 50%, 75% and 90% of individuals of Native American ancestry, respectively, after adjustment for APOE. A similar result was found for Chilean and Argentinian populations: the PGSALZ association weakened as the proportion of individuals with Native American ancestry rose14.
We next checked that we had fully captured the genetic information in the GWAS-defined loci in the non-European populations. To this end, we developed a PGS (PGSALZ+) that included other SNPs associated with AD risk in non-European multiancestry populations (P < 10−3) at the European GWAS-defined loci (Methods). We used the summary statistics generated by Kunkle et al.25, Lake et al.26 and Shigemizu et al.27, and added 30, 13 and 47 variants to the initial 83 PGSALZ variants for Latin American, East Asian and African American ancestries, respectively (Supplementary Table 8). We did not detect any increment in (1) the strength of the PGSALZ+ association with the AD risk or (2) PGSALZ+’s predictive performance, relative to PGSALZ (Supplementary Table 9).
By initially restricting our analyses to the genome-wide significant loci from European ancestry AD GWAS, we probably excluded genetic information associated with AD risk in both European populations and (especially) non-European multiancestry populations (for which ancestry-specific loci may exist). Furthermore, the effect sizes used to construct PGSALZ were extracted from European ancestry populations without taking account of population differences. To deal with these various questions, we used the Bayesian polygenic modeling method PRS-CSx to build a cross-ancestry PRS20. The PRS re-estimates variant effect sizes by coupling various summary statistics with external ancestry-matched allele frequencies and local linkage disequilibrium structure, according to the sparseness of the genetic architecture of AD. We used GWAS summary statistics generated from European (36,569 AD cases and 63,137 controls), African American (2,784 AD cases and 5,222 controls), Latin American (1,088 AD cases and 1,152 controls) and East Asian (3,962 AD cases and 4,074 controls) populations25,26,27. PRSs (all adjusted for the population structure) were generated in multiancestry populations from the Million Veteran Program (MVP; European American, Latin American and African American ancestries), EPIDEMCA (sub-Saharan Africa ancestry) and GARD studies (East Asian ancestry; Supplementary Fig. 13).
We assessed potential increments in the association of PRS with the AD risk and in predictive performance when the summary statistics of the European American, African American, Latin American or East Asian multiancestry populations were applied independently (PRSEUR, PRSAA, PRSLA and PRSEA, respectively) or when the statistics were combined (PRSCOMB) at various sparseness values (10−8, 10−7, 10−6, 10−5, 10−4, 10−2 and 1). We initially excluded the APOE region, to facilitate the comparison with PGSALZ. We did not observe any increases in the association with AD risk or in predictive performance in the different multiancestry populations (Fig. 4, Supplementary Fig. 14 and Supplementary Table 10), with the exception of the Latin American MVP population. However, we cannot rule out overfitting as the reason for this improvement. Next, we included the APOE region when generating the different PRSs. Whereas no impact on European ancestry populations was observed when comparing PRSEUR and PRSCOMB, we detected an increment in both the strength of association with the AD risk and in the predictive performance when comparing PRSEUR and PRSCOMB for all other populations. This indicated that a cross-ancestry PRS is more effective than a PRS constructed solely from European summary statistics when the APOE region is included, whatever the overall shrinkage value used (Fig. 5, Supplementary Fig. 14 and Supplementary Table 10).

All PGSALZ and PRS values were adjusted for interpopulation differences in distribution; PRSEUR were generated by using only European ancestry summary statistics; PRSCOMB were generated by combining European, African American, Latin American and East Asian ancestry summary statistics. The sparseness parameter was set to 10−8, 10−7, 10−6, 10−5, 10−4, 10−2 or 1. OR per s.d. was calculated by logistic regression adjusted for age, sex and specific PCs according to the study (Supplementary Table 2). MVP EUR (4,561 AD cases and 84,587 controls), MVP LA (375 AD cases and 7,166 controls), MVP AA (713 AD cases and 19,405 controls) and South Korea (1,119 AD cases and 1,172 controls).

All PRS were adjusted for interpopulation differences in distribution; PRSEUR were generated by using only European ancestry summary statistics; PRSCOMB were generated by combining European, African American, Latin American and East Asian ancestry summary statistics. The sparseness parameter was set to 10−8, 10−7, 10−6, 10−5, 10−4, 10−2 or 1. OR per s.d. was calculated by logistic regression adjusted for age, sex and specific PCs according to the study (Supplementary Table 2). MVP EUR (4,561 AD cases and 84,587 controls), MVP LA (375 AD cases and 7,166 controls), MVP AA (713 AD cases and 19,405 controls) and South Korea (1,119 AD cases and 1,172 controls).
Finally, we leveraged the MVP data to determine how the association between PGSALZ or PRSCOMB (without the APOE region) and AD risk changed in multiancestry populations as a function of diagnostic specificity. We looked at how a PGSALZ/PRSCOMB derived from AD case/control studies performed when the diagnosis was broadened to dementia. In all the multiancestry population studied, the association between PGSALZ/PRSCOMB and AD risk weakened as the diagnosis became broader (Fig. 6 and Supplementary Table 11).

PGSALZ and PRSCOMB were adjusted for interpopulation differences in distribution; PRSCOMB were generated by combining European, African American and Latin American and East Asian ancestry summary statistics. The sparseness parameter was set to 10−8 and 10−6. OR per s.d. was calculated by logistic regression adjusted for age, sex and specific PCs according to the study (Supplementary Table 2). MVP EUR (4,561 AD, 17,519 ADRD, 26,473 dementia cases and 84,587 controls), MVP LA (375 AD; 1,527 ADRD; 1,981 dementia cases and 7,166 controls), MVP AA (713 AD; 4,016 ADRD; 4,702 dementia cases and 19,405 controls).
Our work produced several important findings. First, the associations between PGSALZ and AD risk in European populations may be influenced slightly by the APOE genotype; this suggests the existence of two independent genetic entities for sporadic AD: one associated with APOE ε4 and the other not, as suggested previously28. Second, the simple PGSALZ (based on the European GWAS-defined loci) seems to be enough to detect an AD genetic risk in most ancestry populations. Our results thus suggest that most of the various ancestry populations are likely to be affected by shared pathophysiological processes that are driven in part by genetic risk factors. Third, in contrast to what has been observed in the genetics of complex traits29 and other multifactorial diseases17,30,31, a cross-ancestry PRS built with a Bayesian polygenic modeling method did not systematically outperform a simple PGSALZ when the APOE locus was excluded. This observation might be due to the small population size of GWAS for the various ancestry populations, which can significantly limit the power of the PRS-CSx approach. However, this might also indicate that a high proportion of AD genetic risk is already accounted for by the European ancestry GWAS-defined loci. Fourth, the APOE region appears to contain additional multiancestry genetic variability, as suggested previously32,33,34,35. Finally, the PGS/PRS associations capture mainly genetic information related to AD because they weakened as the diagnosis was broadened. This observation suggests that the quality of the clinical diagnosis can interfere with the measurement of the association between the PGS/PRS and the AD risk in a given population.
In conclusion, our study of diverse ancestry populations and AD highlights the importance of cross-ancestry analyses for characterizing the genetic complexities of this disease. However, the AD genetics field is still limited by the size of GWASs in these diverse ancestry populations. Furthermore, it is likely that different ancestry populations will differ strongly regarding rare/very rare variants associated with AD risk; this would significantly impact the association of PRSs with AD risk and their predictive abilities36. Better characterization of AD genetics thus requires both GWASs and sequencing studies of more diverse populations.
Methods
Sample and variant quality controls
Written informed consent was obtained from study participants or, for those with substantial cognitive impairment, a caregiver, legal guardian or other proxy. Study protocols for all cohorts were reviewed and approved by the appropriate institutional review boards (Supplementary Information).
To ensure that the β values were completely independent of the summary statistics, all samples from ADGC, CHARGE and FinnGen GWASs were filtered out. Sample overlap was assessed systematically, and there was no sample overlap between any of the non-US studies analyzed. Overlap between Alzheimer's Disease Sequencing Project ADSP and MVP is likely to be negligible—no more than a few cases. For the biomarker analysis, there is a 460-sample overlap between the American samples used in the biomarker analyses and the ADGC (which is included in the summary statistics we used to generate the β values for the PGSALZ). However, this overlap is small (less than 2.5%). Furthermore, we analyzed the association of PGSALZ only with quantitative traits (p-tau, tau and Aβ42 CSF concentrations) in these samples, which limited the risk of inflation.
After each sample had met the conventional GWAS gold standard for quality control, it was included in the analyses16. If a discordance in a variant dose, covariate or APOE status (the difference between the imputation and the genotyping results (if available)) was observed, the sample was discarded. After the quality control, each study’s demographics were described (Supplementary Table 1)37. Genotyped variants had to meet the gold standard for GWAS variant quality control16. All studies containing genotyping data were imputed with the TOPmed reference panel37,38. If the variants were imputed, those with an R2 value below 0.3 were excluded. For whole-genome sequencing data, only variants passing the corresponding quality control were selected (see the Supplementary Information for the ADSP and China samples) (Supplementary Table 2). The global ancestry of each person in the ADSP samples was determined with SNPweights v.2.1 (ref. 39) using a set of ancestry-weighted variants computed on reference populations from the 1000 Genomes Project (as in ref. 40). By applying a global ancestry percentage cutoff of >75%, the samples were assigned to the different ancestry populations. The ancestry of MVP participants was determined using the harmonized ancestry and race/ethnicity (HARE) method41. HARE is like other genotype-based ancestry calling methods, except that concordance between self-reported ancestry and genetically inferred ancestry is checked. Participants with discrepant ancestry calls are not assigned to a HARE category. Within-group principal components (PCs) for ancestry were computed using FlashPCA2 (ref. 42).
Mega-analysis of European populations
We merged samples from five datasets: EADB-core, GERAD, EADI, Demgene and Bonn. To adjust for population structure, we computed PCs using the following procedure. From the list of 146,705 variants used in the PC analysis of EADB-core42, we extracted the TOPMed imputed variants with an imputation quality ≥0.9 in each dataset; this resulted in 91,353 variants. Next, we set a genotype to ‘missing’ if none of the genotype probabilities were greater than 0.8. Finally, all datasets were merged, and variants with a proportion of missing genotypes greater than 0.02 were removed. Ultimately, 90,471 variants were included in the PC analysis (performed with FlashPCA2). The analyses were adjusted for the first 14 PCs, the genotyping chip and the center.
PGS and PRS computations
All codes for PGS and PRS analyses have been made available43. The equation used to calculate the PGSs and the PRSs is as follows:
where the \({{\rm{PGS}}}_{{\rm{sample}}}^{{\rm{ALZ}}}\) PRSsample is the sum per sample of the product of the variant i effect size βi (extracted from GWAS summary statistics) and the number of risk alleles of this variant i (either as a dosage or as a genotype).
PGSALZ includes the 83 independent signals associated with AD13 and listed in Supplementary Table 1. We also calculated another PGSALZ combining the same 83 independent signals and the two SNPs encoding the APOE ε2 (rs7412) and APOE ε4 alleles (rs429358). PGSAPOE includes only these two last SNPs. The stage I meta-analysis of EADB studies13 (without the United Kingdom (UK) Biobank samples) contained 36,659 clinically diagnosed AD cases, and the stage II meta-analysis (including the ADGC, CHARGE and FinnGen data) contained 25,392 (ref. 13). To ensure independence between the samples and the GWAS summary statistics, the European summary statistics used in the PGS analyses were from stage II. In the PGSALZ/PRS analyses adjusted for the difference in distribution between populations, the European more powerful summary statistics (that is, the stage I meta-analysis of EADB) were preferred.
The PGSALZ+ score was developed to include additional SNPs in the GWAS-defined loci, to capture more genetic information in non-European ancestry populations. First, the ‘start and end positions’ of each locus (as specified in the GRCh38 assembly) were defined manually by looking at the regional plots and extracting (1) recombination rate peak positions, (2) chromosome start and end positions, (3) specific variant positions or (4) the start/end positions of regions containing no variants. Next, insertions and deletions were excluded. Variants that were not ambiguous (that is, A/T or C/G) and present in the 1000 Genomes Phase 3 data (1000GP3) and had an imputation quality above 0.3 in the EADB-core TOPMed imputations were selected. To extract information on these variants in non-European ancestry populations, we used the summary statistics generated by Lake et al., Shigemizu et al. and Kunkle et al. to represent Latin American, East Asian and African American ancestries, respectively25,26,27. Since these summary statistics were based on the GRCh37 assembly, we lifted their positions and alleles in the GRCh38 assembly by using the Picard LiftoverVcf tool (v.2.27.5) and restricting the process to variants with a minor allele frequency above 0.01. To remove variants in linkage disequilibrium with the sentinel variant of each locus, we computed the linkage disequilibrium for each sentinel variant versus all the other variants in the locus by using the 1000GP3 data restricted to samples representing European ancestries (the EUR superpopulation), Latin American ancestries (the AMR superpopulation plus the IBS population), Japanese ancestries (the JPT population) and African American ancestries (the AFR superpopulation). Since one of the sentinel variants (chr. 9:104903697:C:G) was not present in the 1000GP3 data, we replaced it with a proxy variant (chr. 9:104903754:G:GC, R2 = 1 in the EUR superpopulation). In each set of summary statistics, we removed variants with R2 > 0.1 in either the European summary statistics or the summary statistics for the corresponding ancestry. Finally, we performed a clumping procedure on the remaining variants in each of the three ancestries by using plink v.1.9, a P value threshold of 1 × 10−3, an R2 of 0.05 (as estimated in the corresponding 1000GP3 data samples, as described above) and a distance of 1 Mb. For the PGSALZ+, this led us to select 30, 13 and 47 variants (in addition to the initial 85 PGS variants) for the Latin American, East Asian and African American ancestries, respectively.
At the time of our analysis, PRS-CSx20,44 was one of the best-performing methods for modeling a cross-ancestry PRS45,46 without a validation dataset and using GWAS summary statistics. With a Bayesian high-dimensional regression framework model based on continuous shrinkage priors, the variant effect sizes were adaptively re-estimated by coupling cross-ancestry GWAS summary statistics13,25,26,27, external ancestry-matched allele frequencies and local linkage disequilibrium structure, according to a global shrinkage parameter. This global shrinkage parameter corresponded to the sparseness of the genetic architecture of AD by avoiding overshrinkage of true signals and by shrinking noisy signals. The sparseness was modeled for the values of 1, 10−2, 10−4, 10−5,10−6, 10−7 and 10−8, with the --meta option and the Strawderman–Berger prior default parameters (a = 1 and b = 0.5). The initial 1,297,432 variants present in the 1000 Genomes reference panel were lifted over in GRCh38. Next, new ancestry-specific or joint-ancestry effect size estimates were obtained with PRS-CSx, leading to a maximum number of 1,292,532 variants in the joint-ancestry summary statistics and potentially included in the PRS computations. The PRSs were computed per chromosome with joint-ancestry, European ancestry and ancestry-specific PRS-CSx-effect size estimates, using PLINK (v.2.0.a) software47 and its --score option. Finally, the PRSs were summed across all chromosomes.
Adjustment for interpopulation differences in the PGSALZ/PRS distribution
To account for the population structure, PRSraw and PGSALZraw were adjusted for interpopulation differences in distribution48. The adjustment was performed with a selection of 84,035 independent and well-imputed (R > 0.8) variants common to all studies. Starting from this list of variants, FlashPCA2 projected the samples into the 1000GP3 PC-space and calculated the projected PCs. For each study, the raw score was fitted into a linear model in controls, according to the first five projected PCs. This model was used to compute a predicted score in all the samples. The resulting adjusted score was the difference between the raw score and the predicted score.
Statistical analyses
The PGSs and PRSs were standardized to a normal distribution, using the mean and s.d. calculated for the samples as a whole. The associations between AD status and the various scores were tested in logistic regressions named according to the score and the covariates used. Hence, the name ‘ALZinclAPOE’ was attributed if the score included variants in the APOE region (from 43 Mb to 47 Mb). The other covariates included age and sex, as well as the covariates specific to each study (Supplementary Table 2).
-
Model PGSALZ: AD ~ PGSALZ + COV
-
Model PGSALZ: AD ~ PGSALZ + COV + the count of APOE ε2 alleles + the count of APOE ε4 alleles (when adjusted for APOE)
-
Model PRS: AD ~ PRS + COV
-
Model PRS: AD ~ PRS + COV + the count of APOE ε2 alleles + the count of APOE ε4 alleles (when adjusted for APOE)
-
Model PRSALZinclAPOE: AD ~ PRSALZinclAPOE + COV
To estimate the proportion of phenotypic variance explained by the variance in the score, we computed Nagelkerke’s Pseudo-R2Full using the Nagelkerke function implemented in the rcompanion package in R49,50. A Pseudo-R2Null was also computed for the covariates only. The adjusted Pseudo-R2 is the difference between Pseudo-R2Full and the tied Pseudo-R2Null. This adjusted Pseudo-R2 corresponds to the phenotypic variance explained by the genetic score only. The adjusted Pseudo-R2 was also transformed into a liability scale for ascertained case–control studies51, using a prevalence value of 0.15. We consider this value of 0.15 to be consistent for populations with a mean age greater than 75 years. However, this prevalence is different in multiethnic populations of the same mean age. Furthermore, the AD prevalence increases with age, so genetic liability is not homogeneous in all age groups. AD heritability cannot be expressed as a single number because it depends on the ages of the cases and controls52.
Quantile and percentile analyses
Depending on the value of the corresponding PGSALZ, the samples were classified into the reference group or into one of the test groups. In the mega-analysis, the reference group corresponded to the 40–60% percentile and was tested across other percentiles (0–2%, 2–5%, 5–10%, 10–20%, 20–40%, 60–80%, 80–90%, 95–98% and 98–100%). In the APOE-stratified analysis and in the multiancestry analyses, the reference group was defined as the 40–60% percentile and was tested across the other quintiles (0–20%, 20–40%, 60–80%, 80–100%). The multiancestry analyses were performed on each population and then meta-analyzed per genetic ancestry by using the inverse variance method, as implemented in METAL53. It should be noted that the Indian, North African and sub-Saharan African populations were excluded because of their small sample size.
-
Model PGSALZ: AD ~ Group0/1(PGSALZ) + COV
-
Model PGSALZ: AD ~ Group0/1(PGSALZ) + COV + number of APOE ε2 alleles + number of APOE ε4 alleles (when adjusted for APOE)
-
Model PGSALZinclAPOE: AD ~ Group0/1(PGSALZinclAPOE) + COV
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The EADB GWAS (without UK biobank) summary statistics used to develop PRS have been deposited with the European Bioinformatics Institute GWAS Catalog under accession no. GCST90565439. Summary statistics from African American multiancestry population used to develop PRS were accessed through NIAGADS under accession number NG00100. Summary statistics from Japan populations were accessed through the National Bioscience Database Center (NBDC) at the Japan Science and Technology Agency (JST) with accession number hum0237.v1.gwas.v1. 1000GP3 data is available at http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/1000_genomes_project/release/20190312_biallelic_SNV_and_INDEL/). GRCh37 assembly data is available at https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/001/405/GCF_000001405.25_GRCh37.p13/GCF_000001405.25_GRCh37.p13_genomic.fna.gz. GRCh38 assembly data is available at https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/001/405/GCF_000001405.39_GRCh38.p13/GCF_000001405.39_GRCh38.p13_genomic.fna.gz. ADSP data is available at https://dss.niagads.org/datasets/ng00067/.
Code availability
All codes developed and shared with collaborators to run PGS and PRS are available via Zenodo at https://doi.org/10.5281/zenodo.15164089 (ref. 43). Based on IRB and protected status of the Latin American population in dbGaP access process for this data, the summary statistics of the Latin American GWAS cannot be shared. The code to generate it as well as the mandated dbGaP link are respectively available here: https://github.com/NIH-CARD/MA_MA_meta and https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000496.v1.p1. SNPweights v.2.1. is available at https://hsph.harvard.edu/research/price-lab/software/. FlashPCA2 is available at https://github.com/gabraham/flashpca. Picard LiftoverVcf tool (v.2.27.5) is available at https://broadinstitute.github.io/picard/. plink v.1.9 is available at https://www.cog-genomics.org/plink2/. PLINK (v.2.0.a) is available at https://www.cog-genomics.org/plink/2.0/. rcompanion package is available at https://cran.r-project.org/web/packages/rcompanion/. METAL v2020–05–05 is available at https://github.com/statgen/METAL.
References
Osterman, M. D., Kinzy, T. G. & Bailey, J. N. C. Polygenic risk scores. Curr. Protoc. 1, e126 (2021).
Kachuri, L. et al. Principles and methods for transferring polygenic risk scores across global populations. Nat. Rev. Genet. 25, 8–25 (2024).
Gatz, M. et al. Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry 63, 168–174 (2006).
Baker, E. & Escott-Price, V. Polygenic risk scores in Alzheimer’s disease: current applications and future directions. Front. Digit. Health 2, 14 (2020).
Desikan, R. S. et al. Genetic assessment of age-associated Alzheimer disease risk: development and validation of a polygenic hazard score. PLoS Med. 14, e1002258 (2017).
Sabuncu, M. R. et al. The association between a polygenic Alzheimer score and cortical thickness in clinically normal subjects. Cereb. Cortex 22, 2653–2661 (2012).
Mormino, E. C. et al. Polygenic risk of Alzheimer disease is associated with early- and late-life processes. Neurology 87, 481–488 (2016).
Xicota, L. et al. Association of APOE-independent Alzheimer disease polygenic risk score with brain amyloid deposition in asymptomatic older adults. Neurology 99, e462–e475 (2022).
Sleegers, K. et al. A 22-single nucleotide polymorphism Alzheimer’s disease risk score correlates with family history, onset age, and cerebrospinal fluid Aβ42. Alzheimers Dement. 11, 1452–1460 (2015).
Hong, S. et al. Genome-wide association study of Alzheimer’s disease CSF biomarkers in the EMIF-AD multimodal biomarker discovery dataset. Transl. Psychiatry 10, 403 (2020).
Clark, K., Leung, Y. Y., Lee, W.-P., Voight, B. & Wang, L.-S. Polygenic risk scores in Alzheimer’s disease genetics: methodology, applications, inclusion, and diversity. J. Alzheimers Dis. 89, 14 (2022).
Sariya, S. et al. Polygenic risk score for Alzheimer’s disease in Caribbean Hispanics. Ann. Neurol. 90, 366–376 (2021).
Jung, S.-H. et al. Transferability of Alzheimer disease polygenic risk score across populations and its association with Alzheimer disease-related phenotypes. JAMA Netw. Open 5, e2247162 (2022).
Dalmasso, M. C. et al. The first genome-wide association study in the Argentinian and Chilean populations identifies shared genetics with Europeans in Alzheimer’s disease. Alzheimers Dement. 20, 1298–1308 (2024).
Kikuchi, M. et al. Polygenic effects on the risk of Alzheimer’s disease in the Japanese population. Alzheimers Res. Ther. 16, 45 (2024).
Bellenguez, C. et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 54, 412–436 (2022).
Koyama, S. et al. Population-specific and trans-ancestry genome-wide analyses identify distinct and shared genetic risk loci for coronary artery disease. Nat. Genet. 52, 1169–1177 (2020).
Lennon, N. J. et al. Selection, optimization and validation of ten chronic disease polygenic risk scores for clinical implementation in diverse US populations. Nat. Med. 30, 480–487 (2024).
Keaton, J. M. et al. Genome-wide analysis in over 1 million individuals of European ancestry yields improved polygenic risk scores for blood pressure traits. Nat. Genet. 56, 778–791 (2024).
Ge, T. et al. Development and validation of a trans-ancestry polygenic risk score for type 2 diabetes in diverse populations. Genome Med. 14, 70 (2022).
Xiang, R. et al. Recent advances in polygenic scores: translation, equitability, methods and FAIR tools. Genome Med. 16, 33 (2024).
Jansen, I. E. et al. Genome-wide meta-analysis for Alzheimer’s disease cerebrospinal fluid biomarkers. Acta Neuropathol. 144, 821–842 (2022).
Logue, M. W., Dasgupta, S. & Farrer, L. A. Genetics of Alzheimer’s disease in the African American population. J Clin. Med. 12, 5189 (2023).
Miyashita, A., Kikuchi, M., Hara, N. & Ikeuchi, T. Genetics of Alzheimer’s disease: an East Asian perspective. J. Hum. Genet. 68, 115–124 (2023).
Kunkle, B. W. et al. Novel Alzheimer disease risk loci and pathways in African American individuals using the African Genome Resources Panel: a meta-analysis. JAMA Neurol. 78, 102–113 (2021).
Lake, J. et al. Multi-ancestry meta-analysis and fine-mapping in Alzheimer’s disease. Mol. Psychiatry 28, 3121–3132 (2023).
Shigemizu, D. et al. Ethnic and trans-ethnic genome-wide association studies identify new loci influencing Japanese Alzheimer’s disease risk. Transl. Psychiatry 11, 151 (2021).
Frisoni, G. B. et al. The probabilistic model of Alzheimer disease: the amyloid hypothesis revised. Nat. Rev. Neurosci. 23, 53–66 (2022).
Wang, Y. et al. Polygenic prediction across populations is influenced by ancestry, genetic architecture, and methodology. Cell Genomics 3, 100408 (2023).
Smith, J. L. et al. Multi-ancestry polygenic risk score for coronary heart disease based on an ancestrally diverse genome-wide association study and population-specific optimization. Circ. Genom. Precis. Med. 17, e004272 (2024).
Khan, A. et al. Genome-wide polygenic score to predict chronic kidney disease across ancestries. Nat. Med. 28, 1412–1420 (2022).
Naslavsky, M. S. et al. Global and local ancestry modulate APOE association with Alzheimer’s neuropathology and cognitive outcomes in an admixed sample. Mol. Psychiatry 27, 4800–4808 (2022).
Rajabli, F. et al. Ancestral origin of ApoE ε4 Alzheimer disease risk in Puerto Rican and African American populations. PLoS Genet. 14, e1007791 (2018).
Bussies, P. L. et al. Use of local genetic ancestry to assess TOMM40-523’ and risk for Alzheimer disease. Neurol. Genet. 6, e404 (2020).
Rajabli, F. et al. A locus at 19q13.31 significantly reduces the ApoE ε4 risk for Alzheimer’s disease in African Ancestry. PLoS Genet. 18, e1009977 (2022).
Nagao, Y. Contribution of rare variants to heritability of a disease is much greater than conventionally estimated: modification of allele distribution model. J. Hum. Genet. 69, 663–668 (2024).
Das, S. et al. Next-generation genotype imputation service and methods. Nat. Genet. 48, 1284–1287 (2016).
Taliun, D. et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed program. Nature 590, 290–299 (2021).
Chen, C.-Y. et al. Improved ancestry inference using weights from external reference panels. Bioinformatics 29, 1399–1406 (2013).
Le Guen, Y. et al. Multiancestry analysis of the HLA locus in Alzheimer’s and Parkinson’s diseases uncovers a shared adaptive immune response mediated by HLA-DRB1*04 subtypes. Proc. Natl Acad. Sci. USA 120, e2302720120 (2023).
Fang, H. et al. Harmonizing genetic ancestry and self-identified race/ethnicity in genome-wide association studies. Am. J. Hum. Genet. 105, 763–772 (2019).
Abraham, G., Qiu, Y. & Inouye, M. FlashPCA2: principal component analysis of Biobank-scale genotype datasets. Bioinformatics 33, 2776–2778 (2017).
Nicolas, A. Transferability of European-derived Alzheimer’s disease polygenic risk scores across multi-ancestry populations. Zenodo https://doi.org/10.5281/zenodo.15164089 (2025).
Ruan, Y. et al. Improving polygenic prediction in ancestrally diverse populations. Nat. Genet. 54, 573–580 (2022).
Ma, Y. & Zhou, X. Genetic prediction of complex traits with polygenic scores: a statistical review. Trends Genet. 37, 995–1011 (2021).
Kurniansyah, N. et al. Evaluating the use of blood pressure polygenic risk scores across race/ethnic background groups. Nat. Commun. 14, 3202 (2023).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015).
Hao, L. et al. Development of a clinical polygenic risk score assay and reporting workflow. Nat. Med. 28, 1006–1013 (2022).
Lee, S. H., Goddard, M. E., Wray, N. R. & Visscher, P. M. A better coefficient of determination for genetic profile analysis. Genet. Epidemiol. 36, 214–224 (2012).
Choi, S. W., Mak, T. S.-H. & O’Reilly, P. F. Tutorial: a guide to performing polygenic risk score analyses. Nat. Protoc. 15, 2759–2772 (2020).
Lee, S. H. & Wray, N. R. Novel genetic analysis for case-control genome-wide association studies: quantification of power and genomic prediction accuracy. PLoS ONE 8, e71494 (2013).
Baker, E. et al. What does heritability of Alzheimer’s disease represent? PLoS ONE 18, e0281440 (2023).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Acknowledgements
We thank all the study participants, researchers and staff for contributing to, or collecting, the data. We also thank the staff at the University of Lille’s high-performance computing service. This work was funded by a grant (European Alzheimer&Dementia DNA BioBank, EADB) from the EU Joint Programme—Neurodegenerative Disease Research (JPND) and La Fondation Recherche Alzheimer. This work was supported by Mécénat Mutuelles AXA. A.N. was funded by La Fondation pour la Recherche Médicale (EQU202003010147) and La Fondation Recherche Alzheimer. UMR1167 is also funded by the INSERM, Institut Pasteur de Lille, Lille Métropole Communauté Urbaine and the French government’s LABEX DISTALZ program (development of innovative strategies for a transdisciplinary approach to Alzheimer’s disease). Full consortium acknowledgements and funding are given in the Supplementary Note. We thank D. Fraser (Biotech Communication SARL, Ploudalmézeau, France) for editorial assistance.
Author information
Authors and Affiliations
Consortia
Contributions
A.N. and J.-C.L. coordinated the project. A.N., Y.LG., J.G., M.D.G., S.v.d.L, E.N.D.M., J.-F.D., H.A., V.E.-P., A. Ruiz, K.H.L., T.I., A. Ramirez, M.L. and J.-C.L. coordinated data collection. A.N., R. Sherva, B.G.-B., Y.K., M.K., J.T., I.D.R., C.D., X.Z., Y.L.G., C.E.A.-B., M.A.C.B., M. Guerchet, S.v.d.L., M. Goss, A.C., C.B. and F.K. analyzed the data. I.d.R., A.C., S.v.d.L., C.B., F.K., O.P., A. Schneider, M.D., D.R., N. Scherbaum, J.D., S.R.-H., L.H., L.M.-P., E.D., T.G., J. Wiltfang, S.H.-H., S. Moebus, T.T., N. Scarmeas, O.D.-I., F.M., J.P.-T., M.J.B., P.P., R.S.-V., V.Á., M.B., P.G.-G., R. Puerta, P. Mir, L.M.R., G.P.-R., J.M.G.-A., J.L.R., E.R.-R., H. Soininen, T.K., A.d.M., S. Mehrabian, J. Hort, M.V., K.L.R., J.Q.T., Y.A.L.P., H.H. J.C.v.S., H. Seelaar, J.A.H.R.C., W.J.S., I. Ramakers, F.V., A.v.d.L. P. Scheltens, S.B., V.F., G.S., C.G., G.P., V.G., G.N., C. Dufouil, F.P., O.H., S.D., A.B., J.-F. Deleuze, E.G., J.P., P. Sachdev, K.A.M., D.G., B. Arosio, P. Mecocci, V.S., L.P., A. Squassina, L.T., B. Borroni, B.N., P.C., D.S., I. Rainero, A. Daniele, J. Williams, C. Masullo, P.A., F.J., P.K., C.V.D., R.F.-S., M.T., P.S.-J., K.S., M.I., G.R., M.H., R. Sims, W.v.d.F., O.A.A., A. Ruiz, A. Ramirez and J-C.L. contributed to EADB sample collection, T.P. and S.M.L. provided the Australian sample. R. Sherva, R.L.H., V.M., M.P., R.Z., J.M.G., C.L.L. and M.L. contributed the MVP sample. M. Goss, C.L.B., B.F., Q.Y., A.J.G., T.F., J. Haines, L.F., A. DeStefano, E.W., R.M., M. P.-V., B.K., A. Goate, G.D.S., B.V., L.-S.W., Y.Y.L., C.L.D., A. Saykin, H.L.L., J.S.Y., M.A.N., S.S. and C. Cruchaga provided US populations. M. Guerchet, P.-M.P., P. Mbelesso, B. Bandzouzi, N.B.S., L. Cherni and J.-F. Dartigues contributed the African sample. Y.K., M.K., X.Z., H.C., N.Y.I., A.K.Y.F., F.C.F.I., A.M., N.H., K.O., S.N., J.G., V.E.-P., K.H.L. and T.I. contributed the East Asia sample. M.C.D., C.E.A.-B., M.A.C.B., N.O., T.J.-C., C. Muchnik, C. Cuesta, L. Campanelli, P. Solis, D.G.P., S.K., L.I.B., J.O.-R., A.G.C.M., M.F.M., R. Pardo, G.A., L.A.d.M., M.A.R.S., B.d.M.V., M.T.G.C., T.J.-C., B. Angel, S.G., M.V.C., R.A., P.O., A. Slachevsky, C.G.-B., C.A., P.F., E.N.d.M., L.M., H.A., A. Ruiz and A. Ramirez contributed the South America sample. The core writing group were A.N., B.G.-B. and J.-C.L.
Corresponding authors
Ethics declarations
Competing interests
S.v.d.L. is a recipient of funding from ABOARD, which is a public–private partnership financed by ZonMW (no. 73305095007) and Health–Holland, Topsector Life Sciences & Health (PPP-allowance; no. LSHM20106). C.C. has received research support from GSK and EISAI. The study’s funders had no role in the collection, analysis or interpretation of data; in the writing of the report or in the decision to submit the paper for publication. C.C. is an advisory board member for Vivid Genomics and Circular Genomics and owns stock. L.M.-P. received personal fees from Biogen for consulting activities unrelated to the submitted work. T.G. received consulting fees from AbbVie, Alector, Anavex, Biogen, Cogthera, Eli Lilly, Functional Neuromodulation, Grifols, Iqvia, Janssen, Noselab, Novo Nordisk, NuiCare, Orphanzyme, Roche Diagnostics, Roche Pharma, UCB and Vivoryon; lecture fees from Biogen, Eisai, Grifols, Medical Tribune, Novo Nordisk, Roche Pharma, Schwabe and Synlab; and has received grants to his institution from Biogen, Eisai and Roche Diagnostics. O.A.A. is a consultant to Cortechs and Precision Health AS, and has received speaker’s honoraria from Lundbeck, Sunovion, Otsuka and Janssen. The other authors declare no competing interests.
Peer review
Peer review information
Nature Genetics thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Association of PGSALZ with the risk of developing AD (a) in 17 European countries and (b) in Men and Women.
Ncases, number of cases; Ncontrols, number of controls; OR, Odds ratio per Standard deviation were calculated using logistic regressions adjusted for age, gender and PCs according to the population studied (Supplementary Table 2). The lines in the Forest plots indicate the 95% confidence interval for the ORs.
Extended Data Fig. 2 Associations between (a) PGSALZ or (b) PGSALZ adjusted for APOE and age at onset of AD in European countries.
Ncases, the number of cases. Since HetP <0.05, the random effect is shown for the meta-analysis results. βs were calculated using a general linear model adjusted for APOE, gender and PCs according to the population studied (Supplementary Table 2).
Extended Data Fig. 3 Distribution and association of APOE ε2/ε3/ε4 alleles with AD risk worldwide.
(a) World map showing the populations analyzed. A color gradient indicates the strength of the association between APOE ε2/ε3/ε4 alleles and the risk of developing AD in different countries (b) frequencies of APOE ε2/ε3/ε4 alleles in case and controls as well association of APOE ε4 alleles with the risk of developing AD in different countries. OR, Odds ratio were calculated using logistic regressions adjusted for age, gender and PCs according to the population studied (Supplementary Table 2). Sample sizes are reported in Supplementary Table 2. The map was generated using ggplot2 and royalty-free data from rnaturalearth (https://www.naturalearthdata.com/about/terms-of-use/).
Extended Data Fig. 4 Association between (a) PGSALZ or (b) PGSALZ (adjusted for APOE) and age at onset of AD in multi-ancestry populations.
Ncases, number of cases. The African-American-ancestry meta-analysis (more than 75% of the population with African-American ancestry) included the MVP and ADSP datasets. The East Asia meta-analysis included datasets from China, Korea, and Japan. The Latin American (LA) ancestry (self-reporting) meta-analysis included the MMVP and ADSP datasets. The South America meta-analysis included the datasets from Argentina, Brazil, Chile, and Colombia. * not used in the meta-analysis. βs were calculated using a general linear model adjusted for gender and PCs according to the population studied (Supplementary Table 2).
Supplementary information
Supplementary Information (download PDF )
Sample description, Supplementary Figs. 1–14, references and Acknowledgements.
Supplementary Tables 1–11 (download XLSX )
Supplementary Tables 1–11.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Nicolas, A., Sherva, R., Grenier-Boley, B. et al. Transferability of European-derived Alzheimer’s disease polygenic risk scores across multiancestry populations. Nat Genet 57, 1598–1610 (2025). https://doi.org/10.1038/s41588-025-02227-w
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41588-025-02227-w
