
Abstract
Metazoan fatty acid (FA) synthases (mFASs) facilitate the de novo synthesis of C16- and C18-FAs through iterative extensions within the FA cycle and hydrolytic release. Here we re-engineer mFAS to fine-tune the interplay between FA extension and FA hydrolytic release for the targeted production of short- and medium-chain fatty acids. Single amino acid exchanges in the ketosynthase domain can redirect FA product profiles from predominantly C8 (G113W) to C8/C10 (G113F) and C12/C14 (G113M). Integration of a thioreductase domain enables the production of medium-chain fatty aldehydes and alcohols. We apply our approach for controlling chain length in FA biosynthesis to the microbial production of C10- and C12-FAs, translate it into a yeast cell factory and achieve C10/C12-FAs titers of 674 mg l−1 and 67% purity of total free FAs. Our work demonstrates a modular platform for programmable FA synthesis and paves the way toward sustainable bioproduction of valuable oleochemicals.

Similar content being viewed by others


Main
The biosynthesis of FAs with specific chain lengths is of high interest to the chemical industry1,2. For example, short-chain FAs (SCFAs) (≤C8) find widespread use in the food, pharmaceutical and cosmetic industries3,4, whereas medium-chain FAs (MCFAs) (C10–C14) can be directly used as lubricants, fragrances, paint additives and pharmaceuticals5. The biotechnological production of short- and medium-chain FAs (SMCFAs) offers a potential sustainable alternative to land-dependent coconut and palm oil extraction. To date, approaches for the microbial production of SMCFAs have been based on the eukaryotic multienzyme FA synthases (FASs)6,7,8,9,10,11 and the bacterial FAS system12,13,14,15,16 composed of separate enzymes. Many approaches harness the substrate-tolerant truncated version of the thioesterase TesA (‘TesA) from Escherichia coli, which is able to hydrolyze short and medium chains17,18,19,20; however, beyond titers lagging behind industrial demands, approaches to date have suffered from a lack of specificity in the production of the desired chain length1,2.
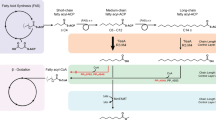
The metazoan FAS (mFAS) forms an open 550 kDa homodimeric X-shaped fold with two reaction clefts (Fig. 1a)21,22,23,24. mFAS naturally synthesizes C16- and C18-FAs through an iterative process, typically starting with acetyl-coenzyme A (Ac-CoA) as the priming substrate and using malonyl-CoA (Mal-CoA) for elongation, with each cycle adding two carbons from malonyl to the growing fatty acyl chain25,26. Each carbon–carbon bond formation by the ketoacyl synthase (KS) domain is followed by reduction of β-ketoacyl by the β-ketoacyl reductase (KR), dehydration to enoyl by the dehydratase (DH) and further reduction by the enoyl reductase (ER) to the saturated fatty acyl. To serve this action, the enzymatic domains KS, KR, DH and ER are substrate-tolerant to the growing chain length of FAs. In contrast, the FA-releasing thioesterase (TE) domain of mFAS is substrate specific, and very precisely intercepts synthesis after seven or eight cycles to release free C16- or C18-FAs, respectively (Fig. 1b)27. FA biosynthesis is assisted by substrate shuttling mediated by the compact four-helical bundle domain ACP, which is flexibly linked to the multienzyme FAS28,29. The ACP covalently binds substrates and intermediates through a post-translationally introduced phosphopantetheine moiety. Their covalent attachment prevents substrates and intermediates from diffusing out of the catalytic compartment, thereby maintaining high local concentrations that enhance enzymatic efficiency30,31. Notably, fungi possess a distinct FAS multienzyme that has evolved along a separate evolutionary trajectory, while following similar synthetic principles (Supplementary Fig. 1a, b)23,24,32.

a, Crystallographic structure of porcine mFAS (PDB ID: 2VZ8). The homodimeric mFAS adopts an extended X-shaped conformation. Condensing and modifying parts are connected by a short linker and form two lateral reaction clefts. The TE and ACP domains are attached flexibly such that they cannot be traced in electron density. Zoom in shows that one reaction cleft is attached to depict substrate shuttling by ACP. Numbers indicate the sequence of reactions (the path of ACP in shuttling substrates and intermediates to the catalytic domains). b, Cycle of metazoan FA biosynthesis. At defined length, TE releases the acyl chain as free FA. Numbers indicate the sequence of reactions as shown in a.
We recently demonstrated that the turnover rate of the KS domain is chain length dependent, with medium-chain substrates being processed more efficiently than longer ones. As the KS domain also serves as the gatekeeper of the FA cycle, the progression of an acyl chain through the cycle is inherently dependent on its length33,34. The following describes the iterative elongation pathway leading to the formation of a C16-FA: the initial elongation of the acetyl moiety, which primes the FA synthesis, proceeds with relatively low efficiency. KS efficiency then increases with chain length, showing a 6.7-fold enhancement for hexanoyl moieties during the third cycle passage, and a further 2.5-fold increase for decanoyl moieties in the fifth cycle. These increases correspond to lowered transition state energies by 2 and 4 kJ mol−1, respectively. As the growing acyl chain approaches the target chain length, the overall rate of the FA cycle declines. At this point, the substrate-specific TE domain terminates the iterative process, releasing the C16-FA27,35,36.
In this work, we engineer mFAS variants that are capable of producing SMCFAs by decreasing the efficiency of the KS in processing acyl moieties of longer chain lengths and installing a TE with broad chain length specificity. In such mFAS variants, the promiscuous TE would preferentially hydrolyze short- and medium-chain length substrates into free FAs, as they are poor substrates for KS. To implement this strategy in mFAS, we generate an mFAS variant that has lost its stringent control over chain length. As a next step, we adjust the kinetic properties of KS and ‘TesA by targeted mutations to decrease efficiency of KS for longer substrates, while enhancing the efficiency of ‘TesA for short and medium chain lengths. This dual optimization enables efficient synthesis of SMCFAs, with tunable enzymatic properties to enable the selective production of specific chain length subsets. We also create an acyl reducing mFAS hybrid by replacing the mFAS TE with a terminating thioreductase domain (TR). By re-tuning KS-mediated elongation versus TR-mediated reductive release, we achieve direct enzymatic synthesis of medium-chain alcohols and aldehydes. Finally, we engineer an Ogataea polymorpha yeast cell factory with tuned β-oxidation for the selective reduction of long-chain FAs (LCFAs) to serve as a chassis for mFAS/‘TesA hybrids. Fed-batch fermentation enables the production of 674 mg l−1 MCFAs (190 mg l−1 C10-FA and 484 mg l−1 C12-FA), highlighting the potential of this host system for sustainable oleochemical production from low-cost feedstocks.
Results
Engineering a generalist variant as production platform
We performed experiments in vitro with full-length mFAS and truncated versions, purified from E. coli37, which allowed us to evaluate the effects of scaffold remodeling and mutations in their impact on protein quality, enzyme kinetics and product output.
As reported earlier, the FA chain length in metazoan FA synthesis is largely determined by the TE domain, which is specific for C16 acyl chains27. To generate a platform for the production of FAs of various chain length, we replaced the native TE domain with ‘TesA from E. coli (Fig. 2a and Supplementary Fig. 2a–d). ‘TesA exhibits broad substrate specificity with a preference for medium-chain acyl moieties (Supplementary Fig. 2a–d)19 and has been widely used for producing SMCFAs12,15,20,38. The replacement of TEmFAS with ‘TesA was informed by previous work swapping mFAS TE with mammalian TEII and Cuphea palustris TE10. We received an mFAS/‘TesA hybrid with rates of FA synthesis that remained essentially unchanged compared to wild-type (WT) mFAS (Fig. 2b), indicating that the replacement of the native TE with ‘TesA does not per se impose a kinetic penalty. At the same time, the TE/‘TesA swap caused a shift in the FA product spectrum toward including medium chain lengths (C12–C18). We refer to this construct as the generalist mFAS, reflecting the loss of stringent chain length control and its suitability as an engineering platform for tailoring FA output.

a, Scheme and domain borders of the TE/‘TesA swap. A cartoon of mFAS is shown for clarity with one protomer colored. b, Exemplary activity assay of mFAS and the mFAS/‘TesA hybrid, monitored by NADPH consumption. The specific activities of WT mFAS and mFAS/‘TesA were 460 and 458 nmol min−1 mg−1, respectively. Inset shows product distributions of mFAS and the mFAS/‘TesA hybrid. The bars represent the means of three biological replicates and the error bars show s.d.
Engineering substrate specificity of KS
The KS performs a two-step reaction comprising the loading of the acyl chain to the active cysteine of KS (transacylation) and the elongation of the bound acyl moiety by a C2-unit (decarboxylative Claisen condensation) (Supplementary Fig. 3). The specificity of the KS-mediated elongation reaction originates from the transacylation of the acyl chain to the active cysteine (Cys161 in murine mFAS)34, such that acyl:KS complex structures provide valuable information for engineering. We utilized structural data of murine mFAS with a KS-bound octanoyl moiety as a basis for engineering (Fig. 3a, b)39. Since acyl chain binding is largely conserved across species, we also harnessed related structural information (Fig. 3c)40,41. Finally, the design of KS variants was further guided by homologous KSs from FAS/PKS systems that are known to produce mainly hexanoyl during olivetol biosynthesis (Supplementary Fig. 4)42,43.

a, Cross section of the KS dimer with the binding tunnel represented by its surface in gray. The ACP binding site is indicated. An mFAS model is attached for overview. b, Active site and acyl binding cavity of the KS domain. Key amino acids and the Cys161-bound octanoyl moiety are highlighted. Gly113 is positioned centrally in the binding tunnel near atom C8 of the substrate. Murine FAS numbering. c, Superposition with E. coli FabB with bound decanoyl and dodecanoyl moieties (PDB IDs 1F91 (light cyan) and 1EK4 (dark cyan)) and S. cerevisiae FAS with bound cerulenin (PDB ID 2VKZ, light pink). d, Radar chart of KS elongation rates for substrates of various chain lengths (C2–C10) of five different KS mutants at position 113. The rates represent three biological replicates and are given in 1 s−1. WT, wild type. e, Normalized activity of mFAS_WT and four different KS mutants at position 113 as monitored by NADPH consumption. The mean enzymatic activity of WT mFAS was 373 nmol min−1 mg−1 (data collected in biological triplicates with three technical replicates each, error bars indicate s.d.). f, Product distributions of mFAS_WT carrying mutated KS (data represents the mean ± s.d. of biological triplicates with three technical replicates each). FAs were monitored as methyl esters (FAMEs) by gas chromatography (GC) after methylation.
We hypothesized that sterically demanding residues replacing Gly113, Ser117, Met132, Ala160 and Thr196 have an influence on chain length control. A second line of evidence for Gly113 as a hotspot for engineering chain length originated from our own work on yeast6 and corynebacterial FAS44 (Fig. 3c). Overall, we generated a library of 14 binding tunnel mutants of the KS (Supplementary Figs. 5–8), which we evaluated in elongation kinetics (Supplementary Fig. 9). Among these variants, Gly113 demonstrated exceptional plasticity to exchanges with sterically demanding residues Ser<Met<Phe<Trp. Low rates for hexanoyl and octanoyl substrates agree with the steric restrictions imposed by phenylalanine and tryptophan, whereas the turnover rates received for acetyl and butyryl substrates remained essentially unchanged in Gly113 mutants (Fig. 3d and Supplementary Fig. 10a, b).
KS mutations (G113S/M/F/W) were then integrated into full-length mFAS and subjected to in vitro activity and product assays. Except for the G113M variant, the activity of the KS-mutated variants decreased as the size of the amino acid at position 113 increased (Fig. 3e). Further, as amino acid size increased, the FA product spectra shifted toward shorter FAs, and yields of free FAs dropped in line with decreased activity (Fig. 3f and Supplementary Fig. 11a, b).
Chain length modulation by combined engineering of KS and ‘TesA
As a next step, we introduced selected KS mutations into the mFAS/‘TesA generalist variant, and additionally modified ‘TesA according to variants with known enzymatic properties from previous studies: ‘TesA_L109P hydrolyzes C8-ACP ten times slower than WT, but retains specificity for shorter chains18,19, and ‘TesA_M141L/E142D/Y145G/L146K (‘TesA_4x) exhibits higher specificity for short chains and a tenfold increased rate for C8-ACP cleavage compared to WT (Supplementary Fig. 10b)18. In the background of WT KS, mFAS/‘TesA_4x and mFAS/‘TesA_L109P had a different impact on the FA synthesis. The mFAS/‘TesA_4x hybrid operated with similar activity to the mFAS/‘TesA (nonmutated ‘TesA) and further broadened the spectrum to release C8 to C16-FAs (Fig. 4a,b; data highlighted by gray background). In contrast, the mFAS/‘TesA_L109P exhibited substantially decreased activity in line with low hydrolysis activities of this ‘TesA variant. The shift in the product spectrum toward C18-FAs correlates with ‘TesA_L109P being ineffective in intercepting the FA cycle driven by the nonmutated KS. Thus, ‘TesA_L109P can only cleave FAs at chain lengths beyond C16, when KS rates have sufficiently dropped (Supplementary Fig. 11c,d).

a, Normalized activity monitored by NADPH consumption. The assay was performed in 20 µl volume. Components were used in final concentrations of 100 μM Acetyl-CoA, 100 μM Malonyl-CoA, 50 μM NADPH and 20–80 nM enzyme. Data collected in biological triplicates with three technical replicates each, and normalized to mFAS_WT activity. Error bars represent s.d. b, Product distributions collected in three biological replicates with three technical replicates each. Data collected for hybrid constructs with nonmutated KS highlighted by a gray background.
We then combined the three ‘TesA variants (‘TesA, ‘TesA_L109P and ‘TesA_4x) with the four selected KS mutations (G113S/M/F/W) (Supplementary Figs. 12 and 13), yielding 12 mFAS/‘TesA hybrids, and recorded catalytic efficiency and product output spectra (Fig. 4a,b and Supplementary Fig. 14a,b). While mFAS/‘TesA hybrids with aromatic amino acids substituting Gly113 (G113F/W) showed consistently decreased activities, as a result of the throttled KS elongation rates, hybrids with KS mutations G113S and G113M varied in activity depending on the ‘TesA variant. In particular, they showed high NADPH consumption rates with ‘TesA (G113S-variant) and ‘TesA_4x (G113S & G113M). The hybrids mFASG113S/‘TesA_4x and mFASG113M/‘TesA even surpassed rates of WT mFAS, which can be attributed to the high elongation rates of G113S and G113M for short acyl chains that excellently align with the specificity profile of ‘TesA and ‘TesA_4x. The data indicate that the produced chain lengths are primarily dictated by the KS mutation and are further fine-tuned by the matching ‘TesA variant.
Production of fatty aldehydes and alcohols
To extend the mFAS product spectrum beyond FAs, we replaced the TE domain with a thioester reductase (TR) domain for production of fatty alcohols and/or aldehydes (Fig. 5a). We focused on two classes of TRs: TRs from carboxylic acid reductases (TRsCAR) and TRs from PKS/NRPS systems (TRsPKS/NRPS) (Supplementary Fig. 15a–c)45,46,47,48,49,50. TRsPKS/NRPS catalyze either 2-electron or nonprocessive (2 + 2) electron reduction to the aldehyde or alcohol, respectively51,52. While earlier studies characterized TRsCAR as exclusively performing two-electron reductions to generate aldehydes48,53, more recent evidence indicates the potential for (2 + 2) electron reductions as well (Supplementary Fig. 16)49. To select a suitable TR to replace the TEmFAS domain, we screened seven TRsCAR and four TRsPKS/NRPS, which represent a broad phylogenetic diversity and are characterized in function and partly in structure (Supplementary Fig. 17)45,46,47,48,49,54.

a, Scheme of aldehyde and alcohol production with mFAS/TR. b, Activity screening of seven TRCAR and four TRPKS/NRPS for the reductive cleavage of the decanoyl group from C10-ACP as decanal and/or decanol. Reductive domains are derived from: Mycobacterium marinum (#1), M. phlei (#2), M. smegmatis (#3), Nocardia iowensis (#4), N. otitidiscaviarum (#5), Tsukamurella paurometabola (#6), Neurospora crassa (#7), Stigmatella aurantiaca (#8), M. tuberculosis (#9), M. smegmatis (#10) and Methanobrevibacter ruminantium (#11). Data represent the mean ± s.d. of technical triplicates. c, AlphaFold model of PCP-TRCAR from M. smegmatis with docked PCP60. NADPH is not part of the PCP-TRCAR M. smegmatis model, but it was superimposed from the PCP-TRCAR structure from Segniliparus rugosus (PDB ID: 5MSV). Selected active site residues in stick representation. Vacuum electrostatics indicated for PCP of modeled M. smegmatis PCP-TRCAR (right/top), and for mFAS ACP modeled to the PCP atomic coordinates with Modeller61 (ACP 24% sequence identical to PCP of M. smegmatis) (right/bottom). d, Activity of mFAS/TR hybrids with WT KS (gray background) and G113W-mutated KS, respectively. NADPH consumption (by KR, ER and TR) during fatty alcohol/aldehyde synthesis was monitored in three technical replicates, error bars represent s.d. e, In vitro production of fatty aldehydes/alcohols with mFAS/TR hybrids with WT KS (gray background) and G113W-mutated KS, respectively. Data from the mFAS/TR hybrids were collected in technical triplicates. Data from the hybrids with G113W mutation were collected in three biological and technical replicates. Error bars represent s.d.
Standalone TRs were tested in their ability to reduce and release saturated C10 acyl chains from mFAS ACP (Fig. 5b and Supplementary Fig. 18). The activities of TRsPKS/NRPS were at least one order of magnitude lower than that of TRsCAR, which also naturally process saturated FAs. Further, we observed that the activity of both TR types remains at least two orders of magnitude lower than the overall mFAS activity, indicating that they will impose a kinetic bottleneck when harnessed in mFAS for fatty alcohol/aldehyde production. Based on structural models, PCP and TRCAR physically interact for the reductive release (Supplementary Fig. 19a)48. An AlphaFold3-modeled Mycobacterium smegmatis PCP:TR complex indicates that the interface is overall little charged, whereas, in contrast, the mFAS ACP displays a more positively charged surface (Fig. 5c and Supplementary Fig. 19b). Thus, we hypothesize that suboptimal, noncomplementary domain–domain interactions at least partially account for the low efficiency of the reductive release of the acyl chain bound to mFAS.
Based on results from the analysis of standalone TR domains, we exchanged the TEmFAS domain with the three best performing TRs, namely the TRsCAR from Mycobacterium phlei, M. smegmatis and Nocardia otitidiscaviarum, termed in the following TR#2, TR#3 and TR#5, respectively (Supplementary Figs. 20–22). For all constructs, the NADPH consumption rates were low (Fig. 5d), and alcohol/aldehyde production could not be detected, except for traces of long-chain fatty aldehydes hexadecanal, octadecanal and eicosanal for the hybrid mFAS/TR#2 (Fig. 5e). In an attempt to match the kinetics of KS and TR, as we did for the kinetic interplay of KS and ‘TesA in SCFAs and MCFAs production, we eventually decreased the flux in the FA cycle with the help of the G113W mutation in KS. In doing so, for all three mFAS/TR hybrids, the activity increased threefold (Fig. 5d), and product amounts for hybrid mFASG113W/TR#2 increased up to eightfold (Fig. 5e).
In vivo production in yeast
We then introduced selected mFAS/‘TesA hybrids into yeast cell factories for the precise biosynthesis of FAs and their derivatives with controlled chain lengths. First, we overexpressed three mFAS/‘TesA constructs carrying KS mutations G113S, G113M and G113F in the thermotolerant yeast Ogataea polymorpha, which has the ability to utilize a wide range of substrates including methanol, glucose and xylose (Fig. 6a)55. Without additional metabolic engineering, the plasmid-based expression enabled the biosynthesis of up to 14.5 mg l−1 SMCFAs (Fig. 6b). For all mFAS/‘TesA hybrids, a slight shift toward shorter chains is observed compared to in vitro results. Since chain length output is substantially influenced by concentrations of acetyl-CoA and malonyl-CoA, shifted spectra may reflect cellular substrate concentrations that do not match our chosen in vitro conditions56.

a, Schematic depiction of the production of free FAs in O. polymorpha (O. p.). The mFAS/‘TesA hybrids directly synthesize free FAs, whereas the O. p. FAS generates fatty acyl-CoAs. The produced free FAs can be coupled to CoAs by Faa1 and then transported into the peroxisome where Pox1 degrades them to acetyl-CoA during β-oxidation. The conversion of LCFAs to MCFAs can be achieved by remodeling the β-oxidation pathway. b, Production of free FAs by engineered O. polymorpha strains without mFAS hybrids (O.p. WT) or one of three mFAS/‘TesA hybrids with different KS mutations. mFAS/‘TesA hybrids were encoded from plasmid. Values shown represent the mean of three biologically independent samples, error bars depict the s.d. of the mean. c, Remodeling β-oxidation for enhancing the production of MCFAs. The latter two strains (XMCFA65 and 69) overexpressed ScFAA1 and YlPOX2-hACOT4 without and with an additional plasmid-based expression of mFASG113S/‘TesA, respectively. Values shown represent the mean of three biologically independent samples, error bars depict the s.d. of the mean. d, Fed-batch fermentation of strain XMCFA69 in 1.5-l bioreactors. Biomass, glucose consumption and MCFA concentration over time in Delft minimal medium containing 20 g l−1 glucose. Values shown represent the mean of three biologically independent samples, error bars depict the s.d. of the mean. e, Free FA distribution in various strains and plants. XCMFA and XCMFA-fed-batch, this study; Xu et al. (Y. lipolytica CnFatB2)9; Rigouin et al. (Y. lipolytica JMY1233_I220W)8; Jindra et al. (E. coli BTE)16; Yan et al. (E. coli PhaG variant Q45R G142V)62; Valencia et al. (P. putida 3KO)13; coconut oil63 and palm kernel oil58. f, Distribution and content of free FAs at the end of fed-batch fermentation.
To further assess the applicability of our chain length control strategy, and given its industrial relevance, we selected C12-FA (lauric acid) as the target chain length for yeast based in vivo production. MCFAs, particularly C12-FA, are widely used in the production of surfactants, detergents, lubricants and personal care products due to their favorable physicochemical properties and antimicrobial activity57. Currently, C12-FA is predominantly obtained from palm kernel oil, which only makes up below 10% of the oil in the fruit and contains about 50% C12-FA58. As a result, large-scale palm cultivation is required to meet demand, contributing considerably to biodiversity loss and greenhouse gas emissions in Southeast Asia59. To this end, the gene encoding mFASG113M/‘TesA was integrated into the genome under the control of the strong constitutive promoter PGAP (promoter of glyceraldehyde-3-phosphate dehydrogenase), which resulted in MCFAs titers comparable to those achieved with the plasmid-based expression (Supplementary Fig. 23). To further enhance free FA titers, we sought to block β-oxidation by deleting the FA oxidase gene POX1 (strain XCMFA03), which increased MCFAs titers by 3.0-fold and improved MCFA selectivity compared to the control strain XCMFA38 (Supplementary Fig. 24). We further aimed at increasing the copy number of mFAS/‘TesA hybrids by parallel expression from plasmids and genome. For variant mFASG113M/‘TesA (XCMFA27), this led to elevated titers of free C12 and C14-FAs by 3.2 and 6.1-fold, respectively. Combining the genomic expression of mFASG113M/‘TesA with the plasmid-based expression of mFASG113S/‘TesA (XCMFA28) increased the production of C12 and C14-FAs by 3.5 and 13.0-fold, respectively (Supplementary Fig. 24).
Additionally, rather than merely blocking β-oxidation, we considered specifically targeting the degradation of LCFAs toward MCFAs through precisely regulating β-oxidation. Feeding experiments revealed the consumption of C12- and C14-FAs by the WT strain (Supplementary Fig. 25a). Deletion of the acyl-CoA ligase encoding gene OpFAA1 led to improved MCFA/LCFA titers (Supplementary Fig. 25b), suggesting that OpFaa1 acts on LCFAs as well as MCFAs. Replacing OpFAA1 with Saccharomyces cerevisiae ScFAA1 reduced LCFA levels while increasing MCFA levels (Supplementary Fig. 25b), demonstrating the selective activity of ScFaa1 toward LCFAs. Notably, a strategy based on differences in acyl-CoA ligase substrate specificity for modulating the FA chain length spectrum has been leveraged earlier for a Pseudomonas putida strain13. For the limited β-oxidation approach, we tested combinations of long-chain acyl-CoA oxidases and medium-chain acyl-CoA thioesterases to enhance the production of free MCFAs. The combinatorial expression of long-chain acyl-CoA oxidase encoding gene YlPOX2 from Yarrowia lipolytica and human thioesterase encoding gene hACOT4 showed the most promising results (Supplementary Fig. 26). We then proceeded to incorporate the limited β-oxidation cascade into the strain with the OpPOX1 deletion expressing the mFASG113M/‘TesA variant from the genome. This resulted in a strain XMCFA65 with a 96% increase in MCFA production and 52% C12 fraction of total free FAs, which is a 33% increase compared to parent strain XMCFA03 (Fig. 6c). Again, increasing the mFAS expression levels through additional plasmid-based expression of mFASG113S/‘TesA (strain XMCFA69) further increased the MCFA titer to 90.8 mg l−1, corresponding to a 4.5-fold increase compared to the control strain XMCFA03 (Fig. 6c). Of note, in strain XMCFA69, the C12-FA fraction accounts for 69% of the total free FAs, which is 77% higher than that of XMCFA03. Furthermore, the strain had a 1.9-fold increase in MCFA production and a 50% increase in the C12 fraction compared to XMCFA28 (Fig. 6c). Thus, remodeling the β-oxidation pathway of LCFAs, combined with multicopy expression of mFAS/‘TesA hybrids in yeast, is an effective strategy to enhance the specific biosynthesis of MCFAs and C12-FA. Finally, to assess the in vivo potential of mFAS/‘TesA hybrids for MCFAs biosynthesis, we performed fed-batch fermentation with strain XMCFA69 in a 1.5-l bioreactor. This approach yielded a total of 708.6 mg l−1 MCFAs, consisting of 189.5 mg l−1 C10-FA, 484.1 mg l−1 C12-FA and 34.0 mg l−1 C14-FA (Fig. 6d and Supplementary Fig. 27). Of the total free FAs produced, 70% are MCFAs, and C12-FA alone accounts for 48% (Fig. 6e). Of note, a portion of 23% of MCFAs was detected in the extracellular medium (Fig. 6f), suggesting partial secretion or a form of FA-leakage. Thus, with a strain subjected to only limited metabolic engineering, we achieved the highest titers of MCFAs reported in yeast to date8,9, with C12-FA produced at a specificity comparable to that found in palm or coconut oil and previously engineered E. coli (Fig. 6e)16.
The expression of mFAS hybrids capable of producing fatty alcohols in vitro did not lead to the desired products in O. polymorpha. In contrast, expression of mFASG113W/TR#2 and mFASG113W/TR#3 in S. cerevisiae resulted in the successful in vivo production of C10 and C12 alcohols (Supplementary Fig. 28).
Discussion
In metazoan de novo FA biosynthesis, the chain length of products is defined by the interplay of the mFAS domains KS and TE, with a saturated acyl chain of a given length being either elongated by the KS or hydrolyzed by the TE. Given equal concentrations of the two enzymatic domains within mFAS, it is ultimately their relative catalytic efficiency that dictates which pathway dominates. In the native biosynthesis, accurate chain length regulation at C16 is enabled by a pronounced increase in the TE’s relative catalytic efficiency at this chain length27, accompanied by a simultaneous efficiency loss of the KS34,39. In this work, we aimed at harnessing this molecular foundation of chain length control of FA biosynthesis. Specifically, we hypothesized that tuning the relative catalytic efficiencies of KS versus TE could serve as a design principle to access distinct FA chain length ranges through just modest mutational impact. As a first step, we constructed a generalist mFAS variant by replacing the highly substrate-specific native TE with the broadly active E. coli thioesterase ‘TesA. Precise kinetic data for ‘TesA hydrolysis of acyl-ACP substrates are lacking; however, we expected that its pronounced substrate promiscuity reduces chain-length selectivity in the mFAS/‘TesA hybrid, yielding a wider and less selective product distribution (for a conceptual representation of the putative kinetic behavior, see Supplementary Fig. 29a–c). Indeed, as documented by the FA output spectra (Fig. 2b), the generalist mFAS only imposed loose chain length control. Using the generalist mFAS as a platform, we subsequently modulated the kinetics of KS and ‘TesA, creating specialized variants for SMCFAs production. Mutations such as G113W, G113F or G113M substantially shifted the product spectrum toward C8 to C14 (Fig. 4b). Our mFAS engineering strategy represents an innovative engineering approach to chain length control. Rather than being dictated by a single dominant factor, chain length regulation emerges from a finely balanced interplay between two catalytic activities, representing a system-level phenomenon rather than the isolated selectivity of a single domain. The broader applicability of this principle is demonstrated by its successful implementation in the production of short and medium-chain fatty aldehydes and alcohols using engineered mFAS/TR hybrids. In these systems, product formation was only observed when the catalytic efficiency of the KS was reduced in the medium chain length range, allowing the terminating TR to effectively compete with the KS for reductive release (Fig. 5e).
For SMCFA production in yeast, three mFAS/‘TesA hybrids were initially selected for integration in O. polymorpha (Fig. 6a). The product spectra of the in vitro measurements are well reflected in the in vivo production, characterizing the mFAS/‘TesA hybrids as robust producers of SMCFAs. The robustness of the mFAS-based chain length regulation in the complex environment of a yeast cytoplasm is remarkable, and can largely be attributed to compartmentalization, which enables synthesis to proceed in a highly autonomous manner (Fig. 6b). We then focused on producing C12-FA in the industrially relevant strain O. polymorpha with the goal of enhancing both, product specificity and titer. To this end, we implemented a strategy involving targeted modulation of β-oxidation as a central concept in our engineering strategy. Specifically, we deleted the POX1 gene, which encodes the rate-limiting acyl-CoA oxidase in peroxisomal β-oxidation, and redirected the degradation of LCFAs toward MCFAs through targeted regulation of β-oxidation. Furthermore, to refine metabolic re-routing, we replaced the endogenous OpFAA1, which acts on both LCFAs and MCFAs, with ScFAA1 from S. cerevisiae. ScFaa1 preferentially activates LCFA and allows MCFA accumulation. Eventually, the strain (XMCFA69) produced 90.8 mg l−1 of MCFAs in high purity of C12 over other chain lengths (Fig. 6c). While β-oxidation is typically viewed as a catabolic process and often suppressed in metabolic engineering strategies for SMCFA production, we demonstrate its utility as a tunable mechanism for refining product chain length profiles in yeast.
MCFAs, particularly C12-FA, are currently primarily derived from palm kernel oil. Microbial production platforms must meet several key criteria to serve as a viable alternative to existing plant-based oleochemical technologies: selective synthesis of short- and medium-chain oleochemicals, industrially relevant titers with low land-use requirements, and the ability to utilize cost-effective, sustainable feedstocks59. In this work, we focused on product specificity with the goal of establishing a robust, broadly applicable and tunable platform for the de novo biosynthesis of SMCFAs. The mFAS/‘TesA hybrid system, presented here, fulfills this role. Without additional metabolic engineering, plasmid-based expression of the mFAS/‘TesA construct enabled selective production of C12-FA (37%, strain XMCFA20) and C8-FA (70%, strain XMCFA06) (Fig. 6b). To explore the potential of our approach, we further optimized C12-FA biosynthesis through metabolic engineering. Under fed-batch fermentation conditions, a minimally engineered O. polymorpha strain yielded 0.674 g l−1 C10/C12-FAs (29% C10-FA, 71% C12-FA), which account for 67% of total FAs (Fig. 6d). Given the industrial relevance of O. polymorpha55, these findings underscore the strong potential of mFAS/‘TesA hybrids for developing efficient cell factories for sustainable SMCFA production.
Methods
Heterologous expression of mFAS constructs
Plasmids containing full-length mFAS constructs and mFAS hybrids were transformed into electrically competent E. coli BAP1 cells and plasmids containing KS-MAT0 were transformed into chemically competent E. coli BL21 gold (DE3). After transformation cells were plated on LB-Agar plates (100 µg ml−1 ampicillin (amp) and 1% (w/v) glucose). Overall, 3–6 colonies were picked and cells were grown overnight at 37 °C in 20 ml LB medium (100 µg ml−1 amp and 1% (w/v) glucose). Pre-cultures were used to inoculate 1 l TB medium (100 µg ml−1 amp). Cultures were grown at 37 °C and 150 rpm until they reached the desired optical density (OD600) of 0.6–0.8. After cooling at 4 °C, cultures were induced with 250 µM IPTG and grown for an additional 16 h at 20 °C and 150 rpm. Cells were collected by centrifugation (5,000g for 15 min). After the supernatant was discarded, the remaining cell pellet was resuspended in 30 ml lysis buffer (200 mM KCl, 30 mM imidazole, 50 mM potassium phosphate, 10% (v/v) glycerol, 1 mM EDTA, pH 7.0), a small amount DNase I (Sigma Aldrich) was added. A French Pressure Cell Press (Amico) was used to disrupt the cells. The resulting suspension was centrifuged using the JA 25.500 rotor at 40,000g for 1 h. The supernatant was mixed with 1 M MgCl2 to a final concentration of 2 mM and then transferred to Ni-NTA columns and washed with five column volumes (CVs) of His-wash buffer (lysis buffer without EDTA). Bound protein was eluted with 5 ml elution buffer (200 mM KCl, 300 mM imidazole, 50 mM potassium phosphate and 10% (v/v) glycerol, pH 7.0). The eluted protein was then transferred to a Strep-Tactin column. After being washed with five CVs of strep-wash buffer (250 mM potassium phosphate, 1 mM EDTA and 10% (v/v) glycerol, pH 7.0), bound protein was eluted with 5 ml strep-elution buffer (strep-wash buffer + 40 mM biotin). Representative SDS–PAGE images can be found in Supplementary Figs. 12 and 15. Afterwards proteins were concentrated to 5–10 mg ml−1, frozen in liquid nitrogen and stored at −80 °C. Next, proteins were thawed at 37 °C for 60 min and further purified by size-exclusion chromatography (SEC). A Superdex 200 GL 10/300 column and strep-wash buffer were used. Representative SEC profiles can be found in Supplementary Figs. 13 and 16. Fractions with dimeric protein were pooled and subsequently concentrated to 2–5 mg ml−1. Aliquots were frozen in liquid nitrogen and stored at −80 °C.
Heterologous expression of mFAS ACP and TR domains
Plasmids containing mFAS ACP and TR domains were transformed into chemically competent E. coli BL21 gold (DE3). They were expressed as the mFAS constructs but no Strep-Tactin purification was performed. Representative SDS–PAGE images can be found in Supplementary Fig. 14. SEC was conducted with a Superdex 200 GL 10/300 column and His-wash buffer was used. Protein fractions containing the correct size were pooled and concentrated to 1–10 mg ml−1 for the TR domains and 40–60 mg ml−1 for the mFAS ACP. Aliquots were frozen in liquid nitrogen and stored at −80 °C.
MabA assay
The KS kinetics were determined as described previously34. In short, condensation activity of the KS domain was monitored indirectly with an enzyme coupled assay using a reductase MabA from Mycobacterium tuberculosis. All solutions were prepared in an assay buffer (50 mM sodium phosphate and 10% glycerol, pH 7.0) as 8× stock or 2× stock (Mal-ACP) and pipetted immediately before monitoring. The components were added to a final concentration of 500 nM enzyme, 20 µM Mal-ACP, 100 µM Acyl-ACP, 50 µM NADPH and 5 µM MabA. The NADPH fluorescence (excited 348–320 nm, detected 476–420 nm) was monitored during the reaction at 25 °C.
mFAS activity assays
Measurements of the mFAS-based constructs were carried out using a CLARIOstar Plus Microplate Reader and 386-well-plates (Greiner). NADPH fluorescence was measured (excited 348–320 nm, detected 470–420 nm) at 25 °C. The assay was performed in 20 µl assay volume. All solutions were prepared in an assay buffer (50 mM potassium phosphate, 10% glycerol (v/v), 5% PEG 400 (v/v), 0.03 mg ml−1 BSA and 1 mM dithiothreitol (DTT), pH 7,0) as 4× stock and pipetted immediately before monitoring. The components were added to a final concentration of 100 μM Acetyl-CoA, 100 μM Malonyl-CoA, 50 μM NADPH and 20–80 nM enzyme (WT and chain length constructs) or 400–800 nM enzyme (TR hybrids). Before the measurements each protein was thawed for 5 min at 37 °C and stored on ice until the measurement. Blank measurements were conducted without acetyl- and malonyl-CoA.
TR activity screening
Measurements of the standalone TR domains were carried out using a CLARIOstar Plus Microplate Reader and 386-well plates (Greiner). NADPH fluorescence was measured (excited 348–320 nm, detected 470–420 nm) at 25 °C. The assay was performed in 21 µl assay volume. All solutions were prepared in an assay buffer (50 mM potassium phosphate, 10 mM MgCl2 and 10% glycerol (v/v), pH 7.0) as 3× stock and pipetted immediately before monitoring. The components were added to a final concentration of 10 μM NADPH, 460 µM C10-mFAS ACP and 1 µM TR. Before the measurements each protein was thawed for 5 min at 37 °C and stored on ice until the measurement. Blank measurements were conducted with apo-mFAS ACP instead of C10-mFAS ACP.
In vitro FA production assay
The in vitro production of FAs was performed similar to the mFAS activity assays. The assay was performed in 500 µl assay volume. All solutions were prepared in an assay buffer (50 mM potassium phosphate, 10% glycerol (v/v), 5% PEG 400 (v/v), 0.03 mg ml−1 BSA and 1 mM DTT, pH 7.0) as 4× stock and added to final concentrations of 500 μM Acetyl-CoA, 500 μM Malonyl-CoA, 500 μM NADPH and 40 nM enzyme concentration. The assay solution was then incubated overnight at room temperature. After incubation 5 µl of heptanoic acid (2 g l−1 in methanol:CHCl3, 3:1) was added as internal standard for the GC measurements.
FA extraction and derivatization
To increase the vapor pressure and decrease the boiling point of the FAs produced in vitro, the FAs were converted to FA methyl esters (FAMEs). The procedure was adapted from rom Bligh and Dyer64. Therefore, the 500 µl test solution was acidified with 50 µl of 8% HCl in methanol to protonate the FAs and make them less polar. Next, 125 µl methanol:CHCl3 (1:1) was added and the solution was vortexed to extract the FAs to the nonpolar phase. The mixture was then centrifuged at 3,000g for 10 min to separate the phases. The lower nonpolar phase was transferred to a fresh Eppendorf tube and the solvent was evaporated under reduced pressure. Then, 20 µl toluene was added and transferred to Duran glass tubes containing 200 µl solution of 8% HCl in methanol and 500 µl methanol. The tubes were sealed tightly and shaken to mix the solutions. The reaction mixture was then heated to 100 °C for 3 h. The reaction was then cooled on ice for 10 min and 100 µl of hexane and 300 µl of H2O were added. The mixture was then vortexed and returned to ice to allow the phases to separate. 70 µl of the hexane phase was transferred to a GC vial with micro inlet.
In vitro fatty aldehyde/alcohol production assay
The in vitro production of FAs was performed similarly to the mFAS activity assays. The assay was performed in 500 µl assay volume. All solutions were prepared in an assay buffer (50 mM potassium phosphate, 10% glycerol (v/v), 5% PEG 400 (v/v), 0.03 mg ml−1 BSA and 1 mM DTT, pH 7.0) as 4× stock and added to final concentrations of 300 μM acetyl-CoA, 300 μM malonyl-CoA, 500 μM NADPH and 1 µM enzyme concentration. The assay solution overlaid with 100 µl hexane and then incubated overnight at room temperature. After incubation 10 µl of heptanoic acid (0.4 g l−1 in hexane) was added as internal standard for the GC measurements.
Fatty aldehyde/alcohol extraction
Extraction was carried out by adding another 50 µl hexane and mixing. The mixture was centrifuged for 10 min at 5,000g for phase separation. Finally, 50 µl of the organic phase was transferred to a GC vial with micro inlet.
Gas chromatography measurements
The measurements of the FAMEs have been conducted with two different devices: a PerkinElmer Clarus 400 gas chromatograph equipped with an Elite 5MS capillary column (30 m × 0.25 mm, film thickness, 0.25 μm) and a flame ionization detector (Software, TotalChom 6.3); And an Agilent 7890B gas chromatograph equipped with a HP-5ms Ultra Inert capillary column (30 m × 0.25 mm, film thickness, 0.25 μm) and an Agilent 5977B MSD (Software MassHunter 12). The injector temperature was 200 °C and the detector temperature was 250 °C. The temperature program used was heating to 50 °C for 5 min; increase of 10 °C min−1 until 120 °C was reached, then hold for 5 min; increase of 15 °C min−1 until 220 °C was reached and then hold for 10 min.
The measurements of FA aldehydes/alcohols have been conducted with an Agilent 7890B GC equipped with an HP-5ms Ultra Inert capillary column (30 m × 0.25 mm, film thickness, 0.25 μm) and an Agilent 5977B MSD. The temperature program used was heating to 50 °C for 7 min; increase of 15 °C min−1 until 220 °C was reached, then hold for 5 min; increase of 20 °C min−1 until 280 °C was reached and then hold for 5 min.
GC data analysis
The peaks at the previously obtained retention times of each FAME were integrated using the program ‘totalchrom’ for the PerkinElmer data, ‘OpenChrom’ was used for the Agilent data. To take losses during extraction and derivatization into account the signals was normalized to the signal of the internal standard. The final concentrations of the FAMEs were calculated using equation (1).
With Asample being the integrated peak area of the respective sample peak, Astandard being the integrated peak area of the standard peak, msample being the slope of the respective sample calibration curve, mstandard being the slope of the standards calibration curve and csample and cstandard the corresponding concentrations.
Strains and cell cultivation
The plasmids, donor DNAs, and strains used to construct the yeast cell factories are detailed in Supplementary Tables 2–4, respectively. All yeast strains for FA production were derived from O. polymorpha NCYC 495, whereas those for fatty alcohol production were derived from S. cerevisiae CEN.PK 113-11 C.
The formulations of LB, SD, YPD and Delft minimal medium with 20 g l−1 glucose are the same as those described by Zhai et al.65, whereas the culture conditions for FA-producing strains are consistent with Zhai et al.65 and the culture conditions for fatty alcohol-producing strains align with Gao et al.66. For the fermentation in a shake bottle, the yeast cells were cultivated in Delft minimal medium at 220 rpm for 96 h, at 37 °C for O. polymorpha and 30 °C for S. cerevisiae, with supplementation of 60 mg l−1 uracil or 20 mg l−1 methionine, respectively, where necessary.
Genetic manipulation by CRISPR/Cas9 system
The oligonucleotides used in construction of donor DNAs shown in Table S3. All guide RNA plasmids refer to the previous descriptions66,67,68. Related reagents and the methods for construction of gRNA expression plasmids and donor DNAs are consistent with Zhai et al.65. DNA transformation into O. polymorpha and S. cerevisiae was conducted according to previous methods66,65.
C12/C14-FA feeding experiment
Following cultivation of the WT O. polymorpha strain JQcr03L in 20 ml of Delft minimal medium with 20 g l−1 glucose to an OD600 of 5, C12-FA and C14-FA were added to concentrations of 800 mg l−1 and 600 mg l−1, respectively. The initial concentrations of these FAs were quantized upon addition and after a 72-h fermentation period by gas chromatography–mass spectrometry (GC–MS).
Qualitative and quantitative analysis of FAs
The total FA extraction procedure was adapted from established methodologies with minor modifications69. In brief, 200 μl of appropriately diluted cell cultures was combined with 10 μl of 40% tetrabutylammonium hydroxide (Sigma, cat. no. 86854). Subsequently, 200 μl of 200 mM iodomethane (Sigma, cat. no. I8507) in dichloromethane (Sigma, cat. no. 650463) was added immediately. The dichloromethane solution contained 50 mg l−1 each of pentacanic acid, tridecanoic acid and nonanoic acid (Sigma, cat. no. P6125, T0502 and 75190) as internal standards. The mixture was vortexed at 1,200 rpm for 30 min and then centrifuged at 1,000g for 10 min to achieve phase separation. About 150 μl of the dichloromethane layer was transferred to a GC vial equipped with a glass insert and evaporated to dryness. The resulting methyl esters were reconstituted in pure hexane and analyzed using a GC–MS system (Thermo Fisher Scientific, ISQ 7610) equipped with a TG-5MS capillary column (30 m × 0.25 mm × 0.25 μm; Thermo Fisher Scientific, cat. no. 26098-1420). The GC–MS temperature program was set as initial temperature of 40 °C, held for 2 min; increased to 180 °C at 30 °C min−1, then raised to 200 °C at 4 °C min−1 and held for 1 min, followed by an increase to 240 °C at 2 °C min−1 and a final hold for 10 min. The injection volume was 1 μl, and the helium carrier gas flow rate was maintained at 1.0 ml min−1. Data were processed using Xcalibur software (v.4.1).
Qualitative analysis of fatty alcohol
Fatty alcohol was directly extracted from the fermentation broth using n-hexane at a solvent-to-broth ratio of 1:4 (v/v). The mixture was shaken at 1,500 rpm for 30 min, followed by centrifugation at 1,000g for 10 min. Subsequently, 200 μl of the n-hexane layer was transferred into a GC vial equipped with a glass insert. Fatty alcohol quantification was conducted on a GC–MS system (Thermo Fisher Scientific, ISQ 7610) fitted with a TG-5MS capillary column (30 m × 0.25 mm × 0.25 μm; Thermo Fisher Scientific, cat. no. 26098-1420). The temperature program for fatty alcohol analysis was set as initial temperature held at 45 °C for 2.5 min, increased to 220 °C at 20 °C min−1 and held for 2 min, then raised to 300 °C at 20 °C min−1 and maintained for 5 min. Data were processed using Xcalibur v.4.1 software.
Fed-batch fermentation
Fed-batch fermentations were conducted in CloudReady Bioreactors System (T&J Bio-engineering) with a 1.5-l bioreactor and 0.5-l working volume. The initial batch phase began by inoculating the pre-cultured strain XMCFA69 (OD600 = 4–5) into a Delft minimal medium with 20 g l−1 glucose, achieving a starting OD600 of 0.5. The process parameters were maintained at 37 °C, pH 5.6 and 30% dissolved oxygen. Agitation started at 400 rpm and aeration at 0.5 VVM, both automatically adjusted (max up to 1,000 rpm and 2 VVM, respectively) to sustain the dissolved oxygen level. Upon glucose depletion, a feeding solution of 5× Delft minimal medium with 500 g l−1 glucose was introduced at 1–3 ml h−1, adjusted according to residual glucose and ethanol concentrations.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data generated in this study are provided within the article and in the Supplementary Information. Further data are available from the corresponding authors upon request. Source data are provided with this paper.
References
Yan, Q. & Pfleger, B. F. Revisiting metabolic engineering strategies for microbial synthesis of oleochemicals. Metab. Eng. 58, 35–46 (2020).
Keasling, J. et al. Microbial production of advanced biofuels. Nat. Rev. Microbiol. 19, 701–715 (2021).
Tomás-Pejó, E. et al. Production of short-chain fatty acids (SCFAs) as chemicals or substrates for microbes to obtain biochemicals. Biotechnol. Biofuels Bioprod. 16, 96 (2023).
Rauf, A. et al. Recent advances in the therapeutic application of short-chain fatty acids (SCFAs): an updated review. Crit. Rev. Food Sci. Nutr. 62, 6034–6054 (2022).
Ho Ahn, J. et al. Recent advances in microbial production of medium chain fatty acid from renewable carbon resources: a comprehensive review. Bioresour. Technol. 381, 129147 (2023).
Gajewski, J., Pavlovic, R., Fischer, M., Boles, E. & Grininger, M. Engineering fungal de novo fatty acid synthesis for short chain fatty acid production. Nat. Commun. 8, 14650 (2017).
Zhu, Z. et al. Expanding the product portfolio of fungal type I fatty acid synthases. Nat. Chem. Biol. 13, 360–362 (2017).
Rigouin, C. et al. Production of medium chain fatty acids by Yarrowia lipolytica: combining molecular design and TALEN to engineer the fatty acid synthase. ACS Synth. Biol. 6, 1870–1879 (2017).
Xu, P., Qiao, K., Ahn, W. S. & Stephanopoulos, G. Engineering Yarrowia lipolytica as a platform for synthesis of drop-in transportation fuels and oleochemicals. Proc. Natl Acad. Sci. USA 113, 10848–10853 (2016).
Leber, C. & Da Silva, N. A. Engineering of Saccharomyces cerevisiae for the synthesis of short chain fatty acids: synthesis of short chain fatty acids in yeast. Biotechnol. Bioeng. 111, 347–358 (2014).
Zhu, Z. et al. Multidimensional engineering of Saccharomyces cerevisiae for efficient synthesis of medium-chain fatty acids. Nat. Catal. 3, 64–74 (2020).
Steen, E. J. et al. Microbial production of fatty-acid-derived fuels and chemicals from plant biomass. Nature 463, 559–562 (2010).
Valencia, L. E. et al. Engineering Pseudomonas putida KT2440 for chain length tailored free fatty acid and oleochemical production. Commun. Biol. 5, 1363 (2022).
Torella, J. P. et al. Tailored fatty acid synthesis via dynamic control of fatty acid elongation. Proc. Natl Acad. Sci. USA 110, 11290–11295 (2013).
Choi, Y. J. & Lee, S. Y. Microbial production of short-chain alkanes. Nature 502, 571–574 (2013).
Jindra, M. A. et al. Evaluation of strategies to narrow the product chain-length distribution of microbially synthesized free fatty acids. Metab. Eng. 77, 21–31 (2023).
Lo, Y.-C., Lin, S.-C., Shaw, J.-F. & Liaw, Y.-C. Crystal structure of Escherichia coli thioesterase I/protease I/lysophospholipase L1: consensus sequence blocks constitute the catalytic center of SGNH-hydrolases through a conserved hydrogen bond network. J. Mol. Biol. 330, 539–551 (2003).
Deng, X. et al. Structure-guided reshaping of the acyl binding pocket of ‘TesA thioesterase enhances octanoic acid production in E. coli. Metab. Eng. 61, 24–32 (2020).
Lo, Y.-C., Lin, S.-C., Shaw, J.-F. & Liaw, Y.-C. Substrate specificities of Escherichia coli thioesterase I/protease I/lysophospholipase L 1 are governed by its switch loop movement. Biochemistry 44, 1971–1979 (2005).
Shin, K. S., Kim, S. & Lee, S. K. Improvement of free fatty acid production using a mutant acyl-CoA thioesterase I with high specific activity in Escherichia coli. Biotechnol. Biofuels 9, 208 (2016).
Maier, T., Leibundgut, M. & Ban, N. The crystal structure of a mammalian fatty acid synthase. Science 321, 1315–1322 (2008).
Maier, T., Leibundgut, M., Boehringer, D. & Ban, N. Structure and function of eukaryotic fatty acid synthases. Q. Rev. Biophys. 43, 373–422 (2010).
Grininger, M. Perspectives on the evolution, assembly and conformational dynamics of fatty acid synthase type I (FAS I) systems. Curr. Opin. Struct. Biol. 25, 49–56 (2014).
Herbst, D. A., Townsend, C. A. & Maier, T. The architectures of iterative type I PKS and FAS. Nat. Prod. Rep. 35, 1046–1069 (2018).
Smith, S., Witkowski, A. & Joshi, A. K. Structural and functional organization of the animal fatty acid synthase. Prog. Lipid Res. 42, 289–317 (2003).
Paiva, P. et al. Animal fatty acid synthase: a chemical nanofactory. Chem. Rev. 121, 9502–9553 (2021).
Chakravarty, B., Gu, Z., Chirala, S. S., Wakil, S. J. & Quiocho, F. A. Human fatty acid synthase: structure and substrate selectivity of the thioesterase domain. Proc. Natl Acad. Sci. USA 101, 15567–15572 (2004).
Bunkoczi, G. et al. Mechanism and substrate recognition of human holo ACP synthase. Chem. Biol. 14, 1243–1253 (2007).
Ploskoń, E. et al. A mammalian type I fatty acid synthase acyl carrier protein domain does not sequester acyl chains. J. Biol. Chem. 283, 518–528 (2008).
Crosby, J. & Crump, M. P. The structural role of the carrier protein – active controller or passive carrier. Nat. Prod. Rep. 29, 1111 (2012).
Buyachuihan, L., Stegemann, F. & Grininger, M. How acyl carrier proteins (ACPs) direct fatty acid and polyketide biosynthesis. Angew. Chem. Int. Ed. 63, e202312476 (2024).
Günenc, A. N., Graf, B., Stark, H. & Chari, A. in Macromolecular Protein Complexes IV (eds Harris, J. R. & Marles-Wright, J.) Vol. 99, 1–33 (Springer International Publishing, 2022).
Heil, C. S., Wehrheim, S. S., Paithankar, K. S. & Grininger, M. Fatty acid biosynthesis: chain-length regulation and control. ChemBioChem 20, 2298–2321 (2019).
Gusenda, C., Calixto, A. R., Da Silva, J. R., Fernandes, P. A. & Grininger, M. The kinetics of carbon-carbon bond formation in metazoan fatty acid synthase and its impact on product fidelity. Angew. Chem. Int. Ed. 64, e202412195 (2025).
Pemble, C. W. I. V., Johnson, L. C., Kridel, S. J. & Lowther, W. T. Crystal structure of the thioesterase domain of human fatty acid synthase inhibited by Orlistat. Nat. Struct. Mol. Biol. 14, 704–709 (2007).
Zhang, W. et al. Crystal structure of FAS thioesterase domain with polyunsaturated fatty acyl adduct and inhibition by dihomo-γ-linolenic acid. Proc. Natl Acad. Sci. USA 108, 15757–15762 (2011).
Rittner, A., Paithankar, K. S., Drexler, D. J., Himmler, A. & Grininger, M. Probing the modularity of megasynthases by rational engineering of a fatty acid synthase type I. Protein Sci. 28, 414–428 (2019).
Lu, X., Vora, H. & Khosla, C. Overproduction of free fatty acids in E. coli: implications for biodiesel production. Metab. Eng. 10, 333–339 (2008).
Rittner, A., Paithankar, K. S., Himmler, A. & Grininger, M. Type I fatty acid synthase trapped in the octanoyl-bound state. Protein Sci. 29, 589–605 (2020).
Olsen, J. G., Kadziola, A., von Wettstein-Knowles, P., Siggaard-Andersen, M. & Larsen, S. Structures of β-ketoacyl-acyl carrier protein synthase I complexed with fatty acids elucidate its catalytic machinery. Structure 9, 233–243 (2001).
Johansson, P. et al. Inhibition of the fungal fatty acid synthase type I multienzyme complex. Proc. Natl. Acad. Sci. USA 105, 12803–12808 (2008).
Okorafor, I. C., Chen, M. & Tang, Y. High-titer production of olivetolic acid and analogs in engineered fungal host using a nonplant biosynthetic pathway. ACS Synth. Biol. 10, 2159–2166 (2021).
Reimer, C. et al. Engineering the amoeba Dictyostelium discoideum for biosynthesis of a cannabinoid precursor and other polyketides. Nat. Biotechnol. 40, 751–758 (2022).
Gajewski, J. et al. Engineering fatty acid synthases for directed polyketide production. Nat. Chem. Biol. 13, 363–365 (2017).
Deshpande, S., Altermann, E., Sarojini, V., Lott, J. S. & Lee, T. V. Structural characterization of a PCP–R didomain from an archaeal nonribosomal peptide synthetase reveals novel interdomain interactions. J. Biol. Chem. 296, 100432 (2021).
Chhabra, A. et al. Nonprocessive [2 + 2]e- off-loading reductase domains from mycobacterial nonribosomal peptide synthetases. Proc. Natl Acad. Sci. USA 109, 5681–5686 (2012).
Barajas, J. F. et al. Comprehensive structural and biochemical analysis of the terminal myxalamid reductase domain for the engineered production of primary alcohols. Chem. Biol. 22, 1018–1029 (2015).
Gahloth, D. et al. Structures of carboxylic acid reductase reveal domain dynamics underlying catalysis. Nat. Chem. Biol. 13, 975–981 (2017).
Daniel, B. et al. Structure of the reductase domain of a fungal carboxylic acid reductase and its substrate scope in thioester and aldehyde reduction. ACS Catal. 12, 15668–15674 (2022).
Hu, Y. et al. Engineering carboxylic acid reductase for selective synthesis of medium-chain fatty alcohols in yeast. Proc. Natl Acad. Sci. USA 117, 22974–22983 (2020).
Little, R. F. & Hertweck, C. Chain release mechanisms in polyketide and non-ribosomal peptide biosynthesis. Nat. Prod. Rep. 39, 163–205 (2022).
Mullowney, M. W., McClure, R. A., Robey, M. T., Kelleher, N. L. & Thomson, R. J. Natural products from thioester reductase containing biosynthetic pathways. Nat. Prod. Rep. 35, 847–878 (2018).
Gahloth, D., Aleku, G. A. & Leys, D. Carboxylic acid reductase: structure and mechanism. J. Biotechnol. 307, 107–113 (2020).
Finnigan, W. et al. Characterization of carboxylic acid reductases as enzymes in the toolbox for synthetic chemistry. ChemCatChem 9, 1005–1017 (2017).
Xie, L., Yu, W., Gao, J., Wang, H. & Zhou, Y. J. Ogataea polymorpha as a next-generation chassis for industrial biotechnology. Trends Biotechnol. 42, 1363–1378 (2024).
Hansen, H. J. M., Carey, E. M. & Dils, R. Fatty acid biosynthesis VII. Substrate control of chain-length of products synthesised by rat liver fatty acid synthetase. Biochim. Biophys. Acta Lipids Lipid Metab. 210, 400–410 (1970).
Matsue, M. et al. Measuring the antimicrobial activity of lauric acid against various bacteria in human gut microbiota using a new method. Cell Transplant. 28, 1528–1541 (2019).
Nainggolan, M. & Sinaga, A. G. S. Characteristics of fatty acid composition and minor constituents of red palm olein and palm kernel oil combination. J. Adv. Pharm. Technol. Res. 12, 22–26 (2021).
Parsons, S., Raikova, S. & Chuck, C. J. The viability and desirability of replacing palm oil. Nat. Sustain. 3, 412–418 (2020).
Gao, Y. et al. Structural insights into catalytic mechanism and product delivery of cyanobacterial acyl-acyl carrier protein reductase. Nat. Commun. 11, 1525 (2020).
Webb, B. & Sali, A. Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinformatics https://doi.org/10.1002/cpbi.3 (2016).
Qiang, Y. et al. Metabolic engineering strategies to produce medium-chain oleochemicals via acyl-ACP:CoA transacylase activity. Nat. Commun. 13, 1619 (2022).
Srivastava, Y., Semwal, A. D. & Majumdar, A. Quantitative and qualitative analysis of bioactive components present in virgin coconut oil. Cogent Food Agric. 2, 1164929 (2016).
Bligh, E. G. & Dyer, W. J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917 (1959).
Zhai, X., Gao, J., Li, Y., Grininger, M. & Zhou, Y. J. Peroxisomal metabolic coupling improves fatty alcohol production from sole methanol in yeast. Proc. Natl Acad. Sci.USA 120, e2220816120 (2023).
Gao, N., Gao, J., Yu, W., Kong, S. & Zhou, Y. J. Spatial–temporal regulation of fatty alcohol biosynthesis in yeast. Biotechnol. Biofuels Bioprod. 15, 141 (2022).
Yu, W., Gao, J., Zhai, X. & Zhou, Y. J. Screening neutral sites for metabolic engineering of methylotrophic yeast Ogataea polymorpha. Synth. Syst. Biotechnol. 6, 63–68 (2021).
Gao, J., Gao, N., Zhai, X. & Zhou, Y. J. Recombination machinery engineering for precise genome editing in methylotrophic yeast Ogataea polymorpha. iScience 24, 102168 (2021).
Gao, J., Li, Y., Yu, W. & Zhou, Y. J. Rescuing yeast from cell death enables overproduction of fatty acids from sole methanol. Nat. Metab. 4, 932–943 (2022).
Acknowledgements
Support for this work was received by intramural funds of the Goethe University Frankfurt, a Kekulé Scholarship awarded to C.G., German Research Foundation (DFG grant GR3854/10-1 to M.G.), the National Natural Science Foundation of China (22425807 to Y.J.Z.) and Unilever research grant (MA-2023-01406N to Y.J.Z.). M.G. and Y.J.Z. received support from the Sino-German Center for Research Promotion (SGC grant M-0246).
Funding
Open access funding provided by Johann Wolfgang Goethe-Universität.
Author information
Authors and Affiliations
Contributions
M.G. and Y.J.Z. designed the research. D.L.L., A.R. and X.Z. conceived and led the project. D.L.L. and A.R. designed the in vitro experiments using the full-length constructs, C.G. designed the KS in vitro experiments and X.Z. designed the in vivo experiments. D.L.L. collected the in vitro data of the full-length constructs for chain length control, M.H. collected the in vitro data of the full-length constructs for the reductive release, C.G. and S.B. collected the in vitro KS data, X.Z. collected the O. polymorpha in vivo data and N.G. collected the S. cerevisiae in vivo data. D.L.L., X.Z., C.G., A.J.J., Y.J.Z. and M.G. analyzed the data. D.L.L. and M.G. wrote the original draft and X.Z., C.G. and Y.J.Z. reviewed and edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
D.L.L., A.R., C.G. and M.G. are inventors of the patent application EP25157462 (filed on 12 February 2025) by Goethe University Frankfurt on FAS variants for SMCFA production. X.Z. and Y.J.Z. are inventors of the patent 202510153331.7 (accepted) by Dalian Institute of Chemical Physics, Chinese Academy of Sciences on yeast cell engineering for SMCFA production. The other authors declare no competing interests.
Peer review
Peer review information
Nature Chemical Biology thanks Clay Semenkovich and the other, anonymous reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information (download PDF )
Supplementary Figs. 1–33, Methods and Tables 1–7.
Supplementary Table 1 (download XLSX )
Lists of oligonucleotides used in this study.
Source data
Source Data Fig. 2 (download XLSX )
Statistical source data.
Source Data Fig. 3 (download XLSX )
Statistical source data.
Source Data Fig. 4 (download XLSX )
Statistical source data.
Source Data Fig. 5 (download XLSX )
Statistical source data.
Source Data Fig. 6 (download XLSX )
Statistical source data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ludig, D.L., Zhai, X., Rittner, A. et al. Engineering metazoan fatty acid synthase to control chain length applied in yeast. Nat Chem Biol (2026). https://doi.org/10.1038/s41589-025-02105-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41589-025-02105-w
