
Abstract
Alzheimer’s disease (AD) and AD-related dementias (AD/ADRD) have a substantial genetic basis, with APOE4 homozygotes increasingly recognized as a distinct genetic subtype. To identify genotype-specific metabolic pathways and modifiable risk factors, we integrated genetic, plasma metabolomic and dietary data from 4,215 women and 1,490 men in prospective cohorts. Here we show that the associations of 57 metabolites with dementia risk varied by APOE4 genotype or other AD/ADRD risk variants. For example, cholesteryl esters and sphingomyelins were most strongly associated with increased dementia risk in APOE4 homozygotes, whereas inverse associations with glycerides were specific to this genotype. Dimethylguanidino-valeric acid was more strongly associated with dementia risk among carriers of the rs2154481-C allele (APP). Adherence to the Mediterranean diet more effectively modulated dementia-related metabolites in APOE4 homozygotes, suggesting targeted prevention strategies. Incorporating metabolomic data modestly improved dementia risk prediction, particularly during early follow-up. Mendelian randomization analysis identified 19 putative causal relationships between metabolites and cognitive outcomes, including protective effects of 4-guanidinobutanoate, carotenoids and N6-carbamoylthreonyladenosine. These findings reveal genotype-dependent metabolic profiles of cognitive health and support precision nutrition approaches for ADRD prevention.
Similar content being viewed by others


Main
Alzheimer’s disease (AD) and AD-related dementias (AD/ADRD) are neurodegenerative disorders characterized by progressive decline in memory, cognitive function and the ability to perform daily activities1. AD/ADRD has a substantial genetic basis, with heritability estimated at up to 80% from twin studies2. The apolipoprotein E (APOE) gene is the strongest genetic risk factor for sporadic AD3; carrying 1 APOE-ε4 (APOE4) allele increases risk 3–4-fold and 2 alleles increase risk 8–12-fold compared to the common APOE-ε3 allele4. APOE4 exacerbates amyloid-β (Aβ) pathology in the brain5 and is strongly linked to dysregulation in lipid metabolism and impaired cerebral glucose metabolism6, highlighting its multifaceted role in the pathology of ADRD. Recent studies further suggest that APOE4 homozygotes exhibit unique clinical, pathological and biomarker changes that begin at younger ages7. Beyond APOE, genome-wide association studies (GWASs) have identified common genetic variants at >80 loci associated with AD/ADRD risk, including ABCA7, BIN1 and CR1, confirming its polygenic basis and implicating key pathogenic pathways such as immune response and endocytosis8,9. However, previous research has predominantly focused on the genetic effects of a limited set of biomarkers that primarily capture Aβ and tau pathologies, often assessed during the prodromal stages of the disease or with limited duration of prospective follow-up. There is a notable lack of evidence about the impact of genetic factors on early stage biomarkers derived from high-throughput omics approaches, such as metabolomics, and their associations with ADRD risk. Furthermore, the potential to leverage identified gene–biomarker interactions to develop individualized prevention and treatment strategies remains largely unexplored.
Metabolomics provides a readout of the combined effects of genetic and environmental factors, offering an expansive snapshot of metabolic states10. Genetic variations, particularly in genes involved in enzyme and transporter functions, directly influence metabolite production, degradation and circulation11. Environmental factors, especially diet, interact with genetics to further shape the metabolome by introducing exogenous metabolites and modulating key metabolic processes, such as inflammation, energy production and oxidative stress12,13. Metabolomic profiles across various tissues, including plasma14, brain15 and cerebrospinal fluid16, have shown associations with ADRD risk and cognitive function. Emerging evidence suggests that the APOE4 genotype may modulate associations between plasma metabolites and dementia risk17,18,19,20. For example, recent findings indicate that females carrying the APOE4 allele have distinct metabolomic profiles, reflecting alterations in lipid and amino acid metabolism, potentially increasing susceptibility to AD17,19. Despite these findings, there is a lack of data from large prospective studies investigating the extent to which associations between the metabolome and cognitive outcomes vary by genetic background. In addition, it remains unclear whether certain modifiable risk factors, such as diet, can mitigate ADRD risk and cognitive decline by targeting specific metabolic pathways across different genetic risk groups.
To examine the interplay of genetics, the plasma metabolome and diet in relation to dementia risk and cognitive function, we conducted a prospective analysis of 4,215 women during a 34-year follow-up in the Nurses’ Health Study (NHS). Notably, we observed widespread variations in associations between metabolites and cognitive outcomes across genotypes, most notably among APOE4 homozygotes. Associations of the Mediterranean diet (MedDiet) with metabolites and dementia risk were also genotype dependent, with metabolites mediating the MedDiet–dementia risk association only among APOE4 carriers. Integration of genetics with metabolomics improved the prediction of cognitive outcomes. Key findings were replicated in 1,490 men in the Health Professionals Follow-Up Study (HPFS). Two-sample Mendelian randomization (MR) further supported causal relationships between plasma metabolites and cognitive outcomes. To our knowledge, this is one of the first studies to demonstrate genotype-dependent associations between metabolites and ADRD risk, with additional findings that inform individualized dietary approaches for ADRD prevention.
Results
Integrating genetics, plasma metabolomics and dietary intakes to study ADRD etiology in long-running prospective studies
We prospectively followed 4,215 women in the NHS from 1989 to 2023 (mean age, at baseline, 57 years; Fig. 1a and Supplementary Table 1), during which 485 participants developed dementia. In addition, we longitudinally assessed objective cognitive function using a telephone-based battery, including the Telephone Interview for Cognitive Status (TICS), in a subset of 1,037 participants (1995–2008). In the replication analyses in the HPFS, 1,490 men (mean age 63 years at baseline) were prospectively followed from 1993 to 2023, with 121 dementia cases documented (Extended Data Fig. 1a and Supplementary Table 2). Details of baseline characteristics in both cohorts are provided in Supplementary Text.

a, Prospective follow-up of 4,215 women in the NHS from 1989 to 2023. Genetic and metabolomic profiles were generated from blood samples collected at baseline. Detailed demographic, lifestyle, dietary, medical history and medication use data were collected via questionnaires. Dementia cases were ascertained through the follow-up as a composite endpoint of incident dementia and death due to dementia. In addition, a telephone-based neuropsychological assessment battery was administered longitudinally from 1995 to 2008 to assess cognitive function in a subset of 1,037 participants. A total of 1,490 men from the HPFS were included as a replication cohort (Extended Data Fig. 1a). b, Distribution of plasma metabolites (n = 401). The outer circle represents the variation of each metabolite, with a gradient in gray indicating the coefficient of variation. The inner circle displays the mean relative abundance of each metabolite, shown as a gradient in blue. The innermost circle color codes represent the different HMDB superclasses defined based on chemical structural similarities. c, Overall genetic structure associated with individual metabolites. Each dot represents an individual and is colored by APOE4 genotype, showing no clear pattern between the overall population substructure and APOE4 genotype. The metabolites with the highest Pearson’s correlations with the top two genetic PCs from each metabolite superclass are included on the plot as arrows, colored by their superclass (see legend for b). The arrowhead coordinates represent the correlation coefficients of the metabolites with genetic PC1 and PC2. d, Associations between established genetic risk factors for AD/ADRD and dementia risk. The lines indicate cumulative incidence across APOE4 genotypes and tertiles of the PRS of ADRD (excluding the APOE region) over the follow-up period, with shaded areas representing 95% CIs and P values from the log-rank test annotated. Consistent with the curves, unadjusted hazard ratios (HRs) were estimated using Cox proportional hazards (PH) model; covariate-adjusted HRs with 95% confidence intervals (CIs) are provided in Supplementary Table 4. Person time was accrued from baseline until the earliest occurrence of an incident dementia case, dementia death or the end of follow-up. No adjustment was made for multiple comparisons, because this was a hypothesis-driven analysis. e, A wide range of adherence to the MedDiet, as assessed by a dietary index and intake levels of food and nutrient components of MedDiet. All analyses and distributions were based on data from 4,215 NHS participants. All statistical tests were two sided. MAG, monoacylglycerol; TAG, triacylglycerol. Panel a created using BioRender.com.
Metabolomic data were generated from plasma samples using a liquid chromatography–mass spectrometry (LC–MS)-based platform in both the NHS and the HPFS; a total of 401 metabolites from 10 Human Metabolome Database (HMDB) superclasses were included in the NHS analyses after quality control (QC), with 254 of the metabolites available in the HPFS (Fig. 1b and Supplementary Table 3). Genotyping data were generated from blood samples, followed by QC and imputation (Methods). We extracted the two APOE variants along with 73 other common variants identified from AD/ADRD GWASs9,21 and calculated two polygenic risk scores (PRSs) for ADRD; one included the two APOE variants and one excluded the APOE region, using weights from published studies9,21. We first investigated the global influence of genetic variations on the plasma metabolome, using principal components (PCs) to capture genetic structure. Strong correlations were observed between genetic PC1 or PC2 and metabolites previously linked to ADRD risk, such as trimethylamine N-oxide22 and lithocholate23 (Fig. 1c and Extended Data Fig. 2a). We next assessed the specific influence of APOE4 genotype on the metabolome. As expected, APOE4 homozygosity was broadly associated with elevated lipid metabolites compared to noncarriers (Extended Data Fig. 2b).
To validate the dementia outcome, we examined plasma phosphorylated tau 217 (p-tau217), an established biomarker for early AD diagnosis24, in 103 NHS participants and found an approximately 3-fold higher dementia risk comparing the highest and lowest quartiles of p-tau217. We further confirmed that carrying APOE4 alleles or having a higher PRS of ADRD was associated with significantly increased risk of dementia and poorer cognitive function in both cohorts (Fig. 1d, Extended Data Fig. 1b, Supplementary Fig. 1, Supplementary Table 4 and Supplementary Text).
We collected long-term dietary data using extensively validated semiquantitative food frequency questionnaires (SFFQs) in both cohorts. To assess dietary quality, we employed the MedDiet index, the only dietary pattern causally linked to delayed cognitive decline in a long-term, randomized controlled trial25. A widespread distribution of MedDiet adherence was observed (Fig. 1e), with higher MedDiet index scores associated with older age, lower body mass index, higher education level and more physical activity (Supplementary Tables 2 and 5).
APOE4 homozygosity exhibited distinct metabolomic profiles of dementia risk
We identified 49 significant interactions of metabolites with APOE4 genotypes in relation to dementia risk at a false discovery rate (FDR) < 0.05 (Fig. 2a,b, Extended Data Fig. 3 and Supplementary Table 6). All significant interactions were specific to APOE4 homozygotes, aligning with recent findings on this isoform in AD pathology7 and suggesting that it exhibits a distinct plasma metabolomic profile associated with ADRD risk, evident even decades before disease onset.

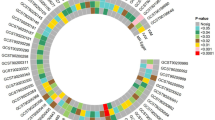
a, Significant variation in the association between metabolites and dementia risk across different genotypes. Left, in the two heatmaps, the color gradient denoting the HR for dementia risk per 1-s.d. increment in metabolite levels among individuals with different genetic predispositions, as defined by APOE4 genotype or PRS of ADRD (including the APOE variants), estimated using Cox PH model. Only metabolites with FDR < 0.05 for their interaction terms with genotype are displayed in the heatmaps. Right, the color gradient in the two heatmaps representing the product of the β coefficient and the −log10(FDR) of the interaction term between each metabolite and genotype, as defined by the APOE4 genotype or other common AD/ADRD genetic variants, from Cox PH model. In all heatmaps, associations or interactions with FDR < 0.05 are indicated by double asterisks (**) and those with FDR < 0.25 by a single asterisk (*). Metabolites are grouped according to HMDB superclasses. The analyses were conducted among 4,215 NHS participants. b, Gene–metabolite interactions related to dementia risk widely distributed across metabolite superclasses and genotypes. The Manhattan plot displays metabolome-wide interaction results, represented by −log10(FDR) values for interaction terms from Cox PH models. Each dot represents a metabolite colored by the direction of interaction and grouped by HMDB superclass. Top, for the APOE4 genotype, the data point with the lower FDR between heterozygous (diamond) and homozygous (square) APOE4 interactions included for each metabolite. Bottom, for other common AD/ADRD variants, the most significant interaction across all 73 variants shown for each metabolite. The analyses were conducted among 4,215 NHS participants. c, Selected associations between metabolites and dementia risk with FDR for interaction <0.05, stratified by genotype. The first row presents stratified HRs and 95% CIs for dementia risk per 1-s.d. increment in metabolite level, categorized by the APOE4 genotype, with FDR values for interaction terms between APOE4 heterozygosity and homozygosity annotated (using the noncarrier as the reference group). The second row displays stratified results (HR and 95% CIs per 1-s.d. increment in metabolite level) by AD/ADRD variants, with FDR values for interaction terms with the variant effect allele dosage annotated. Genotype groups were defined based on rounded allele dosages. Results for the rs1800978 GG genotype group are excluded due to data sparsity. The analyses were conducted across 4,215 NHS participants. d, Consistency of metabolite–APOE4 interaction results across models with dementia risk and cognitive function as dependent variables. Each dot represents a metabolite with significant APOE4 interactions, colored by the HMDB superclass. Pearson’s correlation coefficients in the β coefficients for interaction terms between metabolites and APOE4 carrier status estimated from Cox PH models, with dementia as the dependent variable, and from generalized linear models, with cognitive function scores as the dependent variable, are annotated on each figure. APOE4 carriers were not further divided into heterozygotes and homozygotes due to data sparsity among homozygotes with non-missing values for each metabolite in the cognitive function subset. Dementia risk analyses were conducted among 4,215 NHS participants and cognitive function analyses among 1,037 NHS participants. All statistical tests were two sided. DAG, diacylglycerol; PC, phosphatidylcholine; PE, phosphatidylethanolamine; TAG, triacylglycerol.
Subgroup analyses further confirmed distinct association patterns between metabolites and dementia risk among APOE4 homozygotes compared to others, with variations in both the magnitude and, in some cases, the direction of associations (Fig. 2a, Supplementary Fig. 2 and Supplementary Table 7). For example, a significant positive association between betaine and dementia risk was observed only among APOE4 homozygotes (Fig. 2c). Plasma betaine levels reflect both dietary intake and one-carbon metabolism activity26. The protective, albeit nonsignificant, association observed among APOE4 noncarriers and heterozygotes may primarily reflect dietary betaine’s benefit for cognitive health27, whereas, in APOE4 homozygotes, elevated betaine may indicate methylation imbalance and metabolic dysregulation, contributing to an increased dementia risk26,28. We observed broadly positive associations across cholesteryl esters (CEs), sphingomyelins (SMs) and dementia risk in all genetic risk groups, with the strongest associations in APOE4 homozygotes. The APOE4 allele promotes the accumulation of cholesterol and CEs in the brain29, activating inflammatory pathways that exacerbate neuronal damage and contributing to the formation of amyloid plaques and tau tangles, thereby increasing dementia risk30. We observed inverse associations between glycerides and dementia risk, specifically among APOE4 homozygotes, where elevated glyceride levels likely reflect this reduced delipidation, which may, in turn, limit the aggregation of apolipoprotein E (ApoE) and the formation of amyloid plaques31.
Replication of the findings for dementia risk in objective cognitive function yielded broadly consistent results (Pearson’s r ranging from −0.78 to −0.63 for the interaction effect estimates; Fig. 2d and Supplementary Tables 8 and 9). Sensitivity analyses, excluding family history of dementia from the covariates or modeling dementia case and death separately, showed similar results (Supplementary Figs. 3 and 4). Independent replication of the NHS findings in the HPFS yielded broadly consistent results (Pearson’s r = 0.40), with 32 out of 38 significant interactions (84.2%) in the same direction. Notably, 4 interactions reached P < 0.05 and 10 reached P < 0.10, including inverse interactions with glycerides, although some variation between women and men remained (Extended Data Fig. 4 and Supplementary Table 10).
Common AD/ADRD risk variants modified the associations between plasma metabolites and dementia risk
Although APOE4 is the major contributor to the genetic risk of dementia, recent GWASs have identified many other common variants linked to AD/ADRD risk8,9. We examined how these risk variants, either aggregated into PRSs or as individual variants, may modify the associations between metabolites and dementia risk (Supplementary Tables 11 and 12). Although no significant interaction between PRSs and metabolites was detected after multiple testing correction (Fig. 2a and Supplementary Tables 13 and 14), we identified eight significant interactions between individual AD/ADRD variants, including those mapped to ABCA1, APP, ADAMTS1, CTSH and USP6NL, and metabolites in relation to dementia risk with an FDR < 0.05 (Fig. 2a,b, Supplementary Fig. 5 and Supplementary Table 15).
We observed that the positive association between 1-methylhistamine, a metabolite involved in immune and inflammatory responses in the brain32 (Fig. 2c), and dementia risk was significantly more pronounced in individuals carrying the rs1800978-G allele mapped to ABCA1, a gene that plays a crucial role in clearing Aβ peptide from the brain33. Dimethylguanidino-valeric acid, a metabolite associated with impaired fatty acid and amino acid metabolism, showed a stronger positive association with dementia risk among individuals carrying the C allele at rs2154481, a variant mapped to the APP gene that encodes Aβ precursor protein (APP), suggesting that dysregulated lipid and glucose metabolism may influence the processing of APP, leading to increased Aβ production34. In addition, lipid metabolites showed significant interactions with AD/ADRD genetic variants. For example, C32:2 phosphatidylcholine exhibited a positive interaction with an ADAMTS1-linked variant (rs2830489-T), implicating extracellular matrix remodeling and neuroinflammation in neurodegeneration35 (Supplementary Text).
MedDiet may more effectively modulate metabolites implicated in dementia risk in APOE4 homozygotes
A key distinction between metabolomics and genetics is that metabolites can be modified by exogenous factors and may serve as targets for intervention; in particular, diet significantly impacts the metabolome36. We thus examined whether diet, specifically the MedDiet, which has been implicated in cognitive health25, could modulate metabolite levels in individuals with different genetic predispositions to AD/ADRD.
We found that individuals with greater adherence to the MedDiet had a significantly lower risk of dementia and better cognitive function (Fig. 3a and Supplementary Fig. 6). Notably, these protective associations for dementia risk were more pronounced among APOE4 homozygotes compared to noncarriers and heterozygotes (Fig. 3b), although no clear trend was observed when stratifying by ADRD PRS (Extended Data Fig. 5). The same patterns were observed in the HPFS (Extended Data Fig. 6a,b). Next, to evaluate MedDiet’s impact on the overall metabolomic profile, we used a random Forest (RF) classifier to distinguish individuals with high versus low MedDiet adherence based on plasma metabolite levels. This classifier demonstrated excellent performance, with an area under the receiver operating characteristic (ROC) curve (AUC) of 0.76 in the NHS (Fig. 3c) and 0.72 when replicating in the HPFS (Extended Data Fig. 6c). In addition, individual components of the MedDiet, such as nuts, fruit and monounsaturated fats, were strongly associated with overall metabolomic patterns, as captured by the top two metabolite PCs (Extended Data Fig. 7).

a, Higher adherence to the MedDiet prospectively associated with a lower risk of dementia and enhanced cognitive performance, as assessed by the telephone-based neuropsychological assessment battery (TICS). For dementia risk analysis, a restricted cubic spline Cox PH model estimated HRs and 95% CIs across varying levels of the MedDiet index, using 0 as the reference. The P value from a likelihood ratio test comparing the model without the MedDiet index and the model with its spline term is annotated. For the TICS score analysis, a generalized linear model estimated the adjusted TICS score and corresponding 95% CI across MedDiet index levels, with P values annotated. The analyses were conducted among NHS participants with cognitive and dietary data (n = 86,740 for dementia analysis and n = 16,244 for cognitive function analysis). b, The protective association between adherence to the MedDiet and risk of dementia most pronounced among APOE4 homozygotes. Stratified HR and 95% CIs for dementia risk per a 1-unit increment in the MedDiet index score, categorized by the APOE4 genotype, were estimated from Cox PH models, with stratified P values annotated (unadjusted for multiple comparisons in the hypothesis-driven analysis). The analyses were conducted among NHS participants with genetic, dietary and dementia outcome data (n = 16,497). c, Strong association between adherence to MedDiet and the overall plasma metabolome from an RF model to classify individuals in the top versus the bottom quartile of the MedDiet index based on plasma metabolites. For the RF classification, the dataset was randomly divided into training (60%) and test (40%) sets. The ROC curve for the test set is shown, with the AUC and 95% CI annotated on the plot. The analyses were conducted among 4,215 NHS participants. d, Associations between MedDiet adherence and plasma metabolite levels differing by APOE4 genotype. The heatmap shows β coefficients representing a 1-s.d. increment in the MedDiet index from a generalized linear model, with plasma metabolite levels as the dependent variable. The analyses were conducted among 4,215 NHS participants. e, Select associations between the MedDiet index and plasma metabolite levels with P < 0.05 from the likelihood ratio test for the interaction between APOE4 genotype and MedDiet index in relation to metabolites, using a generalized linear model stratified by APOE4 genotype. Covariate-adjusted residuals of metabolites are shown along with fitted linear regression lines, 95% CIs and P values for interaction. These results were not adjusted for multiple testing. The analyses were conducted across 4,215 NHS participants. All statistical tests were two sided.
To assess whether the metabolic response to MedDiet is genotype dependent, particularly in relation to APOE4, we examined the association between MedDiet adherence and metabolite levels across APOE4 genotypes. As anticipated, the overall association patterns differed among APOE4 homozygotes and heterozygotes compared to noncarriers (Fig. 3d and Supplementary Table 16), with interaction analysis supporting these distinctions (Supplementary Table 17). Similar patterns were observed in the HPFS (Extended Data Fig. 6d, Supplementary Fig. 7 and Supplementary Table 18). Consistent with findings from the PREDIMED trial involving a randomized MedDiet37, greater adherence to the MedDiet was associated with higher levels of unsaturated glycerides and lower levels of saturated glycerides, lipid patterns potentially beneficial for cognitive health, as well as increased levels of established neuroprotective compounds, including piperine, betaine and pantothenic acid38,39 (Extended Data Fig. 8 and Supplementary Table 16). Among the metabolites showing suggestive APOE4–MedDiet interactions (P < 0.05), we observed an inverse association between MedDiet and asparagine levels exclusively among APOE4 homozygotes (Fig. 3e). This may reflect MedDiet-driven changes in amino acid metabolism, including glutaminolysis and the tricarboxylic acid cycle, which are key to asparagine regulation. Given asparagine’s role in protein and nucleotide synthesis, its reduction may signal broader metabolic benefits specific to APOE4 homozygotes40. We also identified nominally significant interactions between MedDiet and the APOE4 genotype in relation to 1,7-dimethyluric acid, a derivative of caffeine metabolism with established antioxidant properties and potential neuroprotective effects41 (Supplementary Text).
Furthermore, we found that 39.5% of the association between MedDiet adherence and dementia risk was mediated by a set of metabolites among APOE4 carriers (P = 0.05), whereas no mediation effect was observed among noncarriers or in the full dataset (Methods). Besides the APOE4 genotype, broad interactions were also observed for ADRD PRS and individual variants (Supplementary Tables 19–21). These findings collectively indicated that the MedDiet’s potential to modulate cognitive health-related metabolites varied by APOE4 genotype, suggesting that this dietary pattern could be an effective strategy to delay dementia onset in APOE4 homozygotes, despite their higher risk profile.
Prediction of dementia risk using genetic, metabolomic and dietary factors
Given that genetics, metabolites and MedDiet adherence are all linked to dementia risk, we further examined how incorporating these factors could enhance the prediction of cognitive outcomes. Compared to a baseline model, which included age, family history of dementia, education level, smoking status, history of depression or regular antidepressant drug use and the MedDiet index, adding APOE4 and ADRD PRS to Cox model moderately improved the performance for predicting dementia risk (Fig. 4a; average AUC improved from 0.75 to 0.77). This also demonstrated that genetic factors capture additional information beyond family history of dementia and that the ADRD PRS adds modest but incremental predictive value beyond APOE4. Adding metabolites predictive of dementia risk to the model further improved the time-specific model performance, indicating that metabolites provide additional predictive value beyond the MedDiet and other major dementia risk factors (average AUC = 0.78; Supplementary Table 22). As expected, these baseline characteristics were better at predicting short-term dementia risk, that is, 15-year risk, compared to long-term risk, which may be attributed to the inherent within-individual variability of metabolomic measurements over longer follow-up periods, potentially introducing random measurement error. Similar patterns were observed for Harrell’s C-index (Extended Data Fig. 9). The performance of different models in APOE4 subgroups did not substantially deviate, likely due to the limited sample size and number of cases within these subgroups, which may have introduced substantial instability. Future studies with larger sample sizes are warranted to more reliably evaluate prediction performance within APOE4 subgroups.

a, The inclusion of genetic factors improving dementia risk prediction using Cox PH model, with an additional modest enhancement when plasma metabolites also included. Time-dependent ROC curve analyses were conducted for dementia risk over both the entire follow-up period and the first 15 years of follow-up. The baseline model predictors included age, family history of dementia, education level, smoking status, history of depression or regular antidepressant use and MedDiet index. The PRS of ADRD excluded variants in the APOE region (see Methods for selection of metabolite predictors). b, Plasma metabolites among the top contributors for predicting dementia risk as quantified by the SHAP value. Feature contributions were evaluated for Cox PH model to predict overall and 15-year dementia risk, including the full list of predictors. SHAP values were calculated for each category of predictors by summing the SHAP value of all predictors in that category. Features were ranked by the SHAP value from the highest to the lowest for predicting the overall and 15-year dementia risk. c, Integration of genetic and metabolomic data enhancing cognitive status prediction within APOE4 subgroups. The heatmap displays AUCs from an RF model classifying participants in the highest versus the lowest tertile of the overall TICS score. In subgroup analyses by APOE4 carrier status, APOE4 genotype was excluded as a predictor. For all analyses, the NHS dataset (n = 4,215) was randomly divided into training (60%) and test (40%) sets; models were fitted on the training set and evaluated on the test set. All results shown are from the test set.
We employed the SHapley Additive exPlanation (SHAP) values to quantify the contributions of individual predictors to dementia risk predictions. As anticipated, age, APOE4 and ADRD PRS were among the top contributors (Fig. 4b and Supplementary Table 23). Plasma metabolites meaningfully contributed to the prediction throughout the entire follow-up period, with their overall contribution ranking just below age and genetic factors. In contrast, for short-term risk prediction, MedDiet emerged as a major predictor. The relatively modest contribution of MedDiet to overall follow-up predictions likely reflects the use of baseline dietary data only, which does not account for changes in dietary behavior over time. We observed similar patterns in the HPFS (Extended Data Fig. 10a,b) and when predicting a dichotomized TICS score (highest versus lowest tertile) within APOE4 subgroups, whereas no improvement was observed in the full dataset (Fig. 4c and Supplementary Fig. 8). The unstable results for cognitive score prediction likely reflect limited sample size in the subset. Larger studies are needed to further assess the predictive utility of these models for cognitive function.
Putative causal relationships between metabolomic features and cognitive outcomes
Last, genetics can offer mechanistic insights by using variants as instrumental variables to test whether associations between metabolites and cognitive outcomes are causal using the MR approach. To maximize the statistical power and mitigate biases, we implemented a two-sample MR design leveraging data from published GWASs and selected genetic instruments for 657 metabolites and 133 ratios of metabolite pairs sharing an enzyme or transporter (reflecting metabolic flux; Supplementary Tables 24 and 25)11. Cognitive outcomes included overall dementia, AD, vascular dementia and cognitive function (Fig. 5a and Methods).

a, Schematic of the two-sample MR and colocalization analyses. Genetic instruments were selected for 657 metabolites and 133 metabolite ratios from a published GWAS. Summary statistics for overall dementia, AD, vascular dementia and cognitive performance were also obtained from published GWASs (Methods). Two-sample MR was performed to identify putative causal relationships between metabolites or ratios and cognitive outcomes, followed by colocalization analysis for the causal associations with an FDR < 0.05. b, Identification of numerous putative causal interrelationships of various metabolites, metabolite ratios and cognitive outcome using genetic instruments. The chord diagram displays causal relationships with an FDR < 0.05 from MR analyses using Wald ratio, inverse variance-weighted or MR Egger methods, where each arc represents an identified link between a metabolite or ratio and a cognitive outcome. Arcs and nodes are color coded by the HMDB superclasses. c, Colocalization analyses strengthening evidence of causality, suggesting that the identified putative causal relationships between metabolites or ratios and cognitive outcomes have potential shared causal variants and biology. Putative causal relationships, represented by the odds ratios (ORs) or β coefficients with 95% CIs, are shown for associations with FDR < 0.05 and colocalization signals. Bayesian colocalization analysis was performed within the ±500-kb region around a genetic instrument (Methods). If multiple instruments were used, a causal association was reported if the metabolite or ratio and cognitive outcomes colocalized at least one genetic locus. Colocalization signals were reported for a locus if the conditional probability of colocalization, PP.H4/(PP.H3 + PP.H4), was >70%, where PP.H3 is posterior probability that the two traits have independent causal variants and PP.H4 is the posterior probability that the two traits share a single causal variant. d, Regional genetic association plots providing evidence of potential shared causal variants affecting both metabolites or ratios and cognitive outcomes at specific genetic loci. The plots display genetic association results for metabolites or ratios and cognitive outcomes at three colocalized loci with PP.H4/(PP.H3 + PP.H4) > 70%. Each plot is annotated with the genetic instrument and dots are color coded according to their linkage disequilibrium with the instrumental variant. The −log10(P) values for both metabolites or ratios and cognitive outcomes were obtained from the original GWASs. The sample sizes for the original GWASs are as follows: metabolites or ratios (n = 8,299), cognitive performance (n = 257,841), dementia (5,933 cases and 166,584 controls), AD (90,338 cases and 1,036,225 controls) and vascular dementia (881 cases and 211,508 controls) (Methods). All statistical tests were two sided. Panel a created using BioRender.com.
We identified 99 significant causal relationships involving 95 metabolites or ratios across all 4 outcomes at an FDR < 0.05 (Fig. 5b and Supplementary Table 26). A colocalization analysis was further performed to prioritize metabolite–outcome pairs with potential shared genetic basis and biology; we identified 19 pairs with a conditional posterior probability of colocalization >70% under a single causal variant assumption (Fig. 5c and Supplementary Table 27).
A notable finding was the protective causal effect of 4-guanidinobutanoate (4-GBA) on dementia, supported by colocalization signals (Fig. 5d); 4-GBA is a gamma-aminobutyric acid (GABA)-related metabolite involved in inhibitory neurotransmission42 and may counteract excitotoxicity, a known contributor to dementia pathogenesis43. The genetic instrument, rs140527149, is mapped to AGMAT, which encodes agmatinase, an enzyme involved in the degradation of agmatine in the brain44. Evidence suggests that agmatine may have therapeutic potential for AD by modulating Aβ production, aggregation and clearance45. Our findings highlight potential shared pathways involving 4-GBA, GABA and agmatine in dementia pathology. Carotene diol (1) and (2), naturally occurring carotenoids with potent antioxidant properties46, showed protective effects against AD, consistent with randomized controlled trial evidence causally linking carotenoid intake to reduced cognitive decline47. Building on studies showing altered glutamine levels in the brain and cerebrospinal fluid of individuals with AD48, our findings also highlighted glutamine’s potentially neuroprotective role in AD (Supplementary Fig. 9). For cognitive function, N6-carbamoylthreonyladenosine exhibited a strong beneficial effect, with colocalization signals allowing for multiple causal variants (Methods and Supplementary Table 28). A number of causal relationships were identified between metabolites involved in ATP metabolism, suggesting a causal role for the interplay between disrupted energy metabolism and oxidative stress in the pathophysiology of cognitive decline (Supplementary Text). Future investigations into these pathways could inform targeted therapeutic strategies.
Discussion
Elucidating the pathogenic mechanisms underlying ADRD and identifying modifiable factors capable of targeting these mechanisms are critical for prevention. Given the strong genetic basis of ADRD, accounting for genetic predisposition is essential when investigating these mechanisms. In this study, we leveraged up to 34 years of follow-up and multiomics data from the NHS, with independent replication in the HPFS, to comprehensively examine gene–metabolite interactions in relation to ADRD risk. We identified distinct plasma metabolomic profiles of dementia risk among APOE4 homozygotes. Beyond APOE4, we found that the metabolite–dementia associations are also modified by other common AD/ADRD risk variants implicated in cholesterol homeostasis, mitochondrial function, APP production and extracellular matrix integrity. Notably, adherence to the MedDiet contributed to cognitive health in an APOE4-dependent manner, potentially mediated by metabolites. We further demonstrated that incorporating metabolites into predictive models alongside traditional and genetic predictors improved the prediction performance. Our two-sample MR analysis further supported the causal effects of specific metabolites, such as 4-guanidinobutanoate, carotene diol and glutamine, on cognitive outcomes.
ADRD has long been considered a metabolic disease, largely due to the central role of APOE4 in lipid transport and metabolism6. Emerging evidence indicates that the metabolomic contributions to ADRD risk are modulated by APOE4 status17,18,19,20. A recent study further highlights that APOE4 homozygosity represents a distinct genetic subtype which exhibits significantly higher levels of AD pathological markers at a younger age7. Our study extends those findings, revealing that APOE4 not only distorts lipid metabolism but also affects other pathways, including betaine and the urea cycle (for example, citrulline) metabolism. These findings suggest a broader role for APOE4, implicating its involvement in neuroinflammation, gut-derived choline metabolism and neurotoxic pathways that elevate ADRD risk5,49. Furthermore, our findings, aligned with the recent study7, indicate that APOE4 homozygosity uniquely alters the plasma metabolome and its associations with ADRD risk, underscoring the necessity of distinguishing homozygotes from heterozygotes in investigations of APOE4’s role in AD pathology. Although APOE4 homozygotes exhibit a high penetrance of AD biology, not all individuals progress to clinical AD6. This resilience presents an opportunity to identify protective mechanisms specific to APOE4 homozygosity. Our findings highlight the potential for preventive strategies targeting specific metabolic pathways in this high-risk group. Beyond APOE4, examination of other common risk variants identified additional findings pointing to broader pathogenic mechanisms.
Consistent with the results from the PREDIMED trial25, we found that long-term adherence to the MedDiet was strongly associated with lower ADRD risk. We further examined whether this association varied by genetic predisposition. A key distinction of our study, compared to previous studies with mixed findings50,51,52, is that we went beyond the binary classification of APOE4 carrier status and found particularly strong associations only among APOE4 homozygotes. The strongest association in APOE4 homozygotes may reflect more effective modulation of ADRD-related metabolic profiles by the MedDiet in this genotype. Thus, our study not only identifies mechanistic insights, but also proposes actionable prevention strategies targeting these pathways. This has important implications for public health messaging, highlighting the overall benefit of adhering to the MedDiet for ADRD prevention, as well as the potential for targeted interventions in genetically vulnerable populations. In addition, our findings underscore the value of metabolites in predicting ADRD risk decades before disease onset. Last, our two-sample MR analysis corroborated established mechanisms in ADRD pathology, such as the role of GABAergic signaling, reinforcing causality derived from mechanistic studies and randomized controlled trials, including evidence of carotenoids’ neuroprotective effects. We also identified new causal biomarkers, including metabolites involved in glutamine, urate and hypotaurine metabolism (Supplementary Text). Collectively, these findings suggest potential druggable targets and inform pathways for more effective and precise prevention and treatment strategies for ADRD.
Our study has limitations. It was conducted in well-educated individuals of European ancestry, which may limit generalizability but enhances internal validity. Ascertainment bias is possible given the exclusion of participants with dementia at baseline, although the baseline age was relatively young and APOE4 homozygotes did not differ significantly in health status, making major bias unlikely. Dementia outcomes were based on self-reported physician diagnoses and death records, which may introduce misclassification, but validity is supported by participants’ professional backgrounds, strong associations with known genetic factors and p-tau217 and consistency across data sources. Although omics-based models are not yet widely implemented clinically, emerging tools such as PRSs and aging clocks show translational promise (Supplementary Text). Despite these limitations, our large, prospective design enabled robust examination of gene–metabolite–diet interactions over decades of follow-up. Importantly, key findings in women were independently validated in men, supporting generalizability across sexes, particularly relevant given the sex-based differences in both metabolite profiles53 and dementia risk54.
In conclusion, our study highlights the substantial influence of genetic variants, particularly APOE4 homozygosity, on plasma metabolites and their associations with ADRD risk. These genetic effects are widespread across the plasma metabolome and our findings identify the MedDiet as a promising approach to mitigate genetically dependent ADRD risk by targeting a broad spectrum of metabolic pathways. Moreover, this work provides a uniquely valuable resource for advancing the early prediction of ADRD risk through emerging omics-based biomarkers and identifying causal mechanisms underlying ADRD risk, which hold potential as prevention targets and druggable pathways.
Methods
Ethics statement
This study included de-identified data from participants who had consented to the use of their anonymized information for research purposes. Participants were not financially compensated for their participation. Approvals for the study protocol of the NHS and the HPFS were granted by the institutional review boards of Brigham and Women’s Hospital and the Harvard T.H. Chan School of Public Health (institutional review board protocol nos. 1999P011114/BWH for the NHS and HSPH 22067-102 for the HPFS).
Study populations
The NHS is a prospective cohort study that enrolled 121,700 US female registered nurses aged 30–55 years in 197655. The participants were followed biennially to collect information on diet, lifestyle, medication use and newly diagnosed diseases through mailed self-administered questionnaires. We included 4,215 women who were aged <75 years and free from dementia, Parkinson’s disease, stroke and cancer at baseline when the blood samples were collected (1989–1992); the blood was assayed for genetics and metabolomics, as described in detail below.
The HPFS is a prospective cohort study that enrolled 51,529 US male health professionals aged 40–75 years in 1986. Similar to the NHS, the participants were followed biennially via mailed questionnaires. We included 1,490 men who met the same inclusion and exclusion criteria at baseline (1993–1996), when the blood samples were collected, as a replication cohort.
Ascertainment of dementia and assessment of objective cognitive function
The participants were followed from the baseline to 2023 for a composite dementia endpoint, which included self-reported dementia and deaths due to dementia. Participants self-reported a physician diagnosis of AD or other forms of dementia (ADRD) every 2 years via questionnaires. Deaths were identified through state vital statistics records, the National Death Index, family reports and the postal system. A study physician reviewed medical records and death certificates to determine whether dementia was listed as the primary or contributing cause of death. Our system for death ascertainment and death cause adjudication is validated and >98% of deaths were identified56.
Objective cognitive function was assessed through a telephone cognitive interview in a subset of 1,037 NHS participants aged ≥70 years, with four telephone interviews conducted between 1995 and 2008. The telephone cognitive battery initially included the TICS, which is a telephone adaptation of the Mini-Mental State Examination57. Five other tests were added later, including (1) immediate recall of the East Boston Memory Test (EBMT), (2) delayed recall of the EBMT, (3) delayed recall of the TICS ten-word list, (4) a test of verbal fluency and (5) the digit span backward test. We assessed three cognitive measures, including the TICS score and the composite scores for global cognition and verbal memory. Global cognition included all six tests. Verbal memory included four tests: immediate recall of the ten-word list in the TICS, delayed recall of the TICS ten-word list and immediate and delayed recalls of the EBMT. To calculate the composite scores for global cognition and verbal memory, we first calculated z-scores for each test at each measurement, based on the baseline mean and the s.d. We then averaged these z-scores across all relevant tests at each measurement and calculated the overall mean value across all measurements.
Genotyping, APOE4 genotype, other AD/ADRD variants and the PRS for ADRD
Details of genotyping, QC and imputation for the NHS and HPFS are described elsewhere58 (https://github.com/cturman15/ChanGWASlab) and in Supplementary Text. Blood samples were genotyped using one of six genotyping platforms. We restricted the samples to inferred European ancestry based on genetic PCs. Post-QC data were imputed using the 1000 Genomes Phase 3 v5 reference panel. APOE4 genotype was determined using two SNPs: rs429358 and rs7412. APOE4 homozygotes carried C/C alleles for both SNPs and APOE4 heterozygotes carried C/T for rs429358 and C/C or C/T for rs7412. All other combinations of alleles were considered APOE4 noncarriers. The average imputation quality score for rs429358 and rs7412 was 0.91 and 0.87 across genotyping platforms, respectively. We calculated PRSs for ADRD using weights from two published studies9,21,59. The PRS developed by Bellenguez et al. (PGS002280) comprised 83 variants and excluded any variants from the APOE region9. The PRS developed by Zhang et al. (PGS000334) comprised 22 variants and included the two APOE variants21. Only variants available in the imputed genetic data were included in the PRS calculation (Supplementary Table 11). The PRS was calculated as a weighted sum of the effect allele dosage across all included variants for each individual and was standardized to have a mean of 0 and an s.d. of 1 within each genotyping platform to maximize comparability. We further extracted individual variants from these two PRSs, which were identified in previous GWASs of AD/ADRD. Duplicated variants and those with a minor allele frequency <0.01 were removed from the combined list, followed by linkage disequilibrium pruning (r2 < 0.1 with the 1000 Genomes European population as the linkage disequilibrium reference) using the SNPclip function in the LDlinkR package in R, leaving a total of 73 variants for subsequent analyses (Supplementary Table 12).
Metabolomic profiling
Plasma metabolomic profiling was performed for nested case–control studies within the NHS and HPFS using high-throughput LC–MS techniques at the Broad Institute of MIT and Harvard (Cambridge, MA, USA). Additional details of metabolomic profiling are provided in Supplementary Text. For metabolites with <25% missing data in an individual study, missing values were imputed with half of the minimum measured value for that metabolite in that study; metabolites with <100 samples were removed. A probit transformation was applied to metabolites within each study to correct for batch effects, reduce the impact of skewed distributions and heavy tails on the results and scale the metabolite values to the same range. After merging the metabolite data with other types of data (n = 4,215 for the NHS and n = 1,490 for the HPFS), we excluded metabolites with ≥90% missing values among dementia cases with genetic data. We further excluded metabolites with an intraclass correlation coefficient <0.4 or coefficient of variation in the top 10 percentiles of all remaining metabolites. A total of 401 metabolites was included in the final analysis for the NHS, of which 254 were available in the HPFS. We created another set of metabolomics data for the NHS by selecting those with ≥100 samples and <25% missing values from the 401 metabolites in the final dataset, followed by RF imputation with 100 trees using the missRanger package in R; 237 metabolites were retained for subsequent analyses of the overall metabolomic profile that required no missing data. Using the same approach, we retained 164 non-missing metabolites in the HPFS.
Dietary assessment and the MedDiet score
Dietary intakes were assessed using SFFQs. The validity and reproducibility of the SFFQs have been demonstrated in previous publications60. We calculated the average dietary intakes from the first dietary assessment (1980 for the NHS and 1986 for the HPFS) to the SFFQ closest to the blood draw time to reflect the long-term diet. The MedDiet was assessed using the Alternate Mediterranean Diet Score, which was calculated based on nine components61. For vegetables, fruit, nuts, whole grains, legumes, fish and the ratio of monounsaturated to saturated fat, a score of 1 was given if the intake was at or above the SFFQ-specific median; otherwise, 0 was given. For red and processed meat consumption, a score of 1 was assigned if the intake was below the SFFQ-specific median; otherwise, a score of 0 was assigned. For alcohol intake, a score of 1 was assigned if the intake was between 5 and 15 g d−1; otherwise, a score of 0 was assigned. The scores of individual components were summed to obtain the overall MedDiet index, which ranged from 0 to 9.
Assessment of global impacts of genetics and MedDiet on plasma metabolome
We assessed the overall correlation of genetic data, the MedDiet index and the plasma metabolome in the NHS. Pearson’s correlation coefficients were calculated for each of the 401 metabolites with genetic PC1 and PC2. Correlation coefficients were then ranked from highest to lowest for PC1 and PC2, respectively. The overall rank was determined by summing the ranks of PC1 and PC2 and ranking them from lowest to highest.
Leveraging the 237 metabolites with no missing values in the NHS (see above), we assessed the overall correlation between metabolites and the MedDiet index, as well as the predictive performance of metabolites on the MedDiet index. Pearson’s correlation coefficients were calculated for the MedDiet index and its individual components (monounsaturated and saturated fat calculated separately) with metabolites PC1 and PC2 calculated from the 237 metabolites. RF regression was performed to evaluate the predictive performance of the metabolites on the MedDiet index. The dataset was first randomly split into training (60%) and test (40%) sets. A dichotomized MedDiet index outcome was derived from the top and bottom quartiles of the continuous score. RF regression with five-fold cross-validation was performed on the training set to tune the parameter using the train and trainControl functions in the caret package in R. The tuned model was then applied to the test set to evaluate performance using the AUC. The same approach was applied to the HPFS to assess the predictive performance of metabolites on the MedDiet index.
Interaction analyses of plasma metabolome and genetic variation in relation to dementia risk and cognitive function
Cox PH models were fitted to assess the associations between each of the 401 metabolites and the time-to-event outcome of dementia among 4,215 women in the NHS using the coxph function in the survival package in R. Details of covariate assessment and adjustment are provided in Supplementary Text. The FDR correction was applied to the P value for metabolites using the Benjamini–Hochberg approach, with an FDR < 0.05 considered statistically significant. The same approach and FDR threshold were used for all other analyses involving multiple testing corrections in this study, including all interaction tests. All statistical tests in this study were two sided. For the interaction analysis of metabolites and APOE4 status, interaction terms between APOE4 carrier status (or APOE4 heterozygote and homozygote) and the metabolite were added to the model, along with ADRD PRS (excluding the APOE region) and its interaction term with the metabolite. For the models of APOE4 heterozygote and homozygote, FDR correction was applied jointly to the interaction P value of the two APOE4 terms. In addition, likelihood ratio tests were performed comparing the model with and without the APOE4 interaction terms. For the interaction analysis of metabolites with ADRD PRS or other AD/ADRD variants, the relevant gene–metabolite interaction terms (PRS or effect allele dosage) were added to the model, along with APOE4 heterozygote and homozygote. All interaction models were additionally adjusted for the top four genetic PCs and genotyping platforms. For the subgroup analyses within APOE4 noncarriers, carriers and heterozygotes, the models were further adjusted for the ADRD PRS (excluding the APOE region), the top four genetic PCs and genotyping platforms. For APOE4 homozygotes, the models were adjusted for only continuous covariates, ADRD PRS and the top four genetic PCs due to the limited sample size. For analyses within ADRD PRS tertiles (defined using genetic PC-adjusted scores), no additional covariate was adjusted.
Generalized linear models (Gaussian family) were fitted to assess the association between each objective cognitive function score and each of the 401 metabolites among a subset of 1,037 women with cognitive function measurements using the glm function of the stats package in R. The models were adjusted for the same covariates as in the dementia risk analysis. Interaction analysis was performed for APOE4 carrier status with the same additional covariates as in the dementia risk analysis. APOE4 carriers were not further stratified into heterozygotes and homozygotes due to data sparsity among homozygotes in this subset. FDR correction was applied to the interaction P value per cognitive outcome. Interaction analysis with other genetic factors or subgroup analysis was not performed due to the limited power.
As a replication, interaction analysis of the 254 available metabolites was conducted in 1,490 men from the HPFS, with additional adjustment for profession. Due to data sparsity in non-missing values for each metabolite, only interactions with APOE4 carrier status were assessed.
Associations of MedDiet adherence with dementia risk and cognitive function
In the analysis of the associations of the MedDiet index score with dementia risk and objective cognitive function, we leveraged data from the full NHS cohort and excluded participants who had dementia, Parkinson’s disease, stroke, cancer or missing components for the MedDiet index at baseline, which was 1980 for the dementia endpoint and 1994 for the objective cognitive function. For the analysis of the objective cognitive function, we further excluded participants aged <70 years at baseline. A total of 86,740 participants were included in the analysis of dementia risk and 16,244 participants in the analysis of objective cognitive function.
Cox PH model was fitted to prospectively assess the association between the continuous MedDiet index and the time-to-event outcome of dementia risk. Details of covariate adjustment are provided in Supplementary Text. A cubic spline regression model was fitted to assess the nonlinear trend. Subgroup analyses were performed by APOE4 genotype and tertiles of ADRD PRSs.
Generalized linear models were fitted to assess the association between the continuous MedDiet index and the objective cognitive function scores. These models adjusted for the same covariates as the dementia risk model. Subgroup analyses were performed by APOE4 genotype and tertiles of ADRD PRSs.
As a replication, a total of 43,500 male participants from the HPFS were included in the analysis of the association between MedDiet adherence and dementia risk, applying the same exclusion criteria as in the NHS. The same Cox PH and cubic spline models were fitted, with additional adjustment for profession. Subgroup analyses were performed by APOE4 genotype.
Associations between MedDiet adherence and plasma metabolome by genetic subgroups
Generalized linear models were fitted to assess the association between the MedDiet index and each of the 401 metabolites among 4,215 women in the NHS, adjusting for the same covariates as the above linear model for cognitive function and metabolites. Subgroup analyses were performed by APOE4 genotype and tertiles of ADRD PRS (excluding APOE region). As a replication, the same models were fitted to assess the association between the MedDiet index and each of the 254 overlapping metabolites among 1,490 men in the HPFS, additionally adjusted for profession.
Mediation effect of metabolites on the association between MedDiet adherence and dementia risk by APOE4 carrier status
We conducted a mediation analysis to quantify the extent to which metabolites mediate the association between MedDiet adherence and dementia risk in the NHS. Metabolites associated with both dementia risk (P < 0.05) and MedDiet adherence (FDR < 0.05) were selected as candidate mediators, followed by orthogonal filtering using a correlation threshold of r < 0.5 to exclude highly correlated metabolites, resulting in seven metabolites: allantoin, C16:1 CE, C18:0 SM, 1-methylguanine, 1,7-dimethyluric acid, C34:5 phosphatidylcholine plasmalogen and piperine. Regression-based mediation analyses were then conducted by comparing a full model (including both MedDiet and selected metabolites) with a reduced model (excluding metabolites), with dementia risk as the dependent variable to quantify the proportion of the effect of MedDiet adherence on dementia risk, explained by these selected metabolites in the full dataset, as well as stratified by APOE4 carrier status.
Prediction of dementia risk and cognitive function
Cox PH models were built for predicting dementia risk among 4,215 women in the NHS. The dataset was first randomly split into training (60%) and test (40%) sets, including within APOE4 subgroups. A 15-year outcome-free attainment was defined by censoring participants without event by year 15. We built four prediction models: the baseline model included age, family history of dementia, educational attainments of nurses, smoking status, history of depression or regular antidepressant drug use and the MedDiet index; the APOE4 model further included the APOE4 heterozygote and homozygote indicators, the PRS model the ADRD PRS (excluding the APOE region) and the metabolite model the selected metabolites, including 12 metabolites for predicting overall outcomes and 4 metabolites for predicting the 15-year outcome-free attainment (Supplementary Text and Supplementary Table 22). APOE4 predictors were not included in the models for APOE4 subgroups. All models were built in the training set and evaluated on the test set. Time-dependent AUC was calculated based on an incident/dynamic version of sensitivity and specificity within distinct risk sets, defined as groups of participants who remained at risk for developing dementia at specified time points, which provides a dynamic view of model performance across the follow-up period62, using the risksetAUC function in the risksetROC package in R. Harrell’s C-index63 was calculated to quantify the overall discriminative ability of the model by assessing how well the model can rank individuals by their risk of the event using the survival package in R. Feature contributions were quantified by the SHAP value64 for Cox PH model in predicting overall and 15-year dementia risk, including the full list of predictors using the fastshap package in R. SHAP values were calculated for each category of predictors by summing the SHAP value of all predictors in that category.
RF models with 500 trees were built for predicting the dichotomized outcomes of the highest versus the lowest tertile of the continuous cognitive function scores, using the randomForest package in R. The same predictors and analysis strategy were used as in the prediction models for dementia risk. The AUC was calculated to evaluate the model performance in the test set.
As a replication, Cox PH models were built to predict dementia risk among 1,490 men in the HPFS. The same modeling approaches were applied, with profession included in the baseline model instead of education level. Due to data sparsity, prediction models were constructed only for the overall dementia risk in the full dataset. The 164 RF-imputed metabolites with no missing values were considered as candidate predictors and 5 metabolites were selected in the final model, following the same procedure used in the NHS (Supplementary Table 22).
Two-sample MR analysis
GWAS sources
Two-sample MR analysis65 was conducted to assess the causal relationships between plasma metabolites and cognitive outcomes using published GWAS summary statistics. GWAS summary statistics for the exposures, including plasma metabolites and metabolite ratios, were obtained from ref. 11 and for the cognitive outcomes were obtained from GWASs of overall dementia66, AD8, vascular dementia66 and cognitive performance67. There was no overlap of the participants between the GWAS cohorts of the exposures and the outcomes. All GWAS populations were of European ancestry.
Genetic instruments
In the original study11, GWASs were performed for 1,091 plasma metabolites and 309 metabolite ratios. In this study, we included all the variant–metabolite and variant–metabolite ratio pairs selected by the original study for MR analyses, with additional selection applied to metabolites and ratios not included in the original set (Supplementary Text). A total of 1,431 variant–metabolite pairs for 657 metabolites and 186 variant–metabolite pairs for 133 metabolites ratios were selected for the MR analysis.
MR analysis
We used three MR methods to assess the causal effect of each metabolite or ratio on each cognitive outcome. Wald ratio method was used when there was only one genetic instrument. The inverse variance-weighted method was used when there were at least two instruments. The MR Egger method was used when there were at least three instruments and potential pleiotropy was detected. MR analysis was performed using the mr function in the TwoSampleMR package in R. Details of the sensitivity analyses are described in Supplementary Text.
Colocalization analysis
Colocalization analysis was performed for exposure–outcome pairs that passed FDR < 0.05 in the MR analysis. For each pair, Bayesian colocalization analysis was performed in the ±500-kb region around each genetic instrument, restricting to variants with minor allele frequency <0.01, using the coloc.abf function with default prior probabilities in the coloc package in R. Colocalization signals were reported for a locus if the conditional probability of colocalization, PP.H4/(PP.H3 + PP.H4), was >70%, where PP.H3 is posterior probability that the two traits have independent causal variants and PP.H4 is the posterior probability that the two traits share a single causal variant. These colocalized exposure–outcome pairs were considered to have putative causal relationships. If there were instruments at multiple loci for a pair, the causal relationship was reported if the exposure and outcome were colocalized at at least one genetic locus. The coloc.abf function assumes that there is a single causal variant for each trait. We further conducted a sensitivity analysis that relaxed the single causal variant assumption using the coloc.susie function in the coloc package in R, which calculates posterior probabilities under the assumption of multiple causal variants.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
As informed consent was gained from the participants, all the individual-level data from the NHS and the HPFS are available through a request for external collaboration and upon approval of a letter of intent and a research proposal. Details on how to request external collaborations with the NHS can be found at https://nurseshealthstudy.org/researchers (contact principal investigator: A.H.E., email: nhahe@channing.harvard.edu) and with the HPFS at https://sites.sph.harvard.edu/hpfs/for-collaborators (contact principal investigator: L. Mucci, email: lmucci@hsph.harvard.edu). Source data are provided with this paper.
Code availability
Analysis-specific programs are publicly available via GitHub at https://github.com/DW-Group/Gene_Metabolites_MedDiet_Dementia.
Change history
02 October 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41591-025-04025-7
References
Emmady, P. D., Schoo, C. & Tadi, P. Major Neurocognitive Disorder (Dementia) (StatPearls Publishing, 2022).
Gatz, M. et al. Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry 63, 168–174 (2006).
Liu, C. C., Liu, C. C., Kanekiyo, T., Xu, H. & Bu, G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat. Rev. Neurol. 9, 106–118 (2013).
Yamazaki, Y., Zhao, N., Caulfield, T. R., Liu, C. C. & Bu, G. Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat. Rev. Neurol. 15, 501–518 (2019).
Parhizkar, S. & Holtzman, D. M. APOE mediated neuroinflammation and neurodegeneration in Alzheimer’s disease. Semin. Immunol. 59, 101594 (2022).
Jackson, R. J., Hyman, B. T. & Serrano-Pozo, A. Multifaceted roles of APOE in Alzheimer disease. Nat. Rev. Neurol. 20, 457–474 (2024).
Fortea, J. et al. APOE4 homozygozity represents a distinct genetic form of Alzheimer’s disease. Nat. Med. 30, 1284–1291 (2024).
Wightman, D. P. et al. A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease. Nat. Genet. 53, 1276–1282 (2021).
Bellenguez, C. et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 54, 412–436 (2022).
Fuller, H. et al. Metabolomic epidemiology offers insights into disease aetiology. Nat. Metab. 5, 1656–1672 (2023).
Chen, Y. et al. Genomic atlas of the plasma metabolome prioritizes metabolites implicated in human diseases. Nat. Genet. 55, 44–53 (2023).
Chen, L. et al. Influence of the microbiome, diet and genetics on inter-individual variation in the human plasma metabolome. Nat. Med. 28, 2333–2343 (2022).
Clemente-Suarez, V. J., Beltran-Velasco, A. I., Redondo-Florez, L., Martin-Rodriguez, A. & Tornero-Aguilera, J. F. Global impacts of western diet and its effects on metabolism and health: a narrative review. Nutrients 15, 2749 (2023).
Niedzwiecki, M. M. et al. High-resolution metabolomic profiling of Alzheimer’s disease in plasma. Ann. Clin. Transl. Neurol. 7, 36–45 (2020).
Huo, Z. et al. Brain and blood metabolome for Alzheimer’s dementia: findings from a targeted metabolomics analysis. Neurobiol. Aging 86, 123–133 (2020).
Panyard, D. J. et al. Large-scale proteome and metabolome analysis of CSF implicates altered glucose and carbon metabolism and succinylcarnitine in Alzheimer’s disease. Alzheimers Dement. 19, 5447–5470 (2023).
Arnold, M. et al. Sex and APOE epsilon4 genotype modify the Alzheimer’s disease serum metabolome. Nat. Commun. 11, 1148 (2020).
Chang, R. et al. Predictive metabolic networks reveal sex- and APOE genotype-specific metabolic signatures and drivers for precision medicine in Alzheimer’s disease. Alzheimers Dement. 19, 518–531 (2023).
Gonzalez-Dominguez, R. et al. Apolipoprotein E and sex modulate fatty acid metabolism in a prospective observational study of cognitive decline. Alzheimers Res. Ther. 14, 1 (2022).
Ye, Z. et al. Contrasting association pattern of plasma low-density lipoprotein with white matter integrity in APOE4 carriers versus non-carriers. Neurobiol. Aging 143, 41–52 (2024).
Zhang, Q. et al. Risk prediction of late-onset Alzheimer’s disease implies an oligogenic architecture. Nat. Commun. 11, 4799 (2020).
Vogt, N. M. et al. The gut microbiota-derived metabolite trimethylamine N-oxide is elevated in Alzheimer’s disease. Alzheimers Res. Ther. 10, 124 (2018).
Marksteiner, J., Blasko, I., Kemmler, G., Koal, T. & Humpel, C. Bile acid quantification of 20 plasma metabolites identifies lithocholic acid as a putative biomarker in Alzheimer’s disease. Metabolomics 14, 1 (2018).
Ashton, N. J. et al. Diagnostic accuracy of a plasma phosphorylated Tau 217 immunoassay for Alzheimer disease pathology. JAMA Neurol. 81, 255–263 (2024).
Valls-Pedret, C. et al. Mediterranean diet and age-related cognitive decline: a randomized clinical trial. JAMA Intern. Med. 175, 1094–1103 (2015).
Ueland, P. M., Holm, P. I. & Hustad, S. Betaine: a key modulator of one-carbon metabolism and homocysteine status. Clin. Chem. Lab. Med. 43, 1069–1075 (2005).
Bhatt, M., Di Iacovo, A., Romanazzi, T., Roseti, C. & Bossi, E. Betaine—the dark knight of the brain. Basic Clin. Pharmacol. Toxicol. 133, 485–495 (2023).
Obeid, R. The metabolic burden of methyl donor deficiency with focus on the betaine homocysteine methyltransferase pathway. Nutrients 5, 3481–3495 (2013).
Blanchard, J. W. et al. APOE4 impairs myelination via cholesterol dysregulation in oligodendrocytes. Nature 611, 769–779 (2022).
van der Kant, R. et al. Cholesterol metabolism Is a druggable axis that independently regulates Tau and amyloid-beta in iPSC derived Alzheimer’s disease neurons. Cell Stem Cell 24, 363–375 (2019).
Kaji, S. et al. Apolipoprotein E aggregation in microglia initiates Alzheimer’s disease pathology by seeding beta-amyloidosis. Immunity 57, 2651–2668.e12 (2024).
Branco, A., Yoshikawa, F. S. Y., Pietrobon, A. J. & Sato, M. N. Role of histamine in modulating the immune response and inflammation. Mediators Inflamm. 2018, 9524075 (2018).
Elali, A. & Rivest, S. The role of ABCB1 and ABCA1 in beta-amyloid clearance at the neurovascular unit in Alzheimer’s disease. Front. Physiol. 4, 45 (2013).
O’Sullivan, J. F. et al. Dimethylguanidino valeric acid is a marker of liver fat and predicts diabetes. J. Clin. Invest. 127, 4394–4402 (2017).
Gottschall, P. E. & Howell, M. D. ADAMTS expression and function in central nervous system injury and disorders. Matrix Biol. 44-46, 70–76 (2015).
Gibney, M. J. et al. Metabolomics in human nutrition: opportunities and challenges. Am. J. Clin. Nutr. 82, 497–503 (2005).
Toledo, E. et al. Plasma lipidomic profiles and cardiovascular events in a randomized intervention trial with the Mediterranean diet. Am. J. Clin. Nutr. 106, 973–983 (2017).
Xu, J. et al. Cerebral deficiency of vitamin B5 (d-pantothenic acid; pantothenate) as a potentially-reversible cause of neurodegeneration and dementia in sporadic Alzheimer’s disease. Biochem. Biophys. Res. Commun. 527, 676–681 (2020).
Snowden, S. G. et al. Association between fatty acid metabolism in the brain and Alzheimer disease neuropathology and cognitive performance: a nontargeted metabolomic study. PLoS Med. 14, e1002266 (2017).
Zhang, J. et al. Asparagine plays a critical role in regulating cellular adaptation to glutamine depletion. Mol. Cell 56, 205–218 (2014).
Fujimaki, M. et al. Serum caffeine and metabolites are reliable biomarkers of early Parkinson disease. Neurology 90, e404–e411 (2018).
McCormick, D. A. GABA as an inhibitory neurotransmitter in human cerebral cortex. J. Neurophysiol. 62, 1018–1027 (1989).
Kim, Y. S. & Yoon, B. E. Altered GABAergic signaling in brain disease at various stages of life. Exp. Neurobiol. 26, 122–131 (2017).
Sastre, M., Regunathan, S., Galea, E. & Reis, D. J. Agmatinase activity in rat brain: a metabolic pathway for the degradation of agmatine. J. Neurochem. 67, 1761–1765 (1996).
Song, J. et al. Agmatine improves cognitive dysfunction and prevents cell death in a streptozotocin-induced Alzheimer rat model. Yonsei Med. J. 55, 689–699 (2014).
Fiedor, J. & Burda, K. Potential role of carotenoids as antioxidants in human health and disease. Nutrients 6, 466–488 (2014).
Grodstein, F., Kang, J. H., Glynn, R. J., Cook, N. R. & Gaziano, J. M. A randomized trial of beta carotene supplementation and cognitive function in men: the Physicians’ Health Study II. Arch. Intern. Med. 167, 2184–2190 (2007).
Madeira, C. et al. Elevated glutamate and glutamine levels in the cerebrospinal fluid of patients with probable Alzheimer’s disease and depression. Front. Psychiatry 9, 561 (2018).
Yang, L. G., March, Z. M., Stephenson, R. A. & Narayan, P. S. Apolipoprotein E in lipid metabolism and neurodegenerative disease. Trends Endocrinol. Metab. 34, 430–445 (2023).
van de Rest, O. et al. APOE epsilon4 and the associations of seafood and long-chain omega-3 fatty acids with cognitive decline. Neurology 86, 2063–2070 (2016).
Shannon, O. M. et al. Mediterranean diet adherence is associated with lower dementia risk, independent of genetic predisposition: findings from the UK Biobank prospective cohort study. BMC Med. 21, 81 (2023).
Migliore, L. & Coppede, F. Gene-environment interactions in Alzheimer disease: the emerging role of epigenetics. Nat. Rev. Neurol. 18, 643–660 (2022).
Lista, S. et al. Integrative metabolomics science in Alzheimer’s disease: relevance and future perspectives. Ageing Res. Rev. 89, 101987 (2023).
Mielke, M. M. Sex and gender differences in Alzheimer’s disease dementia. Psychiatry 35, 14–17 (2018).
Bao, Y. et al. Origin, methods, and evolution of the three Nurses’ Health Studies. Am. J. Public Health 106, 1573–1581 (2016).
Stampfer, M. J. et al. Test of the national death index. Am. J. Epidemiol. 119, 837–839 (1984).
Folstein, M. F., Folstein, S. E. & McHugh, P. R. ‘Mini-mental state’. A practical method for grading the cognitive state of patients for the clinician. J. Psychiatr. Res. 12, 189–198 (1975).
Lindstrom, S. et al. A comprehensive survey of genetic variation in 20,691 subjects from four large cohorts. PLoS ONE 12, e0173997 (2017).
Lambert, S. A. et al. Enhancing the polygenic score catalog with tools for score calculation and ancestry normalization. Nat. Genet. 56, 1989–1994 (2024).
Willett, W. C. et al. Reproducibility and validity of a semiquantitative food frequency questionnaire. Am. J. Epidemiol. 122, 51–65 (1985).
Fung, T. T. et al. Diet quality is associated with the risk of estrogen receptor-negative breast cancer in postmenopausal women. J. Nutr. 136, 466–472 (2006).
Heagerty, P. J. & Zheng, Y. Survival model predictive accuracy and ROC curves. Biometrics 61, 92–105 (2005).
Harrell, F. E., Califf, R. M., Pryor, D. B., Lee, K. L. & Rosati, R. A. Evaluating the yield of medical tests. JAMA 247, 2543–2546 (1982).
Lundberg, S. M. & Lee, S.-I. A unified approach to interpreting model predictions. In Proc. 31st International Conference on Neural Information Processing Systems (eds Guyon, I. et al.) 4768–4777 (Curran Associates Inc., 2017).
Lawlor, D. A. Commentary: two-sample Mendelian randomization: opportunities and challenges. Int. J. Epidemiol. 45, 908–915 (2016).
Kurki, M. I. et al. FinnGen provides genetic insights from a well-phenotyped isolated population. Nature 613, 508–518 (2023).
Lee, J. J. et al. Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nat. Genet. 50, 1112–1121 (2018).
Acknowledgements
We acknowledge the contribution to this study from central cancer registries supported through the Centers for Disease Control and Prevention’s National Program of Cancer Registries and/or the National Cancer Institute’s Surveillance, Epidemiology, and End Results (SEER) program. Central registries may also be supported by state agencies, universities and cancer centers. Participating central cancer registries include the following: Alabama, Alaska, Arizona, Arkansas, California, Colorado, Connecticut, Delaware, Florida, Georgia, Hawaii, Idaho, Indiana, Iowa, Kentucky, Louisiana, Massachusetts, Maine, Maryland, Michigan, Mississippi, Montana, Nebraska, Nevada, New Hampshire, New Jersey, New Mexico, New York, North Carolina, North Dakota, Ohio, Oklahoma, Oregon, Pennsylvania, Puerto Rico, Rhode Island, Seattle SEER Registry, South Carolina, Tennessee, Texas, Utah, Virginia, West Virginia and Wyoming. We are indebted to the participants in the NHS and the HPFS for their continuing outstanding level of cooperation and to the staff of the NHS and the HPFS for their valuable contributions. This work was funded by the National Institutes of Health (NIH) (grant nos. R00DK119412, R01NR019992, R01AG077489, RF1AG083764 and U54AG089325 to D.D.W. and P30DK046200 to F.B.H.). The NHS was supported by the NIH (grant nos. UM1CA186107, P01CA087969, R01HL034594, R01HL088521 and R01HL060712) and also the HPFS (NIH grant nos. U01CA167552, R01HL060712 and R01HL035464). The funding source had no role in the design and conduct of the study, collection, management, analysis and interpretation of the data, preparation, review or approval of the manuscript or the decision to submit the manuscript for publication. The content is solely our responsibility and does not necessarily represent the official views of the NIH. The computations in this paper were run in part on the FAS Research Computing System supported by the FAS Division of Science Research Computing Group at Harvard University.
Author information
Authors and Affiliations
Contributions
Conceptualization and study design: Y.L. and D.D.W. Data analysis: Y.L. Manuscript writing: Y.L. and D.D.W. Study supervision: D.D.W. Sample and data collection and funding acquisition: J.H.K., F.B.H., W.C.W., A.H.E., M.J.S. and D.D.W. All the authors discussed the results, critically reviewed the text and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Medicine thanks Moonil Kang, Oliver Shannon and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Liam Messin, in collaboration with the Nature Medicine team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Overview of the Health Professionals Follow-Up Study as a replication cohort.
(a) We prospectively followed 1,490 men in Health Professionals Follow-Up Study (HPFS) from 1993 through 2023. Genetic and metabolomic profiles were generated from blood samples collected at baseline. Detailed demographic, lifestyle, dietary, medical history and medication use data were collected via questionnaires. Dementia cases were ascertained through the follow-up as a composite endpoint of incident dementia and death due to dementia. (b) Associations between established genetic risk factors for Alzheimer’s disease and related dementias (ADRD) and dementia risk. Lines indicate cumulative incidence across APOE4 genotypes and tertiles of polygenic risk score (PRS) of ADRD (excluding the APOE region) over the follow-up period, with shaded areas representing 95% confidence intervals (CIs) and P from the log-rank test annotated. Consistent with the curves, unadjusted hazard ratios (HRs) were estimated using Cox proportional hazards models; covariate-adjusted HRs with 95% CIs are provided in Supplementary Table 4. Person-time was accrued from baseline until the earliest occurrence of an incident dementia case, dementia death or the end of follow-up. No adjustment was made for multiple comparisons, as this was a hypothesis-driven analysis. The analyses were conducted among 1,490 HPFS participants. All statistical tests were two-sided. Panel a created using BioRender.com.
Extended Data Fig. 2 Associations between genetic factors and 401 metabolites in Nurses’ Health Study.
(a) Correlations between genetic principal components (PCs) and metabolites: PC1 (top panel) and PC2 (bottom panel). The dashed lines denote false discovery rate (FDR) = 0.05, adjusted for multiple comparisons, based on Pearson’s correlation tests. (b) Associations of APOE4 heterozygosity (top panel) and homozygosity (bottom panel) with metabolites, with APOE4 noncarrier as the reference group from Cox proportional hazards models. Models were adjusted for age, the top 4 genetic PCs, and genotyping platforms. The dashed lines denote nominal P = 0.05 (none with FDR < 0.05). All analyses were conducted among 4,215 NHS participants. All statistical tests were two-sided.
Extended Data Fig. 3 Number of metabolites with significant interactions with genetic factors in relation to dementia risk in Nurses’ Health Study by Human Metabolome Database superclass.
(a) Number of metabolites with significant interactions with APOE4 genotype. (b) Number of metabolites with significant interactions with other common AD/ADRD variants. Bars indicate the number of associations that reached nominal significance (P < 0.05) based on the Cox proportional hazards models, and those that remained significant after adjustment for multiple comparisons (false discovery rate <0.05). All statistical tests were two-sided.
Extended Data Fig. 4 Consistency of metabolite-APOE4 interactions in Nurses’ Health Study and Health Professionals Follow-Up Study.
Each dot represents a metabolite with significant APOE4 interactions in Nurses’ Health Study (NHS; n = 38 out of 49 available in Health Professionals Follow-Up Study [HPFS]), colored by the Human Metabolome Database superclass (see Fig. 2 for legend). Pearson’s correlation coefficients in the β coefficients for interaction terms between metabolites and APOE4 carrier status estimated from Cox proportional hazards models with dementia as the dependent variable in NHS and HPFS is annotated on the figure. APOE4 carriers were not further stratified into heterozygotes and homozygotes due to data sparsity among homozygotes with non-missing values for each metabolite in HPFS. Analyses were conducted among 4,215 NHS and 1,490 HPFS participants. All statistical tests were two-sided.
Extended Data Fig. 5 Associations of Mediterranean diet index score with dementia risk and cognitive function by genetic factors in Nurses’ Health Study.
(a) Associations between Mediterranean diet (MedDiet) index score and cognitive function scores by APOE4 genotype from generalized linear models. (b) Associations between MedDiet index score and dementia risk by tertiles of ADRD PRSs from Cox proportional hazards models. (c) Associations between MedDiet index score and cognitive function scores by tertiles of ADRD PRSs from generalized linear models. Stratified β coefficients (for cognitive function) and hazard ratios (HRs; for dementia risk), along with 95% confidence intervals (CIs) per one-unit increase in the MedDiet index score, are shown by APOE4 genotype or PRS categories. The analyses were conducted among NHS participants with genetic, dietary, and cognitive outcome data (n = 16,497 for dementia analyses and n = 3,770 for cognitive function analyses).
Extended Data Fig. 6 Replication of the associations of Mediterranean diet index score with plasma metabolites and dementia risk by APOE4 genotype in Health Professionals Follow-Up Study.
(a) Higher adherence to the Mediterranean diet (MedDiet) was prospectively associated with a lower risk of dementia. A restricted cubic spline Cox proportional hazards (PH) model estimated hazard ratios (HRs) and 95% confidence intervals (CIs) across varying levels of the MedDiet index. The P value from the likelihood ratio test comparing the model without the MedDiet index and the model with its spline term is annotated. This analysis was conducted among Health Professionals Follow-Up Study (HPFS) participants with dementia and dietary data (n = 43,500). (b) The protective association between adherence to the MedDiet and risk of dementia was most pronounced among APOE4 homozygotes. Stratified HR and 95% CIs for dementia risk per one-unit increment in the MedDiet index score, categorized by APOE4 genotype, were estimated from Cox PH models, with stratified P annotated (unadjusted for multiple comparisons in the hypothesis-driven analysis). The analyses were conducted among HPFS participants with genetic, dietary, and dementia outcome data (n = 9,828). (c) Strong association between adherence to MedDiet and the overall plasma metabolome profile identified by a random forest (RF) model to classify individuals in the top versus bottom quartile of the MedDiet index based on plasma metabolites. For the RF classification, the dataset was randomly divided into training (60%) and test (40%) sets. The receiver operating characteristic curve for the test set is shown, with the area under the curve (AUC) and 95% CI annotated on the plot. The analyses were conducted among 1,490 HPFS participants. (d) Associations between MedDiet adherence and plasma metabolite levels are consistent in NHS and HPFS and differ by APOE4 genotype. The heatmap shows β coefficients representing a 1-s.d. increment in the MedDiet index from a generalized linear model, with plasma metabolite levels as the dependent variable, restricting to the 254 metabolites available in both cohorts. In the metabolite analysis, APOE4 carriers were not further stratified into heterozygotes and homozygotes due to data sparsity among homozygotes with non-missing values for each metabolite in HPFS. The analyses were conducted among 4,215 NHS and 1,490 HPFS participants. All statistical tests were two-sided.
Extended Data Fig. 7 Correlation coefficients between principal components of metabolites and the Mediterranean diet index, along with its individual components in Nurses’ Health Study.
Metabolite principal component (PC) plot with correlation coefficients between metabolite PC1 and PC2 and Mediterranean diet (MedDiet) score and its components. Each dot represents an individual and is colored by the MedDiet score. MedDiet score and its components are shown on the plot as arrows; the coordinates of the arrow heads represent their Pearson’s correlation coefficients with metabolite PC1 and PC2. The analyses were conducted among 4,215 participants in Nurses’ Health Study.
Extended Data Fig. 8 Associations of glycerides with the Mediterranean diet index in Nurses’ Health Study.
Each dot represents a metabolite (diglycerides on the left panel and triglycerides on the right panel), color-coded by the direction and significance level of its association with the Mediterranean diet index. The size of each dot represents the effect size of the association. The P value is unadjusted for multiple comparisons given the exploratory nature of the analysis. The analyses were conducted among 4,215 NHS participants. All statistical tests were two-sided.
Extended Data Fig. 9 Harrell’s C index for predicting dementia risk from models with different predictors in Nurses’ Health Study.
APOE4 genotype was not included as a predictor in the subgroup analysis within APOE4 carrier and noncarrier subgroups. The full NHS dataset (n = 4,215) was randomly split into training (60%) and test (40%) sets. All prediction models were fitted in the training set and were evaluated in the test set. All results shown are from the test set.
Extended Data Fig. 10 Integrating genetic variation with plasma metabolites and Mediterranean diet enhances the prediction of dementia risk in the Health Professionals Follow-Up Study.
(a) The inclusion of genetic factors improved dementia risk prediction using Cox proportional hazards (PH) model, with an additional modest enhancement when plasma metabolites were also included. The baseline model predictors included age, family history of dementia, profession, smoking status, history of depression or regular antidepressant use, and Mediterranean diet (MedDiet) index. The polygenic risk score (PRS) of Alzheimer’s disease and related dementias excluded variants in the APOE region. (b) Plasma metabolites are among the top contributors for predicting dementia risk as quantified by the Shapley Additive Explanations (SHAP) value. Feature contributions were evaluated for the Cox PH model for predicting dementia risk including the full list of predictors. SHAP values were calculated for each category of predictors by summing the SHAP value of all predictors in that category. Features were ranked by the SHAP value from the highest to the lowest for predicting dementia risk. We did not assess 15-year dementia risk in the Health Professionals Follow-Up Study (HPFS) due to the limited number of dementia cases. For all analyses, the HPFS dataset (n = 1,490) was randomly divided into training (60%) and test (40%) sets; models were fitted on the training set and evaluated on the test set. All results shown are from the test set.
Supplementary information
Supplementary Information (download PDF )
Supplementary Text and Figs. 1–9.
Supplementary Tables (download XLSX )
Supplementary Tables 1–28.
Supplementary Data 1–9 (download XLSX )
Statistical source data for Supplementary Figs. 1–9.
Source data
Source Data Fig. 1 (download XLSX )
Statistical source data.
Source Data Fig. 2 (download XLSX )
Statistical source data.
Source Data Fig. 3 (download XLSX )
Statistical source data.
Source Data Fig. 4 (download XLSX )
Statistical source data.
Source Data Fig. 5 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 1 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 2 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 3 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 4 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 5 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 6 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 7 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 8 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 10 (download XLSX )
Statistical source data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, Y., Gu, X., Li, Y. et al. Interplay of genetic predisposition, plasma metabolome and Mediterranean diet in dementia risk and cognitive function. Nat Med 31, 3790–3800 (2025). https://doi.org/10.1038/s41591-025-03891-5
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41591-025-03891-5
This article is cited by
-
Immunometabolism Reframes Alzheimer’s Disease: From Systemic Dysmetabolism to Glial Rewiring
Cellular and Molecular Neurobiology (2026)
-
Polygenicity and APOE ε4 shape response to intervention in mild cognitive impairment
Alzheimer's Research & Therapy (2025)
