
Abstract
Risk of death for both mother and fetus following Ebola virus infection is extremely high. In this study, healthy women in Rwanda aged ≥18 years were randomized to two-dose Ebola vaccination (Ad26.ZEBOV, MVA-BN-Filo) during pregnancy (group A) or postpartum (group B). Unvaccinated pregnant group B women served as control. This was a parallel, randomized, controlled, open-label, single-center trial to evaluate the safety (primary endpoint—outcomes of interest and serious adverse events (SAEs)) and immunogenicity (secondary endpoint) of the two-dose Ebola vaccination. Among 3,484 women screened, 2,013 were randomized, and 2,012 women and 1,945 infants born alive were descriptively analyzed. Adverse outcomes of interest occurred in women (5.2% in group A and 7.3% in group B) and infants (26.0% in group A and 25.6% in group B). The most common maternal outcome of interest was pathways to preterm birth (3.2% in group A and 3.4% in group B), and the most common infant outcome of interest was small for gestational age (14.3% in group A and 11.8% in group B). Maternal/fetal and neonatal/infant SAE frequencies were comparable between groups (9.8% in group A, 9.0% in group B and 21.9% in group A, 15.9% in group B, respectively). Anti-Ebola virus glycoprotein-specific binding antibody response (secondary endpoint) was sustained in ≥90% of women at 1 year postdose 1. In group A, binding antibodies were detected in cord blood (99%) and infant serum (95%) samples 14 weeks postbirth. The trial met all primary and secondary objectives. Ad26.ZEBOV, MVA-BN-Filo did not raise concerns regarding adverse maternal/fetal or neonatal/infant outcomes, had no unexpected safety issues, and induced binding antibody responses in women and offspring through passive transfer. ClinicalTrials.gov registration: NCT04556526.
Similar content being viewed by others


Main
Ebola viruses (EBOVs) belong to the Filoviridae family and cause Ebola disease (EBOD). EBOD is transmitted in humans via direct or indirect contact with the bodily fluids or tissues of infected animals or humans. Mortality in pregnant women infected with Ebola is very high, with an estimated case-fatality rate ranging from 89% (before the 2013–2016 EBOD epidemic)1 to 53% (during the 2013–2016 EBOD epidemic). Mortality in foetuses of pregnant women infected with EBOD is also very high; almost all pregnancies are lost, and survival among liveborn infants is extremely low1.
Janssen Vaccines & Prevention BV, a Johnson & Johnson company, has developed a two-dose heterologous Ebola vaccine regimen (Ad26.ZEBOV vaccine followed by MVA-BN-Filo vaccine 56 days later) for EBOD prevention. Ad26.ZEBOV and MVA-BN-Filo are replication-incompetent, viral-vectored vaccines2. Previous studies have found that the Ad26.ZEBOV, MVA-BN-Filo vaccine regimen is well-tolerated and immunogenic, with mild-to-moderate adverse events (AEs) of short duration and no sequelae3,4,5,6. The protective effect of the Ad26.ZEBOV, MVA-BN-Filo vaccine regimen has been inferred from immunobridging studies7. The Ad26.ZEBOV, MVA-BN-Filo vaccine regimen is currently approved for nonpregnant individuals aged ≥1 year.
In September 2019, the Rwanda Food and Drugs Authority (FDA) granted conditional approval under exceptional emergency circumstances for use of the Janssen Ebola vaccine regimen for nonpregnant people ≥2 years of age in a vaccination campaign called UMURINZI. Between 8 December 2019 and 13 September 2021, 216,113 nonpregnant individuals aged ≥2 years were vaccinated in western Rwanda8.
During the 2019–2020 EBOD outbreak in the Democratic Republic of Congo (DRC), a Merck Ebola vaccine (Ervebo) was licensed and used under an expanded access program that included pregnant women, as recommended by the World Health Organization (WHO)9. Because Ervebo is a live-replicating vaccine, the WHO Strategic Advisory Group of Experts prioritized development of nonreplicating Ebola vaccine candidates for use in pregnancy and recommended that lower-risk populations in the DRC beyond outbreak hotspots be vaccinated with the Janssen regimen2.
However, to date, no data are available on the safety, reactogenicity or immunogenicity of Ebola vaccination in pregnant populations; similarly, there are no data from trials of vaccination of infants and no data on vaccination by gestational age2.
The objective of the present study was to evaluate the safety, reactogenicity and immunogenicity of the two-dose Ebola vaccine regimen in healthy pregnant women in Rwanda. This study was intended to guide the administration of the vaccines during pregnancy.
Results
Patient disposition
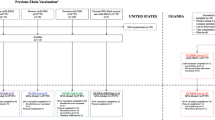
Healthy pregnant women in Rwanda aged ≥18 years were included in this study. Eligibility was initially limited to women in their second or third trimester of pregnancy until the Independent Data Monitoring Committee (IDMC) and the study medical officer reviewed safety data from at least 100 vaccinated pregnant women (group A) and at least 100 nonvaccinated pregnant women (group B), followed for at least 85 days postenrollment. Complete inclusion and exclusion criteria are provided in the Methods. The trial profile is shown in Fig. 1 and Supplementary Fig. 1. Participants were recruited between 6 October 2020 and 29 March 2022. Of the 3,484 women screened, 2,013 were enrolled and randomized (group A, n = 993; group B, n = 1,020). Of these, 992 women in group A received dose 1, composing the complete analysis set (FAS) for group A. The enrolled analysis set (EAS) for group B comprised all 1,020 women randomized to that group. A total of 329 women were enrolled in the immunogenicity subset, of whom 316 were included in the per-protocol immunogenicity (PPI) analysis dataset (group A, n = 161; group B, n = 155). Of those randomized, 959 of 993 (96.6%) group A women and 957 of 1,020 (93.8%) group B women completed study participation. Of the 1,945 infants who were born alive to women enrolled in the study, 964 in group A were in the FAS, and 981 in group B were in the EAS. A total of 75 infants in group A were enrolled in the immunogenicity subset, and all were included in the PPI analysis dataset. Of the liveborn infants, 936 of 964 (97.1%) in group A and 946 of 981 (96.4%) in group B completed study participation.

Infant deaths were captured from birth to 14 weeks. A single asterisk indicates primary analysis datasets for baseline demographic characteristics and safety analyses. Dagger indicates primary analysis datasets for immunogenicity analyses. Main reasons for discontinuation among the randomized women were withdrawal by participant (72/2,013 (3.6%)), loss to follow-up (14/2,013 (0.7%)) and new pregnancy during the study (8/2,013 (0.4%)). Double asterisk indicates data from group C (n = 5) were analyzed separately and are not included in this paper.
Major protocol deviations were reported in 60 of 992 (6.0%) women in group A (FAS) and 46 of 1,020 (4.5%) in group B (EAS). Of these women, 3 (0.1%) entered the study but did not satisfy the inclusion/exclusion criteria and 103 (5.1%) had major protocol deviations for other reasons, which were mainly related to out-of-window visits or not performing certain planned visits or assessments. Major protocol deviations were reported in 29 of 964 (3.0%) infants in group A and 36 of 981 (3.7%) in group B (EAS). All major protocol deviations among infants were for ‘other’ reasons. Main reasons for discontinuation among the infants were withdrawal by parent/guardian (35/1,945 (1.8%)), death (22/1,945 (1.1%)) and loss to follow-up (6/1,945 (0.3%)). Major protocol deviations are described in Extended Data Table 1.
Participant demographic and baseline characteristics are described in Table 1. Median age was 26.0 years (interquartile range (IQR) = 22–30 years). Median gestational age at birth was 40.0 weeks (IQR = 39–40 weeks). Median weight at birth was 3100.0 g (IQR = 2,840–3,400 g) and median height at birth was 49.0 cm (IQR = 48–50 cm). The median Apgar score was 10.0 (IQR = 10–10) at 10 min. The demographic characteristics for mothers and infants were comparable between groups.
Primary and secondary objectives and corresponding outcomes are shown in Table 4; primary outcomes are defined in Extended Data Table 3. Primary objectives were as follows: (1) assess adverse maternal/fetal outcomes in pregnant women who were randomized to receive the two-dose Ebola vaccine regimen (group A) and women in the control group (unvaccinated pregnant women (group B)), and (2) assess adverse neonatal/infant outcomes in neonates/infants born to women who were randomized to receive the two-dose Ebola vaccine regimen (group A) and in neonates/infants born to women in the control group (unvaccinated pregnant women (group B)). Secondary objectives were as follows: (1) assess safety in pregnant women who were randomized to receive the two-dose Ebola vaccine regimen (group A) and women in the control group (unvaccinated pregnant women (group B)), (2) assess safety in neonates/infants born to women who were randomized to receive the two-dose Ebola vaccine regimen (group A) and neonates/infants born to women in the control group (unvaccinated pregnant women (group B)), (3) assess the reactogenicity and unsolicited AEs of the two-dose Ebola vaccine regimen (Ad26.ZEBOV, MVA-BN-Filo) in all vaccinated pregnant women (group A), (4) describe all pregnancy outcomes, (5) assess the immunogenicity of the two-dose Ebola vaccine regimen in 150 pregnant women who were anticipated to receive both vaccine doses within the course of their pregnancy (a subset of the 1,000 vaccinated pregnant women from group A) compared to 150 nonpregnant women who were vaccinated after delivery (a subset of group B) and (6) assess persistence of maternal antibodies in 75 infants born to women from the group A subset. Endpoints from all planned primary and secondary objectives are presented. All endpoints are reported separately.
Primary outcomes and safety
Adverse maternal/fetal outcomes of interest and median days between outcomes and vaccination are described in Table 2 and illustrated in Supplementary Fig. 2, with their definitions in Extended Data Table 3, with 95% confidence interval (CI) half-widths of the frequency of outcomes of interest in Supplementary Table 1. Adverse maternal/fetal outcomes of interest were reported in 51 of 977 (5.2%; 95% CI = 3.9–6.8%) women in group A and 73 of 1,000 (7.3%; 95% CI = 5.8–9.1%) women in group B. One outcome, pathways to preterm birth, occurred with at least 1.0% frequency in both groups (group A—31/977 (3.2%), 95% CI = 2.2–4.5%; group B—34/1,000 (3.4%), 95% CI = 2.4–4.7%). The most common term within this outcome was premature labor (group A—27/977 (2.8%), 95% CI = 1.8–4.0%; group B—28/1,000 (2.8%), 95% CI = 1.9%; 4.0%). In group B, outcomes with at least 1.0% frequency included stillbirth (14/1,000 (1.4%), 95% CI = 0.8–2.3%)), preeclampsia/eclampsia (11/1,000 (1.1%), 95% CI = 0.6–2.0%) and postpartum hemorrhage (11/1,000 (1.1%), 95% CI = 0.6–2.0%)). Although the study was not designed to evaluate adverse maternal/fetal outcomes of interest by trimester, these data are shown in Extended Data Table 4 and Extended Data Fig. 1.
Adverse neonatal/infant outcomes of interest and median days between outcomes and vaccination are described in Table 3 and illustrated in Supplementary Fig. 3, with their definitions in Extended Data Table 3 and 95% CI half-widths of the frequency of outcomes of interest in Supplementary Table 2. Adverse neonatal/infant outcomes of interest were reported in 251 of 964 (26.0%, 95% CI = 23.3–28.9%) infants in group A and 251 of 981 (25.6%, 95% CI = 22.9–28.4%) infants in group B. The most frequently reported outcome was small for gestational age (group A—138/964 (14.3%), 95% CI = 12.2–16.7%), group B—116/981 (11.8%), 95% CI = 9.9–14.0%)). In group A, other adverse outcomes of interest with at least 1.0% frequency included failure to thrive (87/964 (9.0%), 95% CI = 7.3–11.0%)), low birth weight (45/964 (4.7%), 95% CI = 3.4–6.2%)), preterm birth (26/964 (2.7%), 95% CI = 1.8–3.9%)) and neonatal death (11/964 (1.1%), 95% CI = 0.6–2.0%)). In group B, other adverse outcomes of interest with at least 1.0% frequency included failure to thrive (101/981 (10.3%), 95% CI = 8.5–12.4%)), low birth weight (49/981 (5.0%), 95% CI = 3.7–6.6%)), preterm birth (30/981 (3.1%), 95% CI = 2.1–4.3%)) and congenital anomaly (11/981 (1.1%), 95% CI = 0.6–2.0%)). Although the study was not designed to evaluate adverse neonatal/infant outcomes of interest by trimester, these data are shown in Extended Data Table 2 and Extended Data Fig. 2.
Secondary outcomes and safety
Maternal serious AEs (SAEs) are described in Supplementary Table 3. SAEs were reported in 97/992 (9.8%) group A women and 92/1,020 (9.0%) group B women. In group A, maternal SAEs reported with at least 1.0% frequency were attributable to system organ class categories of pregnancy, puerperium and perinatal conditions (56/992 (5.6%)); infections and infestations (37/992 (3.7%)); and reproductive system and breast disorders (15/992 (1.5%)). In group B, SAEs reported with at least 1.0% frequency were attributable to system organ class categories of pregnancy, puerperium and perinatal conditions (75/1,020 (7.4%)) and infections and infestations (13/1,020 (1.3%)). By preferred term, differences in maternal SAEs by group included urinary tract infections (group A—10/992 (1.0%); group B—2/1,020 (0.2%)), postoperative wound infections (group A—9/992 (0.9%); group B—4/1,020 (0.4%)), bacterial vaginosis (group A—7/992 (0.7%); group B—1/1,020 (0.1%)) and vulvovaginal candidiasis (group A—7/992 (0.7%); group B—0/1,020 (0%)).
The majority of SAEs in women were severity grade 1 or 2. Grade 3 SAEs were reported by 16/992 (1.6%) women in group A and 17/1,020 (1.7%) women in group B. One grade 4 SAE was reported by 1 of 1,020 (0.1%) women in group B. Most grade 3 and 4 SAEs were attributable to pregnancy, puerperium and perinatal conditions. Almost all SAEs were considered unrelated to the vaccine; in group A, only 1 of 992 (0.1%) woman experienced a related event of nausea and vomiting postdose 1, 1 of 992 (0.1%) experienced a related event of headache and pyrexia postdose 2 and 2 of 992 (0.2%) experienced related events of fatigue postdose 1.
Infant SAEs are described in Supplementary Table 4. SAEs were reported in 211 of 964 (21.9%) infants in group A and 156 of 981 (15.9%) infants in group B. Deaths of 22 infants were reported (Fig. 1)—12 of 964 (1.2%) infants in group A and 10 of 981 (1.0%) infants in group B. The most frequently reported causes of death were related to hypoxic–ischemic encephalopathy in group A (4/964 (0.4%)) and sudden infant death syndrome in group B (3/981 (0.3%)). In group A, infant SAEs reported with at least 1.0% frequency were attributable to system organ class categories of infections and infestations (110/964 (11.4%)); surgical and medical procedures (73/964 (7.6%)); pregnancy, puerperium and perinatal conditions (56/964 (5.8%)); nervous system disorders (23/964 (2.4%)); and respiratory, thoracic and mediastinal disorders (13/964 (1.3%)). In group B, infant SAEs reported with at least 1.0% frequency were attributable to system organ class categories of surgical and medical procedures (71/981 (7.2%)); infections and infestations (63/981 (6.4%)); pregnancy, puerperium and perinatal conditions (50/981 (5.1%)); and nervous system disorders (14/981 (1.4%)). By preferred term, differences in infant SAEs by group included neonatal infection (group A—52/964 (5.4%); group B—30/981 (3.1%)), neonatal sepsis (group A—23/964 (2.4%); group B—15/981 (1.5%)) and low birth weight (group A—10/964 (1.0%); group B—1/981 (0.1%)).
Most SAEs in infants were severity grade 1 or 2. Grade 3 SAEs were reported for 23 of 964 (2.4%) infants in group A and 13 of 981 (1.3%) in group B. Grade 4 SAEs were reported for 8 of 964 (0.8%) infants in group A and 6 of 981 (0.6%) in group B. Most grade 3 and 4 SAEs were attributable to infections and infestations.
The most frequently reported adverse maternal/fetal outcome not of special interest and not included in the primary endpoints throughout the entire study was cesarean section in both group A and group B (207/977 (21.2%) women in group A and 224/1,000 (22.4%) women in group B).
Solicited AEs were only recorded in group A for 7 days after each vaccination. Solicited AEs were reported in 637 of 992 (64.2%) women during the vaccine regimen period (postdose 1 and postdose 2 combined period)—556 of 992 (56.0%) postdose 1 and 425 of 981 (43.3%) postdose 2. Of the women reporting solicited AEs (local or systemic), the majority (96.7%) reported events that were grade 1 or grade 2 in severity. Unsolicited AEs were recorded in both groups for the entire study duration. Supplementary Table 5 describes both solicited and unsolicited AEs.
Secondary outcomes
The PPI analysis set consisted of 316 women (group A—n = 161; group B—n = 155). As shown in Fig. 2a, anti-EBOV glycoprotein (GP)-specific binding antibody concentrations increased from baseline, with peak responses at 21 days postdose 2. At 365 days postdose 1, responses were durable, with no differences between groups. Anti-EBOV GP-specific binding antibody response was observed in 99.2–100% of women at 21 days postdose 2, regardless of group or trimester at randomization. At 1 year postdose 1, 90.8% of group A and 94.0% of group B women were still classified as responders (Extended Data Table 5). When stratified by trimester, 89.5% of women in group A and 93.0% of women in group B randomized in their second trimester were responders at 1 year postdose 1; 96.4% of group A women and 100% of group B women randomized in their third trimester were still responders at 1 year postdose 1.

a, Anti-EBOV GP-specific binding antibody concentrations in vaccinated women over time. GMCs with 95% CI are shown in the figure. Group A, pregnant women randomized to be vaccinated during pregnancy; group B, pregnant women randomized to be vaccinated after pregnancy. Enrollment of first-trimester pregnant women was opened after the immunogenicity subset was fully enrolled. As a result, no first-trimester pregnant women were included in the immunogenicity subset. b, Correlation between mother anti-EBOV GP-specific binding antibody concentration at postpartum day 1 and mother cord blood anti-EBOV GP-specific binding antibody concentration at postpartum day 1. c, Correlation between mother anti-EBOV GP-specific binding antibody concentration at postpartum day 1 versus infant anti-EBOV GP-specific binding antibody concentration at 14 weeks of age. d, Correlation between mother cord blood anti-EBOV GP-specific binding antibody concentration at postpartum day 1 versus infant anti-EBOV GP-specific binding antibody concentration at 14 weeks of age. For b–d, the Spearman correlation coefficients calculated are partial Spearman correlation coefficients that control for the trimester of the mother at randomization. For a–d, trimester refers to the trimester at randomization. First trimester, 0–12 weeks; second trimester, >12–24 weeks; third trimester, >24 weeks. EU, enzyme-linked immunosorbent assay units. N/A, not applicable.
In group A, anti-EBOV GP-specific binding antibodies were detected in 99.3% of cord blood samples at postpartum day 1 and 94.7% of infant serum samples at 14 weeks postbirth. When stratified by trimester, 99.2% and 100% of cord blood samples from women randomized in trimesters two and three, respectively, were positive for anti-EBOV GP-specific binding antibodies. Infant serum samples at 14 weeks postbirth showed 93.9% positivity when mothers were randomized in their second trimester and 96.2% positivity when mothers were randomized in their third trimester.
Strong correlations were observed between anti-EBOV GP-specific binding antibody concentrations in women at postpartum day 1 and cord blood at postpartum day 1 (partial Spearman correlation coefficient, 0.84; Fig. 2b), in women at postpartum day 1 and infants at 14 weeks of age (partial Spearman correlation coefficient, 0.82; Fig. 2c) and in cord blood at postpartum day 1 and infants at 14 weeks of age (partial Spearman correlation coefficient, 0.87; Fig. 2b–d).
Discussion
To our knowledge, this is the first study to provide a rigorous evaluation of the safety and immunogenicity of a two-dose Ebola vaccine regimen in pregnant women. The Ad26.ZEBOV, MVA-BN-Filo vaccine regimen is well-tolerated, and, overall, no unexpected safety issues were identified. The frequency of maternal/fetal adverse outcomes of interest does not differ in the group with vaccination during pregnancy and the control group of women vaccinated postpartum (group A—5.2%; group B—7.3%). The overall frequency of adverse neonatal/infant outcomes is also similar between the two groups (group A—26.0%; group B—25.6%). Neonatal death and small for gestational age are reported more frequently in group A, but preterm birth and failure to thrive are reported more frequently in group B. Both observed rates of neonatal death and small for gestational age are below background rates for sub-Saharan Africa10,11, and the 95% CI overlap suggests a degree of uncertainty around assignment of these events by group. Furthermore, by 14 weeks of age, the discrepancy in infant deaths disappears.
Slightly higher frequencies of SAEs in group A women (group A—9.8%; group B—9.0%) may be explained by group A women having more study visits before delivery, all of which ascertained unsolicited adverse outcomes. Higher frequencies of nausea, pyrexia and headache in group A women may be partially explained by vaccine reactogenicity.
SAEs are reported more frequently in group A infants (group A—21.9%; group B—15.9%), especially in the neonatal period (birth to day 28). One possible explanation is that group A had scheduled postpartum visits, whereas group B did not.
The Ad26.ZEBOV, MVA-BN-Filo vaccine regimen induces robust anti-EBOV GP-specific binding antibody responses in women, with strong correlations observed between anti-EBOV GP-specific binding antibody concentrations in women, cord blood and infants. Like other studies12, pregnant women have a strong but slightly lower immune response to vaccination than nonpregnant women. The kinetics of the Ad26.ZEBOV, MVA-BN-Filo vaccine-induced immune response indicates an expected steady decline in circulating binding antibody concentrations after 21 days postdose 2 until a plateau is reached at approximately 3 months postdose 2. Passive transfer of anti-EBOV GP-specific binding antibodies from mother to offspring is observed as quantifiable binding antibodies in their offspring. Geometric mean concentrations (GMCs) are higher in women, cord blood and infants born to women in their third trimester than in their second trimester at randomization, which is anticipated given the expected kinetics of the postvaccination binding antibody responses because women in their second trimester experienced a longer time interval between the assessments at 21 days postdose 2 and postpartum day 1.
The immune response of pregnant women by trimester has not been extensively studied. Our study indicates that vaccination in the third trimester yields a greater antibody response, which adds to our overall knowledge about vaccination in pregnancy. A study discussed in ref. 13 does not show evidence that the immune response to influenza vaccination is significantly altered by timing of vaccination in a group of 239 pregnant or postpartum women receiving H1N1 monovalent vaccine, but they observed a trend toward lower immune responses in the first trimester and 6 weeks postpartum. Generally, transplacental antibody transfer gradually increases as pregnancy progresses14, and immunization of pregnant women is often recommended in the second or third trimester to maximize the potential of the passive transfer of maternal antibodies to the fetus and limit potential teratogenicity concerns.
There are several notable strengths of the study. Potential selection bias is minimized in this open-label trial, given the randomization and the clear inclusion and exclusion criteria. The trial has extremely high retention, despite being conducted during the COVID-19 pandemic (baseline characteristics of study completers and noncompleters are provided in Supplementary Table 6, revealing no notable difference), and benefits from the rigorous determination of gestational age and estimated date of delivery. The study design decision to potentially allow enrollment of women during their first trimester of pregnancy after rigorous safety data review by study medical officers and the IDMC was novel; however, the limited sample size of this group precludes conclusions, and we recommend future studies consider evaluation of women at early gestational ages to support protecting women as early in their pregnancies as possible.
The data should be interpreted considering limitations. This is not a masked trial, although it should be noted that the labor and delivery staff do not know which group the women are in, which may have reduced assessment bias. The lack of masking for other staff members may have led to reporting bias for subjective AEs. Additionally, assessment of small for gestational age and failure to thrive used standard WHO charts, which may not be representative of the African/Rwandan context. Finally, solicited AEs are only recorded in group A, limiting the interpretation. Finally, no conclusions can be made regarding the levels of protection against EBOD provided by the vaccine-induced binding antibodies elicited in pregnant women or transferred to their infants, as no correlate or threshold of protection has been established.
The Ad26.ZEBOV, MVA-BN-Filo regimen is well-tolerated, with no unexpected safety issues, and induces robust anti-EBOV GP-specific binding antibody responses in women, as well as quantifiable binding antibodies in their offspring through passive transfer. This regimen may offer protection to pregnant women and their children who have an extremely high risk of death or severe disease following Ebola infection.
Methods
Study design
This is a parallel, randomized, controlled, open-label, single-center clinical trial of the safety, reactogenicity and immunogenicity of the Janssen two-dose Ebola vaccine regimen in healthy adult pregnant women in Rwanda. The study arms compared pregnant women who received the two-dose Ebola vaccine regimen during pregnancy (group A) or after pregnancy completion (group B). Unvaccinated pregnant group B women served as control. The study was conducted between 6 October 2020 (the date the first participant signed an informed consent form) and 2 March 2023 (the date of the last participant visit) at Gisenyi District Hospital in Rubavu and Gihundwe District Hospital in Rusizi, both located in the Western Province of Rwanda bordering the DRC. The study complies with all relevant ethical regulations. The protocol was approved by the Rwanda National Ethics Committee (821/RNEC/2020), the Rwanda FDA (003/CT/RWANDA FDA/2020) and the Emory University Institutional Review Board (STUDY00000367). The study was prospectively registered with ClinicalTrials.gov under NCT04556526 on 21 September 2020. The study protocol has been published15.
Participants
Pregnant women residing in the catchment areas of the District Hospitals were recruited from antenatal care clinics and referred to the two district hospitals. Written informed consent to participate was obtained. Inclusion criteria were as follows:
-
1.
Female (according to their reproductive organs and functions assigned by chromosomal complement).
-
2.
Eighteen years of age or older at the time of written informed consent.
-
3.
Healthy based on physical examination, medical history, obstetric history and vital signs performed at screening.
-
4.
Healthy based on clinical laboratory tests performed at screening. If the results of the clinical laboratory tests are outside of the normal local reference ranges, the participant may be included only if the investigator judges the abnormalities or deviations from normal to be not clinically significant or to be appropriate and reasonable for the population under study. This determination must be recorded in the participant’s source documents and initiated by the investigator. Trace protein in the urine is acceptable if the blood pressure is also normal. Participants must have clinical laboratory test results within the following parameters:
-
a.
Hemoglobin ≥7.0 g dl−1.
-
b.
White blood cells ≥3.4 × 103 cells per µl.
-
c.
Neutrophils ≥1.1 × 109 cells per l.
-
d.
Lymphocytes ≥1.21 × 109 cells per l.
-
e.
Platelets ≥156 × 103 cells per µl.
-
f.
Serum creatinine ≤129 µmol l−1
-
g.
Aspartate aminotransferase within 1.5× the upper limit of the normal range for the laboratory conducting the test.
-
h.
Alanine aminotransferase within 1.5× the upper limit of the normal range for the laboratory conducting the test.
-
i.
Total bilirubin ≤25 µmol l−1.
-
j.
Urine protein <1+ by dipstick.
-
k.
Urine blood ≤1+ by dipstick, without evidence of bacteriuria on microscopy.
-
a.
-
5.
Confirmed singleton pregnancy by positive urine human chorionic gonadotropin and ultrasound at the time of screening and informed consent, and reconfirmed pregnancy via ultrasound at randomization/day 1. Ultrasound not required on randomization/day 1 if ≤10 days have elapsed since the screening ultrasound and an ultrasound is not indicated for other reasons.
-
6.
Capable and willing to give informed consent (signed or thumbprint), which includes compliance with the requirements and restrictions listed in the informed consent form and in this protocol, and willing to give informed consent for the infant to participate in the study.
-
7.
Each potential participant must pass the assessment of understanding by indicating that she understands the purpose, procedures and potential risks and benefits of the study after reading the informed consent and after the investigator or designee has provided detailed information on the study and has answered the potential participant’s questions. Each participant must subsequently sign the informed consent form, indicating that she is willing to participate in the study.
-
8.
Resides within the catchment area of the study site.
-
9.
Able and willing to participate for the duration of the study visits and follow-up.
-
10.
Willing and able to comply with the protocol requirements.
-
11.
Willing to receive standard prenatal care and planning to deliver at a study district hospital.
-
12.
Willing to provide verifiable identification and have a photo taken and an iris scan at study entry and follow-up visits.
-
13.
Evidence of normal progress of gestation before randomization (day 1) based on obstetric evaluation (including obstetric history, obstetric examination and fetal ultrasound).
-
14.
If randomized to group B, must have a negative urine pregnancy test immediately before each study vaccine administration.
Exclusion criteria were as follows:
-
1.
History of EBOV disease (self-declared or laboratory confirmed).
-
2.
Has received any candidate Ebola vaccine (except for women found to be pregnant in the UMURINZI study after they received their first dose of vaccine, who are eligible for enrollment into a nonrandomized group under this protocol).
-
3.
Has received any experimental candidate Ad26- or MVA-based vaccine in the past. Receipt of any approved vaccinia/smallpox vaccine or Ad-based candidate vaccine other than Ad26 at any time before study entry is allowed.
-
4.
Known allergy or history of anaphylaxis or other serious adverse reactions to vaccines or vaccine products (including any of the constituents of the study vaccines (for example, polysorbate 80, ethylenediaminetetraacetic acid or l-histidine for the Ad26.ZEBOV vaccine; tris (hydroxymethyl)-amino methane for the MVA-BN-Filo vaccine)), including known allergies to egg, egg products, chicken proteins and aminoglycosides.
-
5.
Individuals with acute illness (this does not include minor illnesses such as diarrhea or mild upper respiratory tract infection) or body temperature ≥38.0 °C on day 1 will be excluded from enrollment at that time but may be rescheduled for enrollment at a later date.
-
6.
Presence of significant conditions (for example, history of seizure disorders, (auto)immune disease or deficiency, any spleen disease, active malignancy, ongoing tuberculosis treatment and other systemic infections) or clinically significant findings during screening of medical history, obstetric history, physical examination, vital signs or laboratory testing for which, in the opinion of the investigator, it would not be in the best interest of the individual (for example, compromise the safety or well-being) or that could prevent, limit or confound the protocol-specified assessments. Individuals who have recently received treatment for acute uncomplicated malaria are eligible for participation if at least 3 days have elapsed from the conclusion of a standard recommended course of therapy for malaria; participants who are acutely ill with malaria at the time of screening should complete therapy and wait an additional 3 days after completion before screening for the study. Individuals with sickle cell traits can be included.
-
7.
Any of the following during the 6 weeks before screening: (1) confirmed SARS-CoV-2 (COVID-19) infection (positive test), OR (2) suspected SARS-CoV-2 infection (clinical features without documented test results), OR (3) close contact with a person with known or suspected SARS-CoV-2 infection.
-
8.
History of or underlying liver or renal insufficiency or significant cardiac, vascular, pulmonary (for example, persistent asthma), gastrointestinal, endocrine, neurological, hematological, rheumatological, psychiatric or metabolic disturbances.
-
9.
History of positive tests for hepatitis B surface antigen (HBsAg) or hepatitis C virus (HCV) antibodies (anti-HCV), or other clinically active liver disease, or testing positive for HBsAg or anti-HCV at screening.
-
10.
Human immunodeficiency virus (HIV)-infected but not on stable antiretroviral therapy (ART), defined as adherent to the ART regimen for ≥6 weeks before enrollment. Individuals with HIV infection on stable ART may be enrolled.
-
11.
Obstetric history, including the following:
-
a.
≥2 consecutive spontaneous abortions.
-
b.
History of preeclampsia or eclampsia.
-
c.
Rhesus-negative multigravida.
-
d.
Grand multigravida (>5 previous pregnancies).
-
e.
Previous late stillbirth (defined as loss of pregnancy at any time after 28 weeks of gestation).
-
f.
Previous low-birth-weight baby or premature delivery (defined as a delivery before 37 weeks of gestation).
-
g.
Previous neonatal death (defined as death of an infant within the first 28 days of life).
-
h.
Previous delivery of an infant with a known or suspected genetic or chromosomal abnormality.
-
i.
History of other significant pregnancy-related or neonatal complications that are judged as likely to affect the safety of the mother or infant or to significantly compromise the collected endpoint data. Previous cesarean section is not an exclusion criterion.
-
a.
-
12.
Major surgery within the 4 weeks before screening or planned major surgery through the course of the study (from screening until completion of the study).
-
13.
Chronic or recurrent use of immunomodulators/suppressors (for example, cancer chemotherapeutic agents or systemic corticosteroids within 6 months before the planned administration of the first dose of study vaccine). Ocular, topical or inhaled steroids are allowed.
-
14.
Received or planned to receive a licensed nonlive attenuated vaccine (for example, tetanus) within 7 days of a study vaccination (that is, before or after). Received or planned to receive a licensed live attenuated vaccine within 4 weeks of a study vaccination (that is, before or after).
-
15.
Received an investigational drug or investigational vaccine or used an invasive investigational medical device within 3 months before screening, or current or planned participation in another clinical study during the study. Participation in an observational clinical study is allowed.
-
16.
Receipt of blood products or immunoglobulin within 3 months before screening and/or during participation in the study (except for RhoGAM).
-
17.
Current or past abuse of alcohol or recreational or narcotic drugs that, in the investigator’s opinion, would compromise the individual’s safety and/or compliance with the study procedures.
-
18.
History of chronic urticaria (recurrent hives).
-
19.
Individual cannot communicate reliably with the investigator.
-
20.
Employee of the investigator or study site, with direct involvement in the proposed study or other studies under the direction of that investigator or study site, including family members of the employees or the investigator.
-
21.
History of thrombotic thrombocytopenia syndrome (TTS) or heparin-induced thrombocytopenia and thrombosis.
Eligibility was initially limited to women in their second or third trimester of pregnancy until IDMC and the study medical officer reviewed safety data from at least 100 vaccinated pregnant women (group A) and at least 100 nonvaccinated pregnant women (group B), followed for at least 85 days postenrollment. When these data indicated no safety issues, enrollment was extended to women in their first trimester of pregnancy. Trimester at enrollment was categorized as first trimester (0–12 weeks), second trimester (>12–24 weeks) or third trimester (>24 weeks). The estimated gestational age was based on ultrasound for women enrolled in the first trimester. For women enrolled in the second and third trimesters, estimated gestational age was based on ultrasound and the last menstrual period as recommended by the American College of Obstetricians and Gynecologists (ACOG)16.
For women, sex was based on clinical observation—all women participating in the study were pregnant at some point. For infants, sex observed at birth was reported by investigators. The results are described for infants of both sexes together. No difference by sex of the infant was expected.
Randomization and masking
Eligible pregnant women were randomly assigned 1:1 to receive the two-dose Ebola vaccine regimen during pregnancy (group A) or after pregnancy completion (group B). An allocation template was created by coauthor S.A. at Emory University using simple randomization based on the Microsoft Excel random number generator function. Security-type, opaque envelopes containing the randomization assignment cards were prepared by one study team member and inspected by two other team members to ensure correspondence to the allocation table before sealing the envelopes and deploying them to the sites. The envelopes at each site were placed in a single bin, and participants were invited to reach into the bin to select a single sealed envelope containing a randomization assignment at the time of enrollment. Study staff responsible for enrolling participants and overseeing randomization did not have access to the allocation table; therefore, arm assignment within each envelope was unknown to participants and staff until opened. The randomization assignment was complete and irreversible once the envelope was opened. Birth attendants were not formally notified about the vaccine allocation. There was no masking in this study. An open-label design was selected so that group B women could receive the benefits of the vaccine regimen.
Procedures
All study procedures were performed in the district hospitals, including pre-enrollment screening. A schematic overview of study activities and timeline is shown in Supplementary Fig. 1; detailed schedules of activities are published15. A target of 2,000 pregnant women ≥18 years of age was to be randomized on day 1. Women randomized to group A received the two-dose Ebola vaccine regimen on days 1 and 57 during pregnancy. Women randomized to group B received the vaccine regimen from 6 weeks postpartum up to 10 weeks postpartum.
Study vaccines were manufactured and provided under the responsibility of Janssen Vaccines and Prevention B.V. Ad26.ZEBOV is a nonreplicating monovalent vaccine that encodes the full-length GP from EBOV Mayinga. The vaccine is produced in the human cell line PER.C6. A 0.5-ml intramuscular injection of 5 × 1010 viral particles was administered on day 1 (group A) or between 6 and 10 weeks after completion of pregnancy (group B). This vaccine was administered into the deltoid muscle in the upper arm (or thigh if needed). MVA-BN-Filo does not replicate in human cells and is a multivalent vaccine encoding the EBOV Mayinga GP, the Sudan virus Gulu GP, the Marburg virus Musoke GP and the Taï Forest virus nucleoprotein. The EBOV GP expressed by MVA-BN-Filo has 100% homology with the one expressed by Ad26.ZEBOV. A 0.5-ml intramuscular injection of 1 × 108 infectious units of MVA-BN-Filo was administered 56 days (−14 days to +28 days) after the Ad26.ZEBOV dose. This vaccine was administered into the deltoid muscle in the upper arm (or thigh if needed), ideally in the opposite arm/thigh as the first dose (unless the opposite arm/thigh had a condition preventing evaluation of the arm/thigh after injection).
As shown in Supplementary Fig. 1, a target subset of 300 women was to be enrolled and provide blood samples for immunogenicity assessment of the two-dose regimen administered during pregnancy (group A) and after pregnancy (group B).
Group A women were followed until at least 6 weeks after pregnancy completion to capture adverse maternal/fetal outcomes of interest. Group B women were followed until at least 4 weeks after dose 2. Participants in the immunogenicity subset were followed for 12 months after dose 1. Therefore, the duration of individual participation could range from approximately 6 to 23 months. All infants were followed for 14 weeks postdelivery.
Women who tested positive for pregnancy during the UMURINZI Ebola Vaccination Campaign after they received dose 1 were offered screening and enrollment into the present study to receive dose 2 in a controlled setting (group C). These women were not randomized but were followed for safety outcomes for 6 weeks after pregnancy completion (women) or 14 weeks postdelivery (infants).
There were three amendments to the original protocol. Protocol amendment 1 was issued on 8 July 2020. The overall reason for the amendment was to incorporate comments from the ethics committee on the initial protocol, which was never approved by the Health Authority. The study was only initiated under protocol amendment 1. Protocol amendment 2 was issued on 22 July 2021. The overall reason for the amendment was to include information about TTS observed with Janssen’s COVID-19 vaccine and clarification of the safety notification process. Main changes included the exclusion of participants with a known history of TTS or heparin-induced thrombocytopenia with thrombosis, as well as those who planned to or received a COVID-19 vaccine within a disallowed timeframe before study vaccination. Additionally, a 24-h reporting timeframe for TTS events was added, and potential conditions that should be reported within 24 h were listed. Protocol Amendment 3 was issued on 24 November 2021. The overall reason for the amendment was to include additional language around TTS, as recommended by the United States FDA on 17 September 2021. Main changes included an update of the description of TTS observed with Janssen’s COVID-19 vaccine to align with the language recommended by the FDA and inclusion of the recommendation to evaluate any potential risk factors for TTS in participants based on medical history.
Outcomes
Primary and secondary objectives and corresponding outcomes are shown in Table 4. Brighton Collaboration case definitions for primary adverse outcomes of interest are shown in Extended Data Table 3. Outcomes were abstracted from medical records linked across participants using a unique participant identifier and biometric data17. All AEs were recorded using medical terminology in the source documents and case report forms. AEs were coded in accordance with the ‘Medical Dictionary for Regulatory Activities v25.1’ using the lower-level term as the description most closely related to the investigator’s terminology, a preferred term describing a group of closely related lower-level terms, and the system organ class, which is the broad category including related preferred terms. The reporting of safety data included the incidence, severity and relatedness.
Clinical outcomes were collected via hardcopy paper records and captured and validated electronically in RedCap Cloud, with additional validation performed using SAS v9.4. Immunogenicity samples were analyzed using the Filovirus Animal Nonclinical Group (FANG) enzyme-linked immunosorbent assay (ELISA) to assess concentrations of anti-EBOV GP-specific binding antibodies.
Immunogenicity assessments
Venous blood samples were collected for immunogenicity assessments at protocol-defined visits. IgG binding antibodies against EBOV GP were measured using the validated and FDA-endorsed FANG ELISA, performed by Q2 Solutions Vaccine Testing Laboratory.
Statistical analysis
The study is not powered for any formal statistical hypothesis testing. A target sample size of 1,000 participants in group A and 1,000 participants in group B was determined based on feasibility and the estimated background incidence of key pregnancy outcomes. With a sample size of 1,000, the probability of observing at least one AE occurring at a rate of 1 in 1,000 was 63%. If no SAEs or AEs were observed in group A, this would provide 95% confidence that the true incidence was ≤0.3%. The width of the 95% CI for observing an AE at a rate of 1 in 1,000 is 0.006. An overenrollment of ≤10% was permitted. For a 10% overenrollment, the width of the 95% CI for observing an AE at a rate of 1 in 1,000 is 0.005.
Data were analyzed using SAS v9.4. No formal statistical hypothesis testing was planned. To aid understanding of the uncertainty around the estimates, CIs for primary endpoint estimates and the risk differences between groups A and B were computed post hoc. The CI of the risk differences should be interpreted with caution, given the highly inflated type I error due to the numerous primary outcomes. Data from group C (n = 5) were analyzed separately and are not included here. Group C is not expected to influence the conclusions of the study. The unvaccinated pregnant women in group B served as the control group for the vaccinated pregnant women in group A. For group A, the primary analysis dataset for baseline demographic characteristics and safety data was the FAS, which included all randomized participants who received at least one dose of the study vaccine regimen, regardless of protocol deviations, and all infants born to these women. For group B, the primary analysis dataset for baseline demographic characteristics and safety data was the EAS, which included all women enrolled and all infants born to these women.
Immunogenicity assessments were performed on the PPI analysis set, which included all women who provided informed consent for immunogenicity sample collection, received both dose 1 and dose 2 within the protocol-defined window and did not have major protocol deviations that could impact immunogenicity outcomes (for example, another vaccination and immunomodulating medications). Infants in the PPI analysis set were those born to group A women in the PPI analysis set who also provided immunogenicity samples.
The number and percentage of participants screened, enrolled/randomized, vaccinated and completed follow-up were summarized following the Consolidated Standards of Reporting Trials guidance. Major (defined as those that would jeopardize the ability to evaluate study endpoints) and minor protocol deviations were described by group. Baseline demographic characteristics were described by group. The frequency and percentage of primary adverse outcomes of interest were tabulated by group and trimester. Adverse maternal/fetal outcomes of interest were described from randomization until 6 weeks postpartum. Women who discontinued the study without reporting any adverse maternal/fetal outcomes were excluded from the analysis. Additionally, the frequency and percentage of maternal and infant SAEs were tabulated by group; participants were counted only once for any given event, regardless of the number of times they experienced the event.
Missing data were not imputed. Participants who discontinued the study without reporting any maternal/fetal outcomes were excluded from the analyses because of completely missing information on the pregnancy outcome. Efforts were made to query the sites for missing severity of the AE or missing relationship of the AE with the study vaccine. An AE with a missing severity or relationship was considered as an AE reported, but was considered as not reported for the severity or relationship. For example, an AE with missing severity was considered as an AE reported for the analysis of any grade, but was not considered for the analysis of grade 3.
GMCs with corresponding 95% CIs were calculated for the continuous immunological parameter of anti-EBOV GP-specific binding antibody concentration at each time point. Sample positivity (defined as the proportion of samples with a quantifiable response) and the proportion of participants who were responders (defined as the proportion of participants with a greater than 2.5-fold increase in anti-EBOV GP-binding antibody concentration compared to baseline) were calculated at each time point, along with their corresponding Clopper–Pearson exact 95% CIs. All values below the lower limit of quantification (LLOQ) were imputed with half the LLOQ for calculation of GMC, and values greater than the upper limit of quantification (ULOQ) were imputed with the ULOQ. For the calculation of fold changes, the values below LLOQ were imputed with the corresponding LLOQ, and values above the ULOQ were imputed with the ULOQ.
To evaluate potential associations between anti-EBOV GP-specific binding antibody concentrations in women at different time points (predose 1, 21 days postdose 2, postpartum day 1 and 365 days postdose 1), cord blood at postpartum day 1 and infants at 14 weeks of age, a post hoc correlation analysis was performed. Partial Spearman correlation coefficients controlling for the trimester of the mother at randomization were calculated, and correlation plots were generated.
An IDMC consisting of independent experts in obstetrics, pediatrics and statistics evaluated safety data twice at the outset of the study and then three times monthly. The study was prospectively registered with ClinicalTrials.gov under NCT04556526 on 21 September 2020.
Ethics and inclusion statement
Local researchers based at Center for Family Health Research (CFHR) were included throughout the research process, including study design, study implementation, data ownership, intellectual property and authorship of publications. Publications are inclusive of local and regional research relevant to our study. The research is locally relevant and conducted with local leadership and in collaboration with the Rwandan Ministry of Health. Roles and responsibilities were agreed upon among collaborators ahead of the research. The study did not include formal plans for local capacity building. The study was approved by local ethics review committees. The study did not pose any health, safety, security or other risk to researchers. The study could cause risk to participants, and participant safety procedures are described in the manuscript.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data sharing policy of Janssen Pharmaceutical Companies of Johnson & Johnson is available at https://www.janssen.com/clinical-trials/transparency. Janssen prioritizes transparency of clinical trial data, including registration and disclosure of trial results in external registries, publication in peer-reviewed journals, sharing of clinical study reports (CSRs), sharing of analyzable participant-level data and making plain language summaries (written in a nontechnical manner) available. Requests for the study protocol may be made to Janssen Pharmaceutical Companies of Johnson & Johnson. As noted on https://www.janssen.com/clinical-trials/transparency, requests for access to the CSRs and participant-level data, including individual participant-level data, can be submitted through the Yale Open Data Access (YODA) Project site at http://yoda.yale.edu. All procedures for external investigators to access clinical trial data, including project review, due diligence assessments, external review by a Steering Committee and the Data Use Agreement (DUA), are detailed at https://yoda.yale.edu/wp-content/uploads/2022/11/YODA-Project-Data-Release-Procedures-January-2025.pdf. Questions can be submitted to the YODA Project via email yodap@yale.edu. Requesters can expect to hear back within 1 week.
Code availability
Requests for access to the analytic code can be submitted through YODA Project site at http://yoda.yale.edu. Questions can be submitted to the YODA Project via email yodap@yale.edu. Requesters can expect to hear back within 1 week.
References
Gomes, M. F., de la Fuente-Núñez, V., Saxena, A. & Kuesel, A. C. Protected to death: systematic exclusion of pregnant women from Ebola virus disease trials. Reprod. Health 14, 172 (2017).
World Health Organization. Extraordinary meeting of the Strategic Advisory Group of Experts on Immunization on Ebola vaccination, May 2024: conclusions and recommendations. www.who.int/publications/i/item/WER-9927-351-362 (2024).
Barry, H. et al. Safety and immunogenicity of 2-dose heterologous Ad26.ZEBOV, MVA-BN-Filo Ebola vaccination in healthy and HIV-infected adults: a randomised, placebo-controlled phase II clinical trial in Africa. PLoS Med. 18, e1003813 (2021).
Anywaine, Z. et al. Safety and immunogenicity of 2-dose heterologous Ad26.ZEBOV, MVA-BN-Filo Ebola vaccination in children and adolescents in Africa: a randomised, placebo-controlled, multicentre phase II clinical trial. PLoS Med. 19, e1003865 (2022).
Afolabi, M. O. et al. Safety and immunogenicity of the two-dose heterologous Ad26.ZEBOV and MVA-BN-Filo Ebola vaccine regimen in children in Sierra Leone: a randomised, double-blind, controlled trial. Lancet Infect. Dis. 22, 110–122 (2022).
Ishola, D. et al. Safety and long-term immunogenicity of the two-dose heterologous Ad26.ZEBOV and MVA-BN-Filo Ebola vaccine regimen in adults in Sierra Leone: a combined open-label, non-randomised stage 1, and a randomised, double-blind, controlled stage 2 trial. Lancet Infect. Dis. 22, 97–109 (2022).
Roozendaal, R. et al. Nonhuman primate to human immunobridging to infer the protective effect of an Ebola virus vaccine candidate. NPJ Vaccines 5, 112 (2020).
Nyombayire, J. et al. Monitoring of adverse events in recipients of the 2-dose Ebola vaccine regimen of Ad26.ZEBOV followed by MVA-BN-Filo in the UMURINZI Ebola vaccination campaign. J. Infect. Dis. 227, 268–277 (2023).
World Health Organization. Guidelines for the management of pregnant and breastfeeding women in the context of Ebola virus disease. www.ncbi.nlm.nih.gov/books/NBK554100/table/ch4.tab6/ (2020).
Lawn, J. E. et al. Small babies, big risks: global estimates of prevalence and mortality for vulnerable newborns to accelerate change and improve counting. Lancet 401, 1707–1719 (2023).
Lee, A. C. et al. National and regional estimates of term and preterm babies born small for gestational age in 138 low-income and middle-income countries in 2010. Lancet Glob. Health 1, e26–e36 (2013).
Maertens, K., Orije, M. R. P., Van Damme, P. & Leuridan, E. Vaccination during pregnancy: current and possible future recommendations. Eur. J. Pediatr. 179, 235–242 (2020).
Sperling, R. S. et al. Immunogenicity of trivalent inactivated influenza vaccination received during pregnancy or postpartum. Obstet. Gynecol. 119, 631–639 (2012).
Calvert, A. & Jones, C. E. Placental transfer of antibody and its relationship to vaccination in pregnancy. Curr. Opin. Infect. Dis. 30, 268–273 (2017).
Karita, E. et al. Safety, reactogenicity, and immunogenicity of a 2-dose Ebola vaccine regimen of Ad26.ZEBOV followed by MVA-BN-Filo in healthy adult pregnant women: study protocol for a phase 3 open-label randomized controlled trial. Trials 23, 513 (2022).
American College of Obstetricians and Gynecologists Committee on Obstetric Practice opinion no 700: methods for estimating the due date. Obstet. Gynecol. 129, e150–e154 (2017).
Mc Kenna, P. et al. Leapfrogging with technology: introduction of a monitoring platform to support a large-scale Ebola vaccination program in Rwanda. Hum. Vaccin. Immunother. 17, 3192–3202 (2021).
Patwardhan, M. et al. Maternal death: case definition and guidelines for data collection, analysis, and presentation of immunization safety data. Vaccine 34, 6077–6083 (2016).
Rouse, C. E. et al. Spontaneous abortion and ectopic pregnancy: case definition & guidelines for data collection, analysis, and presentation of maternal immunization safety data. Vaccine 35, 6563–6574 (2017).
Da Silva, F. T. et al. Stillbirth: case definition and guidelines for data collection, analysis, and presentation of maternal immunization safety data. Vaccine 34, 6057–6068 (2016).
Harrison, M. S. et al. Pathways to preterm birth: case definition and guidelines for data collection, analysis, and presentation of immunization safety data. Vaccine 34, 6093–6101 (2016).
Rouse, C. E. et al. Hypertensive disorders of pregnancy: case definitions & guidelines for data collection, analysis, and presentation of immunization safety data. Vaccine 34, 6069–6076 (2016).
Prabhu, M. et al. Antenatal bleeding: case definition and guidelines for data collection, analysis, and presentation of immunization safety data. Vaccine 35, 6529–6537 (2017).
Kerr, R. et al. Postpartum haemorrhage: case definition and guidelines for data collection, analysis, and presentation of immunization safety data. Vaccine 34, 6102–6109 (2016).
DeSilva, M. et al. Congenital anomalies: case definition and guidelines for data collection, analysis, and presentation of immunization safety data. Vaccine 34, 6015–6026 (2016).
Schlaudecker, E. P. et al. Small for gestational age: case definition & guidelines for data collection, analysis, and presentation of maternal immunisation safety data. Vaccine 35, 6518–6528 (2017).
Cutland, C. L. et al. Low birth weight: case definition & guidelines for data collection, analysis, and presentation of maternal immunization safety data. Vaccine 35, 6492–6500 (2017).
Quinn, J. A. et al. Preterm birth: case definition & guidelines for data collection, analysis, and presentation of immunisation safety data. Vaccine 34, 6047–6056 (2016).
Pathirana, J. et al. Neonatal death: case definition & guidelines for data collection, analysis, and presentation of immunization safety data. Vaccine 34, 6027–6037 (2016).
Ross, E. et al. Failure to thrive: case definition & guidelines for data collection, analysis, and presentation of maternal immunisation safety data. Vaccine 35, 6483–6491 (2017).
Acknowledgements
This study was funded by the Coalition for Epidemic Preparedness Innovations under the grant FEJA2002 provided to Janssen Vaccines & Prevention BV. The funder had no role in study design, data collection, data analysis, data interpretation, writing of the report, decision to publish or preparation of the manuscript. Johnson & Johnson provided the Ad26.ZEBOV, MVA-BN-Filo regimen doses for the study. Editorial assistance was provided by A. Kahhan, MS, ELS, of Lumanity Communications, and was funded by Johnson & Johnson.
Author information
Authors and Affiliations
Contributions
All authors meet the four criteria for authorship outlined in the International Committee of Medical Journal Editors Recommendations. All authors made substantial contributions to the conception or design of the work, drafting the proposal or revising it critically for important intellectual content, gave final approval of the version to be published, had access to all study data and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. E.K. was the principal investigator. J.N., R.I., A.M., T.S., C.U. and J.M. collected the data. R.P., A.G. and Y.W. directly accessed, verified and analyzed the underlying data reported in the manuscript. S.A., E.K. and M.K. designed the trial. K.M.W. wrote the first draft of the manuscript.
Corresponding author
Ethics declarations
Competing interests
C.R., C.M., A.G., B.K., C.A.F., K.L., Y.W., V.O.-M. and M.K. were full-time employees of Johnson & Johnson at the time of the study. C.M., B.K., C.A.F., V.O.-M. and A.G. hold stock and/or stock options in Johnson & Johnson. The other authors declare no competing interests.
Peer review
Peer review information
Nature Medicine thanks Beate Kampmann, Misaki Wayengera and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Lia Parkin, in collaboration with the Nature Medicine team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Percentage of women with adverse maternal/fetal outcomes of interest with at least 3% frequency in any group by trimester of randomization.
The exact Clopper–Pearson 95% CI is shown for the percentage of each specific outcome in each group. This figure presents select (≥3% for any trimester) of adverse maternal/fetal outcomes of interest by group and by trimester of randomization. The bars represent the percentage of participants. Percentages reflect n/N, where n is the number of participants with at least one of the given outcomes, N is the number of participants with available maternal/fetal outcomes and the error bars denote the 95% Clopper–Pearson exact confidence interval.
Extended Data Fig. 2 Percentage of infants with adverse neonatal/infant outcomes of interest with at least 3% frequency in any group by trimester of randomization.
The exact Clopper–Pearson 95% CI is shown for the percentage of each specific outcome in each group. This figure presents select (≥3% in any trimester) adverse neonatal/infant outcomes of interest by group and by trimester of randomization. The bars represent the percentage of participants. Percentages reflect n/N, where n is the number of participants with at least one of the given outcomes, N is the number of participants available and error bars denote the 95% Clopper–Pearson exact confidence interval.
Supplementary information
Supplementary Information (download PDF )
Supplementary Figs. 1–3, Supplementary Tables 1–6 and CONSORT checklist.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Nyombayire, J., Ingabire, R., Mazzei, A. et al. Heterologous two-dose Ebola vaccine regimen in pregnant women in Rwanda: a randomized controlled phase 3 trial. Nat Med 31, 3899–3906 (2025). https://doi.org/10.1038/s41591-025-03932-z
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41591-025-03932-z
This article is cited by
-
What drugs are safe during pregnancy? There’s a shocking lack of data
Nature (2026)
-
Vaccins contre Ebola, complexité du paludisme, traçabilité du maïs, cartographie des infrastructures et lutte contre la schistosomiase
Nature Africa (2025)
-
Ebola vaccine safe for mothers and infants
Nature Medicine (2025)
-
Ebola vaccines, malaria complexity, tracing maize, mapping infrastructure, and controlling schistosomiasis
Nature Africa (2025)
