
Abstract
The first haplotype-resolved chromosome-scale genome assembly for the bighead catfish (Clarias macrocephalus), a freshwater species native to Thailand and Vietnam, which has importance in aquaculture and conservation, is presented here. The 880 Mb genome was assembled using high-fidelity long-read sequencing from Pacific Biosciences and Oxford Nanopore Technologies, which was scaffolded using high-throughput chromosome conformation capture data, and polished with Illumina short-read sequencing. The assembly, which spans 27 pseudo-chromosomes, demonstrates high completeness and continuity, with a scaffold N50 of 33.48 Mb, 95.5% completeness based on Benchmarking Universal Single-Copy Orthologs (BUSCO), and a quality value (QV) of 50. This genomic resource provides a foundation for studying aquaculture traits, genetic diversity, and structural variation of the bighead catfish and supports efforts in their selective breeding, conservation, and sustainable aquaculture in Southeast Asia.
Similar content being viewed by others


Background & Summary
The bighead catfish (Clarias macrocephalus, Clariidae), a freshwater species belonging to the Siluriformes order, which is one of the most diverse fish orders with over 3,000 extant species, is native to Southeast Asia. Its distribution, which is primarily across the Mekong River Basin, including Cambodia, Thailand, and Vietnam, has been extended to several other countries such as China, Guam, Malaysia, and the Philippines1,2. Phylogenetic analyses, which have indicated that the bighead catfish are most closely related to white-spotted catfish (Clarias fuscus), have shown that they diverged approximately 25 million years ago3, while they are phylogenetically distant from North African catfish (C. gariepinus), a large economic clariid catfish globally, and eel catfish (Channallabes apus)4,5,6. The Bighead catfish, which is a benthopelagic and facultative air-breather that is capable of surviving in hypoxic waters and migrating across land, have adaptability such as aestivation in mud during the dry season. These features, which reflect ecological plasticity, make it a valuable model animal for studying respiratory physiology and environmental adaptation7. The bighead catfish is economically an important aquaculture species in Southeast Asia. However, its wild populations are declining owing to habitat degradation, overfishing, and genetic introgression from hybridization with alien invasive species, such as the North African catfish used for aquaculture breeding and are in a critical situation1,7.
Despite the progress in aquaculture practices and ecological research, genomic resources for the bighead catfish remain limited, and existing data are incomplete. This limitation hampers efforts toward biodiversity conservation and sustainable aquaculture. Genome-wide studies using reduced-representation approaches, such as DArTseq™, lack support from a contiguous reference genome and are inadequate for resolving structural variants and functional elements8,9. By contrast, chromosome-level genome assemblies of other catfish species have greatly advanced our understanding of their physiology, adaptation, and aquaculture traits. For example, in the genome of the channel catfish (Ictalurus punctatus), a key regulator of dermal bone and scale formation provides insights into skeletal evolution in teleosts and information for selection programs10. Similarly, the genome of the Chinese longsnout catfish (Leiocassis longirostris) has provided the information of molecular pathways related to carnivorous feeding and energy metabolism, demonstrating the role of genomic data in ecological adaptation and nutritional studies11. This process, which facilitates genomic selection in breeding programs, has remained unclear for the bighead catfish for which a chromosome-scale assembly is lacking and only a scaffold-level assembly has been made in the past12.
Here, to overcome the longstanding deficiency in genomic resources in bighead catfish and for better conservation and breeding efforts, the first chromosome-scale haplotype-resolved genome assembly was sequenced using a combination of third-generation platforms, including Pacific Biosciences’s High-Fidelity (HiFi) long reads, and Oxford Nanopore Technology ‘s long reads, complemented by Illumina paired-end short reads and High-throughput Chromosome Conformation Capture (Hi-C) paired-end short reads, this new assembly will serve as a foundational resource for genetic and evolutionary research.
Methods
Sample collection and DNA preparation
Broodstocks of the bighead catfish (C. macrocephalus) were obtained from the Faculty of Fisheries at Kasetsart University, Thailand. The bighead catfish was euthanized by severing the spinal cord anterior to the dorsal fin, and the liver tissue was collected and preserved in ethanol for DNA extraction. The sex of bighead catfish was determined by recording the morphology of genital papillae13. Genomic DNA was extracted using standard salting-out method, as described previously by Supikamolseni et al.14.
Quality-control and preparation experiment of sequenced reads
Raw sequencing reads were evaluated for quality using FastQC (http://www.bioinformatics.babraham.ac.uk/ projects/fastqc/) v0.1.12, which was used for short reads, and NanoPlot15 v1.42.0, which was used for long reads. The sequencing depth and Fastq QC (base/quality summary) were calculated by using SeqKit16 and Seqtk (https://github.com/lh3/seqtk). Illumina data, excluding Hi-C data, were trimmed to remove synthetic sequencing adapters by using AdapterRemoval17 v2.3.3. PacBio’s HiFi reads were trimmed by using HiFiAdapterFilt (‘-reward 1 -penalty −5 -gapopen 3 -gapextend 3 -dust no -soft_masking true -value 700 -searchsp 1750000000000)18 v64d1c7b. Cross-species contamination of raw read data was evaluated against the Mash database (‘refseq.genomes + plasmids.k21.s1000.msh’) by using Mash (‘sketch size = 1000, k = 21’)19 v2.3. Reads were filtered for length quality whenever necessary by using NanoFilt (‘-l $length -q $Quality’)15 v2.8.0.
PacBio HiFi long reads
PacBio HiFi reads (library source: genomic, library selection: circular consensus sequencing (CSS)/PCR)) were generated using circular consensus sequencing (CSS), which achieves a base accuracy of >99.9% and maximizes assembly quality by sequencing the same DNA fragment multiple times in a continuous polymerase chain reaction (PCR) without denaturation20. Sequencing on the Sequel II system produced 10.62 Gb of raw data, representing 12.9X genome coverage. The dataset achieved a mean quality score of Q28.7 and a median of Q36, with over 72% of reads that scored above Q30 (Fig. 1C).

Summary of sequencing data of the bighead catfish (Clarias macrocephalus) genome. (A) Summary of sequencing platforms and data types, including PacBio HiFi, Oxford Nanopore Technologies (ONT), and Illumina paired-end sequencing (Hi-C and WGS) and their genome coverage. (B) GenomeScope2.0 profile based on k-mer size of 21, which indicates a haploid genome size of approximately 899 Mb with 0.056% heterozygosity and 1.77% duplication. The x-axis shows k-mer coverage, and y-axis shows the observed frequency of k-mers. (C) Quality and length distribution of PacBio HiFi reads. The x-axis shows read length (bp), and the y-axis shows the mean PHRED score. The difference in GC content is represented by difference in color intensity. (D) Quality and length distribution of ONT reads. The x-axis shows read length (bp, log scale), and the y-axis shows the mean PHRED score. The difference in GC content is represented by difference in color intensity.
Oxford nanopore technologies (ONT) non-UL 1D-long noisy reads
ONT reads (library source: genomic, library selection: size fractionation) provided long and noisy data with a median error rate of approximately 20%. ONT reads have been used to improve assembly continuity and to span complex repeat regions21. These reads were prepared from size-selected, end-polished DNA fragments to which adapters were ligated to double-stranded DNA, and were purified by using AMPure XP beads. Sequencing on a PromethION system generated 29.56 Gb of raw data, equivalent to 36X genome coverage, with a 13 kb N50 read length. The median quality score was Q11.03, and 97.0% of reads were above Q7 (Fig. 1D).
Proximity-ligation Illumina paired-end short reads
Hi-C data (library source: genomic, library selection: DNase) were generated by using proximity ligation techniques to provide long-range linkage information, allowing contig phasing (haplotype separation) and genome scaffolding22,23. The data were generated by using an in situ Hi-C protocol that employed DpnII endonuclease digestion and were sequenced on an Illumina NextSeq. 2000, which yielded 37.19 Gb of paired-end reads and achieving 39.77X genome coverage. The data quality was high, yielding 123.96 M read pairs and 74.77 M unique Hi-C contacts, with > 97.18% that were at Q30.
Illumina paired-end short reads
Illumina paired-end short reads24 (library source: genomic, library selection: PCR) were sequenced on an Illumina NextSeq. 2000 platform, which yielded 31.91 Gb of raw read data (read length = 151 bp) with 40X genome coverage. The average quality score was Q25, and 92% of the reads were above Q30.
Reference-free genome profiling survey
All reference-free genome profiling analyses were performed by using the K-mer Analysis Toolkit (KAT)25 v2.4.1 and GenomeScope26 v2.0. For the genome survey (Table 1), canonical 21-mer k-mer distributions were estimated from trimmed Illumina and HiFi reads using Meryl27 and Jellyfish28 v2.3.1. A haploid sequence of approximately 890 million nucleotides (Mb) and an inter-haplotype heterozygosity rate of approximately 0.549% for bighead catfish were predicted by the genome survey (Fig. 1B).
De novo haplotype-resolved assembly, Hi-C scaffolding, and haplotype phasing
The haplotype-resolved diploid assembly of bighead catfish combined HiFi, ONT, and Hi-C data using Hifiasm (‘-hic-hifi -ul -primary’)29 v0.19.8-r603, resulting in two Hi-C-phased contig sets (.hic.hap1 and.hic.hap2), which represented two separate and complete haploid homologous haplotypes30. Each haplotype genome comprises a random assortment of chromosomes inherited from the maternal and paternal sets. To scaffold haplotype 1 and haplotype 2 with long reads (HiFi and ONT), proximity-ligation (Hi-C), and Illumina paired-end short reads, GreenHill (‘-cph $hap1 $hap2 -p $combined_ont_hifi -IP1 -IP2 -HIC’)31 v1.0.0. was used.
Hi-C maps, manual review, and post-review for obtaining chromosome-scale scaffolds
To build heat maps of Hi-C contacts between pairs of loci and obtain Hi-C scaffolds (post-haplotype resolved assembly), we used the Juicer pipeline (‘-assembly -S early’)32 v2.17.00 and Hi-CCUPS CPU (‘-ignore-sparse -cpu’) v2.17.00, which aligned Hi-C reads to GreenHill scaffolds and assigned a mapping quality score using BWA-MEM33 v0.7.17_r1188. PCR duplicates and near-duplicate mapped reads were removed by using Samtools34 v1.18. Hi-C contacts were generated at various resolutions (2.5 Mb to 5 kb). Visualization of Hi-C maps was performed by running the runassembly_visualizer.sh script from the 3D-DNA pipeline22 v02/12/2018. To manually review Hi-C scaffolds for misjoins or errors, Juicebox Assembly Tools (JBAT)32 v2.16.00 were used, and regions with misjoins or under-collapsed heterozygosity were edited based on off-diagonal signals in the Hi-C read density heatmap, which indicated contig/scaffolding errors. Post-review validation of the Hi-C scaffolds was performed using a 3D-DNA-post-review.sh v180114 (‘-sort-output-c 27’), a module of the 3D-DNA pipeline22. Unplaced scaffolds were filtered by using SeqKit (‘-by-length -reverse -m 2,500,000’)16 v2.7.0, which resulted in 27 chromosome-length scaffolds per haplotype (Fig. 2).

Workflows of haplotype-resolved genome assembly and genome scaffolding for the bighead catfish (Clarias macrocephalus). (A) The results of scaffolding using Juicebox v2.16.0. (B) The strategy of genome assembly using PacBio HiFi, ONT, and Illumina reads, followed by consensus assembly using Flye and haplotype-resolved assembly using Hifiasm (Hi-C UL mode). (C) Genome scaffolding using Hi-C reads, processed through GreenHill, 3D-DNA, and Juicer tools, producing haplotype-resolved and consensus assemblies. (D) Manual curation using JBAT and 3D-DNA post-review to correct scaffolding errors and finalize assemblies. (E) Final assembly improvements include multiple gap-filling rounds with TGS-GapCloser, QuarTeT GapFiller. (F) Polishing with NextPolish2, producing high-quality pseudo-chromosomes and unplaced scaffolds. Various software pipelines and types of sequencing data are used throughout the assembly process, as indicated by the icons. (G) Assembly polishing and QV assessment using Merqury, Pilon, and short-read alignment tools. (H) Mitochondrial genome assembly using Minimap2 and MitoHiFi pipelines.
Haplotype-aware genome polishing with NextPolish2
To increase the QV values and correct small SV errors (SNV/indels), we generated a HiFi mapping file after counting repetitive k-mers in the reads using Meryl (k = 15) for Winnowmap, a version of Minimap2 optimized for read alignment in repetitive regions that leverages a Bloom filter to filter alignments based on k-mer multiplicity35,36. Next, we generated two k-mer databases (21-mer and 31-mer files) from the trimmed short reads using the Yak k-mer analyzer (https://github.com/lh3/yak). The Winnowmap mapping file in BAM format, the target assembly in FASTA format, and the two Yak databases in Yak format were used as inputs for genome polishing using NextPolish237 v0.2.0.
Align-genus and read-homology-based methods for gap-filling and joining of contigs
Homology-based approaches have been used to resolve assembly gaps (i.e., gaps between contigs in pseudochromosomes/scaffolds)38. For this purpose, additional scaffold sets were generated by using Hifiasm (‘-primary’) as a contigger, with variations in the type of read data input (HiFi, ONT, or Hi-C) and haplotype purging options (‘-l0’). Additionally, orthologous sequences were retrieved from NCBI Datasets for closely related species in the family Clariidae (Taxonomy ID: 13012, n = 3 reference sequences): C. fuscus (GCA_030347435.1)39, C. gariepinus (GCA_024256425.2)40, and Channallabes apus (GCA_030522415.1)41, which were last accessed in February 2024. Three rounds of TGS-GapCloser42 v1.2.1, followed by one round of QuarTeT GapFiller, were applied to haplotypes 1 and 2. In the first round, filtered high-quality HiFi reads (Q20, length >10 kb) were used to fill medium-sized gaps (<20,000 bases). In the second round, filtered ONT reads (Q25, length >10 kb) were employed to fill larger gaps (>20,000 bases), whereas in the third round, polished unitigs (. p_utg.fa) from the hifiasm assembly were utilized. The number of gaps was reduced from N = 4015 (haplotype 2) to N = 2501 (haplotype 2), and further to N = 1100 after TGS-GapCloser. Finally, all reference genomes and scaffold sets (p_utg) were used as combined inputs, and the number of gaps was reduced to N = 550 (haplotype 2) by using QuarTeT GapFiller (default parameters: q *. fa’)43 v1.2.1.
Targeted haplotype-aware genome polishing with Pilon
To enhance the accuracy and quality values (QV) of the haplotype assemblies of C. macrocephalus and to precisely polish the genome assembly while correcting error-prone regions, k-mer analysis, read alignments, and sequence polishing were conducted using Pilon44 v1.24. Identification of missing reads at assembly seq-mers error positions: Positions of erroneous k-mers of non-repetitive k-mers found in the genome (i.e., the error seqmers) were identified with Merqury and Meryl, as explained in the T2T-Polish GitHub workflow (“QV estimate with hybrid k-mer db”; https://github.com/arangrhie/T2T-Polish). K-mers were counted using Meryl (‘meryl count k = $k’) for each haplotype assembly, and a combined (hybrid) 21-mers from the Illumina and HiFi data meryl k-mer database was created through union-summing (‘meryl union-sum’). We further filtered k-mers from the hybrid read database, retaining only k-mers with higher multiplicity values, such as greater than 5 or 10 (‘meryl greater-than’), to eliminate low-confidence sequences. Next, the k-mers unique to the reads and absent from the assemblies were isolated using Meryl’s difference operation (‘meryl difference’). These read-only k-mers represent genomic variations that were not captured in the assemblies. These filtered k-mers were used to extract reads containing these unique sequences from both the Illumina and HiFi datasets using the lookup command (‘meryl-lookup’). Mapping of reads: ONT reads were filtered by using NanoFilt to remove low-quality bases, whereas for Illumina paired-end reads (R1 and R2), low-quality sequences and adapter contamination were removed by using Fastp (‘-5 -3 -n 0 -f 5 -F 5 -t 5 -T 5 -q 20’)45 v0.23.4. Next, HiFi reads were aligned with the combined haplotype assemblies using Winnowmap based on k-mers found in repeats (‘-W repetitive_k15.txt’). For ONT reads, we used Minimap2 (‘map-ont’) without secondary alignments (‘-secondary = no’) and the alignments were filtered at MAPQ > 30 (‘-q 30’)46. Illumina short reads were aligned using Bowtie247 end-to-end (sensitive mode) (‘-sensitive’) to minimize spurious alignments, with no mixed read pairs (‘-no-mixed’) and no discordant mappings (‘-no-discordant’) to keep only alignments no farther than the expected insert size (500 bp. + 2 * read length for Illumina data), and PCR duplicates were removed using the SAMtools command (‘markdup’). Running Pilon and calling the consensus: Pilon was used to refine the assemblies by correcting SNPs, indels, and other base-level errors. For each haplotype scaffold, Pilon was run with specific parameters (-genome, -frags, -bam, -targets, -fix all,-vcf, -diploid, -minmq 30, -minqual 30) to incorporate alignments from all data types (HiFi, ONT, and short reads). The output Variant Calling Files (VCFs) containing the detected variants were sorted by position using BCFtools (‘bcftools sort’)48, compressed with bgzip, indexed with (‘bcftools index’), and a consensus sequence was generated for each scaffold using (‘bcftools consensus -f $genome.fa -H 1’). The overall median quality was increased from 41 to approximately 45–47 after the haplotype-aware targeted assembly polishing.
Additional Hi-C scaffolding and sequence integration using quartet
To reintegrate unplaced scaffolds into each haplotype in the pseudo-chromosomes, we used Quartet and HapHiC49 v1.0. First, we aligned the unplaced contigs to the reference genomes C. fuscus (GCA_030347435.1) and C. gariepinus (GCA_024256425.2) using Quartet AssemblyMapper with the following parameters (‘-r $reference -q $contigs -c 50000 -l 2000 -i 90 -a Minimap2’): We identified 53 MB and 33 MB of unplaced scaffold sequences in bighead catfish haplotype 1 and haplotype 2, respectively, with strong homology to C. fuscus pseudo-chromosomes; we filtered bighead catfish haplotype 1 and haplotype 2 with SeqKit to retain pseudo-chromosomes and concatenated them to unplaced scaffolds. For the preparation of Hi-C scaffolding, Hi-C reads were mapped to separate haplotypes using BWA-MEM (‘-5SP”) after making a BWT index for haplotype 1 and haplotype 2 (‘bwa index $genome.fasta’). Alignments were filtered to remove PCR duplicates and secondary alignments using Samblaster (samblaster $BAM | samtools view -@ $threads -S -h -b -F 3340)50 v0.1.26. Hi-C scaffolding was then performed with HapHiC. Hi-C contact maps were visualized for each haplotype in JBAT and with the haphic plot tool (Figs. 3–5). Manual post-review was carried out as described previously. Finally, three rounds of TGS-GapCloser followed by one round of targeted Pilon polishing specifying the new targets resulted in a genome of higher global quality—both in terms of QV metrics and structural accuracy (CRE/CSE) as measured by CRAQ. Duplication artefacts observed in heterozygous peaks of the Merqury k-mer spectra were addressed using haplotype-specific k-mer databases. Erroneous k-mers were identified with meryl difference, and corresponding reads were extracted using meryl lookup. Final consensus sequences were generated using BCFtools with the opposite haplotype reference (-H 1), mitigating most haplotype switch errors.

Genome assembly status of the bighead catfish examined in February 2024 and November 2024. (A) Haplotype 1 shown on the left and Haplotype 2 shown on the right ideograms generated in February, 2024 after manual review with Juicebox. Gaps and telomeres lengths on pseudo-chromosomes are shown by orange rectangles and blue triangles, respectively. (B) The genome assemblies generated in November, 2024.

Hi-C contact maps of scaffolded pseudo-chromosomes in the bighead catfish (Clarias macrocephalus). (A) Contact map of haplotype 1 assembly, visualized in blue. (B) Contact map of haplotype 2 assembly is visualized in purple. The x-axis and y-axis represent genomic coordinates in megabases (Mb), and each axis is segmented according to individual pseudo-chromosome scaffolds, allowing visualization of chromosomal contact patterns. Color intensity indicates the normalized Hi-C contact values (Knight–Ruiz (KR) matrix balancing); h darker colors represent higher interaction (frequencies). Bin size = 50 Kb.

The heat maps of Hi-C contact matrix displaying individual pseudo-chromosome scaffolds in haplotype 1, which were sorted by length. The x-axis and y-axis represent genomic coordinates along each pseudo-chromosome. Darker colors along the diagonal indicate higher contact frequencies, reflecting local chromatin interactions and supporting accurate scaffold continuity and orientation.
For base accuracy and phasing error correction, polishing tools that used various methods were employed. For pileup-based methods, Pilon was used, which corrected pileup based and single-nucleotide polymorphism (SNP) switch errors that had been identified previously with Meryl based on Merqury positioning of assembly errors (hereby referred to as “error seqmers”)51, Inspector52 was used to assess assembly quality and, in some cases, to repair structural errors, pileup errors, and base-level errors through its correction module that was based on HiFi reads. Racon53 with Merfin54 HiFi or Clair355 with Merfin HiFi effectively handled SNPs and other small nucleotide variants (SNVs), especially under low coverage. Polishing based on Nanopore data was always followed by HiFi-based polishing to maintain high base accuracy. A TGS-GapCloser was used to bridge gaps, and CRAQ was applied to break assemblies at read hard-clipping points. JBAT/HiC scaffolding was subsequently applied, followed by a manual review that ensured structural accuracy. Each step was logically dependent on prior outputs. For instance, JBAT scaffolding necessitated subsequent gap closing (e.g., TGS-GapCloser), while iterative refinement employed tools such as Racon or bcftools consensus command from BCFtools. This step was crucial for validating assembly completeness and reducing errors. Non-haplotype-aware tools (e.g., Racon, Clair3) were applied cautiously to minimize errors from parental haplotype switches, while Merfin filtered edits for polishing and BCFtools consensus produced the final polished sequence. For large variants and closing gaps using read alignments we used Sniffles56 for large structural variant (SV) calling, specifically for insertions (INS) and deletions (DEL). The final genome assembly was required to have a high mapping quality, with no MAPQ0 regions and an overall QV > 50. Variant calls were validated visually using IGV57, and the overall quality and structural consistency were assessed using Meryl, Asset/detgaps, CRAQ, Inspector, and VerityMap58.
Additional targeted consensus automated polishing (SVS and SNPs)
The consensus polishing was automated using the T2T-polish GitHub repository https://github.com/arangrhie/T2T-Polish. A HiFi mapping file of repetitive k-mers (k = 15) was generated with Meryl, which was then used in Winnowmap for HiFi read alignment (‘-MD -W. repetitive_k15.txt -ax map-pb’). Alignment filtering was performed using Samtools (‘-Sb’). The tool pb-falconc (‘bam-filter-clipped -t -F 0 × 104 -output-count-fn’) (https://github.com/PacificBiosciences/pbbioconda) v1.15.0 was used to remove clipped reads. Genome polishing was carried out using the liftover branch of the Racon GitHub repository and Racon v1.5.0 with options (‘-L -S’). After polishing, the k-mers present in the genome (seqmers) were counted by using a meryl count (k = 21). Merfin was then employed (‘-readmers Illumina.HiFi.gt1.PCRfree.hybrid.meryl -seqmers’) to evaluate the results by comparing the distributions in the reads and in the polished genome51,54. For consensus generation (i.e., applying polishing edits to the genome assembly), we used BCF tools (‘-H 1’) for two rounds of genome correction. Assembly quality metrics were measured for QV, completeness, and BUSCOs scores, and we found a large increase in QV for most chromosomes (min. increase > + 1-5 QV points) after ONT Racon and Merfin. The median QV was 50, and the progress over time is presented in Fig. 3.
Organelles: Bighead catfish mitochondrial genome
The mitochondrial genome was assembled by aligning reads to the reference (NC_046749.1) bighead catfish mtDNA sequence that is available at NCBI Nucleotide59. Nanopore reads were mapped to the reference using Minimap2 (‘-ax map-ont -secondary = no’), and Illumina reads were mapped using Minimap2 (‘-ax sr’). PCR duplicates in the short reads were removed from the alignments using Samtools, and the results were visualized using IGV. Pilon (‘-fix all -diploid -changes -vcf -tracks -minmq 10’) was used to correct the reference (NC_046749.1), to call SNPs, SVs, gaps, and local variants, and to obtain the consensus mtDNA sequence. Reads were realigned to the consensus and no additional SNPs were visible in the IGV. Subsequently, mapped reads were filtered with Samtools view (‘-F4 -q 20’) and were re-assembled de novo using Unicycler60 for comparison. The results were visualized in Bandage-ng61 v2022.09. The assembly was polished with Pilon, and the two homologous mitochondria were compared using Minimap2 (‘-eqx -x asm5’). Gene annotations were generated using MitoFinder62 v1.4.2. To ensure that the mitochondrial genome was correct and not dissimilar from other mtDNAs in Siluriformes, we downloaded all reference mtDNA sequences for 209 species of Siluriformes catfish from NCBI Nucleotide (last accessed September 2024). All sequences were obtained from NCBI RefSeq and not from NCBI GenBank. All mtDNA nucleotide sequences (N = 210) were renamed using the Pan-SN naming scheme (https://github.com/pangenome/PanSN-spec), concatenated in a single multi-FASTA, including the bighead catfish mtDNA sequence that was generated after Pilon polishing. All-versus-all alignments were performed with wfmash, and ODGI63 was used to filter the alignment graph. Visualizations were made with Bandage and multiQC64. All tools used in the pipeline were integrated within the larger PGGB (pan-genome graph builder) framework65.
Comparative synteny analysis
A comparative synteny analysis was conducted using orthology and alignment tools to investigate the conservation of chromosome structures and identify syntenic regions between the bighead catfish and other teleost fishes. The proteomes of seven representative teleost species, rainbow trout (Oncorhynchus mykiss), medaka (Oryzias latipes), common carp (Cyprinus carpio), Nile tilapia (Oreochromis niloticus), zebrafish (Danio rerio), barramundi (Lates calcarifer), and spotted gar (Lepisosteus oculatus), were retrieved from Ensembl using the latest genome versions to ensure annotation consistency. Orthologous gene clusters were identified using OrthoFinder v2.5.566, followed by reciprocal best-hit filtering with rbhXpress v1.2.3. Syntenic blocks, which enable cross-species comparisons of genome-wide gene orders, were visualized using macrosyntR v0.2.1967.
Transposable element annotation
De novo annotation of transposable elements (TEs), which was performed using the EDTA pipeline (v2.2.0), it integrates multiple tools to detect and classify diverse TE families, including long- terminal tandem repeats (LTRs), terminal inverted repeats (TIRs), helitrons, and other types of repeats68.
Benchmarking of assembly quality metrics
Metrics of continuity, structural accuracy, base accuracy, and functional completeness were used for benchmarking as described in the Vertebrate Genome Project (VGP) paper5. The assembly quality metrics are listed in Table 2, scaffold-wise assembly metrics are listed in Table 3, and the methodology for their calculation is presented below.
-
1.
Continuity and summary statistics: To evaluate the continuity and summary statistics of the assembly, the following measures for scaffolds/contigs (N50, N90, NG50, LG50, and LG90) were computed by using RagTag (ragtag.py asmstats -g)69 v2.1.0.
-
2.
Repeat completeness and continuity of repeats: To measure repeat completeness and continuity to assess assembly quality, the percentage of fully assembled LTR retroelements (LTR-RTs) was estimated, and the long terminal repeat (LTR) assembly index (LAI) was calculated using LTR_retriever70 v2.9.00. Telomeres, which are nucleoprotein complexes located at the ends of eukaryotic chromosomes71, have been identified in all vertebrates studied so far. The DNA component of telomeres contains a tandemly repeated G-rich hexanucleotide sequence (TTAGGG/CCCTAA)n72,73. To assess the assembly quality of complex repeats (centromeres), TandemTools and TandemQUAST58 v1.0 were used (results not shown). Telomere prediction for the presence/absence and orientation of telomeres was performed using TIDK, a Telomere Identification Toolkit, implemented in TeloExplorer (‘-m 50 -c animal’), a module of QuarTeT. To estimate gap length and its location in the genome, detgaps, a script from Asset available at GitHub (https://github.com/dfguan/asset), was used.
-
3.
Structural accuracy (regional and structural errors and reliable blocks): To assess structural accuracy, CRAQ (‘sms_coverage = 6, ngs_coverage = 20, -Minimap2-sensitive’)74 v1.0.9, was used. CRAQ is a method that relies on examining mapped reads, clipped reads, and coverage support from two or more simultaneous sequencing platforms against a reference, to identify supporting regions (i.e., the reliable blocks in the VGP paper). CRAQ metrics consist of global AQI (R-AQI and S-AQI), Small Clip-based Regional Errors (CREs), and Large Clip-based Structural Errors (CSEs), which indicate incorrect assembly breakpoints. The mapping results consisted of loading multiple read-to-genome alignment files in the BAM file format, along with BED and BigWig genome annotation files, which were visualized using the Integrative Genome Viewer (IGV)55 using the command line (‘igv -g $genome.fa $BAM’). To assess cross-species structural correctness, the same references from related catfish species (C. gariepienus and C. fuscus) were used for one-to-one nucleotide-level alignments of orthologous segments using MashMap2 (‘-s 2000000 -pi 90 -c 100000’)75 v3.1.3.
-
4.
Base accuracy and assembly completeness: To assess base accuracy and completeness, specifically Merqury’s QV and 21-mer genome completeness (%), Merqury50 v1.3 was used. It was run three times using different k-mer databases: Illumina, HiFi, and a hybrid 21-mer database combining Illumina and HiFi reads, using the method described in the “Consensus polishing” section above. To assess functional completeness and evaluate gene set completeness, the ray-finned fish lineage and BUSCO (‘-l actinopterygii_odb10’)76 v5.6.1 were used. Finally, mapping rates and inconsistencies were determined by mapping using Minimap2 (‘-ax map-hifi -secondary = no’) and WinnowMap (‘-W repetitive.15. txt’) at MAPQ > 10 for HiFi reads, while for ONT reads, the map-ont default for Minimap2 and the map-pb default in Winnowmap MAPQ > 10 read-to-assembly alignments were used. Visualizations were performed using the IGV (igv-g $genome $BAM(s) $merqury_only_bed_wig_kmer_files).
-
5.
Consensus genome assembly with Flye: To validate the haplotype-resolved assembly, we assembled an additional consensus genome with the assembler Flye77 using default parameters and 10 rounds of minimap2 overlap), which collapses haplotypes and generates approximations of genes by merging divergent alleles into single chimeric alleles.
Data Records
The sequencing datasets and genome assemblies of Clarias macrocephalus have been deposited in multiple public repositories. All raw sequencing data are hosted under NCBI BioProject number PRJNA1132508, with the BioSample accession SAMN41769988 for the diploid bighead catfish isolate CMAM (TaxID: 35657). The sequencing data deposited in the NCBI Sequence Read Archive include: Oxford Nanopore Technologies long-read data (SRR29723575)78; PacBio HiFi sequencing data (SRR29723576)79; Hi-C chromatin conformation data (SRR29723577)80; Illumina paired-end sequencing data from male sample (SRR29723578)81; and Illumina paired-end sequencing data from female sample (additional) (SRR30463128)82. The complete genome assemblies and associated datasets are available through Zenodo (https://doi.org/10.5281/zenodo.14826875)83. The assembled haplotype-resolved genome sequences were deposited in GenBank as whole-genome shotgun sequencing projects, with haplotype 1 available under accession JBLWMO00000000084 and haplotype 2 under accession JBLWMP00000000085.
Technical Validation
Technical validation of bighead catfish genome
The haplotype-resolved genome assembly of the bighead catfish was validated using multiple complementary approaches. The assembly produced 27 Hi-C scaffolds per haplotype with a total size of ~880 Mb per haplotype, consistent with the haploid chromosome number of the diploid karyotype (2n = 54), thereby confirming chromosome-scale structural accuracy. Structural integrity was further supported by visual inspection of Hi-C contact maps (Figs. 4, 5, 6).

The heat maps of Hi-C contact matrix displaying individual pseudo-chromosome scaffolds in haplotype 2. The x-axis and y-axis represent genomic coordinates along each pseudo-chromosome. Darker colors along the diagonal indicate higher contact frequencies, supporting the accuracy of scaffold assembly and orientation.
Pairwise mapping between phased haplotypes, performed with Minimap2 in asm5 mode, revealed 1,968,666 heterozygous single-nucleotide polymorphisms, corresponding to a heterozygosity rate of 0.594%. Structural variant analysis with PlotSR suite86 identified more than 390,000 insertions and deletions spanning ~7.7 Mb in each haplotype. Additional copy number variants were detected, including 114 copy gains (184 kb) and 123 copy losses (416 kb), distributed across highly divergent genomic regions spanning ~57.9 Mb in haplotype 1 and ~56.0 Mb in haplotype 2.
Assembly quality was high, with a scaffold N50 of 35.4 Mb, 95.5% completeness based on Benchmarking Universal Single-Copy Orthologs (BUSCO), and an overall consensus quality value (QV) of 50. RagTag asmstats reported a contig NG50 of 3 Mb and a scaffold N50 of 34 Mb, with LG50 and LG90 values of 11 and 24, respectively (Table 2).
A K-mer–based quality assessment using Merqury showed a substantial increase in consensus accuracy, yielding a final median QV > 46, which exceeds the Vertebrate Genome Project (VGP) standard of QV > 40. Merqury grey peaks indicated the presence of read-only k-mers, supporting the conclusion that some biologically or technically complex regions were not fully recovered. A minor peak at 3× multiplicity was observed, suggesting low-level false duplications; these could be resolved in future versions using tools such as PurgeDups87 or Purge Haplotigs88.
Genome completeness was additionally supported by BUSCO analysis with the actinopterygii_odb10 dataset. For comparison, a collapsed assembly generated with Flye achieved high BUSCO scores (C:97.7%, S:11.2%, D:86.5%, F:0.6%, M:1.7%, n = 3640), with 3,557 complete BUSCOs (408 single-copy and 3,149 duplicated). This collapsed assembly spanned 1.84 Gb across 4,961 contigs and scaffolds with an N50 of 1 Mb. It contained ~2,480 gaps (estimated as half the contig count) but reported 0% gap content by length. Base-level consensus quality was QV 40–50, corresponding to 99.99–99.999% accuracy. Although the assembly from Flye is inherently collapsed, it remains useful for estimating total DNA content and sample-specific gene content:a small fraction of missing orthologs in the Flye assembly likely reflects unresolved complex regions or conservative parameters (Fig 7).

Quality assessment of the genome assembly of bighead catfish. (A) K-mer spectra generated using the Merqury (software). This spectra-asm plot shows k-mer contents for read-only (black), haplotype 1-specific (red), haplotype 2-specific (blue), and shared (green) regions. The x-axis shows k-mer multiplicity, and the y-axis shows the count of k-mers. (B) BUSCO completeness assessment for three assemblies: the two haplotype-resolved assemblies, and a collapsed assembly created using Flye assembler (positive control),. The x-axis shows the percentage of BUSCOs for four categories: complete single-copy (blue), complete duplicated (light blue), fragmented (yellow), and missing (red) genes. (C) K-mer spectra-cn plot displaying copy number multiplicity of k-mers in the diploid genome. The x-axis shows k-mer multiplicity, and the y-axis shows the count of k-mers. Most k-mers fall into the expected diploid peak (copy number = 2, blue). A minor peak at copy number 3 suggests a small degree of homozygous duplication, which may be further reduced by purging redundant haplotypes or trimming contig ends.
Approximately 35.25% of the genome was predicted to consist of transposable elements or other repetitive sequences. Among the identified classes, terminal inverted repeat (TIR) DNA transposons were the most abundant (19.12%), followed by LTR retrotransposons (8.30%) and Helitrons (4.47%). LINE elements were present at low abundance (0.46%), and unclassified repeats contributed 2.82% (Table 4).
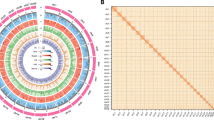
The synteny plots showed strong chromosomal collinearity between bighead catfish, common carp, and Nile tilapia, which share a common ancestor among otophysan teleosts. By contrast, more fragmented or rearranged syntenic signals were observed in zebrafish and spotted gar, suggesting a greater evolutionary divergence (Fig. 8).

Synteny analysis of chromosome-level assemblies for various catfish samples. (A) Whole-genome four-way synteny analysis with 1413 single-copy orthologs shared across Clarias gariepinus (GCA_024256425.2), Clarias macrocephalus (database number), Danio rerio (GRCz11), and Oreochromis niloticus (O_niloticus_UMD_NMBU), reveals conserved chromosome structures and chromosomal rearrangements. (B) Simple phylogeny and geological timescales of C. macrocephalus and related teleosts examined using Time Tree 3 version.
The current assembly contains at most 358 unresolved gaps, substantially fewer than previous efforts12, thus providing a more complete view of the bighead cafish genome. The comparative analysis with related Clarias genomes revealed conserved mtDNA with homology to other catfish species (Fig. 9).

All-against-all pairwise comparisons of mitochondrial DNA sequence alignments in Siluriformes catfishes. The assembled mtDNA sequence of the bighead catfish is represented on the last row (blue color).
Together, these validations demonstrate the high accuracy, biological completeness, and diploid-resolved nature of the bighead catfish genome assembly, supporting its reliability as a reference for future genomic, evolutionary, and applied aquaculture studies.
Usage Notes
The primary genome assembly is well-suited for comparative genomic studies, particularly those involving synteny analysis, due to its high gene set completeness. For studies focused on genetic variation, including single-nucleotide variants or structural variation, either haplotype 1 or haplotype 2 can be employed, depending on the research objective.
Data availability
All sequencing data generated in this study have been deposited in the NCBI Sequence Read Archive under BioProject PRJNA1132508, with the BioSample accession SAMN41769988 (TaxID: 35657) for the diploid bighead catfish (isolate: CMAM). Raw sequencing reads from multiple platforms, including Oxford Nanopore, PacBio HiFi, Hi-C, and Illumina (male and female), are available in the SRA under accession numbers SRR29723575, SRR29723576, SRR29723577, SRR29723578, and SRR30463128. The assembled genome sequence, including haplotypes 1 and 2, has been deposited in GenBank under accession numbers JBLWMO000000000 (haplotype 1) and JBLWMP000000000 (haplotype 2). All assembly data and related resources are also publicly available in the Zenodo repository (https://doi.org/10.5281/zenodo.14826876).
Code availability
No custom software code was developed for this study. All bioinformatics tools and pipelines were utilized according to the manuals and protocols provided by the respective software developers. The software versions used, along with their corresponding parameters, are described.
References
Duong, T.-Y. & Scribner, K. T. Regional variation in genetic diversity between wild and cultured populations of bighead catfish (Clarias macrocephalus) in the Mekong Delta. Fish. Res. 207, 118–125, https://doi.org/10.1016/j.fishres.2018.06.012 (2018).
Lisachov, A. et al. Emerging importance of bighead catfish (Clarias macrocephalus) and North African catfish (C. gariepinus) as a bioresource and their genomic perspective. Aquaculture 573, 739585, https://doi.org/10.1016/j.aquaculture.2023.739585 (2023).
Tian, C. X. et al. A chromosome-level genome assembly of Hong Kong catfish (Clarias fuscus) uncovers a sex-determining region. BMC Genomics 24, 291, https://doi.org/10.1186/s12864-023-09394-2 (2023).
Koepfli, K.-P., Paten, B. & Brien, S. J. The Genome 10K project: A way forward. Annu. Rev. Anim. Biosci. 3, 57–111, https://doi.org/10.1146/annurev-animal-090414-014900 (2015).
Rhie, A. et al. Towards complete and error-free genome assemblies of all vertebrate species. Nature 592, 737–746, https://doi.org/10.1038/s41586-021-03451-0 (2021).
Nguinkal, J. A., Zoclanclounon, Y. A. B., Brunner, R. M. & Goldammer, T. Haplotype-resolved assembly of the African catfish (Clarias gariepinus) provides insights for semi-terrestrial adaptation of airbreathing catfishes. Sci. Data 11, 1095, https://doi.org/10.1038/s41597-024-03906-9 (2023).
Na-Nakorn, U. & Brummett, R. E. Use and exchange of aquatic genetic resources for food and aquaculture: Clarias catfish. Reviews in Aquaculture 1, 214–223, https://doi.org/10.1111/j.1753-5131.2009.01010.x (2009).
Nguyen, D. H. M. et al. Genome-wide SNP analysis suggests male heterogamety in bighead catfish (Clarias macrocephalus, Günther, 1864). Aquaculture 543, 737005, https://doi.org/10.1016/j.aquaculture.2021.737005 (2021).
Chaivichoo, P., Koonawootrittriron, S., Chatchaiphan, S., Srimai, W. & Na-Nakorn, U. Genetic components of growth traits of the hybrid between ♂North African catfish (Clarias gariepinus Burchell, 1822) and ♀bighead catfish (Clarias macrocephalus Günther, 1864). Aquaculture 521, 735082, https://doi.org/10.1016/j.aquaculture.2020.735082 (2020).
Liu, Z. et al. The channel catfish genome sequence provides insights into the evolution of scale formation in teleosts. Nat. Commun. 7, 11757, https://doi.org/10.1038/ncomms11757 (2016).
Liu, Y. et al. The Chinese longsnout catfish genome provides novel insights into the feeding preference and corresponding metabolic strategy of carnivores. Genome Res. 34, 981–996, https://doi.org/10.1101/gr.278476.123 (2024).
Duong, T.-Y., Tan, M. H., Lee, Y. P., Croft, L. & Austin, C. M. Dataset for genome sequencing and de novo assembly of the Vietnamese bighead catfish (Clarias macrocephalus, Günther, 1864). Data Brief 31, 105861, https://doi.org/10.1016/j.dib.2020.105861 (2020).
Viveen, W. J. A. R., Richter, C. J. J., van Oordt, P. G. W. J., Janssen, J. A. L. & Huisman, E. A. Practical Manual for the Culture of the African Catfish (Clarias gariepinus). The Hague: Ministry of Foreign Affairs Cooperation (1986).
Supikamolseni, A. et al. Molecular barcoding of venomous snakes and species-specific multiplex PCR assay to identify snake groups for which antivenom is available in Thailand. Genet. Mol. Res. 14, 13981–13997, https://doi.org/10.4238/2015.October.29.18 (2015).
De Coster, W. & Rademakers, R. NanoPack2: population-scale evaluation of long-read sequencing data. Bioinformatics 39, btad311, https://doi.org/10.1093/bioinformatics/btad311 (2023).
Shen, W., Le, S., Li, Y. & Hu, F. SeqKit: a cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLOS ONE 11, e0163962, https://doi.org/10.1371/journal.pone.0163962 (2016).
Schubert, M., Lindgreen, S. & Orlando, L. AdapterRemoval v2: rapid adapter trimming, identification, and read merging. BMC Res. Notes 9, 88, https://doi.org/10.1186/s13104-016-1900-2 (2016).
Sim, S. B., Corpuz, R. L., Simmonds, T. J. & Geib, S. M. HiFiAdapterFilt, a memory-efficient read processing pipeline, prevents occurrence of adapter sequence in PacBio HiFi reads and their negative impacts on genome assembly. BMC Genomics 23, 691, https://doi.org/10.1186/s12864-022-08375-1 (2022).
Ondov, B. D. et al. Mash Screen: high-throughput sequence containment estimation for genome discovery. Genome Biol. 20, 232, https://doi.org/10.1186/s13059-019-1841-x (2019).
Wenger, A. M. et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat Biotechnol 37, 1155–1162, https://doi.org/10.1038/s41587-019-0217-9 (2019).
Jain, M. et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 36, 338–345, https://doi.org/10.1038/nbt.4060 (2018).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95, https://doi.org/10.1126/science.aal3327 (2017).
Putnam, N. H. et al. Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. Genome Res. 26, 342–350, https://doi.org/10.1101/gr.193474.115 (2016).
Bentley, D. et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456, 53–59, https://doi.org/10.1038/nature07517 (2008).
Mapleson, D., Garcia Accinelli, G., Kettleborough, G., Wright, J. & Clavijo, B. J. KAT: a k-mer analysis toolkit to quality control NGS datasets and genome assemblies. Bioinformatics 33, 574–576, https://doi.org/10.1093/bioinformatics/btw663 (2016).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 11, 1432, https://doi.org/10.1038/s41467-020-14998-3 (2020).
Rhie, A. et al. Meryl: a genomic k-mer counter and analysis toolkit. Bioinformatics 36, 2639–2640, https://doi.org/10.1093/bioinformatics/btaa025 (2020).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770, https://doi.org/10.1093/bioinformatics/btr011 (2011).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175, https://doi.org/10.1038/s41592-020-01056-5 (2021).
Lewin, H. A., Graves, J. A. M., Ryder, O. A., Graphodatsky, A. S. & O’Brien, S. J. Precision nomenclature for the new genomics. GigaScience 8, giz086, https://doi.org/10.1093/gigascience/giz086 (2019).
Ouchi, S., Kajitani, R. & Itoh, T. GreenHill: a de novo chromosome-level scaffolding and phasing tool using Hi-C. Genome Biol. 24, 140, https://doi.org/10.1186/s13059-023-03006-8 (2023).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98, https://doi.org/10.1016/j.cels.2016.07.002 (2016).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760, https://doi.org/10.1093/bioinformatics/btp324 (2009).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26, 589–595, https://doi.org/10.1093/bioinformatics/btp698 (2010).
Jain, C., Rhie, A., Hansen, N. F., Koren, S. & Phillippy, A. M. Long-read mapping to repetitive reference sequences using Winnowmap2. Nat. Methods 19, 705–710, https://doi.org/10.1038/s41592-022-01457-8 (2022).
Jain, C. et al. Weighted minimizer sampling improves long read mapping. Bioinformatics 36, i111–i118, https://doi.org/10.1093/bioinformatics/btaa435 (2020).
Hu, J. et al. NextPolish2: a repeat-aware polishing tool for genomes assembled using HiFi long reads. Genomics Proteomics Bioinformatics 22, qzad009, https://doi.org/10.1093/gpbjnl/qzad009 (2024).
Lischer, H. E. L. & Shimizu, K. K. Reference-guided de novo assembly approach improves genome reconstruction for related species. BMC Bioinformatics 18, 474, https://doi.org/10.1186/s12859-017-1911-6 (2017).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_030347435 (2023).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_024256425 (2022).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_030522415 (2023).
Xu, M. et al. TGS-GapCloser: a fast and accurate gap closer for large genomes with low coverage of error-prone long reads. GigaScience 9, giaa094, https://doi.org/10.1093/gigascience/giaa094 (2020).
Lin, Y. et al. QuarTeT: a telomere-to-telomere toolkit for gap-free genome assembly and centromeric repeat identification. Hortic. Res. 10, uhad127, https://doi.org/10.1093/hr/uhad127 (2023).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9, e112963, https://doi.org/10.1371/journal.pone.0112963 (2014).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890, https://doi.org/10.1093/bioinformatics/bty560 (2018).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100, https://doi.org/10.1093/bioinformatics/bty191 (2018).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359, https://doi.org/10.1038/nmeth.1923 (2012).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. GigaScience 10, giab008, https://doi.org/10.1093/gigascience/giab008 (2021).
Zeng, X. et al. Chromosome-level scaffolding of haplotype-resolved assemblies using Hi-C data without reference genomes. Nat. Plants 10, 1184–1200, https://doi.org/10.1038/s41477-024-01755-3 (2024).
Faust, G. G. & Hall, I. M. SAMBLASTER: fast duplicate marking and structural variant read extraction. Bioinformatics 30, 2503–2505, https://doi.org/10.1093/bioinformatics/btu314 (2014).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 245, https://doi.org/10.1186/s13059-020-02134-9 (2020).
Chen, Y., Zhang, Y., Wang, A. Y., Gao, M. & Chong, Z. Accurate long-read de novo assembly evaluation with Inspector. Genome Biol. 22, 312, https://doi.org/10.1186/s13059-021-02527-4 (2021).
Vaser, R., Sovic, I., Nagarajan, N. & Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27, 737–746, https://doi.org/10.1101/gr.214270.116 (2017).
Formenti, G. et al. Merfin: improved variant filtering, assembly evaluation and polishing via k-mer validation. Nat. Methods 19, 696–704, https://doi.org/10.1038/s41592-022-01445-y (2022).
Zheng, Z. et al. Symphonizing pileup and full-alignment for deep learning-based long-read variant calling. Nat Comput Sci 2, 797–803, https://doi.org/10.1038/s43588-022-00387-x (2022).
Smolka, M. et al. Detection of mosaic and population-level structural variants with Sniffles2. Nat Biotechnol 42, 1571–1580, https://doi.org/10.1038/s41587-023-02024-y (2024).
Thorvaldsdottir, H., Robinson, J. T. & Mesirov, J. P. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 14, 178–192, https://doi.org/10.1093/bib/bbs017 (2012).
Mikheenko, A., Bzikadze, A. V., Gurevich, A., Miga, K. H. & Pevzner, P. A. TandemTools: mapping long reads and assessing/improving assembly quality in extra-long tandem repeats. Bioinformatics 36, i75–i83, https://doi.org/10.1093/bioinformatics/btaa440 (2020).
NCBI RefSeq https://www.ncbi.nlm.nih.gov/nuccore/NC_046749.1 (2023).
Wick, R. R., Judd, L. M., Gorrie, C. L. & Holt, K. E. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13, e1005595, https://doi.org/10.1371/journal.pcbi.1005595 (2017).
Wick, R. R., Schultz, M. B., Zobel, J. & Holt, K. E. Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31, 3350–3352, https://doi.org/10.1093/bioinformatics/btv383 (2015).
Allio, R. et al. MitoFinder: efficient automated large-scale extraction of mitogenomic data in target enrichment phylogenomics. Mol. Ecol. Resour. 20, 892–905, https://doi.org/10.1111/1755-0998.13160 (2020).
Guarracino, A., Heumos, S., Nahnsen, S., Prins, P. & Garrison, E. ODGI: understanding pangenome graphs. Bioinformatics 38, 3319–3326, https://doi.org/10.1093/bioinformatics/btac308 (2022).
Ewels, P., Magnusson, M., Lundin, S. & Käller, M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048, https://doi.org/10.1093/bioinformatics/btw354 (2016).
Garrison, E. et al. Building pangenome graphs. Nat. Methods 21, 2008–2012, https://doi.org/10.1038/s41592-024-02430-3 (2024).
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238, https://doi.org/10.1186/s13059-019-1832-y (2019).
El Hilali, S. & Copley, R. macrosyntR: Drawing automatically ordered Oxford Grids from standard genomic files in R. bioRxiv https://doi.org/10.1101/2023.01.26.525673 (2023).
Ou, S. et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 20, 275, https://doi.org/10.1186/s13059-019-1905-y (2019).
Alonge, M. et al. Automated assembly scaffolding using RagTag elevates a new tomato system for high-throughput genome editing. Genome Biol. 23, 258, https://doi.org/10.1186/s13059-022-02823-7 (2022).
Ou, S. & Jiang, N. LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 176, 1410–1422, https://doi.org/10.1104/pp.17.01310 (2018).
Ocalewicz, K. Telomeres in fishes. Cytogenet. Genome Res. 141, 114–125, https://doi.org/10.1159/000354278 (2013).
Moyzis, R. K. et al. A highly conserved repetitive DNA sequence, (TTAGGG)n, present at the telomeres of human chromosomes. Proc. Natl Acad. Sci. USA 85, 6622–6626, https://doi.org/10.1073/pnas.85.18.6622 (1988).
Meyne, J., Ratliff, R. L. & Moyzis, R. K. Conservation of the human telomere sequence (TTAGGG)n among vertebrates. Proc. Natl Acad. Sci. USA 86, 7049–7053, https://doi.org/10.1073/pnas.86.18.7049 (1989).
Li, K., Xu, P., Wang, J., Yi, X. & Jiao, Y. Identification of errors in draft genome assemblies at single-nucleotide resolution for quality assessment and improvement. Nat. Commun. 14, 4256, https://doi.org/10.1038/s41467-023-42336-w (2023).
Jain, C., Koren, S., Dilthey, A., Phillippy, A. M. & Aluru, S. A fast adaptive algorithm for computing whole-genome homology maps. Bioinformatics 34, i748–i756, https://doi.org/10.1093/bioinformatics/bty597 (2018).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212, https://doi.org/10.1093/bioinformatics/btv351 (2015).
Kolmogorov, M. et al. Assembly of long, error-prone reads using repeat graphs. Nat Biotechnol 37, 540–546, https://doi.org/10.1038/s41587-019-0072-8 (2019).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29723575 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29723576 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29723577 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29723578 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR30463128 (2025).
Andres, Q. L. S. et al. Chromosome-scale, haplotype-resolved genome assembly of Bighead catfish (Clarias macrocephalus). Zenodo https://doi.org/10.5281/zenodo.14826875 (2024).
Andres, Q. L. S. et al. Clarias macrocephalus isolate CMAM, whole genome shotgun sequencing project haplotype 1. GenBank https://identifiers.org/ncbi/insdc:JBLWMO000000000 (2024).
Andres, Q. L. S. et al. Clarias macrocephalus isolate CMAM, whole genome shotgun sequencing project haplotype 2. GenBank https://identifiers.org/ncbi/insdc:JBLWMP000000000 (2024).
Goel, M. & Schneeberger, K. plotsr: visualizing structural similarities and rearrangements between multiple genomes. Bioinformatics 38, 2922–2926, https://doi.org/10.1093/bioinformatics/btac196 (2022).
Guan, D. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 36, 2896–2898, https://doi.org/10.1093/bioinformatics/btaa025 (2020).
Roach, M. J., Schmidt, S. A. & Borneman, A. R. Purge Haplotigs: allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinformatics 19, 460, https://doi.org/10.1186/s12859-018-2485-7 (2018).
Acknowledgements
This research was financially supported in part by a grant from the National Research Council of Thailand (NRCT) (N42A650233) awarded to PS and KS; grants from the High-Potential Research Team Grant Program, NRCT (N42A660605) awarded to WS, TP, NM, PD, JP, PS, YM, KS; the Program Management Unit for Human Resources and Institutional Development and Innovation (PMU-B) (Contract No. B13F670053); the Program Management Unit for Competitiveness (PMU-C) under the Global Partnership Program (Contract No. C23F670224); and the Program Management Unit on Area Based Development (PMU-A) (Contract No. A11F680039), all of which were awarded to WS, TP, NM, PD, JP, and KS. Additional support was provided by grants from the Kasetsart University Research and Development Institute (FF(KU)25.64, FF(S-KU)17.66, and FF(KU)51.67), awarded to WS and KS.; the Kasetsart University Postdoctoral Fellowship (Contract No. 6501.2107/160) awarded to TP; Graduate Scholarship for International Academic Journal Publication (2022), Graduate School, Kasetsart University (Student ID: 6517200011), awarded to QLSA and PS; and the International SciKU Branding (ISB), Faculty of Science, Kasetsart University, awarded to WS and KS. No funding source was involved in the study design, data collection, analysis, or manuscript preparation. We thank the NSTDA Supercomputer Center (ThaiSC) for providing High-Performance Computing resources via the LANTA system. We also acknowledge Zeng Xiaofei for his assistance with HapHiC configuration, Shun Ouchi for support with GreenHill, Olga Dudchenko for guidance on the 3D-DNA pipeline and Juicebox JBAT, and the 3D-DNA community for technical input. We are grateful to the QuarTeT developers for continued tool updates and community support.
Author information
Authors and Affiliations
Contributions
W.S., N.M. and J.P. collected and processed the samples. W.S. and T.P. performed DNA extraction and quality control. Q.L.S.A. conducted genome assembly and annotation. Bioinformatic analyses and data interpretation were carried out by Q.L.S.A., W.S. and T.P. Genome validation and data curation were performed by W.S., P.D., J.P., P.S., Y.M. and K.S. K.S. provided technical input and genomic resources. K.S. supervised the study and coordinated the project. Q.L.S.A., W.S. and K.S. wrote the manuscript. All authors reviewed and approved the final version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Andres, Q.L.S., Singchat, W., Panthum, T. et al. Chromosome-scale, haplotype-resolved genome of Bighead catfish for conservation and aquaculture. Sci Data 13, 8 (2026). https://doi.org/10.1038/s41597-025-06325-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-06325-6
