
Abstract
DNA characterisation in people with tuberculosis (TB) is critical for diagnostic and microbiome evaluations. However, extracellular DNA, more frequent in people on chemotherapy, confounds results. We evaluated whether nucleic acid dyes [propidium monoazide (PMA), PEMAX] and DNaseI could reduce this. PCR [16S Mycobacterium tuberculosis complex (Mtb) qPCR, Xpert MTB/RIF] was done on dilution series of untreated and treated (PMA, PEMAX, DNaseI) Mtb. Separately, 16S rRNA gene qPCR and sequencing were done on untreated and treated sputa before (Cohort A: 11 TB-negatives, 9 TB-positives; Cohort B: 19 TB-positives, PEMAX only) and 24-weeks after chemotherapy (Cohort B). PMA and PEMAX reduced PCR-detected Mtb DNA for dilution series and Cohort A sputum versus untreated controls, suggesting non-intact Mtb is present before treatment-start. PEMAX enabled sequencing-based Mycobacterium-detection in 7/12 (58%) TB-positive sputa where no such reads otherwise occurred. In Cohort A, PMA- and PEMAX-treated versus untreated sputa had decreased α- and increased β-diversities. In Cohort B, β-diversity differences between timepoints were only detected with PEMAX. DNaseI had negligible effects. PMA and PEMAX (but not DNaseI) reduced extracellular DNA in PCR and improved pathogen detection by sequencing. PEMAX additionally detected chemotherapy-associated taxonomic changes that would otherwise be missed. Dyes enhance microbiome evaluations especially during chemotherapy.
Similar content being viewed by others

Introduction
The microbiome plays an essential role in host biological processes1 and is increasingly implicated as an important mediator of human health. Perturbations to the microbiome, particularly after antibiotic exposure, has been found to influence outcomes in diverse disease states2, including tuberculosis (TB)3. First-line TB antibiotics rapidly deplete microbial diversity4,5, and these effects can persist long after chemotherapy is completed6.
Tuberculosis is a leading infectious cause of mortality globally with ~ 1.2 million deaths per year7. Although sensitive tests like Xpert MTB/RIF (Xpert) are available for Mycobacterium tuberculosis complex (Mtb) detection, PCR can be confounded by DNA from non-intact cells, including from previous TB8,9,10. This problem can be significant: in some settings up to 35% of notifications have recurrent TB; requiring special consideration in diagnostic algorithms9. The ability to distinguish Mtb DNA from old versus new episodes is therefore of clinical and epidemiological significance11,12.
Unlike most other infectious diseases with short chemotherapy durations, TB requires at least six months of antibiotics with broad-spectrum rifamycins and potentially fluoroquinolones. These antibiotics have bactericidal effects13,14, resulting in extracellular DNA accumulation that, in the case of Mtb DNA, can persist for years8,10 especially in people with compromised immunological clearance such as people living with HIV15. Characterisations of the microbiome in people receiving (or who have received) TB chemotherapy thus likely measure total bacterial DNA, rather than only the subset within intact (and hence live) cells.
Viability dyes such as ethidium monoazide (EMA), propidium monoazide (PMA), and PEMAX (an EMA and PMA formulation) have been widely used for discriminating intact from non-intact microbial cells16,17,18,19,20 and act by passing through compromised membranes of non-intact cells (e.g., following heat exposure21,22 or repeated freeze-thaw23), after which they covalently cross-link with DNA to irreversibly form complexes not amplifiable by PCR24. As a result, only DNA from intact cells is, in theory, amplified once unbound dye is removed. PMA is more effective than EMA in excluding DNA from non-intact cells due to higher charge25. PEMAX was developed to overcome limitations of PMA and EMA such as the inability to bind DNA from cells that are still intact but no longer metabolically active and thus may improve specificity26.
These dyes have some data when applied to Mtb27,28,29,30,31. For example, the addition of PMA to Xpert shows potential for treatment monitoring, however, specificity remains an issue27,28. More data are needed as dye penetration through Mtb’s waxy cell wall may be suboptimal (especially in specimens with high bacillary load)32 and sputum from diseased individuals contains high levels of host and microbial extracellular DNA that sequesters dye away, leaving insufficient dye to subtract background DNA. Combining viability dyes with 16S rRNA gene sequencing could improve the accuracy of microbial taxonomy by reducing background from non-intact taxa that, by virtue of their lack of viability, are unlikely to play a direct biological role.
We investigated the effects of nucleic acid dyes (PMA, PEMAX) and DNaseI on (1) Mtb PCR detection (16S, Xpert) in a dilution series (with versus without antibiotics, heat kill, freeze-thaw) and sputum from people with presumptive TB as well as (2) changes in the microbiome (compared by TB status and duration of chemotherapy exposure) by comparing sputum treated or untreated with dyes and DNaseI. Our overarching goal was to assess whether usage of dyes and/or DNaseI could enhance the detection of intact Mtb (in vitro and in sputum) and enhance analysis of the sputum microbiome.
Methods
Ethical approval and consent to participate
The study was conducted in accordance with the Declaration of Helsinki. This study was approved by the Stellenbosch University Health Research Ethics Committee (N14/10/136, N16/05/070, M15/10/041). Informed consent was obtained from all participants involved in the study.
In vitro dilution series using Mycobacterium tuberculosis cells
Culture
Mtb H37Rv was cultured (37 °C) to an OD600nm of 1.0 in 20mL Middlebrook 7H9 liquid medium with 10% oleic acid, albumin, dextrose, and catalase (OADC; BD Diagnostics, Johannesburg, South Africa), 0.5% glycerol and 0.05% Tween-80 (Sigma-Aldrich, Modderfontein, South Africa). One millilitre aliquots were frozen (− 80 °C).
Antibiotic exposure (experiment A)
Two hundred microlitres of thawed stock was inoculated into each of six flasks with 20mL 7H9 and incubated until mid-log (OD600nm 0.6–0.8). Isoniazid (0.2 g/mL in nuclease-free water; Thermo Fisher Scientific, Waltham, USA) and rifampicin (1 g/mL in dimethyl sulfoxide; Sigma-Aldrich, Modderfontein, South Africa) were added to three of the 20mL culture flasks (three additional flasks served as non-antibiotic controls) (Fig. 1A). All six flasks were incubated for 24-, 48-, and 72 h at 37 °C. To generate a 1:1 mixture of intact and non-intact cells, 5mL of the non-antibiotic and antibiotic-containing cultures after different incubation periods (24-, 48-, 72 h) were combined in a separate sterile flask and, from each, a 100-fold serial dilution was done (108, 106, 104 CFU/ml) in 7H9 to a total volume of 10 ml. This was done for three biological replicates.

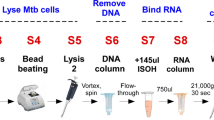
Experimental workflow for in vitro dilution series and sputum. Mycobacterium tuberculosis was cultured in flasks and a dilution series prepared for testing in three separate experiments: antibiotic exposure (Experiment A), heat-kill (Experiment B) and freeze-thaw (Experiment C). Sputum was collected from two distinct cohorts: one comprising patients with presumptive tuberculosis (Cohort A) and the other involving patients treated for drug-resistant tuberculosis (Cohort B). MTBC Mycobacterium tuberculosis complex.
Heat kill (experiment B)
For heat-kill experiments, a single culture was grown to mid-log, 100-fold dilutions (108, 106 CFU/ml) done and 500µL of each dilution done as stated above, heated (100 °C, 30 min) and cooled to room temperature (RT). This was done for three biological replicates.
Freeze-thaw (experiment C)
A freeze-thaw (FT) experiment was done with 106 dilutions after 24-, 48- and 72 h incubation periods that were frozen (− 20 °C) and allowed to thaw at RT. One biological replicate was done due to consumable unavailability.
Treatment of samples to remove background DNA
PMA and PEMAX treatment (experiments A-C)
For Experiments A and B, 500µL of each dilution was aliquoted into (1) an empty DNA LoBind tube (Eppendorf, Hamburg, Germany) without further treatment (hereafter referred to as untreated), (2) a LoBind tube containing 50µM PMA (Biotium, Fremont, USA) and (3) a PEMAX monodose tube (GenIUL, Barcelona, Spain). PMA- and PEMAX-containing tubes were vortexed for 5s before incubation in the dark (30 min, 37 °C). To crosslink dyes with DNA, photoactivation using the Phast Blue system (GenIUL; 60 W, 2 min for PMA, 15 min for PEMAX) was done. PEMAX-treated samples were transferred to new LoBind tubes (PEMAX monodose tubes are hydrophilic and may cause DNA to not be neutralised by the dye16). The same was done for Experiment C except only PEMAX was used. At the end of dye treatment, samples were frozen (− 20 °C) for batched DNA isolation.
DNaseI treatment (experiments A and B)
This was done for the 48 h timepoint only. 500µL of each dilution was aliquoted into an empty LoBind tube (hereafter referred to as untreated) or a LoBind tube containing 1µL of RQ1 RNase-Free DNase 10X Reaction Buffer (400mM Tris-HCl, 100mM MgSO4, 10mM CaCl2; Promega, Madison, USA) and 1µL RQ1 RNase-Free DNaseI (1U/µg DNA; Promega) and incubated (37 °C, 30 min) (hereafter referred to as treated). The treated tube was further incubated (65 °C, 10 min) with 1µL of RQ1 DNase Stop Solution and cooled to RT. Samples were frozen (-20 °C) for batched DNA isolation.
Sputum processing
Cohort A
Induced sputa were collected from 20 people with presumptive TB on the morning they presented at primary healthcare clinics in Cape Town, South Africa. Sputa were decontaminated with a modified decontamination protocol28 omitting sodium-hydroxide (to preserve bacterial viability) and an equal volume of N-acetyl-L-cysteine (NALC; Sigma-Aldrich) solution (0.5% w/v NALC, 2.7% w/v trisodium citrate) for 15 min at RT, followed by neutralisation with a double phosphate buffer volume (BD Diagnostics) and centrifugated (3000xg, 15 min). Pellets were resuspended in 5mL phosphate buffer and bio-banked (-20 °C) for PMA, PEMAX or DNaseI treatment as described earlier (Fig. 1B). People were classified as TB-negative (n = 11) if they were MTBC culture-negative (or, if their culture was contaminated, sputum Xpert negative; n = 1) or TB-positive (n = 9) if they were MTBC culture-positive (or, if culture-contaminated, sputum Xpert-positive; n = 2).
Cohort B
Sputa was collected from 23 people with multidrug resistant (MDR)-TB who were enrolled in a clinical trial33 at Brooklyn Chest Hospital, Cape Town, South Africa. Sputum collection was done at two timepoints, week 0 (before or < 1 week after chemotherapy start) and after 24 weeks chemotherapy (week 24) and were decontaminated and pellets resuspended in 2mL phosphate buffer, from which one 500µL aliquot was immediately PEMAX-treated as described above and the other left as is. Both aliquots were stored at -20 °C for batched DNA isolation (Fig. 1C).
Microbial DNA isolation
DNA was isolated from the Experiments A-C dilution series using the QIAgen DNA Mini Kit (QIAGEN, Hilden, Germany) with modifications. Briefly, aliquots were heat-killed (95 °C, 15 min), resuspended in lysozyme-containing (10 mg/ml) lysis buffer, incubated (1 h, 37 °C), and proteinase K digestion done (56 °C, 30 min) before isolation. DNA was isolated from Cohort A and B sputum using the Purelink Microbiome DNA Purification Kit (Invitrogen, Carlsbad, USA) per manufacturer’s instructions34.
16S rRNA gene characterisation
Quantitative real-time PCR (qPCR) (experiments A-C)
Reactions comprised of 5µL iTaq Universal SYBR Green (Thermo Fisher Scientific, Massachusetts, United States), 0.3µL of each forward and reverse primer (10µM), 1.4µL nuclease-free water and 3µL template DNA were done using the CFX Connect Real-Time PCR Detection System (Bio-Rad, Fremont, USA). Mtb-specific primers were used for the dilution series and universal 16S rRNA primers35 used for sputum (Cohorts A and B) (Supplementary Table 1). Cycling conditions were 95 °C for 5 min followed by 35 cycles of 95 °C for 5s and 60 °C for 30s. All reactions were in three technical replicates and cycle thresholds (CT; number of cycles before product detected) recorded. In each experiment, non-template control CTs were averaged to derive a positivity threshold.
Detection of viable bacilli by Xpert MTB/RIF (experiments A and B)
500µL of each dilution (104, 106, 108 CFU/ml) were Xpert-tested (Fig. 1). Aliquots were either first treated with PMA, PEMAX or DNaseI as described above or were untreated. The manufacturer-recommended input volume of the (un)treated dilution (500 µl) was used for PEMAX Xpert testing36. Therefore, Xpert Sample Reagent (1.5mL) was added to each 500 µl aliquot and incubated (RT, 15 min) with intermittent shaking before 2mL of the mixture tested (version G4; Cepheid, USA). Xpert only reports CTmin for positive results.
Sequencing and analysis
Amplicon library preparation was carried out on an automated platform (Biomek 4000) using a custom liquid handling method as previously described37. Sequences (V4 region, 150 bp paired-end) generated by Illumina MiSeq as described38,39 were demultiplexed (QIIME 2; v2020.2)40 and denoised (DADA2)41. Taxonomic classification was done with the Naïve Bayes classifier trained on Greengenes 13.8 reference database42 (pre-clustered at 99% identity with 515 F/806R primers selected43). BIOM tables were rarefied by random subsampling to equal depth. Potential contaminants (in blank dyes, DNaseI and reagents) were identified using decontam (v1.22.0)44 with a threshold of 0.5 based on their prevalence in sputum vs. background samples. Contaminant amplicon sequencing variants (ASVs) were not removed but highlighted if later identified as discriminatory. α- and β-diversities were calculated using vegan (v2.6-4)45.
Statistical analysis
The Kruskal-Wallis test with Dunn’s multiple comparisons was used to compare untreated and dye-treated CT values (Experiments A-C). The Wilcoxon matched-pairs signed rank test was used for sputum qPCR (Cohort A) and Xpert results. One-sided tests were used because we hypothesised that dyes would improve Mtb detection. P-values are for comparisons between untreated and treated (PMA, PEMAX, or DNaseI) and were not calculated for CTs exceeding that of the non-template control. The Friedman test with Dunn’s multiple comparisons was used to compare α-diversity between untreated and treated sputa (Cohorts A and B). Bray-Curtis distances were compared using Kruskal-Wallis with Dunn’s multiple comparisons. DESeq2 (v1.42.0)46 (with Benjamini–Hochberg multiple comparisons) was used for differential abundance testing. Feature tables were pruned to > 1% relative abundance in > 1% of samples. Analyses were done using GraphPad Prism (v8.0.1) and R (v4.3.2). p ≤ 0.05 and Q-values < 0.2 were significant. Supplementary Table 2 details the figures to which each statistical test applies.
Results
PMA and PEMAX treatment reduce Mtb DNA detection in dilution series (experiments A and B)
16S rRNA qPCR readouts
Dye-treated aliquots had lower detected mycobacterial load (increased CT) than untreated controls (Fig. 2A,B). The same finding occurred using combinations of antibiotic-exposed or heat-killed cells, as well as different incubation periods (48 h, 72 h; Supplementary Fig. 1).

PMA and PEMAX reduce MTBC detection in dilution series (regardless of how aliquots were treated) and in sputum sediments from presumptive TB patients. PCR readouts for non-antibiotic (cultured in absence of antibiotics), antibiotic (cultured with isoniazid and rifampicin), 1:1 mixture (non-antibiotic: antibiotic), and heat-killed (non-antibiotic aliquots heated at 100 °C for 30 min) cultures after 24 h of incubation. Supplementary Fig. 1 has these data for the 48 h and 72 h exposure period. Median CTs (IQR) are shown for (A) 108 and (B) 106 CFU/ml dilutions. Open circles denote CTs that exceed those of the averaged non-template control (dashed line). Median Xpert CTmin (95% CI) are shown for (C) 108 CFU/ml and (D) 106 CFU/ml. Triangles on y-axes indicate decreasing bacterial load. Purple dots: untreated; red dots: PMA; green dots: PEMAX. MTBCMycobacterium tuberculosis complex, CT cycle threshold, CTmin minimum cycle threshold, CI confidence interval, IQR interquartile range.
FT resulted in lower detected mycobacterial load [24 h non-antibiotic: ΔCT 2.13 (95% CI: 1.25–2.99)] compared to pre-FT samples, but when the same FT vs. pre-FT comparison was done for PEMAX-treated aliquots, a larger decrease in detected load occurred [24 h non-antibiotic: ΔCT 6.44 (5.53–7.36)] (Supplementary Fig. 2).
Xpert
Dye reduced load readouts vs. untreated controls regardless of if aliquots underwent antibiotic or heat or no exposure (Fig. 2C,D).
DNaseI treatment does not reduce Mtb DNA detection in dilution series (experiment A and B)
DNaseI had no effect on CT compared to untreated controls. Only the antibiotic-exposed cells showed, after DNaseI treatment, unexpectedly higher detected mycobacterial load [for example, the 108 CFU/ml dilution had median (IQR) CTs of 19.24 (19.02–19.42) vs. 19.32 (19.24–20.96) for untreated; p = 0.014] (Supplementary Fig. 3). There was no significant difference in bacterial load measured in DNaseI-exposed aliquots tested with Xpert (Supplementary Fig. 4).
Sputa from presumptive tuberculosis patients are abundant in extracellular bacterial DNA (cohort A)
PMA, PEMAX, and DNaseI’s effect on PCR quantitation and sputum α- and β-diversity
qPCR
Decontaminated sputum treated with PMA or PEMAX had lower measured load than untreated controls (Fig. 3A) whereas no differences in load resulted from DNaseI-treatment.

In sputa from people with presumptive TB, PMA or PEMAX reduces microbial diversity estimates more than DNaseI in comparison to untreated sputum. (A) PCR readouts for untreated versus treated (PMA, PEMAX, or DNaseI) decontaminated sputum from TB-negatives and TB-positives (Cohort A). Triangles on y-axes indicate decreasing load. (B) α-Diversity decreased after PMA and PEMAX treatment but not DNaseI. (C) β-diversity differences between untreated and treated groups. (D) Bray-Curtis distance to untreated sputum was higher in PMA- and PEMAX-treated sputum versus DNaseI-treated sputum. Volcano plots depicting (E) many differentially abundant taxa in PMA-treated versus untreated sputum and (F) Mycobacterium-enrichment and Neisseria-depletion in PEMAX-treated versus untreated sputa. More discriminatory taxa appear closer to the left or right, and higher above the threshold (red dotted line, false discovery rate of 0.2). Relative abundances correspond to circle size. (G) Use of PEMAX increased the proportion of Mycobacterium reads in TB-positives. ASVs amplicon sequencing variants.
Sequencing
PMA- and PEMAX-treated sputum had lower α-diversity, whereas DNaseI-treated sputum had similar α-diversity to paired untreated controls (Fig. 3B). β-diversity differed between untreated and PMA-treated (p = 0.006), untreated and PEMAX-treated (p = 0.041), and PMA-treated and DNaseI-treated sputum (p = 0.019) (Fig. 3C). Paired β-diversity distances between PMA- or PEMAX-treated and untreated sputum were greater than those between DNaseI-treated and untreated sputum (Fig. 3D). Results remained similar when TB-positive and TB-negative groups were separately analysed (Supplementary Fig. 5A,B).
PMA treatment reveals more taxa (versus untreated controls) than PEMAX, however, only PEMAX permits Mycobacterium detection
PMA permitted 19 more differentially abundant taxa to be identified vs. its untreated controls (seven enriched, 12 depleted; Scardovia most enriched, Corynebacterium most depleted) (Fig. 3E), than PEMAX [one enriched (Mycobacterium), one depleted (Neisseria), one identified as a potential contaminant (Bosea)] (Fig. 3F; Supplementary Figs. 6, 7). None of the seven taxa enriched due to PMA were Mycobacterium. In PEMAX-treated sputum from people with TB, 6/9 (67%) had an increase in detected Mycobacterium relative abundance compared to untreated sputa (p = 0.031) (Fig. 3G) [5/9 (56%) of these would have not had mycobacterial reads detected by 16S rRNA gene sequencing if PEMAX was not used]. No discriminatory taxa were associated with DNaseI treatment. After stratification by TB status, similar results with fewer differential taxa occurred (Supplementary Fig. 8).
PMA and PEMAX improve characterisation of microbiome differences by TB status compared to untreated controls
In untreated sputum, no α- and β-diversity differences occurred by TB status with Gallibacterium being the most discriminatory (enriched in TB-negatives). Upon PMA or PEMAX treatment, similar results occurred with Scardovia additionally enriched in TB-negatives (Fig. 4A–E, Supplementary Fig. 9). Mycobacterium was additionally enriched in TB-positives after PEMAX. A comparison of Mycobacterium relative abundances stratified by TB status and treatment method showed Mycobacterium enriched in PMA- and PEMAX-treated sputa from TB-positives (p = 0.026 and p = 0.002, respectively) (Fig. 4F). No discriminatory taxa were found between patient groups with DNaseI treatment.

PMA and PEMAX enhances Mycobacterium detection in TB-positives versus TB-negatives. In untreated sputum, groups did not differ in (A) α-diversity nor (B) β-diversity. The same was observed for PMA-, PEMAX-, and DNaseI-treated sputum (see Supplement). TB-negatives were Gallibacterium-enriched in (C) untreated, (D) PMA-treated and (E) PEMAX-treated sputum versus TB-positives. More discriminatory taxa appear closer to the left or right, and higher above the threshold (red dotted line, false discovery rate of 0.2). Relative abundances correspond to circle size. (F) Mycobacterium relative abundance was higher in TB-positives than TB-negatives in sputum treated with either PMA or PEMAX, however, abundances were the same in untreated or DNase-treated sputum. In TB-positives, PEMAX permitted greater mycobacterial detection than PMA [0.0015 (0.0001–0.1125) vs. 0.0001(0.0001–0.0009)]. ASVs amplicon sequencing variants.
PEMAX treatment reveals diversity changes from TB chemotherapy otherwise undetected (cohort B)
Comparisons within timepoints (untreated versus treated)
α-Diversity differed only at week 24, not week 0, between treated and untreated sputa, with lower α-diversity observed after PEMAX (Fig. 5A; p = 0.033). There were no β-diversity differences at week 0 nor week 24 (Fig. 5B). No taxonomic differences were found at week 0 in treated versus untreated sputum, however, at week 24, treated sputa were Neisseria-enriched and Mogibacterium-depleted (Fig. 5C).

Microbial changes after TB chemotherapy are only detectable with PEMAX. (A) α-Diversity was only different between week 24 untreated and PEMAX-treated sputum. (B) Unlike in untreated sputum, PEMAX enabled detection of β-diversity changes between weeks 0 and 24. (C) Neisseria-enrichment and Mogibacterium-depletion at week 24 in treated vs. untreated sputum. (D) Only three discriminatory taxa were identified in the absence of PEMAX between weeks 0 and 24, whereas (E) many were identified after PEMAX-exposure. More discriminatory taxa are closer to the left or right, and higher above the threshold (red dotted line, false discovery rate of 0.2). Relative abundances of taxa are indicated by circle size. ASVs amplicon sequencing variants.
Comparisons within treatment groups (week 0 versus week 24)
α-Diversity was unchanged per patient between timepoints (Fig. 5A). β-diversity only changed over time in treated sputa (Fig. 5B; p = 0.007). In untreated sputa, Bifidobacterium and other taxa were enriched at week 0 vs. week 24 (Fig. 5D). However, in treated sputum, these taxa were absent and other differences occurred (Fig. 5E).
Discussion
We demonstrated (1) PMA- and PEMAX-treated samples and sputum have lower detected mycobacterial load compared to untreated counterparts, (2) PMA- and PEMAX treatment of sputa reduces α-diversity and results in β-diversity differences compared to paired untreated sputum, (3) Mycobacterium (detected by 16S rRNA gene sequencing) is enriched in PEMAX-treated sputum from TB-positives compared to TB-negatives, a difference that was not seen in untreated sputum, (4) chemotherapy-associated microbiome changes are clearer (i.e., communities are compositionally dissimilar) in sputum treated with PEMAX versus those not treated, and (5) DNaseI treatment produces inconsistent signals in in vitro experiments and does not influence microbiome estimates. Together, these data have implications for characterising pathogen load and the microbiome, including evaluating the effect of chemotherapy, especially in people with TB.
PMA and PEMAX improve differentiation of live from dead foodborne pathogens such as Salmonella, Campylobacter, and Listeria47. PMA also preferentially excludes dead rather than live Mtb in smear-positive sputa30,32,48, however, no studies have evaluated PEMAX in TB. Lower detected mycobacterial load was found with PMA and PEMAX treatment in PCR (qPCR, Xpert) implying some bacilli even from laboratory-generated dilution series are not intact. The effects of PMA, PEMAX, and DNaseI were not as strong as expected in the presence of increased cellular membrane disruption caused by antibiotics and heat killing (for example, PCR results becoming negative). This corroborates earlier Xpert data showing imperfect specificity and minimal improvement with PMA pre-treatment27.
Compared to paired untreated samples, PMA- and PEMAX-treated samples had lower microbial richness (i.e. suppressed DNA signals from non-intact bacteria). The inclusion of PMA or PEMAX is thus important for removing taxa likely not biologically active. These methods further identified Mycobacterium in people with TB that would otherwise go undetected. Previously, we demonstrated how a nested mycobacteriome approach could help to resolve this49; dye treatment represents an alternative. This is relevant for microbiome studies involving people with TB, as we and others have shown that Mycobacterium reads are often undetectable by 16S rRNA gene sequencing in sputum50,51.
Prolonged exposure to TB chemotherapy, particularly those with broader antimicrobial spectra that are common in treatment regimens for drug-resistant TB, and host immunological responses are likely to produce non-viable bacterial populations15. PEMAX improved detection of microbiome changes in sputum before and after chemotherapy by detecting more discriminatory taxa. It is worth considering if previously documented treatment-related sputum microbiome changes5,52 overlooked any taxonomic changes that could have been detected using a viability dye.
DNaseI performed poorly in dilution series and sputum microbiome evaluations. Prior research had suggested that the presence of antibiotics increased DNaseI sensitivity53, but this was not the case, which may be related to differences in specimen type or bacterial species. While all reagents used for antibiotic and DNaseI preparation was freshly prepared, it cannot be ruled out that the DNaseI was inactivated at some point or that antibiotic preparation contained rRNA contamination, although this is unlikely. We acknowledge that different types of DNaseI exist (including from different suppliers) and that DNaseI may have shown efficacy in other studies; however, the fragile nature of the enzyme prevents it from being used for applications such as those described in our study.
The results must be viewed in the context of its limitations. Xpert assays was done using a dilution series of cells instead of sputum, in which cells are embedded within a matrix, which may affect results. Furthermore, sputum decontamination processes (commonly done as part of TB microbiology) can cause cell death while leaving cell membranes intact, in which case PMA and EMA have lower reduced ability to remove background DNA54,55. This, however, is one reason we used PEMAX, which was designed to overcome this constraint19. Moreover, we used a modified sputum decontamination procedure50 in which sodium hydroxide was omitted to minimise effects on bacterial viability. Some study participants (Cohort B, who had MDR-TB) may have already been on chemotherapy at week 0, however, it is difficult to obtain a true baseline specimen from such people who are often prescribed first-line chemotherapeutics before drug resistance is discovered.
As sputum is an imperfect surrogate for the lower airways, many studies are shifting to bronchoalveolar lavage (BAL) sampling. Pre-treating low biomass specimens, such as BAL fluid or cough aerosols, where background signals from medical equipment or PCR reagents (or “reagent microbiome”) can interfere with biological interpretation56, is likely to be beneficial, but further validation is required.
Our findings highlight the critical role of PMA or PEMAX pre-treatment in respiratory specimens, particularly during chemotherapy, as it provides invaluable insights into bacterial load characterisation and microbiome dynamics, especially when considering chemotherapeutics.
Data availability
The datasets generated and analysed during the current study are available in the Sequence Read Archive under accession number PRJNA1102510. The original R script, metadata, ASV table, and taxonomy file are available in GitHub (https://github.com/segalmicrobiomelab/TB_pemax.git).
References
Bouskra, D. et al. Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature456 (7221), 507–510 (2008).
VanEvery, H. et al. Microbiome epidemiology and association studies in human health. Nat. Rev. Genet.24 (2), 109–124 (2023).
Wen, J. & He, J. Q. The causal impact of the gut microbiota on respiratory tuberculosis susceptibility. Infect. Dis. Ther.12 (11), 2535–2544 (2023).
Wipperman, M. F. et al. Gastrointestinal microbiota composition predicts peripheral inflammatory state during treatment of human tuberculosis. Nat. Commun.12 (1), 1141 (2021).
Musisi, E. et al. Effect of seven anti-tuberculosis treatment regimens on sputum microbiome: a retrospective analysis of the HIGHRIF study 2 and PanACEA MAMS-TB clinical trials. Lancet Microbe4 (11), e913–e922 (2023).
Wipperman, M. F. et al. Antibiotic treatment for tuberculosis induces a profound dysbiosis of the microbiome that persists long after therapy is completed. Sci. Rep.7 (1), 10767 (2017).
Global tuberculosis report 2023. Geneva: World Health Organization (2023).
Theron, G. et al. Xpert MTB/RIF results in patients with previous tuberculosis: can we distinguish true from false positive results? Clin. Infect. Dis.62 (8), 995–1001 (2016).
Mishra, H. et al. Xpert MTB/RIF Ultra and Xpert MTB/RIF for diagnosis of tuberculosis in an HIV-endemic setting with a high burden of previous tuberculosis: a two-cohort diagnostic accuracy study. Lancet Respir. Med.8 (4), 368–382 (2020).
Theron, G. et al. False-positive Xpert MTB/RIF results in retested patients with previous tuberculosis: frequency, profile, and prospective clinical outcomes. J. Clin. Microbiol.56 (3) (2018).
Jacobs, R. et al. Identification of novel host biomarkers in plasma as candidates for the immunodiagnosis of tuberculosis disease and monitoring of tuberculosis treatment response. Oncotarget7 (36), 57581 (2016).
Goletti, D. et al. Can we predict tuberculosis cure? What tools are available? Eur. Respir. J.52 (5) (2018).
Namasivayam, S. et al. Longitudinal profiling reveals a persistent intestinal dysbiosis triggered by conventional anti-tuberculosis therapy. Microbiome5 (1), 1–17 (2017).
Burdet, C. et al. Impact of antibiotic gut exposure on the temporal changes in microbiome diversity. Antimicrob. Agents Chemother.63 (10) (2019).
Rogers, G. et al. The exclusion of dead bacterial cells is essential for accurate molecular analysis of clinical samples. Clin. Microbiol. Infect.16 (11), 1656–1658 (2010).
Agustí, G. et al. Viability determination of Helicobacter pylori using propidium monoazide quantitative PCR. Helicobacter15 (5), 473–476 (2010).
Rawsthorne, H., Dock, C. & Jaykus, L. PCR-based method using propidium monoazide to distinguish viable from nonviable Bacillus subtilis spores. Appl. Environ. Microbiol.75 (9), 2936–2939 (2009).
Vesper, S. et al. Quantifying fungal viability in air and water samples using quantitative PCR after treatment with propidium monoazide (PMA). J. Microbiol. Methods72 (2), 180–184 (2008).
Agustí, G., Fittipaldi, M. & Codony, F. False-positive viability PCR results: an association with microtubes. Curr. Microbiol.74 (3), 377–380 (2017).
Wagner, A. O. et al. Removal of free extracellular DNA from environmental samples by ethidium monoazide and propidium monoazide. Appl. Environ. Microbiol.74 (8), 2537–2539 (2008).
Vondrakova, L. et al. Impact of various killing methods on EMA/PMA-qPCR efficacy. Food Control85, 23–28 (2018).
Kragh, M. L., Thykier, M. & Hansen, L. T. A long-amplicon quantitative PCR assay with propidium monoazide to enumerate viable Listeria monocytogenes after heat and desiccation treatments. Food Microbiol.86, 103310 (2020).
Cuthbertson, L. et al. Implications of multiple freeze-thawing on respiratory samples for culture-independent analyses. J. Cyst. Fibros.14 (4), 464–467 (2015).
Nocker, A. et al. Use of propidium monoazide for live/dead distinction in microbial ecology. Appl. Environ. Microbiol.73 (16), 5111–5117 (2007).
Nocker, A., Cheung, C. Y. & Camper, A. K. Comparison of propidium monoazide with ethidium monoazide for differentiation of live vs. dead bacteria by selective removal of DNA from dead cells. J. Microbiol. Methods67 (2), 310–320 (2006).
Hortelano, I. et al. Optimization of pre-treatments with Propidium Monoazide and PEMAX™ before real-time quantitative PCR for detection and quantification of viable Helicobacter pylori cells. J. Microbiol. Methods185, 106223 (2021).
Kayigire, X. A. et al. Propidium monoazide and Xpert MTB/RIF to quantify Mycobacterium tuberculosis cells. Tuberculosis101, 79–84 (2016).
Nikolayevskyy, V. et al. Utility of propidium monoazide viability assay as a biomarker for a tuberculosis disease. Tuberculosis95 (2), 179–185 (2015).
Kim, Y. J. et al. Evaluation of propidium monoazide real-time PCR for early detection of viable Mycobacterium tuberculosis in clinical respiratory specimens. Ann. Lab. Med.34 (3), 203 (2014).
de Assunção, T. M. et al. Real time PCR quantification of viable Mycobacterium tuberculosis from sputum samples treated with propidium monoazide. Tuberculosis94 (4), 421–427 (2014).
Miotto, P. et al. Early tuberculosis treatment monitoring by Xpert® MTB/RIF. Eur. Respir. J.39 (5), 1269–1271 (2012).
Lu, J. et al. Direct detection from clinical sputum samples to differentiate live and dead Mycobacterium tuberculosis. J. Clin. Lab. Anal.33 (3), e22716 (2019).
Esmail, A. et al. An all-oral 6-month regimen for multidrug-resistant tuberculosis: a multicenter, randomized controlled clinical trial (the NExT study). Am. J. Respir. Crit Care Med.205 (10), 1214–1227 (2022).
Invitrogen. PureLink™ Microbiome DNA Purification Kit. Pub. No. MAN0014267 Rev. A.0.
Caporaso, J. G. et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci.108 (Supplement 1), 4516–4522 (2011).
GenIUL PEMAX 25 Reagent Monodose (TBC-Biomarker kit) INSTRUCTIONS in Doc Code 450000075-00 (2015).
Segal, L. N. et al. Enrichment of the lung microbiome with oral taxa is associated with lung inflammation of a Th17 phenotype. Nat. Microbiol.1 (5), 1–11 (2016).
Segal, L. N. et al. Anaerobic Bacterial Fermentation Products Increase Tuberculosis Risk in Antiretroviral-Drug-Treated HIV Patients (Cell Host & Microbe, 2017).
Tsay, J. C. J. et al. Airway Microbiota is associated with upregulation of the PI3K pathway in lung cancer. Am. J. Respir. Crit Care Med.198 (9), 1188–1198 (2018).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol.37 (8), 852–857 (2019).
Callahan, B. J. et al. DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods13 (7), 581–583 (2016).
McDonald, D. et al. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J.6 (3), 610–618 (2012).
Caporaso, J. G. et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J.6 (8), 1621–1624 (2012).
Davis, N. M. et al. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome6, 1–14 (2018).
Oksanen, J. et al. The vegan package. Community Ecol. Pack.10, 631–637 (2007).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of Fold change and dispersion for RNA-seq data with DESeq2. Genome Biol.15 (12), 550 (2014).
Elizaquível, P., Aznar, R. & Sánchez, G. Recent developments in the use of viability dyes and quantitative PCR in the food microbiology field. J. Appl. Microbiol.116 (1), 1–13 (2014).
Kim, Y. J. et al. Evaluation of propidium monoazide real-time PCR for early detection of viable Mycobacterium tuberculosis in clinical respiratory specimens. Ann. Lab. Med.34 (3), 203–209 (2014).
Sulaiman, I. et al. Evaluation of the airway microbiome in nontuberculous mycobacteria disease. Eur. Respir. J.52(4) (2018).
Naidoo, C. C. et al. Anaerobe-enriched gut microbiota predicts pro-inflammatory responses in pulmonary tuberculosis. EBioMedicine67, 103374 (2021).
Ticlla, M. R. et al. The sputum microbiome in pulmonary tuberculosis and its association with disease manifestations: a cross-sectional study. Front. Microbiol.12, 633396 (2021).
Kateete, D. P. et al. Sputum microbiota profiles of treatment-naïve TB patients in Uganda before and during first-line therapy. Sci. Rep.11 (1), 24486 (2021).
Pezzulo, A. A. et al. Abundant DNase I-sensitive bacterial DNA in healthy porcine lungs and its implications for the lung microbiome. Appl. Environ. Microbiol.79 (19), 5936–5941 (2013).
Nocker, A., Sossa, K. E. & Camper, A. K. Molecular monitoring of disinfection efficacy using propidium monoazide in combination with quantitative PCR. J. Microbiol. Methods70 (2), 252–260 (2007).
Codony, F., Agustí, G. & Allué-Guardia, A. Cell membrane integrity and distinguishing between metabolically active and inactive cells as a means of improving viability PCR. Mol. Cell Probes29 (3), 190–192 (2015).
de Goffau, M. C. et al. Recognizing the reagent microbiome. Nat. Microbiol.3 (8), 851–853 (2018).
Acknowledgements
This work and authors were supported by the European and Developing Countries Clinical Trials Partnership (EDCTP; grant number SF1041), the South African National Research Foundation (NRF; grant number 98948), and the South African Medical Research Council (SAMRC) Intramural Flagship Project. C.C.N and G.T acknowledge funding from the National Institutes of Health under award numbers K43TW012302 and R01AI136894. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health nor of funders. R.V is supported by an NRF PDP Postdoctoral fellowship through the SAMRC, the DSI-NRF Centre of Excellence for Biomedical Tuberculosis Research, and the Stellenbosch University Faculty of Medicine and Health Sciences. Computations were performed using facilities provided by the University of Cape Town’s ICTS High Performance Computing team: https://hpc.uct.ac.za/.
Author information
Authors and Affiliations
Contributions
C.C.N., R.V., R.W., L.N.S., and G.T. conceived and designed the study; C.C.N., R.V., F.C., G.A., N.K., S.N., H.M., and Y.L. performed experiments; C.C.N., R.V., H.M., J.C.C., L.N.S., and G.T. analysed and interpreted the data; C.C.N., R.V., and G.T. wrote the first draft. All authors critically reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
GT acknowledges in-kind donations from Cepheid. GT declares no other competing interests. The other authors each declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Naidoo, C.C., Venter, R., Codony, F. et al. Subtracting the background by reducing cell-free DNA’s confounding effects on Mycobacterium tuberculosis quantitation and the sputum microbiome. Sci Rep 14, 22350 (2024). https://doi.org/10.1038/s41598-024-73497-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-73497-3
