
Abstract
Psoriasis is a complex inflammatory skin disease manifested by altered proliferation and differentiation of keratinocytes with dysfunctional apoptosis. This study aimed to identify regulatory factors and comprehend the underlying mechanisms of inefficient apoptosis to open up promising therapeutic approaches. Incorporating human protein interactions, apoptosis proteins, and physical relationships of psoriasis-apoptosis proteins helped us to generate a psoriasis-apoptosis interaction (SAI) network. Subsequently, topological and functional analyses of the SAI network revealed effective proteins, functional modules, hub motifs, dysregulated pathways and transcriptional gene regulatory factors. Network pharmacology, molecular docking and molecular dynamics simulation methods identified the potential drug-target interactions. RELA, MAPK1, MAPK3, MMP9, IL1B, AKT1 and STAT1 were revealed as effective proteins. The MAPK1-MAPK3-RELA motif was identified as a hub regulator in the crosstalk between 41 pathways. Among all pathways, “lipid and atherosclerosis” was found to be the predominant pathway. Acetylcysteine, arsenic-trioxide, β-elemene, bortezomib and curcumin were identified as potential drugs to inhibit pathway crosstalk. Experimental verifications were performed using the literature search, GSE13355 and GSE14905 microarray datasets. Drug-protein-pathway interactions associated with apoptosis were deciphered. These findings highlight the role of hub motif-mediated pathway-pathway crosstalk associated with apoptosis in the complexity of psoriasis and suggest crosstalk inhibition as an effective therapeutic approach.
Similar content being viewed by others


Introduction
Psoriasis is recognized as an inflammatory disease triggered by both genetic and environmental factors, with unknown exact pathogenesis1. Dysfunctional keratinocyte apoptosis is seen in the psoriatic lesions. In this regard, it has been suggested that targeting the mechanisms involved in the regulation of keratinocyte apoptosis may be a potential therapeutic target in the management and treatment of psoriatic patients2. There are several effective treatments for psoriasis that work by modulating the proliferation and differentiation of keratinocytes, such as vitamin D3 analogs and photochemotherapy2,3,4. Current evidence indicates that focusing on the pathways involved in resistance to apoptosis is necessary to identify the relevant regulatory factors and decipher the detailed underlying mechanisms. The study of protein-protein interaction (PPI) networks is used as a systematic approach to understanding the molecular basis of disease processes. Protein interactions play central roles in cellular processes, and their normal patterns are often disrupted in disease states5. Protein interaction networks can enable us to uncover novel pathways associated with diseases and gain a more comprehensive understanding of their molecular basis6. The complex nature of a disease can be caused by the interactions of proteins in the form of pathways as well as the crosstalk of these pathways7,8,9. Evaluation of protein biomarkers, as a developing field in psoriasis, is recommended for both basic and clinical researchers10. In this study, we focus on discovering the molecular mechanism of apoptosis resistance by systematic and integrative analysis of protein-protein interactions and pathways crosstalk. We use a protein-network-based approach to investigate the physical and functional protein interactions of dysfunctional apoptosis and subsequently identify hub-bottleneck proteins, hub 3-node motifs, modules, dysregulated pathways and pathways crosstalk in psoriasis to depict the disease complexity and provide a promising strategy for overcoming this complexity. We also identify drug-gene interactions of key important genes to improve psoriasis treatment.
Materials and methods
Collection of psoriasis-related data and selection of genes involved in apoptosis
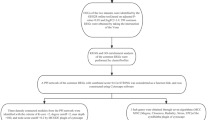
The workflow of this research is summarized in Fig. 1. Firstly, human genes associated with psoriasis were retrieved from public databases including the GeneCards11, DISEASES12, and Disgenet databases13. Thereafter, a comprehensive list of overlapping genes was compiled from the three databases and considered as genes linked to psoriasis. Subsequently, to identify the genes involved in apoptosis, enrichment analysis of the gene list was performed using DAVID Bioinformatics Resources14. The two gene ontology (GO) biological process (BP) categories of “apoptotic process” (GO:0006915) and “regulation of apoptotic process” (GO:0042981) were obtained and merged as a set of genes associated with apoptosis in psoriasis.

Workflow of the study.
Construction of psoriasis-apoptosis interaction network and identification of hub-bottleneck proteins
With the aid of STRING protein interactome database v11.5, human protein-protein interactions (high confidence score (0.700)) of psoriasis-related genes were mined15 and imported to the Cytoscape software to construct the PPI network and assessment of its topological characteristics16. The apoptosis-related genes were also specified in the resulting network. The psoriasis-apoptosis interaction (SAI) network was generated based on connections between all the psoriasis-interacting proteins and the apoptosis proteins. Further, high connectivity proteins (hub nodes) with high betweenness centrality (bottlenecks) were detected in the SAI network by the Network Analyzer plug-in of Cytoscape. To find hub proteins, the mean+(2×standard deviation) of the degree distribution was considered, whereas bottlenecks were characterized as the top 5% nodes with the highest betweenness centrality in the SAI network17,18. Finally, the important nodes that show both features are specified as hub-bottleneck proteins. The hub-bottleneck proteins belonging to the apoptosis-related genes were deemed as apoptosis hub-bottlenecks.
Identification of high‑scoring apoptosis-related modules, sub-modules and key regulators
Modules or communities are closely related proteins that can correspond to specific biological functions using protein interactions19,20. To find the densely connected modules (or clusters) in the SAI network, MCODE (Molecular Complex Detection) was used21 with the predefined parameters of degree cutoff = 2, node score cutoff = 0.2, k-core = 2, and maximum depth = 100. MCODE scores > 5 and numbers of nodes > 10 was set to select the modules. Among them, we screened significant functional modules with the highest number of apoptosis-related proteins, namely apoptosis-related modules. To identify the key regulators or driver genes, apoptosis hub proteins of the selected module were traced up to the motif level. Accordingly, the identified apoptosis-related module was subjected to MCODE for the detection of sub-modules and sub-sub-modules. The clustering coefficient parameter ≤ 1 was considered for this analysis22. Afterward, the functional roles of these modules were further evaluated by comparing the existence of significant apoptosis motifs within them.
Identification of significant apoptosis-related 3-node connected motif patterns
Network motifs are the simplest patterns within a network that have been applied to find key biological information and unravel medical problems, such as exploring essential proteins, identifying protein interactions, and specifying protein functions23,24,25. First, the two kinds of nodes were characterized in the SAI network including apoptosis proteins and non-apoptosis proteins. The relationships between these two types of proteins can be combined in different patterns of 3-node motifs. Towards this end, apoptosis-related 3-node motifs were defined as motifs with at least one apoptosis protein. To detect size-3 network motif types in the SAI network and the identified apoptosis-related module, the FANMOD (Fast Network Motif Detection) software was used26. To evaluate the significance of possible 3-node motifs in the original PPI network, the 1000 random networks were generated and compared with the original input network. When randomizing the network in a local constant model, the same interactions were swapped five times. Finally, we identified the significant apoptosis-related 3-node motifs with threshold values of a Z-score > 2.0 and a p-value < 0.01. The motif z-score is the frequency of motifs in the original network minus their mean of frequency in the random network divided by the standard deviation26,27.
Pathway enrichment analysis and detection of functional protein network motifs
KEGG (Kyoto Encyclopedia of Genes and Genomes)28 pathway enrichment analysis of the apoptosis-related modules was performed using the ClueGO plug-in of Cytoscape29. The p-values were computed using right-sided hypergeometric tests, and the Benjamini-Hochberg (BH) adjustment procedure was implemented for multiple test correction (false discovery rate (FDR) < 0.001). Therefore, choosing this very stringent FDR cutoff along with a high kappa value (0.4) helped to accurately get functional pathways enriched in highly connected genes30. By integrating protein interactions and KEGG pathway information, interactions and crosstalk among the significant pathways were investigated at the motif level. Common significant apoptosis-related 3-node motifs were detected in the interactions among pathways and the top interacting motif was identified.
Prediction of transcription factors regulating the screened module
To predict the regulatory effect of transcription factors (TF) on target genes, the TF-target gene regulatory interactions were extracted for apoptosis-related module. Transcription factors are proteins that regulate the expression of both coding and non-coding genes at the transcriptional level. To find TF-gene pairs, we mined the known regulatory TF-target gene interactions of module 4 from TRRUST v231. Human and mouse transcriptional regulatory networks are manually curated in this database. Overlapped genes ≥ 4 and q-value less than 0.05 were considered for TF enrichment analysis.
Keratinocyte gene expression study
The gene expression of apoptosis-related module was further validated in human lesional psoriatic skin using the literature search and gene expression omnibus (GEO) database. We used the published supplemental table of differentially expressed genes (DEGs) of the keratinocyte transcriptome in psoriasis32. DEGs between keratinocytes from psoriasis lesional skin and healthy control skin were extracted, with adjusted p-value < 0.05 and fold-change < 0.67 or > 1.5. In another study, Weng et al. reported mRNA expression of lesional skin biopsies by qRT-PCR33. Also, the GEO microarray datasets were applied to identify DEGs between skin lesions of patients with psoriasis and normal healthy controls including GSE13355 based on the GPL570 microarray platform (58 skin lesions of psoriasis and 64 normal) and GSE14905 based on the GPL570 microarray platform (33 skin lesions of psoriasis and 21 normal sample). The GEO2R web tool was used to analyze these GEO microarray datasets, with adjusted p-value < 0.05 and > 1.5-fold up/down34.
Drug candidate analysis
The CLUE repurposing database (https://clue.io.) was screened out to find drug-gene relationships of the identified key important genes and key regulators in this study35. The connectivity score analysis was performed using the CLUE touchstone database. Furthermore, a drug-gene interaction network was prepared based on the CLUE database results in the clinical phase “launched” to gain a further understanding of the potential relationships using the STITCH database (combined score value > 0.7)36. This database contains interaction information for drugs, connects them to genes, and predicts the physical and functional contacts between the query genes and drugs.
Molecular docking technology
To gain further insight into the interaction between drug candidates and key proteins, we used molecular docking methodology. The 3D molecular structures of screened compounds were attained from the chemical compound library, PubChem database (https://pubchem.ncbi.nlm.nih.gov) and (https://zinc.docking.org)37,38. The obtained structures were optimized and minimized through the Gaussian09 software package39 at the B3LYP/6-31G** level of theory. The 3D structure of target proteins was downloaded from the RCSB PDB database (https://www.rcsb.org/). The molecular docking calculations were conducted to visualize how drugs bind to the proteins. The method was performed to identify the specific residues involved in the interaction. To perform molecular docking, we used the AutoDock 4.2 molecular docking program, which utilizes an empirical free energy function and the Lamarckian Genetic Algorithm (LGA). The Gastieger charges were added to the macromolecule input files, and AutoGrid was applied to calculate grids40. We also performed a literature search and documented the results of previous articles that have investigated gene expression changes in response to drugs in other diseases through in vitro and in vivo studies.
Molecular dynamics simulation
We conducted all molecular dynamics (MD) simulations using the AMBER 18 package. The FF99SB and GAFF force fields were employed for the protein and its ligands41. The antechamber module of AmberTools was used to calculate the partial charges of the ligands, utilizing the semi-empirical AM1-BCC function as per the standard protocol42. The systems were solvated using TIP3P water models and neutralized by adding Na+ ions via the tLEap input script from the AmberTools package. We modeled long-range electrostatic interactions using the particle-mesh Ewald method43. The SHAKE algorithm44 was used to constrain the length of covalent bonds, including those involving hydrogen atoms. We maintained the system temperature at 310 K using a Langevin thermostat and employed a 2.0-fs time step for all MD setups. For the minimization and equilibration phases (NVT and NPT ensembles), we used 100,000 steps and a 1-ns period, respectively. Following this, we executed 100-ns classical MD simulations without constraints, as an NPT ensemble, for each protein-ligand complex. Additionally, we coupled the MD protocol with the Poisson-Boltzmann (MM-PBSA) or generalized Born (MM-GBSA) molecular mechanics approach, supplemented with the hydrophobic solvent-accessible surface area term45,46.
Results
Psoriasis-related data and genes involved in apoptosis
We obtained 4243, 1308 and 4178 psoriasis-related genes from Diseases, DisGeNet and GeneCards databases, respectively. A total of 790 shared genes for psoriasis were compiled through the three public databases (Fig. 2A). To find apoptosis-related genes, gene ontology analysis was implemented for the psoriasis gene list and mined overlapping genes linked to the two GO terms of “apoptotic process” and “regulation of apoptotic process”, including 81 and 38 enriched genes, respectively (Fig. 2B). Subsequently, we merged the highly enriched categories and listed a set of 106 apoptosis-related genes associated with psoriasis. Figure 2C illustrates the overlap of genes between both GO biological process terms using a Venn diagram (http://bioinformatics.psb.ugent.be/webtools/Venn/).

(A) Venn diagram illustrates the number of shared genes. (B) The enriched genes of psoriasis in the “apoptotic process” and “regulation of apoptotic process” terms are 81 and 38, respectively, with 13 common genes. (C) Properties of the two GO terms are presented. (D) Psoriasis-apoptosis interaction (SAI) network representing the interaction of apoptotic proteins and non-apoptotic proteins in psoriasis (7072 high confidence interactions (score > 0.7) between 717 proteins). 105 apoptotic proteins and their interactions are presented in this network. The hub-bottleneck proteins are represented in ellipse shapes; apoptotic hub-bottlenecks are white with a red border, non-apoptotic hub-bottlenecks are blue.
Construction and analysis of PPI network
To construct the PPI network, protein-protein interactions of the 790 psoriasis-related genes were acquired from the STRING database containing 7072 high-confidence interactions (score > 0.7) between 717 proteins. Figure 2D exhibits the resulting network that was visualized using Cytoscape. The psoriasis-apoptosis interaction (SAI) network indicated a power-law degree distribution r-squared value of 0.844, suggesting a scale-free nature for the constructed network. The 105 apoptosis-related proteins (of 106 genes) were recognized in the SAI network through GO enrichment analysis on the gene list. The proteins related to apoptosis showed 478 interactions with each other and 2311 interactions with 428 non-apoptotic proteins as first neighbors in the SAI network. Structural and functional analysis of the interaction network was implemented to mine apoptosis-related hub-bottleneck nodes. The 28 hub-bottleneck proteins were detected in the SAI network including 11 apoptosis-related hub-bottlenecks (AKT1, HSP90AA1, IL1B, JUN, MAPK1, MAPK3, MMP9, STAT1, TLR2, TP53 and TRAF6) and 17 non- apoptosis-related hub-bottlenecks (ALB, CTNNB1, EGFR, FN1, FOS, IL10, IL2, IL4, IL6, ITGAM, PIK3CA, PTPRC, RELA, STAT3, TLR4, TNF and VEGFA), as shown in Fig. 2D. The name and topological properties of the hub-bottleneck nodes are presented in Table 1.
Identification of key apoptosis-related modules and annotation
A total of 20 protein modules were recognized in the SAI network. Four SAI-specific network modules were identified with cut-off criteria of MCODE scores > 5 and number of nodes > 10. Interestingly, all the 11 apoptotic hub-bottlenecks belonged to these top modules. Module 1 (scoring value 14.769) included 27 nodes and 192 edges with 2 apoptotic hub-bottlenecks (IL1B and TLR2) from 7 apoptotic nodes. Module 2 indicated 56 nodes (score = 14.509) and 399 edges with 1 apoptotic hub-bottleneck (STAT1) from 9 apoptotic nodes, whereas module 3 included 42 nodes (score = 10) and 205 edges with 1 apoptotic hub-bottleneck (AKT1) from 8 apoptotic nodes (Fig. S1). We found the functional module 4 (scoring value 9.86; 212 interactions between 44 nodes) with the 15 significantly enriched apoptosis-related proteins and the largest number of apoptotic hub-bottlenecks as compared to other modules in the SAI network. HSP90AA1, JUN, MAPK1, MAPK3, MMP9, TP53 and TRAF6 apoptotic hub-bottlenecks were seen in functional module 4 (Fig. 3A). Module 4 was called the apoptosis-related module in this study and was considered for additional analysis.

(A) Characteristics of module 4 with the significant and highest number of apoptotic proteins are presented. The apoptosis-related proteins and hub-bottlenecks of the SAI network were detected in module 4 which are shown in pink and yellow, respectively. (B) Identification of sub-modules and sub-sub-modules of module 4 up to the last level of the SAI network to trace hub genes and screen driver genes of the apoptosis-related module. The dashed red arrow shows the hubs transmission to the next level. (C) Hub-module interaction analysis shows the crosstalk among module 4 and other top protein modules through apoptosis-related hubs in the SAI network.
Identification of fundamental regulators and key genes of apoptosis-related module
To provide another level of functional annotation and identify key regulators or driver genes, we screened sub-modules and sub-sub-modules of the functional module 4 by MCODE. Figure 3B represents module 4, sub-modules 4 and sub-sub-modules 4 as level-1, level-2, and level-3, respectively47. Clustering coefficient values were 0.58, 0.745 and 0.933 for module 4, sub-modules 4 and sub-sub-modules 4, respectively. In this analysis, we traced the hub genes of module 4 up to the motif level of the SAI network and identified MMP9 (seed) and MAPK3 as apoptosis-related genes at the last level. The hub proteins, which can control the original network to the motif level are the essential regulators22. Moreover, after detecting the apoptosis-related hub genes of the SAI network in module 4 and other top modules, we investigated crosstalk among module 4 and the apoptosis-related hubs of the top modules through hub-module interaction analysis (Fig. 3C). The hub-bottleneck IL1B in module 1 showed 100 interactions with the four top modules. IL1B interacted with modules 2, 3 and 4 via 26, 27 and 24 proteins. The results revealed that IL1B interacts with one cluster of proteins (modules 1, 2, 3 and 4). The hub-bottleneck STAT1 in module 2 showed 82 interactions with the high-scoring modules. Crosstalk among module 4 and STAT1 hub-bottleneck indicated 22 interactions. The hub-bottleneck AKT1 in module 3 showed 54 interactions with the high-scoring modules. There were 22 interactions between module 4 and AKT1. Therefore, hub-module interaction analysis specified IL1B, STAT1 and AKT1 as key genes and influencing nodes among the hub genes in connecting the functional module 4 with other top modules47. Chemical signals from other modules can quantitatively and qualitatively regulate the function of a module. Connecting the protein modules can make higher-level functions48.
Pathway enrichment analysis and crosstalk analysis of pathways based on overlapping hub motifs
Moreover, functional pathway enrichment of module 4 and crosstalk analysis of pathways enabled us to find functional motifs. To identify significant KEGG pathways, we subjected module 4 to the ClueGO plug-in with a very stringent FDR value (< 0.001) and found functional pathways enriched in highly connected genes. Figure 4 shows the KEGG pathway network analysis of module 4 in the SAI network. The functionally enriched KEGG network groups were visualized using Cytoscape (Fig. 4A). As shown in Fig. 4B, the pie chart illustrates that the most enriched pathways (58.21%) were grouped in the lipid and atherosclerosis pathway. CASP1, HSP90AA1, IFNB1, JUN, MAPK1, MAPK3, MMP9, MYD88, NLRP3, RELA, SELP, STAT3, TNFRSF1A, TP53, TRAF6, and VCAM1 were observed in the lipid and atherosclerosis pathway (KEGG:05417). In addition, motif enrichment analysis revealed the significant existence of key apoptosis-related 3-node motif types in the SAI network (Table 2) and module 4 (Fig. 5A). We estimated that ~ 19% of motifs in module 4 belong to the significant apoptosis-related 3-node patterns, with one or two apoptotic proteins (Fig. 5A). Whereas, this evaluation was not significant for a 3-node pattern containing three apoptotic proteins. Subsequently, we screened apoptotic crosstalk motifs that overlapped between enriched pathways. The crucial apoptosis-related motif (MAPK1-MAPK3-RELA) was identified as a hub motif in the SAI network and with high interaction in crosstalk among the 41 KEGG pathways (Fig. 5B). Moreover, the identified key regulators and hub motifs (MMP9, MAPK1, MAPK3 and RELA) were observed in the enriched gene list of the lipid and atherosclerosis pathway. In this study, the results of functional analysis of the SAI network indicated the lipid and atherosclerosis pathway as the main pathway.

KEGG pathway network analysis of module 4 (FDR < 0.001, kappa value (0.4)) in the SAI network. (A) The functionally grouped network of KEGG pathways. (B) The pie chart shows the respective pathways.

(A) The apoptosis-related 3-node motif analysis (motifs with at least one apoptotic protein) of module 4 in the SAI network. The frequency of each pattern in the original network and its statistical significance are represented. (B) Identification of crucial apoptosis-related motif (MAPK1-MAPK3-RELA) with high interaction in crosstalk among the 41 KEGG pathways.
Gene regulation and gene expression study
To find the regulatory relationship of TF-target gene pairs in module 4, we queried the 44 genes in the TRRUST database. 27 TFs were identified that exhibited 170 interactions. Transcription factors STAT3, TP53, RELA interacted with the largest number of genes (Table S1). The functional relationship of RELA-MMP9 (TF-target gene) was also identified. Moreover, we found significant expression changes in 24 genes in module 4 using the literature search and psoriatic microarray datasets in the GEO database including GSE13355 and GSE14905. q-values less than 0.05 and fold change > 1.5 (up/down) were considered. The results are shown in Table 3.
Identification of potential drugs
The aim was to acquire candidate drugs that could potentially affect crosstalk among pathways. Table S2 provides the total results of drug repurposing and connectivity map analysis including 66 drugs for RELA, MAPK1, MAPK3, MMP9, IL1B, AKT1 and STAT1 using the CLUE repurposing database (connectivity score: 99.97 to 100). A list of drugs in the clinical phase “launched” for candidate key genes by the CLUE repurposing tool is presented in Table 4. Acetylcysteine, arsenic-trioxide, β-elemene, bortezomib and curcumin were identified as drugs that target the important hub proteins and identified hub-motif (RELA, MAPK1, MAPK3 and MMP9) in this study. Angiotensin converting enzyme inhibitors are not shown in the table. Additionally, Fig. 6 includes more information on drug-gene interactions of the four key proteins with the list of drugs in the clinical phase “launched”. This figure shows no interaction for β-elemene. Whereas, it shows potential targets for other drugs, including four interactions for acetylcysteine arsenic-trioxide and curcumin, and three interactions for bortezomib.

Drug-target interaction analysis of four important key proteins and their drugs using the STITCH database.
Molecular docking of screened drugs and target proteins
According to the results of screened drugs and target proteins and the interaction network between them, molecular docking was done between the four target proteins (RELA, MAPK1, MAPK3 and MMP9) and the five small molecule ligands (acetylcysteine, arsenic-trioxide, β-elemene, bortezomib and curcumin). Table S3 presents the grid boxes used to find the best pose and conformation for the drugs, with the boxes centered on the active site of the targets. The grid point spacing was set to 0.375 Å to allow for free rotation of the complex. The docking scores (ΔGbinding), inhibition constant (KI), and involved amino acids in the binding sites of targets are recorded in Table 5. The 3D ribbon structure and 2D scheme of the binding sites are also shown in Fig. S2-S6. As observed in the figures, the drug compounds acetylcysteine, arsenic-trioxide, β-elemene, bortizomib, and curcumin are positioned in the active sites49,50,51. A binding energy greater than 5.0 indicates strong binding activity, while a binding energy greater than 7.0 shows extremely strong binding ability (Table 5)52. Interestingly, β-elemene, bortizomib, and curcumin show docking scores greater than 5.0 or 7.0 with four protein targets. We also provided some in vitro and in vivo gene expression changes in response to drugs based on the literature search (Table S4).
Molecular dynamics simulation for curcumin-MMP9 and β-elemene-MMP9
We refined the results using MD simulations after identifying high-affinity ligands through molecular docking. Curcumin (CUR) and beta-elemene (ELE) emerged as top candidates for MMP9, with binding energies of −11.49 kcal/mol and − 8.29 kcal/mol, respectively. Interestingly, despite a predicted binding energy of -9.16 kcal/mol, bortezomib was excluded due to its incompatibility with the AMBER force field. To understand the dynamic behavior of these ligand-protein complexes, we tracked their structural fluctuations over time using various metrics (Fig. 7). The receptor RMSD (root mean square deviation) in the CUR-MMP9 complex remained below the 3 Å threshold, indicating stability after 40 ns (Fig. 7A). However, the CUR molecule itself showed higher RMSD values (2–3 Å) compared to the ELE molecule, which deviated less (1.0 Å) (Fig. 7B). The receptor RMSF (root mean square fluctuation) revealed greater flexibility in the ELE-MMP9 complex, especially in unstructured loops and the N- and C-termini (Fig. 7C). Similarly, the ligand RMSF showed higher atomic deviations for ELE, exceeding the 3 Å threshold, compared to CUR (Fig. 7D). Interestingly, the Rg function indicated a more compact receptor structure in the CUR-MMP9 complex, suggesting reduced protein flexibility upon ligand binding (Fig. 7E). Further analysis revealed the presence of hydrogen bonds exclusively in the CUR-MMP9 complex, with Asp-74 identified as the key residue contributing to this interaction (Fig. 7F). Finally, MM-PBSA/GBSA calculations based on 100 ns MD trajectories corroborated the 7 trends, confirming significantly higher binding affinity of CUR to MMP9 (Table 6).

RMSD (A, B), RMSF (C, D), Rg (E), and the fraction of H-bonds (F) calculated for CUR-MMP9 and ELE-MMP9 complexes during 100 ns MD simulation. The thresholds are depicted as dotted lines.
Discussion
Psoriasis has been recognized as a complex multifactorial disease characterized by a decrease in keratinocyte apoptosis of skin lesions. The molecular pathophysiology of psoriasis remains unclear and its treatment is still based on controlling the symptoms because of many unknown mechanisms53,54. To date, various studies aim at exploring biomarkers to predict progression and treatment response. However, no particular markers can be used in routine clinical practice53. Disruption of the homeostasis between keratinocyte proliferation and differentiation processes results in the establishment of a self-amplifying loop. Activated keratinocytes also exhibit increased resistance to apoptosis2,55. Accordingly, targeting proteins and protein interactions involved in the dysregulation of apoptosis in keratinocytes may be potential therapeutic targets in psoriasis treatment. In the present study, a total of 790 common genes were identified from the three public databases and considered psoriasis-related genes. Subsequently, we utilized GO term enrichment analysis and recognized 106 significant apoptosis-related genes in the psoriasis gene list. we implemented a network-based approach and generated a psoriasis-apoptosis interaction network (SAI) using PPI of apoptosis-related proteins with non-apoptosis-related proteins, containing 717 nodes and 7072 edges. Protein interactions mediate major biological processes such as immunity, signaling, metabolism, as well as gene expression, thus the protein interactions network as a promising approach is used to unravel the molecular mechanisms of diseases56. The PPI network was constructed and apoptosis-related interactions were characterized. The resulting protein interaction network was analyzed to detect protein hub-bottlenecks, functional modules, significant 3-node motifs, and pathways crosstalk. From 28 identified hub-bottlenecks in the SAI network, 11 of them were specified as apoptosis-related hub-bottlenecks, namely AKT1, HSP90AA1, IL1B, JUN, MAPK1, MAPK3, MMP9, STAT1, TLR2, TP53 and TRAF6. Moreover, ALB, CTNNB1, EGFR, FN1, FOS, IL10, IL2, IL4, IL6, ITGAM, PIK3CA, PTPRC, RELA, STAT3, TLR4, TNF and VEGFA were identified as non-apoptosis-related hub-bottlenecks. Topological and functional analyses of the network at the modular level aided us to attain better insight into the regulation of the apoptosis process. Our findings uncovered high-scored module 4 as an apoptosis-related module in the SAI network. The seven apoptotic hub-bottleneck nodes were observed in functional module 4, including HSP90AA1, JUN, MAPK1, MAPK3, MMP9, TP53 and TRAF6. In this study, MMP9 (matrix metalloproteinase-9) and MAPK3 (Mitogen-Activated Protein Kinase 3) were traced up to the last level of module 4 and identified as key apoptosis-related hub-bottlenecks, and MMP9 was presented as seed node. MMP9 plays a control role in keratinocyte growth11. Overexpression of MMP9 has been shown in psoriatic neutrophils using microarray gene profile data analysis57. Western blot analysis has shown that MMP9 is upregulated in the skin tissue of mouse model samples of psoriasis58. Elevated concentrations of MMP9 in peripheral blood mononuclear cells (PBMC) and the skin of psoriatic patients have been reported59. Moreover, the KEGG pathway enrichment, PPI network motif analysis, and pathway crosstalk analysis of the functional module 4 helped us to detect hub motif-mediated pathway-pathway crosstalk. The MAPK1-MAPK3-RELA hub motif participates in the 41 KEGG pathways, indicating the highest frequency crosstalk pattern. The results of this integrated analysis can help to comprehend the complexity of the disease and can provide information to understand the important effects of targeting multiple pathways instead of targeting a single pathway. Inhibition of pathways crosstalk and identification of pathway-based drug targets can aid therapeutic approaches and drug development to target the entire pathway instead of a single protein. In our study, the RELA hub-bottleneck node is seen as a non-apoptosis-hub-bottleneck node in the SAI network, whereas MAPK1 and MAPK3 hub-bottlenecks are apoptosis-related nodes. MAPK1 and MAPK3 are two MAPKs that play an important role in the MAPK/ERK signaling cascade. They have been implicated in the regulation of keratinocyte proliferation, differentiation and cytokine production60. Abnormal MAPK signaling pathways have also been found in psoriatic keratinocytes61,62. A high RELA level was noticed in psoriatic skin63. RELA (NF-κB p65 subunit) is a transcription factor that participates in multiple biological processes. Elevated mRNA expression of RELA was shown in human psoriatic skin33. In addition, the functional interaction of RELA-MMP9 (TF-target gene) was identified using gene regulatory analysis in this study. Interestingly, through functional analysis, the most influencing nodes of module 4 (MMP9, MAPK1, MAPK3 and RELA) were screened in the lipid and atherosclerosis pathway, as a predominant pathway in this analysis. Studies have shown that specific lipids can promote keratinocyte differentiation and induce apoptosis64,65. Based on our previously published works, psoriatic patients have an altered lipid metabolism and are characterized by abnormal lipid profiles and lipid dysfunction66,67,68. Additionally, psoriasis and atherosclerosis share common pathogenic pathways and overlapping mechanisms66,69.
According to the drug repurposing results, RELA was revealed as a drug target gene for bortezomib. Bortezomib is an NF-kB pathway and proteasome inhibitor that has been marketed for the treatment of multiple myeloma. It is known as the first proteasome inhibitor approved by the US Food and Drug Administration. Bortezomib has inhibited NF-κB activation and proliferation, and promoted apoptosis in head and neck squamous cell carcinoma (HNSCC) cell lines70,71. In a recent study, Chen et al. also highlighted the potential of repurposing bortezomib as a therapeutic drug for psoriasis72. The other key drug-target gene interaction is acetylcysteine-RELA. N-acetylcysteine has been used as a mucolytic agent and antioxidant in various medical fields including dermatology73. The aim of the clinical trial NCT05906498 is to investigate the effect of N-acetylcysteine (NAC) alone or together with vitamin E to improve clinical outcome, oxidative stress and inflammatory response in patients with mild psoriasis vulgaris. Our findings suggest two apoptosis-stimulating substances for repurposing including arsenic-trioxide (As2O3) and β-elemene. MAPK1 and MAPK3 were revealed as drug target genes for As2O3. As2O3 induces the apoptotic process and is used in Chinese medicine to treat psoriasis74. It has also been reported to inhibit cell growth in cultured HaCaT keratinocytes75. Arsenic derivatives were used to treat psoriasis and other skin diseases in Rome and Greece76. β-elemene has been approved by the China Food and Drug Administration to treat different tumors, such as lung, liver, colon, ovary, brain and breast77. According to the results of drug repurposing, it targets MMP9 as a key gene. This antitumor drug can inhibit cell proliferation, arrest the cell cycle, and trigger cell apoptosis78. Curcumin was identified as another candidate drug that can target MMP9. The literature has explained mechanism of actions of curcumin in skin disorders. For example, it can suppress psoriasis by decreasing inflammation and repress melanoma by inhibiting cell proliferation and inducing cell apoptosis79. However, the specific mechanism of it in the treatment of psoriasis is unclear, and its effectiveness and safety have not been affirmed80. Moreover, the molecular docking technique was used to predict the binding affinity of five drug compounds with four target biomacromolecules. The results showed that bortizomib and curcumin had strong binding activity with all four targets, while β-elemene had the best interaction with MMP9. MD simulations were performed to validate the prediction of molecular docking81 and revealed dynamic behavior of curcumin-MMP9 and β-elemene-MMP9 complexes. Overall, the results contribute to the understanding of the potential efficacy of drug compounds in targeting specific proteins for drug design and discovery in psoriasis treatment. However, the presented result has some limitations due to the lack of pharmacological and clinical studies, but it provides directions for further studies.
In conclusion, this study highlighted the role of lipid and atherosclerosis pathway and its protein interactions in the induction of keratinocyte apoptosis in the skin of psoriatic patients. Collectively, MMP9, MAPK1, MAPK3 and RELA were clarified as the most influencing nodes in the apoptosis-related module of the SAI network that were enriched in lipid and atherosclerosis pathway, as predominant pathway in functionally grouped pathway network analysis. The results of this integrated analysis identified hub motif-mediated pathway-pathway crosstalk, which could be important for inhibiting the pathways crosstalk through apoptosis induction as a therapeutic approach. We here also proposed candidate drugs associated with the key proteins for psoriasis treatment. The findings suggest drugs with multiple targets or combinations of drugs in psoriasis treatment. This study further proposes crosstalk analysis of pathways to understand and overcome the complexity of psoriasis and design appropriate drugs to induce apoptosis through crosstalk inhibition.
Data availability
Data is provided within the manuscript or supplementary information files.
Abbreviations
- PPI:
-
protein–protein interaction
- GO:
-
Gene Ontology
- BP:
-
Biological processes
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- DEGs:
-
Differentially expressed genes
- FDR:
-
false discovery rate
- GEO:
-
Gene Expression Omnibus
- TF:
-
Transcription Factor
- MCODE:
-
Molecular Complex Detection
- FC:
-
Fold change
- BH:
-
Benjamini-Hochberg
References
Ortiz-Lopez, L. I., Choudhary, V. & Bollag, W. B. Updated perspectives on keratinocytes and psoriasis: keratinocytes are more than innocent bystanders 73–87 (Targets and Therapy, 2022).
Xu, R. et al.Long-Acting β2 Adrenergic Receptor Agonist Ameliorates Imiquimod-Induced Psoriasis-Like Skin Lesion by Regulating Keratinocyte Proliferation and Apoptosis 13 (Frontiers in Pharmacology, 2022).
Kim, E. S. & Frampton, J. E. Calcipotriol/betamethasone dipropionate foam: a review in plaque psoriasis. Drugs. 76, 1485–1492 (2016).
Morita, A. Current developments in phototherapy for psoriasis. J. Dermatol.45(3), 287–292 (2018).
Kuzmanov, U. & Emili, A. Protein-protein interaction networks: probing disease mechanisms using model systems. Genome Med.5(4), 1–12 (2013).
Gonzalez, M. W. & Kann, M. G. Protein interactions and disease. PLoS Comput. Biol.8(12), e1002819 (2012).
Li, Y., Agarwal, P. & Rajagopalan, D. A global pathway crosstalk network. Bioinformatics. 24(12), 1442–1447 (2008).
Francesconi, M. et al. Reconstructing networks of pathways via significance analysis of their intersections. BMC Bioinform.9, 1–12 (2008).
Wang, J. et al. Pathway crosstalk analysis based on protein-protein network analysis in prostate cancer. Eur. Rev. Med. Pharmacol. Sci.16(9), 1235–1242 (2012).
Yadav, K., Singh, D. & Singh, M. R. Protein biomarker for psoriasis: a systematic review on their role in the pathomechanism, diagnosis, potential targets and treatment of psoriasis. Int. J. Biol. Macromol.118, 1796–1810 (2018).
Safran, M. et al. GeneCards Version 3: the human gene integrator. Database (2010).
Pletscher-Frankild, S. et al. DISEASES: text mining and data integration of disease–gene associations. Methods. 74, 83–89 (2015).
Piñero, J. et al. The DisGeNET knowledge platform for disease genomics: 2019 update. Nucleic Acids Res.48(D1), D845–D855 (2020).
Huang, D. W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc.4(1), 44–57 (2009).
Szklarczyk, D. et al. STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res.47(D1), D607–D613 (2019).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res.13(11), 2498–2504 (2003).
Ray, M., Ruan, J. & Zhang, W. Variations in the transcriptome of Alzheimer’s disease reveal molecular networks involved in cardiovascular diseases. Genome Biol.9, 1–14 (2008).
Safari-Alighiarloo, N. et al. Identification of new key genes for type 1 diabetes through construction and analysis of protein-protein interaction networks based on blood and pancreatic islet transcriptomes. J. Diabetes. 9(8), 764–777 (2017).
Lin, C. Y. et al. Module organization and variance in protein-protein interaction networks. Sci. Rep.5(1), 1–12 (2015).
Newman, M. E. Communities, modules and large-scale structure in networks. Nat. Phys.8(1), 25–31 (2012).
Bader, G. D. & Hogue, C. W. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform.4(1), 1–27 (2003).
Haider, S. et al. Hamiltonian energy as an efficient approach to identify the significant key regulators in biological networks. Plos One. 14(8), e0221463 (2019).
Liu, Z. et al. Evidence for the additions of clustered interacting nodes during the evolution of protein interaction networks from network motifs. BMC Evol. Biol.11(1), 1–12 (2011).
Kim, W. & Haukap, L. NemoProfile as an efficient approach to network motif analysis with instance collection. BMC Bioinform.18(12), 37–45 (2017).
Chen, L. et al. Identification of breast cancer patients based on human signaling network motifs. Sci. Rep.3(1), 1–7 (2013).
Wernicke, S. & Rasche, F. FANMOD: a tool for fast network motif detection. Bioinformatics. 22(9), 1152–1153 (2006).
Farahani, M. et al. Systematic Analysis of Protein–Protein and Gene–Environment Interactions to Decipher the Cognitive Mechanisms of Autism Spectrum Disorder 1–13 (Cellular and Molecular Neurobiology, 2020).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res.28(1), 27–30 (2000).
Bindea, G. et al. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 25(8), 1091–1093 (2009).
Mach, N. et al. Integrated mRNA and miRNA expression profiling in blood reveals candidate biomarkers associated with endurance exercise in the horse. Sci. Rep.6(1), 1–15 (2016).
Han, H. et al. TRRUST v2: an expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res.46(D1), D380–D386 (2018).
Pasquali, L. et al. The keratinocyte transcriptome in psoriasis: pathways related to immune responses, cell cycle and keratinization. Acta Dermato-Venereologica. 99(2), 196–205 (2019).
Weng, Z. et al. Luteolin inhibits human keratinocyte activation and decreases NF-κB induction that is increased in psoriatic skin. PloS One. 9(2), e90739 (2014).
Edgar, R., Domrachev, M. & Lash, A. E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic acids research 207–210 (2002).
Subramanian, A. et al. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell. 171(6), 1437–1452 (2017). e17.
Kuhn, M. et al. STITCH: interaction networks of chemicals and proteins. Nucleic Acids Res.36(suppl_1), D684–D688 (2007).
Irwin, J. J. et al. ZINC20—a free ultralarge-scale chemical database for ligand discovery. J. Chem. Inf. Model.60(12), 6065–6073 (2020).
Kim, S. et al. PubChem 2023 update. Nucleic Acids Res.51(D1), D1373–D1380 (2023).
Zheng, G. et al.Gaussian 09 48 (Gaussian Inc., Wallingford CT, 2009).
Meng, X. Y. et al. Molecular docking: a powerful approach for structure-based drug discovery. Curr. Comput.-Aided Drug Design. 7(2), 146–157 (2011).
Case, D. A. et al. The Amber biomolecular simulation programs. J. Comput. Chem.26(16), 1668–1688 (2005).
Marques, S. M. et al. Screening of natural compounds as P-glycoprotein inhibitors against multidrug resistance. Biomedicines. 9(4), 357 (2021).
Essmann, U. et al. A smooth particle mesh Ewald method. J. Chem. Phys.103(19), 8577–8593 (1995).
Miyamoto, S. & Kollman, P. A. Settle: an analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem.13(8), 952–962 (1992).
Shityakov, S. et al. In silico investigation of propofol binding sites in human serum albumin using explicit and implicit solvation models. Comput. Biol. Chem.70, 191–197 (2017).
Dutta, K. et al. Seq12, Seq12m, and Seq13m, peptide analogues of the spike glycoprotein shows antiviral properties against SARS-CoV-2: an in silico study through molecular docking, molecular dynamics simulation, and MM-PB/GBSA calculations. J. Mol. Struct.1246, 131113 (2021).
Prasad, K. et al. Brain disease network analysis to elucidate the neurological manifestations of COVID-19. Mol. Neurobiol.58, 1875–1893 (2021).
Hartwell, L. H. et al. From molecular to modular cell biology. Nature. 402(Suppl 6761), C47–C52 (1999).
Nuti, E. et al. Development of thioaryl-based matrix metalloproteinase-12 inhibitors with alternative zinc-binding groups: synthesis, potentiometric, NMR, and crystallographic studies. J. Med. Chem.61(10), 4421–4435 (2018).
Wolter, M. et al. Selectivity via cooperativity: preferential stabilization of the p65/14-3-3 interaction with semisynthetic natural products. J. Am. Chem. Soc.142(27), 11772–11783 (2020).
Chaikuad, A. et al. A unique inhibitor binding site in ERK1/2 is associated with slow binding kinetics. Nat. Chem. Biol.10(10), 853–860 (2014).
Hsin, K. Y., Ghosh, S. & Kitano, H. Combining machine learning systems and multiple docking simulation packages to improve docking prediction reliability for network pharmacology. PloS One. 8(12), e83922 (2013).
Sbidian, E. A multistep approach from scoping review to strong evidence: biomarkers in psoriasis. Br. J. Dermatol. ljac101 (2023).
Yadav, K. et al. Preclinical study models of psoriasis: state-of-the-art techniques for testing pharmaceutical products in animal and nonanimal models. Int. Immunopharmacol.117, 109945 (2023).
Albanesi, C. et al. The interplay between keratinocytes and immune cells in the pathogenesis of psoriasis. Front. Immunol.9, 1549 (2018).
Sevimoglu, T. & Arga, K. Y. The role of protein interaction networks in systems biomedicine. Comput. Struct. Biotechnol. J.11(18), 22–27 (2014).
Chen, J. et al. Neutrophils enhance cutaneous vascular dilation and permeability to aggravate psoriasis by releasing matrix metallopeptidase 9. J. Invest. Dermatology. 141(4), 787–799 (2021).
Zhou, Z. et al. Exploration of the potential mechanism of the common differentially expressed genes in psoriasis and atopic dermatitis. BioMed Research International (2022).
Buommino, E. et al. Modification of osteopontin and MMP-9 levels in patients with psoriasis on anti-TNF-α therapy. Arch. Dermatol. Res.304, 481–485 (2012).
Meng, X. et al. MAPK pathway involved in epidermal terminal differentiation of normal human epidermal keratinocytes. Open Med.13(1), 189–195 (2018).
Johansen, C. et al. The mitogen-activated protein kinases p38 and ERK1/2 are increased in lesional psoriatic skin. Br. J. Dermatol.152(1), 37–42 (2005).
Rioux, G. et al. Gene profiling of a 3D psoriatic skin model enriched in T cells: downregulation of PTPRM promotes keratinocyte proliferation through excessive ERK1/2 signaling. Cells. 11(18), 2904 (2022).
Winiarska-Mieczan, A., Mieczan, T. & Wójcik, G. Importance of redox equilibrium in the pathogenesis of psoriasis—impact of antioxidant-rich diet. Nutrients. 12(6), 1841 (2020).
Amen, N. et al. Differentiation of epidermal keratinocytes is dependent on glucosylceramide: ceramide processing. Hum. Mol. Genet.22(20), 4164–4179 (2013).
Obeid, L. M. et al. Programmed cell death induced by ceramide. Science. 259(5102), 1769–1771 (1993).
Robati, R. M. et al. Increased serum leptin and resistin levels and increased carotid intima-media wall thickness in patients with psoriasis: is psoriasis associated with atherosclerosis? J. Am. Acad. Dermatol.71(4), 642–648 (2014).
Villarreal-Martinez, A. et al. Mitochondrial dysfunction: the pathological link between psoriasis and insulin resistance? J. Eur. Acad. Dermatol. Venereol.37(2), 340–347 (2023).
Akhyani, M. et al. The lipid profile in psoriasis: a controlled study. J. Eur. Acad. Dermatol. Venereol.21(10), 1330–1332 (2007).
Su, W. et al. Exploring the pathogenesis of psoriasis complicated with atherosclerosis via microarray data analysis. Front. Immunol.12, 667690 (2021).
Lun, M. et al. Nuclear factor-kappab pathway as a therapeutic target in Head and Neck squamous cell carcinoma: Pharmaceutical and Molecular Validation in Human Cell Lines using Velcade and siRNA/NF-κB. Annals Clin. Lab. Sci.35(3), 251–258 (2005).
Allen, C. et al. Bortezomib-induced apoptosis with limited clinical response is accompanied by inhibition of canonical but not alternative nuclear factor-κB subunits in head and neck cancer. Clin. Cancer Res.14(13), 4175–4185 (2008).
Chen, X. et al. Bortezomib inhibits NLRP3 inflammasome activation and NF-κB pathway to reduce psoriatic inflammation. Biochem. Pharmacol.206, 115326 (2022).
Adil, M., Amin, S. S. & Mohtashim, M. N-acetylcysteine in dermatology. Indian J. Dermatol. Venereol. Leprol.84, 652 (2018).
Wang, J. et al.Potential of natural products in combination with arsenic trioxide: Investigating cardioprotective effects and mechanisms Vol. 162, 114464 (Biomedicine & Pharmacotherapy, 2023).
Tse, W. P. et al. Arsenic trioxide, arsenic pentoxide, and arsenic iodide inhibit human keratinocyte proliferation through the induction of apoptosis. J. Pharmacol. Exp. Ther.326(2), 388–394 (2008).
Binu, P. et al.Studies on curative efficacy of monoterpene eugenol on anti-leukemic drug arsenic trioxide induced cardiotoxicity Vol. 91, 559–566 (Biomedicine & Pharmacotherapy, 2017).
Gonçalves, E. C. et al. Terpenoids, cannabimimetic ligands, beyond the cannabis plant. Molecules. 25(7), 1567 (2020).
Zhai, B. et al. Drug delivery systems for elemene, its main active ingredient β-elemene, and its derivatives in cancer therapy. Int. J. Nanomed.13, 6279–6296 (2018).
Kumar, B. et al. Emerging therapeutic potential of curcumin in the management of dermatological diseases: an extensive review of drug and pharmacological activities. Future J. Pharm. Sci.9(1), 1–10 (2023).
Zhang, S. et al. Efficacy and safety of curcumin in psoriasis: preclinical and clinical evidence and possible mechanisms. Front. Pharmacol.13, 903160 (2022).
Mirzadeh, A. et al. Silico prediction, characterization, docking studies and molecular dynamics simulation of human p97 in complex with p37 cofactor. BMC Mol. Cell. Biology. 23(1), 1–12 (2022).
Acknowledgements
The authors gratefully acknowledge the support of the Skin Research Center, Shahid Beheshti University of Medical Sciences.
Funding
This study was supported by the Skin Research Center, Shahid Beheshti University of Medical Sciences, with grant number 43004529.
Author information
Authors and Affiliations
Contributions
M.F. carried out the scientific research, writing the main draft, data collection and analysis, visualization. R.M.R. provided conceptualization, supervision, project administration roles. M.R.T. provided conceptualization, supervision, project administration roles. F.F. performed data curation, visualization. S.S. provided data curation, visualization. M.R.R., Z.R., M.Z.A. and S.S. researched. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Declaration of competing interest
The authors have no competing interests to declare.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Farahani, M., Robati, R.M., Rezaei-Tavirani, M. et al. Integrating protein interaction and pathway crosstalk network reveals a promising therapeutic approach for psoriasis through apoptosis induction. Sci Rep 14, 22103 (2024). https://doi.org/10.1038/s41598-024-73746-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-73746-5
