
Abstract
Psoriasis is an immune-mediated chronic inflammatory disease. Increasing evidence suggests a close association between ribosome biogenesis (RiboSis) and the pathogenesis of psoriasis. However, the precise mechanisms remain unclear. We first obtained bulk transcriptome and single-cell RNA sequencing datasets from the GEO database. Subsequently, differential expression analysis (DEG) and weighted gene co-expression network analysis (WGCNA) were performed, preliminarily identifying 11 candidate biomarkers. Protein-protein interaction (PPI) analysis revealed that these biomarkers are primarily involved in protein synthesis, regulation of gene expression, and control of the cell cycle and growth. Consensus clustering analysis combined with immune infiltration analysis revealed that the candidate biomarkers were strongly associated with innate immune cells, such as NK cells, mast cells, and monocytes, and were more closely linked to signaling pathways related to cell proliferation, cell cycle, inflammation, and glycolysis. From the 11 candidate biomarkers, we selected MPHOSPH6 and ISG20 (exhibiting the highest fold-changes) for external dataset validation, scRNA-seq analysis, and in vivo expression verification. Subsequently, potential therapeutic compounds targeting these biomarkers were predicted and validated via molecular docking. Collectively, our findings not only substantiate the critical role of RiboSis in psoriasis pathogenesis but also provide a framework for developing targeted therapeutic strategies.
Similar content being viewed by others
Introduction
Psoriasis is a chronic relapsing disease with an average prevalence of 2–4%1, affecting over 125 million people worldwide2. The etiology of psoriasis is very complex and has a genetic background, with multiple factors such as drugs, environmental influences, trauma, and infections contributing to its onset3. The pathogenesis of psoriasis involves the activation of inflammatory pathways in innate and adaptive immune cells4, leading to uncontrolled proliferation of keratinocytes (KCs), hyperplasia of the stratum corneum, neovascularization, and intense infiltration of immune cells into the skin3. Psoriasis is commonly associated with various comorbidities, including psoriatic arthritis, cardiovascular diseases, metabolic syndrome, obesity, inflammatory bowel disease, and mental disorders related to systemic inflammation5. Psoriasis has a significant negative impact on patients’ physical and mental health, as well as a considerable economic burden, making it a major disease that needs urgent attention in dermatology. Therefore, a comprehensive understanding of the pathogenesis of psoriasis and the search for effective therapeutic agents are pressing issues.
Ribosomal biogenesis (RiboSis) is a phenotype that has been extensively studied recently, affecting cell proliferation, differentiation, and apoptosis6. Current research indicates that RiboSis is associated with tumors, viral infections, cardiovascular diseases, neurodegenerative diseases, aging, and hematological disorders7,8. RiboSis is a complex process that is precisely regulated by multiple signaling pathways. The mTORC1 pathway is one of the main pathways that regulate RiboSis, with mTORC1 modulating the synthesis and assembly of all ribosomal components through S6 kinases (S6 Ks) and eukaryotic translation initiation factor 4E-binding proteins (4EBPs)9. In psoriasis, the mTOR pathway is activated10, leading to the activation of downstream ribosomal protein S6, which exacerbates skin inflammation and epidermal proliferation11. This suggests a certain link between psoriasis and RiboSis; however, the mechanism of RiboSis in psoriasis remains unclear and requires further in-depth study.
In this study, we integrated bulk transcriptome and single-cell RNA sequencing data to investigate the potential role of RiboSis-related genes in psoriasis pathogenesis. Through integrated bioinformatics analysis, we systematically identified two pivotal RiboSis-associated biomarkers. These findings were subsequently validated using independent external datasets and in vivo animal models. Furthermore, we predicted potential therapeutic compounds targeting these key biomarkers.
Methods
Acquisition of psoriasis datasets and RiboSis-related genes
Skin bulk transcriptomic and single-cell transcriptomic datasets were sourced from the GEO database (https://www.ncbi.nlm.nih.gov/). In R (version 4.3.2), the “ComBat” function of the “sva” R package was used to eliminate batch effects on the GSE30999, GSE69967, and GSE14905 datasets12. The combined dataset was used for subsequent analysis. The GSE67853 dataset was used as the validation set to validate the key biomarkers. The merged dataset contained a total of 255 samples, including 130 lesional samples (LS) and 125 non-lesional samples (NL). The scRNA-seq dataset GSE151177 contained 6 normal tissue samples and 17 psoriatic tissue samples. Details of the datasets are shown in Table 1. The gene set for RiboSis was derived from a published study with 331 genes13.
Weighted co-expression network analysis
We performed WGCNA14 analysis to access the genes most associated with psoriasis traits. We selected the top 25% of genes with the highest variance from the merged dataset to serve as the input matrix. After removing outlier samples, we conducted topological calculations using a soft-thresholding power range from 1 to 20. Based on the optimal soft-thresholding power, we converted the correlation matrix to an adjacency matrix, and then to a Topology Overlap Matrix (TOM). Utilizing the TOM, we performed average linkage hierarchical clustering to categorize related modules, ensuring that each module contained at least 30 genes. Modules with a similarity above a cutoff height of 0.25 were merged. The correlation between the merged modules and the occurrence of psoriasis was calculated using the Pearson method, with the module exhibiting the highest correlation identified as the core module.
Differential expression analysis
In R (version 4.3.2), the “limma” package15 was utilized to perform differential gene (DEGs) analysis on the merged dataset. Genes with |logFC| > 1 and an adjusted p-value < 0.05 were selected as DEGs.
Protein-protein interaction (PPI) network analysis and functional enrichment analysis
We intersected the core module with the DEGs and RiboSis genes to obtain potential RiboSis-related candidate biomarkers. To elucidate the biological functions of the candidate biomarkers, we initially utilized the GeneMANIA16 online platform (http://genemania.org) to predict genes that are functionally similar or interact with the biomarkers, as well as the primary functional pathways they are involved in. Next, we used the “clusterProfiler”17 R package to perform Gene Ontology (GO) annotation analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis. GO analysis is a common method for large-scale functional enrichment studies18. KEGG is a widely used database that stores information related to genomes, biological pathways, diseases, and drugs19. The filtering criterion for the entries was set at P.adj < 0.05.
Consensus cluster analysis
We utilized the “ConsensuClusterPlus” package20 to conduct a subgroup analysis of disease samples based on candidate biomarkers, aiming to further elucidate the role of RiboSis in the pathogenesis of psoriasis. The parameters were set to use the “pam” clustering method and “Euclidean” distance, with 100 iterations. By comparing the PAC values at different K values, we aimed to determine the optimal number of clusters, thereby achieving relatively stable clustering results.
Immune cell infiltration analysis
We applied the CIBERSORT algorithm21 to perform an immune cell infiltration analysis on the two clusters (C1 and C2) derived from consistent clustering, to assess the expression abundance of 22 immune cell types within the two subgroups. The Wilcoxon test was employed to compare the differences in immune cells between the C1 and C2. The “corrplot” R package was utilized to calculate the correlation between candidate biomarkers and immune cells. We conducted Gene Set Variation Analysis (GSVA)22analysis on disease samples to understand the differences in functional pathways between the two clusters.
Validation of key biomarkers
Among the candidate biomarkers identified above, we selected the two genes with the highest fold-change as key biomarkers and subsequently validated them using external datasets. In the validation dataset GSE67853, we first assessed the expression levels of key biomarkers in LS and NL groups. Subsequently, we evaluated the diagnostic value of the key biomarkers using the area under the receiver operating characteristic (ROC) curve (AUC) with the “pROC” package.
Single-cell analysis
We analyzed the single-cell dataset GSE151177 using the “Seurat” package23. Highly variable genes underwent dimensionality reduction and cell clustering via Principal Component Analysis (PCA). To address batch effects, we applied the “Harmony” R package24. High-dimensional data were further reduced non-linearly using the Uniform Manifold Approximation and Projection (UMAP) algorithm. Cell annotation was performed using “SingleR”25 and cross-validated with established literature references26. Comparative analysis of cell type distribution ratios between disease and normal groups revealed differential expression of key biomarkers across distinct cell populations. Gene set scoring for key biomarkers was conducted using the “AddModuleScore” function27 in Seurat, which calculated the average expression of target genes, binned the expression matrix based on these averages, and derived scores by comparing target genes to 100 randomly selected background genes per bin. Intercellular communication was investigated using the “CellChat” package28, leveraging a ligand-receptor database to identify highly expressed genes, map ligand-receptor interactions, and quantify communication counts and strengths, thereby elucidating signaling dynamics across cell types.
Construction of a psoriasis mouse model
Six-week-old male BALB/c mice of SPF grade were sourced from Chengdu Yaokang Biotechnology Co., Ltd. These mice were housed in cages with adjustable temperatures and a 12-hour light-dark cycle, unrestricted water access, and a standard diet. The dorsal skin was topically treated with 5% imiquimod (IMQ) cream at a dose of 62.5 mg daily for 7 consecutive days. Control group mice received an equivalent amount of petrolatum daily. On the 8 th day, the mice were euthanized by cervical dislocation after anesthesia with 2% pentobarbital, and their dorsal skin was excised for further analysis.
The animal Ethics Committee of Sichuan Scientist Biotechnology Co., Ltd. (approval number: SYST-2024-003) approved all the experimental methodologies employed in this study, and all experiments were performed in compliance with ARRIVE guidelines (https://arriveguidelines.org). All methods were performed in accordance with the relevant guidelines and regulations.
Histopathological analysis
Skin tissues were fixed in 4% paraformaldehyde, dehydrated through a graded series of alcohol, and then embedded in paraffin for sectioning. The sections were deparaffinized in xylene, rehydrated through a graded series of alcohol, stained with hematoxylin and eosin, and finally mounted with xylene. Observations were made under a microscope.
Western blotting
Homogenize the skin tissue and add lysis buffer, followed by centrifugation to collect the supernatant. The protein concentration in the supernatant was determined using the BCA Protein Assay Kit (Biosharp, China). Total proteins were separated by SDS-PAGE (Biosharp, China) and then transferred onto a PVDF membrane (Merck Millipore, USA). After blocking with 5% skim milk for 1 h, the membrane was incubated with primary antibodies against MPHOSPH6 (1:1000, ABClonal, China), ISG20 (1:1000, ABClonal, China), and β-Actin (1:5000, Affinity Biosciences Inc. OH, United States) at 4 °C overnight. Subsequently, the membrane was incubated with horseradish peroxidase (HRP)-conjugated anti-rabbit or anti-mouse IgG secondary antibodies (1:10000; MULTI SCIENCES, China) at room temperature for 1 h. Chemiluminescence imaging analysis was performed using the ChemiScope 6100 system (Clinx, China), and image capture was conducted with ChemiScope Capture (Clinx, China).
Quantitative real‑time polymerase chain reaction (Q‑PCR) analysis
After homogenizing the tissue, total RNA was extracted using the Animal Total RNA Isolation Kit (Foregene, China), and the RNA concentration and purity were assessed using the K2800 Nucleic Acid Analyzer (KAIAO, China). Reverse transcription of RNA to cDNA was performed using the 5×All-In-One MasterMix (abm, Canada). The expression levels of the target genes were quantified in real-time using the StepOne Plus Real-Time PCR System (ABI, USA). GAPDH served as the internal control gene, and the reference sequence for the target gene is: MPHOSPH6: GAGCGCAAGACTAAGTTATCCAA(F), and TCTCCTTCAACTCCGGCAAAT(R). ISG20: TGGGCCTCAAAGGGTGAGT(F), and CGGGTCGGATGTACTTGTCATA (R). The relative changes in mRNA levels were calculated using the 2−ΔΔCt method, with the experiment repeated three times.
Predicting targeted drugs for key biomarkers
We utilized the Drug Signatures Database (DSigDB, http://dsigdb.tanlab.org/DSigDBv1.0/) to predict potential drugs targeting key biomarkers, which may hold significant implications for the treatment of psoriasis. The DSigDB currently encompasses 22,527 gene sets, comprising 17,389 distinct compounds that span 19,531 genes29.
Molecular Docking
We performed molecular docking on the identified biomarkers and corresponding predicted drugs to further explore the druggability of key biomarkers. The 3D structures of drugs were obtained from the PubChem Compound Database (https://pubchem.ncbi.nlm.nih.gov/), while the 3D structures of the protein molecules were obtained from the Protein Data Bank (http://www.rcsb.org/) and the AlphaFold Protein Structure Database30 (https://alphafold.com/). The molecular docking procedures were performed using the AutoDock TOOLs software (version 1.5.6), employing the Vina method. The protein molecules were prepared by removing any ligands and water molecules. Subsequently, polar hydrogen atoms were added to both the protein molecules and the compound ligands. The docking box was configured to include all structures of the protein molecules. Finally, Pymol software (version 2.4.0.)31 was utilized for visualization.
Statistical analysis
Statistical analysis was performed using R software (version 4.3.2) and Graphpad Prism 10.4 (Graphpad, Inc., CA, USA). Unpaired t-test and Wilcoxon rank sum test were used to assess the differences between the two groups. A p-value < 0.05 was considered statistically significant (* indicating P < 0.05, ** indicating P < 0.01, *** indicating P < 0.001, and **** indicating P < 0.0001).
Results
WGCNA and differential expression analysis
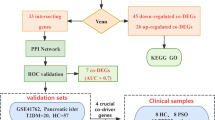
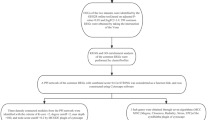
Figure 1 shows the flowchart of this study. We first normalized the three datasets to remove the batch effect (Supplementary Fig. S1). Differential analysis was performed on the combined dataset, and 1715 differentially expressed genes (DEGs) were obtained, including 980 up-regulated genes and 735 down-regulated genes. 2 outlier samples were eliminated from the WGCNA analysis by robust clustering (Supplementary Fig. S2), and topology calculations were carried out using a soft-threshold power ranging from 1 to 20. The optimal soft-threshold power was determined to be 6 (β = 6) (Fig. 2A). Using this soft threshold, the correlation matrix was converted to an adjacency matrix and then to a TOM. The average linkage hierarchical clustering of TOM was utilized to categorize the related gene modules, each containing at least 30 genes (Fig. 2B). Similar modules were merged, and finally 15 modules were obtained, among which the turquoise module was defined as the core module, which contained 1325 genes and showed the highest positive correlation with psoriasis (r = 0.9, p = 2e-94) (Fig. 2C). The intersection of WGCNA core module genes, differentially expressed genes (DEGs), and the RiboSis gene set revealed 11 overlapping genes (Fig. 2D). These 11 genes were identified as candidate RiboSis-ralated biomarkers, among which 10 genes were significantly upregulated in lesional tissues, while one gene was downregulated.

A flowchart of this study. WGCNA: Weighted Gene Co-Expression Network Analysis; PPI: protein-protein interaction; GSVA: Gene Set Variation Analysis.

Acquisition of candidate diagnostic biomarkers. (A) Analysis of scale-free fit indices and mean connectivity values for different soft-threshold powers (β) showed that β = 6 was an appropriate option. (B) The clustering dendrogram displays the modules formed by the co-expression network, with different colors representing different modules. (C) Module-trait relationship plot showing the correlation of different modules with LS and NL. (D) The Venn diagram shows that there are 11 common genes among RiboSis, DEGs, and core module genes. Red labels represent upregulated genes, while blue one indicates downregulated genes.
Functional enrichment analysis of candidate biomarkers
We utilized the GeneMANIA online platform to preliminarily assess the interactions among core genes and predict gene functions, discovering that these core genes are primarily involved in functions such as ribosome biogenesis, nuclease activity, rRNA metabolic processes, and ribonuclease activity (Fig. 3A). These pathways serve crucial biological functions, including protein synthesis, regulation of gene expression, control of the cell cycle and cell growth, as well as response to environmental stress and maintenance of intracellular homeostasis. KEGG enrichment analysis identified only two pathways: RNA degradation and Ribosome biogenesis in eukaryotes (Fig. 3B). GO enrichment analysis encompassed biological process (BP), molecular function (MF), and cellular component (CC). The BP analysis revealed that the core genes primarily regulate ribosome biogenesis, ribonucleoprotein complex biogenesis, rRNA metabolic process, rRNA processing, and RNA phosphodiester bond hydrolysis (Fig. 3B, C). In the CC category, these genes were predominantly located in preribosomes, large subunit precursors, ribonuclease MRP complex, multimeric ribonuclease P complex, and nuclear exosome (RNase complex) (Fig. 3D). The 11 candidate biomarkers were primarily involved in functions such as ribonuclease activity, nuclease activity, 3’−5’-exoribonuclease activity, exoribonuclease activity producing 5’-phosphomonoesters, and exoribonuclease activity (Fig. 3E).

Functional Enrichment Analysis. (A) Interactions and functions of the 11 candidate diagnostic biomarkers. The candidate biomarkers are in the inner ring, the predicted genes are in the outer ring, and the networks and functions formed between the genes are represented in different colors. (B) GO/KEGG enrichment analysis showing the top 5 items per category (enriched to 2 pathways in KEGG). (C) Network diagram of the candidate biomarkers with the top five items in BP. (D) Network diagram of the candidate biomarkers with the top five items in CC. (E) Network diagram of the candidate biomarkers with the top five items in MF.
Consensus cluster analysis and immune infiltration analysis
Consensus cluster analysis was performed on 130 disease samples based on 10 upregulated genes. According to the consensus matrix plot (Fig. 4A), the cumulative distribution function (CDF) curve (Fig. S3 A), and the relative change in the area under the CDF curve (Supplementary Fig. S3B), k = 2 was considered the optimal number of clusters, resulting in two disease subtypes: cluster 1 (C1) = 95 and cluster 2 (C2) = 35. The heatmap revealed high expression of candidate biomarkers in C1 (Fig. 4B). CIBERSORT analysis of C1 and C2 showed significant differences in the immune cell composition between the two subtypes, with activated NK cells (P = 0.043), monocytes (P = 0.041), and activated mast cells (P < 0.001) having a markedly increased proportion in C1. In contrast, CD8 T cells (P = 0.016), neutrophils (P = 0.046), and resting mast cells (P < 0.001) had an increased proportion in C2 (Fig. 4C).

The results of consensus clustering and immune infiltration analysis. (A) The consensus matrix plot shows that 2 clusters are clearly separated. (B) Heatmap of the expression of candidate biomarkers in C1 and C2. (C) Violin Plot of the expression levels of 22 immune cell types in C1 and C2. (D) Heatmap of the correlations between immune cells. (E) GSVA analysis results in C1 and C2.
We employed the “corrplot” package to calculate the correlations between immune cells, as well as between immune cells and candidate biomarkers. The candidate biomarkers all exhibited strong correlations with both activated mast cells and resting mast cells. Moreover, the upregulated biomarkers showed positive correlations with activated mast cells and negative correlations with resting mast cells. Conversely, the downregulated biomarker displayed the opposite pattern. Follicular helper T cells (Tfh), activated NK cells, monocytes, and activated memory CD4 T cells demonstrated positive correlations with the majority of candidate biomarkers. Regulatory T cells (Tregs), gamma delta T cells, resting NK cells, M1 macrophages, M2 macrophages, and resting dendritic cells (DCs) showed no significant correlations with RiboSis-associated candidate biomarkers (Fig. 4D). The results of GSVA analysis indicate that subgroup C1 is predominantly enriched in pathways associated with cell proliferation, cell cycle, inflammation, and glycolysis; whereas subgroup C2 is enriched in pathways primarily related to skeletal muscle generation, maintenance of tissue homeostasis, epithelial-mesenchymal transition, and apoptosis (Fig. 4E).
Validation in external dataset
Based on the magnitude of log2 fold-change (logFC), we identified MPHOSPH6 and ISG20 as key biomarkers (Supplementary Fig. S4). In the validation dataset GSE67853, the gene expression levels of MPHOSPH6 and ISG20 were significantly higher in LS compared to NL (p < 0.001) (Fig. 5A). Subsequently, we applied the “pROC” package to construct the ROC curves for MPHOSPH6 and ISG20 to evaluate their diagnostic efficacy. Both MPHOSPH6 (AUC 0.934, CI 0.848–1.000) and ISG20 (AUC 0.896, CI 0.758–1.000) exhibit a high diagnostic value (Fig. 5B).

Expression levels of MPHOSPH6 and ISG20 in the external validation dataset. (A) Violin plot of MPHOSPH6 and ISG20 expression levels in SL and NL in the GSE67853 dataset. (B) ROC curves of diagnostic efficacy for MPHOSPH6 and ISG20 in the GSE67853 dataset.
Single-cell analysis
GSE151177 comprises 6 normal tissue samples and 17 psoriatic tissue samples. Quality control was performed on all samples: each feature was expressed in at least 3 cells; each cell contained at least 200 features; the number of genes detected per cell ranged from 200 to 10,000; mitochondrial and ribosomal gene expression did not exceed 20%. As a result, 5 normal samples and 17 psoriatic samples passed the quality control, totaling 11,078 cells. Subsequently, the data were normalized, yielding 2,000 highly variable genes. PCA was used for dimensionality reduction of the high-variability genes and cell clustering. The UMAP algorithm was applied for nonlinear dimensionality reduction of high-dimensional data, with a resolution set at 0.5, resulting in 13 cell clusters. We combined “SingleR” and literature to annotate the cells, identifying 9 cell types. These include CD4+ T cells, CD8+ T cells, natural killer cells (NK), macrophages, DCs, endothelial cells, keratinocytes (KCs), fibroblasts, and melanocytes (Fig. 6A).

scRNA-seq analysis. (A) Cell clustering and annotation based on UMAP. (B) Proportion of 9 cell types across different samples. (C) Boxplot comparing the abundance of 9 cell types between psoriasis and normal groups. (D) Violin plot showing differences in gene set (MPHOSPH6 and ISG20) scores across 9 cell types. (E) UMAP plot showing expression of MPHOSPH6 and ISG20 in psoriasis and control groups. (F) Communication frequency and strength among nine cell types. (G) Cell-cell communication of KCs and CD8 + T cells, respectively.
The proportions of different cell types differ significantly between psoriasis and normal groups (Fig. 6B). In the disease group, there is an increase in CD8 + T cells, which is statistically significant compared to the normal group (P = 0.0034), while the proportion of CD4 + T cells decreases (P = 0.047) (Fig. 6C). Gene set scoring for MPHOSPH6 and ISG20 revealed elevated scores in KCs and CD8 + T cells (Supplementary Fig. S5). Specifically, KCs and CD8 + T cells exhibited higher scores, while CD4 + T cells and DCs showed lower scores, with a statistically significant difference observed between high- and low-scoring cell groups (Fig. 6D). Based on UMAP clustering, MPHOSPH6 was predominantly expressed in KCs and DCs, while ISG20 exhibited high expression levels in KCs, CD8 + T cells, macrophages, and DCs (Fig. 6E). Further intercellular communication analysis demonstrated that KCs interacted with all eight other cell types, exhibiting particularly strong communication with fibroblasts and endothelial cells. In contrast, CD8 + T cells showed no ligand-receptor communication patterns with CD4 + T cells or KCs but displayed moderate interactions with DCs and macrophages (Fig. 6F, G).
In vivo validation of key biomarker
In the IMQ-induced psoriasis mouse model, we validated the protein and mRNA levels of MPHOSPH6 and ISG20. Firstly, compared with the control group, the model group exhibited significant hyperkeratosis and parakeratosis in the stratum corneum, a reduction or disappearance of the granular layer, and irregular acanthosis of the spinous layer. In the dermal papillary layer, there was dilation of capillaries and many inflammatory cell infiltrations around the blood vessels (Fig. 7A, B). These histopathological changes indicate the successful establishment of the psoriasis model. Western blot analysis detected a significant increase in the protein levels of MPHOSPH6 and ISG20 in the model group (Fig. 7C). Similarly, qPCR results showed that the mRNA levels of MPHOSPH6 and ISG20 were elevated in the model group with p < 0.0001 (Fig. 7D).

Expression of key biomarkers in mouse models. Compared with the normal group (A), the skin histopathology of the model group (B) showed hyperkeratosis with hyperkeratosis, proliferation, and hypertrophy of the stratum spinosum, prolongation of the epidermal protuberance, dilation of capillaries in the superficial dermis, and many inflammatory cells were seen to be infiltrated (×200). The results of (C) WB and (D) qPCR showed that the expression of MPHOSPH6 and ISG20 was up-regulated in the model group (MPHOSPH6 and β-Actin were derived from different gels; however, the experimental samples and procedures were processed in parallel).
Candidate drug prediction and molecular Docking
We predicted the potential compounds targeting MPHOSPH6 and ISG20 in DSigDB and selected the top five compounds based on the Combined Score (Table 2). Among them, (+)-chelidonine, anisomycin, calcitriol, and acetaminophen are common targeted drugs for MPHOSPH6 and ISG20. We performed molecular docking for each protein-ligand pair to validate the binding of target proteins and drug molecules under natural conditions. The docking results are presented in Table 3. The results showed that most of the protein-ligand bindings were effective, and we visualized the protein-ligand pairs with a free binding energy< −5 kcal/mol. Figure 8 illustrates the hydrogen bonds formed and the corresponding protein residues involved.

Results of molecular docking. (A) Chelidonine with ISG20, (B) Chelidonine with MPHOSPH6, (C) 16,16-Dimethylprostaglandin with ISG20, (D) Anisomycin with ISG20, (E) Calcitrio1with ISG20, and (F) Calcitriol with MPHOSPH6.
Discussion
Ribosomes are primarily responsible for protein synthesis, and research has indicated that the rate of protein synthesis by ribosomes influences cellular functions, including proliferation, development, differentiation, apoptosis, and transformation behaviors7. Mature human ribosomes consist of two subunits: the small 40S subunit and the large 60S subunit32. Each subunit comprises a distinct set of proteins and ribosomal RNA (rRNA). The 60S subunit contains approximately 50 proteins and 5S, 5.8S, and 28S rRNA, while the 40S subunit includes about 30 proteins and 18S rRNA33,34. Eukaryotic ribosome biogenesis is a complex multistep process that encompasses the transcription of ribosomal DNA (rDNA), extensive processing of precursor ribosomal RNA (pre-rRNA) into mature rRNA, and the assembly of these molecules with ribosomal proteins34. Exosomes are complexes of 3’→5’ exoribonucleases, which participate in numerous RNA metabolic processes35. M-phase phosphoprotein 6 (MPP6), encoded by MPHOSPH636, is an essential component for the recruitment of the exosome to pre-rRNA and plays a role as a nucleolar-specific exosome accessory factor during the maturation of 5.8 S rRNA35. Current research on MPHOSPH6 is relatively scarce, but it has been linked to tumors37, coronary artery disease38, and IgA nephropathy36. MPHOSPH6 is highly expressed in KCs, which may contribute to the overproliferation of KCs in psoriasis.
ISG20 is an RNA exonuclease that has been studied for its broad-spectrum antiviral activity and is also involved in rRNA biogenesis39. ISG20 is implicated in the pathogenesis of various diseases, such as lupus nephritis, cancer, and acute kidney injury40,41,42. Additionally, ISG20 is involved in the activation and maturation of DCs, which may be one of the mechanisms by which it modulates innate immune responses43. Single-cell analysis revealed that ISG20 is highly expressed in DCs, a finding that strengthens the link between ISG20 and DCs. Immune infiltration analysis indicated a significant positive correlation between MPHOSPH6 and ISG20 with monocytes and NK cells. However, single-cell analysis showed that MPHOSPH6 and ISG20 are scarcely expressed in NK cells but are highly expressed in DCs, suggesting that MPHOSPH6 and ISG20 may promote psoriasis by modulating the function of DCs.
RiboSis and psoriasis are regulated by common signaling pathways. RiboSis is primarily modulated by the mammalian target of rapamycin (mTOR) and the myelocytomatosis viral oncogene (Myc)7. One form of the mTOR complex, mTORC1, regulates the synthesis and assembly of all ribosomal components through S6 kinases (S6 Ks) and eukaryotic translation initiation factor 4E-binding proteins (4EBPs)9. Research has found that the mTOR pathway is activated in psoriatic lesions10, and the treatment of psoriasis with drugs targeting mTOR-related pathways has made certain progress44,45,46. A recent study has discovered that ribosomes play a significant role in the pathogenesis of psoriasis, and in vitro experiments have confirmed that inhibiting ribosome biogenesis can suppress the proliferation of IL17 A-stimulated HaCaT cells and downregulate inflammatory factors47. Myc is an oncogene that participates in abnormal ribosome biogenesis48, primarily promoting rDNA transcription, regulating rRNA processing and transcription, as well as the translation of ribosomal proteins49,50. In psoriatic keratinocytes, increased expression of MYC has been observed51,52, and inhibition of c-Myc can downregulate the expression of pro-inflammatory cytokines in keratinocytes53.
We conducted drug predictions for MPHOSPH6 and ISG20, identifying several compounds with potential for treating psoriasis. Notably, chelidonine targets both MPHOSPH6 and ISG20, with molecular docking results indicating a strong binding affinity to these proteins. Chelidonine, an important alkaloid extracted from Chelidonium majus, possesses a range of pharmacological activities. This study reveals its potential anti-psoriatic effects, likely mediated through the modulation of ribosome biogenesis. Beyond this, chelidonine also demonstrates anti-inflammatory, pro-apoptotic, immunomodulatory, antiviral, and anticancer properties54,55,56, underscoring its multifaceted therapeutic potential. Calcitriol, the biologically active form of vitamin D3, is a well-established therapeutic agent for psoriasis57. While its anti-psoriatic effects were previously attributed to the activation of the vitamin D receptor (VDR)58,59, this study unveils a novel mechanism by which calcitriol exerts its therapeutic action. Specifically, we demonstrate that calcitriol targets MPHOSPH6 and ISG20, modulating RiboSis to control psoriasis progression, thereby expanding our understanding of its molecular mechanisms in psoriasis treatment.
However, our study has several limitations. Although the identified genes demonstrated associations with psoriasis in external datasets and mouse models, their causal relationship with psoriasis and functional roles require further experimental validation. Further investigation is needed to elucidate the precise mechanisms by which MPHOSPH6 and ISG20 contribute to psoriasis pathogenesis, which will be a key focus of our future research. Additionally, while we performed drug prediction and molecular docking analyses for these genes, these results are preliminary and require experimental validation to confirm their therapeutic potential. Finally, our study integrated multiple publicly available psoriasis datasets, which exhibited inherent heterogeneity in sample sizes and clinical characteristics. Although we mitigated batch effects using ComBat, residual technical biases may persist. Moreover, differences in patient demographics (e.g., age range, treatment history) across datasets could confound the observed associations. Thus, future validation using large-scale, well-characterized clinical cohorts is warranted.
Conclusion
We leveraged bulk transcriptome and single-cell RNA sequencing data, combined with bioinformatics approaches, and animal models, to identify MPHOSPH6 and ISG20 as key RiboSis-related biomarkers. MPHOSPH6 and ISG20 were predominantly expressed in KCs, DCs, and CD8 + T cells, suggesting that RiboSis likely contributes to psoriasis progression by promoting KC proliferation and enhancing the immune activity of both innate immune cells (DCs) and adaptive immune cells (CD8 + T cells).
Data availability
The microarray data supporting the findings of this study are accessible from the GSE30999, GSE69967, GSE14905, GSE67853, and GSE151177 datasets (https://www.ncbi.nlm.nih.gov/geo). Processed data can be obtained from the corresponding author upon request.
References
Pezzolo, E. & Naldi, L. Epidemiology of major chronic inflammatory immune-related skin diseases in 2019. Expert Rev. Clin. Immunol. 16, 155–166 (2020).
Armstrong, A. W., Read, C. & Pathophysiology Clinical presentation, and treatment of psoriasis: A review. JAMA 323, 1945–1960 (2020).
Rendon, A. & Schäkel, K. Psoriasis pathogenesis and treatment. Int. J. Mol. Sci. 20(6), 1475 (2019).
Sieminska, I., Pieniawska, M. & Grzywa, T. M. The immunology of Psoriasis-Current concepts in pathogenesis. Clin. Rev. Allergy Immunol. 66, 164–191 (2024).
Takeshita, J. et al. Psoriasis and comorbid diseases: epidemiology. J. Am. Acad. Dermatol. 76, 377–390 (2017).
Kang, J. et al. Ribosomal proteins and human diseases: molecular mechanisms and targeted therapy. Signal. Transduct. Target. Ther. 6, 323 (2021).
Jiao, L. et al. Ribosome biogenesis in disease: new players and therapeutic targets. Signal. Transduct. Target. Ther. 8, 15 (2023).
Pelletier, J., Thomas, G. & Volarević, S. Ribosome biogenesis in cancer: new players and therapeutic avenues. Nat. Rev. Cancer. 18, 51–63 (2018).
Iadevaia, V., Liu, R. & Proud, C. G. mTORC1 signaling controls multiple steps in ribosome biogenesis. Semin Cell. Dev. Biol. 36, 113–120 (2014).
Buerger, C., Malisiewicz, B., Eiser, A., Hardt, K. & Boehncke, W. H. Mammalian target of Rapamycin and its downstream signalling components are activated in psoriatic skin. Br. J. Dermatol. 169, 156–159 (2013).
Ruf, M. T., Andreoli, A., Itin, P., Pluschke, G. & Schmid, P. Ribosomal protein S6 is hyperactivated and differentially phosphorylated in epidermal lesions of patients with psoriasis and atopic dermatitis. Br. J. Dermatol. 171, 1533–1536 (2014).
Varma, S. Blind Estimation and correction of microarray batch effect. PLoS One. 15, e0231446 (2020).
Zang, Y. et al. Genomic hallmarks and therapeutic targets of ribosome biogenesis in cancer. Brief Bioinform. 25(2), bbae023 (2024).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 9, 559 (2008).
Ritchie, M. E. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Franz, M. et al. GeneMANIA update 2018. Nucleic Acids Res. 46, W60–W64 (2018).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. ClusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012).
Yu, G. Gene ontology semantic similarity analysis using gosemsim. Methods Mol. Biol. 2117, 207–215 (2020).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Wilkerson, M. D. & Hayes, D. N. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics 26, 1572–1573 (2010).
Chen, B., Khodadoust, M. S., Liu, C. L., Newman, A. M. & Alizadeh, A. A. Profiling tumor infiltrating immune cells with CIBERSORT. Methods Mol. Biol. 1711, 243–259 (2018).
Hänzelmann, S., Castelo, R. & Guinney, J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 14, 7 (2013).
Butler, A., Hoffman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018).
Cao, G. et al. Single-cell RNA sequencing reveals the vascular smooth muscle cell phenotypic landscape in aortic aneurysm. Cell. Commun. Signal. 21, 113 (2023).
Aran, D. et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 20, 163–172 (2019).
Kim, J. et al. Single-cell transcriptomics applied to emigrating cells from psoriasis elucidate pathogenic versus regulatory immune cell subsets. J. Allergy Clin. Immunol. 148, 1281–1292 (2021).
Liu, J., Shi, Y. & Zhang, Y. Multi-omics identification of an Immunogenic cell death-related signature for clear cell renal cell carcinoma in the context of 3P medicine and based on a 101-combination machine learning computational framework. Epma J. 14, 275–305 (2023).
Almet, A. A., Cang, Z., Jin, S. & Nie, Q. The landscape of cell-cell communication through single-cell transcriptomics. Curr. Opin. Syst. Biol. 26, 12–23 (2021).
Yoo, M. et al. DSigDB: drug signatures database for gene set analysis. Bioinformatics 31, 3069–3071 (2015).
Varadi, M. et al. AlphaFold protein structure database in 2024: providing structure coverage for over 214 million protein sequences. Nucleic Acids Res. 52, D368–d375 (2024).
Lill, M. A. & Danielson, M. L. Computer-aided drug design platform using PyMOL. J. Comput. Aided Mol. Des. 25, 13–19 (2011).
Khoshnevis, S. et al. RNA 2’-O-methylations regulate translation by impacting ribosome dynamics. Proc. Natl. Acad. Sci. U. S. A. 119, e2117334119, (2022).
Anger, A. M. et al. Structures of the human and Drosophila 80S ribosome. Nature 497, 80–85 (2013).
Tomecki, R., Sikorski, P. J. & Zakrzewska-Placzek, M. Comparison of preribosomal RNA processing pathways in yeast, plant and human cells - focus on coordinated action of endo- and exoribonucleases. FEBS Lett. 591, 1801–1850 (2017).
Schilders, G., Raijmakers, R., Raats, J. M. & Pruijn, G. J. MPP6 is an exosome-associated RNA-binding protein involved in 5.8S rRNA maturation. Nucleic Acids Res. 33, 6795–6804 (2005).
Yang, X. et al. Association between MPHOSPH6 gene polymorphisms and IgA nephropathy risk in a Chinese Han population. Oncotarget 8, 72375–72380 (2017).
Zhang, Y., Wang, S., Wen, X., Zhang, S. & Yang, Y. Association of ACYP2 and MPHOSPH6 genetic polymorphisms with the risk of hepatocellular carcinoma in chronic hepatitis B virus carriers. Oncotarget 8, 86011–86019 (2017).
Song, Y. et al. Association between TNIP1, MPHOSPH6 and ZNF208 genetic polymorphisms and the coronary artery disease risk in Chinese Han population. Oncotarget 8, 77233–77240 (2017).
Alsheikh, H. A. M. et al. Disruption of STAT5A and NMI signaling axis leads to ISG20-driven metastatic mammary tumors. Oncogenesis 10, 45 (2021).
Chen, Z., Yin, M., Jia, H., Chen, Q. & Zhang, H. ISG20 stimulates anti-tumor immunity via a double-stranded RNA-induced interferon response in ovarian cancer. Front. Immunol. 14, 1176103 (2023).
Jia, M. et al. Targeting RNA oxidation by ISG20-mediated degradation is a potential therapeutic strategy for acute kidney injury. Mol. Ther. 31, 3034–3051 (2023).
Karasawa, T. et al. Expression of interferon-stimulated gene 20 (ISG20), an antiviral effector protein, in glomerular endothelial cells: possible involvement of ISG20 in lupus nephritis. Ren. Fail. 45, 2224890 (2023).
Hashimoto, S. I. et al. Identification of genes specifically expressed in human activated and mature dendritic cells through serial analysis of gene expression. Blood 96, 2206–2214 (2000).
Reitamo, S. et al. Efficacy of sirolimus (rapamycin) administered concomitantly with a subtherapeutic dose of cyclosporin in the treatment of severe psoriasis: a randomized controlled trial. Br. J. Dermatol. 145, 438–445 (2001).
Ormerod, A. D., Shah, S. A., Copeland, P., Omar, G. & Winfield, A. Treatment of psoriasis with topical sirolimus: preclinical development and a randomized, double-blind trial. Br. J. Dermatol. 152, 758–764 (2005).
Datta Mitra, A. et al. 1α,25-Dihydroxyvitamin-D3-3-bromoacetate regulates AKT/mTOR signaling cascades: a therapeutic agent for psoriasis. J. Invest. Dermatol. 133, 1556–1564 (2013).
Liu, S. et al. Identification of potential biomarkers and infiltrating immune cells from scalp psoriasis. Gene 893, 147918 (2024).
Popay, T. M. et al. MYC regulates ribosome biogenesis and mitochondrial gene expression programs through its interaction with host cell factor-1. Elife 10, (2021).
Morcelle, C. et al. Oncogenic MYC induces the impaired ribosome biogenesis checkpoint and stabilizes p53 independent of increased ribosome content. Cancer Res. 79, 4348–4359 (2019).
Destefanis, F., Manara, V. & Bellosta, P. Myc as a regulator of ribosome biogenesis and cell competition: A link to Cancer. Int. J. Mol. Sci. 21(11), 4037 (2020).
Elder, L. T. et al. Growth factor and proto-oncogene expression in psoriasis. J. Invest. Dermatol. 95, 7s–9s (1990).
Osterland, C. K., Wilkinson, R. D. & St Louis, E. A. Expression of c-myc protein in skin and synovium in psoriasis and psoriatic arthritis. Clin. Exp. Rheumatol. 8, 145–150 (1990).
Liu, G. et al. Phenformin Down-Regulates c-Myc Expression to Suppress the Expression of Pro-Inflammatory Cytokines in Keratinocytes. Cells 11, (2022).
Jang, H. J. et al. Chelidonine induces apoptosis via GADD45a-p53 regulation in human pancreatic Cancer cells. Integr. Cancer Ther. 20, 15347354211006191 (2021).
Liao, W., He, X., Yi, Z., Xiang, W. & Ding, Y. Chelidonine suppresses LPS-Induced production of inflammatory mediators through the inhibitory of the TLR4/NF-κB signaling pathway in RAW264.7 macrophages. Biomed. Pharmacother. 107, 1151–1159 (2018).
Herrmann, R., Roller, J., Polednik, C. & Schmidt, M. Effect of chelidonine on growth, invasion, angiogenesis and gene expression in head and neck cancer cell lines. Oncol. Lett. 16, 3108–3116 (2018).
Abramovits, W. Calcitriol 3 Microg/g ointment: an effective and safe addition to the armamentarium in topical psoriasis therapy. J. Drugs Dermatol. 8, s17–22 (2009).
Barrea, L. et al. Vitamin D and its role in psoriasis: an overview of the dermatologist and nutritionist. Rev. Endocr. Metab. Disord. 18, 195–205 (2017).
van de Kerkhof, P. C. Biological activity of vitamin D analogues in the skin, with special reference to antipsoriatic mechanisms. Br. J. Dermatol. 132, 675–682 (1995).
Acknowledgements
We are grateful to the GEO database for providing its platforms and to its contributors for uploading valuable datasets.
Funding
This study was supported by the Special Research Project on Traditional Chinese Medicine, Sichuan Provincial Administration of TCM (2023MS390), and Chengdu University of Traditional Chinese Medicine’s “Xinglin” Scholar Academic Talent Program (CCZD2022005).
Author information
Authors and Affiliations
Contributions
J.L. and W.M. conceptualized and designed the study. J.L. and Z.Q. were responsible for statistical analysis and writing the original draft. X. Z. and Z. C. were responsible for visualization and editing the draft. J.L. and N. L. developed the methodology. P. H. was responsible for supervision and acquiring funding. C.L. was responsible for project administration and reviewing. All authors reviewed and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
The animal experimental procedures were approved by the Animal Ethics Committee of Sichuan Scientist Biotechnology Co., Ltd. (approval number: SYST-2024-003). The animal experiments described in this study were performed in compliance with the ARRIVE guidelines (https://arriveguidelines.org). The publicly available human data applied in this study did not require ethical approval, as ethical approval and participant consent were obtained in the original study.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, J., Luo, N., Ma, W. et al. Novel ribosome biogenesis-related biomarkers and therapeutic targets identified in psoriasis. Sci Rep 15, 18525 (2025). https://doi.org/10.1038/s41598-025-03833-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-03833-8
