
Abstract
Idiopathic pulmonary arterial hypertension (IPAH), a rare and devastating pulmonary vascular disorder, is characterized by cellular proliferation and vascular remodeling. Although previous studies have underscored that ferroptosis, an iron-dependent cell death process, plays an important regulatory role in pulmonary artery hypertension, its role remains understudied. Gene expression profiles were downloaded from the Gene Expression Omnibus(GEO). Differentially expressed genes (DEGs) were screened using R software and intersected with a ferroptosis database (FerrDb V1) to identify ferroptosis-related DEGs. GO and KEGG analyses were performed to explore biological functions and potential pathways. LASSO and SVM-RFE algorithms were used to identify optimal gene biomarkers for IPAH. GSVA and GSEA were conducted to explore biological functions and potential pathways associated with these biomarkers. The CIBESORT software was employed to predict immune genes and functions. Of 237 ferroptosis-related genes (FRGs), 27 differentially expressed FRGs (DE-FRGs) showed significant differences between IPAH and normal samples in GSE48149, with 15 downregulated and 12 upregulated genes. Six DE-FRGs, including KEAP1, TNFAIP3, MEG3, NFS1, PRDX1, and BEX1, were identified as predictive diagnostic genes for IPAH. Among these DE-FRGs, PRDX1 and TNFAIP3 were the most promising diagnostic genes for IPAH and may play a corresponding role in IPAH by participating in the cell cycle, lysosomes, immune response, vascular smooth muscle contraction, and various diseases. CIBERSORT analysis revealed a positive correlation between neutrophils and TNFAIP3, whereas macrophages M0 exhibited a negative correlation with PRDX1. Through comprehensive bioinformatics analysis, we identified six differentially expressed ferroptosis-related genes (DE-FRGs)—KEAP1, TNFAIP3, MEG3, NFS1, PRDX1, and BEX1—in idiopathic pulmonary arterial hypertension (IPAH). Among these, PRDX1 and TNFAIP3, which play key roles in IPAH pathogenesis, emerged as the most promising diagnostic biomarkers.
Similar content being viewed by others
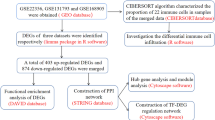

Introduction
Idiopathic pulmonary arterial hypertension (IPAH) is a rare and devastating pulmonary vascular disorder characterized by persistently elevated pulmonary artery pressure, often progressing to right ventricular hypertrophy and heart failure, and it has a relatively low estimated prevalence of 5.9 cases per 1 million individuals1,2. Despite treatment progress, the prognosis of newly diagnosed patients with IPAH remains suboptimal, with 5- and 10-year transplant-free survival rates of 71% and 35%, respectively3. IPAH exhibits intricate pathophysiological alterations driven by genetic, epigenetic, and environmental factors, which result in proliferative remodeling, endothelial dysfunction, fibrosis, inflammation, and cancer-like attributes4,5,6. Although mutations in genes such as BMPR2, ATP13A3, AQP1, ABCC8, KCNK3, SMAD9, Sox17, CAV1, TBX4, and KDR have been established as biomarkers of IPAH, the exact pathogenesis of this disease remains unclear7. Whereas the principles of predictive, preventive, and personalized medicine (PPPM) have been successfully applied in oncology to screen for cancer biomarkers and identify therapeutic targets through genome sequencing8, their application in the realm of IPAH is conspicuously lacking in terms of early prediction and preventative strategies. In fact, PPPM has the potential to expedite diagnosis, alleviate emotional uncertainty in patients, reduce the burden on healthcare resources, and enable treatment initiation at an earlier stage, when interventions are typically more effective9. Therefore, the expedient identification of robust molecular markers is essential for improving the treatment and diagnosis of IPAH.
Iron deficiency is a common occurrence in 43–63% of patients with IPAH and is associated with decreased exercise tolerance and elevated mortality rates10,11, indicating that iron metabolism is crucial in IPAH. Ferroptosis, a regulator of cell death characterized by iron-dependent lipid peroxidation and oxidative stress, is involved in various pathological conditions, including drug-induced liver damage, acute kidney injury, cancer, neurodegenerative disorders, and cardiovascular disease12. Previous research has suggested that ferroptosis is activated and contributes to pulmonary artery hypertension (PAH)13. Dai et al. showed that ferroptosis in pulmonary artery endothelial cells, mediated by peroxiredoxin 6 (PRDX6), plays a role in the development of monocrotaline-induced pulmonary hypertension (MCT)14. For pulmonary artery endothelial ferroptosis, in which the NLRP3 inflammasome is activated via the HMGB1/TLR4 pathway in MCT-affected rats, Lan et al. suggested that a ferroptosis inhibitor could be a novel therapeutic target for PAH15. Moreover, Sun et al. showed that increased levels of NDUFA4L2 are crucial in advancing hypoxia-induced pulmonary hypertension by promoting ROS production and pulmonary arterial smooth muscle cell (PASMC) proliferation, suggesting that targeting NDUFA4L2 could be a promising novel therapeutic approach for PAH16. Furthermore, Hu et al. demonstrated that SLC7A11 suppresses ferroptosis and enhances proliferation in PAH, effectively rebalancing the dynamics between cell death and proliferation in PASMCs13. Additionally, bioinformatic evidence suggests the coexistence of both activation and silencing of ferroptosis in PAH17. In fact, existing studies present some paradoxical phenomena, and the specific mechanism by which ferroptosis aggravates PAH remains unknown18, emphasizing the need for further research in this area.
We thus hypothesize a robust association between ferroptosis-related genes (FRGs) and IPAH, encompassing its intricate immune microenvironment. Our research elucidated the pivotal functional roles of FRGs in accurately predicting IPAH prognosis, providing novel insights into the mechanisms underlying immune regulation within the IPAH patient cohort. The exploration of FRGs as prospective biomarkers for IPAH has the potential to empower high-risk subpopulations with the means to modulate their prognosis via personalized medical interventions firmly situated within the paradigm of PPPM.
Experimental procedure
Data source
We downloaded gene expression profiles and FRGs from the Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/geo/) and the FerrDb online database (FerrDbV1, 2020) (http://www.zhounan.org/ferrdb/), respectively19. GSE48149, consisting of eight IPAH and nine healthy tissue samples, was designated as the training set. Validation sets, including GSE117261 (31 IPAH and 25 healthy tissue samples), GSE84395 (11 IPAH and 18 healthy pulmonary endothelial cells [PEC] samples), GSE33463 (30 IPAH and 41 healthy peripheral blood mononuclear cell [PBMCs] samples), and GSE169471 (3 IPAH and 8 healthy tissue samples), were used to confirm marker gene levels. The Drug Gene Interaction Database (DGIdb, https://www.dgidb.org), and Cytoscape software were used for predicting potential drugs or molecular compounds interacting with marker genes and visualizing the drug–gene interaction network, respectively20,21.
Identification of differentially expressed genes (DEGs)
The expression profiles of 237 FRGs in normal and IPAH samples were extracted from the GSE48149 database. Subsequently, the “limma package” in R software (R version 4.2.2) was employed to identify significant DE-FRGs between the two groups using parameters |Log2fold change| >0.5 and adj.P < 0.05 (Table 1)22. The “pheatmap and ggplot2 packages” were used for constructing the heatmap of DEGs, and the “Corrplot package” was used for analyzing the correlation between DE-FRGs, with p < 0.05 considered significant23.
Functional enrichment analysis
The “clusterProfiler package” was employed to perform the Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses of DE-FRGs. GO analysis explored biological functions in three categories: biological process (BP), cellular component (CC), and molecular function (MF). Simultaneously, KEGG analysis was used to explore potential pathways24,25,26.
Identification of optimal gene biomarkers for IPAH
The “glmnet package” and “e1071 package” were used to identify IPAH-related gene biomarkers using the least absolute shrinkage and selection operator (LASSO) and support vector machine-recursive feature elimination (SVM‐RFE) algorithms, respectively. The “Jvenn package” was employed to construct the Venn diagram. Overlapping biomarkers are considered the most favorable for IPAH27. Receiver Operating Characteristic (ROC) curves, Area Under the Curve (AUC), sensitivity, specificity, and accuracy were calculated to evaluate the diagnostic ability of the selected gene markers. The “glm package” was used to construct a logistic regression model based on the six marker genes to predict GSE48149 dataset sample types and test the diagnostic value of the model using ROC curves28.
Single-gene gene set enrichment analysis (GSEA)
The “GSEA package” was employed to explore pathways or biological functions enriched by those six marker genes. After calculating the associations between the marker genes and all other genes in the GSE48149 dataset, all genes were sorted according to their high-to-low correlations and listed in the gene set for analysis. Meanwhile, the KEGG or Go set was invoked as a predefined set to detect its enrichment in the gene set.
Single-gene gene set variation analysis (GSVA)
The GSVA (version 1.38.0) package in R was used for this analysis29. The KEGG pathway set was adopted as the background gene set for the GSVA of diverse marker genes. Simultaneously, the “limma package” was used to analyze the difference in GSVA score of the marker gene’s high- and low-expression group samples based on the significance levels of |t| >2 and p < 0.05. The pathway was activated in the high-expression group when t > 0; otherwise, it was activated in the low-risk group.
Immune infiltration analysis
CIBERSORT characterizes the cellular composition of complex tissues based on gene expression data30. The GSE48149 dataset was analyzed using CIBERSORT software to estimate the proportions of 22 types of infiltrating immune cells. Additionally, immune genes and functions in each tissue were predicted using CIBESORT software.
mRNA–miRNA–lncRNA CeRNA network construction
The construction of a ceRNA network involves numerous lncRNAs, miRNAs, and mRNAs, along with their interactions. We predicted the binding of mRNA–miRNA nucleic acids using TargetScan, miRDB, and miRanda databases31,32,33. Furthermore, miRNA–lncRNA binding interactions were identified using SpongeScan34 to obtain the mRNA–miRNA–lncRNA ceRNA network.
Validation of marker genes in a single dataset
Single-cell RNA sequencing (scRNA-seq) data analysis
scRNA-seq data, including three IPAH samples and eight control samples, were downloaded from the GEO database (GSE169471). The UMI count matrix was imported into R to generate Seurat objects using the “Seurat package.” Genes detected in fewer than three cells and cells with fewer than 200 genes were excluded after quality control. The SCTransform function was applied for data normalization, variable feature detection, and data scaling. Subsequently, the FindIntegrationAnchors and IntegrateData functions were used to identify anchors for dataset integration, eliminating batch effects between samples. Principal component analysis was conducted for dimensionality reduction using the RunPCA function, and the top 30 principal components were selected to construct a common nearest-neighbor graph in low-dimensional subspaces. Cell clustering was executed with the FindClusters function, setting the resolution to 0.4, and nonlinear dimensionality reduction was performed using the RunUMAP function.
Cell type identification and gene expression analysis
Cell types were annotated based on the expression of canonical gene markers in the lung tissue from previously published articles and assigned to the corresponding clusters. Expression analysis of target genes was presented as dot, violin, box, and feature plots using functions from the Seurat and ggplot2 packages.
Statistical analysis
All statistical analyses were performed using R software (V4.2.2). The Wilcoxon rank-sum test was used to compare the two groups, and Pearson’s correlation analysis was used to evaluate the relationship between the DE-FRGs. Statistical significance was set at p < 0.05.
Results
Identification of DE-FRGs from GSE48149
Of 237 FRGs, 27 DE-FRGs exhibited a significant difference between IPAH and normal samples in GSE48149, with 15 downregulated and 12 upregulated genes (Table 1), visualized using a heatmap (Fig. 1A). The correlation plots demonstrated an association among DE-FRGs, with no correlation between ADAM23 and SRC and any other DE-FRGs (Fig. 1B).

Identification of DE-FRGs from the GSE48149. (A) Visualization of 15 downregulated and 12 upregulated genes between IPAH and normal samples using a heatmap. (B) Associations between DE-FRGs, with no correlation between ADAM23 and SRC and any other DE-FRGs. C and D. GO enrichment and Reactome pathway analyses were employed to elucidate DE-FRG-associated biological functions and pathways. E and F. Reactome pathway analyses revealed remarkable enrichment in fluid shear stress, atherosclerosis, hepatocellular carcinoma, and chemical carcinogenesis reactive oxygen species.
Functional annotation for DE-FRGs
GO enrichment and Reactome pathway analyses were conducted to elucidate the biological functions and pathways associated with DE-FRGs. GO enrichment analyses revealed significant associations of DE-FRGs with CC such as “focal adhesion,” “cell–substrate junction,” and “membrane raft.” BP annotation indicated a notable relation of DE-FRGs with “cellular response to chemical stress,” “cellular response to peptidyl-serine phosphorylation,” and “peptidyl-serine modification.” Additionally, MF annotation suggested a close association between DE‐FRGs and “RNA polymerase II-specific DNA-binding transcription factor binding,” “DNA-binding transcription factor binding,” and “phosphoprotein binding”(Fig. 1C and D). Reactome pathway analyses revealed marked enrichment in pathways such as fluid shear stress, atherosclerosis, hepatocellular carcinoma, and chemical carcinogenesis reactive oxygen species (Fig. 1E and F). Based on this evidence, DE‐FRGs may play a crucial role in IPAH by participating in the regulation of autophagy, cytokines, kinases, and immune cells.
Identification of six DE-FRGs as diagnostic genes for IPAH
To assess the potential utility of predicted DE-FRGs as diagnostic genes for IPAH, we explored variations between patients with IPAH and healthy individuals. Initially, two machine learning algorithms (LASSO and SVM-RFE) were employed to identify significant DE-FRGs capable of distinguishing patients with IPAH from normal individuals in the GSE48149 dataset. Using LASSO logistic regression with penalty parameter tuning through 10-fold cross-validation, eight IPAH-related features were selected. The SVM-RFE algorithm further filtered 27 DE‐FRGs to identify the optimal gene combination, resulting in the identification of 13 genes (minimal RMSE = 0 and maximal accuracy = 1) as the best feature genes. Intersection of marker genes from LASSO and SVM-RFE models led to the identification of six DE‐FRGs—KEAP1, TNFAIP3, MEG3, NFS1, PRDX1, and BEX1—as potential diagnostic genes for IPAH. These genes were then used in subsequent analyses. A logistic regression model, implemented using the R “glnn package” based on the six marker genes, achieved an AUC value of 1, indicating perfect discriminatory ability. Subsequently, an ROC curve was generated to illustrate the efficacy of the six marker genes in distinguishing patients with IPAH from normal individuals, with AUC values exceeding 0.8 for all genes. Consequently, our logistic regression model demonstrated superior accuracy and specificity compared to individual marker genes. Further analysis revealed distinct expression patterns in IPAH tissues relative to normal tissues, with decreased expression levels of KEAP1 and PRDX1 and increased expression of BEX1, MEG3, NFS1, and TNFAIP3 in IPAH tissues (Fig. 2).

LASSO and SVM-RFE were used to screen for significant DE-FRGs to distinguish patients with IPAH from normal individuals in the GSE48149 dataset. A and B. Eight IPAH-related features were selected using LASSO logistic regression with penalty parameter tuning by 10-fold cross-validation. C and D. Twenty-seven DE-FRGs were filtered to identify the best feature gene combination using the SVM‐RFE algorithm. Finally, 13 genes (minimal RMSE = 0 and maximal accuracy = 1) were identified as the best feature genes. E. Marker genes were obtained from the LASSO and SVM-RFE models. F. A logistic regression model was used to identify the AUC of the disease samples. G. ROC curves for the six marker genes.
Marker gene associations with IPAH-related pathways
We utilized single-gene GSEA-KEGG pathway analysis to explore the potential roles of marker genes in distinguishing patients with IPAH from normal controls, highlighting six pathways linked to these markers. Significantly, the marker genes demonstrated associations with the cell cycle, lysosomes, immune response (including natural killer cell-mediated cytotoxicity, T cell receptor signaling pathway, cytokine-receptor interaction, complement, intestinal immune network for IgA production, and Fc gamma R-mediated phagocytosis), vascular smooth muscle contraction, and various diseases (primary immunodeficiency, carcinoma, dilated cardiomyopathy, hypertrophic cardiomyopathy, and systemiclupuserythematosus). Furthermore, we observed tight associations of marker genes with xenobiotic metabolism by cytochrome p450, chemokine signaling pathway, MAPK signaling pathway, Nod-like receptor signaling pathway, Notch signaling pathway, WNT signaling pathway, JAK-STAT signaling pathway, and insulin signaling pathway (Fig. 3).

Single-gene GSEA–KEGG pathway analysis was employed to explore the potential function of marker genes in distinguishing IPAH from normal controls: A. BEX1, B. KEAP1, C. MEG3, D. NFS1, E. PRDX1, F. TNFAIP3.
Differential pathway activation levels were analyzed between patients with IPAH and normal individuals based on GSVA and marker gene expression levels (Fig. 4). Notably, PRDX1 downregulation was implicated in IPAH pathogenesis by activating pathways such as “purine metabolism”, “alanine aspartate and glutamate metabolism”, “fructose and mannose metabolism”, “lysosome”, “other glycan degradation” and “other glycan degradation”, whereas its overexpression activated “ABC_transporters”. TNFAIP3 upregulation was associated with the activation of “maturity onset diabetes of the young”, whereas its downregulation correlated with “glycerolipid metabolism” and “nature killer cell mediated cytotoxicity”. In addition, MEG3 overexpression activated pathways such as “pantothenate and coa biosynythesis”, “homologous recombination”, “propanoate metabolism” and “valine leucine and isoleucine biosynthesis”, whereas its downregulation activated pathways such as “glycosaminoglycan biosynthesis keratan sulfate”, “dilated cardiomypathy”,“arrhythmogenic right ventricular cadiomyopathy”, “WNT signaling pathway”, “NOTCH signaling pathway”, “basal cell carcinoma” and “hypertrophic cardiomyopathy”. Moreover, BEX1 upregulation was associated with “pantothenate and coa biosynthesis”, “homologous rcombination” and “propanoate metabolism,” whereas its downregulation may activate “arrhythmogenic right ventricular cadiomyopathy”, “dilated cardiomypathy”, “glycosaminoglycan biosythesis keratan sulfate”, “hypertrophic cardiomyopathy”, “WNT signaling pathway” “NOTCH signaling pathway” and “basal cell carcinoma”. NFS1 downregulation significantly activated “starch and sucrose metabolism”, “nicotinate and nicotinamide metabolism” and “amino sugar and nucleotide sugar metabolism,” whereas its overexpression was associated with “ABC_transporters” and “glycosaminoglycan biosynthesis_heparan sulfate”. Interestingly, no significant difference in pathway activation levels of KEAP1 was observed between the two groups.

The differential activation levels of the pathway were analyzed between IPAH and normal controls according to GSVA and marker gene expression levels: A. TNFAIP3, B. NFS1, C. PRDX1, D. MEG3, E. KEAP1, F. BEX1.
Immune microenvironment analysis
Based on the results, the marker genes were closely associated with immunity. Previous research has confirmed the close relationship between the immune response and IPAH35. The CIBERSORT algorithm was employed to investigate differences in the immune microenvironment between patients with IPAH and normal individuals. We observed overexpression of activated NK cells, macrophages M1, and CD4 memory T cells in patients with IPAH compared with normal individuals. Pearson’s correlation analysis suggested a positive correlation between neutrophils and TNFAIP3, whereas macrophages M0 showed a negative correlation with PRDX1. Subsequently, we compared different immune-related genes (IRGs) and functions between IPAH and normal samples. Immunostimulatory genes, including TNFSF9, LDHA, JAK1, and FGL1, were highly expressed in normal samples, whereas LAG3, IFNG, and CTLA4 were upregulated in patients with IPAH. We compared the different immune functions in IPAH and normal controls and found that most immune functions, including Th1_cells, Tfh, T_helper_cells, T_cell_co-stimulation, T_cell_co-inhibition, pDCs MHC_class_I, Inflammation-promoting, Cytolytic_activity, Check point, CD8+_T_cells, Macrophages down, and CCR, were significantly enriched in IPAH compared with normal samples. These results suggest that complex immune components are involved in the pathology of IPAH (Fig. 5).

Immune microenvironment analysis: A. Pearson’s correlation analysis showing positive correlation between neutrophils and TNFAIP3 and negative correlation between macrophages M0 and PRDX1. B. The CIBERSORT algorithm was employed to investigate the differences in the immune microenvironment between IPAH and normal controls. C. ssGSEA was used to calculate the difference in immune cell infiltration between the IPAH and normal control groups. D. Heatmap showing the differences in immune cell infiltration.
Competitive endogenous RNA (ceRNA) networks based on marker genes
Next, a ceRNA network with 166 nodes (5 marker genes, 81 miRNAs, and 86 lncRNAs) was constructed based on the 6 marker genes (Fig. 6A). We observed that TNFAIP3 was regulated by the most predicted miRNAs and lncRNAs. PRDX1 was regulated by hsa-miR-3148, hsa-miR-581, hsa-miR-155-3p, hsa-miR-485-3p, and hsa-miR-1283 without lncRNAs, whereas seven lncRNAs binding to hsa-miR-600, hsa-miR-10a-3p, hsa-miR-320e, hsa-miR-662, hsa-miR-542-3p, and hsa-miR-4273 may regulate the expression of BEX1. In addition, 11 lncRNAs could regulate the expression of KEAP1 by competitively binding to hsa-miR-1292-5p, hsa-miR-141-3p, hsa-miR-423-5p, hsa-miR-626, hsa-miR-200a-3p, hsa-miR-3065-5p, hsa-miR-4292, and hsa-miR-26b-3p. Conversely, eight lncRNAs could competitively bind hsa-miR-4282, hsa-miR-16-5p, hsa-miR-944, hsa-miR-548v, hsa-miR-581, hsa-miR-15b-5p, hsa-miR-491-3p, hsa-miR-922, hsa-miR-1253, hsa-miR-195-5p, hsa-miR-15a-5p, hsa-miR-149-5p, hsa-miR-133b, hsa-miR-424-5p, hsa-miR-3121-3p, hsa-miR-497-5p, hsa-miR-607, hsa-miR-582-3p, hsa-miR-3132, and hsa-miR-570-3p to regulate the expression of NFS1. Furthermore, both TNFAIP3 and NFS1 expression were regulated by hsa-miR-607 and hsa-miR-570-3p, which could also regulate the expression of BEX1.

A. A ceRNA network, including 166 nodes (5 marker genes, 81 miRNAs and 86 lncRNAs), was constructed based on the 6 marker genes. B. The DGIdb was used to identify drugs possibly targeting marker genes, showing that only two genes have the target drugs.
Prediction of marker gene-targeted drugs
The DGIdb was used to identify drugs targeting marker genes, revealing that only two genes had associated target drugs (Fig. 6B). Bardoxolone methyl and dimethyl fumarate are inhibitors of KEAP1, whereas ustkinumab and stekinumab are unidentified inhibitors of TNFAIP3. Notably, drugs targeting BEX1, PRDX1, NFS1, or MEG3 have not been identified to date.
Marker gene expression in the validation set
Validation was performed using GSE117261, GSE33463, and GSE84395 to confirm the expression patterns of marker genes. Initially, we assessed marker gene expression trends in the GSE117261 dataset, including 31 IPAH and 25 healthy tissue samples. KEAP1, BEX1, PRDX1, TNFAIP3, and MEG3 in the GSE117261 dataset exhibited expression trends similar to those observed in the GSE48149 dataset. Subsequently, compared to the GSE48149 dataset, the GSE33463 dataset, consisting of 30 IPAH and 41 healthy PBMC samples, was analyzed. It revealed a similar expression level trend for BEX1, contrasting with the patterns of KEAP1, PRDAX1, and TNFAIP3. Additionally, we examined marker gene expression levels in GSE84395, including 11 IPAH and 18 healthy PEC samples. The expression trends of PRDX1 remained consistent with the GSE48149 dataset, whereas the expression of TNFAIP3 was lower in IPAH (Fig. 7).

A. GSE33463, B. GSE117261, C. GSE84395 were employed as validation sets to verify the expression of marker genes.
Validation in a single-cell dataset
The single-cell dataset GSE169471, which included three patients with IPAH and eight healthy tissue samples, was used to assess marker gene expression in various cell types. Following quality control, normalization, data scaling, and dimensionality reduction, cells were classified into 18 clusters: AT1, AT2, B cell, Basal, Ciliated cell, Club, Dendritic cell, EC arterial, EC capillary, EC venous, Fibroblasts, Lymphatic EC mature, Macrophages, Mast cells, Monocytes, NK cells, SMC, and T cells. Subsequently, six marker genes—KEAP1, BEX1, PRDX1, TNFAIP3, NFS1, and MEG3—were identified in the respective cell types. Notably, compared to normal tissues, smooth muscle cells in IPAH exhibited high expression of MEG3 and KEAP1. Furthermore, KEAP1 was predominantly expressed in macrophages. Among immune cells, T cells, macrophages, NK cells, and B cells exhibited associations with TNFAIP3, which was highly expressed in IPAH. Similar to GSE84395, PRDX1 displayed low expression levels in capillary endothelial cells (Fig. 8).

The single-cell dataset GSE169471 was employed to test the marker gene expression in different cells. A. Expression levels of six marker genes—KEAP1, BEX1, PRDX1, TNFAIP3, NFS1, and MEG3—were marked in respective cell types. Dot plot (B), violin plot and boxplot (C) showing the expression levels of the six marker genes in respective cell types. D. Expression levels of the six marker genes in immune cells. E. Expression levels of the six marker genes in other cells.
Discussion
IPAH is a rare and life-threatening condition. Despite advancements in understanding the disease, its precise molecular mechanisms remain unclear due to the multifaceted interplay between genetic, environmental, and cellular factors. Previous research has underscored the intricate association between ferroptosis and various cardiovascular diseases, including cardiomyopathy, myocardial infarction, ischemia/reperfusion injury, and heart failure36.Ferroptosis in the systemic vasculature leads to endothelial cell dysfunction and promotes the development of atherosclerosis in mice. Notably, this effect can be attenuated by ferrostatin-1 treatment37 This association extends to the pulmonary domain, where the pulmonary vasculature plays a pivotal role in ferroptosis due to the unique composition of oxygenated blood, which is rich in polyunsaturated fatty acids and iron ions. Initially elucidated within oncological frameworks, ferroptosis has emerged as a significant contributory factor in the pathogenesis of various pulmonary disorders, including chronic obstructive pulmonary disease, acute lung injury, and pulmonary fibrosis. Current scientific inquiries are vigorously directed towards identifying and characterizing ferroptosis biomarkers across morphological, biochemical, proteomic, and genomic domains. However, identifying reliable gene biomarkers and the precise regulatory intricacies linked to ferroptosis in IPAH remain a formidable challenge in medical research. Our comprehensive analysis explored potential ferroptosis-related biomarkers and therapeutic targets in IPAH, offering valuable insights into its complex pathophysiology and presenting novel diagnostic and therapeutic avenues.
In our study, we identified 27 DE-FRGs that exhibited significant differences between IPAH and normal samples, comprising 15 downregulated and 12 upregulated genes. The GO enrichment analysis highlighted significant associations of DE-FRGs with CC (“focal adhesion,” “cell–substrate junction,” “membrane raft”), BP (“cellular response to chemical stress,” “cellular response to peptidyl-serine phosphorylation,” “peptidyl-serine modification”), and MF (“RNA polymerase II-specific DNA-binding transcription factor binding,” “DNA-binding transcription factor binding,” “phosphoprotein binding”). Notably, focal adhesion, which is critical for cell adhesion and migration within the matrix, may regulate interactions between pulmonary vascular endothelial cells and their environment during remodeling38. Maintenance of cell–substrate junctions is crucial for intercellular communication and tissue integrity, influencing communication between pulmonary vascular endothelial cells and subsequently affecting vascular wall structure, function, remodeling, and narrowing. These cellular alterations likely contribute significantly to pulmonary vascular pathology, influencing the process of vascular remodeling significantly39. Concurrently, Reactome pathway analyses indicated a notable enrichment in pathways such as fluid shear stress, atherosclerosis, hepatocellular carcinoma, and chemical carcinogenesis, specifically related to reactive oxygen species. The restructuring of blood vessels caused by fluid shear stress may be associated with the generation of detrimental substances that are potentially linked to atherosclerosis via iron-mediated pathways. These pathways may induce lipid damage, affect protein carbonylation, and contribute to pulmonary vascular remodeling40. This evidence suggests that DE-FRGs play pivotal roles in the regulation of autophagy, cytokines, kinases, and immune cells, potentially contributing to critical effects on IPAH pathogenesis.
This analysis identified six DE-FRGs as potential diagnostic markers of IPAH using robust machine learning. Validation across independent datasets (GSE117261 and GSE84395) generally supported their potential, although discrepancies surfaced in GSE33463, particularly evident in the varied expression of KEAP1, PRDX1, and TNFAIP3. Among the DE-FRGs, we suggest that PRDX1 is linked to endothelial dysfunction and that TNFAIP3 is associated with immune responses, serving as a potential diagnostic marker for IPAH.
Pulmonary vascular remodeling, a hallmark pathological feature of PAH, arises from the dysregulated interactions of multiple vascular cell populations, with dysfunctional endothelial cells (ECs) serving as both initiators and amplifiers of disease progression41. Factors such as hypoxia, inflammation, and shear stress can damage pulmonary vascular ECs, especially pulmonary artery ECs (PAECs) and pulmonary arterial endothelial cell (PAEC) dysfunction, characterized by mitochondrial impairment and altered iron and lipid metabolism, creates a pro-ferroptotic microenvironment in PAH42. In studies on monocrotaline (MCT)-induced PAH in rats, Xie et al. found weakened PAEC activity, along with increased levels of ferroptosis-related proteins in PAECs15. The ferroptosis inhibitor, ferristatin-1, was able to mitigate the progression of MCT-induced pulmonary vascular remodeling by downregulating these markers42. This hypothesis is supported by the observation that small molecule and genetic interventions that suppress ferroptosis reduce PAH severity in rodents14,37,42. Furthermore, in vitro studies suggest that ferroptosis in PAECs can modulate the progression of PAH through the HMGB1/TLR4/NLRP3 inflammasome signaling pathway, indicating a relationship between ferroptosis and EC dysfunction13. Its pathogenicity primarily originates from a cascade of oxidative stress-induced lipid peroxidation, ultimately compromising the structural integrity of the pulmonary vasculature43.These studies suggest that inhibiting ferroptosis may reduce pulmonary vascular remodeling due to EC dysfunction, serving as a potential target for PAH treatment. In our study, PRDX1, consistent downregulation was observed in the IPAH group compared with healthy controls, except for GSE33463. This trend was reinforced by findings from single-cell sequencing analysis (GSE169471), which demonstrated diminished PRDX1 expression in IPAH capillary endothelial cells compared with healthy tissue, potentially implicating its involvement in endothelial dysfunction, a key aspect of IPAH pathology.
The Peroxiredoxins (PRDXs) are a family of six thiol-dependent peroxidases (PRDX1-PRDX6)44. PRDXs can catalyze the reduction of hydroperoxide and hydrogen peroxide and have been implicated as antioxidant signaling proteins45,46.Previous research has shown that PRDXs are important modulators of physiological and pathophysiological cardiovascular events. PRDX3 overexpression prevents cardiac hypertrophy and HF by reducing oxidative stress and restoring mitochondrial function. PRDX2 decressed expression during cardiac hypertrophy and may be a potential therapeutic target for the treatment of cardiac hypertrophy47,48. Ping Dai et al. Found that PRDX6 was expressed in PAECs and was significantly decreased in MCT-induced PAH. They suggested that PRDX6 mediates PAEC ferroptosis through HMGB1 release and subsequent activation of TLR4/NLRP3 inflammasome signalling14. PRDX1is a typical 2-cysteine (Cys) peroxiredoxin. Its primary function is to catalyze reduction reactions, including the conversion of hydrogen peroxide (H2O2) to water, and to act as a multifunctional antioxidant49. PRDX1 functions as an antioxidant factor and redox signaling protein that regulates various metabolic pathways and lysosomal processes to mitigate oxidative stress and influence oxidant sensitivity50,51. PRDX1 plays an important role in countering increased endothelial activation52. Previous studies have shown that PRDX1 could resist oxidation, protect cells from oxidative damage, and improve cardiac hypertrophy49. This evidence suggests a potential protection role for PRDX1 in IPAH pathology.
The intricate pathogenesis of PAH remains under ongoing investigation without definitive clarity. Recent years of research in both preclinical and clinical studies within the PAH field have increasingly emphasized the involvement of inflammation and immunity in its progression53. Notably, reports have highlighted the presence of circulating autoantibodies targeting endothelial cells and fibroblasts in a substantial percentage (10–40%) in patients diagnosed with IPAH and SSc-PAH. These findings suggest a potential association between autoimmunity and the development of pulmonary vascular lesions54,55. We used CIBERSORT to detect elevated expression of activated NK cells, macrophages M1, and activated T cells CD4 memory in patients with IPAH. Moreover, Pearson’s correlation analysis revealed positive correlations between neutrophils and TNFAIP3 and negative correlations between macrophages M0 and PRDX1. In our single-cell validation dataset, immune cell types, such as T cells, macrophages, NK cells, and B cells, were associated with TNFAIP3, which exhibited high expression levels in IPAH. Similar to PRDX1, TNFAPI3 exhibited elevated expression levels in the IPAH cohort, except in the validation dataset GSE33463. TNFAIP3, an anti-inflammatory protein, plays a significant role in the immune response and cellular apoptosis56. In contrast to other organ systems, the lungs demonstrate comparatively elevated levels of TNFAIP3 mRNA expression, with detectable protein levels observed in whole-lung homogenates of rodents57. Dysfunctions in TNFAIP3 have implications in various autoimmune conditions, such as Crohn’s disease, asthma, and chronic obstructive pulmonary disorder58. Subsequent pathway analysis demonstrated that TNFAIP3 upregulation influenced diabetes-related pathways and immune responses. Most chronic inflammatory autoimmune diseases are associated with reduced TNFAIP3 expression and continuous NF-kB activation58. Paradoxically, high TNFAIP3 levels have been associated with disease promotion, suggesting a cell- and organ-specific approach to TNFAIP3 augmentation to prevent adverse effects and induce metabolic benefits59. GSE33463, containing 30 IPAH and 41 healthy PBMC samples, showed lower expression of TNFAIP3. Moreover, in neurodegenerative diseases associated with systemic inflammation, basal TNFAIP3 mRNA expression in peripheral blood lymphocytes and monocytes is significantly lower than in healthy individuals60. These findings are consistent with our expression analysis of TNFAIP3 in IPAH, where upregulation in macrophages was observed, indicating its potential role in inflammatory processes that drive iron dysregulation and ferroptosis in IPAH.
In addition, KEAP1, NFS1, MEG3, and BEX1, though not as prominently featured in the single-cell analysis, are known to be involved in oxidative stress regulation, iron metabolism, and cell cycle regulation, all of which are processes critical to ferroptosis. KEAP1, for instance, regulates oxidative stress responses by controlling NRF2 activity. In the context of IPAH, downregulation of KEAP1 could lead to enhanced oxidative stress, further exacerbating ferroptosis in endothelial cells and smooth muscle cells, thereby contributing to vascular remodeling. Similarly, NFS1 is involved in the synthesis of iron-sulfur clusters, and its downregulation in IPAH may disrupt iron homeostasis, promoting ferroptosis in susceptible cells. MEG3 and BEX1 are also involved in regulating cell cycle and apoptosis, processes that are crucial in maintaining vascular integrity in IPAH. As ferroptosis is tightly linked with these cellular processes, alterations in the expression of these genes may have profound implications for disease progression.
The establishment of a comprehensive ceRNA network centered on marker genes sheds light on the intricate regulatory interactions among miRNAs and lncRNAs. TNFAIP3 has emerged as a central node influenced by numerous miRNAs and lncRNAs. However, PRDX1 is specifically regulated by distinct miRNAs, excluding lncRNA involvement. Furthermore, the identification of drugs targeting marker genes has uncovered limited options, with no drugs identified for targeting PRDX1, highlighting the scarcity of pharmaceutical options addressing these genes in the context of IPAH treatment.
Despite the comprehensive nature of our analysis, several limitations warrant consideration. Firstly, the sample sizes in individual datasets were relatively small, potentially influencing the robustness and generalizability of our findings. Additionally, the inherent heterogeneity across IPAH subtypes may introduce variability in the observed gene expression patterns. Furthermore, the use of diverse cell and tissue sources across datasets could contribute to variations in the results. While our bioinformatic approach provides valuable insights, experimental validation is necessary to confirm the functional relevance of the identified biomarkers. Lastly, the need for further research is emphasized to validate the potential drug targets for their clinical applicability. These limitations underscore the importance of cautious interpretation and future investigations to refine our understanding of ferroptosis-related mechanisms in IPAH.
Conclusions
In summary, our exploration, facilitated by a comprehensive bioinformatic analysis, delved into the ferroptosis genes within IPAH. We identified six DE-FRGs—KEAP1, TNFAIP3, MEG3, NFS1, PRDX1, BEX1—among which PRDX1 and TNFAIP3 exhibited stable expression across multiple datasets. These genes are implicated in critical pathways, including cell cycle regulation, immune response modulation, and vascular constriction, indicating their involvement in IPAH development. Furthermore, the identification of potential drug targets, such as the TNFAIP3-targeting ustkinumab, offers promising avenues for therapeutic interventions in IPAH. These findings propose PRDX1 and TNFAIP3 for future use in predictive diagnostics, prevention, patient stratification, and ustkinumab as the personalized medicine for IPAH. Nevertheless, further research must be conducted to validate potential drug targets for clinical applicability.
Data availability
The datasets generated and/or analysed during the current study are available in GSE48149 repository, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE48149, GSE117261 repository, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE117261, GSE84395 repository, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE84395, GSE33463 repository, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE33463, GSE169471 repository, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE169471.
References
Kasavi, C. Idiopathic Pulmonary Arterial Hypertension: Network-Based Integration of Multi-Omics Data Reveals New Molecular Signatures Candidate Drugs OMICS 27(7): 315–326. (2023).
Galie, N. et al. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: the joint task force for the diagnosis and treatment of pulmonary hypertension of the European society of cardiology (ESC) and the European respiratory society (ERS): endorsed by: association for European paediatric and congenital cardiology (AEPC), international society for heart and lung transplantation (ISHLT). EUR. HEART J. 37 (1), 67–119 (2016).
Hendriks, P. M. et al. The evolution of survival of pulmonary arterial hypertension over 15 years. PULM CIRC. 12 (4), e12137 (2022).
Li, Q., Meng, L. & Liu, D. Screening and identification of therapeutic targets for pulmonary arterial hypertension through microarray technology. FRONT. GENET. 11, 782 (2020).
Zeng, Y. et al. Screening of key biomarkers and immune infiltration in pulmonary arterial hypertension via integrated bioinformatics analysis. BIOENGINEERED 12 (1), 2576–2591 (2021).
Boucherat, O. et al. The cancer theory of pulmonary arterial hypertension. PULM CIRC. 7 (2), 285–299 (2017).
Humbert, M. et al. 2022 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension. EUR. RESPIR J. 61(1). (2023).
Grech, G. et al. EPMA position paper in cancer: current overview and future perspectives. EPMA J. 6 (1), 9 (2015).
Kiely, D. G., Lawrie, A. & Humbert, M. Screening strategies for pulmonary arterial hypertension. EUR. HEART J. SUPPL. 21 (Suppl K), K9–K20 (2019).
Soon, E. et al. Unexplained iron deficiency in idiopathic and heritable pulmonary arterial hypertension. THORAX 66 (4), 326–332 (2011).
Ruiter, G. et al. Iron deficiency is common in idiopathic pulmonary arterial hypertension. EUR. RESPIR J. 37 (6), 1386–1391 (2011).
Wu, X., Li, Y., Zhang, S. & Zhou, X. Ferroptosis as a novel therapeutic target for cardiovascular disease. THERANOSTICS 11 (7), 3052–3059 (2021).
Hu, P. et al. The mechanism of the imbalance between proliferation and ferroptosis in pulmonary artery smooth muscle cells based on the activation of SLC7A11. EUR. J. PHARMACOL. 928, 175093 (2022).
Liao, J. et al. PRDX6-mediated pulmonary artery endothelial cell ferroptosis contributes to monocrotaline-induced pulmonary hypertension. MICROVASC RES. 146, 104471 (2023).
Xie, S. S. et al. Endothelial cell ferroptosis mediates monocrotaline-induced pulmonary hypertension in rats by modulating NLRP3 inflammasome activation. Sci. Rep. 12 (1), 3056 (2022).
Liu, Y. et al. NDUFA4L2 in smooth muscle promotes vascular remodeling in hypoxic pulmonary arterial hypertension. J. CELL. MOL. MED. 25 (2), 1221–1237 (2021).
Zhang, F. & Liu, H. Identification of ferroptosis-associated genes exhibiting altered expression in pulmonary arterial hypertension. MATH. BIOSCI. ENG. 18 (6), 7619–7630 (2021).
Wang, E., Zhou, S., Zeng, D. & Wang, R. Molecular regulation and therapeutic implications of cell death in pulmonary hypertension. Cell. Death Discov. 9 (1), 239 (2023).
Zhou, N. & Bao, J. FerrDb: a manually curated resource for regulators and markers of ferroptosis and ferroptosis-disease associations. Database (Oxford). 2020. (2020).
Liu, Y. et al. Integrative analyses of biomarkers and pathways for adipose tissue after bariatric surgery. ADIPOCYTE 9 (1), 384–400 (2020).
Freshour, S. L. et al. Integration of the Drug-Gene interaction database (DGIdb 4.0) with open crowdsource efforts. NUCLEIC ACIDS RES. 49 (D1), D1144–D1151 (2021).
Ritchie, M. E. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. NUCLEIC ACIDS RES. 43 (7), e47 (2015).
Zhang, H. et al. Comprehensive analysis of gene expression changes and validation in hepatocellular carcinoma. Onco Targets Ther. 14, 1021–1031 (2021).
Li, Z. et al. Diagnostic and predictive values of Ferroptosis-Related genes in child Sepsis. FRONT. IMMUNOL. 13, 881914 (2022).
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: biological systems database as a model of the real world. NUCLEIC ACIDS RES. 53 (D1), D672–D677 (2025).
Kanehisa, M. Toward Understanding the origin and evolution of cellular organisms. PROTEIN SCI. 28 (11), 1947–1951 (2019).
Wu, M. N. et al. Genetic analysis of potential biomarkers and therapeutic targets in ferroptosis from psoriasis. FRONT. IMMUNOL. 13, 1104462 (2022).
Wu, X. et al. Genetic analysis of potential biomarkers and therapeutic targets in ferroptosis from coronary artery disease. J. CELL. MOL. MED. 26 (8), 2177–2190 (2022).
Hanzelmann, S., Castelo, R. & Guinney, J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 14, 7 (2013).
Newman, A. M. et al. Robust enumeration of cell subsets from tissue expression profiles. NAT. METHODS. 12 (5), 453–457 (2015).
Agarwal, V., Bell, G. W., Nam, J. W. & Bartel, D. P. Predicting Effective microRNA Target Sites in Mammalian mRNAs4 (ELIFE, 2015).
Chen, Y. & Wang, X. MiRDB: an online database for prediction of functional MicroRNA targets. NUCLEIC ACIDS RES. 48 (D1), D127–D131 (2020).
Betel, D., Koppal, A., Agius, P., Sander, C. & Leslie, C. Comprehensive modeling of MicroRNA targets predicts functional non-conserved and non-canonical sites. GENOME BIOL. 11 (8), R90 (2010).
Furio-Tari, P., Tarazona, S., Gabaldon, T., Enright, A. J. & Conesa, A. SpongeScan: A web for detecting MicroRNA binding elements in LncRNA sequences. NUCLEIC ACIDS RES. 44 (W1), W176–W180 (2016).
Zeng, H., Liu, X. & Zhang, Y. Identification of potential biomarkers and immune infiltration characteristics in idiopathic pulmonary arterial hypertension using bioinformatics analysis. Front. Cardiovasc. Med. 8, 624714 (2021).
Chen, Y., Li, X., Wang, S., Miao, R. & Zhong, J. Targeting Iron Metabolism and Ferroptosis as Novel Therapeutic Approaches in Cardiovascular Diseases. NUTRIENTS. 15(3). (2023).
Bai, T., Li, M., Liu, Y., Qiao, Z. & Wang, Z. Inhibition of ferroptosis alleviates atherosclerosis through attenuating lipid peroxidation and endothelial dysfunction in mouse aortic endothelial cell. Free Radic Biol. Med. 160, 92–102 (2020).
Wang, R., Xu, J., Wu, J., Gao, S. & Wang, Z. Angiotensin-converting enzyme 2 alleviates pulmonary artery hypertension through Inhibition of focal adhesion kinase expression. EXP. THER. MED. 22 (4), 1165 (2021).
Junhui, Z. et al. Reduced number and activity of Circulating endothelial progenitor cells in patients with idiopathic pulmonary arterial hypertension. Respir Med. 102 (7), 1073–1079 (2008).
Rashdan, N. A., Zhai, B. & Lovern, P. C. Fluid shear stress regulates placental growth factor expression via Heme Oxygenase 1 and iron. Sci. Rep. 11 (1), 14912 (2021).
Ruopp, N. F. & Cockrill, B. A. Diagnosis and treatment of pulmonary arterial hypertension: A review. JAMA 327 (14), 1379–1391 (2022).
Song, J. et al. Ferrostatin-1 blunts right ventricular hypertrophy and dysfunction in pulmonary arterial hypertension by suppressing the HMOX1/GSH signaling. J. Cardiovasc. Transl Res. 17 (1), 183–196 (2024).
Lin, J., Lin, S., Zhang, Y. & Liu, W. Identification of Ferroptosis-related potential biomarkers and immunocyte characteristics in chronic thromboembolic pulmonary hypertension via bioinformatics analysis. BMC Cardiovasc. Disord. 23 (1), 504 (2023).
Park, M. H., Jo, M., Kim, Y. R., Lee, C. K. & Hong, J. T. Roles of Peroxiredoxins in cancer, neurodegenerative diseases and inflammatory diseases. Pharmacol. Ther. 163, 1–23 (2016).
Rhee, S. G., Woo, H. A. & Kang, D. The role of Peroxiredoxins in the transduction of H(2)O(2) signals. Antioxid. Redox Signal. 28 (7), 537–557 (2018).
Veal, E. A., Underwood, Z. E., Tomalin, L. E., Morgan, B. A. & Pillay, C. S. Hyperoxidation of peroxiredoxins: gain or loss of function?? Antioxid. Redox Signal. 28 (7), 574–590 (2018).
Matsushima, S. et al. Overexpression of mitochondrial peroxiredoxin-3 prevents left ventricular remodeling and failure after myocardial infarction in mice. CIRCULATION 113 (14), 1779–1786 (2006).
Su, H., Pistolozzi, M., Shi, X., Sun, X. & Tan, W. Alterations in NO/ROS ratio and expression of Trx1 and Prdx2 in isoproterenol-induced cardiac hypertrophy. Acta Biochim. Biophys. Sin (Shanghai). 49 (11), 1022–1028 (2017).
Tang, C. et al. Peroxiredoxin-1 ameliorates pressure overload-induced cardiac hypertrophy and fibrosis. BIOMED. PHARMACOTHER. 129, 110357 (2020).
McCann, M. S., Fernandez, H. R., Flowers, S. A. & Maguire-Zeiss, K. A. Polychlorinated biphenyls induce oxidative stress and metabolic responses in astrocytes. NEUROTOXICOLOGY 86, 59–68 (2021).
Guo, W. et al. Prdx1 alleviates cardiomyocyte apoptosis through ROS-activated MAPK pathway during myocardial ischemia/reperfusion injury. INT. J. BIOL. MACROMOL. 112, 608–615 (2018).
Kisucka, J. et al. Peroxiredoxin1 prevents excessive endothelial activation and early atherosclerosis. CIRC. RES. 103 (6), 598–605 (2008).
Hu, Y., Chi, L., Kuebler, W. M. & Goldenberg, N. M. Perivascular inflammation in pulmonary arterial hypertension. CELLS-BASEL 9(11). (2020).
Tamby, M. C. et al. Anti-endothelial cell antibodies in idiopathic and systemic sclerosis associated pulmonary arterial hypertension. THORAX 60 (9), 765–772 (2005).
Tamby, M. C. et al. Antibodies to fibroblasts in idiopathic and scleroderma-associated pulmonary hypertension. EUR. RESPIR J. 28 (4), 799–807 (2006).
Wu, Y., He, X., Huang, N., Yu, J. & Shao, B. A20: a master regulator of arthritis. ARTHRITIS RES. THER. 22 (1), 220 (2020).
Wu, D. Q., Wu, H. B., Zhang, M. & Wang, J. A. Effects of zinc finger protein A20 on lipopolysaccharide (LPS)-Induced pulmonary Inflammation/Anti-Inflammatory mediators in an acute lung injury/acute respiratory distress syndrome rat model. Med. Sci. Monit. 23, 3536–3545 (2017).
Momtazi, G., Lambrecht, B. N., Naranjo, J. R. & Schock, B. C. Regulators of A20 (TNFAIP3): new drug-able targets in inflammation. Am. J. Physiol. Lung Cell. Mol. Physiol. 316 (3), L456–L469 (2019).
Lee, J. H. et al. A20 promotes metastasis of aggressive basal-like breast cancers through multi-monoubiquitylation of Snail1. NAT. CELL. BIOL. 19 (10), 1260–1273 (2017).
Perga, S. et al. A20 in multiple sclerosis and parkinson’s disease: clue to a common dysregulation of Anti-Inflammatory pathways?? NEUROTOX. RES. 32 (1), 1–7 (2017).
Acknowledgements
We would like to thank Editage (www.editage.com) for the English language editing.
Funding
This work was supported by the Major Scientific and Technological Projects for collaborative prevention and control of birth defects in Hunan Province (Grant numbers :2019SK1010).This work was supported by the Major Research Program of National Key Clinical Specialty (Hunan Province Health Commission)(Grant numbers :20230314).
Author information
Authors and Affiliations
Contributions
Can Huang and Yuhao Zhang contributed to the study conception and design. Material preparation, data collection were performed by Ting Lu and Haoyong Yuan. Data analysis were performed by Tao Qian, Wei Jiang and Ni Yin. The first draft of the manuscript was written by Yuhao Zhang. Can Huang, Zhongshi Wu, Ting Lu and Ni Yin commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, Y., Qian, T., Jiang, W. et al. Integrated bioinformatics identifies ferroptosis biomarkers and therapeutic targets in idiopathic pulmonary arterial hypertension. Sci Rep 15, 25187 (2025). https://doi.org/10.1038/s41598-025-11066-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-11066-y
