
Abstract
GM1-gangliosidosis is a lysosomal storage disease characterized by the accumulation of GM1 ganglioside in neurons, including retinal ganglion cells (RGCs). Although vision impairment and retinal degeneration are well-known manifestations in humans, knowledge about the underlying mechanism of these lesions is limited. Pathological retinal changes in a Glb1 knockout (Glb1−/−) mouse model were assessed using immunohistochemistry, immunofluorescence and transmission electron microscopy in 4- and 7-month-old wild type and Glb1 knockout (Glb1−/−) mice. Increased numbers of glial fibrillary acidic protein (GFAP) positive Müller cells (MCs) were detected in Glb1−/− mice both at 4 and 7 months of age, indicating glial reactivity. A transient increase in glutamine synthetase (GS) positive MCs at 4 months of age with a subsequent decrease by 7 months, most likely related to reduced expression of GS, was also observed. Immunohistochemistry revealed loss of RGCs, storage material accumulation and decreased BRN3A expression in 7-month-old Glb1−/− mice. Increasing numbers of Iba1-positive microglia/macrophages were detected in Glb1−/− mice at both time points. Microglia/macrophages showed migration towards the inner retinal layers and acquired a morphological phenotype that indicates activation. The present study shows that the murine GM1 model used in this study is suitable for investigating ocular changes in lysosomal storage diseases.
Similar content being viewed by others

Introduction
GM1-gangliosidosis is a rare inherited metabolic neurodegenerative disease caused by a deficiency of β-galactosidase (β-gal)1. The enzyme deficiency leads to progressive storage of GM1 ganglioside (GM1) and its derivates, particularly within lysosomes of neuronal tissue2,3,4. In humans, the disease is classified into three clinical subtypes: the infantile (type 1, OMIM: 230500), the late infantile (juvenile; type 2, OMIM: 230600), and the adult form (adult; type 3, OMIM: 230650). The clinical symptoms in human GM1-gangliosidosis vary depending on the age of onset and the specific disease phenotype4.
Various naturally occurring and genetically modified animal models have been utilized for GM1 research5,6,7. A comparable naturally occurring genetic defect for GM1-gangliosidosis in animals has also been reported in cats, dogs, cattle, sheep, emus, and American black bears8,9,10,11,12,13. The first transgenic mice were reported in 1997 by Hahn et al. and Matsuda et al. (1997)14,15. These models closely resemble the infantile/juvenile form of the disease, exhibiting pronounced neuronal loss and neuroinflammation14,15. More recent murine models have been developed using TALEN and CRISPR/Cas9 technologies. In 2019, Przybilla et al. introduced a knockout model with a deletion in exon 8 of the Glb1 gene, which resulted in neurocognitive impairment16. In 2020, Eikelberg et al. reported a new Glb1−/− mouse model that mimics the adult form of GM1-gangliosidosis with delayed onset of observable symptoms and limited lesion distribution17. More recently, Liu et al. generated a transgenic mouse model characterized by a human missense mutation in exon 14 that mimics the late-infantile subtype. This model exhibits pronounced motor function impairment and extensive microgliosis18.
Despite the extensive research conducted on the pathomechanism of GM1-gangliosidosis, ocular pathology associated with GM1 accumulation remains insufficiently characterized19,20. Ocular manifestations in human patients may include visual impairment, corneal opacities, tortuous retinal vessels, retinal haemorrhages and optic atrophy19. Notably, a cherry-red macula is detected in about half of the cases19. This significant fundoscopic finding represents a hallmark feature in numerous ganglioside catabolism disorders21. Pathological changes in ocular structures during GM1-gangliosidosis have already been described in cats, dogs, and calves22,23,24,25. However, retinal changes have not yet been investigated in rodent models of GM1 gangliosidosis.
Pathological processes within the retina, including neuronal damage, are often accompanied by glial cell activation and reactive gliosis26,27. The mammalian retina contains two main categories of macroglial cells, astrocytes and Müller cells (MCs), alongside resident microglia28. MCs belong to a special subgroup of glial cells of the central nervous system (CNS), also known as aldynoglia. They appear as radially oriented cells whose processes extend across the entire thickness of the retina from the inner limiting membrane (ILM) to the distal end of the outer nuclear layer (ONL)26. In the ILM, they form structures called endfeet that wrap around axons and somas of retinal ganglion cells (RGCs)26. Consequently, MCs provide structural support and play a pivotal role in maintaining retinal homeostasis, including involvement in the organization of the blood-retinal barrier29,30,31. Upon activation, MCs exhibit reaction patterns similar to other glial cells of the CNS and peripheral nervous system (PNS). Under pathological conditions, they show an upregulation of glial fibrillary acidic protein (GFAP), which is also observed in astrocytes as well as PNS satellite glial cells (SGCs) of the dorsal root ganglia (DRG)32,33,34. Another shared feature of MCs, astrocytes and SGCs is the continuous expression of glutamine synthetase (GS) under physiological circumstances30,35. The expression of this enzyme is crucial for the regulation and recycling of excess glutamate from neuronal synapses, ensuring effective neurotransmitter recovery36,37. Retinal microglia are predominantly located in the inner and outer plexiform layers (IPL, OPL) in the physiological state38. Comparable to brain microglia, their cellular phenotypes can change depending on their activation status39,40. Resting, inactivated microglia show ramified processes39,40,41. Upon activation, a change in morphology towards enlarged, partly amoeboid cell bodies with shortening and thickening of cellular processes towards small pseudopodia can be observed39,40,42. A profound characterization of these morphological and functional changes is not only critical for understanding retinal disease mechanisms but also for the development of potential therapeutic approaches43.
The present study investigates the phenotypic and morphological changes of retinal ganglion cells and retinal glial cells in a late-onset Glb1−/− mouse model during the course of GM1-gangliosidosis. The aim is to characterise neurodegenerative changes and the subsequent reaction pattern of retinal glial cells, with a focus on MCs and microglial cells.
Results
Müller cells upregulate GFAP in 4- and 7-months-old Glb1 −/− mice
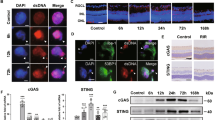
Immunofluorescence (IF) staining of retinal tissue for GFAP (Fig. 1A–D) demonstrated positive signals in WT animals only within innermost retinal layers, most likely detecting MC end feet and perivascular astrocytes (Fig. 1A,C). Increasing GFAP-positive signals in MCs, which also stain vertically orientated cell bodies (Fig. 1B,D), could be detected in Glb1−/− mice with significant differences to WT animals from the age of 4 months. An increasing number of GFAP-positive MCs was detected in knockout mice (Fig. 1I) until the end of the investigation period, indicating an upregulation of GFAP in response to ongoing RGC damage in this Glb1−/− mouse model17,34. This raised the question, whether MCs also showed changes in other cell type specific markers.

Immunofluorescence (A–D) and immunohistochemistry (E–H) targeting glial fibrillary acidic protein (GFAP, green) and glutamine synthetase (GS) in Müller cells (MCs) in 4- and 7-month-old wild type (WT) and Glb1 knockout (Glb1−/−) mice. WT mice show weak GFAP positive signals only within uppermost retinal layers at both time points investigated, most likely in the endfeet of MCs and perivascular astrocytes (A, C). Within 4 and 7-month-old Glb1−/− mice, strong GFAP positive signals can be detected also labeling the cell bodies of MCs, extending into deeper retinal layers (B, D). Nuclei are counterstained with bisbenzimide (blue). 4 and 7-month-old WT and Glb1−/− mice show high numbers of MCs immunopositive for the MC specific marker GS. Quantification of GS positive MCs reveals an increase of GS positive cells in Glb1−/− mice at 4 months of age (F) compared to WT animals (E). No difference was detected between WT and Glb1−/− mice in 7-month-old animals (H), probably due to downregulation of GS in MCs as result of progressive neuronal loss. Scale bars: 20 μm. Statistical analysis of GFAP and GS expression in retinal MCs (I,J). Graphs display box and whisker plots. Significant differences between groups were detected by Kruskal-Wallis test followed by Dunn’s-Bonferroni procedure; *p ≤ 0.05; n = 5. GCL, ganglion cell layer; IPL, inner plexiform layer; INL, inner nuclear layer; OPL, outer plexiform layer.
Early increase and subsequent loss of glutamine synthetase positive Müller cells in Glb1 −/− mice
Immunohistochemistry (IHC) targeting GS (Fig. 1E–H) and subsequent quantification of positive cells revealed significant differences in the number of GS-positive MCs between WT and Glb1−/− mice starting at 4 months of age (Fig. 1J). A significant increase was also detected between 4-month-old and 7-month-old WT mice, indicating age-related changes. Interestingly, 7-month-old Glb1−/− mice showed no significant difference in GS-positive MCs compared to their age-matched WT counterparts but a significant decrease when compared to 4-month-old Glb1−/− mice. In order to determine the possible interactions of these partly transient phenotypic changes with neuronal changes in more detail, the RGCs were examined more closely.
Neuronal loss is accompanied by increased accumulation of storage material and decreased expression of transcription marker BRN3A in 7-month-old Glb1 −/− mice
IHC targeting βIII-tubulin, a pan-neuronal marker, was used for overall quantification of neuronal cells of the ganglion cell layer (Fig. 2A–D). Data (Fig. 2E) indicate a significant loss of neurons in 7-month-old Glb1−/− mice when compared with all other experimental groups, including WT mice at 4 and 7 months of age but also Glb1−/− mice at 4 months. This indicates a progressive loss of neuronal cells, especially at late time points at an advanced stage of the disease.

Immunohistochemistry (IHC) targeting βIII-tubulin for visualization of retinal ganglion cells (RGCs) (A–D) and Caspase 3 for quantification of neuronal apoptosis (F–I) in wild type (WT) mice as well as Glb1 knockout (Glb1−/−) mice at 4- and 7-month-old. Positive cytoplasmic staining can be detected in retinal ganglion cells (RGCs) with prominent intracytoplasmic vacuoles especially in 7-month-old Glb1−/− mice (A–D). Quantification of RGCs revealed that 7-month-old Glb1−/− mice show a significant reduction of neuronal cells compared to all other groups, including 4-month-old Glb1−/− mice (E). Using IHC targeting cleaved caspase-3 a weak intranuclear signal was detected especially in 7-month-old Glb1−/− RGCs (I, arrows). Within 4-month-old WT (F) and Glb1−/− mice (H) no immunopositive reaction was observed. Within the retina of 7-month-old WT mice (G, arrows), a mildly increased number of ganglion cells with intranuclear positive results (arrow) could be detected (J). 7-month-old Glb1−/− mice show a significant increase in caspase 3-positive RGCs (arrows; insert) compared to all other investigation groups (J). Scale bars: 20 μm; Insert (C,D,I): 10 μm. Graphs display box and whisker plots. Significant differences between groups were detected by Kruskal–Wallis test followed by Dunn’s–Bonferroni procedure; *p ≤ 0.05; n = 5. GCL, ganglion cell layer; IPL, inner plexiform layer; INL, inner nuclear layer.
In addition, IHC targeting cleaved caspase-3 to screen for neuronal apoptosis (Fig. 2F–I) was used. No caspase 3-positive cells were detected in 4-month-old WT and Glb1−/− mice (Fig. 2F,H). Within WT mice at 7 months of age, few immunopositive neurons were detected (Fig. 2G), while comparable staining results were frequently seen in neurons of 7-month-old Glb1−/− mice (Fig. 2I). RGCs showed weak, brownish, punctate-like, nuclear staining44. Cells appeared partially shrunken with condensed chromatin, suggesting apoptosis based on IHC staining and morphology. 7-month-old WT and Glb1−/− mice showed significantly increased numbers of positive cells when compared to 4-month-old animals. Moreover, Glb1−/− mice at 7 months of age showed a significant increase in apoptotic ganglion cells compared to age-matched WT animals (Fig. 2J).
In parallel, GCL neurons with intracytoplasmic accumulation of GM1 storage material were quantified as percentage of all GCL neurons examined (Fig. 3). WT animals showed no positive signals (Fig. 3A,C) while neurons with granular to vacuolated signals were detected in 4- and 7-month-old Glb1−/− mice (Fig. 3B,D). Statistical analysis revealed significant differences between both WT groups and 7-month-old knockout mice (Fig. 3G). Interestingly, cells within the apical layer of the INL showed positive signals in Glb1−/− mice, indicating storage material accumulation also within this cell population, while no signals were detected within the ONL.

Representative images of immunohistochemistry targeting storage material (GM1) within the retinas of 4- (A) and 7-month-old (C) wild type (WT) as well as 4- (B) and 7-month-old (D) Glb1 knockout (Glb1−/−) mice as well as BRN3A-positive neurons of the GCL in 7-month-old WT (E) and Glb1−/− (F) mice. Storage material is not detected in neurons of the ganglion cell layer in WT mice at both time points (A, C; thick arrows). Several neurons with brownish, granular, intracytoplasmic signals can be observed in 4- and 7-month old Glb1−/− mice (B,D; thin arrows). Some neurons without obvious intracytoplasmic signals can be observed, especially within 4-month-old Glb1−/− mice (B; thick arrow). In 7-month-old animals there is loss of visible neuronal nuclei (D; open arrowhead). Moreover, within the inner nuclear layer, scant (B; closed arrowheads) and more pronounced (D; closed arrowheads) accumulation of storage material can be detected. No positive signals were detected within the outer nuclear layer. A-F: scale bars: 20 μm. G,H: Graphs display box and whisker plots. Significant differences between groups were detected by Kruskal–Wallis test followed by Dunn’s–Bonferroni procedure; * p ≤ 0.05; n = 5. GCL, ganglion cell layer; IPL, inner plexiform layer; INL, inner nuclear layer; OPL, outer plexiform layer; ONL, outer nuclear layer.
To further investigate RGC integrity, IHC for BRN3A, a transcription factor specifically expressed in RGCs, was conducted. BRN3A-positive cells were clearly detectable in the GCL of both WT and Glb1−/− mice (Fig. 3E,F). Quantification and statistical analysis revealed a significant reduction of BRN3A positive RGCs in 7-month-old Glb1−/− mice (Fig. 3F,H). BRN3A cell counts remained stable in WT mice between 4 and 7 months, indicating a progressive loss of BRN3A positivity in Glb1−/− mice.
Disease progression is associated with increased numbers, directed migration and morphologic changes in Iba1-positive cells in Glb1 −/− mice
Whether these degenerative changes are associated with a secondary reaction of phagocytic cells was addressed by studying the role of ionized calcium-binding adapter molecule 1 (Iba1) positive cells (microglia/macrophages). Iba1-positive cells were detected in both WT and Glb1−/− mice at 4 and 7 months of age (Fig. 4A–D). Statistical analysis of Iba1-positive cells within the whole retina (Fig. 4E) showed a significant increase in Glb1−/− mice compared to WT mice. Interestingly, no significant difference was observed between 4- and 7-month-old Glb1−/− mice, indicating an early and consistent microglial response. Moreover, no significant difference was detected between young and old WT mice. Analysis of the distribution of Iba1-positive cells across the different retinal layers (Fig. 4F) revealed an increase of microglia/macrophages especially affecting the inner retinal layers (ganglion cell layer: GCL, inner plexiform layer: IPL) of Glb1−/− mice with 7-month-old animals showing highest cell numbers, especially within the GCL. Only few Iba1-positive cells were detected in the outer retinal layers in all groups. In addition, the morphological phenotype of Iba1-positive cells was investigated (Fig. 5A–G). In Glb1−/− mice, a significant increase in spiky and amoeboid Iba1-positive cells was observed (Fig. 5I,J). Interestingly, in contrast to the amoeboid morphology predominantly seen in the IPL adjacent to the GCL in 7-month-old animals, the OPL exhibited a significant increase in spiky Iba1-positive cells in 4 and 7-month-old Glb1−/− mice (Fig. 5I,J), which suggests a gradual activation and migration of Iba1-positive cells towards the GCL. In contrast, ramified Iba1-positive cells were more abundant in the IPL and OPL of WT mice (Fig. 5H).

Immunohistochemistry targeting Iba1-positive cells within retinal layers. Wild type (WT) animals show low numbers of Iba1-positive cells in 4- (A) and 7-month-old mice (B). Increased numbers of Iba1-positive cells can be detected in 4- (C) and 7-(D) month-old Glb1 knockout (Glb1−/−) mice, especially in the ganglion cell layer (GCL) and inner plexiform layer (IPL). Scale bars: 50 μm. Quantification of total numbers of retinal Iba1 positive cells revealed significant differences between Glb1−/− and WT mice (E). Quantification of Iba1-positive cells according to different retinal layers shows a predominant increase in innermost retinal layers (GCL, IPL) compared to, for example, the outer plexiform layer (OPL) (F). Graphs display box and whisker plots. Significant differences between groups were detected by Kruskal-Wallis test followed by Dunn’s-Bonferroni procedure; * p ≤ 0.05; n = 5. INL, inner nuclear layer. Folds within the inner layers of retinal tissue have been digitally lightened.

Representative images of immunohistochemistry targeting Iba1 demonstrating distinct microglia/macrophage morphologies within different retinal layers. Ramified microglia, characterized by long and thin cellular processes around a small, distinct soma, were mostly located within OPL and IPL of wild type (WT) mice. A ramified morphology indicates a non-activated, resting state (A, B), which is most abundant in the OPL and IPL of WT mice (H). Activation of microglia/macrophages in Glb1 knockout (Glb1−/−) mice leads to shortening and thickening of processes resulting in a spiky morphology and enlarged, more irregular cell bodies. Spiky Iba1-positive cells (C, E: arrow, G) were mostly detected in the OPL of Glb1−/− mice (I). Amoeboid microglia/macrophages display a roundish morphology with very short or even absent processes as well as an increased diameter of the soma (D, E: arrowhead, F). This phenotype is mostly detected in the IPL layers of Glb1−/− mice (J). Scale bars: 10 μm. Graphs display box and whisker plots. Significant differences between groups were detected by Kruskal-Wallis test followed by Dunn’s-Bonferroni procedure; *p ≤ 0.05; n = 5. IPL, inner plexiform layer; OPL, outer plexiform layer.
Müller glial cells are not affected by intracytoplasmic storage of GM1-ganglioside
To further analyze the impact of GM1 ganglioside storage material accumulation on MCs reactivity, immunofluorescence double stainings combining anti-GM1 with GFAP or GS-staining were performed. The analysis confirmed the absence of detectable GM1 immunoreactivity in MCs, as no colocalisation of GM1 and either GFAP or GS was observed (Fig. 6A,B). In contrast, neurons in the GCL and INL displayed immunopositivity for GM1 (Fig. 6, arrows), indicating that storage material is primarily localized within neurons. In addition, transmission electron microscopic investigation of the retina of 7-month-old Glb1−/− mice also revealed no indications for intracytoplasmic accumulation of GM1 storage material in MCs (Fig. 7). However, typical electron-dense, lamellated, concentric or whorled membranous inclusions of storage material, resembling myelin figures or ”zebra bodies” were clearly observed in neurons of RGC (Fig. 7A) and INL, as well as in both bipolar and amacrine cells in the INL (Fig. 7B,C). These findings emphasise the dominant susceptibility of neurons in the retina to GM1 accumulation.

Immunofluorescence (IF) double staining of murine retinal tissue from a 7-month-old Glb1 knockout mouse. IF targeting GM1-1 shows an accumulation of GM1-positive lysosomal storage material (green; arrows) in ganglion cells (A,B). IF targeting glutamine synthetase (red, A) and GM1 (green) as well as glial fibrillary acidic protein (red, B) and GM1 (green) does not show colocalisation. This suggests that, in contrast to neurons of the RGC and INL (arrows), Müller cells are not affected by an excessive accumulation of storage material. Nuclei are counterstained with bisbenzimide (blue). Scale bars: 20 μm. GCL, ganglion cell layer; IPL, inner plexiform layer; INL, inner nuclear layer; OPL, outer plexiform layer.

Transmission electron microscopic images illustrating the ultrastructural anatomy of the inner retinal tissue of a 7-month-old Glb1 knockout (Glb1−/−) mouse. Müller cells (MCs) appear morphologically intact, with no evidence of storage material accumulation, while neurons show characteristic membranous inclusions of GM1 ganglioside. (A) The inner limiting membrane (ILM; outlined in purple) is formed by the “endfeet” of MCs. Müller cell endfeet (MCef) show mitochondrial (Mi) swelling (arrow). Retinal ganglion cells (RGC) and macrophages/microglia (M) show large phagolysosomes (PLs; outlined in green) with membranous inclusions (asterisk); bar: 1000 nm. (B) Amacrine cells (ACs) and bipolar cells (BCs) in the inner nuclear layer are tightly enveloped by the cell body of MCs (purple overlay ) and exhibit cytoplasmic membranous inclusions of GM1 gangliosides; bar: 2500 nm; insert: higher magnification in (C). (C) Higher magnification of a large phagolysosome with accumulation of electron dense, laminar, membranous inclusions of storage material resembling myelin figures or “zebra bodies” (white arrows); bar: 500 nm.
Discussion
The present study is the first to describe retinal changes in a late-onset murine model mimicking the adult form of GM1-gangliosidosis. Already at the age of 4 months, significant changes were found in the retina of knockout mice, especially with regard to the glial cell populations examined. Reactive glial changes within the retina can be categorized into “stereotypic” non-specific and specific responses28,45. The non-specific response is characterized by MC hypertrophy, proliferation, and upregulation of the intermediate filaments vimentin and GFAP32,46,47. In the present study, a significant and continuous increase of GFAP-positive MCs in Glb1−/− mice was noted. This finding suggests activation in response to neuronal damage, especially since damage to the MCs themselves by intracytoplasmic accumulation of storage material could be excluded by immunofluorescence double staining and electron microscopy. Furthermore, an increase in GFAP-staining due to astrogliosis was excluded based on the cellular morphology of the counted cells and the localization of their nuclei within the INL. Interestingly, the upregulation of GFAP in MCs is consistent with previous observations in astrocytes and SGCs in animals of the same age in the same mouse model17,34. Similar to astrocytes in the brain and SGCs within DRG, MCs in the retina are able to upregulate GFAP expression in response to neuronal injury and dysfunction26,48,49. This indicates a universal mechanism of reactive gliosis, emphasizing a conserved glial response to neuronal ganglioside accumulation across diverse nervous compartments49,50,51. MC gliosis, as a common phenomenon in the pathogenesis of non-degenerative and degenerative retinopathies, is also considered a double-edged sword28,47. Initially, it supports neuroprotection and the release of neurotrophic factors and antioxidants, which favour neuronal survival and limit the extent of tissue damage. However, excessive gliosis can proceed into exuberant, long-lasting gliosis, which results in retinal scarring as well as increased neurotoxicity and neurodegeneration27,52. Few GFAP-positive MCs were also detected in 7-month-old WT mice, most likely indicating a certain degree of reactivity connected to age-related changes.
The specific response of MC activation is more targeted and can involve changes in GS expression, as demonstrated in the present study. Among retinal macroglia, MCs exhibit the highest levels of GS expression36,53. GS has a crucial function in the glutamate-glutamine cycle. GS regulates neurotransmitter metabolism by converting glutamate to glutamine, thereby maintaining a stable microenvironment for RGCs and preventing neuronal excitotoxicity. It is therefore continuously expressed in healthy retinal tissue28,54,55,56. In Glb1−/− mice, the number of GS-positive MCs in 4-month-old Glb1−/− mice was significantly increased compared to age-matched WT controls, most likely along with MC gliosis, as also indicated by GFAP-staining. However, in 7-month-old Glb1−/− mice, the amount of GS-positive cells was comparable to WT counterparts. Notably, GS-positive MCs in 7-month-old Glb1−/− mice were reduced compared to 4-month-old knockout mice. This result suggests a reduced expression of GS in MCs, which is most likely linked to the ongoing degeneration of RGC. Downregulation of GS expression by MCs has been previously reported after loss of major glutamate-releasing neurons, e.g., after inherited photoreceptor degeneration, retinal detachment, inflammatory and traumatic conditions, as well as in glaucoma in humans or other species28,33,45. The expression of GS in MCs is another common feature with astrocytes in the brain and SGC of the DRG, where GS also plays a key role in the removal of excess extracellular excitatory glutamate57,58,59. However, the exact pathogenic mechanism behind GS dysregulation in MCs in retinopathies and whether these changes contribute to visual impairment in the Glb1−/− model require further investigation. The results obtained suggest that GFAP and GS represent suitable indicators for reactive, potentially noxious changes in MC activity and could therefore also be interesting pharmaceutical targets for intervention strategies.
Retinal changes in a transgenic mouse model of juvenile/early-onset GM1-gangliosidosis were characterized by the detection of storage material within ganglion cells60. Intracytoplasmic accumulation of lamellated storage material in swollen RGCs was also detected in affected calves and cats using electron microscopy, comparable to the results of the present study11,61. A significant reduction in RGCs was observed in 7-month-old Glb1−/− mice, most likely as a result of excessive intracytoplasmic accumulation of GM1, as demonstrated using IHC. Loss of RGCs has also been observed in human patients with GM1-gangliosidosis as well as in various animal species, including cats, dogs and calves19,22,23,24,25. Within the context of GM1-gangliosidosis, ganglioside-mediated activation of the unfolded protein response could trigger RGC apoptosis62. Using IHC targeting cleaved caspase-3, a robust marker of apoptosis63,64,65, increasing numbers of cells with faint intranuclear labelling were observed. While procaspase-3 is located within the cytoplasm, active caspase-3 has been shown to be translocated into the nucleus66. A mildly increased number of cleaved caspase-3 positive cells was also detected in 7-month-old WT mice, suggesting an age-related phenomenon. Further insight into RGC integrity was gained through BRN3A immunostaining, a specific marker for RGCs67. Quantitative analysis revealed a significant reduction in BRN3A-positive cells in 7-month-old Glb1−/− mice compared to all other groups. While BRN3A, a transcription factor, is not consistently and uniformly expressed in all RGC, its expression is known to decline in response to stress or early degeneration67,68. Studies have shown that it is downregulated in injured or degenerating neurons and that this precedes overt cell loss in some models. A decrease in BRN3A is also connected to sensory neuronal apoptosis [66, 68]. In the context of lysosomal storage disorders, transcriptional dysregulation due to substrate accumulation and cellular stress may contribute to neuronal vulnerability and apoptosis [62, 68]. Therefore, the observed reduction in BRN3A positive RGCs in Glb1−/− mice likely reflects a combination of actual cell loss and downregulation of survival-promoting transcriptional programs. The nuclear caspase-3 staining, together with the decrease in BRN3A positive RGCs and the increase in amoeboid Iba-1 positive cells in vicinity of the RGCs, suggests apoptosis of retinal neurons in Glb1−/− mice66. These findings further underscore the degenerative nature of retinal involvement in GM1-gangliosidosis.
Resident microglia in the retina also play a pivotal role in maintaining neuronal homeostasis. They are involved in immune surveillance and react quickly to retinal damage. However, a comprehensive characterization of the functions and tasks of retinal microglia is still incomplete in many areas [69]. In the present study, a significant increase in the total number of Iba1-positive cells was detected in Glb1−/− mice at 4 and 7 months of age. This finding suggests an early and consistent activation of Iba1-positive cells during GM1-gangliosidosis. Under physiological conditions, Iba1-positive cells are predominantly located in the synaptic inner and outer plexiform layers (IPL and OPL) and are less frequently found within nuclear layers and around RGC and their axons38. Interestingly, in Glb1−/− mice, not only an increase but also a spatial redistribution of Iba1-positive cells was detected. Previous studies in mouse and rat models for retinal degeneration reported migration of activated microglia throughout the retinal layers after photoreceptor loss69,70. In the present study, targeted migration towards the inner retinal layers was demonstrated, indicating a specific immune response towards neuronal damage and loss. In addition, morphological changes were observed, with microglia shifting from a ramified morphology to spiky and amoeboid phenotypes. A ramified to hyper-ramified morphology is thought to correlate with an inactivated functional state of microglia. Thin ramified processes allow continuous monitoring of the neuronal environment. Upon activation, the cells quickly adapt their morphology by shortening their processes and adopting a spiky or amoeboid phenotype with small pseudopodia, which enables rapid and dynamic migration to injury sites26,39,40,73. Compatible with this, higher numbers of spiky and amoeboid Iba1-positive cells were detected in the IPL and OPL of Glb1−/− mice, again indicating a specific response towards neuronal damage and loss. Iba1-positive cells in retinal tissue of WT mice were primarily detected in IPL and OPL and predominantly exhibited a ramified morphology at both time points investigated.
The present study demonstrates that the Glb1−/− mouse strain used in this investigation is suitable for characterizing the pathologic changes within the retina in a model of the adult form of GM1-gangliosidosis. The obtained results are consistent with changes described in bovine, feline, canine, and also human retinas22,23,24,25. Rodent models are highly valuable for research of retinal changes in GM1-gangliosidosis, especially due to wide range of possibilities of genetic manipulation74,75. However, species-specific differences and limitations, such as the sheer size of the eyeball in mice, must be considered when selecting suitable models. The cherry-red macula in GM1-gangliosidosis patients cannot be depicted in the listed animal models, as no comparable structure exists in animals other than primates.
Further studies including an assessment of visual capacity and a correlation with pathohistological and functional changes in retinal cells will be highly valuable. Furthermore, aditional research is needed to investigate the interactions between neurons and glial cells in order to explore the key pathophysiological mechanisms and develop ways to effectively alleviate the effects of the disease.
Materials and methods
Animals and tissue processing
WT and Glb1−/− mice were sacrificed as part of an unrelated study. Briefly, Glb1−/− mice were generated via insertion of a 636 bp fragment of the lacZ gene into the Glb1 exon 1517. Mice were kept in ventilated cages on a 12 h light/dark cycle with ad libitum access to food and water. Breeding and genotyping was performed as described previously17. Ocular tissue samples were harvested from 5 male and female, 4- and 7-month-old WT and Glb1−/− mice, respectively. During previous own studies using this mouse line, no sex-specific differences were obtained17,76. Mice were initially anaesthetized via intraperitoneal injection of medetomidine (Domitor®, Pfizer 1.0 mg/ml) and ketamine (Ketamin 10%, WDT, 100 mg/ml) diluted in NaCl at a dosage of 0.5 mg/kg medetomidine and 100 mg/kg ketamine, and subsequently euthanized by an intraperitoneal injection of an overdose of undiluted medetomidine and ketamine. Afterwards animals were transcardially perfused with phosphate-buffered saline (PBS), and the eye globes were carefully removed and fixed in 10% neutrally-buffered formalin for a maximum of 24 h. To provide comparable section levels for the subsequent investigations, fixed globes were sectioned vertically, with one half containing the optic nerve. Subsequently, tissue was embedded in paraffin wax (formalin-fixed and paraffin-embedded material; FFPE) with the cut surface facing downwards. The tissue was cut into approximately 3 μm thick serial sections using a microtome and subsequently mounted on SuperFrost-Plus® slides (Thermo Fisher Scientific Inc., Fisher Scientific GmbH, Schwerte, Germany). For electron microscopy, eyes were fixed in 2.5% glutaraldehyde for at least 24 h.
Ethics declaration
All animal experiments were performed in accordance with the German Animal Welfare Law and were approved by the local authorities (Niedersächsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit (LAVES), Oldenburg, Germany, permission number 33.8-42502-21/3632). This study is reported in accordance with the ARRIVE guidelines and conforms to its principles.
Light and electron microscopy
Ocular sections were stained with hematoxylin and eosin (H&E) and examined using routine light microscopy. For transmission electron microscopy, samples were post-fixed with 1% osmium tetroxide and embedded in EPON 812 (Serva, Heidelberg, Germany). Semi-thin sections (ca. 0.5–1 μm-thick) were stained with toluidine blue and again investigated with a routine light microscope. 70 nm ultra-thin sections were cut with diamond knife (Diatome, Hatfield, USA) from selected regions of interest (ROI; from ILM till the end of the INL) and contrasted with uranyl acetate followed by lead citrate. Ultrastructural analyses were performed using a transmission electron microscope (Zeiss EM 906, Oberkochen, Germany) as previously described77.
Immunohistochemistry and immunofluorescence
IHC on retinal sections was performed using primary antibodies targeting GS, GFAP, Iba1, βIII-tubulin (pan-neuronal marker), BRN3A (POU4F1; specific for GCL-neurons), cleaved caspase-3 and GM1-1. Briefly, IHC on FFPE tissue sections was performed using 0.5% H2O2 for 30 min to block endogenous peroxidase. For all antibodies applied, antigen retrieval was achieved by boiling for 20 min in citrate buffer (pH 6) or Tris-EDTA (BRN3A). To block non-specific binding sites, slides were incubated with 20% goat or horse (Iba1) serum diluted in PBS for 30 min. Subsequently, sections were incubated with the respective primary antibody (Table 1) overnight at 4 °C. The negative controls were incubated with inactivated serum of the host species of the primary antibody. Biotinylated goat-anti-rabbit IgG (BA-1000) or goat-anti-mouse IgG (BA-9200) diluted 1:200 (Vector Laboratories, Burlingame, CA, USA) were used as secondary antibodies. After visualization of the antigen-antibody reaction using the avidin-biotin-peroxidase complex (ABC) method (Vector Laboratories) and the chromogen 3,3′-diamino-benzidine (DAB), sections were counterstained with Mayer’s hematoxylin. For immunofluorescence (IF) staining, sections were also treated with H2O2 and incubated in citrate buffer (pH 6) for 20 min in a microwave. Afterwards, they were blocked with 20% goat serum in PBS containing 1% bovine serum albumin (BSA) and 0.1% Triton-X (Triton® X-100, Merck Millipore, Merck KGaA). For single staining procedure, primary antibody targeting GFAP was incubated overnight at 4 °C. Negative controls were incubated with inactivated serum as described above. Secondary antibody (goat anti-rabbit Alexa Fluor 488; 1:200; 111-545-003; Jackson ImmunoResearch Europe Ltd) was applied at room temperature for 1 h. Specific markers were selected for double staining procedures (GFAP/GM1; GS/GM1). The first primary antibodies (GS; GFAP) followed by incubation of the second first antibody (GM1-1) were incubated overnight at 4 °C. Afterwards, an appropriate secondary antibody (goat anti-rabbit Alexa Fluor 488; 1:200; 111-545-003 and goat anti-mouse Alexa Fluor 488; 1:200; 115-545-003; Jackson ImmunoResearch Europe Ltd) was applied at room temperature for 1 h. To visualize the nuclei, bisbenzimide (diluted in double distilled water; bisbenzimide H 33258, Merck KGaA) was used, followed by mounting sections with a fluorescence mounting medium (Dako North America Inc.). Immunofluorescence images were taken with a Keyence BZ9000 fluorescent microscope (Keyence) with Nikon Plan Apo λ objectives (Nikon Europe BV)77.
Tissue analysis, evaluation of cell density, and statistical analysis
Slides were digitalized using the Olympus VS200 slidescanner (Olympus Deutschland GmbH, Hamburg, Germany) to quantify immunopositive cells within different retinal tissue layers. The scanned slides were analyzed using QuPath for digital pathology image analysis77. Only intact, full-thickness retinal sections of good quality were chosen. For quantification of neurons with positive nuclear/cytoplasmic reaction for BRN3A or GM1-1, respectively, neurons of the GCL were counted manually on digitalized images. For evaluation of overall ganglion cell density, as well as quantification of GS and GFAP positive MCs, regions of interest (ROI; retinal tissue from ILM to the end of photoreceptor outer segment) were outlined manually for both immunostaining methods. Specifically, MCs immunopositively labelled for GFAP and GS were identified by their cell body in the INL and their radial processes extending the entire retinal thickness. Cell counts were determined manually in all ROIs, and only immunopositive cells with identifiable nuclei in the INL were included.
Analysis of Iba1 cell distribution and morphological characteristics
For further quantification and examination of the distribution pattern of microglia/macrophages, Iba1 immunopositive cells were evaluated in all retinal layers. Only cells with clearly identifiable nuclei/soma were included. Additionally, the morphology of positive cells in each retinal layer was determined according to a previously published evaluation scheme39,40. Cells were categorized as ramified (”resting”), spiky (“activated”) or amoeboid (“activated”) according to their shape39,40. Cells categorized as ramified Iba1 immunolabelled cells were characterized by spider-like morphology with numerous linear processes and thin ramifications as well as clear borders of the soma. In contrast, Iba1-positive cells displaying shortened processes (larger near the soma and smaller in the periphery) and different diameters were interpreted as spiky cells. In addition, cells with round morphology and very short processes or even without processes, displaying an increased diameter of the soma larger than that of the spiky cells, were categorized as amoeboid cells.
Statistical analysis
Statistical data analysis was performed using SPSS for Windows (version 29.0.1.0; IBM® SPSS® Statistics, SPSS Inc., Chicago, IL, USA). Normal distribution was tested via Kolmogorov–Smirnov and Shapiro–Wilk tests. Differences between individual groups were analyzed via the Kruskal–Wallis test followed by Dunn’s post-hoc tests and adjustment for multiple testing using the Bonferroni procedure. Statistical significance was accepted at a p-value of < 0.05.
Data availability
The data are available from the corresponding author upon request.
References
Dacremont, G. & Kint, J. A. GM1-ganglioside accumulation and beta-galactosidase deficiency in a case of GM1-gangliosidosis (Landing disease). Clin. Chim. Acta. 21, 421–425 (1968).
Jatzkewitz, H. & Sandhoff, K. On a biochemically special form of infantile amaturotic idiocy. Biochim. Biophys. Acta. 70, 354–356 (1963).
Caciotti, A. et al. GM1 gangliosidosis: molecular analysis of nine patients and development of an RT-PCR assay for GLB1 gene expression profiling. Hum. Mutat. 28, 204 (2007).
Jarnes Utz, J. R. et al. Infantile gangliosidoses: mapping a timeline of clinical changes. Mol. Genet. Metab. 121, 170–179 (2017).
Baker, H. J. & Lindsey, J. R. Animal model: feline GM1 gangliosidosis. Am. J. Pathol. 74, 649–652 (1974).
Nicoli, E. R. et al. GM1 gangliosidosis-a mini-review. Front. Genet. 12, 734878 (2021).
Rha, A. K., Maguire, A. S. & Martin, D. R. GM1 gangliosidosis: mechanisms and management. Appl. Clin. Genet. 14, 209–233 (2021).
Uddin, M. M. et al. Molecular epidemiology of canine GM1 gangliosidosis in the Shiba Inu breed in japan: relationship between regional prevalence and carrier frequency. BMC Vet. Res. 9, 132 (2013).
Uddin, M. M. et al. Mutation analysis of GM1 gangliosidosis in a Siamese Cat from Japan in the 1960s. J. Feline Med. Surg. 14, 900–902 (2012).
Ryder, S. J. & Simmons, M. M. A lysosomal storage disease of Romney sheep that resembles human type 3 GM1 gangliosidosis. Acta Neuropathol. 101, 225–228 (2001).
Sheahan, B. J., Donnelly, W. J. & Grimes, T. D. Ocular pathology of bovine GM1 gangliosidosis. Acta Neuropathol. 41, 91–95 (1978).
Bermudez, A. J. et al. Heritability and biochemistry of gangliosidosis in emus (Dromaius novaehollandiae). Avian Dis. 41, 838–849 (1997).
Muthupalani, S. et al. GM1-gangliosidosis in American black bears: clinical, pathological, biochemical and molecular genetic characterization. Mol. Genet. Metab. 111, 513–521 (2014).
Hahn, C. N. et al. Generalized CNS disease and massive GM1-ganglioside accumulation in mice defective in lysosomal acid beta-galactosidase. Hum. Mol. Genet. 6, 205–211 (1997).
Matsuda, J. et al. Beta-galactosidase-deficient mouse as an animal model for GM1-gangliosidosis. Glycoconj. J. 14, 729–736 (1997).
Przybilla, M. J. et al. Examination of a blood–brain barrier targeting beta-galactosidase-monoclonal antibody fusion protein in a murine model of GM1-gangliosidosis. Mol. Genet. Metab. Rep. 27, 100748 (2021).
Eikelberg, D. et al. Axonopathy and reduction of membrane resistance: key features in a new murine model of human GM1-Gangliosidosis. J. Clin. Med. 9, (2020).
Liu, S. et al. A GM1 gangliosidosis mutant mouse model exhibits activated microglia and disturbed autophagy. Exp. Biol. Med. 246, 1330–1341 (2021).
Chen, H. et al. Beyond the cherry-red spot: ocular manifestations of sphingolipid-mediated neurodegenerative and inflammatory disorders. Surv. Ophthalmol. 59, 64–76 (2014).
Cairns, L. J., Green, W. R. & Singer, H. S. GM1 gangliosidosis, type 2: ocular clinicopathologic correlation. Graefes Arch. Clin. Exp. Ophthalmol. 222, 51–62 (1984).
Matmat, K. et al. Ocular manifestations in patients with inborn errors of intracellular cobalamin metabolism: a systematic review. Hum. Genet. 141, 1239–1251 (2022).
Baker, H. J. Jr. et al. Neuronal GM1 gangliosidosis in a Siamese Cat with beta-galactosidase deficiency. Science. 174, 838–839 (1971).
Donnelly, W. J., Sheahan, B. J. & Kelly, M. Beta-galactosidase deficiency in GM1 gangliosidosis of Friesian calves. Res. Vet. Sci. 15, 139–141 (1973).
Murray, J. A., Blakemore, W. F. & Barnett, K. C. Ocular lesions in cats with GM1-gangliosidosis with visceral involvement. J. Small Anim. Pract. 18, 1–10 (1977).
Muller, G. et al. GM1-gangliosidosis in Alaskan huskies: clinical and pathologic findings. Vet. Pathol. 38, 281–290 (2001).
Vecino, E. et al. Glia-neuron interactions in the mammalian retina. Prog. Retin Eye Res. 51, 1–40 (2016).
Bringmann, A. & Wiedemann, P. Muller glial cells in retinal disease. Ophthalmologica. 227, 1–19 (2012).
Bringmann, A. et al. Muller cells in the healthy and diseased retina. Prog. Retin Eye Res. 25, 397–424 (2006).
Coorey, N. J. et al. The role of glia in retinal vascular disease. Clin. Exp. Optom. 95, 266–281 (2012).
Reichenbach, A. & Bringmann, A. New functions of Muller cells. Glia 61, 651–678 (2013).
Balaratnasingam, C. et al. Studies of the retinal microcirculation using human donor eyes and high-resolution clinical imaging: insights gained to guide future research in diabetic retinopathy. Prog. Retin Eye Res. 94, 101134 (2023).
Bignami, A. & Dahl, D. Radial glia of Muller in the rat retina and their response to Injury - Immunofluorescence study with antibodies to the glial fibrillary acidic (Gfa) protein. Exp. Eye Res. 28, 63–69 (1979).
Grosche, J., Hartig, W. & Reichenbach, A. Expression of glial fibrillary acidic protein (GFAP), glutamine synthetase (GS), and Bcl-2 protooncogene protein by Muller (glial) cells in retinal light damage of rats. Neurosci. Lett. 185, 119–122 (1995).
Huang, B. et al. Phenotypical changes of satellite glial cells in a murine model of G(M1) -gangliosidosis. J. Cell. Mol. Med. 26, 527–539 (2022).
Hanani, M. Satellite glial cells in sensory ganglia: from form to function. Brain Res. Brain Res. Rev. 48, 457–476 (2005).
Linser, P. J., Sorrentino, M. & Moscona, A. A. Cellular compartmentalization of carbonic anhydrase-C and glutamine synthetase in developing and mature mouse neural retina. Brain Res. 315, 65–71 (1984).
Xu, Q. A. et al. Muller cell degeneration and microglial dysfunction in the alzheimer’s retina. Acta Neuropathol. Commun. 10, 145 (2022).
Guo, L. et al. Microglia: key players in retinal ageing and neurodegeneration. Front. Cell. Neurosci. 16, 804782 (2022).
Schwabenland, M. et al. Analyzing microglial phenotypes across neuropathologies: a practical guide. Acta Neuropathol. 142, 923–936 (2021).
Prinz, M. et al. Microglia and central nervous System-Associated Macrophages-From origin to disease modulation. Annu. Rev. Immunol. 39, 251–277 (2021).
Fan, W. et al. Retinal microglia: functions and diseases. Immunology. 166, 268–286 (2022).
Davis, E. J., Foster, T. D. & Thomas, W. E. Cellular forms and functions of brain microglia. Brain Res. Bull. 34, 73–78 (1994).
Devoldere, J. et al. Muller cells as a target for retinal therapy. Drug Discov. Today. 24, 1483–1498 (2019).
Zeiss, C. J., Neal, J. & Johnson, E. A. Caspase-3 in postnatal retinal development and degeneration. Investig. Ophthalmol. Vis. Sci. 45, 964–970 (2004).
de Hoz, R. et al. Retinal macroglial responses in health and disease. Biomed. Res. Int. 2016, 2954721 (2016).
Eisenfeld, A. J., Bunt-Milam, A. H. & Sarthy, P. V. Muller cell expression of glial fibrillary acidic protein after genetic and experimental photoreceptor degeneration in the rat retina. Investig. Ophthalmol. Vis. Sci. 25, 1321–1328 (1984).
Bringmann, A. & Reichenbach, A. Role of Muller cells in retinal degenerations. Front. Biosci. 6, 72–92 (2001).
Pfeiffer, R. L., Marc, R. E. & Jones, B. W. Muller cell metabolic signatures: evolutionary conservation and disruption in disease. Trends Endocrinol. Metab. 31, 320–329 (2020).
Hanani, M. & Verkhratsky, A. Satellite glial cells and astrocytes, a comparative review. Neurochem. Res. 46, 2525–2537 (2021).
Middeldorp, J. & Hol, E. M. GFAP in health and disease. Prog. Neurobiol. 93, 421–443 (2011).
Sullivan, S.M., GFAP variants in health and disease: stars of the brain… and gut.J. Neurochem. 130, 729–732 (2014).
Peña, J. S. & Vazquez, M. Harnessing the neuroprotective behaviors of Müller glia for retinal repair. Front. Biosci. 27, 169 (2022).
Riepe, R. E. & Norenburg, M. D. Muller cell localisation of glutamine synthetase in rat retina. Nature. 268, 654–655 (1977).
Izumi, Y. et al. Müller cell swelling, glutamate uptake, and excitotoxic neurodegeneration in the isolated rat retina. Glia. 25, 379–389 (1999).
Rauen, T. & Wiessner, M. Fine tuning of glutamate uptake and degradation in glial cells: common transcriptional regulation of GLAST1 and GS. Neurochem. Int. 37, 179–189 (2000).
Pow, D. V. & Robinson, S. R. Glutamate in some retinal neurons is derived solely from glia. Neuroscience. 60, 355–366 (1994).
Huang, B. et al. Phenotypical peculiarities and species-specific differences of canine and murine satellite glial cells of spinal ganglia. J. Cell. Mol. Med. 25, 6909–6924 (2021).
Fernández-Montoya, J., Avendaño, C. & Negredo, P. The glutamatergic system in primary somatosensory neurons and its involvement in sensory Input-Dependent plasticity. Int. J. Mol. Sci. 19, (2017).
Verkhratsky, A. & Nedergaard, M. Physiology of astroglia. Physiol. Rev. 98, 239–389 (2018).
Denny, C. A. et al. Neurochemical, morphological, and neurophysiological abnormalities in retinas of Sandhoff and GM1 gangliosidosis mice. J. Neurochem. 101, 1294–1302 (2007).
De Maria, R. et al. Beta-galactosidase deficiency in a Korat cat: a new form of feline GM1-gangliosidosis. Acta Neuropathol. 96, 307–314 (1998).
Tessitore, A. et al. GM1-ganglioside-mediated activation of the unfolded protein response causes neuronal death in a neurodegenerative gangliosidosis. Mol. Cell. 15, 753–766 (2004).
Santagostino, S. F. et al. Mechanisms of regulated cell death: current perspectives. Vet. Pathol. 58, 596–623 (2021).
Kamada, S. et al. Nuclear translocation of caspase-3 is dependent on its proteolytic activation and recognition of a substrate-like protein(s). J. Biol. Chem. 280, 857–860 (2005).
Nadal-Nicolás, F. M. et al. Brn3a as a marker of retinal ganglion cells: qualitative and quantitative time course studies in Naive and optic nerve-injured retinas. Investig. Ophthalmol. Vis. Sci. 50 (8), 3860–3868 (2009).
Badea, T. C. et al. Distinct roles of transcription factors brn3a and brn3b in controlling the development, morphology, and function of retinal ganglion cells. Neuron. 61 (6), 852–864 (2009).
Wiggins, A. K. et al. Interaction of Brn3a and HIPK2 mediates transcriptional repression of sensory neuron survival. J. Cell. Biol. 167 (2), 257–267 (2004).
Murenu, E. et al. More than Meets the eye: the role of microglia in healthy and diseased retina. Front. Immunol. 13, 1006897. https://doi.org/10.3389/fimmu.2022.1006897 (2022).
Zeng, H. Y. et al. Identification of sequential events and factors associated with microglial activation, migration, and cytotoxicity in retinal degeneration in Rd mice. Investig. Ophthalmol. Vis. Sci. 46, 2992–2999 (2005).
Liu, Y. et al. Correlation of cytokine levels and microglial cell infiltration during retinal degeneration in RCS rats. PLoS One. 8, e82061. https://doi.org/10.1371/journal.pone.0082061 (2013).
Rathnasamy, G. et al. Retinal microglia—A key player in healthy and diseased retina. Prog. Neurobiol. 173, 18–40 (2019).
Pennesi, M. E., Neuringer, M. & Courtney, R. J. Animal models of age related macular degeneration. Mol. Aspects Med. 33, 487–509 (2012).
Winkler, P. A., Occelli, L. M. & Petersen-Jones, S. M. Large animal models of inherited retinal degenerations: A review. Cells. 9 https://doi.org/10.3390/cells9040882 (2020).
Wannemacher, R. et al. Sinbaglustat ameliorates disease pathology in a murine model of G(M1) gangliosidosis without affecting CNS ganglioside levels. Neurobiol. Dis. 210, 106917 (2025).
Gerhauser, I. et al. Dynamic changes and molecular analysis of cell death in the spinal cord of SJL mice infected with the bean strain of theiler’s murine encephalomyelitis virus. Apoptosis. 23, 170–186 (2018).
Ulrich, R. et al. Limited remyelination in theiler’s murine encephalomyelitis due to insufficient oligodendroglial differentiation of nerve/glial antigen 2 (NG2)-positive putative oligodendroglial progenitor cells. Neuropathol. Appl. Neurobiol. 34, 603–620 (2008).
Bankhead, P. et al. QuPath: open source software for digital pathology image analysis. Sci. Rep. 7 https://doi.org/10.1038/s41598-017-17204-5 (2017).
Acknowledgements
The authors thank Kerstin Schöne, Danuta Waschke, Caroline Schütz, Kerstin Rohn, Jana-Svea Harre and Julia Baskas for their excellent technical support.We acknowledge financial support by the Open Access Publication Fund of the University of Veterinary Medicine Hannover, Foundation. Anastasiia Ulianytska received a scholarship from the University of Veterinary Medicine Hannover, Foundation.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
L.J.: Conceptualization; investigation; methodology; visualization; writing-original draft; R.W.: Investigation; methodology; writing-original draft; A.U.: Methodology; I.G.: Methodology; W.B.: Conceptualization; writing-original draft; E.L.: Conceptualization; investigation; methodology; writing-original draft. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jubran, L., Wannemacher, R., Ulianytska, A. et al. Novel insights into pathomechanisms of retinal neuronal degeneration and reactive gliosis in a murine model of GM1-gangliosidosis. Sci Rep 15, 29674 (2025). https://doi.org/10.1038/s41598-025-15639-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-15639-9
