
Abstract
Acute myeloid leukemia (AML) exhibits significant heterogeneity in disease progression and therapeutic response, highlighting the urgent need for novel biomarkers to improve risk stratification and therapeutic targeting. In this study, we integrated multi-omics data from The Cancer Genome Atlas (TCGA, n = 151) and Genotype-Tissue Expression (GTEx, n = 337) cohorts to systematically analyze dynamic expression patterns of exosome-related genes in AML. Using univariate Cox regression and least absolute shrinkage and selection operator (LASSO) algorithms, we identified 13 exosome-associated genes (EXOSC4, TMEM109, THBS1, MYH9, HLA-DRA, CAPZB, ITGA4, MYL6, CYB5R1, PSMA2, MPO, NDST2, and CANX) and constructed a prognostic risk model. The model demonstrated superior predictive accuracy compared to traditional clinical parameters, with area under the curve (AUC) values of 0.819, 0.825, and 0.832 for 1-, 2-, and 3-year survival predictions in the training set, and 0.909 in the independent GEO validation cohort (GSE71014). Kaplan-Meier analysis revealed significantly shorter overall survival in the high-risk group (log-rank P < 0.001, hazard ratio = 0.22, 95% CI = 0.13–0.36). Immune microenvironment characterization using CIBERSORTx identified increased infiltration of regulatory T cells (Tregs, P < 0.01) in high-risk patients. Functional enrichment analysis revealed enrichment of PI3K-Akt signaling pathways and TP53 transcriptional networks in high-risk groups. Molecular docking studies confirmed strong binding affinity of verteporfin (ITGA4 inhibitor, docking score=-16.0 kcal/mol) and ebselen (MPO inhibitor) to their respective targets, suggesting potential therapeutic strategies to overcome chemotherapy resistance. This study establishes a robust 13-gene exosome-based prognostic signature for AML risk stratification and identifies novel immunomodulatory mechanisms mediated by exosome-driven Treg polarization.
Similar content being viewed by others

Introduction
Acute myeloid leukemia (AML) is an aggressive hematological malignancy arising from the malignant clonal evolution of myeloid hematopoietic stem cells. It is pathologically characterized by abnormal proliferation of bone marrow blasts, failure of normal hematopoiesis, and infiltration of extramedullary tissues. AML exhibits significant heterogeneity, and clinical management faces dual challenges of chemotherapy resistance and relapse1,2. In particular, elderly patients bear a substantial clinical burden due to complex comorbidities and poor tolerance to chemotherapy, resulting in induction treatment failure rates as high as 40%−50%3,4.
The pathogenesis of AML involves dysregulation at multiple hierarchical levels, including aberrant epigenetic regulation (e.g., DNA methylation disorders caused by TET2/DNMT3A mutations), high-frequency driver mutations (such as FLT3-ITD and NPM1 mutations), and immune evasion mediated by the bone marrow microenvironment5,6,7. In recent years, exosomes have garnered widespread attention as core signaling carriers within the tumor microenvironment. These 30–150 nm nanovesicles play pivotal roles in AML clonal competition, stromal remodeling, and drug resistance transmission by delivering functional molecules, including non-coding RNAs (e.g., miR-155, lncRNA HOTAIR), oncogenic proteins (e.g., FLT3-ITD mutants), and immunomodulatory molecules8,9,10. Recent studies have underscored the critical functions of exosomes as key mediators in the tumor microenvironment, influencing AML clonal evolution, bone marrow stromal remodeling, and immune evasion11,12,13. For instance, exosomes secreted by AML cells can induce the expansion of regulatory T cells by delivering TGF-β signaling pathway activators, thereby suppressing anti-leukemic immune responses14. Moreover, exosomes released by chemotherapy-resistant AML cells are enriched with drug efflux pump proteins (e.g., MDR1/P-gp), which can confer a multidrug-resistant phenotype to sensitive cells through horizontal transfer mechanisms15. Although strategies targeting exosome biogenesis (e.g., Rab27a inhibitors) or cargo loading have advanced to preclinical studies16, the molecular heterogeneity and spatiotemporal dynamic regulation of exosomes remain poorly understood, hindering their translation into precision diagnostic and therapeutic tools.
Increasing evidence highlights that the tumor immune microenvironment (TME) plays a pivotal role in AML progression and treatment resistance, with factors such as immune checkpoint expression, immunosuppressive cell infiltration, and TME remodeling attracting considerable attention. For instance, the IOBR tool enables multi-omics dissection of the TME landscape, providing valuable insights into immune dynamics and their correlation with treatment outcomes, akin to our approach using GTEx and TCGA datasets17. Immune profiling studies also reveal that molecular subtypes exhibit distinct immunological features, suggesting that TME heterogeneity may contribute to differential therapeutic responses18. Notably, recent integrative models such as iMLGAM further highlight the prognostic importance of immune cell infiltration, identifying CEP55 as a critical regulator of immune evasion, and raising the possibility that exosome-mediated pathways may play similar roles19.
Addressing this context, this study transcended the limitations of single-cohort analyses by integrating multi-omics data from healthy individuals (GTEx, n = 337) and AML patients (TCGA, n = 151) to systematically dissect the dynamic expression profiles of exosome-related genes and their prognostic associations. A risk scoring model incorporating 13 exosome genes (including EXOSC4, ITGA4, MPO, etc.) was constructed using LASSO-Cox regression. A nomogram integrating age and pathological features was developed, achieving an overall AUC of 0.924 in predicting 1-, 2-, and 3-year survival, significantly outperforming traditional clinical indicators. Further analysis using the CIBERSORTx algorithms revealed molecular characteristics of increased regulatory T cells infiltration (P < 0.05). Potential targeted drugs, such as verteporfin (an ITGA4 inhibitor with a docking score of −16.0), were identified through molecular docking. This study not only elucidated novel mechanisms by which exosomes remodel the immune microenvironment but also provided translational evidence for AML risk stratification and combination therapy strategies.
Materials and methods
Data collection and preprocessing
In this study, gene expression data from 337 healthy whole blood samples (formatted as log2(FPKM + 0.001), calculated by RSEM) were downloaded from the Genotype-Tissue Expression (GTEx) project via the UCSC Xena website (https://xena.ucsc.edu/). Transcriptomic and clinical data of 151 acute myeloid leukemia (AML) patients were obtained from The Cancer Genome Atlas (TCGA-LAML) dataset. Notably, the TCGA expression data were provided in the format of log2(FPKM + 1), resulting in a scale difference between the two datasets. To ensure consistency, GTEx expression values were first subjected to inverse log2 transformation, followed by re-transformation using log2(FPKM + 1), aligning the format with TCGA data. The human reference genome annotation file (human.gtf, version GRCh38.p14) was downloaded from the Ensembl database to extract gene-level annotation information. Perl (v5.30.2) was used to parse the GTF file and establish a mapping between Ensembl gene IDs and standard HUGO gene symbols (Gene Symbol). If an Ensembl ID corresponded to multiple gene symbols, only the first non-empty symbol was retained. The dataset GSE71014 (platform GPL10558-50081), containing gene expression and survival data for AML from the Gene Expression Omnibus (GEO) database, was downloaded as a test set. After probe ID matching, merging of redundant genes (avereps), and removal of missing values, a standardized gene expression matrix was obtained for validating prognostic features. To identify genes associated with exosome biogenesis, secretion, and function, we initially queried the GeneCards database (https://www.genecards.org/) using the keyword “exosome.” Genes with a relevance score greater than 2 were selected to ensure a high degree of functional relevance. To enhance the robustness of the gene set, we supplemented the list by reviewing published literature and incorporating genes with well-characterized roles in exosome biology. After removing duplicate entries and excluding non-coding genes, a curated list of 922 protein-coding exosome-related genes was obtained for subsequent analyses.
Batch correction and dataset merging
The preprocessed GTEx and TCGA AML gene expression matrices were converted into numerical matrices. The avereps function from the limma package was used to average the expression levels of duplicate genes, ensuring that each gene uniquely corresponded to one row in the expression profile. Common genes between GTEx and TCGA were extracted, and the two datasets were merged horizontally to generate a combined expression matrix. The normalizeBetweenArrays function from the limma package in R was employed to perform quantile normalization on the GTEx and TCGA data to eliminate technical biases and standardize the expression value distributions across datasets.
Screening of exosome-related genes and differential expression analysis
Low-expression genes (average expression value > 0 across all samples) were filtered from the merged expression matrix, and the gene expression profiles overlapping with the exosome gene list were extracted. A Venn diagram was drawn using the ggvenn package. The Wilcoxon rank-sum test was used to perform differential expression analysis on the exosome-related gene expression data between the healthy control group and AML patients, identifying differentially expressed genes (FDR < 0.05, |logFC| ≥ 2). A heatmap of the differentially expressed genes was plotted using the pheatmap package.
Construction and validation of exosome-related prognostic features
Datasets from the TCGA database and the GSE71014 dataset from the GEO database were used as the training and validation cohorts, respectively. Exosome-related gene sequencing data for each sample were integrated with corresponding survival data. Univariate Cox regression analysis (p < 0.01) was performed to screen key genes associated with AML prognosis in the TCGA cohort. A forest plot was drawn using the bioForest function (p-value < 0.001, HR(95% CI)). The glmnet package was used to select prognostic genes via LASSO-Cox regression (10-fold cross-validation, λ.min criterion). Based on the gene expression values (Expi) and their regression coefficients (βi) from the LASSO-Cox model, an individual risk score was calculated using the following formula (Eq. (1)):
where n, βi, and Expi represent the number of feature genes, the coefficient value of the gene in LASSO analysis, and the gene expression, respectively. The surv_cutpoint function was used to automatically determine the optimal cutoff value for the risk score based on the maximum survival difference criterion, thereby dividing the samples into high- and low-risk groups. This method selects the cutoff point by maximizing the chi-square value (χ2) of the Log-rank test, thereby maximizing the survival difference between groups. The formula is as follows (Eq. (2)):
Kaplan-Meier curves were used to evaluate the overall survival (OS) differences between groups. Principal component analysis (PCA) was also performed to estimate the clustering ability of the risk features. The prognostic performance was further validated in the validation set.
Comprehensive analysis of prognostic risk scores and clinicopathological parameters in aml patients
The risk scores of each AML sample were combined with corresponding clinicopathological features. To assess the clinical relevance of the risk score, we used the Wilcoxon rank-sum test in R to analyze its associations with clinicopathological characteristics including gender, age, and previous treatment. Statistical significance was defined as P < 0.05. Univariate and multivariate Cox regression analyses were conducted to identify independent prognostic factors among these variables. A p-value < 0.05 was used as the selection criterion. ROC curves were plotted to assess the prognostic prediction accuracy of the risk score model and clinical variables.
Construction and evaluation of a nomogram for aml patients
To further explore individual prognosis, a novel prognostic nomogram incorporating independent prognostic indicators was constructed using the “nomogram” package in R. Additionally, ROC curve analysis and Cox regression analysis were performed to evaluate the predictive ability of the nomogram from different perspectives.
Characterization of immune cell infiltration
CIBERSORTx (https://cibersortx.stanford.edu/), an advanced algorithm based on support vector regression, was used to estimate the relative abundance of 22 immune cell subsets in each AML sample by deconvoluting bulk transcriptomic data against the LM22 leukocyte gene signature matrix. The analysis was performed with 500 permutations to ensure robust estimation. The Wilcoxon rank-sum test was applied to compare differences in immune cell infiltration between the high- and low-risk groups.
Functional enrichment analysis
Differentially expressed genes (DEGs) were screened between the high- and low-risk groups, with cutoff values set at |log2FC| ≥ 1 and raw p-value < 0.05, and further corrected for false discovery rate (FDR) using the Benjamini-Hochberg method (q-value < 0.05). Based on the significant DEGs, gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were performed using the clusterProfiler package. GO analysis covered three categories: biological process (BP), molecular function (MF), and cellular component (CC). KEGG analysis focused on metabolism and signal transduction. Significant enrichment results were visualized using ggplot2, with a significance threshold of q-value < 0.05.
Construction and analysis of protein-protein interaction (PPI) networks
Based on the STRING online platform, a human protein-protein interaction (PPI) network was constructed for the DEGs screened between the high- and low-risk groups, with a particular focus on Homo sapiens data. High-confidence protein interactions were filtered based on a combined score ≥ 0.4 from the STRING database to explore the functional synergies of DEGs in the tumor microenvironment.
Screening of core exosome prognostic differential genes and construction of transcription factor regulatory networks
Core exosome prognostic differential genes were screened through univariate Cox regression analysis (P < 0.001). In the TRRUST (Transcriptional Regulatory Relationships Unraveled by Sentence-based Text mining) database, the species was set to human, and relevant transcription factors for the core genes were searched, resulting in a list of related transcription factors. Subsequently, a core gene-transcription factor regulatory network was further constructed using Cytoscape software.
Drug enrichment analysis and construction of drug regulatory networks for core exosome prognostic-related genes
The DSigDB (Drug Signature Database) was selected as the reference database. The enricher function from the clusterProfiler package was used, and the core gene list and drug-gene interaction data from the DSigDB database were input for drug enrichment analysis (p < 0.05 and q < 0.05). To ensure information richness and result accuracy, the minimum and maximum sizes of gene sets were set to 10 and 500, respectively. A drug enrichment model and regulatory network based on core genes were constructed, providing valuable clues for subsequent drug development and therapeutic target discovery.
Molecular docking
To enhance clinical translation potential, a hierarchical screening strategy was adopted in molecular docking studies. Candidate compounds were selected from a drug library with p < 0.05 in enrichment analysis. Priority was given to FDA-approved first-line drugs for AML and emerging targeted inhibitors with well-defined mechanisms. Non-drug entries and highly toxic compounds were excluded, focusing on compounds with good safety profiles. Compounds that potentially regulate exosome secretion, packaging, or signaling pathways were retained. High-quality three-dimensional structures of drug targets were screened from the PDB database to ensure the reliability of molecular docking. Based on the results of drug enrichment analysis for core genes, suitable drugs or compounds were selected as docking drugs, and the core exosome prognostic-related genes with the closest enrichment were determined as docking targets. During the docking process, high-precision 3D molecular structure data for the docking drugs or compounds were obtained from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/), and detailed protein structure data for the core exosome prognostic-related genes were downloaded from the RCSB PDB database (https://www.rcsb.org/). The CB-Dock2 website (https://cadd.labshare.cn/cb-dock2/index.php) was selected as the docking tool.
Statistical methods, software, and tools
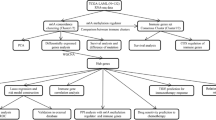
Perl (v5.30.2) was used to assist in data processing, including large-scale text parsing and format conversion. Statistical analysis, modeling, and visualization were performed using the R language (v4.4.3) environment. The Benjamini-Hochberg method was used for multiple testing correction to control the FDR. The Wilcoxon rank-sum test was used for differential expression analysis. Cytoscape software (version 3.10.1) was used for drawing. The experimental design and workflow are illustrated in Fig. 1.

Flow chart of research design and analysis.
Results
Data collection and preprocessing
Gene expression data from 337 healthy whole blood samples were obtained from the GTEx database, stored in the format of log2(FPKM + 0.001). The TCGA database contained transcriptomic and clinical data for 151 AML patients. The GTEx data underwent an inverse log2 transformation followed by re-normalization via log2 transformation, resulting in a standardized expression matrix that included expression profiles for 56,754 genes. In the TCGA AML data, 60,660 Ensembl IDs were successfully converted to HUGO symbols, and after removing unannotated genes, 58,697 genes were retained. Following data cleaning and ID conversion, the integrated dataset of GTEx and TCGA AML retained a total of 55,150 common genes. The GEO dataset GSE71014 (platform GPL10558-50081) was processed through probe matching, merging of redundant genes (avereps), and removal of missing values, ultimately retaining a standardized expression matrix for 104 AML samples (covering 34,681 genes) for independent validation. Among the 922 exosome-related genes extracted from the GeneCards database, 878 genes were effectively expressed (average expression value > 0) in the combined GTEx and TCGA expression matrix and were included in subsequent differential expression analysis and modeling.
Differential expression analysis of exosome-related genes
After overlapping the combined GTEx and TCGA expression matrix with the list of 922 exosome-related genes, a total of 836 exosome-related genes were extracted. A Venn diagram was used to illustrate the intersection between the exosome genes and the combined GTEx and TCGA expression matrix (Fig. 2A). Based on the Wilcoxon rank-sum test (FDR < 0.05, |logFC| ≥ 2), a total of 174 exosome-related genes were identified as differentially expressed between healthy controls and AML patients. A heatmap revealed distinct clustering patterns of the differentially expressed genes between the two groups (Fig. 2B).

(A) Venn Diagram Identifying Differential Genes Associated with Exosomes from AML. (B) Heatmap of exosome-related DEGs in AML and normal samples (red for high, blue for low).
Construction of an exosome-related prognostic signature
In the TCGA dataset, univariate Cox regression analysis (P < 0.01) was employed to screen 20 exosome-related genes that were significantly associated with the overall survival (OS) of AML patients (Fig. 3 A). Subsequently, LASSO-Cox regression (with 10-fold cross-validation and λ = min) was applied to further determine the optimal prognostic genes. Eventually, 13 exosome-related genes were identified (Table 1) and incorporated into a prognostic risk model (Fig. 3B, C).
The risk score for each patient was calculated using the following formula: Risk Score = (Expression of EXOSC4 × 0.489131757414089) + (Expression of TMEM109 × 0.122385896623323) + (Expression of THBS1 × 0.0315566929862512) + (Expression of MYH9 × 0.291033480323073) + (Expression of HLA-DRA × 0.107916651629561) + (Expression of CAPZB × 0.269733384075537) + (Expression of ITGA4 × −0.164132610693896) + (Expression of MYL6 × 0.405708399356843) + (Expression of CYB5R1 × 0.150359117251523) + (Expression of PSMA2 × −0.297116568474912) + (Expression of MPO × −0.244293996471651) + (Expression of NDST2 × −0.658043797748103) + (Expression of CANX × −0.0644590533948854). Based on the criterion of maximum survival difference, the optimal cutoff value for the risk score was determined to be 3.991441. Patients were then divided into high-risk (71 cases) and low-risk groups (71 cases). Kaplan-Meier analysis revealed a significantly worse OS in the high-risk group (log-rank P < 0.001, HR = 0.22, 95% CI = 0.13–0.36)(Fig. 3D).
Furthermore, principal component analysis (PCA) was conducted to compare the gene expression levels between high-risk and low-risk patients based on the 13 exosome-related genes included in the prognostic signature. The results demonstrated that the risk signature exhibited the best discriminatory ability between the two groups (Fig. 3E).

Construction of the prognostic risk model. (A) forest plot displays 20 exosome-related genes identified through univariate Cox regression analysis as being associated with prognosis. (B, C) LASSO Cox regression analysis was employed to determine exosome-related genes closely associated with the prognosis of acute myeloid leukemia (AML). (D) Kaplan–Meier curve of the gene signature. (E) Principal component analysis (PCA) was conducted based on the exosome risk score to distinguish tumor samples from normal samples. The group marked in blue represents low-risk patients, while the group marked in red represents high-risk patients.
Independent validation and clinical relevance of the prognostic model
In the independent validation set GSE71014, the high-risk group also exhibited a significantly shorter overall survival (OS) (log-rank P < 0.001, Fig. 4A). The receiver operating characteristic (ROC) curve demonstrated that the area under the curve (AUC) values for the risk score in predicting 1-year, 2-year, and 3-year OS were 0.819, 0.825, and 0.832, respectively (Fig. 4B). When comparing the risk score with all clinical variables in predicting OS, the risk score achieved the highest AUC value of 0.909 (Fig. 4C). Both univariate and multivariate Cox regression analyses confirmed that the risk score was an independent prognostic factor for AML (univariate HR = 4.10, P < 0.001; multivariate HR = 3.40, P < 0.001) (Figs. 4D, E).

Validation of the prognostic risk signature. (A) Kaplan–Meier survival curves in the GEO cohort. (B) Receiver Operating Characteristic (ROC) curves for predicting 1-year, 3-year, and 5-year survival rates in the GEO cohort. (C) ROC curves comparing the prognostic performance of the risk score versus clinical characteristics. (D, E) Univariate and multivariate Cox regression analyses of clinical parameters in acute myeloid leukemia (AML) patients.
Association between risk score and clinicopathological features
Given the accuracy of survival risk prediction, we further investigated the role of the risk signature in predicting AML progression and explored the associations between the risk score and clinicopathological features. The risk score was significantly correlated with an age of 65 years or older (P = 0.00022) (Fig. 5A). However, no significant correlations were observed between the risk score and gender or previous treatment (p > 0.05)(Figs. 5B, C). Moreover, the ROC curve shown in Sect. 4 further supported the prognostic predictive power of the risk score compared to traditional clinical variables (Fig. 4C).

(A-C) Box plots illustrating the correlations between risk score and age, gender, as well as prior treatment.
Construction of a nomogram for AML patients
A prognostic nomogram incorporating independent risk factors (age and risk score) was constructed to predict the 1-, 2-, and 3-year survival probabilities of AML patients (Fig. 6 A). The area under the curve (AUC) analysis demonstrated that the nomogram (AUC = 0.924) exhibited higher sensitivity and specificity compared to other single prognostic factors, such as age (AUC = 0.775), gender (AUC = 0.460), previous treatment (AUC = 0.641), and the risk score alone (AUC = 0.910) (Fig. 6B). Furthermore, the calibration curves indicated excellent predictive performance, with our model closely resembling the ideal model in estimating 1-, 2-, and 3-year overall survival (OS) (Fig. 6 C). Additionally, Cox regression analysis was conducted to confirm that the nomogram score was an independent predictor of AML prognosis among all participants (Fig. 6D and E). Overall, we have validated the outstanding predictive capability of the disease prognostic signature and revealed its high potential for clinical application from multiple perspectives.

Development and evaluation of a nomogram for patients with AML. (A) Nomogram for predicting the 1-year, 2-year, and 3-year overall survival rates in AML patients. (B) Calibration plot analysis to evaluate the predictive power of the nomogram. The x-axis is the survival rate predicted by the nomogram, and the y-axis is the actual survival rate. (C) Nomogram for predicting prognosis, risk score, and ROC curve for clinical characteristics. (D, E) Univariate and multivariate Cox regression analysis was performed to determine whether nomogram score was an independent predictor of AML patients. Green square: danger is higher than HR < 1; Red squares: HR > 1.
Landscape of tumor microenvironment (TME) and immune cell infiltration in AML patients
CIBERSORTx analysis revealed that the proportion of regulatory T cells (Tregs) was significantly elevated in the high-risk group (P < 0.05), whereas the proportions of resting mast cells and resting dendritic cells were significantly reduced (P < 0.05) (Fig. 7).

Box plot of the proportion of 22 immune cells in the high-risk group and low-risk group. * p < 0.05; ** p < 0.01; and *** p < 0.001.
Functional enrichment and regulatory network analysis
To distinguish the biological functions and networks associated with the risk signature, we screened a total of 43 differentially expressed genes (DEGs) (|log2FC|≥1, FDR < 0.05) between the high-risk and low-risk groups. GO/KEGG enrichment analysis revealed that these genes were significantly enriched in biological processes such as “collagen-containing extracellular matrix”, “extracellular matrix organization”, and “DNA-binding transcription activator activity”, as well as in signaling pathways including PI3K-Akt signaling pathway, Cytokine-cytokine receptor interaction, Focal adhesion, and ECM-receptor interaction (Fig. 8A and C). Functional clustering of the DEGs was visualized through principal component dimension reduction, showing the mutually exclusive distribution of MHC class II antigen presentation (right quadrant) and angiogenesis (left quadrant), suggesting parallel mechanisms of immune escape and microenvironment remodeling (Fig. 8B). The protein-protein interaction (PPI) network (combined score ≥ 0.4) identified 12 hub genes (such as HOX family proteins), which were primarily involved in functional modules related to embryonic development and cell differentiation (Fig. 8D).

(A, C) GO and KEGG pathway analyses of exosome-related core differentially expressed genes (DEGs) in the high-risk and low-risk groups of acute myeloid leukemia (AML), adapted from the KEGG database (https://www.kegg.jp/), Kanehisa Laboratories. Accessed 21 April 2025. © Kanehisa Laboratories20. (B) Gene Ontology (GO) functional clustering distribution of the differentially expressed genes (DEGs). (D) Protein-protein interaction (PPI) network.
Construction of a transcription factor regulatory network
Based on univariate Cox regression analysis, seven core exosome-related genes significantly associated with the overall survival (OS) of acute myeloid leukemia (AML) patients were identified (p < 0.001) (Fig. 9A). To further explore the expression characteristics and synergistic relationships of these genes, box plots were first generated to illustrate their expression differences between the AML group and the normal control group (Fig. 9B). Subsequently, Pearson correlation coefficients were calculated among the seven core genes to construct a co-expression relationship matrix (Fig. 9C). Next, the selected seven core exosome-related prognostic differential genes (P < 0.001) were analyzed in TRRUST (Transcriptional Regulatory Relationships Unraveled by Sentence-based Text mining), a database focusing on the interactions between mammalian transcription factors and their target genes. By setting the species to human, we searched for the transcription factors of these seven core exosome-related differential genes in the TRRUST database and obtained a list of transcription factors associated with the THBS1 and MVP genes. A transcription factor regulatory network was then constructed using Cytoscape software (Fig. 9D).

(A) Forest plot showing the screening of core exosome-related prognostic difference genes by univariate Cox regression analysis with P < 0.001. (B) Box plot of core exosome-related prognostic difference genes. (C) Correlation matrix of core exosome-related prognostic difference genes. (D) The regulatory relationship diagram of transcription factors and core exosome-related prognostic differential gene regulation, where yellow represents transcription factors and red represents differential genes.
Drug enrichment analysis and construction of a drug regulatory network for core exosome-related prognostic differentially expressed genes
The enrichment results revealed that the seven core exosome-related prognostic differentially expressed genes were primarily enriched in drugs and compounds such as “5-Aminosalicylic acid”, “CHLOROGENIC ACID”, “verteporfin”, “MAGNESIUM”, and “ebselen” (p < 0.05 and q < 0.05)(Fig. 10A). Based on these enrichment results, a drug regulatory network was constructed (Fig. 10B).

(A) Drug Enrichment Analysis Network Diagram. Gray nodes represent genes or proteins, while orange nodes represent chemical substances. Edges of different colors indicate different types of interactions or associations between genes or proteins and chemical substances. The size of the nodes reflects their importance. (B) Drug Regulation Network Diagram. Genes are marked with purple diamond, and drugs are represented by blue circles.
Molecular docking
Drug enrichment analysis (DSigDB, P < 0.05) identified 221 drugs (such as verteporfin, ebselen, and Ginsenoside Rh1) that potentially target the core exosome prognostic genes. Ultimately, three drugs were selected for further study. Molecular docking results demonstrated that Verteporfin exhibited a strong affinity for the ITGA4 protein, with a docking score as low as −16.0 calculated using AutoDock Vina software (Table 2; Fig. 11A). Similarly, Ebselen showed a strong affinity for the MPO protein, with a docking score of −7.6 (Table 3; Fig. 11B). Ginsenoside Rh1 also displayed a strong affinity for the ITGA4 protein, with a docking score of −10.2 (Table 4; Fig. 11C).

(A) Schematic diagram of molecular docking between Verteporfin and ITGA4. (B) schematic diagram of molecular docking between ebselen and MPO. (C) schematic diagram of molecular docking between ginsenoside Rh1 and ITGA4.
Discussion
In this study, we constructed and validated a robust 13-gene exosome-related prognostic signature that effectively stratifies AML patients by survival risk and reveals immunological distinctions within the tumor microenvironment (TME). By integrating transcriptomic, survival, immune infiltration, and molecular docking data, our findings suggest that exosome-associated genes critically contribute to immune suppression and therapeutic resistance in AML, offering potential avenues for precision therapy.
Exosomes, as intercellular communicators, influence leukemic progression through bioactive cargoes such as proteins, RNAs, and lipids21,22. In our analysis, the high-risk group defined by the exosome-based signature exhibited a distinct immunosuppressive TME, characterized by elevated regulatory T cells (Tregs), and reduced resting mast cells and dendritic cells. This immune landscape reflects a shift toward an immune-evasive phenotype, consistent with prior studies showing that AML-derived exosomes promote Treg expansion via TGF-β and IL-10 signaling, and impair dendritic cell maturation and antigen presentation14,23. Such immunomodulatory effects may partly underlie the reduced immunotherapy responsiveness observed in high-risk patients.
Functional enrichment analysis revealed that key oncogenic pathways, such as PI3K-Akt and TP53 signaling, were significantly upregulated in the high-risk cohort. These pathways are not only essential for leukemogenesis but also intersect with immunosuppressive signaling and therapy resistance mechanisms11,13. Notably, TP53 has been shown to regulate THBS1, a poor prognostic gene in our model, and may modulate tumor immune tolerance through downstream effects on TGF-β signaling24.
Among the 13 exosomal genes in our signature, several exhibit mechanistic relevance to AML biology. ITGA4, known for mediating AML cell adhesion and homing, contributes to chemotherapy resistance through the METTL3-m6A-ITGA4 axis25. Paradoxically, our model identified high ITGA4 expression as associated with better prognosis, suggesting subtype-specific or context-dependent regulatory effects of exosome-associated ITGA4. MPO, a myeloperoxidase involved in oxidative stress and cytarabine sensitivity, showed protective effects in our cohort, aligning with its role in chemotherapy response26. NDST2 exhibited a favorable association with survival, in contrast to its tumor-promoting role in hepatocellular carcinoma27,28, highlighting tissue-specific expression dynamics.
Conversely, overexpression of genes such as EXOSC4, THBS1, MYH9, and CYB5R1 correlated with poor outcomes, consistent with their roles in promoting stemness, immunosuppression, and ferroptosis resistance in other malignancies29,30,31,32,33. CAPZB and HLA-DRA, though better characterized in lymphomas and FUS-ERG–positive AML respectively, may similarly impact immune cell infiltration and antigen presentation in high-risk AML patients34,35.
To identify potential therapeutic agents that could target the high-risk signature, molecular docking analyses were performed. Verteporfin (VP), a YAP pathway inhibitor, demonstrated strong binding affinity with ITGA4 and may disrupt leukemic cell adhesion and survival via dual regulation of the Hippo/YAP and AKT/MAPK pathways36,37,38. Similarly, Ebselen, a GPx mimic, targets MPO to inhibit ROS-mediated DNA damage and resistance, while simultaneously activating the caspase cascade and promoting myeloid differentiation39,40,41,42. Importantly, both compounds have been shown to modulate immune and oxidative stress pathways, underscoring their potential for exosome-guided therapeutic repurposing.
Another promising agent is Ginsenoside Rh1, a natural compound with pro-apoptotic and anti-invasive properties. Recent studies have suggested that Rh1 may target ITGA4-mediated cell–matrix interactions, potentially disrupting leukemia stem cell niche maintenance43,44. Given its redox regulatory effects and anti-MAPK activity, Rh1 may complement existing therapies in ITGA4-high AML subtypes.
Clinically, the exosome-based risk model demonstrated superior predictive performance compared to conventional parameters, with AUCs exceeding 0.8 across both TCGA and GEO cohorts. The nomogram combining the risk score and age (AUC = 0.924) further enhances prognostic accuracy. By linking exosomal gene expression to immune infiltration patterns and druggable molecular targets, this study proposes a dual-pronged strategy—simultaneously targeting exosome-mediated immune dysfunction and guiding individualized drug selection.
Nevertheless, this study has limitations. All results are based on publicly available transcriptomic datasets, and the bulk RNA-seq data may obscure cell-type-specific exosomal gene expression. Functional validation through in vitro and in vivo experiments is necessary to verify the causal roles of the identified genes and therapeutic compounds. Additionally, although molecular docking provided mechanistic clues, further pharmacological assessments are required before clinical translation.
In conclusion, our work highlights the prognostic and therapeutic relevance of exosome-associated genes in AML. By uncovering their immunosuppressive functions and therapeutic vulnerabilities, we provide a foundation for biomarker-guided, immune-informed treatment strategies to overcome drug resistance in AML.
Conclusion
In this study, through multi-omics integrative analysis and leveraging the dynamic regulatory network of exosomes, we successfully constructed an AML risk scoring system incorporating 13 key genes, including EXOSC4, ITGA4, and MPO. This system demonstrated excellent predictive performance in independent validation cohorts. The integration of age, risk stratification, and the ExoRiskScore into a nomogram enables individualized survival rate predictions, providing a quantitative basis for adjusting treatment intensity. Molecular docking studies validated the potential of verteporfin and ebselen to reverse exosome-mediated chemotherapy resistance, suggesting the initiation of multi-center Phase II clinical trials. The clinical application of this model will usher in an era of “exosome-based molecular subtyping” for AML, offering a new paradigm for precision immunotherapy combinations.
Data availability
Raw data are directly accessible via the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/), the UCSC Xena website (https://xena.ucsc.edu/).
References
Wachter, F. & Pikman, Y. Pathophysiology of acute myeloid leukemia. Acta Haematol. 147, 229–246. https://doi.org/10.1159/000536152 (2024).
Hackl, H., Astanina, K. & Wieser, R. Molecular and genetic alterations associated with therapy resistance and relapse of acute myeloid leukemia. J. Hematol. Oncol. 10, 51. https://doi.org/10.1186/s13045-017-0416-0 (2017).
Venugopal, S. & Sekeres, M. A. Contemporary management of acute myeloid leukemia: a review. JAMA Oncol. 10, 1417–1425. https://doi.org/10.1001/jamaoncol.2024.2662 (2024).
Thol, F. & Heuser, M. Treatment for relapsed/refractory acute myeloid leukemia. HemaSphere 5, e572. https://doi.org/10.1097/hs9.0000000000000572 (2021).
Conway O’Brien, E., Prideaux, S. & Chevassut, T. The epigenetic landscape of acute myeloid leukemia. Adv. Hematol. 2014, 103175. https://doi.org/10.1155/2014/103175 (2014).
Daver, N., Venugopal, S. & Ravandi, F. FLT3 mutated acute myeloid leukemia: 2021 treatment algorithm. Blood Cancer J. 11, 104. https://doi.org/10.1038/s41408-021-00495-3 (2021).
Krause, D. S. et al. Differential regulation of myeloid leukemias by the bone marrow microenvironment. Nat. Med. 19, 1513–1517. https://doi.org/10.1038/nm.3364 (2013).
Tkach, M. & Théry, C. Communication by extracellular vesicles: where we are and where we need to go. Cell 164, 1226–1232. https://doi.org/10.1016/j.cell.2016.01.043 (2016).
Maas, S. L. N., Breakefield, X. O. & Weaver, A. M. Extracellular vesicles: unique intercellular delivery vehicles. Trends Cell. Biol. 27, 172–188. https://doi.org/10.1016/j.tcb.2016.11.003 (2017).
Bobrie, A., Colombo, M., Raposo, G. & Théry, C. Exosome secretion: molecular mechanisms and roles in immune responses. Traffic 12, 1659–1668. https://doi.org/10.1111/j.1600-0854.2011.01225.x (2011).
Qi, Y. et al. Discovery of Cmpd D6 (FH-001) as a efficiency enhancement and myelosuppression degradation small-molecule fms-like tyrosine kinase 3 inhibitor for the treatment of FLT3-ITD positive acute myeloid leukemia. Eur. J. Pharmacol. 996, 177541. https://doi.org/10.1016/j.ejphar.2025.177541 (2025).
Amiri, V., Mirzaeian, A. & Noroozi-Aghideh, A. Non-mutational changes of autophagy marker LC3A in patients with acute myeloid leukemia; effect of DNA methylation and expression level of LncRNA-GAS5 and miRNA-155-5p, a case control study. Indian J. Hematol. Blood Transfus. 40, 621–628. https://doi.org/10.1007/s12288-024-01765-3 (2024).
Whiteside, T. L. Immune modulation of T-cell and NK (natural killer) cell activities by TEXs (tumour-derived exosomes). Biochem. Soc. Trans. 41, 245–251. https://doi.org/10.1042/bst20120265 (2013).
Dong, Q. et al. Levels and clinical significance of regulatory B cells and T cells in acute myeloid leukemia. Biomed. Res. Int. 2020, 7023168. https://doi.org/10.1155/2020/7023168 (2020).
Zhang, X. et al. Inside-out extracellular vesicles-like biomimetic magnetic nanoparticles for efficient screening P-glycoprotein inhibitors to overcome cancer multidrug resistance. Colloids Surf. B Biointerfaces. 222, 113134. https://doi.org/10.1016/j.colsurfb.2023.113134 (2023).
Kumar, B. et al. Acute myeloid leukemia transforms the bone marrow niche into a leukemia-permissive microenvironment through exosome secretion. Leukemia 32, 575–587. https://doi.org/10.1038/leu.2017.259 (2018).
Fang, Y. et al. Systematic investigation of tumor microenvironment and antitumor immunity with IOBR. Med. Res. 1, 136–140. https://doi.org/10.1002/mdr2.70001 (2025).
Zhou, H. et al. Optimized dynamic network biomarker Deciphers a high-resolution heterogeneity within thyroid cancer molecular subtypes. Med. Res. 1, 10–13. https://doi.org/10.1002/mdr2.70004 (2025).
Li, W. et al. iMLGAM: integrated machine learning and genetic algorithm–driven multi-omics analysis. Brief. Bioinform. https://doi.org/10.1093/bib/bbae276 (2024).
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: biological systems database as a model of the real world. Nucleic Acids Res. 53, D672–D677 (2025).
Ghosh, S. et al. The theragnostic advances of exosomes in managing leukaemia. J. Cell. Mol. Med. 28, e70052. https://doi.org/10.1111/jcmm.70052 (2024).
Hornick, N. I. et al. Serum exosome MicroRNA as a minimally-invasive early biomarker of AML. Sci. Rep. 5, 11295. https://doi.org/10.1038/srep11295 (2015).
Xu, Z. J. et al. The M2 macrophage marker CD206: a novel prognostic indicator for acute myeloid leukemia. Oncoimmunology 9, 1683347. https://doi.org/10.1080/2162402x.2019.1683347 (2020).
Kadia, T. M. et al. TP53 mutations in newly diagnosed acute myeloid leukemia: clinicomolecular characteristics, response to therapy, and outcomes. Cancer 122, 3484–3491. https://doi.org/10.1002/cncr.30203 (2016).
Li, M. et al. METTL3 mediates chemoresistance by enhancing AML homing and engraftment via ITGA4. Leukemia 36, 2586–2595. https://doi.org/10.1038/s41375-022-01696-w (2022).
Hosseini, M. et al. Targeting myeloperoxidase disrupts mitochondrial redox balance and overcomes cytarabine resistance in human acute myeloid leukemia. Cancer Res. 79, 5191–5203. https://doi.org/10.1158/0008-5472.CAN-19-0515 (2019).
Khalil, M. M. I. M., Sohaib, A., Mansour, M. M. & Genena, E. S. R. The role of ZNF384, DNAH17, and NDST2 gene expression in non-Hodgkin’s lymphoma patients. Gene Rep. 25, 101354. https://doi.org/10.1016/j.genrep.2021.101354 (2021).
Wan, W. et al. Incomplete thermal ablation-induced FOXP4-mediated promotion of malignant progression in liver cancer via NDST2. J. Hepatocell Carcinoma. 11, 1945–1959. https://doi.org/10.2147/jhc.S476612 (2024).
Wang, N. et al. RUNX3 exerts tumor-suppressive role through inhibiting EXOSC4 expression. Funct. Integr. Genomics. 24, 103. https://doi.org/10.1007/s10142-024-01363-6 (2024).
Zhu, L. et al. THBS1 is a novel serum prognostic factor of acute myeloid leukemia. Front. Oncol. 9, 1567. https://doi.org/10.3389/fonc.2019.01567 (2019).
Chen, S. et al. Bioinformatics analysis identifies key genes and pathways in acute myeloid leukemia associated with DNMT3A mutation. Biomed. Res. Int. 2020, 9321630. https://doi.org/10.1155/2020/9321630 (2020).
Yu, M. et al. Prognostic impact of MYH9 expression on patients with acute myeloid leukemia. Oncotarget 8, 156–163. https://doi.org/10.18632/oncotarget.10613 (2017).
Lan, H., Gao, Y., Zhao, Z., Mei, Z. & Wang, F. Ferroptosis: redox imbalance and hematological tumorigenesis. Front. Oncol. 12, 834681. https://doi.org/10.3389/fonc.2022.834681 (2022).
Buteyn, N. J. et al. EZH2-mediated MHC class II Silencing drives immune evasion in AML with t(16;21) (FUS-ERG). Blood 138, 374. https://doi.org/10.1182/blood-2021-153256 (2021).
Wang, Y., Tsukamoto, Y., Hori, M. & Iha, H. Disulfidptosis: a novel prognostic criterion and potential treatment strategy for diffuse large B-cell lymphoma (DLBCL). Int. J. Mol. Sci. 25, 7156. https://doi.org/10.3390/ijms25137156 (2024).
Chen, M. et al. Verteporfin inhibits cell proliferation and induces apoptosis in human leukemia NB4 cells without light activation. Int. J. Med. Sci. 14, 1031–1039. https://doi.org/10.7150/ijms.19682 (2017).
Xiu, Y. et al. Stabilization of NF-κB-inducing kinase suppresses MLL-AF9-induced acute myeloid leukemia. Cell. Rep. 22, 350–358. https://doi.org/10.1016/j.celrep.2017.12.055 (2018).
Song, H. et al. Single-cell analysis of hepatoblastoma identifies tumor signatures that predict chemotherapy susceptibility using patient-specific tumor spheroids. Nat. Commun. 13, 4878. https://doi.org/10.1038/s41467-022-32473-z (2022).
Maraldi, T. et al. Induction of apoptosis in a human leukemic cell line via reactive oxygen species modulation by antioxidants. Free Radic Biol. Med. 46, 244–252. https://doi.org/10.1016/j.freeradbiomed.2008.10.027 (2009).
Bi, C. L. et al. Selenium inhibits Staphylococcus aureus-induced inflammation by suppressing the activation of the NF-κB and MAPK signalling pathways in RAW264.7 macrophages. Eur. J. Pharmacol. 780, 159–165. https://doi.org/10.1016/j.ejphar.2016.03.044 (2016).
Martini, F. et al. A multifunctional compound Ebselen reverses memory impairment, apoptosis and oxidative stress in a mouse model of sporadic alzheimer’s disease. J. Psychiatr Res. 109, 107–117. https://doi.org/10.1016/j.jpsychires.2018.11.021 (2019).
Wei, W. et al. Targeting Peroxiredoxin I potentiates 1,25-dihydroxyvitamin D3-induced cell differentiation in leukemia cells. Mol. Med. Rep. 13, 2201–2207. https://doi.org/10.3892/mmr.2016.4787 (2016).
Kim, J. H. & Choi, J. S. Effect of ginsenoside Rh-2 via activation of caspase-3 and Bcl-2-insensitive pathway in ovarian cancer cells. Physiol. Res. 65, 1031–1037 (2016).
Choi, Y. J., Yoon, J. H., Cha, S. W. & Lee, S. G. Ginsenoside Rh1 inhibits the invasion and migration of THP-1 acute monocytic leukemia cells via inactivation of the MAPK signaling pathway. Fitoterapia 82, 911–919. https://doi.org/10.1016/j.fitote.2011.05.005 (2011).
Funding
National Natural Science Foundation of China (82300209).
Author information
Authors and Affiliations
Contributions
Conceptualization: Peng-fei Han, Yan-hui Yu.Data curation: Chang-sheng Liao, Min-xiao Wang.Formal analysis: Chang-sheng Liao.Investigation: Chang-sheng Liao, Min-xiao Wang, Yu-qin Xie, Xue-qin Wei.Methodology: Chang-sheng Liao, Min-xiao Wang.Resources: Min-xiao Wang, Yu-qin Xie, Xue-qin Wei.Software: Chang-sheng Liao, Min-xiao Wang.Supervision: Peng-fei Han, Yan-hui Yu.Validation: Chang-sheng Liao, Min-xiao Wang.Visualization: Chang-sheng Liao, Min-xiao Wang.Writing – original draft: Chang-sheng Liao, Min-xiao Wang.Writing – review & editing: Chang-sheng Liao, Min-xiao Wang.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
This study is based entirely on publicly available datasets from the Gene Expression Omnibus (GEO) (accession: GSE71014) and multi-omics data from The Cancer Genome Atlas (TCGA) and Genotype-Tissue Expression (GTEx) projects. All data were anonymized and aggregated prior to analysis, in strict compliance with the Declaration of Helsinki.
Consent for publication
All data used in this study are publicly accessible and anonymized. No individual patient data or identifiable information were collected, ensuring full compliance with privacy regulations.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, Mx., Liao, Cs., Xie, Yq. et al. Construction of a prognostic risk model for acute myeloid leukemia based on exosomal genes and analysis of immune microenvironment characteristics. Sci Rep 15, 32140 (2025). https://doi.org/10.1038/s41598-025-17845-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-17845-x
