
Abstract
Mitochondrial autophagy (mitophagy) in macrophages is crucial yet poorly understood within the lung adenocarcinoma (LUAD) tumor microenvironment. This study aimed to identify key macrophage mitophagy-related genes and develop a robust prognostic model for LUAD patients. By integrating single-cell transcriptomics with machine learning algorithms, including LASSO, SVM, and random forest, we identified TUBB6 and CAT as core prognostic genes. A novel risk model based on these genes (RiskScore = 0.225 × TUBB6 − 0.19 × CAT) was constructed and validated, demonstrating that patients in the high-risk group had significantly shorter overall survival (P < 0.001). The high-risk score also correlated with an altered immune microenvironment and increased sensitivity to chemotherapies like cisplatin and gemcitabine. Furthermore, in vitro experiments confirmed that macrophage-specific overexpression of TUBB6 significantly enhanced LUAD cell proliferation, migration, and invasion through secreted factors. Our findings establish a reliable, macrophage mitophagy-based prognostic model and highlight TUBB6 and CAT as novel biomarkers and potential therapeutic targets, offering new avenues for precision medicine in LUAD.
Similar content being viewed by others


Introduction
Lung cancer remains one of the most critical global health challenges, ranking first in both incidence and mortality worldwide. In the United States, lung cancer (LC) is the leading cause of cancer-related deaths, accounting for 20.5% of all cancer fatalities, with about 340 deaths per day—2.5 times higher than those caused by colorectal cancer, which ranks second in mortality1. According to global cancer statistics, there are approximately 2 million new cases of LC diagnosed annually, with nearly half occurring in Asia2. Among these, LUAD is the most common histological subtype, representing approximately 40% of all LC cases. Data from the American Cancer Society in 2023 suggest the 5-year survival rate for LC patients is only 23.6%1,3. Although LUAD tends to progress more slowly than other subtypes, its unfavorable prognostic features significantly impact long-term survival outcomes4. Currently, clinical management of LUAD includes a variety of treatment strategies such as surgical resection, chemotherapy, immune checkpoint inhibitors, and targeted therapies. However, despite ongoing optimization of these multimodal approaches, the overall prognosis for LUAD patients remains suboptimal. Therefore, elucidating the molecular pathogenesis of LUAD and identifying disease-specific and prognostically relevant biomarkers are of paramount scientific and clinical importance. Such efforts will contribute to improving early detection strategies and advancing the development of personalized targeted therapies.
Mitochondria are essential organelles within cells, often referred to as the “powerhouses” of eukaryotic cells. They primarily generate cellular energy in the form of ATP through aerobic respiration and oxidative phosphorylation5. Dysregulation of mitochondrial morphology and function has been closely linked to the pathogenesis of various diseases6. Mitophagy, a selective mechanism for the removal of damaged mitochondria, has a critical role in maintaining mitochondrial homeostasis and function through pathways such as PINK1/Parkin and BNIP3/NIX7. In recent years, mitophagy has attracted considerable attention as a key regulatory process in a wide range of clinical conditions, encompassing neurodegenerative diseases, cardiovascular diseases, cancer, and metabolic disorders8. Increasing evidence suggests mitophagy-related genes are closely associated with the initiation and progression of LUAD. For instance, deletion of the mitophagy-related gene PINK1 alters energy metabolism in lung cancer cells and increases the sensitivity of non-small cell lung cancer (NSCLC) to glycolysis inhibitors9. Additionally, the pro-apoptotic compound fluorizoline has been found to inhibit PRKN-dependent mitophagy, thereby reducing the viability of lung cancer cells10. Dai et al. conducted a systematic analysis of key mitophagy genes involved in LUAD progression and confirmed that their expression levels are significantly associated with LUAD prognosis and may influence therapeutic strategies11. Therefore, in-depth investigation of mitophagy holds promise as a novel therapeutic avenue for LUAD.
TME has a central role in tumor initiation, progression, and therapeutic response12. Among its key components, macrophages are pivotal in shaping the immune landscape of tumors13. Based on their functional states, macrophages are broadly categorized into M1 and M2 phenotypes. M1 macrophages exert anti-tumor effects by secreting pro-inflammatory cytokines, whereas M2 macrophages are associated with tumor progression14,15. The polarization of macrophages is regulated by various factors, among which mitophagy plays a crucial role in metabolic reprogramming and phenotype determination. Studies have shown that mitophagy promotes a metabolic shift toward glycolysis, thereby facilitating M1 polarization; notably, Nix-dependent metabolic regulation has been demonstrated to be essential for this process16,17. In contrast, histone lactylation has been reported to regulate PRKN-mediated mitophagy, promoting M2 macrophage polarization and facilitating immune evasion in bladder cancer18. Emerging evidence suggests that macrophages serve as a critical link between mitophagy and the TME19,20. For example, the IL-33/ST2 axis has been shown to regulate mitophagy in macrophages, inducing a metabolic shift from oxidative phosphorylation (OXPHOS) to glycolysis (Warburg effect), which reprograms macrophage polarization and ultimately affects tumor growth within the TME21. Moreover, a study by Liu et al. demonstrated polyamines modulate mitophagy in tumor-associated macrophages (TAMs), enhancing OXPHOS and promoting M2 polarization in hepatocellular carcinoma (HCC)22. Collectively, mitophagy in TAMs not only influences macrophage polarization but also shapes the TME through functional regulation. Therefore, elucidating the mechanistic role of TAM mitophagy in the LUAD microenvironment holds significant scientific and clinical value for optimizing immunotherapeutic strategies in LUAD.
The TME is not only compositionally diverse but also analytically tractable with standardized multi-omics workflows. Recent toolkits such as IOBR provide an end-to-end framework—from transcriptomic preprocessing and deconvolution (e.g., CIBERSORT/ESTIMATE) to TME patterning, ligand–receptor inference, genome–TME association, and modeling—thereby enabling consistent, reproducible TME profiling across cohorts23 static characterization, disease trajectories can exhibit non-linear “tipping points.” Using an optimized Dynamic Network Biomarker approach, Zhang et al. identified a critical transition in thyroid cancer and derived the TCPSLevel score capturing progression risk, illustrating how dynamic systems methods decode state shifts in tumor evolution24. In parallel, integrative machine-learning models that fuse gene-pairing, feature selection, and ensemble learners have achieved superior performance for immunotherapy-response prediction across multiple cohorts; notably, the iMLGAM score links low scores to inflamed immune microenvironments and identifies actionable targets (e.g., CEP55) with functional validation—underscoring the clinical utility of data-driven TME readouts25.
In this study, genome-wide expression profiles and corresponding clinical data of LUAD were obtained from multiple public databases. Single-cell transcriptomic analysis was employed to delineate gene expression patterns across distinct cellular subpopulations within LUAD, with a particular focus on the functional roles of macrophages and their contributions to TME during LUAD progression. Mitophagy-related genes (MRGs) were retrieved from the GeneCards database. Three machine learning (ML) algorithms—LASSO regression, SVM and random forest—were used in an integrative approach to identify two hub genes through cross-validation. Subsequently, univariate analysis combined with Cox regression was conducted to build a risk score formula and prognostic model based on macrophage-specific MRGs. The model’s predictive performance was systematically evaluated by comparing survival outcomes and immune microenvironment characteristics between high-risk group (HRG) and low-risk groups (LRG). The results demonstrated that this model effectively predicts patient survival, delivering novel insights into possible therapeutic strategies targeting the tumor immune microenvironment. In addition, drug sensitivity analysis was carried out to validate the model’s clinical relevance in guiding chemotherapeutic drug selection. Overall, this study proposes a novel prognostic model centered on TAM mitophagy, offering a new theoretical framework and practical direction for personalized therapy and immune modulation in LUAD. The overall analytical workflow of this study is illustrated (Fig. 1).
Results
Single-cell transcriptomic analysis
Dimensionality reduction, clustering, and annotation of single-cell data
To investigate the gene expression profiles of LUAD cell populations, we utilized the GSE131907 dataset, extracting data from 11 tumor tissue samples and 11 matched distant normal lung tissue samples. t-SNE analysis identified 19 distinct cell clusters, which were annotated based on cell-specific markers and expression patterns, ultimately identifying 11 major cell types, including macrophages, epithelial cells, and smooth muscle cells (Fig. 2A). Furthermore, analysis of the AUC scores for mitophagy-related genes revealed elevated expression of these genes in macrophages, T cells, and epithelial cells (Fig. 2B). Statistical analysis further confirmed that AUC scores differed significantly between the high- and low-anoikis groups (Wilcoxon test, p < 2.2e − 16), as shown in Fig. 2B. The robustness of this difference was illustrated by both boxplot and density distribution visualizations with statistical annotation (Supplementary Figure S3).

Technical roadmap.

Single-cell transcriptomic landscape of LUAD. (A) t-SNE plot showing clustering of single cells into 19 clusters, with 11 major cell types identified based on canonical markers, including epithelial cells, macrophages, and T cells. (B) t-SNE visualization of mitophagy-related gene activity (AUC scores), highlighting enriched expression in epithelial cells and macrophages. (C) Global cell-cell communication network illustrating intercellular interactions across all annotated cell types. (D) Macrophage-centered communication network showing bidirectional interactions between macrophages and other cell populations. (E) Heatmap representing the strength of cell-cell communication between distinct cell types. (F) Bubble plot showing key ligand-receptor pairs mediating intercellular signaling, particularly involving macrophages. (G) Pseudotime trajectory revealing dynamic cell fate transitions during tumor evolution. (H) Hierarchical pseudotime trajectories under different conditions showing transcriptional reprogramming patterns. (I) Macrophage-specific pseudotime trajectory outlining differentiation paths within the tumor microenvironment. (J) Distribution of exogenous signaling pathway activity across cell types, reflecting immune modulation potential. (K) Distribution of endogenous signaling pathway activity, revealing differential pathway responsiveness among cell types.
Pseudotime trajectory analysis
Within the identified macrophage population, we performed pseudotime trajectory analysis to explore key gene expression programs involved in LUAD progression. This analysis successfully reconstructed the pseudotime trajectory of macrophages, revealing distinct transcriptional states and potential cell fate decisions (Fig. 2G–I).
Cell-cell communication analysis
To further elucidate the integrated roles of various cell types within lung cancer tissues, we conducted cell-cell communication analysis (Fig. 2C,E), with a particular focus on macrophage-centered interactions (Fig. 2D). The results indicated that macrophages acted as major signal senders, exhibiting strong interactions with other cell types through ligand-receptor pairs such as FN1–SDC4 and FN1–CD44 (Fig. 2F). Macrophages demonstrated bidirectional signaling behavior, actively participating in immune responses through outgoing signals and responding robustly to incoming signals, thereby contributing to intercellular interactions. Notably, macrophages exhibited strong signaling activity in both outgoing and incoming pathways within the GALECTIN (involved in the recognition of damaged organelles, such as exposed glycans, potentially marking targets for mitophagy), COMPLEMENT (likely regulating autophagy indirectly through inflammatory signaling), and THBS (thrombospondin, which modulates apoptosis and autophagy via the CD36 receptor and may indicate metabolic stress or the need for mitochondrial repair) signaling networks (Fig. 2J and K).
Cross-cohort harmonization and batch correction
A CONSORT-style flowchart summarizing sample inclusion and exclusion is shown in Supplementary Figure S1. Prior to correction, PCA demonstrated distinct separation between TCGA and GEO datasets, reflecting strong batch effects. Following ComBat correction, PCA plots revealed that samples from different cohorts became more intermixed, indicating substantial attenuation of inter-dataset variation (Supplementary Figure S2). Quantitative assessment further confirmed that the fraction of variance attributable to batch was markedly reduced after correction. These results validated the robustness of the harmonized preprocessing strategy, enabling reliable cross-cohort modeling and validation.
Identification of macrophage-intrinsic mitophagy-related genes associated with lung adenocarcinoma
By screening DEGs between normal lung tissue and early-stage LUAD tissue, a total of 14,953 DEGs were identified, encompassing 11,657 upregulated and 3,296 downregulated genes (Fig. 3A). Further single-cell differential expression analysis of LUAD tissues yielded 1,682 macrophage-associated genes. In parallel, 5,140 MRGs were retrieved from the GeneCards database. The intersection of these three gene sets resulted in the identification of 56 macrophage-intrinsic mitophagy-related genes (Fig. 3B). These intersecting genes were subjected to protein-protein interaction (PPI) analysis using the STRING database (https://cn.string-db.org/) (Fig. 3C), heatmap analysis based on TCGA data(Fig. 3D), and GO/KEGG enrichment analysis. The enrichment results suggested these genes are involved in a wide range of biological processes, encompassing: Inflammation and immune regulation: GO:0051092 (positive regulation of NF-kappaB transcription factor activity), GO:0070372 (regulation of ERK1 and ERK2 cascade), GO:0072567 (chemokine [C-X-C motif] ligand 2 production), GO:0050729 (positive regulation of inflammatory response); Oxidative stress and metabolic reprogramming: GO:0072593 (reactive oxygen species metabolic process), GO:0042304 (regulation of fatty acid biosynthetic process), GO:0045820 (negative regulation of glycolytic process); Phagocytosis and organelle interactions: GO:0006909 (phagocytosis), GO:0045335 (phagocytic vesicle), GO:0008305 (integrin complex), GO:0005925 (focal adhesion); Cell death and homeostasis regulation: GO:0032717 (negative regulation of interleukin-8 production), GO:0042771 (intrinsic apoptotic signaling pathway in response to DNA damage by p53 class mediator), among others. These enrichment findings provide critical insights for further investigation into how macrophages respond under different physiological or pathological conditions (Fig. 3E).

Identification of macrophage-intrinsic mitophagy genes in lung adenocarcinoma. (A) Volcano plot of DEGs from the TCGA dataset. The volcano plot displays differentially expressed genes identified from the TCGA dataset, highlighting significantly upregulated and downregulated genes. (B) Venn diagram of macrophage-related genes, mitophagy-related genes, and TCGA DEGs. The Venn diagram illustrates the intersection of macrophage-associated genes, mitophagy-related genes, and DEGs from the TCGA dataset, identifying 56 overlapping genes. (C) PPI network analysis of the 56 overlapping genes via the STRING database. The PPI network reveals the functional interactions among the 56 overlapping genes, providing insight into their potential regulatory roles. (D) Heatmap of the 56 overlapping genes. The heatmap shows the expression patterns of the overlapping genes associated with macrophages and mitophagy, indicating their differential expression and potential involvement in tumor progression. (E) GO and KEGG enrichment analysis of the 56 overlapping genes. The enrichment analysis demonstrates that these DEGs are significantly involved in multiple biological pathways, particularly those related to inflammation, immune regulation, phagocytosis, and oxidative stress.
Machine learning-based identification of key mitophagy-related genes in LUAD-associated macrophages
U-COX analysis of the 56 intersecting genes identified 15 filtered genes with statistically significant prognostic value (p < 0.05) (Fig. 4A). Expression analysis based on the TCGA dataset further confirmed that all 15 filtered genes exhibited significant differential expression between LUAD tissues and adjacent normal tissues (p < 0.05) (Fig. 4B). To identify key genes, three ML algorithms—LASSO, SVM, and Random Forest—were applied to the 15 filtered genes. LASSO logistic regression selected 8 predictive genes with statistical significance from the univariate analysis (Fig. 4C,D). The SVM algorithm identified 5 optimal feature genes (Fig. 4E,F). Similarly, Random Forest analysis, combined with feature selection based on the number of decision trees and error rates, excluded 5 genes and retained the most informative ones (Fig. 4G,H). The intersection of genes selected by all three machine learning methods yielded two hub genes: TUBB6 and CAT (Fig. 4I).

Identification of prognosis-related genes and diagnostic biomarkers. (A) Univariate Cox regression analysis of 56 overlapping genes identified 15 significantly associated with prognosis. (B) Expression differences of these 15 genes between lung adenocarcinoma (LUAD) and normal tissues. (C) LASSO regression narrowed down to 8 key genes. (D) Cross-validation curve determining the optimal lambda in the LASSO model. E. 5-fold cross-validation accuracy of SVM-RFE feature selection. (F) 5-fold cross-validation error rate during SVM-RFE. (G) Classification error curve of the Random Forest model with varying tree numbers. (H) Feature importance scores from the Random Forest model, ranking gene contributions. (I) Venn diagram integrating machine learning results, identifying two hub genes: TUBB6 and CAT.
Expression profiles of hub genes
The expression profiles of the hub genes were visualized using t-SNE plots derived from single-cell transcriptomic analysis. The results suggested TUBB6 and CAT were highly expressed in macrophages and epithelial cells, suggesting their potential roles in macrophage-mediated and tumor-associated immune functions. Their expression levels were relatively low in other cell types, indicating intercellular heterogeneity and highlighting the potential significance of these genes in specific cell populations (Fig. 5A). Pseudotime heatmap analysis further illustrated the dynamic expression patterns of TUBB6 and CAT across cell populations, reflecting their temporal or condition-dependent regulation. Clustering patterns in the heatmap revealed both similarities and differences in gene expression among distinct cell groups, suggesting that these genes may play key roles in specific biological processes (Fig. 5B). Pan-cancer differential expression analysis using TCGA data from 33 common tumor types demonstrated that TUBB6 and CAT were significantly differentially expressed between tumor and normal tissues in LUAD and several other cancers (Fig. 5C,D). Additionally, pan-cancer immune infiltration analysis indicated that high and low expression groups of TUBB6 and CAT were associated with distinct patterns of immune cell infiltration—including T cells and B cells—across multiple malignancies, including LUAD (Fig. 5E). Pan-cancer survival analysis using TCGA datasets revealed that both heatmap and Cox regression analyses identified TUBB6 and CAT as being remarkably associated with overall survival (OS) in various cancers, including lung adenocarcinoma (Fig. 5F,G).

Expression and prognostic significance of hub genes. (A) t-SNE plots illustrating the spatial expression distribution of the hub genes across various cell types in the LUAD tumor microenvironment, highlighting their potential cellular specificity and functional relevance. (B) Pseudotime heatmaps showing dynamic expression changes of hub genes during cell state transitions, indicating their involvement in lineage differentiation and fate decisions. (C) Bar plots comparing the expression levels of hub genes between LUAD tissues and adjacent normal tissues based on TCGA data. (D) Network plots showing differential expression of hub genes across tumor and normal tissues in 33 TCGA cancer types. (E) Heatmap depicting the connection between hub gene expression and immune cell infiltration levels across 33 cancer types in TCGA. (F) Prognostic heatmap analyzing the connection between hub gene expression and overall survival across pan-cancer datasets. (G) Forest plot of M-Cox analysis for hub genes across 33 cancers in the TCGA dataset.
Macrophage TUBB6 overexpression promotes proliferation, migration, and invasion of lung adenocarcinoma cells
To study the impact of TUBB6 expression in macrophages on the behavior of lung adenocarcinoma cells, a TUBB6-overexpressing macrophage model was established, and its CM was collected for co-culture experiments. Western blot analysis confirmed successful overexpression, showing a significant increase in TUBB6 protein levels in macrophages infected with the lentiviral vector compared to controls (Fig. 6A,B). CCK-8 assay results demonstrated that NCI-H1975 and A549 cells treated with CM from TUBB6-overexpressing macrophages exhibited significantly higher OD450 values compared to those treated with control CM (P < 0.05), indicating enhanced cell proliferation (Fig. 6C). Transwell invasion assays further confirmed this observation, as the number of cells migrating through the membrane was markedly increased in the TUBB6-overexpression CM group compared to the control CM one (P < 0.05), indicating enhanced invasive ability (Fig. 6D,F). In the wound healing assay, the scratch closure rate at 24 h was remarkably higher in the TUBB6-overexpression CM group than in the control group (P < 0.05), suggesting increased migratory capacity (Fig. 6E and G). Collectively, these findings suggest that macrophages overexpressing TUBB6 promote the proliferation, migration, and invasion of lung adenocarcinoma cells through the secretion of soluble factors.

TUBB6 overexpression in macrophages enhances proliferation, migration, and invasion of LUAD cells. (A) Schematic map of the lentiviral overexpression vector pLVX-TUBB6-Flag (9380 bp), used to construct macrophage TUBB6 overexpression models. (B) Western blot analysis confirming elevated TUBB6 expression in TUBB6-OE-Mac compared to WT-Mac. GAPDH served as a loading control. (C) Bar plots of CCK-8 assays showing that TUBB6_OE_CM significantly promotes proliferation of A549 and NCI-H1975 cells compared to WT_CM. (D) Bar graphs of Transwell assays quantifying the increased migratory capacity of A549 and NCI-H1975 cells upon treatment with TUBB6_OE_CM versus WT_CM. (E) Bar plots of wound healing assays revealing enhanced migration in A549 and NCI-H1975 cells exposed to TUBB6_OE_CM. (F) Representative images of Transwell assays visualizing increased invasion of LUAD cells in the TUBB6_OE_CM group. (G) Representative images of wound healing assays demonstrating faster wound closure in cells treated with TUBB6_OE_CM compared to WT_CM. WT-Mac wild-type macrophages, TUBB6-OE-Mac TUBB6-overexpressing macrophages, WT_CM conditioned medium from WT-Mac, TUBB6_OE_CM conditioned medium from TUBB6-OE-Mac.
TUBB6 overexpression promotes mitophagy in macrophages
Western blot analysis demonstrated that TUBB6 overexpression significantly altered the expression of mitophagy-related proteins in macrophages (Fig. 7A). Compared with WT-Mac, TUBB6-OE-Mac exhibited a markedly increased LC3-II/LC3-I ratio (Fig. 7B) and significantly decreased TOM20 and P62 expression (Fig. 7C,D), indicating enhanced mitophagy activity. These results suggest that TUBB6 overexpression directly promotes mitophagy in macrophages.

TUBB6 overexpression alters mitophagy-related protein expression. (A) Western blot analysis of LC3 I, LC3 II, TOM20, and P62 in WT_Mac and TUBB6_OE_Mac conditions. (B) Quantitative analysis of LC3 II/LC3 I ratio in the indicated groups. (C) Quantitative Western blot analysis of TOM20 expression in the indicated groups. (D) Quantitative Western blot analysis of P62 expression in the indicated groups.
Establishment and validation of a prognostic risk model
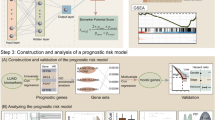
M-COX analysis identified TUBB6 and CAT as independent risk factors for lung adenocarcinoma (Fig. 8A). Based on their expression levels, a risk score formula was constructed: RiskScore = (TUBB6 × 0.225) + (CAT × −0.19). The prognostic performance of this model was evaluated in the TCGA training cohort, and in two independent validation cohorts (GSE68465 and GSE13213). Time-dependent ROC curves, along with risk scores and survival status analyses, demonstrated robust prognostic predictive ability across all datasets (Fig. 8B–D).

Construction and validation of the LUAD prognostic risk model. (A) M-Cox analysis confirming TUBB6 and CAT as independent prognostic indicators, and used to build a LUAD risk scoring model. (B) TCGA-LUAD training set: Kaplan-Meier survival analysis, risk score distribution, gene expression heatmap, and time-dependent ROC curves evaluating model performance. (C) GSE68465 validation cohort: Survival curves, risk stratification, expression profiles, and ROC analysis demonstrating model robustness. (D) GSE13213 validation cohort: Time-dependent ROC and survival analysis confirming the prognostic value of the model in an independent dataset.
Correlation between clinical features and riskscore
Integrating clinical data from patients with lung adenocarcinoma, we analyzed the distribution of RiskScores across various clinical parameters. The results demonstrated significant differences in RiskScores based on sex, survival status, clinical stage, and TNM classification (Fig. 9A–E). Additionally, a heatmap depicting the relationship between RiskScore and clinical characteristics was generated to further explore its clinical relevance (Fig. 9F).

Correlation between RiskScore and clinical features. (A–E). Violin and box plots illustrating the distribution of RiskScore across different clinical subgroups, encompassing age, gender, TNM stage, overall stage, and survival status. Significant differences in RiskScore suggest its strong association with key clinical parameters in LUAD. (F) Heatmap displaying the correlations between RiskScore and clinical characteristics, further supporting the potential of RiskScore as a prognostic indicator linked to disease progression.
Construction of a nomogram
U-COX analysis identified clinical factors associated with patient prognosis (Fig. 10A). Based on these factors, a nomogram was built to predict OS (Fig. 10B). Calibration curves and time-dependent ROC curves suggested the nomogram had good predictive accuracy for 1-, 3-, and 5-year OS (Fig. 10C), with AUC values of 0.746, 0.759, and 0.708 (Fig. 10D).

Construction and validation of the prognostic nomogram. (A) U-Cox identifying clinical variables significantly associated with overall survival in LUAD patients. (B) Prognostic nomogram integrating RiskScore and independent clinical features to predict 1-, 3-, and 5-year survival probabilities. (C) Calibration curves showing the consistency between predicted and observed survival outcomes, indicating the nomogram’s accuracy. (D) Time-dependent ROC curves evaluating the discrimination ability of the nomogram, with AUC values supporting strong predictive performance. (E) Decision curve analysis (DCA) comparing the net clinical benefit of the full model, base model, and "treat all/none" strategies. (F) Net reclassification index (NRI) of the full model versus the base model at 1-, 3-, and 5-year time points. (G) Integrated discrimination improvement (IDI) of the full model compared with the base model at 1-, 3-, and 5-year time points.
DCA demonstrated that the nomogram integrating RiskScore with clinical factors consistently achieved a higher net benefit than the base model and default “treat-all/none” strategies, with the advantage being most pronounced at the 3-year time point (Fig. 10E). Furthermore, NRI and IDI analyses revealed that the addition of RiskScore significantly improved risk reclassification and discrimination at 1-, 3-, and 5-year time points (all P < 0.001), confirming the incremental value of the full model over the base model (Figs. 10F,G). These findings collectively support the robust clinical applicability of our prognostic nomogram.
TME analysis
The results from the ESTIMATE algorithm indicated that the high-risk group had a lower ImmuneScore, but higher StromalScore and ESTIMATEScore. Using the “CIBERSORT” and “ssGSEA” algorithms, we found remarkable differences in the proportions of immune cells between the HRG and LRG, particularly in macrophages, eosinophils, and mast cells (Fig. 11A–F). Additionally, we calculated the correlation between RiskScore and key genes and immune cell types, further revealing the connection between RiskScore and the immune characteristics of the tumor microenvironment (Fig. 11G).

Association between RiskScore and the tumor microenvironment (TME). (A–C) Box plots showing differences in StromalScore, ImmuneScore, as well as ESTIMATEScore between HRG and LRG, indicating distinct TME composition patterns. (D) ssGSEA analysis of immune cell infiltration showing significant variation across RiskScore groups. (E) CIBERSORT analysis validating differential infiltration of immune cells between HRG and LRG. (F) Correlation heatmap based on CIBERSORT data illustrating associations between RiskScore, hub genes, and immune cell types. (G) ssGSEA-based correlation analysis further demonstrating links between RiskScore and infiltration of specific immune cell subsets and hub gene expression.
GSEA analysis, GO/KEGG analysis, and immune checkpoint analysis
In the HRG, higher cytotoxicity (CYT), higher tumor mutational burden (TMB), and lower Tertiary Lymphoid Structures (p < 0.05) suggest the HRG may have a stronger immune response, which could be related to enhanced immune activity in the tumor microenvironment (Fig. 12A–C). Immune checkpoint gene expression analysis suggested the HRG had lower expression of immune checkpoint genes BTLA and CD47 (p < 0.05) (Fig. 12D), indicating a potential for better response to immunotherapy in this group.

Functional enrichment and immune checkpoint analysis. (A) Violin plots comparing TMB between HRG and LRG, suggesting a link to immunogenicity and tumor aggressiveness. (B) Distribution of cytolytic activity (CYT) scores across RiskScore groups, with elevated CYT in high-risk patients indicative of enhanced immune activation. (C) Violin plots depicting differences in tertiary lymphoid structure (TLS) scores, reflecting heterogeneity in adaptive immune responses. (D) Expression analysis of immune checkpoint genes revealed lower BTLA and CD47 expression in high-risk patients, suggesting potential responsiveness to immunotherapy. (E) GO and KEGG pathway enrichment analysis of hub genes highlighting roles in oxidative stress, immune signaling, and mitophagy. (F) GSEA results showing differential activation of immune-related and metabolic pathways between HRG and LRG.
GO/KEGG enrichment analysis identified multiple functions related to oxidative stress and mitochondrial quality control, including: GO:0034614 (cellular response to reactive oxygen species), GO:0072593 (reactive oxygen species metabolic process), GO:0098869 (cellular oxidant detoxification), and GO:0034599 (cellular response to oxidative stress). Additionally, pathways related to energy metabolism and autophagy regulation were enriched, such as: GO:0015980 (energy derivation by oxidation of organic compounds), GO:0014068 (positive regulation of phosphatidylinositol 3-kinase signaling). Enriched functions related to organelle structure and function included GO:0005758 (mitochondrial intermembrane space), and key signaling pathways such as hsa04068 (FoxO signaling pathway), hsa05208 (Chemical carcinogenesis - reactive oxygen species), and hsa04145 (Phagosome) (Fig. 12E).
GSEA analysis revealed upregulation of several immune activation and antigen presentation pathways, and metabolic and immune regulation pathways, while immune suppression, tolerance-related pathways, mitochondrial functions, and tumor progression pathways were downregulated (Fig. 12F).
Chemotherapy and targeted drug IC50 analysis
The IC50 analysis revealed differences in drug sensitivity between the HRG and LRG for commonly used chemotherapy and targeted drugs. The results indicated the HRG was more sensitive to DNA-damaging agents (cisplatin, gemcitabine) and kinase inhibitors (dasatinib, cabozantinib), whereas the LRG showed better responses to targeted therapies (osimertinib, KRAS inhibitors) and oxaliplatin (Fig. 13).

IC50 analysis of chemotherapeutic and targeted drugs. The HRG exhibited greater sensitivity to cisplatin, gemcitabine, dasatinib, and savolitinib, whereas the LRG responded more favorably to osimertinib, KRAS inhibitors, and oxaliplatin.
Discussion
Autophagy plays a crucial role in maintaining cellular homeostasis, regulating metabolism, and responding to oxidative stress. Specific types of autophagic processes may influence tumor progression and immune responses26. As key immune cells within the TME, the functional plasticity of macrophages is regulated by various metabolic signals. Their mitophagy may modulate LUAD progression by affecting energy metabolism, inflammation, and immune suppression9,10,11. In this study, single-cell transcriptomic analysis was used to systematically investigate the functions of macrophages in LUAD and their roles within the TME. Based on macrophage mitophagy-related genes TUBB6 and CAT, we constructed a risk score formula and prognostic model. This model effectively stratifies LUAD patients into HRG and LRG and demonstrates strong predictive ability in independent cohorts, providing a reliable tool for individualized survival assessment and prognosis in LUAD patients. Moreover, by integrating clinical data, TME immune characteristics, immune checkpoint expression, and chemotherapy drug IC50 data, this study further explored the immune microenvironment features of patients in different risk groups and their potential therapeutic responses. These findings offer new theoretical insights for precision treatment and personalized intervention strategies for LUAD.
Cell-cell communication analysis revealed macrophages play a crucial role in both outgoing and incoming signaling, further emphasizing their complex regulatory functions within TME. TAMs are one of the most prominent infiltrating immune cell types in the solid TME27. In recent years, targeting TAMs has emerged as one of the most promising immune therapeutic strategies28. However, current research on TAMs primarily focuses on their modulation of mitophagy within tumor tissues to influence the TME, and there remains a lack of systematic exploration regarding how mitophagy within TAMs specifically regulates the TME and its impact on tumor prognosis. This study further utilized AUC score analysis, which showed that MRGs were highly expressed in smooth muscle cells and macrophages, providing further evidence of the close association between macrophages and mitophagy. Therefore, investigating the role of mitophagy within TAMs in TME remodeling and LUAD prognosis will offer new insights into the dynamic mechanisms of TME changes and provide potential intervention strategies for the precise treatment of LUAD.
This study performed PPI analysis and GO/KEGG enrichment analysis on 56 mitophagy-related genes identified in macrophages. The results showed that these genes are extensively involved in multiple key biological processes, encompassing inflammation and immune regulation, oxidative stress and metabolic reprogramming, phagocytosis and organelle interactions, and cell death and homeostasis regulation, further revealing the important roles of macrophage-intrinsic MRGs in the initiation, progression, and prognosis of LUAD. Using three ML algorithms—LASSO, SVM, and Random Forest—the MRGs in macrophages were further screened, ultimately identifying TUBB6 and CAT as hub genes. Analysis suggested the expression levels of these two genes were significantly higher in macrophages compared to other cell types. Moreover, pan-cancer analysis using TCGA data from 33 common cancers revealed significant expression differences of TUBB6 and CAT between tumor and adjacent normal tissues, and these genes were closely associated with immune cell infiltration and prognosis. Based on these results, TUBB6 and CAT may play a crucial role in macrophage-mediated immune functions. Further M-COX analysis demonstrated both TUBB6 and CAT are independent risk factors for LUAD. Using these two hub genes along with related clinical prognostic factors, we constructed a nomogram-based prognostic risk prediction model, which accurately predicts 1-, 3-, and 5-year OS and demonstrates strong clinical applicability. Notably, this is the first study to integrate macrophage-intrinsic mitophagy with DEGs and successfully establish a prognostic prediction model for LUAD. The findings suggest TUBB6 and CAT serve as key prognostic predictors for LUAD, offering new potential targets for risk stratification and precision treatment in LUAD.
Beyond conventional metrics such as ROC curves and calibration, we further evaluated the clinical utility of the prognostic model using DCA, NRI, and IDI. The results demonstrated that integrating RiskScore into the nomogram provided meaningful clinical benefit, consistently outperforming the base model and standard treatment strategies in decision curve analysis. Moreover, the significant improvements observed in NRI and IDI at multiple time points confirmed that the inclusion of RiskScore enhanced both patient risk stratification and discrimination ability. These findings underscore that our model not only achieves robust statistical performance but also offers tangible value for guiding individualized prognostic assessment and clinical decision-making in LUAD.
TUBB6 is a structural subunit of microtubule α/β-heterodimers, with distinct isoforms exhibiting different expression and distribution patterns in mitosis and tumor tissues29,30. Microtubules are essential for key cellular functions, encompassing cell division, intracellular transport, neuronal morphology, and the movement of cilia and flagella31. TUBB6 is closely associated with cancer invasion and metastasis in aggressive malignancies32. Recent studies have shown that TUBB6 promotes the growth of various solid tumors and serves as an important prognostic factor33,34. Research by Wen et al.35 identified TUBB6 as a macrophage-related gene associated with prognosis and immunotherapy efficacy in NSCLC patients, a conclusion supported by our study. Additionally, Luo et al.36 demonstrated that TUBB6 may influence the progression of non-alcoholic fatty liver disease by regulating mitophagy and immune activation. Furthermore, TUBB6 has been shown to induce macrophage polarization from the M0 to M2 phenotype, thereby promoting tumor proliferation35. As a critical structural scaffold for intracellular material transport, microtubules play an essential role in the process of mitophagy, where damaged mitochondria rely on microtubules for transport to lysosomes for degradation. Therefore, we hypothesize that TUBB6 not only induces macrophage polarization but also potentially regulates macrophage-intrinsic mitophagy, thereby promoting immune escape and progression in LUAD.
This study, through co-culture experiments, for the first time reveals that the overexpression of TUBB6 in macrophages significantly enhances the proliferation, migration, and invasion of lung adenocarcinoma cells. Previous studies have suggested TUBB6 is closely associated with tumor progression and metastasis in various cancers, and its overexpression can increase the invasiveness of tumor cells33,34. This study further extends the understanding of TUBB6 function, indicating that TUBB6 expression in macrophages can also exert a remote regulatory effect on lung cancer cell behavior through secreted factors.
Our study further revealed that TUBB6 overexpression directly enhances mitophagy in macrophages, as evidenced by increased LC3-II/LC3-I ratios and reduced TOM20 and P62 expression. These findings suggest that TUBB6 may promote mitochondrial quality control, sustaining macrophage metabolic activity and survival, thereby strengthening their tumor-promoting role within the microenvironment. Previous studies have shown that macrophage mitophagy facilitates M2 polarization and supports tumor immune evasion. Our results provide protein-level evidence linking TUBB6 to macrophage mitophagy.
Studies have shown that, compared to homologous normal tissues, catalase (CAT) expression is generally reduced in cancer tissues. However, elevated levels of CAT expression have been observed in tumor tissues of gastric cancer, skin cancer, and chronic myeloid leukemia patients37. In LUAD tissues, CAT mRNA levels are downregulated compared to normal lung tissues38. This finding is consistent with our study. TCGA data analysis indicates that LUAD patients with high CAT expression have a longer survival, and the gene polymorphism at the CAT rs769217 locus is remarkably associated with progression-free survival in LC patients receiving platinum-based chemotherapy39. Furthermore, recent studies have further suggested that CAT is an independent prognostic marker for LUAD40. Abnormal expression or reduced activity of CAT causes increased intracellular reactive oxygen species (ROS) levels, which directly or indirectly promote tumorigenesis41,42. Studies have shown that knockdown of CAT results in elevated ROS levels, inhibiting cell proliferation and migration, while overexpression of CAT reduces ROS levels, promoting cell growth and migration. The upregulation of Parkin partially reverses these effects43. Therefore, as a binding partner and substrate of Parkin, CAT plays a role in cancer progression by regulating intracellular ROS levels. ROS can cause mitochondrial DNA damage, reduce mitochondrial membrane potential, and induce protein and lipid oxidation44. Mitophagy may function more broadly to limit the harmful effects of ROS on cellular function45. Under hypoxic conditions, high abundance of pro-tumor M2 macrophages can induce tumor progression and drug resistance46. Studies show that catalase and its mimetic compounds can effectively participate in the polarization of TAMs and hypoxia alleviation within TME46. Therefore, in summary, CAT may influence LUAD development, drug resistance, and prognosis by regulating macrophage-intrinsic mitophagy, reducing ROS production, and altering the hypoxic environment in the TME.
To explore the potential molecular mechanisms underlying the poor prognosis in HRG patients, we conducted GSEA analysis, GO/KEGG analysis, and immune checkpoint analysis on the sequencing data of HRG and LRG. The results indicated the HRG may have a stronger immune response. GO/KEGG analysis revealed that, within the LUAD microenvironment, both tumor cells and immune cells generate a large amount of ROS. Macrophages activate mitophagy through cellular oxidant detoxification and the hydrogen peroxide catabolic process to eliminate dysfunctional mitochondria, preventing excessive ROS accumulation and cell apoptosis. Additionally, through the positive regulation of phosphatidylinositol 3-kinase signaling, the mTOR pathway is inhibited, promoting autophagosome formation and enhancing mitophagy. Immune checkpoint analysis showed the low expression of BTLA and CD47, along with the high expression of CD274, suggests that while tumor cells may escape immune surveillance through PD-L1, the immune system may still be able to respond to the tumor to some extent due to the low expression of CD47 and BTLA. We also found remarkable differences in the immune cell proportions between the HRG and LRG, particularly in macrophages, eosinophils, and mast cells. TME ESTIMATEScore analysis further confirmed that patients with high-risk scores are in an immune-suppressive state, with immune scores being lower. All of these findings provide a comprehensive explanation for the poor prognosis observed in HRG patients.
Another important finding is the differential sensitivity to common targeted and chemotherapy drugs between the HRG and LRG. The HRG showed greater sensitivity to DNA-damaging agents (cisplatin, gemcitabine) and kinase inhibitors (dasatinib, cabozantinib), while the LRG had a better response to targeted therapies (osimertinib, KRAS inhibitors) and oxaliplatin. This may be attributed to the genomic instability, activation of pro-cancer signaling pathways, and an immune-suppressive microenvironment in the high-risk group, which collectively contribute to their heightened sensitivity to DNA-damaging agents (cisplatin, gemcitabine) and kinase inhibitors (dasatinib, cabozantinib). In contrast, the low-risk group’s stronger immune activity, specific mutations (such as KRAS G12C), and metabolic characteristics contribute to a better response to targeted therapies (osimertinib, KRAS inhibitors) and oxaliplatin.
Although our study provides novel insights and identifies significant prognostic biomarkers (TUBB6 and CAT) for lung adenocarcinoma (LUAD) through an integrative analysis of single-cell transcriptomics and machine learning, several limitations warrant consideration. Firstly, the primary data utilized in our analyses originated from TCGA and GEO databases, predominantly representing Western populations, and the discovery single-cell cohort was mainly based on GSE131907 (22 samples). Although we screened available single-cell datasets, challenges such as heterogeneous sequencing platforms, inconsistent preprocessing pipelines, and pronounced batch effects limited reliable cross-cohort integration. These factors, together with the absence of a fully independent, large-scale external cohort (e.g., multi-center Asian samples), may restrict the generalizability of our findings. Future work will focus on validating this prognostic model in prospective, real-world cohorts and expanding multi-center single-cell datasets to further enhance its translational applicability. Secondly, despite the robust in silico predictions and preliminary in vitro validations provided, further comprehensive animal model studies and large-scale clinical validations are essential to confirm the precise biological mechanisms by which TUBB6 and CAT regulate macrophage mitophagy and their roles in the tumor microenvironment. Lastly, the exact molecular mechanisms underlying how TUBB6 mediates microtubule-dependent mitophagy and how CAT modulates immune cell functions via reactive oxygen species (ROS) remain incompletely elucidated, necessitating more in-depth mechanistic investigations.
In conclusion, this study integrates single-cell transcriptomics with advanced machine learning methodologies to identify TUBB6 and CAT as key macrophage MRGs associated with prognosis in LUAD. We successfully developed a reliable prognostic model based on these genes, effectively distinguishing between HRG and LRG patient groups with robust predictive accuracy. Moreover, this model highlights potential therapeutic targets and provides valuable insights into the tumor immune microenvironment and drug sensitivity profiles, thus offering promising avenues for precision therapy and personalized management in LUAD.
Materials and methods
Acquisition and processing of bulk transcriptomic data
In this study, genome-wide transcriptomic data and corresponding clinical information for LUAD were obtained from The Cancer Genome Atlas (TCGA) database via the Bioconductor package “TCGAbiolinks” (version 2.25.0). The data were provided in TPM (Transcripts Per Million) format. The TCGA-LUAD dataset (n = 600) included 59 normal control samples and 541 tumor samples. Additionally, two independent LUAD cohorts were retrieved from the Gene Expression Omnibus (GEO) database: the GSE13213 dataset comprising 117 LUAD samples, and the GSE68465 dataset containing 462 LUAD samples. During data preprocessing, samples lacking survival information or with a survival time of zero were excluded to ensure valid prognostic analysis47,48.
To ensure comparability across datasets, expression matrices from TCGA and the two GEO cohorts were standardized to a common scale. Expression values were transformed to log2-TPM (or log2-transformed intensities for applicable microarray platforms). Gene identifiers were normalized by removing version suffixes and whitespace, and duplicated symbols were resolved by retaining the entry with the highest mean expression within each dataset. Genes with > 50% missing values or zero expression were excluded, and residual missing values were imputed with the gene-wise median. Only genes present in all three cohorts were retained for subsequent analyses.
Batch effects were assessed by PCA using the most variable genes, which revealed clear separation between datasets prior to correction. To mitigate these effects, ComBat (sva) was applied with “dataset” specified as the batch factor, using empirical Bayes adjustment (with non-parametric fallback when required). Batch-effect attenuation was further confirmed by ANOVA-based variance decomposition, which quantified the proportion of variance attributable to batch before and after correction. All procedures were performed in R (sva/limma/ggplot2).
Download and processing of single-cell RNA sequencing data
We obtained the single-cell RNA sequencing dataset GSE131907 from the GEO database (https://www.ncbi.nlm.nih.gov/geo/). The dataset encompasses 11 tumor tissue samples and 11 matched adjacent normal lung tissue samples. Data preprocessing and analysis were carried out using the Seurat package (5.1.0) in R. Normalization was conducted using the NormalizeData function. Highly variable genes were selected based on their average expression and dispersion. Cell clustering was carried out using the FindClusters function, which applies a Shared Nearest Neighbor modularity optimization algorithm. A total of 19 cell clusters were identified based on the top 30 principal components (PCs), with a resolution parameter set to 1.0. Then, t-distributed Stochastic Neighbor Embedding (t-SNE) was performed using the RunTSNE function to visualize the distribution of cell populations. Differentially expressed genes (DEGs) across clusters were identified using the FindAllMarkers function in Seurat, which also facilitated cell-type annotation and assessment of cell-type proportions.
AUC score analysis of MRGs
We retrieved a total of 5,140 MRGs from the GeneCards database (https://www.genecards.org/). GSEA was conducted using the AUCell R package to calculate the Area Under the Curve (AUC) value for each individual cell, thereby assessing the expression level of mitophagy-associated genes. Cells with higher AUC scores were considered to exhibit elevated expression of MRGs. The distribution of AUC scores was visualized using a t-SNE plot, which illustrated the spatial distribution of cells across different AUC score groups. To evaluate the statistical significance of AUC score differences between high- and low-anoikis groups, a Wilcoxon rank-sum test was performed.
Cell–cell communication analysis
We utilized the CellChat package (1.6.1) to analyze intercellular communication among different cell types, with a particular focus on interactions between macrophages and other cell populations. CellChat infers the cell-cell communication network by calculating ligand-receptor pairs and their associated signaling pathways. The results were visualized using heatmaps and bubble plots, illustrating the enrichment and communication strength of ligand-receptor interactions among different cell types. Additionally, CellChat was employed to examine the relative contribution of both incoming and outgoing signaling pathways across different cell types, uncovering how distinct cellular populations respond to exogenous signals in the tumor microenvironment, such as immune and inflammatory responses.
Pseudotime trajectory analysis
We performed pseudotime analysis using the Monocle 2 toolkit, focusing on genes with high expression variability and substantial expression levels (dispersion ≥ 1, mean expression ≥ 0.1). This analysis enabled the construction of a pseudotime trajectory map, revealing the developmental trajectory of LUAD cells and illustrating dynamic changes in cellular states. To further investigate gene expression dynamics involved in cell fate decisions, we applied branch expression analysis modeling (BEAM). Differential gene expression across distinct trajectory branches was visualized using heatmaps. Notably, an independent pseudotime trajectory analysis was conducted for macrophages, uncovering their developmental progression and differentiation within the LUAD tumor microenvironment.
Identification of DEGs
We used the “limma” package to identify DEGs between lung adenocarcinoma and normal tissue samples. Multiple testing correction was performed using the Benjamini-Hochberg method to calculate adjusted p-values (adj. p-values). DEGs were defined as those with a false discovery rate (FDR) < 0.05 and an absolute log2 fold change (|log2FC|) ≥ 1. Subsequently, enrichment analysis of the identified DEGs was conducted via the ‘clusterProfiler’ package, with a significance threshold of adj. p-value < 0.05.
Screening of macrophage-specific mitophagy-related genes in lung adenocarcinoma
Through Venn diagram analysis, we identified the intersecting genes among differentially expressed genes between LUAD and normal tissues, macrophage-related genes, and mitophagy-related genes. This approach led to the identification of differentially expressed mitophagy-related genes specifically present in macrophages within LUAD.
PPI network construction using the STRING database
The intersecting genes were input into the STRING database, with “Homo sapiens” selected as the reference species to ensure biological relevance of the PPI analysis.
A minimum confidence score threshold of 0.7 (medium confidence) was applied to exclude low-confidence interactions and enhance the reliability of the network. The “network analysis” mode was selected, and the option to “hide disconnected nodes in the network” was enabled to ensure that all nodes included in the PPI network represent biologically meaningful interactions.
Construction and validation of a prognostic risk model
To identify prognostic genes, we applied three ML algorithms—LASSO, SVM, and Random Forest—using R packages including e1071, randomForest, and glmnet. Genes identified by all three algorithms were intersected to obtain candidate prognostic genes. UnivariateCox regression (U-COX) and multivariate Cox regression (M-COX) analyses were performed via the survminer package, and a prognostic risk score (RiskScore) was calculated according to the regression coefficients: RiskScore = β₁X₁ + β₂X₂ + … + βnXn (where β represents the regression coefficient and X denotes gene expression levels). Based on the calculated RiskScore, samples from the TCGA training set and GEO validation set were stratified into HRG and LRG. Kaplan-Meier survival curves were used to assess the prognostic value of the model, while time-dependent ROC (timeROC) curves were applied to evaluate its predictive performance.
Correlation analysis between clinical characteristics and riskscore
We integrated clinical data from patients with lung adenocarcinoma and analyzed the distribution differences of the RiskScore across various clinical characteristics, including age, TNM stage, gender, and smoking history.
Construction of a nomogram
By integrating clinical characteristic data with the RiskScore, U-COX and M-Cox analyses were performed to identify independent prognostic factors. These factors were then used to build a nomogram model. Using the rms package for analysis and visualization, a nomogram was developed to predict 1-, 3-, and 5-year OS in patients with early-stage lung adenocarcinoma. The predictive performance of the nomogram was evaluated using calibration curves and time-dependent ROC (timeROC) curves.
To further assess the clinical utility of the RiskScore and nomogram, we performed decision curve analysis (DCA), net reclassification index (NRI), and integrated discrimination improvement (IDI) analyses. DCA was conducted to evaluate the net clinical benefit of the full model (RiskScore + independent clinical variables) compared with the base clinical model and the “treat-all” or “treat-none” strategies. NRI and IDI were calculated at 1-, 3-, and 5-year time points to quantify the improvement in risk reclassification and discrimination ability provided by the full model over the base model. These analyses were carried out using the “rms,” “stdca,” and “survIDINRI” packages in R.
TME analysis
We used the CIBERSORT and ssGSEA algorithms to analyze differences in immune cell infiltration between the HRG and LRG. In addition, the ESTIMATE package was used to calculate immune scores, stromal scores, and ESTIMATE scores for each group, allowing further investigation into the connection of the RiskScore with the tumor immune microenvironment. This analysis helps to elucidate the potential impact of the RiskScore on the tumor immune landscape, especially in relation to immune cell infiltration and immune evasion mechanisms.
GSEA and GO/KEGG enrichment analyses
We performed GSEA to explore hallmark gene sets (c2.all.v2024.1.Hs.symbols) between different RiskScore groups, and GO and KEGG pathway enrichment analyses related to hub genes. Enrichment results were considered significant based on a p-value < 0.05 and an absolute normalized enrichment score (|NES|) > 1. These analyses enabled us to further investigate functional and pathway-level differences between RiskScore groups, revealing pathways closely associated with tumorigenesis, immune responses, and cellular metabolism.
Immunotherapy response analysis
We evaluated the differences in tumor mutational burden (TMB) between patients in different RiskScore groups and calculated the cytolytic activity (CYT) score based on the expression of cytotoxic effector genes. Additionally, we analyzed the expression levels of immune checkpoint-related genes. These indicators were used to investigate the association of the RiskScore with immunotherapy response, with a particular focus on the potential role of immune evasion mechanisms in the high-risk group. This analysis provides important insights into the potential efficacy of immunotherapy.
Drug sensitivity analysis
Using the GDSC database, we evaluated the sensitivity of patients in different RiskScore groups to commonly used targeted therapies and chemotherapeutic agents. The OncoPredict package was utilized to estimate the half-maximal inhibitory concentration (IC50) values for each drug, and compounds showing significant differences between the HRG and LRG were identified. This analysis provides a valuable reference for personalized treatment strategies in LUAD patients, particularly in guiding the clinical selection of targeted and chemotherapeutic agents.
Cell culture
NCI-H1975 (CL-0298, Procell, CN) and A549 (CL-0016, Procell, CN) cells were cultured in RPMI-1640 medium (10–040-CVR, Corning, CN) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin at 37 °C in a humidified incubator with 5% CO₂. When cell confluence reached approximately 80%–90%, the cells were washed with PBS and digested with 0.25% trypsin-EDTA (1 min for A549 and 2 min for H1975). Enzymatic digestion was terminated by adding complete medium, and the cells were either passaged or harvested for subsequent experiments.
TUBB6 overexpression in macrophages
THP-1 monocytes (CL-0233, Procell, CN) were differentiated into macrophages by treatment with 100 ng/mL phorbol 12-myristate 13-acetate (PMA) for 24 h, followed by a 24-hour resting period in fresh medium. TUBB6 overexpression was achieved by infection with a lentiviral vector encoding human TUBB6 cDNA (see Supplementary Table 1 for the transfection sequence). Cells transduced with an empty vector or non-infected cells served as controls. After 48 h of infection, cells were harvested for verification of TUBB6 expression via Western blot analysis (described below). For collection of conditioned medium (CM), macrophages were washed with PBS and cultured in serum-free medium for 24 h. The supernatant was then collected by centrifugation at 300 × g for 5 min and stored at − 80 °C for further use.
Western blot
Total protein was extracted using RIPA lysis solution supplemented with protease and phosphatase inhibitors. subjected to SDS-PAGE and then transferred to a PVDF membrane. After blocking the membrane with 5% non-fat milk for an hour at room temperature, primary antibodies against TUBB6 (Invitrogen, PA5-98948, 1:1000), and internal controls GAPDH (Abcam, ab8245, 1:5000) were added, and the membrane was incubated overnight at 4 °C. The membrane was incubated with HRP-conjugated secondary antibodies (1:5000) for one hour at room temperature following TBST washing. Enhanced chemiluminescence (ECL) was used to visualize protein bands, and densitometry was used to measure the intensity of the bands.
Co-culture experiment
Conditioned medium (CM) from TUBB6-overexpressing or control macrophages was mixed with fresh culture medium at a 1:1 ratio and used to treat NCI-H1975 or A549 cells for 24–48 h. Control groups were treated with CM from control macrophages mixed with fresh medium in the same ratio. Functional assays were performed concurrently.
Cell proliferation assay (CCK-8)
LUAD cells were seeded into 96-well plates at a density of 2 × 10³ to 5 × 10³ cells per well and treated with either experimental or control CM. After 24–48 h of incubation, 10 µL of CCK-8 reagent was added to each well and incubated for an additional 2 h. The absorbance at 450 nm (OD450) was then measured. Each group was set up in five replicates and the experiment was independently repeated three times.
Wound healing assay
Cells were seeded into 6-well plates and allowed to reach about 90% confluence before a scratch was made using a sterile pipette tip. After washing with PBS to remove detached cells, culture medium containing 10% FBS (control) or experimental CM was added. Wound areas were imaged at 0 and 24 h using a microscope, and the migration rate was calculated. Each group included three replicates, and the experiment was independently repeated three times.
Transwell migration assay
LUAD cells pretreated for 24 h were seeded into the upper chamber of Transwell inserts (8 μm pore size) at a density of 5 × 10⁴ cells in 100 µL serum-free medium. The lower chamber was filled with 600 µL of medium containing 10% FBS or macrophage-CM. After 24 h of incubation, migrated cells on the lower surface of the membrane were fixed with 4% paraformaldehyde and stained with crystal violet. Migrated cells were counted in five randomly selected fields under a microscope. Each group was set up in triplicate, and the experiment was independently repeated three times.
Ethics declarations
All data utilized in this study were obtained from publicly available, anonymized databases, including The Cancer Genome Atlas (TCGA) and the Gene Expression Omnibus (GEO). Therefore, institutional review board approval and informed consent were not required for the analysis of these datasets. The human cell lines (NCI-H1975, A549, THP-1) used for in vitro experiments were obtained from commercial sources (Procell, CN) and did not require separate ethics approval for their use.
Detection of mitophagy-related proteins in macrophages
To evaluate the effect of TUBB6 overexpression on mitophagy in macrophages, total protein was extracted from WT and TUBB6-overexpressing macrophages (WT-Mac and TUBB6-OE-Mac) and subjected to Western blot analysis. Primary antibodies included LC3A/B (CST, #12741, 1:1000), TOM20 (Abcam, ab186735, 1:1000), P62 (Abcam, ab109012, 1:1000), and GAPDH. Band intensities were quantified using ImageJ software, with LC3-II/LC3-I ratios calculated and TOM20 and P62 expression compared between groups.
Data availability
All datasets analyzed during the current study are publicly available. The transcriptomic and clinical data for Lung Adenocarcinoma (LUAD) were obtained from The Cancer Genome Atlas (TCGA) repository (https://portal.gdc.cancer.gov/). The single-cell RNA sequencing dataset and additional LUAD cohorts are available in the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/) under accession numbers GSE131907, GSE13213, and GSE68465. The list of mitophagy-related genes was retrieved from the GeneCards database (https://www.genecards.org/). Drug sensitivity data were derived from the Genomics of Drug Sensitivity in Cancer (GDSC) database (https://www.cancerrxgene.org/). Further details on data access and processing are provided in the Methods section of the manuscript.
References
Siegel, R. L., Giaquinto, A. N. & Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 74 (1), 12–49 (2024).
Fan, Z. A two–sample Mendelian randomization study of lipidome and lung cancer. J. Pharm. Biomed. Anal. 252, 116514 (2025).
Sayeeram, D. et al. Identification of potential biomarkers for lung adenocarcinoma. Heliyon 6 (11), e05452 (2020).
Yang, X. et al. m(6) A-Dependent modulation via IGF2BP3/MCM5/Notch axis promotes partial EMT and LUAD metastasis. Adv. Sci. (Weinh). 10 (20), e2206744 (2023).
Palikaras, K., Lionaki, E. & Tavernarakis, N. Mitophagy: in sickness and in health. Mol. Cell. Oncol. 3 (1), e1056332 (2016).
Perez Ortiz, J. M. & Swerdlow, R. H. Mitochondrial dysfunction in alzheimer’s disease: role in pathogenesis and novel therapeutic opportunities. Br. J. Pharmacol. 176 (18), 3489–3507 (2019).
Li, Y., Zheng, N. & Ding, X. Mitophagy Disequilibrium, a prominent pathological mechanism in metabolic heart diseases. Diabetes Metab. Syndr. Obes. 14, 4631–4640 (2021).
Lu, Y. et al. Cellular mitophagy: Mechanism, roles in diseases and small molecule Pharmacological regulation. Theranostics 13 (2), 736–766 (2023).
Movahedi, K. et al. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res. 70 (14), 5728–5739 (2010).
Esteban-Martínez, L. et al. Programmed mitophagy is essential for the glycolytic switch during cell differentiation. EMBO J. 36 (12), 1688–1706 (2017).
Naik, P. P., Birbrair, A. & Bhutia, S. K. Mitophagy-driven metabolic switch reprograms stem cell fate. Cell. Mol. Life Sci. 76 (1), 27–43 (2019).
Boutilier, A. J. & Elsawa, S. F. Macrophage polarization States in the tumor microenvironment. Int. J. Mol. Sci. 22 (13), 6995 (2021).
Dai, K., Radin, D. P. & Leonardi, D. PINK1 depletion sensitizes non-small cell lung cancer to glycolytic inhibitor 3-bromopyruvate: involvement of ROS and mitophagy. Pharmacol. Rep. 71 (6), 1184–1189 (2019).
Núñez-Vázquez, S. et al. The prohibitin-binding compound fluorizoline inhibits mitophagy in cancer cells. Oncogenesis 10 (9), 64 (2021).
Dai, D., Liu, L., Guo, Y., Shui, Y. & Wei, Q. A comprehensive analysis of the effects of key mitophagy genes on the progression and prognosis of lung adenocarcinoma. Cancers (Basel). 15 (1), 57 (2022).
Mao, X. et al. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: new findings and future perspectives. Mol. Cancer. 20 (1), 131 (2021).
Wang, Y. et al. Spatial transcriptomics delineates molecular features and cellular plasticity in lung adenocarcinoma progression. Cell. Discov. 9 (1), 96 (2023).
Deng, X. et al. Histone lactylation regulates PRKN-Mediated mitophagy to promote M2 macrophage polarization in bladder cancer. Int. Immunopharmacol. 148, 114119 (2025).
Xu, H. et al. The IL-33/ST2 axis affects tumor growth by regulating mitophagy in macrophages and reprogramming their polarization. Cancer Biol. Med. 18 (1), 172–183 (2021).
Liu, Q. et al. Polyamine signal through HCC microenvironment: A key regulator of mitochondrial preservation and turnover in TAMs. Int. J. Mol. Sci. 25 (2), 996 (2024).
Liu, S. et al. Regulator of cell death. Cell. Death Dis. 14 (10), 648 (2023).
Hu, Z. et al. IL6-STAT3-C/EBPβ-IL6 positive feedback loop in tumor-associated macrophages promotes the EMT and metastasis of lung adenocarcinoma. J. Exp. Clin. Cancer Res. 43 (1), 63 (2024).
Fang, Y., Rong QL, Y. K. G. & Liao, W. Systematic investigation of tumor microenvironment and antitumor immunity with IOBR. Med. Res. https://doi.org/10.1002/mdr2.70001 (2025).
Zhang, H. et al. Optimized Dynamic Network Biomarker Deciphers a High-Resolution Heterogeneity Within Thyroid Cancer Molecular Subtypes. Med. Res. https://doi.org/10.1002/mdr2.70004 (2025).
Ye, B. et al. iMLGAM: integrated machine learning and genetic Algorithm-driven multiomics analysis for pan-cancer immunotherapy response prediction. Imeta 4 (2), e70011 (2025).
Xu, Z., Hang, J., Hu, J. & Gao, B. Gefitinib, an EGFR tyrosine kinase inhibitor, activates autophagy through AMPK in human lung cancer cells. J. BUON. 19 (2), 466–473 (2014).
Tailor, D. et al. Novel Aza-podophyllotoxin derivative induces oxidative phosphorylation and cell death via AMPK activation in triple-negative breast cancer. Br. J. Cancer. 124 (3), 604–615 (2021).
Cheng, K. et al. Tumor-associated macrophages in liver cancer: from mechanisms to therapy. Cancer Commun. (Lond). 42 (11), 1112–1140 (2022).
Janke, C. & Magiera, M. M. The tubulin code and its role in controlling microtubule properties and functions. Nat. Rev. Mol. Cell. Biol. 21 (6), 307–326 (2020).
Lopes, D. & Maiato, H. The tubulin code in mitosis and cancer. Cells 9 (11), 2356 (2020).
Leandro-García, L. J. et al. Tumoral and tissue-specific expression of the major human beta-tubulin isotypes. Cytoskeleton (Hoboken). 67 (4), 214–223 (2010).
Kim, B. et al. Quantitative proteomics identifies TUBB6 as a biomarker of muscle-invasion and poor prognosis in bladder cancer. Int. J. Cancer. 152 (2), 320–330 (2023).
Pisanic, T. R. et al. Methylomic analysis of ovarian cancers identifies Tumor-Specific alterations readily detectable in early precursor lesions. Clin. Cancer Res. 24 (24), 6536–6547 (2018).
Lin, X. et al. Prognostic significance of pyroptosis-related factors in lung adenocarcinoma. J. Thorac. Dis. 14 (3), 654–667 (2022).
Wen, S. et al. Identification of macrophage-related genes correlated with prognosis and immunotherapy efficacy in non-small cell lung cancer. Heliyon 10 (6), e27170 (2024).
Luo, Z., Yan, S., Chao, Y. & Shen, M. Unveiling the mitophagy puzzle in non-alcoholic fatty liver disease (NAFLD): six hub genes for early diagnosis and immune modulatory roles. Heliyon 10 (7), e28935 (2024).
Glorieux, C., Zamocky, M., Sandoval, J. M., Verrax, J. & Calderon, P. B. Regulation of catalase expression in healthy and cancerous cells. Free Radic Biol. Med. 87, 84–97 (2015).
Wang, Z. et al. Establishment and validation of a prognostic signature for lung adenocarcinoma based on metabolism-related genes. Cancer Cell. Int. 21 (1), 219 (2021).
Liu, J. S. et al. Association of variations in the CAT and prognosis in lung cancer patients with platinum-based chemotherapy. Front. Pharmacol. 14, 1119837 (2023).
Chen, P. M., Huang, Y. H., Chen, H. H. & Chu, P. Y. Catalase expression is an independent prognostic marker in lung adenocarcinoma. Anticancer Res. 44 (1), 287–300 (2024).
Hasegawa, Y. et al. Decreased expression of catalase mRNA in thyroid anaplastic carcinoma. Jpn J. Clin. Oncol. 33 (1), 6–9 (2003).
Glorieux, C. et al. Catalase overexpression in mammary cancer cells leads to a less aggressive phenotype and an altered response to chemotherapy. Biochem. Pharmacol. 82 (10), 1384–1390 (2011).
Zhang, R. et al. Parkin inhibits proliferation and migration of bladder cancer via ubiquitinating catalase. Commun. Biol. 7 (1), 245 (2024).
Suski, J. M. et al. Relation between mitochondrial membrane potential and ROS formation. Methods Mol. Biol. 810, 183–205 (2012).
Ma, K. et al. Mitophagy, mitochondrial Homeostasis, and cell fate. Front. Cell. Dev. Biol. 8, 467 (2020).
Najafi, A. et al. Catalase application in cancer therapy: simultaneous focusing on hypoxia Attenuation and macrophage reprogramming. Biomed. Pharmacother. 153, 113483 (2022).
Yang, K. et al. Methylation modification is a poor prognostic factor in non-small cell lung cancer and regulates the tumor microenvironment: mRNA molecular structure and function. Int. J. Biol. Macromol. 282 (Pt 4), 137214 (2024).
Ji, T. et al. Single-cell transcriptomics and machine learning unveil ferroptosis features in tumor-associated macrophages: Prognostic model and therapeutic strategies for lung adenocarcinoma. Front. Pharmacol. https://doi.org/10.3389/fphar.2025.1598756 (2025).
Acknowledgements
The authors gratefully acknowledge the TCGA, GEO, and GeneCards database platforms for providing open-access datasets that enabled the present study. We also extend our appreciation to colleagues for their valuable discussions and constructive feedback throughout the research process.
Author information
Authors and Affiliations
Contributions
T.R.Y, Q.L, W L, contributed equally to this work. T.R.Y: Methodology, Data Curation, Validation, Writing—Original Draft. Q.L: Software Development, Data Curation, Visualization, Writing—Original Draft. W L: software analysis, validation, visualization, and contributed to writing—original draft. B.P: Conceptualization, Methodology, Writing—Original Draft. J.F.J: Supervision, Funding Acquisition, Writing—Review & Editing. H.M.T: Project Administration, Data Analysis, Conceptualization, Writing—Review & Editing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yang, T., Liu, Q., Li, W. et al. Macrophage mitophagy-related genes predict prognosis and therapeutic response in lung adenocarcinoma. Sci Rep 15, 41010 (2025). https://doi.org/10.1038/s41598-025-24769-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-24769-z
