
Abstract
Wall-broken Ganoderma lucidum spore powder (BGLSP), a traditional Chinese medicinal substance, contains bioactive triterpenoids with anti-tumor potential. However, its efficacy and mechanisms against papillary thyroid cancer (PTC) remain unclear. This study investigated the anti-tumor effects of BGLSP on PTC using three human PTC cell lines (TPC-1, K1, KTC-1) treated with BGLSP (0.5–4 mg/mL). We assessed cell viability (CCK-8 assay), proliferation (colony formation, Ki-67 immunofluorescence), apoptosis (flow cytometry, caspase activation), and migration (wound healing, transwell assays). Western blot analyzed the key Epithelial-Mesenchymal Transition (EMT) markers, including E-cadherin, N-cadherin, Vimentin, Zonula occludens-1 (ZO-1), Snail and Zinc-finger E-box binding homeobox 1 (ZEB1), while a nude mouse xenograft model assessed BGLSP efficacy (2 mg/kg/day). Results showed that BGLSP significantly inhibited PTC cell proliferation and colony formation while inducing apoptosis via caspase-8/caspase-3 activation. It also suppressed migration by modulating EMT, upregulating epithelial markers (E-cadherin, ZO-1), and downregulating mesenchymal markers (N-cadherin, Vimentin) and EMT-transcription factors (Snail, ZEB1). In vivo, BGLSP reduced tumor growth and Ki-67 expression, consistent with in vitro findings. These findings demonstrate that BGLSP exerts anti-tumor effects in PTC by targeting proliferation, apoptosis, and EMT, supporting its traditional use and suggesting potential as a natural adjuvant therapy worthy of further studies.
Similar content being viewed by others


Introduction
Over the past four decades, the global incidence of thyroid cancer has increased by approximately 313%, largely due to advancements in imaging technologies and improved diagnostic capabilities1. As the ninth most common cancer worldwide, thyroid cancer predominantly affects women and is associated with a relatively high five-year survival rate2. Histologically, thyroid cancer is classified into five subtypes: papillary, follicular, poorly differentiated, anaplastic, and medullary carcinomas. Among these, papillary thyroid carcinoma (PTC) accounts for nearly 80% of all cases3. Current clinical management strategies for thyroid cancer include thyroidectomy, radioactive iodine therapy, and TSH suppression. However, these conventional treatments are often limited by drug resistance and significant adverse effects4, underscoring the urgent need for novel therapeutic approaches for PTC.
Compared to chemical drugs, traditional Chinese medicine (TCM) offers distinct advantages in tumor treatment, including low toxicity, multi-target effects, and reduced likelihood of drug resistance. In TCM theory, the synergistic interactions among herbs—guided by the principles of “the ruler, the minister, and the enabler”—play a critical role in achieving therapeutic efficacy. However, this complex synergy also poses significant challenges in elucidating the mechanistic underpinnings of TCM, which has hindered its acceptance by regulatory bodies such as the U.S. FDA and limited its global recognition. Consequently, the standardization and modernization of TCM, particularly through the identification of therapeutic targets, are essential steps toward its international integration.
Ganoderma lucidum, a natural medicinal fungus, is listed as a first-class precious medicinal material in various collections of traditional Chinese medicine. Ganoderma lucidum spore powder (GLSP) comprises the reproductive spores (5–8 μm in diameter) released upon maturation of the Ganoderma lucidum fungus. These microscopic spores are rich in bioactive constituents including proteins, polysaccharides, triterpenes, nucleosides, and essential trace elements5,6. However, the therapeutic potential of GLSP is limited by its indigestible chitinous spore wall, which impedes bioavailability of these bioactive compounds. Modern processing techniques such as superfine grinding and low-temperature wall disruption have enabled production of wall-broken Ganoderma lucidum spore powder (BGLSP)5. This advanced formulation enhances nutrient absorption by effectively fracturing the rigid spore wall and liberating encapsulated active components. Current evidence indicates that BGLSP exerts immunomodulatory and antitumor effects primarily through its triterpenoid constituents, which mediate these activities via lymphocyte subpopulation regulation7,8,9. Notably, BGLSP has shown clinical potential against various malignancies including ovarian, lung, and rectal cancers7,10,11. However, its therapeutic potential for thyroid malignancies remains unexplored, and the exact molecular mechanisms and cellular targets responsible for its antitumor properties require further elucidation.
Epithelial-Mesenchymal Transition (EMT) is a critical biological process in tumor progression, characterized by the transformation of epithelial cells into a mesenchymal phenotype. This transition involves the loss of epithelial markers such as E-cadherin, disruption of cell polarity, and acquisition of migratory and invasive properties, which collectively promote metastasis and drug resistance. Key hallmarks of EMT include the downregulation of E-cadherin and Zonula occludens-1 (ZO-1), leading to the loss of epithelial morphology, and the upregulation of N-cadherin and Vimentin, which confers a spindle-shaped mesenchymal phenotype conducive to cancer cell dissemination12. The activation of key EMT-transcription factors, such as Snail and Zinc-finger E-box binding homeobox 1 (ZEB1), further drives this phenotypic transition and collectively facilitates cancer cell dissemination. Whether BGLSP exerts anti-tumor effects by modulating the EMT pathway in PTC is the primary focus of this study.
In this study, we investigated the effects of BGLSP on the proliferation, apoptosis, migration, and EMT pathway in three PTC cell lines (TPC-1, K1, and KTC-1) through in vitro experiments. Additionally, we explored the impact of BGLSP on tumor growth in vivo.
Results
Anti-proliferative effects of BGLSP in PTC cells
To evaluate the anti-proliferative effects of BGLSP, we performed comprehensive analyses using CCK-8 viability assays, colony formation tests, and Ki-67 immunofluorescence staining across three PTC cell lines (TPC-1, K1, and KTC-1).
In TPC-1 cells, CCK-8 experiments revealed that BGLSP treatment induced dose-dependent growth inhibition after 24 h (Fig. 1A). Specifically, 1 mg/mL BGLSP significantly reduced colony formation capacity and inhibited clones by approximately 90%, 84%, and 65% at inoculations of 200, 400, and 600, respectively (Fig. 1B, C, P < 0.001 for all groups). Cellular immunofluorescence assays further indicated that 1 mg/mL BGLSP decreased the expression of Ki-67 protein (a proliferation marker) by approximately 28% (Fig. 1D,E, P < 0.01).

Anti-proliferative effects of BGLSP on TPC-1 cells in vitro. (A) Dose-dependent effects of BGLSP (0.5, 1, 2, and 4 mg/mL, 24 h) on TPC-1 cell viability assessed by CCK-8 assay (n = 3). (B, C) Colony formation capacity of TPC-1 cells treated with 1 mg/mL BGLSP at different seeding densities (200, 400, 600 cells/well) (n = 3). (D, E) Immunofluorescence staining of Ki-67 proliferation marker in TPC-1 cells following 1 mg/mL BGLSP treatment (24 h). Scale bar: 50 μm (n = 3). Data represent mean ± SD. Statistical significance was determined by one-way ANOVA (A) and unpaired Student’s t-test (C, E). *P < 0.05, **P < 0.01, ***P < 0.001.
In K1 cells, CCK-8 analysis revealed differential responses to BGLSP treatment after 24-hour exposure, a clear dose-response relationship was evident (Fig. S1A). Low concentrations (0.5–1.5 mg/mL) exhibited modest growth inhibition, while higher concentrations (2–4 mg/mL) produced significant anti-proliferative effects. Furthermore, the inhibition rate of 1 mg/mL BGLSP was approximately 66%, 36%, and 50% against cell clones of different inoculum numbers (Fig. S1B, C, P < 0.001 for all groups). The fluorescence intensity of Ki-67 inhibition by 1 mg/mL BGLSP was approximately 26% (Fig. S1D, E, P < 0.01).
KTC-1 cells displayed pronounced sensitivity to BGLSP, with 24-hour treatment causing dose-dependent viability reduction (Fig. S2A). At 1 mg/mL, BGLSP diminished colony formation by approximately 60% (P < 0.05), 39% (P < 0.01), and 35% (P < 0.001), respectively (Fig. S2B,C). The expression of Ki-67 was reduced by 25% compared with the control group (Fig. S2D,E, P<0.01).
Pro-apoptotic activity of BGLSP in PTC cells
Flow cytometry apoptosis assays were conducted to evaluate the apoptotic effects of BGLSP on PTC cells. In these assays, quadrant Q3 represents early apoptosis, while quadrant Q2 indicates late apoptosis. After 24 h of treatment with BGLSP, a dose-dependent increase in apoptosis was observed in TPC-1, KTC-1, and K1 cells (Fig. 2A, D, and G). Western blot analysis revealed that 1 mg/mL of BGLSP significantly promoted apoptosis in TPC-1, K1, and KTC-1 cells. A significant increase in the activation of cleaved caspase-3 protein, a key marker of apoptosis, was detected. Additionally, BGLSP also activated caspase-8, demonstrating that apoptosis was initiated via the extrinsic apoptotic pathway (Fig. 2B,C,E,F,H,I, P < 0.05). The original blots are presented in Supplementary Fig. 5A.

BGLSP induces apoptosis in PTC cell lines. (A, D, G) Flow cytometric quantification of apoptotic populations in TPC-1, K1 and KTC-1 cells treated with BGLSP (0.5–4.5 mg/mL, 24 h; n = 3). (B, C, E, F, H, I) Western blot analysis of caspase cascade activation (caspase-8 and cleaved caspase-3) following 1 mg/mL BGLSP treatment (n = 3). Data shown as mean ± SD. Statistical significance was determined by unpaired Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001. Original blots are presented in Supplementary Fig. 5A.
Anti-migratory effects through EMT modulation
To investigate the impact of BGLSP on the migration of PTC cells, a comprehensive series of assays, including scratch healing, transwell, cytoskeletal fluorescence, and western blot, were employed. Our migration assays consistently demonstrated that 1 mg/mL BGLSP treatment for 24 h significantly impaired the migratory capacity of all these three PTC cell lines, though with varying degrees of efficacy.
In TPC-1 cells, wound healing assays revealed an 80% reduction in migration distance (Fig. 3A,B, P < 0.001), while Transwell assays showed a 55% decrease in cell penetration (Fig. 3C,D, P < 0.01). Examination of the cytoskeleton by phalloidin staining revealed that this migratory inhibition was accompanied by significant morphological alterations. As shown in Fig. 3E, control cells displayed abundant filopodia and actin-rich protrusions at their periphery, which are characteristic of actively migrating cells. In contrast, BGLSP treatment substantially reduced these structures, resulting in a more stable cellular morphology. This cytoskeletal remodeling provides direct morphological evidence for the suppression of cell migration by BGLSP. Western blot analysis showed that the proteins in the EMT pathway were significantly altered, with E-cadherin and ZO-1 upregulated, while N-cadherin, vimentin and the key EMT-transcription factors Snail and ZEB1 were downregulated (Fig. 3F,G, P < 0.05). The original blots are presented in Supplementary Fig. 5B.

Anti-migratory effects of 1 mg/mL BGLSP on TPC-1 cells for 24 h. (A, B) Wound healing assay. Scale bar: 500 μm (n = 3). (C, D) Transwell invasion assay. Scale bar: 200 μm (n = 3). (E) Cytoskeletal fluorescence assay. Scale bar: 50 μm. (F, G) Western blot analysis of EMT-related markers (n = 3). Data represent mean ± SD. Statistical significance was determined by unpaired Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001. Original blots are presented in Supplementary Fig. 5B.
K1 cells demonstrated moderate sensitivity, with migration inhibition rates of 41% in the wound healing assay (Fig. S3A,B, P < 0.05) and 64% in the cell transwell assay (Fig. S3C,D, P < 0.05). Additionally, cytoskeletal remodeling was observed, characterized by a significant reduction in filopodia (Figure S3E). The relatively weaker response correlated with less pronounced EMT marker changes, with upregulation of E-cadherin (P < 0.001) and ZO-1 (P < 0.05), downregulation of ZEB1 and Snail. The expression levels of N-cadherin and Vimentin showed no statistically significant differences, but an upward trend was observed in the Fig. S3F,G, the original blots are presented in Supplementary Fig. 5C. The disparity in migratory capacity potentially due to the stronger basal adhesion properties of K1 cell line.
KTC-1 cells showed sensitivity comparable to TPC-1, with 89% wound closure inhibition (Figure S4A, B, P < 0.001), 55% transwell invasion reduction (Figure S4C, D, P < 0.01), and marked cytoskeletal alterations (Figure S4E). Western blot confirmed modulation of EMT, including elevated expression of E-cadherin (P < 0.05), ZO-1 (P < 0.001), along with decreased expression of N-cadherin, Vimentin, ZEB1 and Snail (P < 0.05) (Fig. S4F,G). The original blots are presented in Supplementary Fig. 5D.
BGLSP inhibits tumor growth in vivo
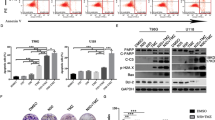
To investigate the antitumor efficacy of BGLSP in vivo, we established a mouse tumor model via subcutaneous inoculation of K1 cells. The results demonstrated that BGLSP treatment significantly reduced tumor volume and weight compared to the control group (Fig. 4A−C, P < 0.05). Ki-67 immunohistochemical analysis revealed a marked decrease in the proliferation marker’s positive staining area in the treatment group (Fig. 4D,E, P < 0.05). HE staining showed that tumor cells in the BGLSP-treated group exhibited smaller nuclei, reduced nucleolar size and number, and lower nucleoplasmic ratios and mitotic indices compared to controls (Fig. 4F). To determine whether BGLSP exerts its antitumor effects by suppressing the EMT pathway, we assessed EMT-related protein expression via western blot. Shown in Fig. 4G and H (original blots are presented in Supplementary Fig. 6), BGLSP significantly upregulated epithelial markers [e.g., E-cadherin and ZO-1 (P < 0.01)] while downregulating mesenchymal markers [(e.g., N-cadherin (P < 0.01) and Vimentin (P < 0.001)] and the key EMT-transcription factors [(e.g., ZEB1 and Snail (P < 0.001)]. Additionally, apoptosis-related proteins in the caspase cascade [e.g., caspase-8 (P<0.001) and cleaved caspase-3 (P<0.01)] were elevated in the treatment group. Collectively, these findings demonstrate that BGLSP effectively inhibits tumor growth in vivo.

In vivo antitumor efficacy of BGLSP. (A) Tumor specimens from control and BGLSP-treated (2 mg/kg/day) groups at day 21. (B) Tumor growth kinetics. (C) Final tumor weights. (D, E) Immunohistochemical staining and expression of Ki-67 proliferation index. Scale bar: 50 μm. (F) H&E-stained tumor histopathology. Scale bar: 50 μm. (G, H) Western blot analysis of EMT and apoptosis-related protein expression in tumor tissues. Data presented as mean ± SD (n = 6 per group). Statistical comparisons were performed by two-way ANOVA (B) and unpaired Student’s t-test (C, E, H). *P < 0.05, **P < 0.01, ***P < 0.001. Original blots are presented in Supplementary Fig. 6.
Discussion
PTC, the most prevalent histological subtype of thyroid cancer, has shown a steadily increasing incidence worldwide. BGLSP, a bioactive compound derived from mechanically disrupted Ganoderma lucidum spores, has demonstrated therapeutic potential in various malignancies. Previous studies have established its role as an adjuvant agent in ovarian cancer therapy, where it effectively mitigates the adverse effects of conventional chemotherapy13. Furthermore, BGLSP exhibits significant antitumor activity against cervical carcinoma, cholangiocarcinoma, and breast cancer cells12,14,15. The present study was designed to systematically evaluate the therapeutic effects of BGLSP on PTC through comprehensive in vitro and in vivo investigations. Our in vitro experiments revealed that BGLSP treatment significantly suppressed cellular proliferation and migration in multiple PTC cell lines (TPC-1, K1, and KTC-1), while simultaneously inducing apoptosis. These antitumor properties were further confirmed in our animal studies, where BGLSP administration markedly inhibited tumor growth.
Uncontrolled proliferation is a fundamental hallmark of cancer. The eukaryotic cell cycle comprises five distinct phases: G0 (quiescence), G1 (gap 1), S (DNA synthesis), G2 (gap 2), and M (mitosis). Normal proliferation involves precisely regulated checkpoints that initiate senescence or apoptosis when needed. However, malignant transformation frequently involves dysregulation of these checkpoint controls, resulting in aberrant proliferative capacity. The nuclear protein Ki-67 has been established as a robust biomarker of cellular proliferation. This antigen exhibits cell cycle-dependent expression patterns, being detectable throughout all active phases (G1, S, G2, M) while absent in quiescent (G0) cells. Its distinctive expression profile makes Ki-67 an invaluable diagnostic tool for differentiating normal proliferative states from neoplastic growth. Clinically, Ki-67 immunohistochemical staining has become a standard procedure for tumor evaluation, with the Ki-67 proliferation index (percentage of positive cells) serving as a prognostic indicator for disease progression16,17. In our investigation, BGLSP demonstrated significant anti-proliferative effects across three papillary thyroid carcinoma cell lines (TPC-1, K1, KTC-1). Treatment with BGLSP not only inhibited cancer cell proliferation but also substantially downregulated Ki-67 expression, further corroborating its growth-inhibitory properties.
Induction of apoptosis in cancer cells represents a pivotal strategy in current anticancer therapeutics. Apoptosis plays crucial roles in physiological processes, such as endometrial shedding and normal cell renewal during the female menstrual cycle. Under pathological conditions, however, apoptosis can be triggered by various intrinsic or extrinsic factors, including neurodegenerative disorders (e.g., Alzheimer’s disease), physical stimuli, or viral infections18,19. Diminished apoptotic capacity constitutes a hallmark feature of cancer cells, and most contemporary anticancer agents exert their therapeutic effects by targeting apoptotic pathways20. The apoptotic machinery primarily consists of three major pathways: the intrinsic mitochondrial pathway, extrinsic death receptor pathway, and endoplasmic reticulum stress pathway. Notably, caspase-dependent pathways serve as central executors of apoptosis, with both intrinsic and extrinsic pathways ultimately converging on caspase-3 to implement the apoptotic program21,22. Cleaved caspase-3, the activated form of caspase-3, serves as a definitive marker of apoptosis execution. In this study, flow cytometric analysis demonstrated that BGLSP induced apoptosis in papillary thyroid carcinoma cells (TPC-1, K1, and KTC-1) in a concentration-dependent manner. Western blot analysis further revealed that BGLSP treatment significantly upregulated the expression of both initiator caspase-8 and effector cleaved caspase-3 in the extrinsic apoptotic pathway.
Cancer cell metastasis represents a critical determinant of disease progression in patients with advanced malignancies. The EMT process plays a pivotal role in this context, wherein epithelial cells, characterized by strong intercellular connections, acquire mesenchymal traits with enhanced motility and reduced adhesion. While EMT participates in physiological processes such as wound healing, its dysregulation is fundamentally implicated in cancer progression. During malignant transformation, tumor cells undergo profound cytoskeletal reorganization that significantly enhances their invasive potential. E-cadherin serves as a canonical marker of epithelial phenotype. The downregulation of E-cadherin expression, frequently observed in tumor cells compared to their normal counterparts, directly contributes to increased cellular motility and subsequent metastatic dissemination. Consequently, E-cadherin expression levels have emerged as important indicators of tumor cell invasiveness23. Furthermore, various transcription factors (including ZEB1 and Snail) regulating epithelial and mesenchymal genes (including N-cadherin, vimentin, and ZO-1) participate in EMT activation across diverse tumor types24. Our investigation demonstrated that BGLSP effectively suppressed metastatic potential in three papillary thyroid carcinoma cell lines (TPC-1, K1, and KTC-1). Western blot analysis revealed that BGLSP treatment modulated the expression profile of EMT-associated transcription factors in these cell lines, promoting a shift toward epithelial phenotype characteristics and consequently attenuating their metastatic capacity.
In contemporary clinical practice, histopathological examination remains the gold standard for tumor staging. The assessment primarily relies on morphological evaluation through H&E staining and quantification of proliferation markers, particularly Ki-67. Our in vivo studies demonstrated the tumor growth inhibitory effects of BGLSP. Immunohistochemical analysis revealed a significant reduction in Ki-67-positive areas in the treatment group compared to controls (p < 0.05), indicating potent antiproliferative activity.
The results demonstrated that BGLSP significantly inhibited the proliferation and migration of PTC cell lines (TPC-1, K1, and KTC-1), although its efficacy varied among different cell lines due to their distinct molecular subtypes and genetic backgrounds despite all being derived from PTC patients. Notably, BGLSP consistently exhibited potent anti-migratory and anti-proliferative effects across all tested cell lines, confirming its broad-spectrum inhibitory activity against PTC progression. These findings suggest BGLSP likely exerts its antitumor effects through multi-target mechanisms and emphasize the need for further investigation into its molecular actions, which could provide a foundation for developing personalized treatment strategies based on tumor molecular profiling and highlight the importance of establishing predictive biomarkers to optimize clinical outcomes. However, several limitations should be acknowledged: (1) the specific bioactive constituents responsible for its antitumor effects remain to be identified; (2) the precise molecular mechanisms underlying its activity require further elucidation; (3) the differential responses among PTC subtypes warrant additional investigation.
In summary, our study demonstrates that BGLSP exhibits significant inhibitory effects on PTC progression. As a bioactive extract derived from Ganoderma lucidum, BGLSP contains multiple pharmacologically active components including polysaccharides, triterpenoids, alkaloids, polypeptides, and essential trace elements, all of which have been documented to possess antitumor properties in numerous preclinical studies. Compared to conventional chemotherapeutic agents, this traditional Chinese medicine preparation offers distinct advantages including cost-effectiveness and favorable safety profile. This investigation represents the first systematic evaluation of BGLSP’s anticancer efficacy against PTC, thereby expanding its potential therapeutic applications in oncology. From a translational perspective, BGLSP shows promise as either a standalone therapeutic option or an adjuvant to enhance conventional anticancer regimens, particularly in the context of personalized medicine approaches. Furthermore, purification, structural modification, or synthetic derivation of its active components may significantly potentiate its anticancer efficacy, representing an important direction for future pharmaceutical development.
Methods
Reagents and cell lines
The human PTC cell lines TPC-1, K1 and KTC-1 were obtained from ATCC. Wall-broken Ganoderma lucidum spore powder was provided by Zhejiang Shouxiangu Pharmaceutical Co. (China). Primary antibodies against Ki-67 (Abcam, UK), E-cadherin, ZO-1, N-cadherin, Vimentin, ZEB1, Snail, Caspase-8, Cleaved Caspase-3 and Caspase-3 (WanClass Bio-technology, China) and GAPDH (Proteintech, China) were commercially acquired.
Preparation of BGLSP solution and cell culture
BGLSP stock solution was prepared by dissolving BGLSP in complete cell culture medium, sonication at 80 kHz for 1 h, and filtration through a 0.22 μm filter. TPC-1 and KTC-1 cells were maintained in high-glucose DMEM (Gibco, New Zealand), while K1 cells were cultured in F12K medium, supplemented with 10% FBS (Gibco, New Zealand) and 1% penicillin-streptomycin at 37 °C in a 5% CO₂ humidified incubator. In the CCK8 and flow cytometry experiments, cellular administration groups were subjected to concentration gradients of 0.5, 1, 2, and 4 mg/mL, respectively. Following a comprehensive evaluation of the CCK8 experimental results across three distinct cell types, the minimal effective concentration of the drug was established at 1 mg/mL. This concentration was subsequently adopted for the administration in all other experiments.
Cell viability assay (CCK-8)
Cells (2 × 10³/well) were seeded in 96-well plates (Corning, USA). After 24 h attachment, cells were treated with various concentrations of BGLSP (0, 0.5, 1, 2, and 4 mg/mL) for 24 h. Cell Counting Kit-8 (Beyotime, China) was added (10 µL/well), incubated for 2 h, and absorbance measured at 450 nm.
Colony formation assay
Cells in logarithmic growth phase were harvested and inoculated into 6-well plates at densities of 200, 400, and 600 cells per well. Following cell attachment, the experimental group was treated with complete medium containing 1 mg/mL BGLSP. Colonies were allowed to form for 10–14 days until individual clones contained > 50 cells. Cells were then fixed with 4% paraformaldehyde (room temperature, 15 min), stained with 0.1% crystal violet solution (Beyotime, Shanghai, China) for 20 min, and gently washed with distilled water. After air-drying, colony images were captured and quantified using Image J software (version: ImageJ 1.52a, URL: https://imagej.net/ij/download.html) with the following criteria : only colonies containing ≥ 50 cells were counted as positive clones.
Immunofluorescence staining
Cells were seeded on sterile glass coverslips and allowed to adhere overnight before treatment with 1 mg/mL BGLSP for 24 h. Following treatment, cells were fixed with 4% paraformaldehyde (15 min, RT), permeabilized with 0.5% Triton X-100 (10 min, RT). After blocking with 1% BSA (1 h, RT), samples were incubated with anti-Ki-67 primary antibody (1: 500 dilution in 1% BSA; 37 °C, 2 h). Cells were then incubated with Alexa Fluor 488-conjugated goat anti-rabbit IgG secondary antibody (1: 1000; Abcam, UK; 1 h, RT, light-protected). Nuclei were counterstained with DAPI-containing antifade mounting medium (Beyotime, China). Images were acquired at 400× magnification using a Leica DMi8 inverted fluorescence microscope equipped with the appropriate filter set and analyzed for cellular fluorescence intensity using Image J software.
Flow cytometry assay
Cells were treated with varying concentrations of BGLSP (0, 0.5, 1, 2, and 4 mg/mL) for 24 h. Following treatment, both floating cells in the supernatant and adherent cells (detached using EDTA-free trypsin) were collected by centrifugation (1000 × g, 3 min). After three washes with cold PBS, cell pellets were resuspended in 1× binding buffer. Apoptosis was assessed within 1 h according to the procedure of Annexin FITC/PI Apoptosis Detection Kit (Yeasen, Shanghai, China). Apoptosis assay was performed using a flow cytometric analyzer (BD, FACS Calibur, USA) and apoptosis rates were analyzed with FlowJo Software v10.
Wound-healing assay
Cells were cultured to 80% confluence in complete growth medium. A wound was created vertically with a sterile 1 mL pipette tip, followed by several washes with PBS to remove detached cells. The experimental group was treated with 1 mg/mL BGLSP. Images were captured at identical positions for 0 h and 24 h time points, and migration distances were quantified using Image J software. The migration area was calculated as: [(0 h wound area − 24 h wound area) µm2]
Transwell invasion assay
Cells in logarithmic growth phase were harvested and resuspended in serum-free medium at a density of 5 × 10^4 cells/mL. The upper chambers of 8 μm pore-size Transwell inserts (Corning, USA) were pre-coated with 100 µL of Matrigel matrix (1: 8 dilutions in serum-free medium) and allowed to polymerize at 37 °C for 4 h. After hydration with serum-free medium (30 min, 37 °C), 200 µL of cell suspension was added to the upper chamber, while the lower chamber contained 750 µL of complete medium with 10% FBS as chemoattractant, with or without 1 mg/mL BGLSP for experimental and control groups, respectively. After 24 h incubation at 37 °C with 5% CO2, non- invading cells on the upper membrane surface were removed with a cotton swab, while invaded cells on the lower surface were fixed with 4% paraformaldehyde (15 min, RT), stained with 0.1% crystal violet (20 min, RT), and washed three times with PBS. Five representative fields (center and four quadrants) were imaged at 100× magnification using an inverted microscope. Invaded cells were quantified using Image J software. by threshold-based particle analysis, with results expressed as mean number of invaded cells per field ± standard deviation (SD) from three independent experiments.
Cytoskeletal fluorescence staining
Cells were seeded on sterile glass coverslips at an appropriate density and allowed to adhere overnight. Experimental group was treated with 1 mg/mL BGLSP for 24 h, fixed with 4% paraformaldehyde (15 min, room temperature) and permeabilized with 0.5% Triton X-100 in PBS (10 min, room temperature). After three washes with PBS (5 min each), actin filaments were stained with rhodamine-conjugated phalloidin (TRITC-Phalloidin, 1: 200 dilutions; Yeasen, Shanghai, China) for 1 h at room temperature (light-protected), nuclei were counterstained with DAPI (5 µg/mL; Beyotime, Shanghai, China) for 10 min. Coverslips were mounted using anti-fade mounting medium and imaged by fluorescence microscope equipped with appropriate filter set at a magnification of 400x (excitation/emission: 550/570 nm for TRITC, 358/461 nm for DAPI).
Western blotting analysis
Protein samples were separated by SDS-PAGE (10–12% gels) and electrophoretically transferred onto polyvinylidene difluoride (PVDF) membranes (0.22 μm pore size; Millipore, USA). Membranes were blocked with 5% non-fat milk in TBST (Tris-buffered saline with 0.1% Tween-20) for 2 h at room temperature with gentle agitation, then washed three times with TBST (10 min each). PVDF membranes were incubated with primary antibody overnight at 4 °C. After three additional TBST washes (10 min each), membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (1: 5000 dilution) for 1 h at room temperature. Protein bands were detected using Enhanced Chemiluminescence reagent (ECL Kit; Advansta, USA) according to the manufacturer’s protocol, and images were captured using a ChemiDoc Imaging System (Bio-Rad, USA). Band intensities were quantified using Image J software with normalization to appropriate loading controls.
Tumor xenograft assay
Male BALB/c nude mice (4–6 weeks old) were obtained from Charles River Laboratories (Beijing, China) and housed under standard specific pathogen-free (SPF) conditions with a 12-hour light/dark cycle at controlled temperature (22 ± 1 °C) and humidity (50 ± 5%), with free access to autoclaved food and water. The mice were subcutaneously injected with K1 cells (1 × 10⁷ cells in 100 µL PBS) in the right flank. When the tumor volume reached about 150 mm3 (volume = \(\:\frac{\text{a}\times\:\text{b}^2}{2}\), where a represents the long axis and b represents the short axis), mice were randomized into control (saline) and treatment groups (n = 6/group). The treatment group received BGLSP at a dose of 2 mg/kg/day (dissolved in saline, with a gavage volume of 10 mL/kg body weight), administered daily by oral gavage for 21 days. The control group received an equivalent volume of saline. Tumor dimensions were measured every 3 days using calipers throughout the study. After gavage administration, the mice were anesthetized by isoflurane and executed by decapitation, and the tumors were stripped and weighed and subjected to the next step of analysis. Animal experiments in this study have been approved by the Ethics Committee of the Sir Run Run Shaw Hospital (No. SRRSH202107216). All methods were carried out in accordance with relevant guidelines and regulations. This study was conducted as recommended by the ARRIVE guidelines.
Histopathological and immunohistochemical analysis
Tumor tissues were fixed in 4% paraformaldehyde for 24 h, alcohol dehydration and paraffin embedding, and sectioned at 5 μm thickness. For hematoxylin and eosin (H&E) staining, sections were deparaffinized and prepared for staining, the nuclei were stained with hematoxylin and the cytoplasm with eosin. and mounted with neutral resin.
For Ki-67 immunohistochemistry, antigen retrieval was performed using citrate buffer (pH 6.0) followed by blocking with 3% BSA. Sections were incubated with anti-Ki-67 primary antibody (1: 500 dilutions in 3% BSA) overnight at 4 °C. Detection was performed using the Mouse/Rabbit Enhanced Polymer Detection System (ORIGENE) according to the manufacturer’s protocol, with DAB as the chromogen. Sections were re-stained with hematoxylin for nuclei and mounted with neutral resin.
All slides were imaged by inverted microscope equipped with a high-resolution digital camera. Magnification was 400x and Ki-67 positivity was quantified using Image J software.
Statistical analysis
All quantitative data are presented as mean ± SD. Statistical analyses were performed using SPSS software (version 19.0; IBM, USA). For comparisons between two groups, Student’s t-test was employed, while one-way analysis of variance (ANOVA) with appropriate post-hoc tests was used for comparisons among three or more groups. All statistical tests were two-tailed, and a p-value < 0.05 was considered statistically significant.
Data availability
All data generated in this study that support our findings are included in this paper and its supplemental data file.
References
Lim, H., Devesa, S. S., Sosa, J. A., Check, D. & Kitahara, C. M. Trends in thyroid cancer incidence and mortality in the united States, 1974–2013. Jama 317, 1338–1348. https://doi.org/10.1001/jama.2017.2719 (2017).
Miller, K. D. et al. Cancer statistics for adolescents and young adults, 2020. CA: a cancer j. clin. 70, 443–459. https://doi.org/10.3322/caac.21637 (2020).
Chmielik, E. et al. Heterogeneity of thyroid cancer. Pathobiol. J. ImmunoPathol Mol. Cell. Biol. 85, 117–129. https://doi.org/10.1159/000486422 (2018).
Nabhan, F., Dedhia, P. H. & Ringel, M. D. Thyroid cancer, recent advances in diagnosis and therapy. Int. J. Cancer. 149, 984–992. https://doi.org/10.1002/ijc.33690 (2021).
Liu, M. T., Chen, L. X., Zhao, J. & Li, S. P. Ganoderma spore powder contains little triterpenoids. Chin. Med. 15, 111. https://doi.org/10.1186/s13020-020-00391-1 (2020).
Song, M. et al. Ganoderma lucidum Spore Polysaccharide Inhibits the Growth of Hepatocellular Carcinoma Cells by Altering Macrophage Polarity and Induction of Apoptosis. J. Immunol. Res. 2021, 6696606. https://doi.org/10.1155/2021/6696606 (2021).
Jin, X., Ruiz Beguerie, J., Sze, D. M. & Chan, G. C. Ganoderma lucidum (Reishi mushroom) for cancer treatment. Cochrane Database Syst. Rev. https://doi.org/10.1002/14651858.CD007731.pub3 (2016).
Liu, X., Yuan, J. P., Chung, C. K. & Chen, X. J. Antitumor activity of the sporoderm-broken germinating spores of ganoderma lucidum. Cancer Lett. 182, 155–161. https://doi.org/10.1016/s0304-3835(02)00080-0 (2002).
Yue, G. G., Fung, K. P., Leung, P. C. & Lau, C. B. Comparative studies on the Immunomodulatory and antitumor activities of the different parts of fruiting body of ganoderma lucidum and ganoderma spores. Phytother. Res. 22, 1282–1291. https://doi.org/10.1002/ptr.2478 (2008).
Zhao, S. et al. Ganoderma lucidum exerts anti-tumor effects on ovarian cancer cells and enhances their sensitivity to cisplatin. Int. J. Oncol. 38, 1319–1327. https://doi.org/10.3892/ijo.2011.965 (2011).
Liu, X. et al. Ganoderma lucidum fruiting body extracts inhibit colorectal cancer by inducing apoptosis, autophagy, and G0/G1 phase cell cycle arrest in vitro and in vivo. Am. J. Translational Res. 12, 2675–2684 (2020).
Zhu, H. S., Yang, X. L., Wang, L. B., Zhao, D. X. & Chen, L. Effects of extracts from sporoderm-broken spores of ganoderma lucidum on HeLa cells. Cell Biol. Toxicol. 16, 201–206. https://doi.org/10.1023/a:1007663006548 (2000).
Cen, K. et al. Sporoderm-Broken spores of ganoderma lucidum sensitizes ovarian cancer to cisplatin by ROS/ERK signaling and attenuates Chemotherapy-Related toxicity. Front. Pharmacol. 13, 826716. https://doi.org/10.3389/fphar.2022.826716 (2022).
Li, L., Guo, H. J., Zhu, L. Y., Zheng, L. & Liu, X. A supercritical-CO2 extract of ganoderma lucidum spores inhibits cholangiocarcinoma cell migration by reversing the epithelial-mesenchymal transition. Phytomedicine: Int. J. Phytotherapy Phytopharmacology. 23, 491–497. https://doi.org/10.1016/j.phymed.2016.02.019 (2016).
Jiao, C. et al. Ganoderma lucidum spore oil induces apoptosis of breast cancer cells in vitro and in vivo by activating caspase-3 and caspase-9. J. Ethnopharmacol. 247, 112256. https://doi.org/10.1016/j.jep.2019.112256 (2020).
Scholzen, T. & Gerdes, J. The Ki-67 protein: from the known and the unknown. J. Cell. Physiol. 182, 311–322. https://doi.org/10.1002/(sici)1097-4652(200003)182 (2000).
Gerdes, J., Schwab, U., Lemke, H. & Stein, H. Production of a mouse monoclonal antibody reactive with a human nuclear antigen associated with cell proliferation. Int. J. Cancer. 31, 13–20. https://doi.org/10.1002/ijc.2910310104 (1983).
Zhang, Y. & Weinberg, R. A. Epithelial-to-mesenchymal transition in cancer: complexity and opportunities. Front. Med. 12, 361–373. https://doi.org/10.1007/s11684-018-0656-6 (2018).
Lu, W. & Kang, Y. Epithelial-Mesenchymal plasticity in cancer progression and metastasis. Dev. Cell. 49, 361–374. https://doi.org/10.1016/j.devcel.2019.04.010 (2019).
Pfeffer, C. M., Singh, A. T. K. & Apoptosis,. A Target for Anticancer Therapy. Int. J. Mol. Sci. https://doi.org/10.3390/ijms19020448 (2018).
Mustafa, M. et al. Apoptosis: A Comprehensive Overview of Signaling Pathways, Morphological Changes, and Physiological Significance and Therapeutic Implications. Cells https://doi.org/10.3390/cells13221838 (2024).
Carneiro, B. A. & El-Deiry, W. S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 17, 395–417. https://doi.org/10.1038/s41571-020-0341-y (2020).
Wong, R. S. Apoptosis in cancer: from pathogenesis to treatment. J. Experimental Clin. Cancer Research: CR. 30, 87. https://doi.org/10.1186/1756-9966-30-87 (2011).
Ghobrial, I. M., Witzig, T. E. & Adjei, A. A. Targeting apoptosis pathways in cancer therapy. Cancer J. Clin. 55, 178–194. https://doi.org/10.3322/canjclin.55.3.178 (2005).
Funding
This study was supported by the Co-construction science and technology program of Zhejiang Traditional Chinese Medicine Administration (GZY-ZJ-KJ-24032) and the Science and Technology Program of Zhejiang Province (2025C02133).
Author information
Authors and Affiliations
Contributions
L.S. and Y.W. designed the study and wrote the manuscript; L.S., J.C., L.F., X.L. and J.Z. performed the experiments; L.X., J.Y. and Y.W. analyzed the data; X.Y. and H.Y. acquisited Fundings and revised the manuscript. All authors read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Shi, LH., Xie, L., Yu, JG. et al. Study on the anti-tumor effect of Wall-broken ganoderma lucidum spore powder on papillary thyroid cancer. Sci Rep 15, 43798 (2025). https://doi.org/10.1038/s41598-025-27791-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-27791-3
