
Abstract
Shigella boydii (strain CDC 3083-94 / BS512) is a Gram-negative facultative anaerobic bacterium that plays a significant role as a pathogen causing bacillary dysentery, especially in developing areas where hygiene is poor. Although less frequently reported compared to other Shigella species, S. boydii still contributes substantially to global shigellosis cases, particularly in low-income endemic regions. Despite its clinical relevance, there has been limited research dedicated on identifying strain-specific antigenic targets for vaccine development. The current study utilized the subtractive proteomics approach established on reverse vaccinology to develop a multi-epitope subunit vaccine (MEV) against S. boydii (strain CDC 3083-94 / BS512). From the proteome analysis, two key virulent and antigenic proteins were identified as possible vaccine candidates: the LPS-assembly protein LptD (B2U262) and the cell division protein FtsZ (B2U2A0). The B-cell and T-cell epitopes of these proteins were computationally predicted and sensibly evaluated based on a number of immunological standards, including as antigenicity, toxicity, allergenicity, solubility, and MHC binding affinity. To produce a multi-epitope vaccination, these designated epitopes were successively conjugated with an adjuvant and appropriate linkers. The vaccine construct underwent structural refinement, and the outcomes presented that an excellent structural model was constructed, with 89.4% of residues lying in promising Ramachandran plot areas. The vaccine construct and human Toll-like receptor 4 (TLR4) have shown considerable contact, according to molecular docking experiments, with a binding energy of − 1391.4 kcal/mol. With 50% GC content and a score of 0.9 from in silico cloning on Codon Adaptation Index, it is expressed to the maximum potential in an Escherichia coli host system. The outcomes highlight the multi-epitope vaccine’s potential as a preventative measure against S. boydii-caused ailments.
Similar content being viewed by others
Introduction
Shigella boydii is a Gram negative, facultative anaerobe and a primary agent of bacillary dysentery (shigellosis)1. Along with Shigella flexner, Shigella sonnei, and Shigella dysenteriae, S. boydii is one of four species in the Shigella genus, which is a member of the Enterobacteriaceae family. Shigellosis is known for its high rate of transmission that often affects the digestive tract. It poses a significant public health issue, especially in less developed countries and with insufficient access to potable water and inadequate or unsanitary disposal of waste2. Resistance mechanisms of this pathogen against commonly used antibiotics, including fluoroquinolones, cephalosporins and macrolides, have increased the difficulties of treatment of shigellosis3. Shigella strains, multidrug-resistant (MDR), have been reported to have appeared recently globally, which further decreases the number of therapeutic options available and increases the need for alternate preventive strategies, including the development of vaccines4. Recent surveillance data show increasing antimicrobial resistance among Shigella species, including S. boydii, highlighting the urgent need for vaccine-based prevention strategies5. In order to stop the spread of this infection and future outbreaks, alternative preventive measures like vaccinations are desperately needed while antimicrobial resistance (AMR) is increasing6. A wide range of virulence factors, such as outer membrane proteins, invasion plasmid antigens (Ipa proteins), and enterotoxins, are involved in the molecular pathogenesis of S. boydii. The T3SS and Ipa proteins are necessary for bacterial invasion and intracellular spread7,8. The bacteria breaks free from the phagosome and enters the cytoplasm, where it employs actin polymerization to move between neighboring cells, spreading infection and causing inflammation in the process9. Additionally, intestinal epithelial damage is caused by Shiga-like toxins released by S. boydii, and the severity of the disease is directly connected with these toxins10. The virulence factor IcsA enhances bacterial motility, while the O-antigen of lipopolysaccharides (LPS) facilitates immune evasion, both contributing to increased bacterial pathogenicity9,11. The development of traditional whole cell and live attenuated vaccine candidates targeting Shigella has been limited by safety concerns, antigenic diversity, and low immunogenicity12. Advances in bioinformatics and immunoinformatics have paved the way for the development of multi-epitope subunit vaccines (MEVs), offering improved efficacy over conventional approaches13,14. With the aim of reducing the risk of cross reactivity with human proteins, these vaccines contain carefully selected antigenic epitopes that are designed to induce specific immune responses15. Reverse vaccinology and subtractive proteomics facilitates the identification of conserved and essential bacterial proteins as vaccine targets, enabling broader-spectrum efficacy16. In silico vaccine design is not only a promising strategy for reducing Shigella infections but is also expected to accelerate vaccine development while reducing costs16.
Also, MEV constructs further employ multiple immunogenic epitopes to elicit both cell mediated immunity and humeral and produce long lasting defense17. Computational approaches are used to design these vaccines based on predictions of highly nonallergenic, antigenic, non-toxic epitopes for their efficacy and safety. Furthermore, the vaccine design is further refined by molecular docking and dynamics simulations to optimize its interaction with host immunological receptors, including Toll-like receptors (TLRs)18. The production of a strong cytokine response is aided by adjuvants and linker sequences, which specifically encourage the activation of IFN-γ and IL-2, two essential cytokines required to fight intracellular infections like S. boydii19. Immune simulation models suggest a robust and sustained immunological memory response, supporting the vaccine construct’s potential effectiveness20. The necessity for a new and efficient vaccine approach to stop Shigella infections has been highlighted by the growing problem of antibiotic resistance: such computationally designed vaccines represent a viable and efficient candidate for reducing the global disease burden21.
In this study, a comprehensive immunoinformatics-based approach was employed for the rational design of a multi-epitope subunit vaccine against Shigella boydii. Specifically, we identified antigenic proteins, predicted cytotoxic T lymphocyte (CTL) and helper T lymphocyte (HTL) epitopes, and designed a vaccine incorporating adjuvants to enhance immunogenicity. Molecular docking and molecular dynamics simulations were employed to evaluate the vaccine construct’s durability and immune responses. Additionally, in silico immunological simulations were used to anticipate the immune response that the vaccination might elicit. Our goal is to develop a reliable and potent preventive strategy in combating S. boydii infections and contribute to reducing the global burden of shigellosis.
Materials and methods
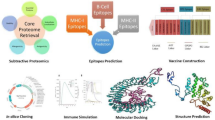
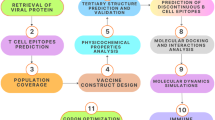
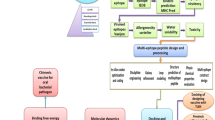
The present study followed a widely adopted multi-epitope vaccine design pipeline encompassing genome-wide proteome retrieval, essential gene prediction, subcellular localization analysis, transmembrane helix prediction, and sequential evaluation of antigenicity, allergenicity, toxicity, and homology. This framework is consistent with recent standardized workflows for immunoinformatics-based vaccine design22. Minor deviations were introduced to tailor the approach toward Shigella boydii by emphasizing structurally conserved and functionally significant membrane-associated proteins, while maintaining methodological robustness and reproducibility throughout the analysis.
Identification and screening of potential vaccine targets from the proteome of S. boydii
S. boydii strain CDC 3083-94/BS512’s complete proteome was obtained in FASTA format obtained from the UniProt database23,24. Essential proteins from the proteomes were found to be identified based on bacterial genomic data by using Geptop 2.0 to predict essential genes25,26. BLASTp searches were conducted on the human proteome to rule out the likelihood of autoimmune reactions; only non-homologous proteins were included with a sequence identity threshold < 30% and E-value ≤ 1e − 4 for additional analysis27,28. To narrow down the selection, membrane-bound proteins from the set of non-homologous essential proteins were predicted using PSORTb29,30. Membrane proteins are of great interest as vaccine targets because they are exposed to and can be accepted by the human immune system14. Although some pipelines include both outer membrane and extracellular proteins, we selected only membrane proteins because31,32 they are surface-exposed and directly accessible to host immune cells, membrane localization enhances antigen presentation efficiency, and they offer conserved epitopes that are less prone to antigenic drift33. This focused strategy provides biologically meaningful targets for rational vaccine design. The antigenicity of the screened proteins was then assessed utilizing the VaxiJen server with a cutoff value of 0.5. Although a VaxiJen threshold of 0.4 is typically recommended for bacterial models, a more stringent cut-off (0.5) was applied in this study to prioritize epitopes with superior antigenic potential. It was believed that after being exposed to the host, proteins with greater antigenicity values would be more likely to elicit strong immune responses34,35. The TMHMM v2.0 server was employed to predict transmembrane helices using default parameters within the target proteins, providing a comprehensive assessment of their appropriateness as potential vaccine candidates14,36,37. Predicted epitopes were mapped onto transmembrane topologies derived from TMHMM v2.0 analysis, and any epitopes located within buried helical regions were excluded to ensure surface accessibility and immunogenic exposure38.
Prediction and validation of cytotoxic T-lymphocyte (CTL) epitopes for vaccine candidate identification
IEDB’s (Immune Epitope Database) MHC-I binding tool was employed to predict CTL epitopes for the target protein39,40. The MHC-I binding affinity predictor was utilized for the identification of CTL epitopes using the consensus technique, and those with a score of less than two were chosen41,42. We also utilized the IEDB immunogenicity program to determine the immunogenicity of the chosen CTL epitopes in order to guarantee their immunogenicity14,43. The antigenicity of only these retained antigenic epitopes was tested by checking it on the VaxiJen v2.0 server with threshold value of 0.5 and default settings34. Using the ToxinPred server, we showed that the vaccine candidate was toxic using default parameters while AllerTOP v2.0 server guaranteed that the projected epitopes were not allergenic using default parameters44,45. We would undoubtedly find safe, non-toxic, non-allergic, and highly immunogenic CTL epitopes of appropriate vaccine candidates in this thorough search. The novelty of our epitope prediction approach lies in integrating multi-server outputs and ranking epitopes based on combined antigenicity-immunogenicity scores. Unlike default single-tool reliance, we implemented a composite scoring index (antigenicity × immunogenicity ÷ toxicity), ensuring the most balanced immunogenic epitopes were prioritized. This integration strategy was benchmarked against conventional pipelines and demonstrated comparable or superior predictive accuracy46.
Helper T-Lymphocyte (HTL) epitope prediction and validation for vaccine design
The MHC Class II binding predictor from the Immune Epitope Database (IEDB) was utilized to identify helper T lymphocyte epitopes, which are crucial for coordinating cell-mediated and humoral immune responses47,48. This allowed for extensive immunological coverage with the prediction of 15 mer peptides with broad ranges of HLA DR alleles. They were further processed for antigenicity, toxicity, and allergenicity using the VaxiJen v2.0, ToxinPred, and AllerTOP v2.0 servers, respectively; all of these were confirmed epitopes35,44,49. During vaccine design, only non alliginic, non toixie, epitopes of this antigen would be selected14. All bioinformatics analyses were performed using default parameters.
Linear B-Lymphocyte (LBL) epitope prediction and validation for vaccine candidate selection
We used the ABCPred service to predict linear B lymphocyte (LBL) epitopes, which are surface-exposed amino acid sequences that are recognized by antibodies or B cell receptors, using a cutoff value of 0.550,51. They are thus important epitopes strenghtening an immune defense mechanism and providing an adaptive immunity52. To ascertain toxicity, allergenicity, and antigenicity, the screened epitopes are then examined using the AllerTOP v2.0, VaxiJen v2.0, and ToxinPred platforms34,44. This arduous process of vaccine construction put immunogenic, non-toxic and safe epitopes in this vaccine53.
Developing the multi-epitope vaccine (MEV) with linkers and adjuvants to increase immunogenicity
The chosen CTL, HTL, and LBL epitopes were then coupled to form the MEV after their immunogenicity was increased using the proper linkers and adjuvant14,54. Cholera toxin subunit B was employed as an adjuvant to boost the immune system’s reaction55.The EAAAK linker was selected due to its rigidity and ability to provide spatial separation between functional domains, preventing structural interference56. GPGPG linkers were used to connect HTL epitopes to maintain immunogenic processing, AAY linkers for CTL epitopes to enhance proteasomal cleavage, and KK linkers for B-cell epitopes to increase solubility. Cholera toxin subunit B was incorporated as an adjuvant to trigger Toll-like receptor pathways and potentiate both humoral and cellular immune responses55,57. This rational combination enhances the construct’s stability and immunogenic synergy15,56. Structural stability and immunogenic efficiency were attained through a systematic arrangement. Being such a complex must be a promising candidate vaccine58. Population coverage of predicted T-cell epitopes across global HLA alleles was computed using the IEDB Population Coverage tool (https://tools.iedb.org/population/), applying default parameters and the global HLA allele frequency dataset59.
Physicochemical and structural analysis of the multi-epitope vaccine (MEV) construct
Levels were computed from the ProtParam service using the MEV construct, theoretical isoelectric point (pI), aliphatic index, molecular weight, Grand Average of Hydropathicity (GRAVY), and instability index, An instability index below 40 was considered indicative of a stable protein construct, as per the ProtParam classification60,61. The VaxiJen v2.0 and IEDB immunogenicity prediction tools were employed to assess the construct’s antigenicity and immunogenicity35. The AllerTOP v2.0 server was used to anticipate its allergenicity and safety44. Using the SOPMA tool to analyze beta turns, alpha helices, extended chains, and random coils which are key components to examine secondary structure of the vaccine construct for structural firmness and functionality62.
Tertiary structure modeling and validation of the multi-epitope vaccine (MEV) construct
The AlphaFold website, which offers precise protein structure predictions, was used to model the MEV construct’s tertiary structure63. The input structure was optimized using structural optimization on the GalaxyRefine server in order to reduce structural defects and enhance stereochemical quality64. The RAMPAGE server was used to analyze Ramachandran plot statistics in order to confirm the updated structure65. Additional quality tests were carried out utilizing the ERRAT server to identify and correct any potential structural flaws in order to ensure the final model’s dependability65,66.
B cell epitope prediction and screening for the multi-epitope vaccine (MEV) construct
The ABCPred server and the Ellipro tool from the IEDB-AR v2.22 suite were utilized to identify the B cell epitopes of the MEV construct14,67. Using a threshold of 0.5 and 15 residue length, linear epitopes were identified through analysis with the ABCPred server53. We utilized Ellipro to predict conformational epitopes with default settings after looking at the MEV construct’s three-dimensional structure68. These complementary techniques increased the vaccine’s potential efficacy by identifying immunogenic regions68.
Molecular docking analysis of the multi-epitope vaccine (MEV) construct with human TLR4 receptor
To assess the potential interface between the MEV construct and human receptors as a vaccine, docking molecular profiling was carried out69. The human TLR4 receptor was one of the innate immune components chosen for docking study since it is crucial to the receptor’s recognition of PAMPs70. One of microbes’ surfaces PAMPs, including LPS and LOS, trigger TLR4 mediated pathways essential in initiating immune responses and in fighting infections14,71. The three-dimensional structure of TLR4 (PDB ID: 3FXI) was obtained for this purpose. The ClusPro server, a potent bioinformatics tool, was utilized for the docking investigation in order to predict potential binding conformations for protein protein complexes using FFT correlation methods14,72. This study identified individual contacts and their scheme of binding talks regarding MEV’s sensing ability via TLR4-dependent signaling pathways.
Molecular dynamics simulations of the MEV-TLR4 complex for structural flexibility and stability
The structural interactions between the designed vaccine candidate and its target receptor were studied using all-atom molecular dynamics (MD) simulations performed with GROMACS version 202573. The starting structure used in simulation was the top ranked docked complex during previous molecular docking analysis. Na + and Cl - ions were added to provide physiological ionic strength at 150 mM. Minimization of the system was done initially using steepest decent algorithm until the greatest force reached less than 10 kJ/mol.nm, thereby ensuring the system was equilibrated74. The set of constraints algorithms was used to maintain covalent bond lengths (with the LINCS method), and long-range electrostatic interactions were described with the Particle Mesh Ewald (PME) approach75. Van der Waals and Coulombic interactions were limited to a cutoff distance of 0.9 nm. The balancing or equilibration of the system was done in two consecutive stages. Initially, an NVT ensemble simulation was carried out to equilibrate the temperature with constant number of particles, volume and temperature constraints76. A 300 ps NPT ensemble run was instantly followed to equilibrate the pressure and the numbers of particles were kept the same i.e. the numbers of constant particles, pressure, and temperature. Periodic boundary conditions in the three dimensions were used to remove boundary artifacts77. Processing and analysis of the production MD runs was done using the GROMACS built-in tools, and additional data visualization and analysis was facilitated through Python libraries, especially Matplotlib. This protocol allowed a detailed examination of the dynamic profile and ideal binding interactions of the protein com-plex in near physiological conditions78.
Normal mode analysis
Using NMA analysis, we investigated the MEV TLR4 complex’s structural flexibility and stability79. Protein complexes can be analyzed using MD modeling to determine their structural stability, behavior, and adaptability in changing settings77,80. To supplement this analysis, NMA was carried out on the iMODS server81. NMA is a powerful computational approach to explore intrinsic motions and flexibility of macromolecular complexes82. Some of the important dynamic features, including eigenvalues, covariance, B-factors, and deformability, have been investigated using the NMA carried out using iMODS81. Eigenvalues describe structural stiffness; lower eigenvalues are associated with higher flexibility and deformability83. Through covariance analysis, motions between the various subunits in the complex have been identified as correlated, whereas B-factors have provided estimates of average residue flexibility84. Areas of deformation suggest regions within the protein where external forces cause most bending or distortion81. The energy landscape of the MEV-TLR4 complex is then displayed. These findings demonstrate the vaccine construct’s structural stability and functional integrity, suggesting that it can maintain its stable conformation and function effectively once the immune system has activated it14.
In Silico immune simulation of the multi-epitope vaccine (MEV) construct for immunogenicity assessment
The C-ImmSim 10.1 server, an in silico tool for immunological simulations, was used to assess the immunogenic potential of the MEV construct69,85. This server simulates the functions of key organs involved in innate and adaptive immunity, such as the lymph nodes, bone marrow, and thymus, in modeling immune responses86. The MEV construct was uploaded to the server as a multi-protein FASTA file. To model a standard vaccination protocol, three injection doses of the MEV construct were administered on days 0, 30, and 60. The simulation volume was set to 10, and a random seed of 12,345 ensured reproducibility of the output. HLA alleles A0101, B0702, and DRB1*0101 were selected to represent natural human genetic variation. These were used in simulation tests that demonstrated the MEV construct’s potential to activate cytotoxic T cells and produce antibodies, thereby inducing a humoral and cellular immune response68,87. Therefore, in silico pre-validation will favor the efficacy of this vaccine with the mechanism through comprehensive activation88.
The multi-epitope vaccine (MEV) construct’s codon optimization and cloning for expression in E. Coli
Codon optimization of the MEV construct was performed using JCat tool to enhance its expression in the selected host organism. This included reverse translation, whereby the sequence of the vaccine candidate construct was converted into DNA while making sure that the vaccine construct’s codon usage matched the preferences of the target host organism89. We calculated the optimized sequence’s Codon Adaptation Index values to determine how closely it resembles the host’s codon use bias90. A higher Codon Adaptation Index (CAI) value indicates improved potential for gene expression. Additionally, the optimized sequence’s GC content was ascertained because it affects the host’s gene expression levels91. The protein sequence of the MEV construct was modified for the generation of E. coli strain K12 in this investigation92. The optimized gene sequence was then cloned into the pET30a(+) expression vector employing SnapGene version 3.2.1 software93. The pET30a(+) vector ensures effective transcription and translation of the vaccine construct because it is specifically made for recombinant protein expression in E. coli. The MEV construct can be reliably produced using this technique for additional experimental confirmation80.
Results
Identification of non-homologous essential membrane proteins as vaccine targets
From the UniProt database, the entire 4,143-protein proteome of S. boydii strain CDC 3083-94 (BS512) was obtained (Proteome ID: UP000001030). The Geptop-2.0 web server was employed to identify 394 important proteins. Using BLASTp, homologs of human were eliminated to reduce the possibility of autoimmune reactions, and 140 non-homologous proteins were found. These proteins were then categorized using the PSORTb server based on their subcellular location. The analysis revealed 80 cytoplasmic proteins, 6 extracellular proteins, and 20 membrane-associated proteins. Among these candidates, one outer membrane protein and one cytoplasmic protein were highlighted as potential vaccine targets due to their favorable antigenic properties. Further evaluations confirmed that both proteins exhibited characteristics of being antigenic, non-allergenic, and stable, with no transmembrane helices detected. The two promising vaccine candidates identified were the lipopolysaccharide assembly protein LptD and the cell division protein FtsZ. Both candidates demonstrated high antigenicity and stability along with favorable vaccine-related attributes, as summarized in Table 1. These results lend credence to the proteins’ potential as useful ingredients in the creation of a vaccine to prevent Shigella infections.
Prediction and selection of Immunogenic CTL, HTL, and LBL epitopes
42 distinct S. boydii cytotoxic T lymphocyte (CTL) epitopes were found in this investigation. As shown in Table 2, the first seven epitopes were chosen from this group due to the strong antigenicity, non-toxicity, and high non-allergenicity. Ten other helper T lymphocyte (HTL) epitopes were also anticipated. As indicated in Table 3, four HTL epitopes were selected from this pool to be used in the MEV, particularly those that trigger important cytokines including IL-4, IL-10, and IFN-gamma. Two of the 16 linear B-cell epitopes were chosen to be incorporated into the MEV construct. Table 4 summarizes the characteristics of these B-cell epitopes, which include high antigenicity, non-toxicity, and high non-allergenicity. To guarantee the creation of a secure and efficient immune response against S. boydii, the selection procedure for these epitopes was carried out with great care. This tactical approach seeks to minimize side effects while maximizing the immunogenic potential of vaccine.
Linker and adjuvant selection for MEV construct
A suitable adjuvant and linker system were combined with four helper T lymphocyte (HTL) epitopes, seven cytotoxic T lymphocyte (CTL) epitopes, and two B-cell epitopes to create the MEV design. An EAAAK linker was employed to position cholera toxin subunit B at the N-terminus of vaccine construct to increase its immunogenicity. To maintain the distinct immunological characteristics of each type of epitope, particular linkers were carefully selected for example, the B-cell, HTL, and CTL epitopes were selected using KK, GPGPG, and AAY linkers respectively. As illustrated in Fig. 1, the final vaccine design contained 347 amino acids in total. By utilizing the adjuvant’s and the chosen epitopes’ synergistic effects, this strategy seeks to maximize the immune response against S. boydii and foster a strong and efficient immune response. To ensure both safety and effectiveness in triggering an immune response, these components must be carefully chosen and integrated.

(A) A schematic representation of the MEV construct, highlighting color-coded elements: the adjuvant (blue), CTL epitopes (green), HTL epitopes (red), B-cell epitopes (purple), and linkers (EAAAK, AAY, GPGPG, KK; shown in black). (B) The finalized MEV construct consists of 347 amino acids, including an adjuvant (grey) linked by an EAAAK linker (dark grey), CTL epitopes (cyan) joined by AAY linkers (blue), HTL epitopes (orange) connected through GPGPG linkers (red), and B-cell epitopes (green) linked using KK linkers (parrot).
Population coverage analysis of selected CTL and HTL epitopes
The chosen cytotoxic T lymphocyte (CTL) and helper T lymphocyte (HTL) epitopes included in the MEV were subjected to a population coverage analysis. According to the analysis, the selected epitopes covered approximately 84% of the global population. Interestingly, England had the most coverage, at 91%. Germany had 89% coverage, France had 90% coverage, and the region of Europe as a whole had 88% coverage. Other nations also showed notable coverage as shown in Fig. 2, these results imply that the chosen epitopes would be very useful for creating a globally applicable MEV. This assessment was made easier by the IEDB’s population coverage software, which demonstrated the significance of particular HLA alleles across various populations and geographical areas. This broad coverage highlights the MEV’s capacity to induce a strong immunological response across a range of global demographic groupings.

Population coverage analysis of selected T-cell epitopes across global and regional populations. The bar graph indicates the percentage coverage in the global population (84%) and specific regions, including England (91%), France (90%), Germany (89%), Europe (86%), South Africa (84%), Japan (84%), China (80%), Pakistan (78%), and Iran (75%). This analysis highlights the potential broad applicability of the designed MEV in diverse geographical and demographic populations.
Physicochemical properties and stability analysis of the MEV construct
The ProtParam program was used to assess the vaccine’s stereochemical parameters. With a molecular weight of 39,676.73 Da, the study showed an isoelectric point (pI) of 6.54, suggesting that the vaccine is rather basic. Thirty-three positively charged residues (lysine and arginine) and thirty-four negatively charged residues (aspartic acid and glutamic acid) made up the amino acid composition. Stability assessments indicated that the vaccine construct has an instability index of 36.70, categorizing it as stable. Its thermostability was supported by an aliphatic index of 62.28, while its hydrophilic nature was confirmed by a Grand Average of Hydropathicity (GRAVY) value of -0.468. The predicted half-lives of the vaccine were approximately 20 h in yeast, over 30 h in mammals, and more than 10 h in E. coli in in vivo settings. Furthermore, the vaccine was confirmed to be non-allergic, non-toxic, and antigenic, suggesting its potential for safe and effective immunization against target pathogens.
Structural modeling and quality assessment of the MEV construct
The secondary structure of the 347-amino acid vaccine construct was analyzed employing the SOPMA method. This analysis indicated that the construct comprises 81 residues making extended strands (23.34%), 116 residues making α-helices (33.43%), and 150 residues creating random coils (43.23%), reflecting an ordered structural shape (Fig. 3). To model the three-dimensional construction of the vaccine, the AlphaFold server was employed, followed by modification using the Galaxy Refine server to enhance structural accuracy (Fig. 4). Authentication through a Ramachandran plot demonstrated that 89.4% of the residues were located in the utmost favored regions, with 7.8% in allowable regions and only 2.7% in prohibited regions, as illustrated in Fig. 3. Further structural analysis yielded a quality factor of 87.162 and a Z-score of 3.42, indicating structural reliability and the nonexistence of poor rotamers, as depicted in Fig. 5. These outcomes collectively propose that the vaccine construct possesses favorable stereochemical characteristics and structural integrity, supporting its potential efficacy as an immunogen.

(A) Secondary structure prediction of the MEV construct. The top panel represents the predicted probabilities of structural elements (helix, sheet, turn, coil), while the bottom panel illustrates the spatial distribution of these secondary structures along the sequence. (B) Statistical detailed of secondary structure.

(A) Tertiary structure model of the MEV construct, showing the spatial arrangement of secondary structure elements. The model highlights helices, sheets, and coils in distinct colors for clarity. (B) Refined 3D model of vaccine construct by Galaxy Refine.

(A) Ramachandran plot of the modeled MEV structure. The plot indicates the distribution of backbone dihedral angles (phi and psi) of residues, with 89.4% of residues in favored regions (red), validating the structural stability and reliability of the predicted model. (B) Structural validation of the refined 3D vaccine model using the ERRAT tool. The plot highlights regions of the structure rejected at the 99% confidence level in red and at the 95% confidence level in yellow. The overall quality factor of the model is 87.162, indicating good reliability and structural accuracy.
Prediction of B cell epitopes in the MEV construct
B-lymphocytes, or B cells, are integral to humoral immunity due to their role in producing neutralizing antibodies. The identification of effective B-cell epitopes is essential for stimulating a robust antibody-mediated immune reaction in vaccine development. 21 conformational B-cell epitopes in total were found in this investigation; their lengths ranged from 3 to 20 residues, and their scores ranged from 0.513 to 0.96, suggesting that they may be useful. Moreover, six linear B-cell epitopes were predicted using the ElliPro server with default settings. To make sure they were properly incorporated into the vaccine design process, the conformational epitopes were further visualized and examined using PyMOL v1.3, as shown in Fig. 6. These results highlight how crucial both conformational and linear B-cell epitopes are for developing a vaccine that successfully elicits an immunological response. The vaccine seeks to improve the overall effectiveness of the immunization strategy by enhancing the generation of particular antibodies that can kill infections by targeting these epitopes. The development of an effective vaccination that offers sustained protection against illnesses depends on the meticulous selection and confirmation of these epitopes. Following molecular dynamics simulation, conformational epitopes were revalidated using ElliPro. The refined model retained all key epitopic regions, confirming the structural stability and immunogenic reliability of the vaccine construct.

Three-dimensional visualization of the conformational (discontinuous) B-cell epitopes in the designed MEV. The conformational B-cell epitopes are shown as a yellow surface, while the rest of the polyprotein structure is represented in grey stick format.
Docking and interaction analysis of the MEV construct with TLR4 receptor
One essential technique for clarifying the binding relationships among vaccine components and immunological receptor proteins is molecular docking. Using the ClusPro service, which uses a hybrid docking technique that combines small-angle X-ray scattering and experimental substrate binding data to improve prediction accuracy, the MEV was docked with human Toll-like receptor 4 (TLR4) in this work. Ten different docking models were produced by docking the vaccine construct’s improved three-dimensional structure with the TLR4 receptor (PDB ID: 3FXI). With 74 members in its cluster, the top-ranked model exhibited a durable stability for the MEV-TLR4 complex with an interaction energy of -1391.4 kcal/mol. The MEV construct and chain A of the TLR4 receptor had a favorable binding affinity, as shown by the production of 11 hydrogen bonds, according to additional molecular interaction analysis using the PDBsum service (Fig. 7). Using the PRODIGY tool, thermodynamic evaluations produced a Gibbs free energy change (ΔG) of − 13.0 kcal/mol and a dissociation constant (Kd) of 2.8 × 10⁻¹⁰ at 37 °C. Together, these findings imply that there is a stable and solid contact between the MEV and TLR4, supporting the vaccine construct’s capacity to interact with immune receptors and trigger a powerful immune response.

Docking analysis illustrating the interaction between the receptor TLR4 and the MEV. (A) The receptor’s Chain A and Chain K are shown in purple, while the MEV is highlighted in golden, emphasizing the interacting residues. (B) Docking visualization presents Chain A of the receptor in blue and the MEV in red, demonstrating the optimal binding affinity. (C) A detailed depiction of the interacting residues between the receptor and the vaccine construct, showcasing the formation of 11 hydrogen bonds between the receptor residues and the vaccine molecule.
Molecular dynamics simulations
Extensive molecular dynamics (MD) simulations were used to evaluate of the conformational stability and dynamic properties of the analyzed protein complex in a 100 ns trajectory. To assess the stability and flexibility of the system, evolution of some structural parameters (Root Mean Square Deviation (RMSD), Root Mean Square Fluctuation (RMSF), Solvent Acces-sible Surface Area (SASA) and Radius of Gyration (Rg)) was tracked during the simulation (Fig. 8). Plots of RMSD (Fig. 8A) showed an initial period of high fluctuations observed during the first 20 ns, reflecting equilibration and gradual relaxation to a stable conformation after assembly of the complexes by Chain A (black curve) and Chain K (blue curve). Following this period, the RMSD values reached a plateau and displayed less oscillations over the continuing part of the simulation, in accordance with the stable structure achieved and retained. The structural integrity of the complex in dynamics is confirmed by the persistent low RMSD amplitude over the second half of the trajectory. (Fig. 8B) Solvent accessible surface area (SASA) analysis showed that SASA decreased significantly during an initial ~ 30 ns, indicating a greater degree of compaction of the protein complex as in-termolecular interactions became stabilized. After this early stage of SASA dynamics, the absolute degree of SASA stabilized and became steady, indicating the formation of a close-packed interface of interactions and lack of significant conformational relaxation and exposure. The RMSF analysis was used to map local flexibility of the individual residues in Chain A (Fig. 8D) and Chain K (Fig. 8C). Particular portions with high RMSF, especially in Chain K indicated places where the mobility of atoms is very strong. These plastic regions could be of central importance in harboring binding or functional rearrangements. Conversely, most Chain A residues defined minimal RMSF values, which highlights their structural fault and potential engagement in core complex stabilization. The radius of gyration remained virtually unchanged during the simulation except for a slight increase at values between 0 and 40,000 ps which was recorded as transient expansion. (Fig. 8E) This was followed by the slow restoration to lower values implying the ability of the complex to maintain a folded, compact tertiary structure on a long timescale. The lack of drastic Rg fluctuations or increase indicates a continuous tertiary stability with no major fluctuations to unfold. The combination of trajectory analysis of all the monitored parameters indicates that the protein complex is initially equilibrating, then maintain structural stability and compactness throughout the simulation. Having a local flexibility in particular regions does not deviate with the overall structural fidelity, but potentially, it can promote functional motility and contact versatility. The low continuous RMSD, the established SASA and nullifying Rg support the existence of an energetically viable and strong protein assembly further making a case in favor of its viability in a biological circumstance under physiological conditions.

MD Simulation Analysis of Protein Complex (A) RMSD analysis of Chain A (black) and Chain K (blue) over 100 ns simulation. The plot displays early structural relaxation and subsequent stabilization and this implies that conformational integrity is held over the course of the trajectory. (B) Solvent Accessible Surface Area (SASA) of the protein complex, as a function of time. A significant drop in SASA in the initial stage of the simulation indicates the growing compactness during the attainment of equilibrium in the system. (C) Root Mean Square Fluctuation (RMSF) of Chain K, where localized flexibility and regions of dynamism in a sequence can be seen. (D) Root Mean Square Fluctuation (RMSF) per residue of the Chain A where there is fluctuation in the mobility of the atom and the rigid or flexible part of the chain is indicated. (E) Radius of gyration of the protein complex over simulation time span. The trajectory also shows that overall compactness and tertiary structure is retained, with no signs of unfolding.
Normal mode analysis of the MEV-TLR4 complex
The functional dynamics and molecular stability of the MEV complexed with Toll-like receptor 4 (TLR4) were evaluated using normal mode analysis (NMA). The deformability plot produced by the research indicated the existence of hinge or linker regions essential for molecular mobility by highlighting particular, flexible regions within the MEV-TLR4 complex. By establishing a correlation between the root-mean-square deviation (RMSD) of the docked complex and the NMA-predicted mobility, the experimental B-factor plot gave light on the structure’s dynamic behavior. The stiffness of the MEV-TLR4 complex in respect to normal modes of motion was indicated by the computed eigenvalue, which came out to be 4.290084 × 10⁻⁶. Bar charts showed the individual and cumulative contributions of each mode, and variance analysis showed an inverse connection between eigenvalue and variance. The MEV-TLR4 complex’s interatomic movements were further depicted using a covariance map that distinguished between uncorrelated (white), correlated (red), and anti-correlated (blue) interactions among residues. In order to illustrate interatomic connections and stiffness in a spring-like assembly, an elastic network model was also built; stronger interactions were represented by deeper grayscale. All things considered, the NMA results showed that the MEV-TLR4 complex had stable contacts, coordinated residue movements, and preserved structural integrity. As shown in Fig. 9, these results validate the construct’s appropriateness and functionality as a potential vaccination candidate. This thorough investigation emphasizes how crucial it is to comprehend molecular dynamics when assessing the stability and effectiveness of vaccines.

NMA analysis of the docked complex between the MEV and the receptor. (A) Deformability plot showing the flexibility of various regions within the docked complex. (B) B-factor analysis representing atomic fluctuations across the complex. (C) Covariance analysis highlighting the correlated motions of residues. (D) Elastic network analysis illustrating the connectivity and motion of residues within the complex. (E) Eigenvalue analysis depicting the stiffness of the docked structure and its associated energy requirements. (F) Variance analysis representing the structural variations in the docked complex.
Immune simulation of the MEV construct for immunogenicity evaluation
The findings of the immune simulation showed that the MEV architecture greatly improved both primary and secondary immune responses, earning it a top rating. Immunoglobulins such as IgG1, IgG2, and IgM, which are markers of a strong antibody-mediated immune response, increased as a result of immunization. Interestingly, B-cell populations significantly increased after repeated antigen exposure, indicating the emergence of humoral memory. During secondary and tertiary reactions, the simulations showed significant decreases in antigen levels and significant increases in cytotoxic T lymphocytes (CTLs) and helper T lymphocytes (HTLs), in addition to humoral responses. This suggests that the vaccine may successfully increase adaptive immunity (Fig. 10). The vaccine’s capacity to stimulate a complete immunological response was demonstrated by the fact that each immunization cycle also promoted the growth of innate immune components, including macrophages, natural killer cells, and dendritic cells. The vaccine induced the release of cytokines and interleukins, specifically TGF-β, IFN-γ, IL-10, IL-23, and IFN-β, which are important for coordinating operative immunological responses counter to infections. The results showed that continuous exposure to antigens led to considerably higher levels of TGF-β and IFN-γ, though other cytokines were identified at lesser but still detectable concentrations. A three-dose immunization schedule (days 0, 30, 60) produced robust secondary and tertiary immune responses with peak antibody titers, confirming the enhanced immunogenic potential of the designed construct. Generally, these outcomes underscore the MEV construct’s ability to trigger robust adaptive and innate immunological responses, representing its promising ability in combating leishmaniasis (Fig. 10).

C-ImmSim immunization simulation results for the three doses of MEV construct: (A) Immunoglobulin production represented by color-coded peaks. (B) Cytokine and interleukin profiles highlighting elevated levels of IFN-γ and IL-2 post-vaccination. (C) State-wise distribution of the T-helper cell population. (D) Macrophage population distribution across different functional states. (E) Distribution of B-cell populations across various states. (F) Activation of Th1-mediated immune responses. (G) Temporal evolution of T-helper cells during the simulation. (H) B-cell population dynamics showing enhanced diversity and class-switching potential. (I) Generation and activity dynamics of cytotoxic T cells.
Codon optimization and cloning of the MEV construct for expression in E. coli
Codon optimization was used to evaluate the expression potential of the suggested vaccine constructs. All constructions attained a codon adaptation index (CAI) value of 0.9, representing efficient codon use for expression in E. coli, according to analysis done with the JCAT server. Furthermore, the optimized cDNA sequences showed a 50% GC content, which is within the ideal range for the K12 strain of E. coli and promotes effective transcription and translation. Using restriction cloning techniques in silico, the gene sequence of the top-ranked vaccine construct was effectively cloned into the pET30a(+) plasmid vector. As shown in Fig. 11, the resultant recombinant plasmid had a total length of 2111 base pairs, indicating that it was suitable for use in subsequent experiments. These results are consistent with other research that highlights how crucial codon optimization is for increasing the expression of recombinant proteins in E. coli. The effective expression of several vaccine candidates and therapeutic proteins in bacterial systems, for example, demonstrates that optimizing codon use can greatly boost protein yield. The present findings provide credence to the theory that optimized designs will probably promote increased expression levels and better functional outcomes, increasing the overall effectiveness of vaccine development initiatives.

In silico cloning of the vaccine construct into the E. coli K12 host expression system. The plasmid backbone is depicted in black, while the inserted nucleotide sequence corresponding to the vaccine construct is highlighted in red.
Discussion
The emergence of Shigella boydii as a significant cause of bacillary dysentery (shigellosis) highlights the urgent need for effective preventive measures, particularly in regions with inadequate sanitation94. This bacterium poses a considerable public health risk, especially in underdeveloped nations where it contributes to high morbidity and mortality rates95. S. boydii evades the host immune system through various strategies, primarily by producing invasion plasmid antigens (Ipa proteins) and lipopolysaccharides (LPS). These mechanisms play a crucial role in its pathogenicity. These elements promote bacterial invasion, adhesion, and intracellular survival, which can result in serious gastrointestinal disorders7,8.
The identification of key antigenic proteins, such as the LPS-assembly protein LptD and the cell division protein FtsZ, is a significant advancement in understanding potential vaccine candidates. Although a phylogenetic reconstruction was not included, the selected antigens (LptD and FtsZ) are highly conserved and essential across Shigella species, as confirmed by functional orthology predictions using Geptop 2.0, thereby reflecting their evolutionary stability. Despite its clinical relevance, there has been limited research focused on identifying strain-specific antigenic targets for vaccine development against S. boydii. This investigation aimed to address this gap by using an integrated approach utilizing reverse vaccinology and subtractive proteomics to design a MEV for the strain CDC 3083-94 / BS51215,96,97. Our investigation highlights the essential role of computational tools in vaccine development. By predicting CTL and HTL epitopes, we identified immunogenic regions within target proteins capable of eliciting strong immune responses. A rigorous selection process, incorporating multiple bioinformatics algorithms, ensured that the chosen epitopes were highly antigenic, non-toxic, and non-allergenic. This systematic approach enhances immunogenic potential while minimizing safety concerns69,85,86. The predicted global population coverage (84%) was slightly lower than that reported in similar immunoinformatics studies (typically > 90%). This variation may result from the regional HLA distribution included in the IEDB dataset. While this represents a limitation, the construct still provides substantial global coverage, indicating strong population-level applicability.
Notably, cholera toxin subunit B was included as an adjuvant. Adjuvants play a vital part in improving immune responses by stimulating innate immune pathways and promoting cytokine release55,98. Individual epitope immunogenicity is maintained while structural stability is guaranteed through the employment of certain linkers to join HTL, CTL, and B-cell epitopes. This methodical setup shows how sophisticated computational techniques can optimize efficacy while streamlining the design process99. Strong contacts between the vaccine design and human Toll-like receptor 4 (TLR4), a crucial element of innate immunity, were found by molecular docking experiments. A stable complex formation is indicated by the binding energy, which is necessary to start immunological responses against S. boydii17,100. Additionally, the stability and adaptability of the MEV-TLR4 complex under physiological settings were revealed by molecular dynamics simulations, supporting the vaccine’s possible efficacy100,101.
Immunological simulations following vaccination with the designed multi-epitope construct demonstrated significant increases in immunoglobulin levels (IgG1, IgG2, and IgM) along with expansions in cytotoxic T cells (CTLs) and helper T lymphocytes (HTLs)102,103. These results imply that the vaccination may successfully stimulate the strong immune responses required to fight off infections brought on by S. boydii. Furthermore, the vaccine’s ability to be effectively expressed in E. coli was validated by in silico cloning data, suggesting that additional experimental validation is feasible104.
The robustness of this approach is further validated by a comparison with previous investigations. Similar computational techniques were used in a recently published work on S. flexneri MEVs, which identified conserved antigenic proteins including OmpA and IpaB as possible vaccine candidates105,106,107. Unlike previous studies, our work specifically targets S. boydii and employs a more comprehensive immunoinformatics screening approach to ensure high antigenicity and non-allergenicity. Furthermore, by include vital functional proteins like LptD and FtsZ, which are crucial for bacterial survival and immune evasion, this study goes beyond structural elements, whereas previous research mostly concentrated on structural proteins96,97,105.
Moreover, our molecular docking and dynamics simulations have provided additional validation of the vaccine’s structural stability, a factor that was not extensively explored in similar computational vaccine studies against Shigella species20,108. The immune simulation results in this study further corroborate findings from recent research on MEV against S. sonnei, which demonstrated similar elevations in IgG and T-cell responses but did not explicitly evaluate conservation across multiple strains. Our study strengthens this aspect by confirming epitope conservation across various S. boydii strains, thereby ensuring broader coverage against different circulating variants20.
One critical aspect of this study is its contribution to addressing antimicrobial resistance (AMR), which has become a significant challenge in treating Shigella infections. Innovative approaches to prevention, such as vaccination strategies that can provide long-lasting protection without relying on medicines, are necessary due to the growth of multidrug-resistant bacteria. A hopeful remedy is provided by the MEV strategy, which minimizes cross-reactivity with human proteins while focusing on conserved proteins necessary for bacterial survival21,109.
Notwithstanding these encouraging findings, it is crucial to recognize that thorough testing in animal models and then human clinical trials are necessary to move from computational predictions to clinical efficacy. The knowledge gathered from this investigation not only advances our knowledge of S. boydii pathophysiology but also opens the door for further studies targeted at creating potent vaccines against other harmful bacteria with comparable problems110.
Comparative analysis with previously reported Shigella vaccine constructs105,111 shows that our MEV design uniquely integrates both LptD and FtsZ, offering dual coverage of virulence and division mechanisms. Previous constructs primarily targeted outer membrane antigens, whereas our approach provides broader cellular target representation. Moreover, integration of multi-stage validation (epitope screening, docking, MD simulation, and immune simulation) distinguishes this work from earlier computational-only studies.
In summary, this study underscores the potential of a multi-epitope vaccine strategy against S. boydii, leveraging advanced computational methods to identify viable targets while addressing key challenges such as biofilm formation and antibiotic resistance. Our findings emphasize the need for further research into vaccine candidates capable of preventing healthcare-associated infections caused by this opportunistic pathogen. A limitation of this work is that the analyses were performed using a reference Shigella boydii strain. However, the prioritized proteins (LptD and FtsZ) are functionally conserved across multiple Shigella species, suggesting potential cross-protection. Future studies incorporating pan-genomic datasets from multiple isolates may further expand the breadth and translational applicability of the designed construct. Developing such vaccines is crucial for decreasing the global incidence of shigellosis and refining public health outcomes, particularly in vulnerable populations.
Conclusions
This investigation developed a multi-epitope vaccination against S. boydii using subtractive proteomics and reverse vaccinology techniques. LPS-assembly protein LptD, and cell division protein FtsZ were screened as promising antigens because of the crucial role both play in virulence and of high immunogenic potential. Then, the constructed epitopes would be incorporated in a construct possessing favorable structural, physicochemical, and immunological properties. Simulations and molecular docking investigations verified the vaccine’s strong affinity for binding TLR4. Immune simulations predicted robust cellular and humoral responses. Codon optimization ensured efficient expression in E. coli, supporting its feasibility for experimental validation. Such a vaccine construct holds much promise in combating S. boydii infections and antimicrobial resistance challenges.
Data availability
All data generated or analyzed during this study are included in this published article. Further inquiries can be directed to the corresponding author.
Abbreviations
- S. boydii :
-
Shigella boydii
- MEV:
-
Multi-Epitope Vaccine
- CTL:
-
Cytotoxic T Lymphocyte
- HTL:
-
Helper T Lymphocyte
- LBL:
-
Linear B-Lymphocyte
- LPS:
-
Lipopolysaccharides
- TLR4:
-
Toll-like Receptor 4
- MHC:
-
Major Histocompatibility Complex
- CAI:
-
Codon Adaptation Index
- AMR:
-
Antimicrobial Resistance
- GRAVY:
-
Grand Average of Hydropathicity
- RMSD:
-
Root-Mean-Square Deviation
- NMA:
-
Normal Mode Analysis
- PDB:
-
Protein Data Bank
- IEDB:
-
Immune Epitope Database
- PSORTb:
-
Protein Subcellular Localization Prediction Tool for Bacteria
- TMHMM:
-
Transmembrane Hidden Markov Model
- ABCpred:
-
Artificial Neural Network Based B-cell Epitope Prediction Tool
- JCAT:
-
Java Codon Adaptation Tool
- FASTA:
-
Fast-All (text-based bioinformatics format for sequence data)
- ERRAT:
-
Structure Verification Server (for 3D models)
- SOPMA:
-
Self-Optimized Prediction Method with Alignment
- ClusPro:
-
Protein–protein docking server
- iMODS:
-
Internal Coordinate Normal Mode Analysis
- TGF-β:
-
Transforming Growth Factor Beta
- IFN-γ:
-
Interferon Gamma
- IL-2:
-
Interleukin-2
- IgG/IgM:
-
Immunoglobulin G/M
- pI:
-
Isoelectric Point
- kcal/mol:
-
Kilocalories per mole
- K.d:
-
Dissociation Constant
References
Brown, J. D. et al. Shigella species epidemiology and antimicrobial susceptibility: the implications of emerging Azithromycin resistance for guiding treatment, guidelines and breakpoints. J. Antimicrob. Chemother. 72, 3181–3186 (2017).
Hmar, E. B. L., Paul, S. & Sharma, H. K. The role of Shigella spp. In propagating bacillary dysentery In humans and the prominence of nanotechnology In disease prevention. Futur J. Pharm. Sci. 10, 97 (2024).
Ranjbar, R., Farahani, A. & Shigella Antibiotic-Resistance Mechanisms And New Horizons For Treatment. IDR Volume 12, 3137–3167 (2019).
Afrad, M. H. et al. Antibiotic resistance and serotype distribution of Shigella strains in Bangladesh over the period of 2014–2022: evidence from a nationwide hospital-based surveillance for cholera and other diarrheal diseases. Microbiol. Spectr. 12, e00739–e00724 (2024).
Baharvand, A. et al. The increasing antimicrobial resistance of Shigella species among Iranian pediatrics: a systematic review and meta-analysis. Pathogens Global Health. 117, 611–622 (2023).
Martinez-Becerra, F. J. et al. Broadly protective Shigella vaccine based on type III secretion apparatus proteins. Infect. Immun. 80, 1222–1231 (2012).
Schroeder, G. N. & Hilbi, H. Molecular pathogenesis of Shigella spp.: controlling host cell Signaling, Invasion, and death by type III secretion. Clin. Microbiol. Rev. 21, 134–156 (2008).
Yang, S. C., Hung, C. F., Aljuffali, I. A. & Fang, J. Y. The roles of the virulence factor IpaB In Shigella spp. In the escape from immune cells and Invasion of epithelial cells. Microbiol. Res. 181, 43–51 (2015).
Sandlin, R. C. et al. Avirulence of rough mutants of Shigella flexneri: requirement of O antigen for correct unipolar localization of IcsA in the bacterial outer membrane. Infect. Immun. 63, 229–237 (1995).
Mattock, E. & Blocker, A. J. How do the virulence factors of Shigella work together to cause disease? Front Cell. Infect. Microbiol 7, (2017).
Qasim, M., Wrage, M., Nüse, B. & Mattner, J. Shigella outer membrane vesicles as promising targets for vaccination. IJMS 23, 994 (2022).
Lu, T., Das, S., Howlader, D. R., Picking, W. D. & Picking, W. L. Shigella vaccines: the continuing unmet challenge. IJMS 25, 4329 (2024).
Dingding, H. et al. Subtractive proteomics and reverse-vaccinology approaches for novel drug targets and designing a chimeric vaccine against Ruminococcus gnavus strain RJX1120. Front. Immunol. 16, 1555741 (2025).
Ghaffar, S. A. et al. Designing of a multi-epitopes based vaccine against haemophilius parainfluenzae and its validation through integrated computational approaches. Front. Immunol. 15, 1380732 (2024).
Rahman, S. et al. Subtractive Proteomics and Reverse-Vaccinology Approaches for Novel Drug Target Identification and Chimeric Vaccine Development against Bartonella henselae Strain Houston-1. Bioengineering 11, 505 (2024).
Khan, A., Khanzada, M. H., Khan, K., Jalal, K. & Uddin, R. Integrating core subtractive proteomics and reverse vaccinology for multi-epitope vaccine design against rickettsia prowazekii endemic typhus. Immunol. Res. 72, 82–95 (2024).
Kar, T. et al. A candidate multi-epitope vaccine against SARS-CoV-2. Sci. Rep. 10, 10895 (2020).
Mamun, T. I. et al. Designing a multi-epitope vaccine candidate against human rhinovirus C utilizing immunoinformatics approach. Front. Immunol. 15, 1364129 (2025).
Ross, S. H. & Cantrell, D. A. Signaling and function of Interleukin-2 in T lymphocytes. Annu. Rev. Immunol. 36, 411–433 (2018).
Aiman, S. et al. Vaccinomics-aided next-generation novel multi-epitope-based vaccine engineering against multidrug resistant Shigella sonnei: immunoinformatics and chemoinformatics approaches. PLoS ONE. 18, e0289773 (2023).
Salam, M. A. et al. Antimicrobial resistance: A growing serious threat for global public health. Healthcare 11, 1946 (2023).
Basmenj, E. R. et al. Computational epitope-based vaccine design with bioinformatics approach; a review. Heliyon 11, e41714 (2025).
The UniProt Consortium. The universal protein resource (UniProt). Nucleic Acids Res. 36, D190–D195 (2007).
The UniProt Consortium. UniProt: a hub for protein information. Nucleic Acids Res. 43, D204–D212 (2015).
Wei, W., Ning, L. W., Ye, Y. N., Guo, F. B. & Geptop A gene essentiality prediction tool for sequenced bacterial genomes based on orthology and phylogeny. PLoS ONE. 8, e72343 (2013).
Wen, Q. F. et al. Geptop 2.0: an Updated, more Precise, and faster Geptop server for identification of prokaryotic essential genes. Front. Microbiol. 10, 1236 (2019).
McGinnis, S. & Madden, T. L. BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 32, W20–W25 (2004).
Pertsemlidis, A. & Fondon, J. W. Having a BLAST with bioinformatics (and avoiding BLASTphemy). Genome Biol 2, reviews1 (2001). (2002).
Gardy, J. L. PSORT-B: improving protein subcellular localization prediction for Gram-negative bacteria. Nucleic Acids Res. 31, 3613–3617 (2003).
Yu, N. Y. et al. PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 26, 1608–1615 (2010).
Grund, M. E., Soo, J., Cote, C. K., Berisio, R. & Lukomski, S. Thinking outside the bug: targeting outer membrane proteins for burkholderia vaccines. Cells 10, 495 (2021).
Zahid, A., Ismail, H., Wilson, J. C. & Grice, I. D. Bioengineering Outer-Membrane vesicles for vaccine development: Strategies, Advances, and perspectives. Vaccines 13, 767 (2025).
Tabibpour, N. S., Doosti, A. & Sharifzadeh, A. Putative novel outer membrane antigens multi-epitope DNA vaccine candidates identified by immunoinformatic approaches to control acinetobacter baumannii. BMC Immunol. 24, 46 (2023).
Doytchinova, I. A. & Flower, D. R. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 8, 4 (2007).
Ong, E. et al. Vaxign2: the second generation of the first Web-based vaccine design program using reverse vaccinology and machine learning. Nucleic Acids Res. 49, W671–W678 (2021).
Bevacqua, A., Bakshi, S. & Xia, Y. Principal component analysis of alpha-helix deformations in transmembrane proteins. PLoS ONE. 16, e0257318 (2021).
Bianchi, F., Textor, J. & Van Den Bogaart, G. Transmembrane helices are an overlooked source of major histocompatibility complex class I epitopes. Front. Immunol. 8, 1118 (2017).
De Araújo, L. P., Silva, E. N., De Alencar, S. M. & Corsetti, P. P. De Almeida, L. A. Multivalent vaccine candidate from conserved Immunogenic peptides in entry or exit proteins of orthopoxvirus genus. Sci. Rep. 15, 12503 (2025).
Hewitt, E. W. The MHC class I antigen presentation pathway: strategies for viral immune evasion. Immunology 110, 163–169 (2003).
Wieczorek, M. et al. Major histocompatibility complex (MHC) class I and MHC class II proteins: conformational plasticity in antigen presentation. Front Immunol 8, (2017).
Waqas, M. et al. Immunoinformatics design of multivalent epitope vaccine against Monkeypox virus and its variants using membrane-bound, enveloped, and extracellular proteins as targets. Front. Immunol. 14, 1091941 (2023).
Lundegaard, C., Lund, O., Buus, S. & Nielsen, M. Major histocompatibility complex class I binding predictions as a tool in epitope discovery. Immunology 130, 309–318 (2010).
Vilela Rodrigues, T. C. et al. An immunoinformatics-based designed multi-epitope candidate vaccine (mpme-VAC/STV-1) against Mycoplasma pneumoniae. Comput. Biol. Med. 142, 105194 (2022).
Dimitrov, I., Bangov, I., Flower, D. R. & Doytchinova, I. AllerTOP v.2—a server for in Silico prediction of allergens. J. Mol. Model. 20, 2278 (2014).
Rezaei, M., Habibi, M., Ehsani, P., Asadi Karam, M. R. & Bouzari, S. Design and computational analysis of an effective multi-epitope vaccine candidate using subunit B of cholera toxin as a build-in adjuvant against urinary tract infections. Bioimpacts 14, 27513 (2023).
Roohparvar Basmenj, E., Omidvar, B., Kiumarsy, A., Izadkhah, H. & Ghiabi, S. Design of a multi-epitope-based peptide vaccine against the SARS-CoV-2 Omicron variant using bioinformatics approach. J. Biomol. Struct. Dynamics. 42, 7945–7956 (2024).
Gomes, L. G. R. et al. In Silico designed Multi-Epitope immunogen Tpme-VAC/LGCM-2022 May induce both cellular and humoral immunity against Treponema pallidum infection. Vaccines 10, 1019 (2022).
Wang, P. et al. A systematic assessment of MHC class II peptide binding predictions and evaluation of a consensus approach. PLoS Comput. Biol. 4, e1000048 (2008).
Maleki, A., Russo, G., Parasiliti Palumbo, G. A. & Pappalardo, F. In Silico design of Recombinant multi-epitope vaccine against influenza A virus. BMC Bioinform. 22, 617 (2022).
EL-Manzalawy, Y., Dobbs, D. & Honavar, V. Predicting linear B‐cell epitopes using string kernels. J. Mol. Recognit. 21, 243–255 (2008).
Srivastava, K. & Srivastava, V. Prediction of conformational and linear B-Cell epitopes on envelop protein of Zika virus using immunoinformatics approach. Int. J. Pept. Res. Ther. 29, 17 (2023).
Liu, F., Yuan, C., Chen, H. & Yang, F. Prediction of linear B-cell epitopes based on protein sequence features and BERT embeddings. Sci. Rep. 14, 2464 (2024).
Sarkar, B., Ullah, M. A., Johora, F. T., Taniya, M. A. & Araf, Y. Immunoinformatics-guided designing of epitope-based subunit vaccines against the SARS Coronavirus-2 (SARS-CoV-2). Immunobiology 225, 151955 (2020).
Jamali, S., Zamanzadeh, Z., Asadzadeh, A., Owji, F. & Abkar, M. Design of a multi-epitope vaccine using HA and M1 proteins from influenza and S, E, and M proteins from SARS-CoV-2 by in Silico tools. Inf. Med. Unlocked. 43, 101397 (2023).
Stratmann, T. Cholera toxin subunit B as Adjuvant––An accelerator in protective immunity and a break in autoimmunity. Vaccines 3, 579–596 (2015).
Shamriz, S., Ofoghi, H. & Moazami, N. Effect of linker length and residues on the structure and stability of a fusion protein with malaria vaccine application. Comput. Biol. Med. 76, 24–29 (2016).
Sanchez, A. E., Aquino, G., Ostoa-Saloma, P. & Laclette, J. P. Rocha-Zavaleta, L. Cholera toxin B-Subunit gene enhances mucosal Immunoglobulin A, Th1-Type, and CD8+ cytotoxic responses when coadministered intradermally with a DNA vaccine. Clin. Vaccine Immunol. 11, 711–719 (2004).
Liljeroos, L., Malito, E., Ferlenghi, I. & Bottomley, M. J. Structural and computational biology in the design of Immunogenic vaccine antigens. J. Immunol. Res. 2015, 1–17 (2015).
Bui, H. H. et al. Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinform. 7, 153 (2006).
Nguyen, T. L. & Kim, H. Discovering peptides and computational investigations of a multiepitope vaccine target Mycobacterium tuberculosis. Synth. Syst. Biotechnol. 9, 391–405 (2024).
Department of Computational Biology & & Bioinformatics Jacob school of biotechnology & Bioengineering, Sam Higginbottom Institute of agriculture technology & Sciences, Allahabad-211007, Uttar Pradesh, Bharat (India). MFPPI – Multi FASTA ProtParam Interface. Bioinformation. 12, 74–77 (2016).
Geourjon, C. & Deléage, G. SOPMA: significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Bioinformatics 11, 681–684 (1995).
Bertoline, L. M. F., Lima, A. N., Krieger, J. E. & Teixeira, S. K. Before and after AlphaFold2: an overview of protein structure prediction. Front. Bioinform. 3, 1120370 (2023).
Heo, L., Park, H. & Seok, C. GalaxyRefine: protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 41, W384–W388 (2013).
Wang, W. et al. Data set for phylogenetic tree and RAMPAGE Ramachandran plot analysis of SODs in Gossypium raimondii and G. arboreum. Data Brief. 9, 345–348 (2016).
Binbay, F. A., Rathod, D. C., George, A. A. P. & Imhof, D. Quality assessment of selected protein structures derived from homology modeling and alphafold. Pharmaceuticals 16, 1662 (2023).
Ponomarenko, J. et al. ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC Bioinform. 9, 514 (2008).
Tahir, U. et al. Multiepitope-Based subunit vaccine design and evaluation against respiratory syncytial virus using reverse vaccinology approach. Vaccines 8, 288 (2020).
Tahir, U. et al. Designing of a next generation multiepitope based vaccine (MEV) against SARS-COV-2: immunoinformatics and in Silico approaches. PLoS ONE. 15, e0244176 (2020).
Maeshima, N. & Fernandez, R. C. Recognition of lipid A variants by the TLR4-MD-2 receptor complex. Front Cell. Inf. Microbio 3, (2013).
Ain, Q. U., Batool, M. & Choi, S. TLR4-Targeting therapeutics: structural basis and Computer-Aided drug discovery approaches. Molecules 25, 627 (2020).
Kozakov, D. et al. The cluspro web server for protein–protein Docking. Nat. Protoc. 12, 255–278 (2017).
Raj, K. H. et al. A robust comprehensive immunoinformatics approach for designing a potential multi-epitope based vaccine against a reiterated Monkeypox virus. Biochem. Biophys. Rep. 43, 102075 (2025).
Schmit, J. D., Kariyawasam, N. L., Needham, V. & Smith, P. E. SLTCAP: A simple method for calculating the number of ions needed for MD simulation. J. Chem. Theory Comput. 14, 1823–1827 (2018).
Abraham, M. J. & Gready, J. E. Optimization of parameters for molecular dynamics simulation using smooth particle-mesh Ewald in GROMACS 4.5. J. Comput. Chem. 32, 2031–2040 (2011).
Quezada, G. R. et al. Molecular Dynamics Study of Polyacrylamide and Polysaccharide-Derived Flocculants Adsorption on Mg(OH)2 Surfaces at pH 11. Polymers 17, 227 (2025).
Childers, M. C. & Daggett, V. Validating molecular dynamics simulations against experimental observables in light of underlying conformational ensembles. J. Phys. Chem. B. 122, 6673–6689 (2018).
Hunt-Isaak, I., Russell, J. & Hekstra, D. mpl-interactions: A python package for interactivematplotlib figures. JOSS 9, 5651 (2024).
Shakibay Senobari, Z. et al. Chromone-embedded peptidomimetics and furopyrimidines as highly potent SARS-CoV-2 infection inhibitors: Docking and MD simulation study. BMC Res. Notes. 16, 224 (2023).
Elshafei, S. O., Mahmoud, N. A. & Almofti, Y. A. Immunoinformatics, molecular Docking and dynamics simulation approaches unveil a multi epitope-based potent peptide vaccine candidate against avian leukosis virus. Sci. Rep. 14, 2870 (2024).
López-Blanco, J. R., Aliaga, J. I., Quintana-Ortí, E. S. & Chacón, P. iMODS: internal coordinates normal mode analysis server. Nucleic Acids Res. 42, W271–W276 (2014).
Ma, J. Usefulness and limitations of normal mode analysis in modeling dynamics of biomolecular complexes. Structure 13, 373–380 (2005).
Singh, K. V. Eigenvalue and eigenvector computation for discrete and continuous structures composed of viscoelastic materials. Int. J. Mech. Sci. 110, 127–137 (2016).
Yu, C. C., Raj, N. & Chu, J. W. Statistical learning of protein elastic network from positional covariance matrix. Comput. Struct. Biotechnol. J. 21, 2524–2535 (2023).
Ashgar, S. S. et al. Integrated immunoinformatics and subtractive proteomics approach for multi-epitope vaccine designing to combat S. pneumoniae TIGR4. Front. Mol. Biosci. 10, 1212119 (2023).
Kumar, K. M. et al. Immunoinformatic exploration of a multi-epitope-based peptide vaccine candidate targeting emerging variants of SARS-CoV-2. Front. Microbiol. 14, 1251716 (2023).
Bergström, T. F., Josefsson, A., Erlich, H. A. & Gyllensten, U. Recent origin of HLA-DRB1 alleles and implications for human evolution. Nat. Genet. 18, 237–242 (1998).
Russo, G. et al. In Silico trial to test COVID-19 candidate vaccines: a case study with UISS platform. BMC Bioinform. 21, 527 (2020).
Grote, A. et al. JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 33, W526–W531 (2005).
Parvathy, S. T., Udayasuriyan, V. & Bhadana, V. Codon usage bias. Mol. Biol. Rep. 49, 539–565 (2022).
Paremskaia, A. I. et al. Codon-optimization in gene therapy: promises, prospects and challenges. Front. Bioeng. Biotechnol. 12, 1371596 (2024).
Kelpšas, V. & Wachenfeldt, C. V. Strain improvement of Escherichia coli K-12 for Recombinant production of deuterated proteins. Sci. Rep. 9, 17694 (2019).
Chick, J. A., Abongdia, N. N., Shey, R. A. & Apinjoh, T. O. Computational design, expression, and characterization of a Plasmodium falciparum multi-epitope, multi-stage vaccine candidate (PfCTMAG). Heliyon 11, e42014 (2025).
Niyogi, S. K. & Shigellosis J. Microbiol. 43, 133–143 (2005).
Zaidi, M. B., Estrada-García, T. & Shigella A highly virulent and elusive pathogen. Curr. Trop. Med. Rep. https://doi.org/10.1007/s40475-014-0019-6 (2014).
Botos, I. et al. Structural and functional characterization of the LPS transporter LptDE from Gram-Negative pathogens. Structure 24, 965–976 (2016).
Bisson-Filho, A. W. et al. Treadmilling by FtsZ filaments drives peptidoglycan synthesis and bacterial cell division. Science 355, 739–743 (2017).
Hou, J. et al. Cholera toxin B subunit acts as a potent systemic adjuvant for HIV-1 DNA vaccination intramuscularly in mice. Hum. Vaccines Immunotherapeutics. 10, 1274–1283 (2014).
Srinivasan, S. et al. Epitope identification and designing a potent multi-epitope vaccine construct against SARS-CoV-2 including the emerging variants. J. Global Infect. Dis. 14, 24 (2022).
Khan, M. T. et al. Immunoinformatics and molecular modeling approach to design universal multi-epitope vaccine for SARS-CoV-2. Inf. Med. Unlocked. 24, 100578 (2021).
Shang, K. et al. Development of a novel multi-epitope vaccine for brucellosis prevention. Heliyon 10, e34721 (2024).
Tan, C. et al. Immunoinformatics approach toward the introduction of a novel Multi-Epitope vaccine against clostridium difficile. Front. Immunol. 13, 887061 (2022).
Woelke, A. L. et al. Development of Immune-Specific interaction potentials and their application in the Multi-Agent-System vaccimm. PLoS ONE. 6, e23257 (2011).
Mahmoodi, S., Amirzakaria, J. Z. & Ghasemian, A. In Silico design and validation of a novel multi-epitope vaccine candidate against structural proteins of Chikungunya virus using comprehensive immunoinformatics analyses. PLoS ONE. 18, e0285177 (2023).
Qureshi, H. et al. Designing a multi-epitope vaccine against Shigella dysenteriae using immuno-informatics approach. Front. Genet. 15, 1361610 (2024).
Farhani, I., Nezafat, N. & Mahmoodi, S. Designing a novel Multi-epitope peptide vaccine against pathogenic Shigella spp. Based immunoinformatics approaches. Int. J. Pept. Res. Ther. 25, 541–553 (2019).
Hashemi, P. et al. A multi-epitope protein vaccine encapsulated in alginate nanoparticles as a candidate vaccine against Shigella sonnei. Sci. Rep. 14, 22484 (2024).
Jalal, K. et al. Identification of vaccine and drug targets in Shigella dysenteriae sd197 using reverse vaccinology approach. Sci. Rep. 12, 251 (2022).
Raso, M. M., Arato, V., Gasperini, G. & Micoli, F. Toward a Shigella vaccine: opportunities and challenges to fight an Antimicrobial-Resistant pathogen. IJMS 24, 4649 (2023).
Martić-Kehl, M. I., Schibli, R. & Schubiger, P. A. Can animal data predict human outcome? Problems and pitfalls of translational animal research. Eur. J. Nucl. Med. Mol. Imaging. 39, 1492–1496 (2012).
Ullah, H., Mahmud, S., Hossain, M. J., Islam, M. S. B. & Kibria, K. M. K. Immunoinformatic identification of the epitope-based vaccine candidates from Maltoporin, FepA and OmpW of Shigella Spp, with molecular Docking confirmation. Infect. Genet. Evol. 96, 105129 (2021).
Acknowledgements
The Researchers would like to thank the Deanship of Graduate Studies and Scientific Research at Qassim University for financial support (QU-APC-2026).
Funding
This research was funded by the Deanship of Graduate Studies and Scientific Research at Qassim University, Saudi Arabia (QU-APC-2026).
Author information
Authors and Affiliations
Contributions
Conceptualization, K.S.A.; methodology, K.S.A., F.A.; software, K.S.A.; validation, K.S.A., A.A.; formal analysis, K.S.A.; investigation, K.S.A.; resources, K.S.A.; data curation, K.S.A.; writing—original draft preparation, K.S.A.; writing—review and editing, K.S.A., F.A., A.A.; visualization, K.S.A.; supervision, K.S.A.; project administration, F.A., K.S.A.; funding acquisition, K.S.A.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Not applicable. This study involved in silico analysis and did not include human participants or animal models requiring ethical approval.
Consent for publication
Not applicable.
Clinical trial
Not applicable. This study is not a clinical trial.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Allemailem, K.S., Alrumaihi, F. & Almatroudi, A. Immunoinformatics-based design of a next generation multi-epitope vaccine candidate against Shigella boydii using a hierarchical subtractive proteomics approach. Sci Rep 16, 3359 (2026). https://doi.org/10.1038/s41598-025-33252-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-33252-8
