
Abstract
Sexual dimorphism (SDM) is regulated by sex chromosomes, yet the specific contribution of individual genes remains unclear. To address this, we conducted a comprehensive phenotyping analysis to investigate the roles of Y chromosome-linked genes in SDM formation in mice. Using C57BL/6J mice, we identified 49 SDM traits across 14 biological systems. To assess gene-specific effects, we generated knockout (KO) mice for 10 unique Y-linked genes using CRISPR/Cas9. As expected, Sry KO resulted in feminization of most SDM traits, including gonadal sex. However, certain body size-related traits remained male-like, suggesting the involvement of additional Y-linked genes. Consistently, KOs of Uty and Usp9y significantly altered traits related to body and organ size. We further applied a dimensionality reduction approach to quantitatively capture SDM variation at the individual level, enabling visualization of phenotypic shifts in each KO mouse. Our findings demonstrate that non-Sry Y-linked genes contribute to SDM and introduce a generalizable framework for quantifying sex differences across individuals and species.
Similar content being viewed by others


Introduction
Sexual dimorphism (SDM) is defined as the traits that show differences between males and females in the same species. SDM is prevalent in a wide range of species including humans1, other animals2, and plants3 and covers morphology, behavior, metabolism, and responses to disease and aging4,5,6,7,8,9. The SDM is thought to be ultimately under the regulation of sex chromosomes, X or Y, in mammals10,11. In the central dogma of mammalian sex differentiation, the sex determining gene, Sry, on the Y chromosome is expressed in undifferentiated male gonads to induce their differentiation into testes12,13. Testosterone, secreted from the testes, induces the differentiation of tissues and organs into male type throughout the body11. Therefore, it is believed that sex hormones secreted from the gonads, but not the Y chromosome, play critical roles in regulating SDM formation in nongonadal organs.
However, numerous lines of evidence suggest that, in addition to hormonal regulation, sex chromosomal genes could have a direct contribution in SDM formation. For example, it has been shown that Sry is expressed in the XY brain to mediate the function of dopaminergic neurons14,15. In addition to Sry, mouse Y chromosomes possess about 700 protein-coding genes16,17. Although most of them are multicopy, there are 11 genes that are unique (one or two copies): Kdm5d, Eif2s3y, Ddx3y, Usp9y, Uty, Uba1y, Rbm31y, H2al2y, Zfy1, Zfy2, and Sry. Whereas many of these unique Y-linked genes show apparent testis-specific expression to support spermatogenesis, some genes, including Ddx3y, Eif2s3y, Kdm5d, and Uty, are clearly expressed in a wide range of adult tissues16, and some are expressed even from the very early stages of embryogenesis18. These facts raise the possibility that Y-linked genes might play a direct role in forming SDM phenotypes in many nongonadal organs10,19. However, it remains unclear whether and to what extent the Y-linked genes contribute to SDM formation.
Approaches that analyze whole-body phenotypes in a large pipeline, such as the International Mouse Phenotyping Consortium (IMPC)20, which aims to identify the function of every protein-coding gene in the mouse genome, have revealed previously undiscovered relationships between phenotypes and genotypes21,22,23. By comparing the data of wild-type (WT) mice obtained by such phenotypic analysis, it is possible to analyze sex differences comprehensively. In fact, a number of SDM parameters have been detected between females and males of WT mice in IMPC datasets22. To quantitatively understand the impact of a given genetic modification of sex chromosomal genes on SDM, a new setting is required to capture the extent of sex differences at the individual level.
In this study, we analyzed the extent to which the SDM phenotype is detectable in the mouse model by a pipeline consisting of comprehensive phenotype screenings. We also generated KO mice for 10 unique Y-linked genes by CRISPR/Cas9 to determine the extent to which Y chromosomal gene KO affects the formation of SDM phenotypes by applying a statistical compression method to the SDM parameter values from these KO mice. Our results indicate that non-Sry genes on the Y chromosome may contribute to the formation of SDM in mice.
Results
Detection of SDM parameters in adult mice by comprehensive phenotyping screens
First, to determine the phenotypic traits that show SDM in adult mice, we designed a systematic and comprehensive phenotype analysis based on a phenotyping pipeline that has been used in the IMPC project at RIKEN BRC. The detailed protocols for the analysis followed those used in the IMPC24 (see "Methods"). We used seven WT male and eight WT female mice (maximum seven females for each test) in the C57BL/6J (B6J) strain to detect SDM parameters by the pipeline (Supplementary Tables 1 and 2). We chose the B6J strain because it was used to generate the initial Y chromosome reference sequence16. For more accurate comparisons with Y-linked gene KO mice described below, we generated all WT males and females by in vitro fertilization (IVF) followed by Cas9 mRNA alone injection, embryo transfer, and cesarean section. These B6J WT mice were subjected to 14 phenotyping tests covering a wide range of biological systems (functions) such as behavior, morphology, metabolism, and immunity from 9 to 19 weeks of age (Table 1; Fig. 1A). The 14 phenotyping tests consisted of 398 parameters (223 quantitative and 175 qualitative parameters; Supplementary Data 1) covering 20 of the 28 biological systems defined by mammalian phenotyping (MP) ontology25 (Table 1). The measured values for these 398 phenotypic traits were tested for statistical significance between males and females. Welch’s t-test was applied to continuous variables, and Fisher’s exact test to categorical variables. As a result, 49 traits (45 quantitative and 4 qualitative) showed statistically significant sex differences (uncorrected p < 0.05) and were defined as SDM traits (Fig. 1B; Supplementary Table 3). The primary objective of this study was to explore and quantify the multivariate structure of phenotypic sexual dimorphism using representative traits. Therefore, in this initial screening step, we adopted an exploratory analysis strategy and did not apply multiple comparison correction, using the uncorrected p-value threshold to identify candidate SDM traits. Thus, we focused on these 49 SDM traits to quantitatively analyze the sexual differences of individuals at either a whole-body or ontology-annotated biological system level in the following analyses and experiments.

Characterization of sexual dimorphism (SDM) traits. (A) Overview of the comprehensive phenotyping tests for the wild-type strain. See Table 1 for the explanation of the three-letter alphabetical abbreviations of phenotyping test names. (B) (i, ii) Proportion of measured parameters (traits) where the SDM was detected (i: quantitative datasets, ii: qualitative datasets). (iii, iv) Distribution of classifications by procedure (phenotyping test) (iii: quantitative datasets, iv: qualitative datasets). (C) (i, ii) Distribution of sexual difference degree for the SDM traits (i: higher in males, ii: higher in females). SDM traits are arranged in descending order of sexual difference degree from left to right. For the parameter names (x-axis), see the ‘SDM ID’ or ‘Parameter id’ columns in Table 2. For the degree of sexual difference (y-axis: − log10(p-value)), see the rightmost column in Table 2. (D) Distribution of sexual difference degree of the 53 SDM traits by biological function and sex.
Characteristics of SDM parameters
For clarity, we classify the SDM traits into two descriptive groups. ‘External traits’ include anatomical, morphological, and organ-weight measurements, whereas ‘internal traits’ comprise physiological, hematological, metabolic, and immunological parameters. These categories are used solely for interpretative purposes and do not correspond to formal IMPC ontology terms. The selected 49 SDM parameters consisted of 14 biological systems of the 20 MP biological systems surveyed (Supplementary Table 3), and were categorized into 34 external phenotypes, such as external morphology and organ weight, and 15 internal phenotypes, such as blood and immunity (Supplementary Table 3). These parameters included not only known SDM traits such as body weight, body length, liver weight, and glucose/cholesterol metabolism, but also previously underappreciated SDM parameters such as adrenal gland weight26,27, pituitary gland weight28, and immune cell type populations. This wide variation in the biological systems covered by the 49 SDM parameters allows a more accurate, comprehensive, and quantitative understanding of sex differences.
For 45 quantitative traits among the 49 SDM traits, 18 were relatively high in females (Fig. 1B-i, iii; Supplementary Fig. 1A) and 27 were relatively high in males (Figs. 1B-i, iii; Supplementary Fig. 1B). The remaining four qualitative traits (Fig. 1B-ii, iv; Supplementary Fig. 1C) were treated as eight traits after one-hot encoding to treat them quantitatively; therefore, the 49 SDM traits apparently became 53 (e.g., a categorical parameter ‘CSD_013: Head morphology’ was divided into two categories, ‘CSD_013_M_type: Head morphology_M_type’ and ‘CSD_013_F_type: Head morphology_F_type’) (Table 2). These 53 SDM traits were classified into two groups of 22 and 31 traits with high expression in females or males, respectively, and were ranked by log p-value (− log10(p-value)) for the significance test in each group (Welch’s t-test for quantitative data, Fisher’s exact test for qualitative data; Fig. 1C; Table 2). Note that this rank corresponds to the rank of the magnitude of influence for each sex in the sex spectrum representation (described below). The group of 22 SDM traits highly expressed in females included significantly more phenotypic traits belonging to the endocrine/exocrine gland phenotype biological system (n = 4 of 53 SDM traits) (Bonferroni-corrected p = 1.7 × 10−3, one-tailed Fisher’s exact test; Supplementary Table 4). On the other hand, in the group of 31 SDM traits highly expressed in males, phenotypes belonging to the growth/size/body region phenotype (n = 12) and homeostasis/metabolism phenotype (n = 9) biological systems were significantly more abundant (Bonferroni-corrected p = 1.0 × 10−5 and 3.7 × 10−3, respectively, one-tailed Fisher’s exact test; Supplementary Table 5).
Next, to show which SDM traits have more significant effect on sex differences at the biological system level in each sex, the distribution of mutation degree (− log10(p-value)) for the 53 SDM traits is presented by biological system (Fig. 1D). The results show that phenotypic traits belonging to the endocrine/exocrine gland phenotype were highly represented in females, whereas the growth/size/body region phenotype and homeostasis/metabolism phenotype were highly represented in males. These results suggest that the endocrine/exocrine gland phenotype in females, and the growth/size/body region phenotype and homeostasis/metabolism phenotype in males have a significant impact on differential sexual phenotypic expression, both qualitatively (numerous numbers of constituting phenotypes) and quantitatively (high mutability of constituting phenotypes).
Generation of KO founder mice for 10 unique Y-linked genes by CRISPR
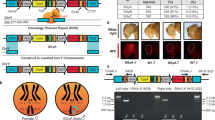
Having successfully extracted SDM traits in the WT B6J strain, we next investigated the degree to which Y chromosome-linked genes contribute to the expression of such sexual phenotypes by focusing on these SDM traits. First, we generated KO mice for each of the 10 unique genes (only 1 or 2 copies) on the Y chromosome (Y gene KO; YKO) including Sry KO, using the CRISPR/Cas9 system29,30 (Fig. 2A,B). We were concerned that for some genes, such as Sry, Eif2s3y, and Zfy2, KO mouse lines would not be established because the KO mice are known to be infertile or subfertile31,32,33. Therefore, we adopted a Triple CRISPR-based method that can produce whole-body KO mice with nearly perfect efficiency even for autosomal genes that have two target alleles34,35,36. The founder mice were directly used for the phenotyping analysis. We confirmed that injection of these CRISPR complexes had no detrimental effect on embryonic development (Fig. 2C, Supplementary Table 6) but induced insert-or-deletions (indels) at the target genes with very high efficiencies (Supplementary Fig. 2, Supplementary Table 7). Importantly, Sry KO mice possessed female-type external and internal genitalia (Fig. 2D-i), Eif2s3y KO mice showed severe testicular hypoplasia (Fig. 2D-ii), and Zfy2 KO mice showed abnormal sperm head morphology (Fig. 2D-iii), consistent with the previously reported KO mice31,32,33, and thereby validating the reliability of our approach to generate KO mice.

Generation of YKO mice. (A) Schematic illustration of the mouse production process through in vitro fertilization (IVF) and embryo transfer. Control XY and XX mice were generated by injecting Cas9 mRNA alone at the zygote stage. The Y chromosome gene knockout (KO) mice were generated by injecting Cas9 mRNA and sgRNA mixture at the zygote stage. (B) Three single-guide (sg) RNAs targeting protein-coding exons were designed for nine Y chromosome genes. For Sry, two sgRNAs were designed. (C) Bar graphs showing the implantation rate (i), pup rate (ii), and XY ratio (iii) of the wild-type and Y gene KO (YKO) mice (see Supplementary Table 6). (D) Phenotypes of YKO mice. (i) Sry KO mice showed sex reversal of the external and internal genitalia. (ii) Eif2s3y KO mice showed severe testicular hypoplasia. (iii) Zfy2 KO mice showed abnormal sperm head morphology.
Effects of Y chromosome gene disruption on the 53 SDM parameters
Next, we performed comprehensive phenotypic analysis of the 10 YKO mouse lines generated above from 9 to 19 weeks of age (398 parameters, n = 7 for each gene; Supplementary Tables 1 and 2). To understand the changes in expression of SDM traits caused by YKO, we focused on the phenotypic expression of the 53 SDM traits detected in the WT sex comparisons (Supplementary Data 2 and 3). The values (− log10(p-value): measure of mutation degree) of these parameters for each of the 10 YKO lines compared with WT males and females were placed on a two-dimensional scatterplot (SDM Identification Map; SDM-IM) (Fig. 3A, Supplementary Data 4). On this SDM-IM, each SDM parameter shows a vertical distribution pattern when its value is close to that of WT males (see the B6J_M panel in Fig. 3A) and a horizontal distribution pattern when its value is close to that of WT females (see the B6J_F panel in Fig. 3A). As a result, it is possible to understand the dynamic variation of SDM traits caused by each YKO according to where each parameter is distributed in the nine divided regions (Fig. 3A). For example, in the Sry KO, 36 (67.9%) of the 53 total phenotypes became closer to the WT female values than to the WT male values, whereas ‘39: Body length 09w’, ‘50: Tail length’, and ‘47: Lactate dehydrogenase (LDH)’ were retained as male type (Fig. 3B). ‘2: Lean mass’ and ‘21: Adrenal gland weight’ of Sry KO were found to be statistically significantly different, in opposite directions, for both groups of WT females and WT males, indicating that they are intermediate traits showing sufficient phenotypic differences for both sexes (in most cases, KO traits showing intermediate values between males and females show no statistical significance to both sexes: category #5). Interestingly, ‘15: Body mass index (BMI)’ (blue plot in #7) and ‘13: Alkaline phosphatase (ALP)’ (red plot in #3) were significantly lower in the former and significantly higher in the latter relative to the WT female phenotype, indicating that Sry KO mice lie beyond the WT female range for these traits in our composite sex-associated space. When we examined, for each of the 10 YKO lines, how many of the 53 sex-associated phenotypes shifted from the wild-type male level toward the wild-type female level (Fig. 3C), such female-shifted patterns (red plots in #4 or blue plots in #6; pink shade) were observed in 1, 3, 2, 5, and 4 parameters in Ddx3y KO, Rbm31y KO, Uba1y KO, Uty KO, and Usp9y KO, respectively. Specifically, in Ddx3y KO male mice, ‘44: NKT cells (panel A)’ shifted into the WT female-typical range, whereas in Rbm31y KO males, ‘28: Spleen weight/BW’, ‘44: NKT cells (panel A)’, and ‘46: Blood glucose concentration (30 min)’ shifted from the WT male-typical range toward the WT female-typical range. Uba1y KO affected ‘53: Spleen weight’ and ‘28: Spleen weight/BW’. Uty and Usp9y appeared to have broader functions as Uty KO affected ‘15: Body mass index (BMI)’, ‘43: Fat mass’, ‘22: Liver weight (LIW)’, ‘28: Spleen weight/BW’, ‘14: Spleen weight to body weight ratio (SPW BW)’, and Usp9y KO affected ‘23: Body length 14w’, ‘24: Body weight 18w_01’, ‘22: Liver weight (LIW)’, and ‘13: Alkaline phosphatase (ALP)’ (Supplementary Data 4). Thus, by constructing the SDM-IM, we were able to capture a snapshot of the change of SDM traits in 10 different YKO lines.

Examination of phenotypic variations for the 53 SDM traits in the 10 YKO strains. (A) Distribution of the degree of phenotypic differences (defined as mutation degree) in YKO lines relative to the WT male group (B6J_M, x-axis) and the WT female group (B6J_F, y-axis) shown on two-dimensional scatterplot named SDM Identification Map (SDM-IM). Red and blue plots represent statistically female-higher and male-higher phenotypic expressions compared with those between opposite sexes in WT lines, respectively. Red dashed lines represent the phenotypic call threshold (±log10(0.05) = ±1.3: p-value = 0.05). Each SDM-IM was separated into nine categories (#1 to #9) by the red dashed lines. Categories in pale red (#4 and #6) and blue (#2 and #8) on each SDM-IM represent female-specific and male-specific phenotypic expressions, respectively. The numbers assigned to plots correspond to those in the ID column of Table 2. (B) Enlarged example of SDM-IM for Sry KO mice. (C) The number of SDM traits in each of the nine categories (#1 to #9) shown in (A). F and M in each #3 and #7 category represent the traits that showed higher phenotypic expressions in WT female and male groups, respectively.
Setting weights of the SDM parameters for quantitative detection of sex differences
To quantitatively maximize sex differences at the individual level and identify the degree to which each of the 53 SDM parameters is related to the sex differences, we performed principal component analysis (PCA) on the dataset of all 15 WT individuals (eight females and seven males) × 53 SDM traits. The four qualitative SDM traits consisted of multi-category descriptors, and each category was converted into an independent binary variable (one-hot encoding) before PCA. Because some encoded categories were imbalanced between sexes, all variables were z-score transformed before PCA, which performs trait-wise centering and scaling and therefore reduces the influence of unequal category frequencies. As expected, the first principal component (PC1) represents the axis that clearly discriminates between males and females (Fig. 4A). Accordingly, the PC1 factor loadings for each phenotypic trait were used as weights for differentially classifying males and females. In this case, missing value imputation for analysis groups with small sample sizes (e.g., Immunophenotyping [IMM]) leads to overestimation of the resulting PC1 factor loadings relative to the corresponding mutation degree (− log10(p-values)) from the statistical test for the sex difference using the original data before the imputation (Fig. 4B). To avoid this overestimation, bias reduction by a robust nonlinear regression37 was performed on all acquired PC1 factor loadings (Fig. 4C-i, ii). A weighted dataset, in which the value of each phenotypic trait (z-scores) with missing value completion was multiplied by the corrected PC1 factor loading, was used for SDM representation in the following analyses to enable quantitative detection of sex differences.

Setting of weights for the 53 SDM traits (parameters) to differentially detect sexual differences. (A) Principal component analysis (PCA) of the 53 SDM traits. All traits, including one-hot encoded categorical variables, were z-score transformed prior to PCA. (B) Differences in the degree of sexual dimorphism in WT mice before and after missing-value imputation. The x-axis shows the 53 SDM traits, and the y-axis indicates the statistical significance of the sex differences, presented as − log10(p-value). Colored regions highlight traits whose − log10(p-value) changed markedly following imputation. For measured parameters (SDM traits) with small sample sizes within the same analysis group (typically n = 3–4 per sex), missing-value imputation can artificially exaggerate the apparent sex differences and inflate the corresponding − log10(p-value). For the parameter names shown on the x-axis, see the “SDM ID” or “Parameter ID” columns in Table 2. (C) Correction of PC1 factor loadings distorted by missing-value imputation. Because variance distortion introduced by missing-value imputation propagated to the PCA and produced artificially large PC1 factor loadings for affected traits, we applied robust nonlinear regression (MM estimation) to obtain stabilized loading values. For each SDM trait, PC1 loadings that were exaggerated relative to the actual degree of abnormality (− log10(p-value)) were corrected by this model (panel (i), before correction; panel (ii), after correction).
One-dimensional SDM visualization for YKO mice
For a simple one-dimensional representation of the SDM degree of individual YKO mice, we utilized the developed statistical method (Fig. 4) on the data of these YKO mice. First, we created a dataset of 53 SDM parameters (z-scores) of all 86 examined individuals (‘WT females and males’ + ‘YKO males’) multiplied by the corresponding corrected PC1 factor loadings (Fig. 5A, Supplementary Data 5). The mean for all 53 traits in each individual, mean PC1 scores, using this 86 × 53 dataset allows quantitative representation of individuals of each YKO group depending on the degree of cumulative SDM level (Fig. 5B). In the following analyses, we use the term ‘feminization’ as an operational description for a decrease in the PCA-based sex score—i.e., a shift of the composite phenotype toward the WT female centroid in the multivariate space—without implying any direct inference about hormonal status or developmental ‘femaleness’. A comparison test (Dunnett’s test) of cumulative SDM with WT males found a statistically significant difference in the Sry KO group (corrected p = 1.1 × 10−16). Other comparisons against the Usp9y KO and Uty KO groups detected significant differences at a threshold of uncorrected p < 0.05. These results are consistent with those that indicated Sry, Usp9y, or Uty KO lines showed a greater number of SDM traits shifted toward female type than other YKO lines (Fig. 3C). Furthermore, by performing the cumulative SDM analysis for each biological system (function), we quantified the degree to which feminization progressed in each biological system of each mutant line (Fig. 5C,D). This analysis revealed more detailed drifts of the cumulative SDM in each line. For example, the Rbm31y KO group showed significant change of the cumulative SDM position at the biological system level, specifically in hematopoietic system phenotype and immune system phenotype (Fig. 5C,D), although this group did not show significant change by the statistical test using all SDM traits (Fig. 5B). In contrast, whereas the Uty KO group showed a significant shift toward the female type by the test using all SDM traits, at the biological system level, there was a slight trend toward the female type in multiple systems such as limb/digit/tail phenotype and liver/biliary system phenotype. Only immune system phenotype showed a significant change (Fig. 5C,D). This result indicates that the change of the Uty KO group by the test using all SDM traits (Fig. 5B) is predominantly through the cumulative effect of a series of subtle changes. Conversely, there was a significant difference in the analysis using all SDM traits in the Usp9y KO group (Fig. 5B), but not at the biological system level (Fig. 5C,D). By constructing a workflow for quantifying the degree of female/male sexuality in the individual mice using a weighted dataset, in which the phenotypic values of the SDM traits were multiplied by the corresponding corrected PC1 factor loadings, we succeeded in describing the effect of each mutation as cumulative SDM at the individual level.

One-dimensional cumulative SDM representation for YKO mice. (A) Heat map of the dataset used for cumulative scoring (see Supplementary Data 5). Each SDM trait was z-score transformed and multiplied by its corrected PC1 factor loading, providing sex-axis–aligned weights. (B) Cumulative SDM scores for all WT and YKO groups. Higher values indicate a shift toward the WT-female end of the sex axis. Statistical comparisons with WT males were performed using Dunnett’s test. (C )Summary of cumulative SDM values aggregated by biological system (MP ontology). Patterns reveal gene-specific and system-specific sensitivity to Y-linked gene disruption. *corrected p < 0.05 versus WT males (B6J_M). (D) Deviation of each biological system from the WT male reference. Significant whole-body shifts (e.g., Sry KO) and distributed mild effects (e.g., Usp9y KO) are distinguished. *Corrected p < 0.05 versus WT males (B6J_M).
Two-dimensional visualization of SDM changes for YKO mice
For a more accurate understanding of the SDM changes and the multifaceted nature of variation, we attempted to visualize the two-dimensional representation of the cumulative SDM using the dataset in which the values of each of the 49(53) phenotypic traits (z-scores) of all 86 tested individuals were multiplied by the absolute values of the corrected PC1 factor loadings (Supplementary Fig. 3, Supplementary Data 6).
First, to intuitively understand the relationships between individuals as a continuous progression (trajectory), using the weighted dataset, we performed a tree-shaped visualization for all 86 individuals based on the minimum spanning tree (MST) algorithm38 (Fig. 6A). On the trajectory leading to WT females (B6J_F) and Sry KO individuals, we found not only individuals of the Uty KO and Usp9y KO lines that showed statistically significant differences from the WT male line (B6J_M) (Fig. 5B), but also three individuals of the Ddx3y KO line that did not significantly differ from the B6J_M line. We also found a branch that differed from that leading to the B6J_F line, allowing the detection of trait variation patterns in a trajectory (projection) different from feminization to WT female and Sry KO.

Two-dimensional cumulative SDM representation for YKO mice. (A) Minimum spanning tree (MST) visualization of weighted SDM vectors (z-scores multiplied by |corrected PC1 loadings|) for all 86 mice. The MST reveals a tree-like structure in which the main trajectory traces a phenotypic continuum from WT males through the various YKO lines toward WT females. (B) PHATE embedding of the same dataset, showing smooth multivariate transitions among groups. Sry KO individuals cluster near the WT-female region, whereas Uty and Usp9y KO occupy intermediate positions. Collectively, the MST and PHATE visualizations demonstrate that Y-linked gene knockouts induce graded, multidimensional shifts along a continuous sex-associated phenotypic spectrum.
Next, for the weighted dataset, we performed Potential of Heat-diffusion for Affinity-based Trajectory Embedding (PHATE)39, which preserves both local and global distances of high-dimensional data, and visualized (plotted) relationships between individual mice on two-dimensional coordinates (Fig. 6B). By the PHATE visualization, we succeeded in capturing the continuous progression of phenotypic variation of the individual Y mutants relative to the WT males as a smooth trajectory, without assuming a tree-like data structure in advance. Thus, the application of the MST and the PHATE visualizations to the SDM trait data in mice led to advancement of the cumulative SDM representation.
Discussion
Despite the prevailing wide presence of SDM in mammalian species, the degree to which SDM could be detected (or modeled) in mice remains unclear and a method to describe the comprehensive phenotypes into statistically comparable values has not been achieved. In this study, we conducted large-scale phenotyping analysis of WT males and females using B6J mice as a model and succeeded in comprehensively detecting 49 traits with SDM. These SDM phenotypes covered a variety of biological systems in both external and internal traits. Furthermore, based on these SDM traits, we successfully quantified the cumulative SDM at the individual level using whole (all) phenotypic traits or only phenotypic traits with the same biological functions by weighting each data value through statistical analysis. By applying this method to KO mice for Y chromosome genes, we were able to quantify the effect of the Y gene KO on cumulative SDM at the individual level as a transition, which we term ‘sex spectrum’ representation. This unique approach to quantitatively redefining sexual differences may not only be applicable to other model organisms, but it also may allow us to describe the cumulative SDM of each individual from human phenotypic data, which are abundantly available as clinical datasets.
We found that most of the SDM traits were converted to the female type in Sry KO mice, consistent with the previous report31,40, but some traits such as body length, tail length, and blood LDH level remained male type. This suggests that genes on the Y chromosome, other than Sry, might be involved in the expression of the male-type phenotype. In support of this idea, a previous report of Sry KO mice described that the ventromedial preoptic area of Sry KO mouse brain remained the male type31. Our results consistently demonstrated that some phenotypes such as body mass index, body length, and liver weight underwent spectral conversion to female type in Uty and Usp9y KO mice, whereas most phenotypes including internal and external genitalia remain male type in these mice. Because at least some of the founders are likely to be mosaic, the line-consistent phenotypic shifts observed here probably underestimate, rather than overestimate, the full impact of Y-linked gene disruption. Our preliminary serum steroid profiling revealed no major changes in testosterone or 17β-estradiol levels in most YKO mice compared with WT males, with the exception of Ddx3y KO, which showed an increase in testosterone, and Sry KO, which showed reduced testosterone consistent with gonadal sex reversal (Supplementary Fig. 4). This suggests that Usp9y and Uty may be expressed in a variety of tissues to induce the expression of male-type phenotype in a cell- or tissue-autonomous manner. Indeed, whereas many Y-linked genes show germline-specific expression, Uty expression has been detected in a wide range of tissues16. Uty encodes a protein that belongs to the JMJD family that shares histone demethylase activities to regulate the epigenetic landscape. Although Uty does not possess histone demethylase activity, it can still mediate transcription of target genes to ensure proper heart development41. It has recently been shown that Uty confers protection against the development of pulmonary arterial hypertension by suppressing a proinflammatory pathway in male mouse lungs42. Thus, Uty might play an important role in forming SDM in various tissues. Usp9y encodes a ubiquitin-specific peptidase, but its physiological function has not yet been analyzed. Although Usp9y is mainly expressed in the testis in adult mice, it is detectable in other tissues including submandibular gland, skin, lung, and brain43,44. Importantly, both UTY and USP9Y are widely expressed in human tissues45, which suggests conserved roles of these genes in SDM formation in humans. How these genes mechanistically contribute to the formation of SDM during development awaits further investigation. It should be noted that subtle endocrine effects cannot be ruled out, even though our preliminary endocrine and gonadal data do not indicate major systemic hormonal disruption by Y-linked gene knockouts.
To advance the cumulative SDM representation, we presented it not only in one dimension at the individual level, but also in two dimensions. The MST method of visualization, which assumes a tree structure to represent the continuity of the cumulative SDM, enables the detection of branches (clusters) that differ from the progression (trajectory) between WT females and males (Fig. 6A). Furthermore, PHATE visualization, which is more suitable for hypothesis generation and data exploration because it does not require a priori assumptions such as the tree structure of the MST method, succeeded in capturing the natural progression and trajectory between sexes at the individual level (Fig. 6B). The advanced cumulative SDM representation method (visualization procedures) presented in this study is expected to be of secondary use for cumulative SDM representation in other species because there is no established methodology for their representation, and the method is not only explainable, interpretable, and transparent in content, but also intuitive and understandable.
Accumulating evidence across taxa shows that other Y-linked genes may contribute to male-specific traits, particularly in body size and growth regulation. For example, in the fish Poecilia parae, Y chromosomes carry male morph-determining loci affecting body size and coloration46. Similarly, in the cichlid Lamprologus callipterus, Y-linked variation affects male body size, which is crucial for their reproductive strategy47. In the seed beetle Callosobruchus maculatus, Y chromosome effects on body size and development time have been reported48. Moreover, in humans, Y chromosome haplogroups have been associated with variation in adult height49. The effects we observed in Uty and Usp9y KO mice—specifically on body mass index, liver weight, and body length—may reflect such a conserved function of Y-linked genes in shaping SDM traits. Notably, recent advances in long-read sequencing technologies are now enabling near-complete assemblies of individual human Y chromosomes50, opening new avenues to explore how person-to-person variation in Y-linked gene structure and function may underlie trait diversity in men.
In summary, we have successfully detected 49(53) SDM traits in C57BL/6J mice by using a comprehensive phenotyping pipeline. By applying this approach to 10 different YKO mouse lines, we revealed that, in addition to Sry, other Y-linked genes such as Uty and Usp9y could contribute to the formation of SDM traits. Furthermore, we established a statistical method to represent these SDM transitions at an individual level, with which we successfully delineated the transition of sexual phenotypes as a spectrum in one- and two-dimensional representation. Further refinement of the phenotyping system and expansion of the analysis to other KO mice for autosomal and X chromosomal genes may reveal the detailed and unexpected picture of molecular cascade of SDM formation in mammals. These findings pave the way for further research into the genetic and molecular basis of SDM, with potential applications in medicine and evolutionary biology.
Methods
Mice
C57BL/6JJcl (B6J) and ICR mice were purchased from CLEA Japan. Mice were housed under controlled lighting conditions (daily light from 07:00 to 21:00). Mice were euthanized by cervical dislocation, performed by trained personnel in accordance with the guidelines approved by the Institutional Animal Care and Use Committee (IACUC) of RIKEN Tsukuba Institute. When required, anesthesia was administered using a combination of medetomidine (0.3 mg/kg, Domitor; Nippon Zenyaku Kogyo), midazolam (4.0 mg/kg, Dormicum Injection 10 mg; Maruishi Pharmaceutical), and butorphanol (5.0 mg/kg, Vetorphale 5 mg; Meiji Animal Health), delivered intraperitoneally. All animal experiments were approved by the IACUC of RIKEN Tsukuba Institute and were conducted and reported in accordance with the relevant guidelines and regulations including ARRIVE guidelines (https://arriveguidelines.org).
IVF
B6J mice were used for IVF. Briefly, spermatozoa were collected from epididymis of adult B6J males and incubated in human tubal fluid (HTF) drops for 1 h before insemination for their activation. Cumulus oocyte complexes (COCs) were collected from the oviducts of B6J females that were superovulated by injecting 7.5 IU of pregnant mare serum gonadotropin (PMSG; Millipore # 367222) plus anti-inhibin serum (AIS) and 7.5 IU of human chorionic gonadotropin (hCG; Millipore # 230734). Then, 15–17 h after the hCG injection, the isolated COCs were incubated in HTF containing 1.25 mM reduced glutathione (GSH) for 1 h before insemination. After preincubation, the activated spermatozoa were introduced to the COC-containing HTF drops to initiate insemination. The fertilized zygotes were washed in KSOM drops 5–6 h after insemination,
Triple CRISPR to generate YKO mice
Triple CRISPR was performed as described previously35,36. Briefly, three single-guide (sg) RNAs (two for Sry) targeting different sites of protein-coding regions were designed using MIT sgRNA design tool ver.1. Complementary sequences of sgRNAs were cloned into px330 vector and DNA was synthesized by PCR adding T7 promoter. Then sgRNA was synthesized by using PCR products as templates using the MEGAshort script kit. The primer sequences used for sgRNA synthesis are listed in Supplementary Table 8. Synthesized sgRNAs were purified using the MEGAclear kit and the concentration was adjusted to 500 ng/µL. Aliquots were frozen at − 80 °C until use. The mixture of two or three distinct sgRNAs (50 ng/µL each) targeting coding exons of target single Y-linked gene and Cas9 mRNA (100 ng/µL) were injected into zygotes by using piezo-driven micromanipulator 5–6 h postinsemination (hpi). For WT control mice, Cas9 mRNA alone (without sgRNA) was injected into zygotes. The injected embryos were cultured in KSOM until the 2-cell stage.
Embryo transfer
Two-cell stage embryos were transferred to the oviducts of pseudopregnant (E0.5) ICR females. The pups were recovered by cesarean section on the day of delivery (E19.5) and nursed by lactating ICR females.
Genotyping PCR
Genomic DNA was purified from tails of all YKO mice using a Wizard Genomic DNA Purification Kit. The purified DNA was used as a template for genotyping PCR. The primers used for genotyping including SX primer51 for sex chromosome composition were listed in Supplementary Table 9. The PCR products were analyzed by Sanger sequencing as shown in Supplementary Table 7.
Phenotyping analysis
Comprehensive phenotyping analyses were conducted for both WT female/male and YKO male mice, based on an IMPC-compliant pipeline concept whereby a mouse is characterized by a series of standardized tests underpinned by standard operating procedures (SOPs: https://www.mousephenotype.org/impress/PipelineInfo). The pipeline is composed of 14 phenotyping tests, consisting of 398 measured parameters (223 quantitative and 175 qualitative parameters; Supplementary Data 1), covering 20 biological systems such as behavior, morphology, metabolism, and immunity (Table 1; Fig. 1A). Cohorts of seven mice of each sex per pipeline were basically applied to the 14 tests from 9 to 19 weeks (Supplementary Tables 2 and Fig. 1A). Finally, we used 86 mice (eight WT mice in each sex, seven males in each of 10 YKO lines) for the analysis (Supplementary Table 1).
Metabolome analysis
Blood was collected from mice at 19 weeks of age. After collection, the blood was incubated at 25 °C for 30 min and then kept at 4 °C for approximately 16 h to allow complete clot formation. The samples were subsequently centrifuged at 1,200×g for 20 min at 4 °C, taking care not to disturb the precipitated clot. The clear supernatant (serum) was gently transferred to a new tube.
For metabolomic analysis, 100 µL of mouse serum was mixed with 300 µL of 1% formic acid–acetonitrile containing internal standards at a final concentration of 4 µM, and the mixture was vortexed. After centrifugation at 2,300×g for 5 min at 4 °C, the supernatant was collected and phospholipids were removed by solid-phase extraction. The resulting filtrate was dried and reconstituted in 100 µL of 50% isopropanol in water (v/v) for LC–TOFMS analysis. In total, 36 serum samples were analyzed by LC–TOFMS in both positive and negative ion modes. All metabolomic measurements were performed by Human Metabolome Technologies (Tsuruoka, Japan).
Statistical analysis
All data processing and analysis in this study were carried out using R programming language52. The SDM traits were extracted by the statistical significance test on each of the 398 measured parameters (traits) between WT males and WT females (Welch’s t-test for quantitative data (223), Fisher’s exact test for qualitative data (175), uncorrected p < 0.05 in both tests) (Fig. 1B). Four of the resulting 49 SDM traits were categorical variables; one-hot encoding was performed to convert these into quantitative inputs for analysis. Because of this encoding, the 49 SDM traits were represented as 53 variables in downstream analyses. Although one-hot encoding itself does not substantially distort variance after z-score transformation, several SDM traits originated from analysis groups with very small sample sizes (typically n = 3–4 per sex), making these traits particularly sensitive to subsequent missing-value imputation. Enrichment analyses were conducted to extract functional features (biological systems) of two SDM subgroups (22 and 31 SDM traits with high expression in females or males, respectively) (Fig. 1C). Because we only wished to detect significant positive enrichment, p-values were calculated only for fold-enrichment ≥1 (one-tailed Fisher’s exact test). The variation of 53 SDM traits in the 10 YKO lines we generated was visualized on a two-dimensional plot named SDM-IM, where the resulting p-values (− log10(p-value)) of the statistical significance test between YKO males and WT males/females were plotted on the x- and y-axes, respectively (Fig. 3). Missing values were complemented for the following PCA analysis using the R package missForest. To effectively discriminate sex differences at the individual level, PCA analysis was performed for 53 SDM trait data from WT male and female individuals using the R package prcomp (Fig. 4A). The resulting PC1 factor loadings for each of the 53 SDM traits were used as weights for quantitative representation of the degree of female/male sexuality in each of the WT and YKO mice. Note that because missing value complementation for measured parameters with small sample sizes (e.g., Immunophenotyping [IMM]) leads to overestimation of the p-values (− log10(p-value)) before complementation (Fig. 4B), the factor loading in each of the 53 SDM traits was adjusted by MM estimation53 for robust nonlinear regression using the R package robustbase (Fig. 4C). For the one-dimensional cumulative SDM visualization, z-scores of the 53 SDM parameters in all 86 individuals examined were multiplied by the corresponding corrected PC1 factor loadings, and the resulting dataset was used for that quantification (Fig. 5A). Using the weighted dataset, comparison tests (Dunnett’s test) of the cumulative SDM with the WT male group (B6J_M) for each analysis group were conducted for both whole (Fig. 5B) and biological system levels (Fig. 5C,D). By contrast, for the two-dimensional SDM visualization, the z-scores noted above were multiplied by the corresponding |corrected PC1 factor loadings|, and the resulting dataset was used to represent more precisely the relationships between individuals based on the degree of female/male sexuality (Supplementary Fig. 3). For the two-dimensional visualization, the MST and PHATE algorithms were used to elucidate the relationships between individuals as a continuous progression (trajectory) (Fig. 6). For graphs using the MST and PHATE algorithms, cytotree and phate packages were used, respectively.
Data availability
All data supporting the findings of this study are available within the article and in the Supplementary Information files. All source data and R codes for analysis are available as a Zenodo repository at https://doi.org/10.5281/zenodo.13347177.
References
Frayer, D. W. & Wolpoff, M. H. Sexual dimorphism. Annu. Rev. Anthropol. 14, 429–473 (1985).
Williams, T. M. & Carroll, S. B. Genetic and molecular insights into the development and evolution of sexual dimorphism. Nat. Rev. Genet. 10, 797–804 (2009).
Barrett, S. C. H. & Hough, J. Sexual dimorphism in flowering plants. J. Exp. Bot. 64, 67–82 (2013).
Sampathkumar, N. K. et al. Widespread sex dimorphism in aging and age-related diseases. Hum. Genet. 139, 333–356 (2020).
Chi, L. et al. Sexual dimorphism in skin immunity is mediated by an androgen-ILC2-dendritic cell axis. Science. 384, eadk6200 (2024).
Goossens, G. H., Jocken, J. W. E. & Blaak, E. E. Sexual dimorphism in cardiometabolic health: the role of adipose tissue, muscle and liver. Nat. Rev. Endocrinol. 17, 47–66 (2021).
Anderson, S. T. & FitzGerald, G. A. Sexual dimorphism in body clocks. Science 369, 1164–1165 (2020).
Viveiros, A. et al. Sex differences in COVID-19: candidate pathways, genetics of ACE2, and sex hormones. Am. J. Physiol. Circ. Physiol. 320, H296–H304 (2020).
Wells, J. C. K. Sexual dimorphism of body composition. Best Pract. Res. Clin. Endocrinol. Metab. 21, 415–430 (2007).
Arnold, A. P., Chen, X., Link, J. C., Itoh, Y. & Reue, K. Cell-autonomous sex determination outside of the gonad. Dev. Dyn. 242, 371–379 (2013).
Arnold, A. P. A general theory of sexual differentiation. J. Neurosci. Res. 95, 291–300 (2017).
Miyawaki, S. et al. The mouse Sry locus harbors a cryptic exon that is essential for male sex determination. Science 370, 121–124 (2020).
Koopman, P., Gubbay, J., Vivian, N. & Goodfellow, P. Lovell-Badge, R. Male development of chromosomally female mice Transgenic for Sry. Nature 351, 117–121 (1991).
Dewing, P. et al. Direct regulation of adult brain function by the male-specific factor SRY. Curr. Biol. 16, 415–420 (2006).
Czech, D. P. et al. The human testis-determining factor SRY localizes in midbrain dopamine neurons and regulates multiple components of catecholamine synthesis and metabolism. J. Neurochem. 122, 260–271 (2012).
Soh, Y. Q. S. et al. Sequencing the mouse Y chromosome reveals convergent gene acquisition and amplification on both sex chromosomes. Cell 159, 800–813 (2014).
Yano, Y., Chiba, T. & Asahara, H. Analysis of the mouse Y chromosome by single-molecule sequencing with Y chromosome enrichment. Front. Genet. 11, 406 (2020).
Kobayashi, S. et al. Comparison of gene expression in male and female mouse blastocysts revealed imprinting of the X-linked gene, Rhox5/Pem, at preimplantation stages. Curr. Biol. 16, 166–172 (2006).
Arnold, A. P. Rethinking sex determination of non-gonadal tissues. Curr. Top. Dev. Biol. 134, 289–315 (2019).
Brown, S. D. M. & Moore, M. W. The international mouse phenotyping consortium: past and future perspectives on mouse phenotyping. Mamm. Genome. 23, 632–640 (2012).
Rozman, J. et al. Identification of genetic elements in metabolism by high-throughput mouse phenotyping. Nat. Commun. 9, 288 (2018).
Karp, N. A. et al. Prevalence of sexual dimorphism in mammalian phenotypic traits. Nat. Commun. 8, 15475 (2017).
Hagihara, H. et al. Large-scale animal model study uncovers altered brain pH and lactate levels as a transdiagnostic endophenotype of neuropsychiatric disorders involving cognitive impairment. Elife 12, (2024).
Brown, S. D. M. & Moore, M. W. Towards an encyclopaedia of mammalian gene function: the international mouse phenotyping consortium. Dis. Model. Mech. 5, 289–292 (2012).
Smith, C. L. & Eppig, J. T. The mammalian phenotype ontology as a unifying standard for experimental and high-throughput phenotyping data. Mamm. Genome. 23, 653–668 (2012).
Levasseur, A., Dumontet, T. & Martinez, A. Sexual dimorphism in adrenal gland development and tumorigenesis. Curr. Opin. Endocr. Metab. Res. 8, 60–65 (2019).
Bielohuby, M. et al. Growth analysis of the mouse adrenal gland from weaning to adulthood: time- and gender-dependent alterations of cell size and number in the cortical compartment. Am. J. Physiol. Endocrinol. Metab. 293, E139–E146 (2007).
MacMaster, F. P. et al. Development and sexual dimorphism of the pituitary gland. Life Sci. 80, 940–944 (2007).
Ran, F. A. et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154, 1380–1389 (2013).
Wang, H. et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/cas-mediated genome engineering. Cell 153, 910–918 (2013).
Kato, T. et al. Production of Sry knockout mouse using TALEN via oocyte injection. Sci. Rep. 3, 3136 (2013).
Matsubara, Y. et al. TALEN-mediated gene disruption on Y chromosome reveals critical role of EIF2S3Y in mouse spermatogenesis. Stem Cells Dev. 24, 1164–1170 (2015).
Nakasuji, T. et al. Complementary critical functions of Zfy1 and Zfy2 in mouse spermatogenesis and reproduction. PLoS Genet. 13, e1006578 (2017).
Sunagawa, G. A. et al. Mammalian reverse genetics without crossing reveals Nr3a as a Short-Sleeper gene. Cell. Rep. 14, 662–677 (2016).
Matoba, S. et al. Paternal knockout of Slc38a4/SNAT4 causes placental hypoplasia associated with intrauterine growth restriction in mice. Proc. Natl. Acad. Sci. U S A. 116, 21047–21053 (2019).
Akter, M. S. et al. CRISPR/Cas9-based genetic screen of SCNT-reprogramming resistant genes identifies critical genes for male germ cell development in mice. Sci. Rep. 11, 15438 (2021).
Yohai, V. J. High breakdown-point and high efficiency robust estimates for regression. Ann Stat 15, (1987).
Prim, R. C. Shortest connection networks and some generalizations. Bell Syst. Tech. J. 36, 1389–1401 (1957).
Moon, K. R. et al. Visualizing structure and transitions in high-dimensional biological data. Nat. Biotechnol. 37, 1482–1492 (2019).
Wang, H. et al. TALEN-mediated editing of the mouse Y chromosome. Nat. Biotechnol. 31, 530–532 (2013).
Shpargel, K. B., Sengoku, T., Yokoyama, S. & Magnuson, T. UTX and UTY demonstrate histone demethylase-independent function in mouse embryonic development. PLoS Genet. 8, e1002964 (2012).
Cunningham, C. M. et al. Y-Chromosome gene, Uty, protects against pulmonary hypertension by reducing Proinflammatory chemokines. Am. J. Respir Crit. Care Med. 206, 186–196 (2022).
Xu, J., Burgoyne, P. S. & Arnold, A. P. Sex differences in sex chromosome gene expression in mouse brain. Hum. Mol. Genet. 11, 1409–1419 (2002).
Lizio, M. et al. Update of the FANTOM web resource: Expansion to provide additional transcriptome atlases. Nucleic Acids Res. 47, D752–D758 (2019).
Fagerberg, L. et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteom. MCP. 13, 397–406 (2014).
Sandkam, B. A. et al. Extreme Y chromosome polymorphism corresponds to five male reproductive morphs of a freshwater fish. Nat. Ecol. Evol. 5, 939–948 (2021).
Singh, P., Taborsky, M., Peichel, C. L. & Sturmbauer, C. Genomic basis of Y-linked dwarfism in cichlids pursuing alternative reproductive tactics. Mol. Ecol. 32, 1592–1607 (2023).
Kaufmann, P., Wolak, M. E., Husby, A. & Immonen, E. Rapid evolution of sexual size dimorphism facilitated by Y-linked genetic variance. Nat. Ecol. Evol. 5, 1394–1402 (2021).
Ellis, J. A., Stebbing, M. & Harrap, S. B. Significant population variation in adult male height associated with the Y chromosome and the aromatase gene. J. Clin. Endocrinol. Metab. 86, 4147–4150 (2001).
Jobling, M. A. & Tyler-Smith, C. Human Y-chromosome variation in the genome-sequencing era. Nat. Rev. Genet. 18, 485–497 (2017).
McFarlane, L., Truong, V., Palmer, J. S. & Wilhelm, D. Novel PCR assay for determining the genetic sex of mice. Sex. Dev. 7, 207–211 (2013).
R Core Team. R: A language and environment for statistical computing. at (2015). https://www.r-project.org/
Alsalem, A. K. & Altaher, A. M. A comparative study of some robust nonlinear regression methods. J Pure Appl. Sci 18, (2019).
Acknowledgements
We thank Toshiko Tomishima, Yukiko Dodo, Ayano Tsukahara, Tomoko Kushida, Kyoko Ikeda, Akiko Shinogi, Mao Ozaki, Jin Shundan, and Tomona Oikawa for technical assistance.
Funding
This study was supported by JSPS KAKENHI Grant Numbers JP21K05993 (NT), JP18H04891 (SM), JP20H04934 (SM), and JP20H03159 (SM).
Author information
Authors and Affiliations
Contributions
SM conceived the project. SM and KM generated control and CRISPR founder mice. SM, KM, AiO, YK, and MT performed phenotypic analyses. MT supervised AiO and YK. NT performed statistical analysis of the obtained datasets. NT and SM wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tanaka, N., Miura, K., Ozaki, A. et al. Quantitative dissection of sexual dimorphism in mice through Y-linked gene knockouts and multivariate phenotyping. Sci Rep 16, 2190 (2026). https://doi.org/10.1038/s41598-025-33814-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-33814-w
