
Abstract
Asthma, allergic rhinitis, and pollinosis are prevalent respiratory conditions that often co-occur, suggesting common genetic and environmental causes. While significant progress has been made in identifying genetic loci in European populations, the genetic architecture in East Asian populations remains poorly understood. Using GWAS summary statistics from BioBank Japan, we performed multi-trait genome-wide association studies (MTAG) to quantify the genetic overlap among asthma, allergic rhinitis, and pollinosis in East Asians. Genetic correlation analysis revealed significant positive correlations among the three conditions, and stratified LDSC (Linkage Disequilibrium Score Regression) analysis identified heritability enrichments in Blood/Immune and Digestive tissues. We discovered novel pleiotropic loci at 9q32 and 10q25.2 specific to East Asians, with candidate gene expression analysis highlighting differential expression of AKNA, ATP6V1G1, and GPAM. These findings provide new insights into the shared genetic architecture and biological mechanisms underlying these conditions, advancing our understanding of their genetic determinants and identifying potential therapeutic targets.
Similar content being viewed by others

Introduction
Asthma, allergic rhinitis, and pollinosis are widespread respiratory conditions that pose a substantial public health challenge by significantly impacting the quality of life and placing a heavy burden on healthcare systems worldwide1. These conditions often occur together, indicating that they may share common genetic and environmental causes2. Although significant progress has been made in identifying the genetic bases of these diseases, particularly through GWAS in European populations that have uncovered hundreds of associated genetic loci, the genetic architecture in non-European populations remains poorly understood3,4. This highlights a critical need for more research to characterize the genetic determinants of these conditions in non-European populations and to understand the shared genetic factors contributing to their prevalences.
Recent advancements in genome-wide association studies (GWAS) have facilitated the identification of numerous genetic loci implicated in complex traits and diseases. Traditional single-trait GWAS approaches, while valuable, may lack the statistical power necessary to detect loci that are common to related traits. This limitation arises due to the analysis being restricted to individual traits, potentially overlooking loci with pleiotropic effects.
To address these limitations, several advanced cross-trait analytical methods have been developed. These methodologies allow for the joint analysis of multiple related traits, thereby enhancing the detection of shared genetic variants and novel loci. Among these approaches, Multi-Trait Analysis of GWAS (MTAG) stands out as a powerful tool5. MTAG leverages the correlations between related traits to boost statistical power, thereby uncovering shared genetic underpinnings that contribute to multiple conditions. This approach not only facilitates the discovery of common genetic variants but also enhances our understanding of the biological pathways involved in these interrelated diseases.
In this study, we aim to utilize MTAG to investigate the shared genetic architecture of asthma, allergic rhinitis, and pollinosis in East Asian populations. Our goal is to identify common genetic variants that contribute to the susceptibility of these interrelated respiratory conditions. Through this comprehensive analysis, we seek to advance our understanding of the genetic determinants of these diseases and pave the way for the development of more effective prevention and treatment strategies.
Methods
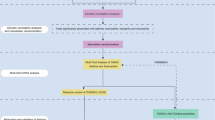
The analytical workflow, summarizing the key steps from data preparation to genetic correlation analyses, MTAG, and gene expression validation, is presented in Fig. 1.

Overview of Analytical Workflow. This figure summarizes the analytical workflow for identifying shared genetic loci for asthma, allergic rhinitis, and pollinosis in East Asians. It includes data preparation, genetic correlation analysis, MTAG to detect pleiotropic loci, and differential gene expression analysis of key loci (e.g., 9q32 and 10q25.2) to uncover underlying biological mechanisms.
GWAS data
GWAS summary statistics of asthma, allergic rhinitis, and pollinosis from East Asians were downloaded from GWAS Catalog6, information of all included GWAS data from East Asians is summarized in Table S1.
Global genetic correlation analysis
Genetic correlations rgamong asthma, allergic rhinitis, and pollinosis was estimated by LD (Linkage Disequilibrium) score regression (LDSC v 1.0.0) using GWAS summary statistic7. Quality control steps included filtering SNPs with INFO score < 0.9 and minor allele frequency (MAF) < 1%. The LDSC method is described by the following equation: \(E\left[{\beta }_{j}{\gamma }_{j}\right]=\frac{\sqrt{{N}_{1}{N}_{2}}{r}_{g}}{M}{l}_{j}+\frac{{N}_{s}r}{\sqrt{{N}_{1}{N}_{2}}}\) , where \({\beta }_{j}\) and \({\gamma }_{j}\) denote the effect size of SNP j on the two tested traits, \({N}_{1}\) and \({N}_{2}\) are the sample sizes of two tested traits, \({N}_{s}\) is the number of overlapping samples between two tested traits, r is the phenotypic correlation in overlapping samples and \({l}_{j}\) is the LD score. Pre-computed linkage disequilibrium scores for HapMap3 SNPs calculated based on East-Asian-ancestry data from the 1000 Genomes Project were used in the analysis.
Cell-type-specific enrichment of SNP heritability
Stratified LDSC (s-LDSC) was used to identify functional categories or cell types disproportionately contributing to trait heritability8. s-LDSC divides the genome into non-overlapping sets of regions based on functional annotations, then estimates heritability for each subset. The observed heritability in each stratum is compared to the expected heritability based on SNP proportion, assessing enrichment within specific genomic annotations. Significant enrichment indicates that variants in those regions play a key role in influencing the trait, offering insights into biological mechanisms and prioritizing regions for further study. In this study, annotation data from the Roadmap project for six chromatin marks (DHS, H3K27ac, H3K36me3, H3K4me1, H3K4me3, H3K9ac) across 88 cell types or tissues were used to partition SNP heritability. These annotations were further classified into nine broad groups: adipose, central nervous system, digestive system, cardiovascular, musculoskeletal and connective tissue, immune and blood, liver, pancreas, and other. Annotation-specific enrichment values for each trait were visualized using hierarchical clustering with a color scale.
Local genetic correlation analysis
Given that the genetic correlation estimated by LDSC aggregates information across all variants in the genome, we proceeded to estimate the pairwise local genetic correlation using ρ-HESS (heritability estimation from summary statistics, v 1.0.1)9. The analysis was performed as follows:
Step 1: Segmentation of the Genome.
The genome was divided into 1,703 pre-specified LD-independent segments (average 1.6 Mb each) based on the 1000 Genomes East Asian reference panel to minimize LD confounding and reduce spurious correlations from long-range LD. This segmentation balances resolution and statistical power by ensuring sufficient SNP density for reliable local genetic correlation estimates while avoiding inflation from residual LD. Following the standard ρ-HESS approach established by Shi et al.9, this method maintains comparability with previous studies.
Step 2: Estimation of Local Genetic Correlation.
We used the ρ-HESS software to estimate the local genetic correlation for each pair of traits (e.g., asthma and allergic rhinitis, asthma and pollinosis) within these segments, quantifying the shared genetic architecture between traits.
Step 3: Statistical Significance.
Statistical significance was determined by applying a Bonferroni correction for multiple testing (P < 0.05/1703), following the standard approach established by Shi et al.9. While Bonferroni correction is conservative and may increase the likelihood of false negatives, it provides robust control against false positives, maintaining the reliability of our findings.
Step 4: Visualization and Interpretation.
Significant results were visualized using custom R scripts to generate heatmaps and regional plots, facilitating interpretation of genomic regions contributing to the shared genetic basis of asthma, allergic rhinitis, and pollinosis.
Multi-trait GWAS analysis
The multi-trait analysis of GWAS (MTAG) framework, a generalized meta-analysis method that outputs trait-specific SNP associations, was employed in this study5. MTAG enhances the power to detect loci from correlated traits by jointly analyzing GWAS summary statistics. It effectively addresses sample overlap and incomplete genetic correlation, offering advantages over conventional inverse-variance weighted meta-analysis. The analysis was performed as follows:
Step 1: Variant Filtering.
MTAG begins by filtering variants, excluding non-common SNPs, duplicated SNPs, and SNPs with strand ambiguity to ensure data quality.
Step 2: Genetic Correlation.
Estimation Pairwise genetic correlations between traits were estimated using LDSC10. These estimates were then used to calibrate the variance–covariance matrix of the random effect component, accounting for genetic correlation between traits.
Step 3: Random-Effect Meta-Analysis.
MTAG conducts a random-effect meta-analysis to generate SNP-level summary statistics, leveraging the shared genetic architecture of traits to improve detection power.
Step 4: Prioritization of Pleiotropic SNPs.
In this study, we prioritized pleiotropic SNPs that achieved genome-wide significance (P < 5 × 10–8) in the multi-trait analysis and suggestive significance (P < 0.01) in the original single-trait GWAS. This dual-threshold approach allowed us to identify and highlight SNPs with potential pleiotropic effects across asthma, allergic rhinitis, and pollinosis.
Results
Global genetic correlations among asthma, allergic rhinitis, and pollinosis
To investigate the shared genetic mechanisms among asthma, allergic rhinitis, and pollinosis in East Asian populations, we first estimated the heritability of these conditions using GWAS summary statistics from the BioBank Japan (BBJ) project11. SNP-based heritability estimates were calculated using LDSC to assess the proportion of phenotypic variance explained by the tested variants. Heritability estimates on the observed scale using GWAS summary statistics of East Asians were 3.01%, 1.14%, and 2.18% for asthma, allergic rhinitis, and pollinosis, respectively (Table S2).
Subsequently, we estimated the genetic correlation between pairs of these conditions using LDSC7. Consistent with epidemiological findings, we observed a significant and strong positive genetic correlation between asthma and allergic rhinitis in East Asian populations (rg = 0.52, P = 2 × 10–4, Table 1). In contrast, the genetic correlation between asthma and pollinosis was lower but still highly significant (rg = 0.29, P = 5 × 10–4, Table 1). Additionally, the genetic correlation between allergic rhinitis and pollinosis was found to be 1, indicating an almost complete genetic overlap between these two closely related conditions. We conducted sensitivity analyses to assess the robustness of our findings by performing LDSC excluding variants in the major histocompatibility complex (MHC) region. The results remained highly consistent with those obtained when including MHC variants, indicating that the genetic correlations observed were not driven by MHC effects.
Cell-type-specific enrichment of SNP heritability
To further investigate the genetic similarities and differences among asthma, allergic rhinitis, and pollinosis, we partitioned SNP heritability based on six chromatin marks and nine cell types. Overall, the three diseases displayed significant heritability enrichments in tissues or cell types classified as Blood/Immune and Digestive (Figure S1). However, when examining specific chromatin marks, asthma, allergic rhinitis, and pollinosis each showed unique enrichment patterns within distinct tissue classifications. Notably, compared to asthma and pollinosis, allergic rhinitis demonstrated a smaller number of significantly enriched tissues or cell types (Figure S1). These findings highlight both the shared and unique genetic components of asthma, allergic rhinitis, and pollinosis, providing insights into their complex etiologies.
Local genetic correlations among asthma, allergic rhinitis and pollinosis
We next conducted a comprehensive genome-wide scan to identify specific genomic regions that contribute to the shared heritability of asthma, allergic rhinitis, and pollinosis. Our analysis revealed significant local genetic correlations among these conditions (Figure S2 and Table S3). Specifically, we observed that the regions 2q37.3, 10p14, 12q24.1, and the major histocompatibility complex (MHC) region exhibit significant genetic correlations between asthma and allergic rhinitis. Furthermore, the regions 10p14 and 17q21 were also found to be correlated between asthma and pollinosis. Additionally, the regions 3p21 and the MHC region showed significant correlations between allergic rhinitis and pollinosis. These findings suggest that these genomic regions play a critical role in the shared genetic basis of these allergic conditions.
Multi-trait analysis among asthma, allergic rhinitis and pollinosis
Building on the insights from our genetic correlation analysis, we conducted a more detailed investigation to identify shared genetic loci. We performed a MTAG analysis for each pairwise combination of asthma, allergic rhinitis, and pollinosis. MTAG capacity to enhance the statistical power of GWAS by incorporating information from effect estimates across genetically correlated traits, and generate trait-specific associations of each SNP. To assess this increased detection power, we compared the average χ2 test statistic for asthma, allergic rhinitis, and pollinosis derived from the multi-trait GWAS with those from the initial GWAS conducted in East Asian populations. In the MTAG analysis between asthma and allergic rhinitis, we observed varying degrees of increase in the effective sample size for these conditions. For the single trait GWAS, the mean χ2 statistics for asthma and allergic rhinitis were 1.129 and 1.031, respectively. In contrast, the mean χ2 statistics for the MTAG analysis were 1.129 for asthma and 1.036 for allergic rhinitis. Consequently, the effective sample size for allergic rhinitis increased from 161,563 to 189,167, while for asthma, it remained almost unchanged, increasing slightly from 175,948 to 175,962. To evaluate potential inflation, we assessed QQ plots (Figure S3) and genomic inflation factor (λGC). The λGC values for MTAG-derived test statistics remained within acceptable ranges, indicating no substantial systematic inflation. Correspondingly, we identified six pleiotropic loci for allergic rhinitis, none of which had been previously reported in allergic rhinitis studies in Asian populations. Five of these loci had been reported in asthma studies, but one locus had not been discovered in either allergic rhinitis or asthma studies (Table 2). For asthma, we also discovered six pleiotropic loci, but all of them were known from previous studies. Similarly, in the MTAG analysis between asthma and pollinosis, the mean χ2 statistics for asthma remained virtually unchanged at 1.129, with the effective sample size increasing slightly from 175,948 to 176,292. For pollinosis, the mean χ2 statistics showed a marginal increase from 1.071 to 1.072, with the effective sample size increasing from 161,563 to 163,767. In pollinosis, we identified three pleiotropic loci, one of which is novel but has been previously reported in asthma. In asthma, we discovered four shared loci, including one novel locus that has not been reported in either pollinosis or asthma studies (Table 3). In the MTAG analysis between allergic rhinitis and pollinosis, the mean χ2 statistics for pollinosis showed little change, moving from 1.07 to 1.071, with the effective sample size increasing from 161,563 to 163,767. For allergic rhinitis, the mean χ2 statistics increased from 1.03 to 1.046, with the effective sample size rising significantly from 161,563 to 248,624. We identified two pleiotropic loci between allergic rhinitis and pollinosis, which had not been reported in allergic rhinitis but were already known in pollinosis (Table 4).
Based on the above results, we noticed two loci that had never been reported in Asian populations for asthma, allergic rhinitis, and pollinosis, and selected them for further investigation. In the MTAG analysis of asthma and pollinosis, we discovered a signal specific to asthma (9q32, rs57656777) located around the TNFSF15 gene (Fig. 2A). We then examined the association of the top SNP rs57656777 in the GWAS of asthma in Europeans. Our results indicated that rs57656777 was not significant (P= 0.68), suggesting that this signal is specific to Asian populations (Figure S4). However, further investigation of this region revealed another signal in Europeans (Figure S4)4, implying that this region may contain potential risk genes involved in the development of asthma across different populations. In the MTAG analysis of asthma and allergic rhinitis, we discovered a signal specific to allergic rhinitis at 10q25.2, covering multiple genes (Fig. 2B). According to the gnomAD database, the lead SNP rs139174066 is an Asian-specific variant with a frequency of 3% in Asians but is absent in Europeans. Correspondingly, rs139174066 is also missing in European allergic rhinitis GWAS12, indicating its specificity to Asian populations. Additionally, when we examined this region in European AR GWAS, we did not find any significant signals (Figure S5), further confirming the Asian-specific nature of this association.

Regional plots for novel pleiotropic loci at 9q32 and 10q25.2. This figure shows regional plots (LocusZoom plots) for two newly identified genetic loci at 9q32 and 10q25.2, which are associated with multiple conditions and are specific to East Asian populations. Panel A shows the genetic associations at the 9q32 locus. The most significant genetic variant, rs57656777, is marked with a purple diamond. The y-axis indicates the strength of the genetic associations (measured by the -log10 of the p-value, which is a statistical measure of significance), while the x-axis shows the location of this variant along chromosome 9 (in megabases, Mb). The blue line represents the recombination rate, which shows how often nearby genetic variants are inherited together. Panel B shows the genetic associations at the 10q25.2 locus. The top variant here, rs139174066, is also marked with a purple diamond. As in Panel A, the y-axis represents the strength of the associations, and the x-axis shows the position on chromosome 10. The blue line again indicates the recombination rate in this region. The circles represent other genetic variants in this area, with the color of the circles showing how strongly each variant is linked to the main variant (lead SNP) – stronger links are indicated by darker colors.
To further explore the potential regulatory mechanisms underlying these associations, we examined GTEx eQTL data but found that neither rs57656777 at 9q32 nor rs139174066 at 10q25.2 were identified as eQTLs. Given that the majority of individuals in GTEx are of European ancestry, with limited representation of African and Latino populations and almost no East Asian data, it is possible that population differences in gene regulation contribute to the lack of eQTL signals in this dataset. To address this limitation, we referenced a recently published mQTL study in a Chinese population and found that both rs57656777 and rs139174066 act as cis-mQTLs, suggesting potential epigenetic regulatory effects in East Asians (Table S4)13. These findings provide further evidence that these loci may influence asthma and allergic disease susceptibility through DNA methylation. The identification of these loci as cis-mQTLs suggests that they may have regulatory functions, potentially affecting gene expression or chromatin accessibility in a population-specific manner.
Gene expression analysis of candidate genes at 9q32 and 10q25.2
To further investigate the potential asthma risk genes located in the 9q32 and 10q25.2 regions, we analyzed the gene expression data from individuals with asthma and allergic rhinitis, from a previous study (GSE141661)14. At the 9q32 locus, we identified significant differential expression of several genes in individuals with asthma. Specifically, AKNA, ATP6V1G1, and C9orf91 exhibited notable differences in expression levels in the blood samples of asthma patients compared to controls. The expression levels for these genes were analyzed using the Wilcoxon test, and the results are visualized in Fig. 3. AKNA showed a marked increase in expression in asthma patients (P = 3.9 × 10–5), highlighting its potential role in immune response regulation. ATP6V1G1, involved in proton transport, also demonstrated significant upregulation (P = 0.00035), suggesting its impact on cellular homeostasis and immune cell function. C9orf91, although less characterized, showed significant differential expression (P = 0.0031), indicating a possible novel role in asthma pathophysiology. In the 10q25.2 region, our analysis revealed significant differential expression of GPAM in individuals with allergic rhinitis. The expression levels for GPAM were higher in allergic rhinitis patients compared to controls (P = 0.0019), as shown in Fig. 4. Additionally, TECTB also exhibited a significant difference in expression levels (P = 0.043), pointing to their potential involvement in the genetic susceptibility and molecular mechanisms of allergic rhinitis.

Differential gene expression levels of candidate genes at the 9q32 locus in asthma patients compared to controls. This figure shows the expression levels of candidate genes located at the 9q32 locus, including LOC100505478, TNC, TNFSF15, TNFSF8, AKNA, ATP6V1G1, C9orf91, DFNB31, in asthma patients and healthy controls. Each panel displays the expression data for one gene. The X-axis represents the gene expression levels, and the Y-axis divides the samples into two groups: asthma patients and controls. The dots represent the data points for individual samples, while the lines show the average (mean) expression levels for each group. This allows for comparison of gene expression between asthma patients and controls, highlighting differences that may be associated with the condition.

Differential gene expression levels of candidate genes at the 10q25.2 locus in allergic rhinitis patients compared to controls. This figure illustrates the expression levels of candidate genes located at the 10q25.2 locus (ACSL5, GPAM, GUCY2GP, TECTB, VTI1A, ZDHHC6) in individuals with allergic rhinitis compared to healthy controls. Each panel focuses on a single gene, with the X-axis showing the expression levels and the Y-axis categorizing the samples into two groups: allergic rhinitis patients and controls. The dots represent individual data points from each sample, while the lines indicate the mean expression levels for each group. This visualization highlights differences in gene expression that may be associated with allergic rhinitis.
Discussion
This study aimed to elucidate the shared genetic architecture of asthma, allergic rhinitis, and pollinosis in East Asian populations by leveraging advanced multi-trait analysis methods. Our findings provide valuable insights into the genetic determinants of these interrelated respiratory conditions, highlighting both shared and unique genetic components and contributing to a better understanding of potential treatment strategies.
Our genetic correlation analysis revealed significant positive genetic correlations among asthma, allergic rhinitis, and pollinosis, aligning with epidemiological observations and underscoring the interconnectedness of these respiratory diseases. These findings suggest that common genetic variants contribute to the susceptibility of these conditions, supporting the hypothesis of shared etiological pathways. Moreover, our stratified LDSC analysis identified significant heritability enrichments in specific cell types and tissues, particularly those classified as Blood/Immune and Digestive. This indicates that genetic variants within these regions play a disproportionate role in influencing the traits of interest. The distinct enrichment patterns observed for asthma, allergic rhinitis, and pollinosis within different tissue classifications highlight the complex genetic underpinnings of these diseases. The significant enrichments observed in Blood/Immune tissues align with the known roles of immune dysregulation in these conditions, providing further evidence for the involvement of immune-related pathways in their pathogenesis.
We subsequently conducted a detailed genome-wide exploration to identify specific genomic regions responsible for the genetic associations observed. The identification of specific genomic regions with significant local genetic correlations among asthma, allergic rhinitis, and pollinosis provides crucial insights into the shared genetic basis of these conditions. Regions such as 2q37.3, 10p14, 12q24.1, and the MHC region exhibited significant genetic correlations between asthma and allergic rhinitis, while regions like 10p14 and 17q21 were significant for both asthma and pollinosis. Additionally, the MHC region and 3p21 showed significant correlations between allergic rhinitis and pollinosis. These findings suggest that these genomic regions harbor genetic variants that contribute to the co-occurrence of these conditions, highlighting potential targets for further investigation.
To further pinpoint pleiotropic genes and variants within the genome, we utilized MTAG, which enhanced the detection power of GWAS by incorporating information from multiple related traits. This approach allowed us to identify pleiotropic loci that were missed in traditional single-trait analyses. Our MTAG analysis identified several novel loci linked to asthma, allergic rhinitis, and pollinosis, highlighting the effectiveness of this method in uncovering shared genetic factors that may contribute to the co-occurrence of these conditions. The identification of pleiotropic SNPs that were significant in multi-trait but not single-trait GWAS emphasizes the value of exploring genetic correlations, as it reveals common genetic pathways that influence multiple traits simultaneously. From a biological perspective, these shared genetic factors suggest potential molecular mechanisms underlying the overlapping features of asthma, allergic rhinitis, and pollinosis, which could guide the development of targeted therapies. Notably, the loci at 9q32 and 10q25.2 were found to be novel pleiotropic regions specific to East Asian populations, indicating that genetic factors influencing these diseases may differ across populations. These findings suggest that further investigation is warranted to explore how these loci contribute to disease susceptibility and to assess their potential as biomarkers for risk prediction or therapeutic targets in East Asian populations. Here, our findings highlight genetic loci associated with asthma in East Asians; however, differences in disease prevalence across populations are likely influenced by polygenic effects rather than single loci alone. A broader polygenic risk score (PRS) approach incorporating multiple variants would be necessary to fully assess genetic contributions to prevalence disparities. Additionally, environmental and lifestyle factors, such as air pollution, diet, early-life microbial exposures, and healthcare access, may interact with genetic predisposition to influence asthma risk. While our study is based on publicly available genetic data and does not include direct environmental measures, future research integrating both genetic and environmental factors across ancestries will be essential to fully elucidate population differences in asthma susceptibility.
The gene expression analysis of candidate genes at 9q32 and 10q25.2 provided further evidence for their involvement in asthma and allergic rhinitis. The significant differential expression of AKNA, a gene known to regulate immune cell development and inflammatory responses, highlights its potential role in modulating airway inflammation in asthma. Similarly, ATP6V1G1 and C9orf91 at the 9q32 locus may contribute to cellular homeostasis and immune regulation, although their precise mechanisms in asthma pathogenesis require further study. At the 10q25.2 locus, the differential expression of GPAM, a key regulator of lipid metabolism, suggests a potential link between metabolic pathways and immune responses in allergic rhinitis. These findings indicate the importance of these genes in immune system regulation and cellular processes, supporting their potential as therapeutic targets. Further research is warranted to explore the pathways of these genes and their roles in disease pathophysiology. For instance, investigating GPAM’s role in lipid signaling could clarify the link between metabolic regulation and immune responses in allergic rhinitis, while examining AKNA’s involvement in T-cell development and inflammatory regulation may shed light on its contribution to airway inflammation in asthma.
This study has several limitations that should be considered when interpreting the results. First, the analysis relied on publicly available GWAS summary statistics, which limited our ability to access raw genotyping data. As a result, we could not assess potential heterogeneity within East Asian subpopulations, such as differences in allele frequencies or environmental exposures, which may influence genetic associations. This limitation highlights the need for future studies with diverse cohorts to better account for population-specific effects. Second, while MTAG significantly enhanced the statistical power to detect shared genetic loci, the marginal increase in effective sample sizes for certain traits suggests that some findings may be influenced by the correlated structure of the traits analyzed rather than independent genetic signals. Further validation in independent cohorts is essential to confirm the robustness of these results. Third, while gene expression data provided supporting evidence for the identified loci, these findings are associative rather than causal. Consequently, experimental studies are needed to validate the biological roles of these genes and clarify their mechanisms.
In conclusion, this study provides an analysis of the genetic correlations between asthma, allergic rhinitis, and pollinosis in East Asian populations. Our findings underscore the potential of multi-trait analysis methods to identify shared genetic loci and enhance our understanding of the genetic architecture of these conditions. These insights may inform future research on the genetic determinants of respiratory diseases in East Asian populations.
Data availability
Data is provided within the manuscript or supplementary information files.
References
Dierick, B. J. H. et al. Burden and socioeconomics of asthma, allergic rhinitis, atopic dermatitis and food allergy. Expert Rev. Pharmacoecon Outcomes Res. 20, 437–453. https://doi.org/10.1080/14737167.2020.1819793 (2020).
Iordache, A. et al. A Review Regarding the Connections between Allergic Rhinitis and Asthma - Epidemiology, Diagnosis and Treatment. Curr. Health Sci. J. 49, 5–18. https://doi.org/10.12865/chsj.49.01.5 (2023).
Chang, X. et al. Genetic architecture of asthma in African American patients. J. Allergy Clin. Immunol. 151, 1132–1136. https://doi.org/10.1016/j.jaci.2022.09.001 (2023).
Tsuo, K. et al. Multi-ancestry meta-analysis of asthma identifies novel associations and highlights the value of increased power and diversity. Cell Genom. 2, 100212. https://doi.org/10.1016/j.xgen.2022.100212 (2022).
Turley, P. et al. Multi-trait analysis of genome-wide association summary statistics using MTAG. Nat. Genet. 50, 229–237. https://doi.org/10.1038/s41588-017-0009-4 (2018).
Buniello, A. et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 47, D1005-d1012. https://doi.org/10.1093/nar/gky1120 (2019).
Bulik-Sullivan, B. K. et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295. https://doi.org/10.1038/ng.3211 (2015).
Finucane, H. K. et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 47, 1228–1235. https://doi.org/10.1038/ng.3404 (2015).
Shi, H., Mancuso, N., Spendlove, S. & Pasaniuc, B. Local Genetic Correlation Gives Insights into the Shared Genetic Architecture of Complex Traits. Am. J. Hum. Genet. 101, 737–751. https://doi.org/10.1016/j.ajhg.2017.09.022 (2017).
Bulik-Sullivan, B. et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet. 47, 1236–1241. https://doi.org/10.1038/ng.3406 (2015).
Sakaue, S. et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat. Genet. 53, 1415–1424. https://doi.org/10.1038/s41588-021-00931-x (2021).
Waage, J. et al. Genome-wide association and HLA fine-mapping studies identify risk loci and genetic pathways underlying allergic rhinitis. Nat. Genet. 50, 1072–1080. https://doi.org/10.1038/s41588-018-0157-1 (2018).
Peng, Q. et al. Analysis of blood methylation quantitative trait loci in East Asians reveals ancestry-specific impacts on complex traits. Nat. Genet. 56, 846–860. https://doi.org/10.1038/s41588-023-01494-9 (2024).
Lemonnier, N. et al. A novel whole blood gene expression signature for asthma, dermatitis, and rhinitis multimorbidity in children and adolescents. Allergy 75, 3248–3260. https://doi.org/10.1111/all.14314 (2020).
Funding
This work is supported by the Special Funds of Taishan Scholar Project, China (tsqn202211224).
Author information
Authors and Affiliations
Contributions
DW, SL and XC made a substantial contribution to the conception or design of the paper, as well as to the acquisition, analysis, or interpretation of the data.QFW and YJ calculated and organized the experimental data and verified the results.All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, D., Liu, S., Wu, Q. et al. Identification of shared genetic loci for asthma, allergic rhinitis, and pollinosis in East Asians. Sci Rep 15, 6068 (2025). https://doi.org/10.1038/s41598-025-90443-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-90443-z
