
Abstract
Aloe vera is a popular medicinal plant in the cosmetic, pharmaceutical, and food industries. Acemannan (ACE), a β-(1,4)-acetylated mannan, is one of the bioactive compounds isolated from the A. vera gel. The pharmacological effects of ACE have been reported regarding digestive disease protection, antimicrobia, and prebiotic activity. Here, we used human HaCaT cells as a model to uncover the potential biological functions of ACE in keratinocytes. ACE increased cell growth in a concentration-dependent manner, and a higher incorporation of BrdU was detected in ACE-treated cells than in vehicle-treated cells, indicating ACE promotes cell proliferation. Furthermore, ACE concentration-dependently promoted cell migration in the wound scratch model. ACE regulated cell differentiation by transiently decreasing p63α expression, but increasing the expression of involucrin, loricrin, and transglutaminase 1 (TGase 1). These effects were non-additive to those induced by phorbol myristate acetate (PMA), but additive to epidermal growth factor (EGF), which are complete and incomplete differentiation agents of keratinocytes, respectively. Moreover, ACE activated EGF receptor (EGFR), protein kinase C (PKC), and protein kinase B (AKT/PKB). PKC inhibitor Ro320432 enhanced cell growth and migration, while EGFR inhibitor osimertinib blocked both responses. In summary, ACE is a potential therapeutic agent in wound healing. ACE activates PKC, leading to keratinocyte differentiation and activates EGFR, contributing to keratinocyte proliferation and migration.
Similar content being viewed by others

Introduction
Aloe species (family Asphodelaceae) have been used as medicinal plants for centuries. Most commercially related products are derived mainly from the A. vera of the Aloe genus. A. vera has been reported to have pharmacological activities including digestive disease protection, antidiabetic, antimicrobial, prebiotic, anticancer, cardioprotection, bone protection, wound healing, and skin protection1,2. These pharmacological activities are mainly due to the rich content of bioactive phytochemicals in A. vera such as aloesin, aloin, emodin, aloe-emodin, and acemannan1. Nevertheless, some phytochemicals of A. vera have also been reported to be cytotoxic. For example, aloin is associated with diarrhea due to increased gastric motility, leading to pathological changes in the mucosa3,4. Another aloe compound emodin has been reported to induce hepatotoxicity in zebrafish by activating nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB)-mediated inflammatory pathway and p53-mediated apoptosis5. In addition, emodin can cause nephrotoxicity6, induce DNA fragmentation that leads to genotoxicity in a mouse model7, and phototoxicity by generating singlet oxygen when cells are irradiated with UV8.
Acemannan (ACE), one of the main bioactive polysaccharides of A. vera, has been reported to be of great value in applications in medical and industrial fields9,10. ACE can prevent pulp inflammation and oral ulceration11,12 and has beneficial antimicrobial and prebiotic activities13. A report on dentistry demonstrates the immunomodulatory ability of ACE14. ACE binds to Toll-like receptor 5 (TLR5) to enhance the binding of NF-κB to DNA, leading to an increase of interleukin-6 (IL-6) and interleukin-8 (IL-8) gene expression in human gingival fibroblasts14. In addition, ACE positively affects cutaneous wound healing in the mouse skin wound model15. A further study in cultured skin primary fibroblasts indicates that ACE promotes the expression of cyclin D1 via activation of protein kinase B (AKT)/mammalian target of rapamycin (mTOR)-eukaryotic translation initiation factor 4 F (eIF4F) signaling pathways15. With this finding, recent studies focus on the feasibility of applying ACE-based films in wound dressings16.
Keratinocytes are the major cell types in the skin and normal proliferation and differentiation of keratinocytes play a crucial role in skin barrier function. Aberration of keratinocytes by excessive proliferation and poor differentiation leads to psoriasis, skin inflammation, and impaired epidermal barrier integrity17,18. In contrast, keratinocyte growth and migration loss lead to impaired wound healing in diabetic patients17,19. It is well-recognized that epidermal growth factor (EGF) is the key growth factor in mediating keratinocyte growth and migration20,21. Although aloe extract can promote wound healing, proliferation, and differentiation of keratinocytes via the upregulation of growth factors such as keratinocyte growth factor-1, transforming growth factor-β, insulin-like growth factor 1, vascular endothelial growth factor, and basic fibroblastic growth factor2,22, and ACE has a high potential for skin wound healing15,16, the direct action of ACE on keratinocytes remains unclear. Therefore, in this study, we used human HaCaT cells as a model to explore the effects of ACE on the growth, migration, and differentiation of HaCaT cells and deciphered the underlying mechanism by which ACE regulates the cell physiology of keratinocytes. HaCaT is a spontaneously transformed immortal keratinocyte from adult human skin, and the preserved features allow it to be widely used in the characterization of human keratinocytes in scientific research23. The information provided in this study would help us pave the way for the therapeutic development of ACE in skin diseases.
Results
Quantification of ACE and O-acetyl content in A. vera polysaccharide (I50)
There are no commercially available A. vera polysaccharide reference materials with known acetyl content. Hence, an industrial sample from Dazzeon Biotech. Co., Ltd of purified aloe polysaccharide (internal ACE standard) isolated from A. vera inner leaf powder and prepared by various gradient ethanol precipitation processes was used to prepare the standard curve. Fig. S1 illustrates the elution profiles of Aloe powder (200X, Fig. S1A), and isolated aloe polysaccharide (I50, Fig. S1B) obtained by high-performance size exclusion chromatography-refractive index detection (HPSEC-RI). Aloe powder exhibited four distinct peaks (heterogeneous polydisperse profile) of major intensities at 13.0 (11.8 to 16.8-broad peak), 17.3, 19.6, and 21.5 min by the RI detector (Fig. S1A). The I50 presents broadband only at the retention of 16.7 min (Fig. S1B) in agreement with that observed in Aloe powder (200X). The ACE content of Aloe powder was estimated to be 87.21% by comparing with the calibration curve (Fig. S2A) using Dazzeon’s internal ACE standard as a reference compound. The O-acetyl content of I50 was quantified by measuring the complex’s absorbance at 540 nm and comparing it with a calibration curve (Fig. S2B) using acetylcholine chloride (C7H16ClNO2) as a reference standard24. The results indicated that the level of O-acetyl content in I50 was 590.43 mg/g (59.04%).
Molecular weight and monosaccharide composition analysis of I50
The average molecular weight of the isolated polysaccharide I50 was determined using HPSEC-RI. Pullulan standards were used for plotting a trendline, and the relative molecular weight of I50 was 37 kDa based on the highest detected peak point inside the trendline range (Fig. S3).
Monosaccharide composition analysis of I50 was examined by high-performance anion-exchange chromatography-pulsed amperometric detection (HPAEC-PAD). Trifluoroacetic acid (TFA)-solubilized I50 and standards were hydrolyzed in duplicate and then analyzed by HPAEC-PAD. The results showed that the main monosaccharide observed for I50 was mannose, with lower amounts of arabinose, glucose, and galactose (Table 1).
Characterization of I50
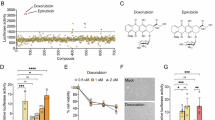
The 13C-NMR spectrum of I50 showed six main signals, one in the anomeric and five in the skeletal regions for mannose moieties, at δC 100.1 (C-1), 70.0 (C-2), 71.3 (C-3), 76.6 (C-4), 75.3 (C-5), and 60.6 (C-6) (Fig. 1, left y-axis). The anomeric major signal at δC 100.1 was attributed to C-1 of β-mannopyranosyl units. In the 1H-NMR spectrum (Fig. 1, top x-axis), the presence of anomeric signals at δH 4.67 and 4.64 were in agreement with β-glycosidic linkage. The 1H- and 13C-NMR spectrums of I50 are consistent with our previous study25. From the heteronuclear single quantum coherence (HSQC) experiment, the correlations at 100.12/4.67, 4.64, 70.04/5.44, 71.26/5.36, 76.59/3.74, 75.32/3.49, and 60.61/3.83, 3.70 ppm were assigned to C-1/H-1-C-5/H-5 and C-6/H-6a, b, respectively of the 4-linked β-mannopyranosyl units. In the 13C-NMR spectrum of I50, the signals of low intensity at δC 102.5 (correlated with the H-1 at δH 4.44 ppm in the HSQC experiment) designated the presence of β-glucopyranosyl units. Furthermore, other carbon signals at δC 73.3 (C-2), 77.1 (C-3), 77.9 (C-4), and 73.5 (C-5) were assigned for glucopyranosyl units from the HSQC spectrum. These chemical shifts were in good agreement with the 13C-NMR data of the acetylated galactoglucomannan and indicated the presence of β-mannosyl and β-glucosyl residues in the isolated polysaccharide. The cross peak of H-1/C-1 atoms of very low intensity in the HSQC spectrum at 4.98/98.5 ppm indicated the presence of terminal α-D-galactose units linked to the acetylated mannose, which was consistent with the monosaccharide composition results26.

The analysis of functional groups in I50 by NMR. The 1H-NMR spectra (top x-axis), the 13C-NMR spectra (left y-axis), and the HSQC spectra of I50 are shown. Peaks and spots representing different carbons, protons, and monosaccharides are labeled. In the 1H-NMR spectrum, the O-acetyl groups of I50 displayed a characteristic signal in the range of 2.03 to 2.13 ppm, considered the fingerprint of A. vera. Three signals correspond to methyl groups at 2.08, 2.10, and 2.13 ppm indicating that O-acetyl groups are located at different positions. These results were further confirmed by the 13C-NMR spectrum which showed the presence of methyl carbon signals at 20.09, 20.30, and 20.57 ppm, as well as the presence of carboxyl carbons at 172.56, 173.38, and 173.91 ppm.
Therefore, the data of NMR together with chemical analysis (Table 1) demonstrated that the major structure of I50 appears to be β-(1→4) linked mannopyranosyl-glucopyranosyl backbone with a single branched galactose at C-6 of second acetylated mannose residue and there are abundant acetyl groups randomly distributed in the O-2, O-3, and O-6 positions of mannose (Fig. S4) with a relative molecular weight of 37 kDa. Although, the isolated polysaccharide I50 showed a similar backbone structure as those of ACE polymer proposed by Chokboribal et al.27, it differed in molecular weight and proportion of percentage of monosaccharide constituents.
ACE promotes HaCaT cell proliferation
Functionally we first analyzed whether ACE is cytotoxic to HaCaT cells. We treated cells with different concentrations of ACE dissolved in deionized H2O and then performed MTT assays to determine cell viability. The results showed that ACE at concentrations up to 3 mg/ml did not cause cytotoxicity after 48 h incubation. Furthermore, cells treated with 5 mg/ml ACE dissolved in H2O for 24 and 48 h also did not cause cytotoxicity as indexed by MTT assay (Fig. 2A) or by Annexin V/PI staining (Fig. 2B). When the ACE concentration was 10 mg/ml, the cell viability indexed by MTT assay was reduced to about 40% after 24 h of treatment (data not shown). To avoid unnecessary interference resulting from cell death, we used ACE at concentrations not higher than 3 mg/ml for analysis in subsequent experiments. We examined whether ACE affects cell growth. After 24 h of starvation, the cells were cultured with DMEM containing 10% FBS, and treated with different concentrations of ACE. We found that cell growth was promoted by ACE in a concentration-dependent manner (Fig. 2C). The data from the BrdU assay also showed that ACE-treated cells had a higher BrdU uptake, indicating that ACE promotes the DNA synthesis of cells at the S-phase of the cell cycle (Fig. 2D).

The effects of ACE on cytotoxicity and proliferation of HaCaT cells. ACE dehydrate was dissolved in sterilized water in a concentration of 50 mg/ml before use. (A) Cells (5 × 103) were seeded on a 24-well plate and incubated overnight at 37 °C with 5% CO2, and different concentrations of ACE or vehicle agent were added followed by incubation for 24–48 h. The MTT assay was conducted to determine the effect of ACE on cell viability. (B) The cell viability was also determined by flow cytometry with Annexin V-FITC/PI staining after the treatment with 5 mg/ml ACE for 48 h. (C) Cells were seeded on a 12-well plate and treated with different concentrations of ACE. The live cells were counted by trypan blue staining every 24 h. (D) Cells seeded on a 96-well plate were starved for 24 h, then cultured with fresh DMEM containing 10% FBS and treated with indicated concentrations of ACE. Cells were collected after 48 h-incubation to determine BrdU signal intensity according to the manufacturer’s protocol. * p < 0.05 compared to vehicle-treated cells.
ACE enhances HaCaT cell migration
Next, we determined the effect of ACE on keratinocyte migration. We used a wound healing model by seeding cells in a 2-well silicone culture insert to create a gap between cells. Because ACE can increase cell growth as shown in Fig. 2, we treated cells with mitomycin C during the cell migration assays to avoid cell growth interfering with cell migration results. We first determined the proper concentration of mitomycin C to inhibit cell growth but not to induce cell death. As shown in Fig. 3A (left panel), mitomycin C at 2 µg/ml and 5 µg/ml blocked cell growth and reduced cell number. The reduction of cell number occurred at 48 h- and 24 h-treatment of mitomycin C at 2 µg/ml and 5 µg/ml, respectively. Based on the purpose of using mitomycin C to maintain cell number at the original level but not to exert a suppression which affects cell migration assay, mitomycin C at 2 µg/ml might be proper in the migration assay within 24 h. To further confirm this issue, we tested the effects of mitomycin C (2 µg/ml) together with ACE. We found ACE-induced cell growth was abrogated by mitomycin C (2 µg/ml) (Fig. 3A, right panel). Based on the above results, we measured cell migration in the condition with mitomycin C (2 µg/ml) and ACE co-treatment. As shown in Fig. 3B, cells treated with ACE (3 mg/ml) had higher migration than cells treated with vehicle.

ACE accelerates HaCaT cell migration. (A) Cells were treated with mitomycin C at the indicated concentrations in the absence (left panel) or presence (right panel) of ACE (1 and 3 mg/ml, respectively). Cell number was counted every 24 h by trypan blue staining. (B) Cells cultured in mitomycin C (2 µg/ml) containing medium were treated with vehicle or ACE (1 and 3 mg/ml), and cell migration was determined at the indicated time point under a microscope. The effect of ACE on cell migration was evaluated using Image J software to quantify the gap recovery area as shown (A, right panel). * p < 0.05 compared to vehicle-treated cells at the same migration time.
ACE induces the expression of keratinocyte differentiation markers in an EGFR/PKC-dependent manner
We also explored whether ACE affects the differentiation of HaCaT cells. To this end, we treated cells with ACE and then determined the expression levels of specific differentiation marker proteins. In parallel, cells were also treated with the differentiation-inducing agent phorbol myristate acetate (PMA, 100 nM) as the standard response. Similar to the effect of PMA, ACE (3 mg/ml) could increase the expressions of involucrin and loricrin, but not keratin 10 (K10), within 6–36 h (Fig. 4A). After treatment for 36 h, we found ACE at 1 mg/ml was sufficient to increase involucrin, loricrin, and transglutaminase 1 (TGase1) as the effects at 3 and 5 mg/ml (Fig. 4B). Moreover, p63α is a key regulator of keratinocyte differentiation, and its downregulation is a prerequisite28,29,30. We found ACE can downregulate p63α within 12 h, but after 12 h the effect was no longer observed (Fig. 4A). The biphasic changes (i.e. early downregulation) of p63α are consistent with the previous notion showing the dual roles of p63α in keratinocyte differentiation and proliferation28,29. Moreover, these results suggest a positive role of ACE in inducing cell differentiation.

ACE induces the expressions of keratinocyte differentiation protein markers as PMA. (A) The time-dependent effect of ACE on the expressions of involucrin, loricrin, and p63α. (B) After treatment with ACE at indicated concentrations and PMA (100 nM) for 36 h, the expressions of involucrin, loricrin, and TGase 1 were determined. The relative protein expression level was normalized by β-actin, and the phosphor-protein expression level was shown with the normalization by their endogenous protein level. The line charts representing quantifications of protein expression levels in each panel were shown in Figure S5.
EGFR and protein kinase C (PKC) are important regulators for keratinocyte differentiation; however, the differentiation markers resulting from EGFR and PKC activation are not all the same20,21,31,32,33. Unlike PMA whose activation of PKC is regarded to induce full terminal differentiation, incomplete keratinocyte differentiation is induced by EGF. Herein, we determined the effects of ACE on EGFR and PKC activities. Like PMA, ACE could activate EGFR and PKCβ, as well as EGFR downstream molecule AKT, as indexed by increasing their phosphorylation (Fig. 5A). We found ACE-induced EGFR phosphorylation was weaker in extent and shorter in duration than PMA (Figs. 4A and 5A). Of note, we observed the rapid upregulation of EGFR protein expression by PMA and ACE (Figs. 4A and 5A). In addition, ACE induced higher PKCβ activation than PMA (Fig. 5A). These findings suggest the roles of EGFR and PKC in the action of ACE.

ACE activates EGFR, AKT, and PKC as PMA. (A) HaCaT cells were treated with PMA (100 nM) or ACE (3 mg/ml) for indicated times to determine EGFR, AKT, and PKC phosphorylations. (B) HaCaT cells were pretreated with TH1020 (2 µM) or osimertinib (1 µM) for 30 min and followed by ACE treatment. The effects of TH1020 and osimertinib on ACE-induced phosphorylation of AKT and PKC were determined. (C) NHEK cells were pretreated with osimertinib or DMSO (vehicle) for 30 min, then treated with ACE (3 mg/ml) for 1 and 3 h. The relative protein expression level was normalized by β-actin, and the phosphor-protein expression level was shown with the normalization by their endogenous protein level. The line charts representing quantifications of protein expression levels in each panel were shown in Figure S6.
To decipher if EGFR activation contributes to the action of ACE and PMA, we treated cells with an EGFR inhibitor osimertinib. As shown in Fig. 5B, we found osimertinib (1 µM) abolished ACE-induced activations of AKT and PKCβ. In addition, to understand whether ACE also activates both molecules through TLR5, which has been reported to bind to ACE selectively through its ectodomain flagellin recognition sites14, we treated cells with a TLR5 inhibitor TH1020 before the addition of ACE. We found that TH1020 (2 µM) cannot affect ACE-induced activation of AKT and PKCβ (Fig. 5B), suggesting that ACE induces AKT and PKCβ activation through EGFR but not TLR5.
To further confirm that ACE triggers the AKT/mTOR signaling pathway through EGFR activation, we analyzed the effect of ACE on primary normal human epidermal keratinocytes (NHEK). As shown in Fig. 5C, we observed EGFR, AKT, mTOR, and PKCβ activations by increasing their phosphorylations in ACE-treated NHEK cells compared to vehicle-treated cells. Pretreatment of EGFR inhibitor osimertinib suppressed ACE-induced phosphorylation of these proteins, which is consistent with our finding in HaCaT cells.
To further understand whether ACE induces keratinocyte differentiation through EGFR and PKC activation, we compared the expression of differentiation marker proteins in cells treated with ACE alone or co-treated with PMA or EGF. The results showed that ACE treatment could increase the expressions of involucrin, loricrin, and TGase 1 compared to the vehicle group. Co-treatment with PMA could not further increase the expressions of these molecules compared to ACE treatment alone, suggesting the non-additive effects of ACE and PKC. On the other hand, the combination effects of ACE and EGF on loricrin and TGase 1 displayed the additivity (Fig. 6A). In addition, the expression of involucrin caused by ACE and PMA was significantly inhibited when co-treated with osimertinib (Fig. 6B). Based on these findings, ACE might induce keratinocyte differentiation via EGFR-PKC activation, and EGFR transactivation is essential for keratinocyte differentiation.

ACE induces keratinocyte differentiation through activation of PKC and EGFR. (A) Cells were treated with ACE (3 mg/ml), PMA (100 nM), and EGF (50 ng/ml) for 6, 12, 18, and 24 h, respectively. (B) Alternatively, cells were treated with ACE, PMA, and osimertinib for 24 and 36 h. The expressions of differentiation markers including involucrin, loricrin, and TGase 1 were determined. The relative protein expression level was normalized by β-actin. The line charts representing quantifications of protein expression levels in each panel were shown in Figure S7.
Osimertinib inhibits ACE-induced proliferation and migration in HaCaT cells
We further investigated whether ACE promotes cell growth via activation of EGFR and PKC. As described above, ACE increased cell growth compared to the vehicle group, and we found that osimertinib itself inhibited cell growth as well as that induced by ACE. Treatment with PKC inhibitor Ro320432 alone significantly increased cell growth, and co-treatment with PMA or ACE decreased Ro320432-induced cell growth (Fig. 7A). These findings suggest that EGFR and PKC are involved in regulating the cell proliferative action of ACE in different manners, where PKC is a negative regulator, while constitutive EGFR activation is a positive regulator.

Effects of PKC and EGFR inhibitors on ACE-induced proliferation and migration of HaCaT cells. (A) Cells were starved for 24 h and then cultured with fresh DMEM containing 10% FBS and the indicated agents including PMA (100 nM), ACE (3 mg/ml), Ro320432 (30 µM), and osimertinib (1 µM). The cell number was counted every 24 h. * p < 0.05 compared to vehicle-treated cells at the same migration time. # p < 0.05 compared to Ro320432-treated cells at the same migration time. (B) The effects of PMA, Ro320432, EGF, and osimertinib on ACE-induced migration of HaCaT cells. The gap recovery area was quantified using Image J software (right panel). * p < 0.05 compared to vehicle-treated cells at 0 h migration time. # p < 0.05 compared to ACE-treated cells.
Next, we examined the roles of EGFR and PKC on ACE-induced cell migration. As shown in Fig. 7B, cells treated with Ro320432 or EGF exhibited faster migration than vehicle-treated cells, whereas PMA and osimertinib themselves alone inhibited cell migration as well as that induced by ACE. Accordingly, as we suggest for cell growth, EGFR and PKC oppositely regulated the keratinocyte migration regardless of the presence or not of ACE.
Discussion
Aloe extract has been reported to promote wound healing, cell proliferation, and differentiation of keratinocytes22, suggesting the therapeutic potential of this herb medicine in skin injury. Therefore, exploring the direct effect of ACE, the major bioactive component of Aloe, in keratinocyte biology is interesting. In this study, we for the first time found that ACE promotes HaCaT cell proliferation and migration, and induces the expression of differentiation markers like involucrin, loricrin, and TGase1, indicating that ACE should be one of the components that contribute to the beneficial effects of Aloe extract in wound healing. It has been reported that deacetylation deactivates ACE bioactivity27 and in our previous work, we found ACE with higher O-acetyl group content exhibited better biological activity against colorectal cancer cell lines25. The mechanistic study further shows the ACE’s effect in activating EGFR and PKC in keratinocytes, which differentially contribute to the impact of ACE in cell proliferation, migration, and differentiation.
EGFR is the most important growth factor in keratinocytes and it can be easily activated via autocrine/paracrine and transactivation manners. For example, UVB and PKC activator PMA have been shown to induce the rapid and sustained activation of EGFR in keratinocytes34,35,36. Our results showed that ACE like PMA activates EGFR and PKC, and increases EGFR protein expression. Both EGFR and PKC have been documented to play important roles in regulating keratinocyte proliferation34,37, migration20,36, and differentiation20,28,31,32,33. However, their effects on these events are not all the same.
Unlike EGFR activation which increases growth34, migration36, and partial/incomplete differentiation in keratinocytes32, PMA-mediated PKC activation leads to complete differentiation33, but growth and migration inhibition37,38,39. As to cell growth, our data support the previous notion that PKC activation induces an inhibition of keratinocyte proliferation37,39, while EGFR can promote cell growth. We observed the increased cell growth in PKC inhibitor-treated keratinocytes, and PMA reverses this effect of PKC inhibitor. The reason for PKC-mediated inhibition of keratinocyte proliferation might be partially related to the PKC-dependent promotion of keratinocyte differentiation. PKCδ has been reported to suppress keratinocyte proliferation by increasing KLF4-mediated p21Cip1 gene expression37. Another isoform PKCη binds and activates Fyn, a c-Src kinase, to cause keratinocyte growth arrest by decreasing DNA synthesis in a Fyn-dependent pathway39. Likewise, our data also support the inhibitory effect of PKC activation on keratinocyte migration. There are currently no relevant reports on the regulatory mechanism of how PKC inhibits cell migration in keratinocytes. However, in endothelial cells, activation of PKCδ negatively regulates cell migration by binding to phospho-syndecan-4, blocking the binding of syndecan-4 to PKCα, thereby inhibiting the positive regulation of cell migration by PKCα40. In addition, inhibition of endothelial cell migration by PMA results from the upregulation of Thy-1 expression via activating the PKCδ/Syk/NF-κB pathway41.
As to differentiation marker expression, there are differences between the actions of PMA and EGF. Many studies using selective PKC activator PMA as the standard in vitro keratinocyte differentiation model indicate that PKC activation is required for expressions of the differentiation markers at late stage including involucrin, loricrin, filaggrin, keratin 10, and the terminal marker keratinocytes-specific TGase 131,33. However, EGF cannot upregulate keratin 10 and loricrin32. Moreover, the downregulation of EGFR by miR-146a promotes terminal differentiation of keratinocytes42. The effect of EGFR signaling indicating the incomplete differentiation feature of keratinocytes contributes to the pathogenesis of psoriasis, i.e. overactivation of keratinocytes but defect of terminal and complete differentiation of keratinocytes in epidermis43,44. Another evidence to support the negative role of EGFR in terminal differentiation is that during keratinocyte differentiation in the epidermis or in the standard cell models using PMA, cell confluence, and calcium, EGFR expression and activity are downregulated31,32,45. Aberrant and excess activation of EGFR leads to overproliferation of keratinocytes, contributing to psoriasis, while EGFR inhibitors switch keratinocytes from a proliferative to a differentiative phenotype, thus affecting epidermal development and barrier function46. All these findings suggest the need to tightly balance the biological functions of PKC and EGFR in keratinocytes. In this study, our data reveal a non-additive effect between ACE and PMA, but an additive effect between ACE and EGF in the upregulation of differentiation markers, suggesting ACE can induce PKC activation independent of EGFR. This suggestion is confirmed by the higher PKC phosphorylation caused by ACE than PMA. The major reason for the action of ACE beyond EGFR remains elusive. Taking these findings together, we suggest ACE can activate PKC and transactivate EGFR. The net outcomes of both actions lead to increased keratinocyte growth, migration, and differentiation.
Not only in regulating cell growth, EGFR activation displays the antiapoptotic response in keratinocytes after UVB irradiation, which requires the PI3K-AKT signaling pathway activated by EGFR47. In this study, we did not observe a protective effect of ACE on UVB-induced cell death in HaCaT cells (data not shown) even though ACE can activate EGFR. One possible explanation is that although ACE can induce a rapid activation of EGFR, this effect is short-acting. Of course, we cannot rule out other action mechanisms of ACE that coordinately regulate cell viability together with EGFR.
Taken together, ACE can increase proliferation, differentiation, and migration of HaCaT cells possibly through the activation of EGFR and PKC. The schematic diagram of this study was shown in Fig. 8. Our data strengthen the therapeutic potential of ACE in skin wound healing and possibly in psoriasis. Further animal study in psoriasis disease model is interesting to explore in the future.

A schematic representation showing the pharmacological properties of ACE in HaCaT cells.
Materials and methods
Cell culture, antibodies, reagents, and compounds
Human keratinocyte cell line HaCaT purchased from American Type Culture Collection (ATCC; Manassas, VA, USA) was cultured in DMEM (Gibco™, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS) and 10% penicillin-streptomycin solution. NHEK cell line purchased from ScienCell Research Laboratories (Carlsbad, CA, USA) was cultured in Keratinocyte-SFM basal medium (Gibco™) supplemented with bovine pituitary extract (50 µg/ml) and EGF (5 ng/ml). All the cells were incubated at 37 °C with 5% CO2. Primary antibodies against involucrin, K10, TGase 1, PKCβII, mTOR, and β-actin were purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). Primary antibodies against p63α, phospho-EGFR Y1068, phospho-PKCβII S660, phospho-AKT T308, AKT, phospho-mTOR S2448, and peroxidase-conjugated anti-rabbit/mouse IgG secondary antibodies were purchased from Cell Signaling Technology, Inc (Danvers, MA, USA). The primary antibody against EGFR was purchased from EMD Millipore Corp. The primary antibody against loricrin was purchased from GeneTex (Irvine, CA, USA). PMA and EGF were purchased from Sigma-Aldrich (St. Louis, MO, USA). PKC inhibitor Ro320432 was purchased from Tocris Bioscience (a brand of In Vitro Technologies, Melbourne, VIC, Australia). Osimertinib and TH1020 were purchased from MedChemExpress LLC (Monmouth Junction, NJ, USA). The ACE dehydrate was prepared and provided by Dazzeon Biotech Co., Ltd. (New Taipei, Taiwan).
Isolation and purification of Aloe polysaccharides
The raw material used in this study was a 200X A. vera concentrate produced by a proprietary processing method from mature leaves of A. barbadensis Miller. Aloe polysaccharide I50 was made according to a modified protocol27,48. Briefly, A. vera powder was dissolved in deionized H2O, and the filtrate was then subjected to the column containing activated nonpolar resin (HP-20 DIAION™, Mitsubishi Chemical Co., Japan). We used an ethanol-H2O gradient (0-100%) to elute the column and obtained three major fractions: 100% H2O, 50% ethanol, and 100% ethanol at a rate of 1 l/min. The eluents obtained from the 50% ethanol fraction were freeze-dried and subsequently subjected to 80% ethanol wash to obtain I50.
High-performance liquid chromatography (HPLC)
The ACE contents in I50 were quantified using HPLC (Agilent 1260-Infinity II, Agilent Technologies Inc., USA). A 2.5 mg/ml I50 solution was prepared using a mobile phase composed of 0.5% sodium azide (NaN3) (Thermo Fisher Scientific) in H2O (wt/wt). This solution was then subjected to a chromatographic column following shaking for 8 h and subsequently filtered through a 0.2 μm filter. The HPLC consists of an ultra-hydrogel TM 1000 column (7.8 × 300 mm, Waters, Japan) and a Shodex KS-804 column (8 × 300 mm, Shodex, Japan) with a refractive index (RI) detector, operating at a flow rate of 1.0 ml/min at 50 °C. The ACE content of I50 was detected. The ACE standard was used to create the calibration curve for quantifying ACE content.
High-performance size exclusion chromatography (HPSEC)
ACE content of I50 was analyzed using HPLC equipped with ultra-hydrogel TM 1000 column and Shodex KS-804 series SEC column, (8 × 300 mm, 1.0 ml/min at 50 °C, Japan) attached to a refractive index detector (RID). Briefly, samples were dissolved with the mobile phase and filtered through a 0.45 μm syringe filter. The mobile phase consisted of 0.5% NaN3 in H2O (wt/wt.) at a flow rate of 1 ml/min. Accurately weighed standard ACE (internal standard) and made it 3 mg/ml standard stock solution with the mobile phase. Measured 300, 400, 500, 600, 700, and 800 µl, of the standard stock solution and diluted to 1 ml with the mobile phase. Prepared a 2.5 mg/ml I50 solution with the mobile phase, adjusted the concentration if necessary to meet the linear range, and performed HPLC analysis. For the 6 standard linear solutions (STD 1–6), performed HPLC analysis in order from the lowest concentration to the highest concentration, analyzing each solution 3 times. Using concentration as the x-axis, the ratio of the average peak area/height of the sample to the average peak area/height of ACE as the y-axis, performed linear regression to obtain the best-fit linear equation y = ax + b and its coefficient of determination R2 > 0.995. The ACE content of the aloe polysaccharide was calculated by using the formula,
Ru: ACE chromatographic peak area in the sample solution; b: the y-intercept of the calibration curve; a: the slope of the calibration curve; V: constant volume of the sample solution tested, in ml; D: dilution ratio (determined by sample concentration; W: Weight of the solid sample, in mg.
Ultraviolet-visible (UV-Vis) spectroscopy
The O-acetyl group content of I50 was quantified using UV-Vis spectrometry (Epoch 2, Agilent Technologies Inc., USA). The sample solution was prepared at a concentration of 10 mg/ml. Mix 1 ml of the sample solutions with 2 ml of 2 M hydroxylamine hydrochloride solution (HONH2·HCl) which was added with 3.5 M NaOH (Nippon Shinyaku Co., Ltd., Japan), and then incubate the mixture for 4 min at room temperature. Added 1 ml of 4 M HCl (Honeywell International Inc, USA) and 0.37 M ferric chloride-hydrochloric acid solution (Sigma-Aldrich) into the mixture, and measured the absorbance at 540 nm wavelength using UV-Vis spectrometry. The standard curve to quantify the O-acetyl group was established utilizing the acetylcholine chloride (C7H16ClNO2, Sigma-Aldrich) solutions. A standard curve was built up based on the 540 nm absorbance obtained at various concentrations of the standards, and the level of O-acetyl groups (mg/kg) in the samples was calculated by interpolating from the standard curve.
Monosaccharide composition analysis
The monosaccharide composition of I50 was analyzed by high-performance anion-exchange chromatography-pulsed amperometric detection (HPAEC). Polysaccharide samples (5 mg) were hydrolyzed with 2 M TFA at 120 °C for 3 h. After hydrolysis, the residual TFA was removed by blow drying with nitrogen gas. Then the hydrolysates were completely dissolved in 5 ml of ultrapure water and centrifuged. The supernatant was collected for ion-chromatography analysis. The mobile phases were 0.1 M NaOH (mobile phase A) and 0.1 M NaOH plus 0.2 M NaAc (mobile phase B). The following parameters were performed: the flow rate of 0.2 mg/ml, injection volume of 2 µl, and column temperature of 30℃. Monosaccharide compositions were identified by comparison with the retention times of the monosaccharide standards.
Nuclear magnetic resonance spectroscopy (NMR)
The 1H-NMR, 13C-NMR, and 2D HSQC experiments were recorded on an AVANCE III HD 600 MHz spectrometer (Bruker, MA, USA) which was equipped with a BBFO probe. The freeze-dried powder of I50 was dissolved in 99.9% deuterium oxide (Sigma-Aldrich), at 100 mg/ml and transferred to Wildman Economic 5 mm NMR tubes for analysis. 2D HSQC experiments were carried out using the pulse programs supplied with the Bruker manual.
MTT assay
The cells were seeded in 24-well plates in 500 µl DMEM at 37 °C with 5% CO2. When the cell confluence reached 60%, the cells were treated with vehicle and different concentrations of ACE for the indicated time. The 1 mg/ml MTT [3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (Sigma-Aldrich) solution was added into the medium for 1 h to form crystals by mitochondrial dehydrogenases of living cells. The medium was removed and replaced with 200 µl DMSO to dissolve the purple crystal. The cell viability was determined by the OD value (OD550nm-OD630nm) of the DMSO solution containing the crystal product.
Flow cytometry analysis
Cells were harvested and re-suspended in the staining buffer containing Annexin V-FITC/PI according to the manufacturer’s protocol (BioLegend, San Diego, CA, USA), After the 20-min incubation at 37 °C, the cells were measured by flow cytometry (FACSCalibur, BD, Franklin Lakes, NJ, USA). FlowJo software was used for data analysis.
Cell migration assay
Cells were seeded at 2 × 104 cells/well on 24-well plates with a 2-well silicone culture insert in each well. After seeding overnight, the silicone culture insert was removed and fresh DMEM containing 2 µg/ml mitomycin C was added. Cells were treated with different concentrations of ACE, or other agents including PMA, Ro320432, EGF, and osimertinib. At the indicated time points, the cell migration was observed with a microscope and recorded by taking photos. The percentage of gap recovery area was analyzed and quantified using Image J software.
Cell proliferation assay
Cells (2 × 104/well) were seeded on 12-well plates in DMEM without FBS, and incubated for 24 h in a CO2 incubator. The next day, fresh DMEM with 10% FBS in the absence or presence of ACE was added. The cell proliferation was determined by counting the cell number every 24 h with trypan blue staining. For the BrdU incorporation assay, cells (1 × 104/well) were seeded on a black 96-well plate with a clear and flat bottom in DMEM without FBS, and incubated for 24 h. Remove the medium and add fresh DMEM with 10% FBS in the presence or absence of ACE for 46 h. Add BrdU labeling solution for an additional 2 h incubation. Cells were fixed, incubated with anti-BrdU-peroxidase for 90 min, and washed before conducting substrate reaction according to the manufacturer’s protocol (Cell proliferation ELISA, BrdU, Roche, Switzerland). Chemiluminescence was measured using a Synergy H1 microplate reader (BioTeK, Winooski, VT, USA).
Western blot
Cells were lysed using RIPA buffer (50 mM Tris at pH 7.6, 150 mM NaCl, 0.1% SDS, 0.1% sodium deoxycholate, 1% Triton X-100, 2 mM NaF, 2 mM EDTA, 2 mM Na3VO4, and 1 mM PMSF). After homogenization by a sonicator, the cell extract was diluted in 5X sample buffer (50 mM Tris at pH 6.8, 5% SDS, 37.5% glycerol, 36 mM β-mercaptoethanol, and 0.25% bromophenol blue), heated for 10–15 min at 95 °C, and then subjected to SDS-polyacrylamide gel electrophoresis (PAGE). The running conditions of the PAGE are 70 V for 25 min at step 1, then 130 V for 85 min at step 2. Proteins on the gel were transferred to a polyvinylidene difluoride membrane (PVDF, GE Healthcare Life Sciences, USA) at 250 mA for 1 h, and then blocked for 30 min with TBS-T (20 mM Tris-HCl pH 7.4, 150 mM NaCl, and 0.1% Tween 20) containing 5% skim milk. The membrane was rinsed three times consecutively with TBS-T buffer, followed by incubation with 1:1000–2000 dilutions of primary antibodies in TBS-T buffer for 4–8 h. The membrane was washed three times with TBS-T buffer for 15 min and then incubated for 1 h with horseradish peroxidase-conjugated anti-goat IgG (1:10000) or anti-rabbit/mouse IgG secondary antibody (1:10000) in TBS-T buffer. Development was carried out using an enhanced chemiluminescence solution (Western Lightning Plus-ECL, PerkinElmer, Inc. MA, USA). The protein signals were acquired using a ChemiDoc™ MP Image System (Bio-Rad, CA, USA), and analyzed using Image Lab software (Bio-Rad).
Statistical analysis
Values were expressed as the mean ± S.E.M. of at least three independent experiments. Student’s t-test or one-way ANOVA was performed to analyze the statistical significance of the differences, and a p-value < 0.05 was statistically significant.
Data availability
Data is provided within the manuscript or supplementary information files.
References
Sánchez, M., González-Burgos, E. & Iglesias, I. Gómez-Serranillos, M. P. Pharmacological update properties of Aloe vera and its major active constituents. Molecules 25, 1324 (2020).
Lee, Z. M., Goh, B. H. & Khaw, K. Y. Aloe vera and the proliferative phase of cutaneous wound healing: status quo report on active principles, mechanisms, and applications. Planta Med. 91, 4–18 (2025).
Akao, T. et al. A purgative action of Barbaloin is induced by Eubacterium Sp. strain BAR, a human intestinal anaerobe, capable of transforming Barbaloin to aloe-emodin anthrone. Biol. Pharm. Bull. 19, 136–138 (1996).
Boudreau, M. D. et al. From the cover: Aloin, a component of the aloe vera plant leaf, induces pathological changes and modulates the composition of microbiota in the large intestines of f344/N male rats. Toxicol. Sci. 158, 302–318 (2017).
Quan, Y. et al. Aloe Emodin induces hepatotoxicity by activating NF-κB inflammatory pathway and p53 apoptosis pathway in zebrafish. Toxicol. Lett. 306, 66–79 (2019).
Panigrahi, G. K. et al. Activity-guided chemo toxic profiling of Cassia occidentalis (CO) seeds: detection of toxic compounds in body fluids of CO-exposed patients and experimental rats. Chem. Res. Toxicol. 28, 1120–1132 (2015).
Nesslany, F., Simar-Meintières, S., Ficheux, H. & Marzin, D. Aloe-emodin-induced DNA fragmentation in the mouse in vivo comet assay. Mutat. Res. 678, 13–19 (2009).
Vath, P., Wamer, W. G. & Falvey, D. E. Photochemistry and phototoxicity of aloe Emodin. Photochem. Photobiol. 75, 346–352 (2002).
Sierra-García, G. D., Castro-Ríos, R., González-Horta, A., Lara-Arias, J. & Chávez-Montes, A. Acemannan, an extracted polysaccharide from Aloe Vera: a literature review. Nat. Prod. Commun. 9, 1217–1221 (2014).
Liu, C. et al. Extraction, purification, structural characteristics, biological activities and Pharmacological applications of Acemannan, a polysaccharide from aloe Vera: A review. Molecules 24, 1554 (2019).
Bhalang, K., Thunyakitpisal, P. & Rungsirisatean, N. Acemannan, a polysaccharide extracted from Aloe Vera, is effective in the treatment of oral aphthous ulceration. J. Altern. Complement. Med. 19, 429–434 (2013).
Songsiripradubboon, S. et al. Stimulation of dentin regeneration by using Acemannan in teeth with lipopolysaccharide-induced pulp inflammation. J. Endod. 43, 1097–1103 (2017).
Quezada, M. P., Salinas, C., Gotteland, M. & Cardemil, L. Acemannan and Fructans from Aloe vera (Aloe barbadensis Miller) plants as novel prebiotics. J. Agric. Food Chem. 65, 10029–10039 (2017).
Thunyakitpisal, P., Ruangpornvisuti, V., Kengkwasing, P., Chokboribal, J. & Sangvanich, P. Acemannan increases NF-κB/DNA binding and IL-6/-8 expression by selectively binding Toll-like receptor-5 in human gingival fibroblasts. Carbohydr. Polym. 161, 149–157 (2017).
Xing, W. et al. Acemannan accelerates cell proliferation and skin wound healing through AKT/mTOR signaling pathway. J. Dermatol. Sci. 79, 101–109 (2015).
Rodrigues, L. C. et al. Physicochemical features assessment of acemannan-based ternary blended films for biomedical purposes. Carbohydr. Polym. 257, 117601 (2021).
Werner, S. & Smola, H. Paracrine regulation of keratinocyte proliferation and differentiation. Trends Cell. Biol. 11, 143–146 (2001).
Wang, J., Li, J., Zhou, L., Hou, H. & Zhang, K. Regulation of epidermal barrier function and pathogenesis of psoriasis by Serine protease inhibitors. Front. Immunol. 15, 1498067 (2024).
Smith, J. & Rai, V. Novel factors regulating proliferation, migration, and differentiation of fibroblasts, keratinocytes, and vascular smooth muscle cells during wound healing. Biomedicines 12, 1939 (2024).
Chang, H. C. et al. BLIMP1 transcriptionally induced by EGFR activation and post-translationally regulated by proteasome and lysosome is involved in keratinocyte differentiation, migration and inflammation. J. Dermatol. Sci. 92, 151–161 (2018).
Wu, N. L. et al. EGFR-driven up-regulation of decoy receptor 3 in keratinocytes contributes to the pathogenesis of psoriasis. Biochim. Biophys. Acta. 1832, 1538–1548 (2013).
Moriyama, M. et al. Beneficial effects of the genus Aloe on wound healing, cell proliferation, and differentiation of epidermal keratinocytes. PLoS One. 11, e0164799 (2016).
Schoop, V. M., Fusenig, N. E. & Mirancea, N. Epidermal organization and differentiation of HaCaT keratinocytes in organotypic coculture with human dermal fibroblasts. J. Invest. Dermatol. 112, 343–353 (1999).
Metcalfe, C. Quantitation of Aloe vera polysaccharides by O-Acetyl and UV-Vis spectrophotometry: first action 2018.14. J. AOAC Int. 102, 1091–1094 (2019).
Shih, P. C. et al. The Aloe vera Acemannan polysaccharides inhibit phthalate-induced cell viability, metastasis, and stemness in colorectal cancer cells. Ecotoxicol. Environ. Saf. 288, 117351 (2024).
Capek, P., Alföldi, J. & Lišková, D. An acetylated Galactoglucomannan from Picea abies L. Karst. Carbohydr. Res. 337, 1033–1037 (2002).
Chokboribal, J. et al. Deacetylation affects the physical properties and bioactivity of Acemannan, an extracted polysaccharide from Aloe vera. Carbohydr. Polym. 133, 556–566 (2015).
Wu, N. L., Lee, T. A., Tsai, T. L. & Lin, W. W. TRAIL-induced keratinocyte differentiation requires caspase activation and p63 expression. J. Invest. Dermatol. 131, 874–883 (2011).
Truong, A. B. & Khavari, P. A. Control of keratinocyte proliferation and differentiation by p63. Cell. Cycle. 6, 295–299 (2007).
Koster, M. I. & Roop, D. R. The role of p63 in development and differentiation of the epidermis: Tanioku Kihei memorial lecture. J. Dermatol. Sci. 34, 3–9 (2004).
Wu, N. L., Huang, D. Y., Hsieh, S. L., Dai, Y. S. & Lin, W. W. Decoy receptor 3 is involved in epidermal keratinocyte commitment to terminal differentiation via EGFR and PKC activation. Exp. Mol. Med. 54, 542–551 (2022).
Wu, N. L. et al. Spleen tyrosine kinase mediates EGFR signaling to regulate keratinocyte terminal differentiation. J. Invest. Dermatol. 136, 192–201 (2016).
Papp, H. et al. Protein kinase C isozymes regulate proliferation and high cell density-mediated differentiation in HaCaT keratinocytes. Exp. Dermatol. 12, 811–824 (2003).
Chiu, L. Y. et al. PARP-1 involves in UVB‐induced inflammatory response in keratinocytes and skin injury via regulation of ROS‐dependent EGFR transactivation and p38 signaling. FASEB J. 35, e21393 (2021).
Liu, Q. et al. Depletion of Gsdma1/2/3 alleviates PMA-induced epidermal hyperplasia by inhibiting the EGFR–Stat3/Akt pathway. J. Mol. Cell. Biol. 16, mjad080 (2024).
Lee, H. et al. Blimp-1 upregulation by multiple ligands via EGFR transactivation inhibits cell migration in keratinocytes and squamous cell carcinoma. Front. Pharmacol. 13, 763678 (2022).
Chew, Y. C., Adhikary, G., Wilson, G. M., Reece, E. A. & Eckert, R. L. Protein kinase C (PKC) δ suppresses keratinocyte proliferation by increasing p21Cip1 level by a KLF4 transcription factor-dependent mechanism. J. Biol. Chem. 286, 28772–28782 (2011).
Ando, Y., Lazarus, G. S. & Jensen, P. J. Activation of protein kinase C inhibits human keratinocyte migration. J. Cell. Physiol. 156, 487–496 (1993).
Cabodi, S. et al. A PKC-η/Fyn-dependent pathway leading to keratinocyte growth arrest and differentiation. Mol. Cell. 6, 1121–1129 (2000).
Chaudhuri, P., Colles, S. M., Fox, P. L. & Graham, L. M. Protein kinase Cδ-dependent phosphorylation of syndecan-4 regulates cell migration. Cir. Res. 97, 674–681 (2005).
Wen, H. C., Huo, Y. N., Chou, C. M. & Lee, W. S. PMA inhibits endothelial cell migration through activating the PKC-δ/Syk/NF-κB-mediated up-regulation of Thy-1. Sci. Rep. 8, 16247 (2018).
Lee, A. Y. The role of MicroRNAs in epidermal barrier. Int. J. Mol. Sci. 21, 5781 (2020).
Wang, S., Zhang, Z., Peng, H. & Zeng, K. Recent advances on the roles of epidermal growth factor receptor in psoriasis. Am. J. Transl Res. 11, 520 (2019).
Dai, X. et al. EGFR ligands synergistically increase IL-17A‐induced expression of psoriasis signature genes in human keratinocytes via IκBζ and Bcl3. Eu J. Immunol. 52, 994–1005 (2022).
King, L. E. Jr., Gates, R. E., Stoscheck, C. M. & Nanney, L. B. Epidermal growth factor/transforming growth factor alpha receptors and psoriasis. J. Invest. Dermatol. 95, S10–S12 (1990).
Joly-Tonetti, N., Ondet, T., Monshouwer, M. & Stamatas, G. N. EGFR inhibitors switch keratinocytes from a proliferative to a differentiative phenotype affecting epidermal development and barrier function. BMC Cancer. 21, 1–10 (2021).
Canguilhem, B. et al. RhoB protects human keratinocytes from UVB-induced apoptosis through epidermal growth factor receptor signaling. J. Biol. Chem. 280, 43257–43263 (2005).
Chow, J. T. N., Williamson, D. A., Yates, K. M. & Goux, W. J. Chemical characterization of the Immunomodulating polysaccharide of Aloe vera L. Carbohydr. Res. 340, 1131–1142 (2005).
Acknowledgements
We would like to acknowledge the service provided by the Immune Research Core of the Department of Medical Research at the National Taiwan University Hospital and the First Core Labs at the National Taiwan University College of Medicine. We thank Dr. Nan-Lin Wu for the discussion of the manuscript. This research was funded by the National Taiwan University Hospital Yunlin Branch (NTUHYL112-S013) and Dazzeon Biotechnology Co., Ltd.
Author information
Authors and Affiliations
Contributions
Conceptualization: S.-H. H., Y.-S. D., P. E., and W.-W. L.; Funding acquisition: S.-H. H. and P. E.; Designed the experiments and analyzed the results: C.-Y. C., P. E. and W.-W. L.; Performed the experiments and analyzed the results: C.-Y. C., U. C. and P.-C. S; Writing-original draft preparation: C.-Y. C. and U. C.; Writing-review and editing: C.-Y. C., S.-H. H., Y.-S. D., and W.-W. L. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cheng, CY., Hsu, SH., Chokkalingam, U. et al. Aloe polysaccharide promotes keratinocyte proliferation, migration, and differentiation by upregulating the EGFR/PKC-dependent signaling pathways. Sci Rep 15, 8196 (2025). https://doi.org/10.1038/s41598-025-91201-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-91201-x
Keywords
This article is cited by
-
Effects of Ultrasonic Processing on the Structural Properties and Chain Scission of the Acemannan Biopolymer
Journal of Polymers and the Environment (2025)
