
Abstract
Malaria remains a critical global health concern, especially in tropical and subtropical regions, where it causes substantial morbidity and mortality. Current diagnostic methods, such as microscopy and PCR-based assays, are reliable but often impractical in resource-limited settings due to their dependency on complex equipment and skilled personnel. This study developed a novel malaria diagnostic platform by combining the Chelex-100/boiling DNA extraction method with a Loop-mediated Isothermal Amplification-MicroScanner (LAMP-MS) assay. The Chelex-100/boiling method is simpler and more cost-effective than conventional DNA extraction processes, making it suitable for use in resource-limited settings. The LAMP-MS assay enables multiplex detection through a microchip design with four chambers. Each chamber of the microchip is preloaded with specific primers targeting Pan, Plasmodium falciparum (Pf), Plasmodium vivax (Pv), and an internal control, minimizing non-specific amplification in multiplex LAMP reactions. In a clinical evaluation of 260 samples, the assay demonstrated a sensitivity of 97.5% for the Pan target and 100% for the Pf-specific target in the 80 Plasmodium falciparum (Pf) clinical samples. Similarly, for the 80 Plasmodium vivax (Pv) clinical samples, the assay achieved a sensitivity of 95% for the Pan target and 94% for the Pv-specific target. Notably, in the 100 non-infected clinical samples, the assay exhibited 100% specificity, with no false positives observed. These findings suggest that LAMP-MS is a rapid and reliable alternative to PCR-based methods, especially in resource-limited environments.
Similar content being viewed by others

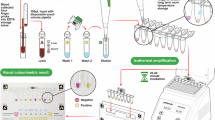
Introduction
Malaria is a febrile illness transmitted by the bite of infected Anopheles mosquitoes carrying Plasmodium protozoa (Plasmodium spp.), predominantly found in tropical regions worldwide1. Among the five types of malaria that can infect humans, P. falciparum and P. vivax are the most common2. P. falciparum is primarily found in Africa, while P. vivax occurs more frequently in Southeast Asia and the Western Pacific3. Malaria is a significant global public health issue, with approximately 241 million cases reported in 2020, leading to around 627,000 deaths, most of whom were children under the age of five in sub Saharan Africa4,5. The burden of malaria continues to place significant pressure on healthcare systems, economies, and communities, particularly in regions with limited access to healthcare6.
Early and accurate diagnosis of malaria is critical for effective treatment and reducing the disease burden, especially in resource-limited settings7. Malaria is a disease that requires immediate attention, as delayed diagnosis can result in severe complications or death8. Traditional malaria diagnostic methods include microscopy, rapid diagnostic tests (RDTs), and polymerase chain reaction (PCR)9,10,11. Each of these diagnostic methods has its own unique benefits and limitations, making it necessary to adapt diagnostic strategies according to the available resources and specific local needs. Microscopy is a well-established and widely available method that requires basic laboratory equipment, reagents, and supplies12. Blood smears are prepared and stained with Giemsa to identify malaria parasites13. This technique provides valuable information for treatment decisions14. However, microscopy is labor-intensive and requires a skilled microscopist, and its accuracy can be limited by the quality of staining and the ability of the technician15. Rapid Diagnostic Tests (RDTs) have gained popularity as an alternative means to establish a diagnosis quickly16. These tests detect specific malaria antigens in a patient’s blood using immunochromatographic methods17. RDTs are particularly useful in remote areas where microscopy is not feasible due to a lack of trained personnel or laboratory infrastructure18. The use of RDTs allows for a fast turnaround, often within 15–20 min, making them ideal for point-of-care testing. However, the sensitivity of RDTs can be compromised in cases of low parasitemia19. PCR is a molecular diagnostic tool that offers high sensitivity and specificity for detecting Plasmodium DNA20. This technique is capable of differentiating between malaria species and even identifying mixed infections, making it an invaluable tool for research and case confirmation21. However, PCR requires specialized equipment and trained personnel, limiting its applicability in remote or resource-poor settings22. The main limitation of PCR in acute settings is that it requires a longer processing time and sophisticated laboratory infrastructure, which are often unavailable in areas most affected by malaria23.
Given the limitations of traditional diagnostic methods, several emerging techniques have been developed to improve the speed, accuracy, and accessibility of malaria diagnostics. Loop-Mediated Isothermal Amplification (LAMP) has emerged as a promising alternative for malaria diagnosis, particularly in areas with limited healthcare infrastructure24. LAMP is an isothermal nucleic acid amplification technique that allows DNA or RNA to be amplified at a constant temperature, eliminating the need for the thermal cycling equipment used in PCR25. This simplification makes LAMP more suitable for field use, as it requires only minimal laboratory infrastructure26. LAMP provides results in less than an hour and has been shown to have high sensitivity and specificity, comparable to PCR27.
LAMP-based technology is well-suited for point-of-care diagnostics, and various detection methods have been developed in recent years. Colorimetric LAMP allows for easy visual confirmation of DNA amplification by detecting pH changes, which results in a color shift, eliminating the need for specialized equipment. This makes it highly advantageous in resource-limited settings28. Additionally, LAMP-amplified products can be detected using lateral flow dipsticks (LFD), which enables rapid visualization of results, making it a practical solution for environments with limited laboratory infrastructure29. Real-time fluorescence LAMP, which uses a portable isothermal amplification device and fluorescent dyes to monitor the amplification process in real-time, is particularly useful for diagnostic research30.
Recently, we developed a novel LAMP-based diagnostic method that enables visual detection of magnesium pyrophosphate, a byproduct of LAMP amplification, using either a microscope or a microscanner31. Based on this technology, in this study, we developed a malaria diagnostic platform by combining the Chelex-100 extraction method with LAMP-MS (Loop-Mediated Isothermal Amplification-MicroScanner) (Fig. 1). Specifically, we optimized the Chelex-100/boiling nucleic acid extraction method for malaria blood samples and designed a microchip for detecting malaria, which includes Pan, Pf, Pv, and internal controls (IC). The performance of the platform was evaluated by comparing its sensitivity, specificity, detection limit, and field applicability with those of a commercial qPCR kit.

Schematic illustration of the Malaria Pan/Pf/Pv/IC LAMP-MS assay.
Results
Optimization of the Chelex-100/boiling method for nucleic acid extraction
To determine the optimal conditions for DNA extraction from blood samples using the Chelex-100/heat method with newly developed custom-made extraction tube (Fig. S1), experiments were conducted by combining different diluted whole blood concentrations (2.5%, 5%, 10%) in 10% Chelex-100 resin solution (Tris-EDTA buffer) and different Chelex-100 resin concentrations (5%, 10%, 20%) in Tris-EDTA buffer with 2.5% whole blood. The experimental results were compared and analyzed using the Malaria Pan qPCR and Malaria Pan LAMP assays (Fig. 2). First, in experiments with varying blood concentrations, DNA was extracted and analyzed from a 10% Chelex-100 solution mixed with whole blood at concentrations of 2.5%, 5%, and 10%. The Malaria Pan qPCR results showed Ct values of 19.23, 19.57, and 19.80, with RFU values of 3163, 1767, and 865, respectively. The Malaria Pan LAMP results showed nucleic acid amplification times of 12.94 min, 13.06 min, and 13.55 min, with RFU values of 3020, 2010, and 1209, respectively. In both assays, the 2.5% blood sample demonstrated the fastest Ct values and the highest RFU values. Therefore, a blood concentration of 2.5% was determined to be the optimal blood concentration for DNA extraction. Next, experiments with varying Chelex-100 resin concentrations (5%, 10%, 20%) in Tris-EDTA buffer with 2.5% whole blood were conducted for DNA extraction. In the Malaria Pan qPCR assay, Ct values were 24.94, 22.23, and 21.99, with RFU values of 6390, 6645, and 5846, respectively. In the Malaria Pan LAMP assay, nucleic acid amplification times were 13.13 min, 12.08 min, and 12.12 min, with RFU values of 4918, 4654, and 4146, respectively. While the qPCR results indicated the fastest Ct value with the 20% Chelex-100 solution, the LAMP assay results were used to determine the optimal conditions. Based on the LAMP assay, the 10% Chelex-100 solution exhibited the fastest Ct value and the highest RFU value, making it the optimal Chelex-100 resin solution concentration. In conclusion, the optimal conditions for DNA extraction were identified as a blood concentration of 2.5% and a Chelex-100 resin solution concentration of 10%.

Optimization of the Chelex-100/boiling method for nucleic acid extraction from blood. Comparison of blood sample concentrations (2.5%, 5%, and 10%) for nucleic acid extraction was performed using both the Malaria Pan qPCR assay (A) and the Malaria Pan LAMP assay (B). Similarly, Chelex-100 resin concentrations (5%, 10%, and 20%) were tested for nucleic acid extraction using both the Malaria Pan qPCR assay (C) and the Malaria Pan LAMP assay (D).
Development of malaria Pan/Pf/Pv/IC LAMP-MS assay
Here, we developed the Pan/Pf/Pv/IC LAMP-MS assay for the detection of malaria and evaluated its performance in comparison with the multiplex malaria Pan/Pf/Pv/IC LAMP assay32 using P. falciparum samples. The results of the multiplex malaria Pan/Pf/Pv/IC LAMP assay (Fig. 3A) showed nucleic acid amplification for the Pan, Pf, and IC targets, but not for the Pv target. The newly developed LAMP-MS assay (Fig. 3B) yielded diagnostic outcomes consistent with those of the LAMP assay. Furthermore, the LAMP-MS assay allowed for the direct visualization of amplified nucleic acids using a portable microscope. Small granules, indicative of nucleic acid amplification were observed in positive samples, while no granules were detected in negative samples. These results confirm the reliability of the LAMP-MS assay in providing accurate diagnostic outcomes with clear visual differentiation between positive and negative samples.

Detection of Malaria Using the Newly Developed LAMP-MS Assay. (A) Results of the Pan/Pf/Pv/IC LAMP assay for a Plasmodium falciparum sample. The graph shows the fluorescence amplification curves for Pan (①), Pf (②), Pv (③), and IC (④) targets. (B) Results of the Pan/Pf/Pv/IC LAMP-MS assay for a Plasmodium falciparum sample. Microscopic images show the presence of amplified DNA products for Pan (①), Pf (②), and IC (④) targets, while no amplification is observed for the Pv (③) target.
Detection limit of the malaria Pan/Pf/Pv/IC LAMP-MS assay
The limit of detection (LoD) for the Malaria Pan/Pf/Pv/IC LAMP-MS assay was evaluated using Plasmodium falciparum (Pf) and Plasmodium vivax (Pv) clinical samples that were serially diluted in 10-fold increments. Two DNA extraction methods, Chelex-100/boiling and the Qiagen QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany), were used, and the resulting data were compared with those obtained from both the Malaria Pan qPCR assay and BinaxNOW™ Malaria rapid diagnostic test (RDT) (Binax, Inc., Scarborough, Maine, USA). For P. falciparum, the LAMP-MS assay consistently demonstrated an LoD of 1.1 × 102 parasites/µL with both extraction methods (Table 1, Fig. S2). By contrast, the qPCR assay was able to detect as few as 1.1 × 101 parasites/µL, reflecting higher sensitivity than the LAMP-MS assay, whereas the RDT showed a LoD of 1.1 × 103 parasites/µL (Table 1, Fig. S4), indicating comparatively lower sensitivity. For P. vivax, the LAMP-MS assay showed a LoD of 2 × 102 parasites/µL (Table 2, Fig. S3). However, the qPCR assay exhibited different sensitivities depending on the extraction method: the Qiagen kit enabled detection of as few as 2 × 100 parasites/µL, representing the highest sensitivity, while the Chelex-100 method yielded a LoD of 2 × 101 parasites/µL, comparable to that of the LAMP-MS assay. In the RDT results for P. vivax, detection was possible at 2 × 103 parasites/µL (Table 1, Fig. S4), indicating lower sensitivity relative to both LAMP-MS and qPCR. Overall, the LAMP-MS assay demonstrated robust and consistent LoD values irrespective of the DNA extraction method, confirming its reliability as a diagnostic tool. Although the qPCR assay displayed slightly higher overall sensitivity, the LAMP-MS assay proved more sensitive than the RDT.
Clinical performance of the malaria Pan/Pf/Pv/IC LAMP-MS assay
The clinical performance of the Malaria Pan/Pf/Pv/IC LAMP-MS assay was evaluated using 260 clinical samples, comprising 80 Plasmodium falciparum (Pf) samples, 80 Plasmodium vivax (Pv) samples, and 100 non-infected samples (Table 3). All DNA was extracted from clinical samples using the Chelex-100/boiling DNA extraction method. The results were analyzed in comparison with the commercial RealStar® Malaria Screen & Type PCR kit. For P. falciparum samples, the LAMP-MS assay demonstrated a sensitivity of 97.5% for the Pan target (95% CI: 91.3–99.7) and 100% for the Pf-specific target (95% CI: 95.5–100.0), showing equivalent performance to the RealStar® PCR kit. Both assays exhibited 100% specificity for P. falciparum detection, with no false positives observed for any target. For P. vivax samples, the LAMP-MS assay recorded a sensitivity of 95% for the Pan target (95% CI: 87.7–98.2) and 94% for the Pv-specific target (95% CI: 86.0–97.9). In contrast, the RealStar® PCR kit demonstrated 100% sensitivity for the Pv-specific target, indicating a slight difference in performance. Nevertheless, both assays achieved 100% specificity for P. vivax detection, with no false positives observed across all targets. Finally, for the 100 non-infected samples, the LAMP-MS assay exhibited 100% specificity across all diagnostic targets (Pan, Pf, and Pv), while achieving 100% sensitivity for internal control (IC). These results were consistent with the RealStar® PCR kit. These findings highlight the comparable performance of the Malaria Pan/Pf/Pv/IC LAMP-MS assay to the RealStar® PCR kit in detecting P. falciparum. Although the LAMP-MS assay showed slightly lower sensitivity for P. vivax, its ability to deliver accurate results with high specificity underscores its reliability as a diagnostic tool for malaria.
Cross-reactivity test
The cross-reactivity of the Malaria Pan/Pf/Pv/IC LAMP-MS assay was evaluated using samples infected with other viruses. The assay was tested against Zika virus, Chikungunya virus, Japanese encephalitis virus, and Dengue virus (types 1, 2, 3, and 4). The results showed that the Malaria Pan/Pf/Pv/IC LAMP-MS assay exhibited no cross-reactivity with any of these viruses, with negative results for all targets (Pan, Pf, Pv, and the internal control [IC]). Additionally, when tested with human whole blood DNA and distilled water, the assay showed positive results only for the internal control (IC), while Pan, P. falciparum, and P. vivax targets all returned negative results (Table 4). These findings indicate that the Malaria Pan/Pf/Pv/IC LAMP-MS assay has excellent specificity and does not cross-react with other viruses, making it a highly reliable tool for malaria diagnosis.
Discussion
Malaria is a disease that poses serious health problems worldwide, infecting hundreds of millions of people and causing hundreds of thousands of deaths each year33. This disease is prevalent mainly in tropical and subtropical regions, becoming a significant obstacle to economic development and public health34. In such a situation, rapid and accurate point-of-care diagnosis of malaria is essential for early treatment of patients and prevention of disease spread35. However, conventional malaria diagnostic methods, especially PCR-based molecular diagnostics, offer high sensitivity and specificity but have limitations in resource-limited settings due to the need for complex equipment and skilled personnel36,37. To overcome these limitations, we developed a malaria diagnostic platform by combining LAMP-MS assay with the Chelex-100/boiling extraction method. In particular, the Chelex-100/boiling extraction method is simpler and more cost-effective compared to traditional complex nucleic acid extraction processes, making it applicable even in resource-limited environments. Furthermore, the LAMP-MS assay utilizes the formation of magnesium pyrophosphate during the LAMP reaction as a visual marker, allowing direct observation of nucleic acid amplification through a microscope or a microscanner. This approach eliminates the need for additional chemical indicators or probes, thereby simplifying the detection process, reducing costs, and enhancing the assay’s accessibility for point-of-care testing.
One of the most significant strengths of the LAMP-MS assay is its ability to perform multiplex detection using a microchip composed of four chambers, each preloaded with specific primers. Traditional multiplex LAMP assays that utilize probes often require multiple primers and probes in a single tube, necessitating meticulous design to prevent non-specific amplification38. In contrast, our LAMP-MS assay assigns one LAMP primer set per target and distributes different target primers across separate chambers on the microchip. This design allows for the simultaneous detection of various genes without interference between target primers, thereby simplifying the primer design process and increasing the robustness of the assay. In our study, we successfully detected Pan, Plasmodium falciparum (Pf), Plasmodium vivax (Pv), and an internal control simultaneously. The assay’s multiplexing capability can be significantly expanded by fabricating chips with additional chambers, meaning more targets can be included in a single test. This flexibility enhances the assay’s utility in detecting multiple pathogens simultaneously, which is particularly beneficial in regions where mixed infections are common. In a clinical evaluation of 260 samples, the assay demonstrated a sensitivity of 97.5% for the Pan target and 100% for the Pf-specific target in the 80 Plasmodium falciparum (Pf) clinical samples. Similarly, for the 80 Plasmodium vivax (Pv) clinical samples, the assay achieved a sensitivity of 95% for the Pan target and 94% for the Pv-specific target. Notably, in the 100 non-infected clinical samples, the assay exhibited 100% specificity, with no false positives observed. Additionally, the LAMP-MS assay showed 100% specificity in cross-reactivity tests with other tropical disease pathogens. These results are comparable to those of the commercial RealStar® Malaria Screen & Type PCR kit, indicating that the LAMP-MS assay is a reliable alternative to PCR-based methods. Furthermore, the LAMP-MS assay can provide results within 30 min of incubation at 60 °C, making it well-suited for point-of-care settings where rapid decision-making is crucial.
The LAMP-MS platform provides a cost-effective alternative to conventional RT-PCR, with minimal instrumentation requirements and affordable per-sample costs. As previously reported31, the LAMP-MS assay offers significant advantages in terms of cost, time, and ease of use, requiring only heating blocks and micro-scanners instead of the sophisticated and expensive equipment used in RT-PCR. For sample preparation, the Chelex-based extraction method further reduces costs by eliminating the need for commercial nucleic acid extraction kits. This allows for a per-sample extraction cost of less than $1, making it an attractive option for low-resource settings. Combined with the $4–6 per-test cost of LAMP-MS31, this approach ensures that the overall diagnostic process remains highly cost-effective.Moreover, the cost-effectiveness of LAMP-MS does not come at the expense of diagnostic accuracy. Clinical validation has shown that the assay achieves high sensitivity and specificity, comparable to established molecular diagnostic methods31. Given its robust performance, reproducibility, and simplified workflow, LAMP-MS is particularly well-suited for field applications and decentralized testing, where access to sophisticated laboratory infrastructure is limited.
One of the primary limitations of this assay is the risk of cross-contamination during sample loading into the microchip channels, particularly in field settings where controlled laboratory conditions may be lacking. Since pipetting is an essential step in this method, the potential for aerosol contamination and carryover between samples must be considered. Additionally, the requirement for multiple pipetting steps increases the likelihood of human error, which may affect the reproducibility and reliability of the assay. Future research should focus on the development of a fully enclosed cartridge system, which would minimize manual pipetting and further enhance field applicability by reducing contamination risks and improving usability.
In conclusion, the combination of the Chelex-100 extraction method and the micro-chip-based multiplex LAMP-MS platform offers a highly promising approach for malaria diagnosis in terms of speed and accuracy. However, while the platform demonstrates encouraging results, further validation is necessary to confirm its feasibility in real field conditions. Specifically, additional studies should evaluate the assay’s performance when conducted entirely in field settings, including considerations for pipetting, multistep processes, and contamination control. Furthermore, its applicability to dried blood spots and capillary blood samples should be assessed to ensure practical usability in remote or resource-limited areas. Although there may be some sensitivity limitations compared to qPCR, the assay is particularly useful in resource-limited settings and can contribute significantly to the effective management and eradication of malaria. Future improvements in sensitivity for P. vivax and the inclusion of additional malaria species could further expand the assay’s diagnostic capabilities.
Methods
Clinical samples
Malaria residual blood samples were collected from patients suspected of malaria infection at Korea University Guro Hospital between 2013 and 2022. These clinical blood samples were originally obtained for routine diagnostic testing and stored at -80 °C after testing. Malaria infection was confirmed through various methods, including Giemsa staining, blood smear microscopy, rapid antigen tests, and PCR tests. A total of 260 clinical samples were used in this study, including 80 P. falciparum, 80 P. vivax, and 100 negative clinical samples. To confirm the cross-reactivity of Malaria pan/Pf/Pv/IC LAMP-MS assay, Zika virus, Chikungunya virus, and Japanese encephalitis virus, provided by the Korea Disease Control and Prevention Agency (KDCA), were used. This study was approved by the Medical Ethics Committee of Korea University’s Guro Hospital (2021GR0337). All experiments were performed in accordance with relevant guidelines and regulations.
DNA extraction
DNA extraction was performed using two methods: the Qiagen QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany) and a custom-designed Chelex-100/boiling nucleic acid extraction method. The Qiagen kit was used as the standard to evaluate the performance of the Chelex-100/boiling method. For the Qiagen protocol, 200 µL of blood samples were lysed with a proprietary buffer and proteinase K to break down the cells, allowing the DNA to bind to a silica membrane in a spin column. After several washing steps to remove impurities, the DNA was eluted in 100 µL of elution buffer. In the Chelex-100/boiling method, a custom-made extraction tube (Fig. S1) was used. This tube contained 195 µL of a 10% Chelex-100 resin solution (10 mM Tris-HCl, 1 mM EDTA, pH 8.0). The sample tube is made of a soft plastic material, allowing the solution inside to be expelled when pressed. The tube cap consists of two parts: a blue part with a hole and a transparent white cover that seals the hole. A 0.45 μm filter is built inside the blue cover, enabling the removal of Chelex-100 resin from the sample solution. The 10% (w/v) Chelex-100 resin solution was prepared by suspending 1 g of Chelex-100 resin powder in 10 ml of Tris-EDTA buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0). The solution was thoroughly vortexed to ensure homogeneity, and 195 µL of the prepared 10% Chelex-100 resin solution was dispensed into the custom-made extraction tube. For DNA extraction, 5 µL of blood was added to the tube, and the cap was securely closed. The sample was heated at 100 °C for 10 min to promote blood sample lysis. After heating, the tube was compressed, filtering out the Chelex-100 resin, and about 100 µL of the nucleic acid containing solution was collected in a new tube through the filter. The optimization experiment for the Chelex-100 extraction method from blood was conducted using Malaria Pan qPCR (Table S1) and the multiplex malaria Pan/Pf/Pv/IC LAMP assays32.
Malaria pan/Pf/Pv/IC LAMP-MS assay
The LAMP primer sets targeting Malaria Pan, Plasmodium falciparum, Plasmodium vivax, and an internal control (IC) used in this study were previously developed by our research group (Table 5)32. These LAMP primers were designed to amplify conserved gene regions specific to each target: the 18 S rRNA gene for Malaria Pan, the LDH gene for P. falciparum, the 16 S rRNA gene for P. vivax, and the actin beta gene for internal control. All primers were synthesized by Macrogen Inc. (Seoul, Korea). For the LAMP-microscanner assay, each primer mix contained 4 µM of two outer primers (F3 and B3), 32 µM of two inner primers (FIP and BIP), and 10 µM of two loop primers (FLP and BLP). Then, 0.5 µL of each LAMP primer set for Malaria Pan, P. falciparum, P. vivax, and IC was loaded into separate channels on a microchip (Biozentech, Seoul, Korea). The microchip was dried in an oven at 60 °C for 1 h and stored at room temperature until use. The reaction mixture of the Malaria pan/Pf/Pv/IC LAMP-MS assay was prepared with 25 µL of Mmiso® DNA amplification kit Master Mix (Mmonitor, Daegu, Korea), 4 µL of Mmiso® DNA amplification kit Enzyme (Mmonitor, Daegu, Korea), 15 µL of distilled water and 6 µL of DNA sample (final reaction volume: 50 µL). Next, 10 µL of the prepared reaction mixture (containing test DNA and LAMP reagents) was loaded into each channel of the microchip, which was subsequently placed on a heating block (Beijing HiYi Technology, Beijing, China) at 62 °C for 30 min. After the LAMP reaction, amplification was confirmed using a microscanner (Biozentech, Seoul, Korea) or an optical RV400TMCL microscope (OMAX OMB microscope, Gyeonggi-do, Korea) (Fig. 1).
Quantitative real-time PCR
For clinical performance of the Malaria pan/Pf/Pv/IC LAMP-MS assay was compared and evaluated against that of the RealStar® Malaria Screen & Type PCR Kit 1.0 (Altona Diagnostics, Hamburg, Germany) using the CFX96 Touch Real-Time PCR Detection System (Bio-Rad Laboratories) following a manufacture’s protocol. The thermocycling parameters of the RealStar® Malaria Screen & Type PCR kit was as follows: inactivation at 95 °C for 2 min, 45 cycles of denaturation at 95 °C for 15 s, annealing with fluorescence detection at 58 °C for 45 s, and extension at 72 °C for 15 s. For the optimization of the Chelex-100/boiling DNA extraction method and comparative performance testing of the Malaria Pan/Pf/Pv/IC LAMP-MS assay, an in-house Malaria Pan qPCR (Table S1) was performed. The PCR conditions were as follows: 95 °C for 3 min, followed by 45 cycles of 95 °C for 15 s and 60 °C for 30 s. The reaction mixture (20 µL) consisted of 10 µL of EzAmp™ HS qPCR 2X Master Mix (ELPIS Biotech, Daejeon, Korea), 1 µL of primer mix, 5 µL of template DNA, and the remaining volume was adjusted with nuclease-free water.
Limit of detection tests of the malaria Pan/Pf/Pv/IC LAMP-MS assay
The limit of detection (LoD) for the Malaria Pan/Pf/Pv/IC LAMP-MS assay was determined using actual clinical samples of Plasmodium falciparum (Pf) and Plasmodium vivax (Pv). Parasite density in each sample was estimated by reviewing hospital records and patient data, and by counting parasites in Giemsa-stained blood smears in conjunction with white blood cell (WBC) counts. From these calculations, parasite densities were determined to be 114,511 parasites/µL for Pf and 20,832 parasites/µL for Pv. For the LoD determination, the Pf and Pv clinical samples were serially diluted in phosphate-buffered saline (PBS), and DNA extraction was performed using both the Qiagen QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany) and the Chelex-100/boiling method. The extracted DNA was then tested using both the LAMP-MS assay and the Malaria Pan qPCR assay, whereas the BinaxNOW™ Malaria rapid diagnostic test (RDT) (Binax, Inc., Scarborough, Maine, USA) was performed with the serially diluted whole blood samples. The LoD was defined as the lowest parasite concentration at which consistent positive results were obtained. The final LoD values were confirmed by consistent results across three independent experiments, and were then compared with those obtained from the RDT and the Malaria Pan qPCR assay to evaluate the performance of the LAMP-MS assay.
Statistics
The confidence intervals (CI) for sensitivity and specificity were set at 95%. The sensitivity, specificity and 95% CI for the assays were calculated using a diagnostic test evaluation calculator program (https://www.medcalc.org/calc/diagnostic_test.php, accessed on 21 December 2021).
Data availability
Data will be made available on request (plasmid18@korea.ac.kr).
References
Paton, D. G. et al. Exposing Anopheles mosquitoes to antimalarials blocks plasmodium parasite transmission. Nature 567, 239–243 (2019).
Naing, C., Whittaker, M. A., Nyunt Wai, V. & Mak, J. W. Is plasmodium Vivax malaria a severe malaria? A systematic review and Meta-Analysis. PLoS Negl. Trop. Dis. 8, e3071 (2014).
Gore-Langton, G. R. et al. Global estimates of pregnancies at risk of plasmodium falciparum and plasmodium Vivax infection in 2020 and changes in risk patterns since 2000. PLOS Global Public. Health. 2, e0001061 (2022).
Rosenthal, P. J. Malaria in 2022: challenges and progress. Am. J. Trop. Med. Hyg. 106, 1565–1567 (2022).
Roberts, D. & Matthews, G. Risk factors of malaria in children under the age of five years old in Uganda. Malar. J. 15, 246 (2016).
Stratton, L., O’Neill, M. S., Kruk, M. E. & Bell, M. L. The persistent problem of malaria: addressing the fundamental causes of a global killer. Soc. Sci. Med. 67, 854–862 (2008).
Landier, J. et al. The role of early detection and treatment in malaria elimination. Malar. J. 15, 363 (2016).
Trampuz, A., Jereb, M., Muzlovic, I. & Prabhu, R. M. Clinical review: severe malaria. Crit. Care. 7, 315 (2003).
Kudisthalert, W., Pasupa, K. & Tongsima, S. Counting and classification of malarial parasite from Giemsa-Stained thin film images. IEEE Access. 8, 78663–78682 (2020).
Dietze, R. et al. The diagnosis of plasmodium falciparum infection using a new antigen detection system. Am. J. Trop. Med. Hyg. 52, 45–49 (1995).
Erdman, L. K. & Kain, K. C. Molecular diagnostic and surveillance tools for global malaria control. Travel Med. Infect. Dis. 6, 82–99 (2008).
Ishengoma, D. R. S. et al. The performance of health laboratories and the quality of malaria diagnosis in six districts of Tanzania. Ann. Trop. Med. Parasitol. 104, 123–135 (2010).
Iqbal, J., Hira, P. R., Al-Ali, F., Khalid, N. & Sher, A. Modified Giemsa staining for rapid diagnosis of malaria infection. Med. Principles Pract. 12, 156–159 (2003).
Siahaan, L. Laboratory diagnostics of malaria. IOP Conf. Ser. Earth Environ. Sci. 125, 012090 (2018).
Yoon, J., Jang, W. S., Nam, J., Mihn, D. C. & Lim, C. S. An automated microscopic malaria parasite detection system using digital image analysis. Diagnostics 11, 527 (2021).
Ishengoma, D. S. et al. Accuracy of malaria rapid diagnostic tests in community studies and their impact on treatment of malaria in an area with declining malaria burden in north-eastern Tanzania. Malar. J. 10, 176 (2011).
Moody, A. Rapid diagnostic tests for malaria parasites. Clin. Microbiol. Rev. 15, 66–78 (2002).
Kyabayinze, D. J. et al. Programme level implementation of malaria rapid diagnostic tests (RDTs) use: outcomes and cost of training health workers at lower level health care facilities in Uganda. BMC Public. Health. 12, 291 (2012).
McMorrow, M. L., Aidoo, M. & Kachur, S. P. Malaria rapid diagnostic tests in elimination settings—can they find the last parasite? Clin. Microbiol. Infect. 17, 1624–1631 (2011).
Cunha, M. G. et al. Development of a polymerase chain reaction (PCR) method based on amplification of mitochondrial DNA to detect plasmodium falciparum and plasmodium Vivax. Acta Trop. 111, 35–38 (2009).
Kimura, M. et al. Identification of the four species of human malaria parasites by nested PCR that targets variant sequences in the small subunit rRNA gene. Parasitol. Int. 46, 91–95 (1997).
Mouatcho, J. C. & Goldring, J. P. D. Malaria rapid diagnostic tests: challenges and prospects. J. Med. Microbiol. 62, 1491–1505 (2013).
Britton, S., Cheng, Q. & McCarthy, J. S. Novel molecular diagnostic tools for malaria elimination: a review of options from the point of view of high-throughput and applicability in resource limited settings. Malar. J. 15, 88 (2016).
Lucchi, N. W., Ndiaye, D., Britton, S. & Udhayakumar, V. Expanding the malaria molecular diagnostic options: opportunities and challenges for loop-mediated isothermal amplification tests for malaria control and elimination. Expert Rev. Mol. Diagn. 18, 195–203 (2018).
Abdul-Ghani, R., Al-Mekhlafi, A. M. & Karanis, P. Loop-mediated isothermal amplification (LAMP) for malarial parasites of humans: would it come to clinical reality as a point-of-care test? Acta Trop. 122, 233–240 (2012).
Cuadros, J. et al. Field evaluation of malaria microscopy, rapid malaria tests and Loop-Mediated isothermal amplification in a rural hospital in South Western Ethiopia. PLoS One. 10, e0142842 (2015).
Marti, H., Stalder, C. & González, I. J. Diagnostic accuracy of a LAMP kit for diagnosis of imported malaria in Switzerland. Travel Med. Infect. Dis. 13, 167–171 (2015).
Zen, L. P. Y. et al. End-point detection of loop-mediated isothermal amplification (LAMP) on malaria by direct observation with colorimetric dyes. Exp. Parasitol. 239, 108310 (2022).
Kongkasuriyachai, D., Yongkiettrakul, S., Kiatpathomchai, W. & Arunrut, N. Loop-Mediated isothermal amplification and LFD combination for detection of Plasmodium falciparum and Plasmodium vivax, 431–443 (2017). https://doi.org/10.1007/978-1-4939-6911-1_28
Lucchi, N. W. et al. Real-Time fluorescence loop mediated isothermal amplification for the diagnosis of malaria. PLoS One. 5, e13733 (2010).
Choi, M., Lee, E., Park, S., Lim, C. S. & Jang, W. S. Enhanced Point-of-Care SARS-CoV-2 detection: integrating RT-LAMP with microscanning. Biosens. (Basel). 14, 348 (2024).
Jang, W. S. et al. Development of a multiplex Loop-Mediated isothermal amplification assay for diagnosis of plasmodium spp., plasmodium falciparum and plasmodium Vivax. Diagnostics 11, 1950 (2021).
Patel, P., Bagada, A. & Vadia, N. Epidemiology and current trends in malaria. In Rising Contagious Diseases, 261–282 (Wiley, 2024). https://doi.org/10.1002/9781394188741.ch20.
Recht, J. et al. Malaria in Brazil, Colombia, Peru and Venezuela: current challenges in malaria control and elimination. Malar. J. 16, 273 (2017).
Rei Yan, S. L., Wakasuqui, F. & Wrenger, C. Point-of-care tests for malaria: speeding up the diagnostics at the bedside and challenges in malaria cases detection. Diagn. Microbiol. Infect. Dis. 98, 115122 (2020).
Farcas, G. A., Zhong, K. J. Y., Mazzulli, T. & Kain, K. C. Evaluation of the RealArt malaria LC Real-Time PCR assay for malaria diagnosis. J. Clin. Microbiol. 42, 636–638 (2004).
Lyimo, B. M. et al. Potential opportunities and challenges of deploying next generation sequencing and CRISPR-Cas systems to support diagnostics and surveillance towards malaria control and elimination in Africa. Front. Cell. Infect. Microbiol. 12 (2022).
Sharma, S., Singh, J., Sen, A. & Anvikar, A. R. Multiplex loop mediated isothermal amplification (m-LAMP) as a point of care technique for diagnosis of malaria. J. Vector Borne Dis. 59, 29–36 (2022).
Acknowledgements
We thank the Korea Disease Control and Prevention Agency (KDCA) for providing Zika virus, Chikungunya virus, and Japanese encephalitis virus samples.
Funding
This work was supported by a grant from the Korea Health Technology R&D Project, through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health and Welfare, Republic of Korea (grant number: HR20C0021); a grant from the National Research Foundation of Korea (NRF), funded by the Korean Government, MSIP (RS-2023-00207833); the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (grant number: 2022R1I1A1A01063976). The authors declare no conflict of interest.
Author information
Authors and Affiliations
Contributions
W.S. Jang: Conceptualization, Methodology, Writing-Original Draft, Writing-Review & Editing, Funding acquisition. M.K. Choi: Writing-Original Draft, Investigation, Visualization. Y.L. Choe: Methodology, Validation, Resources. C.S. Lim: Supervision, Writing-Review & Editing, Funding acquisition.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
This study was approved by the Medical Ethics Committee of Korea University’s Guro Hospital (2021GR0337).
Informed consent
Patient informed consent was waived by the Institutional Review Board of Korea University Guro Hospital, as the identities of the subjects were completely anonymous, and there was minimal risk involved in the study.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jang, W.S., Choi, M.K., Choe, Y.L. et al. Development of a rapid malaria LAMP-MS assay for diagnosis of malaria infections. Sci Rep 15, 8547 (2025). https://doi.org/10.1038/s41598-025-92935-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-92935-4
