
Abstract
The relationship between LRRK2 mutations and susceptibility to synuclein pathology in Parkinson’s disease (PD) is still unclear. We here investigate whether the mice carrying the D1994S kinase-dead (KD) mutation of LRRK2 show enhanced susceptibility to synucleinopathy. Twelve-month-old LRRK2 KD and WT mice were injected with AAV2/9 carrying human A53T α-synuclein (AAV-h-A53Tα-syn) or AAV2/9-GFP as a control. Three months after injection, α-synuclein pathology and nigrostriatal dopaminergic neuron degeneration were assessed along with motor behaviour. AAV-h-A53Tα-syn-injected LRRK2 KD mice showed a decline in stepping activity in the drag test compared to baseline levels and AAV-GFP-injected controls. This was associated with higher transgene levels and Serine129 α-syn phosphorylation in striatum and substantia nigra measured by immunohistochemistry. Total α-synuclein levels were also elevated in the substantia nigra but not striatum of AAV-h-A53Tα-syn LRRK2 KD mice compared to AAV-h-A53Tα-syn controls. Stereological counting of nigral dopaminergic neurons and densitometric analysis of striatal dopaminergic terminals did not reveal overt nigrostriatal degeneration. We conclude that silencing of kinase activity results in greater α-syn load due to greater viral transduction and/or defective α-syn clearance, possibly related to autophagy-lysosomal pathway impairment, however, with no consequence upon dopaminergic neuron survival in the mouse.
Similar content being viewed by others

Introduction
Deposition of α-synuclein aggregates in the central nervous system (CNS) is a common feature of synucleinopathies, a class of age-dependent neurodegenerative disorders featuring Parkinson’s Disease (PD). The neuropathological hallmarks of PD are the loss of dopaminergic neurons in substantia nigra pars compacta (SNpc) and the accumulation of intracytoplasmic inclusions, called Lewy Bodies (LB) and Lewy neurites, mainly composed of α-synuclein phosphorylated at Serine129 (pSer129 α-syn). Mutations in the LRRK2 gene cause autosomal dominant forms of PD1,2, whereas LRRK2 itself is considered a risk factor of idiopathic PD3. LRRK2 is a multidomain protein containing kinase and GTPase domains surrounded by several protein-protein interacting motifs4. The most prevalent G2019S mutation occurs in the kinase domain and enhances the kinase activity of the protein5,6,7. Among the possible mechanisms linking the increase in kinase activity associated with LRRK2 G2019S to PD pathogenesis is the modulation of α-syn function and/or turnover. Transgenic G2019S overexpressors and G2019S knock-in (KI) mice do not spontaneously develop typical LBs or LB-like a-syn inclusions8,9. However, immunoblot analysis has revealed increased levels of high-molecular weight α-syn species and pSer129 α-syn in conditional hG2019S transgenic mice10 or increased pSer129 α-syn levels in G2019S KI mice11,12. Instead, it was originally reported in double transgenic mice that overexpression of WT LRRK2 or G2019S LRRK2 worsened A53T α-syn pathology13 although in later studies, this effect was either less robust14 or absent15. Conversely, overexpression of human α-syn via adeno-associated viral (AAV) vectors resulted in larger nigrostriatal dopaminergic neurodegeneration and α-syn accumulation in BAC G2019S LRRK2 rats16 or G2019S KI mice12 compared to wild-type (WT) controls. This was also evident in BAC G2019S mice injected with preformed fibrils (PFFs) of α-syn, suggesting a role of LRRK2 in α-syn turnover and spreading17. Moreover, the larger synuclein pathology reported in G2019S KI mice became evident at 12 months but not younger ages12 further suggesting that it could rely on a progressive deficit in autophagy-lysosomal pathway (ALP), that is responsible for α-syn clearance in physio-pathological conditions18. In addition, ALP function deteriorates over aging19, the main risk factor for PD. However, despite several in vitro studies inconsistently showed ALP dysfunction in cells or neurons overexpressing LRRK2 G2019S20,21,22, in vivo studies failed to prove major ALP changes in G2019S KI mice21,23,24,25. However, when focusing upon the striatum and not the entire brain, we found that 12-month-old LRRK2 kinase-dead (KD) mice had impaired ALP21, suggesting that silencing and not hyperactivation of LRRK2 kinase activity negatively impacts on basal striatal autophagy in vivo in disease-relevant brain structure. We thus sought to investigate whether the ALP impairment observed in the striatum and SNpc of LRRK2 KD mice21 is associated with dysfunction of α-syn handling, leading to its accumulation. Recombinant AAV2/9 vector carrying h-A53T-α-syn (AAV-h-A53Tα-syn)12,26,27 was injected in the SNpc of 12-month-old LRRK2 KD and WT mice. Control mice were injected with AAV2/9 vector carrying GFP (AAV-GFP). Motor performance was evaluated before and 1, 2 and 3 months after AAV injection. Immunohistochemistry (IHC) was performed to assess α-syn and pSer129 α-syn levels in both the striatum and the SNpc, as well as degeneration of nigral dopaminergic cell bodies and striatal dopaminergic terminals.
Results
Based on our recent finding that 12-month-old LRRK2 KD mice have impaired striatal autophagic flux21, we investigated whether overexpression of h-A53Tα-syn in these mice could result in greater α-syn accumulation and neuro degeneration and worsened parkinsonian phenotype.
Assessment of motor activity with the drag test (Fig. 1) revealed no difference across genotypes in the number of steps at baseline (WT 8.33 ± 0.51 n = 18, LRRK2 KD 8.27 ± 0.29 n = 18). Three-way ANOVA showed an overall effect of treatment (F1,128 = 86.12, p < 0.0001), time (F3,128 = 3.675, p = 0.0140) and genotype (F1,128 = 5.389, p = 0.0218) but no interactions between these variables. Mice injected with AAV-h-A53Tα-syn showed a reduced performance compared to AAV-GFP-injected mice, irrespective of genotype. Interestingly, however, LRRK2 KD mice injected with AAV-h-A53Tα-syn showed a reduced performance compared to their baseline 3 months after injection, whereas WT mice injected with AAV-h-A53Tα-syn did not (Fig. 1). To test the efficiency of transgene expression upon AAV-h-A53Tα-syn injections, h-A53Tα-syn levels were measured in SNpc and striatum (Fig. 2). Aligned rank transform (ART) ANOVA revealed high levels of h-A53Tα-syn staining in the SNpc of mice injected with AAV-h-A53Tα-syn (Fig. 2A) (treatment F1,32 = 96.78 p < 0.0001, genotype F1,32 = 33.42 p < 0.0001, treatment X genotype F1,32 = 31.02 p < 0.0001). Moreover, a ~ 50% greater nigral h-A53Tα-syn load was detected in AAV-h-A53Tα-syn-injected LRRK2 KD mice compared to AAV-h-A53Tα-syn-injected WT mice. Consistently, ART ANOVA revealed high levels of h-A53Tα-syn staining in the striatum of mice injected with AAV-h-A53Tα-syn compared to those injected with the GFP construct, and also a stronger h-A53Tα-syn staining in LRRK2 KD mice (84%) compared to WT mice (51%) (Fig. 2B, treatment F1,32 = 96.7 p < 0.0001, genotype F1,32 = 27.32 p < 0.0001, treatment X genotype F1,32 = 27.21 p < 0.0001). Next, we wondered whether h-A53Tα-syn overexpression increased α-syn phosphorylation at Ser129 and this effect was larger in LRRK2 KD mice (Figs. 3 and 4). pSer129 α-syn levels were evident in AAV-h-A53Tα-syn-injected mice but not AAV-GFP mice. In both SNpc (Fig. 3A) and striatum (Fig. 4A), pSer129 α-syn staining after AAV-h-A53Tα-syn injection was about twice larger in LRRK2 KD mice compared to WT mice (SNpc, t = 2.92 df = 14 p = 0.011; striatum, t = 1.74 df = 17 p = 0.098). Consistently, two-way ANOVA on total α-syn levels in SNpc (Fig. 3B) revealed a significant effect of treatment (F1,32 = 32.6 p < 0.0001), genotype (F1,32 = 11.54 p = 0.0018), and a significant treatment X genotype interaction (F1,32 = 13.00 p = 0.0010). An increase in total α-syn overload was observed in AAV-h-A53Tα-syn-injected LRRK2 KD mice compared to AAV-GFP-injected controls (Fig. 3B) and AAV-h-A53Tα-syn-injected WT counterparts. In striatum (Fig. 4B), the pattern was slightly different since similar elevations of total α-syn were observed in both WT and LRRK2 KD mice injected with AAV-h-A53Tα-syn (treatment F1,33 = 25.77 p < 0.0001, genotype F1,33 = 0.08 p = 0.77, treatment X genotype F1,33 = 1.51 p = 0.22).

LRRK2 KD mice injected with AAV-h-A53Tα-syn show mild motor deficits. Stepping activity was evaluated in 12-month-old WT and LRRK2 KD mice injected with AAV-h-A53Tα-syn or AAV-GFP using the drag test. Motor tests were carried out before (baseline, time 0) and 1, 2, 3 months after surgery. Data are expressed as number of steps and are mean ± SEM of 9 mice per group. *p < 0.05 (three-way ANOVA followed by Tukey’s test for multiple comparisons).

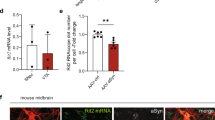
AAV-h-A53Tα-syn injection results in greater transgene levels in LRRK2 KD mice compared to WT mice. h-A53Tα-syn transgene expression was evaluated by immunohistochemistry in 12-month-old WT and LRRK2 KD mice 3 months after AAV-h-A53Tα-syn or AAV-GFP injection. Representative images and quantification of h-A53Tα-syn in SNpc (A) and striatum (B). Data are expressed as mean percentage ± SEM of: panel (A), n = 9 mice (KD AAV-h-A53Tα-syn, KD AAV-GFP, WT AAV-h-A53Tα-syn) or 10 mice (WT AAV-GFP) per group; panel (B), n = 9 mice per group. **p < 0.01 (ART ANOVA followed by the Bonferroni’s test for multiple comparisons).

AAV-h-A53Tα-syn injection results in greater pSer129 α-syn levels in the SN of LRRK2 KD mice compared to WT mice. Immunohistochemistry analysis of pSer129 α-syn and total α-syn staining in the SNpc of 12-month-old WT and LRRK2 KD mice 3 months after AAV-h-A53Tα-syn or AAV-GFP injection. Representative images and quantification of nigral pSer129 α-syn (A) and total α-syn (B) staining. Data are expressed as immunopositive surface of the structure of interest ± SEM of: panel (A), n = 8 mice per group; panel (B), n = 9 (WT AAV-h-A53Tα-syn, KD AAV-GFP) or n = 10 (WT AAV-GFP, KD AAV-h-A53Tα-syn) mice per group. **p < 0.01 (Student’s t test two-tailed for unpaired data or two-way ANOVA followed by the Tukey’s test for multiple comparisons).

AAV-h-A53Tα-syn injection results in greater pSer129 α-syn levels in the striatum of LRRK2 KD mice compared to WT mice. Immunohistochemistry analysis of pSer129 α-syn and total α-syn staining in the striatum of 12-month-old WT and LRRK2 KD mice 3 months after AAV-h-A53Tα-syn or AAV-GFP injection. Representative images and quantification of striatal pSer129 α-syn (A) and total α-syn (B) staining. Data are expressed as immunopositive surface of the structure of interest ± SEM of: panel (A), n = 9 (WT AAV-h-A53Tα-syn) or n = 10 (KD AAV-h-A53Tα-syn) mice per group; panel (B), n = 9 (WT AAV-h-A53Tα-syn, KD AAV-h-A53Tα-syn, KD AAV-GFP) or n = 10 (WT AAV-GFP) mice per group. *p < 0.05, **p < 0.01 (Student’s t test two-tailed for unpaired data or two-way ANOVA followed by the Tukey’s test for multiple comparisons).
Finally, the integrity of the nigrostriatal pathway was investigated (Fig. 5). Stereological analysis revealed no differences in the number of nigral TH+ neurons between AAV-h-A53Tα-syn and AAV-GFP-injected mice, regardless of genotype (Fig. 5A). Consistent with the lack of nigral degeneration, no loss of striatal TH+ staining was observed in WT and LRRK2 KD mice injected with AAV-h-A53Tα-syn compared to their respective AAV-GFP-injected controls (Fig. 5B).

AAV-h-A53Tα-syn injection does not result in degeneration of the nigrostriatal dopaminergic pathway. Stereological count of nigral neurons and optical density of striatal TH + terminals were carried out in 12-month-old WT and LRRK2 KD mice 3 months after AAV-h-A53Tα-syn or AAV-GFP injection. Representative images and quantification of TH+ neurons in SNpc (A) and terminals in striatum (in grey scale arbitrary units; B). Data are expressed as mean percentage ± SEM of n = 6 mice per group (A) or n = 9 mice per group (B).
Discussion
We previously showed an age-dependent impairment of autophagic flux in LRRK2 KD mice, with accumulation of striatal autophagic markers emerging at 12 months of age21. We now report that 12-month-old LRRK2 KD mice develop mild motor decline, higher striatal and nigral h-A53Tα-syn and pSer129 α-syn levels compared to WT counterparts in response to viral-mediated h-A53Tα-syn overexpression, suggesting that ALP impairment in LRRK2 KD mice is associated with greater susceptibility to α-syn pathology in vivo while not resulting into degeneration. As expected, AAV-h-A53Tα-syn-injected WT and LRRK2 KD mice showed increases in transgene levels both in SN (injection area) and the anatomically connected striatum, likely indicating the axonal transport of h-A53Tα-syn from the cell bodies to the projection area26,28,29. Consistently, α-syn phosphorylation at Ser129 was doubled in both areas indicating an aggregation-prone modification of the endogenous protein in response to pathogenic h-A53Tα-syn overexpression. However, α-syn neuropathology was not associated with any loss of nigral dopaminergic neurons or striatal dopaminergic terminals. This was quite surprising since in a previous study we reported a marked nigrostriatal degeneration 3 months after bilateral AAV-h-A53Tα-syn injection12. The discrepancy might be due to variability across AAV batches. Despite the lack of nigrostriatal degeneration, α-syn neuropathology was quite robust and more evident in LRRK2 KD mice than WT counterparts, possibly supporting a facilitating role of the silenced LRRK2 kinase activity. Consistently, double transgenic mice expressing human LRRK2 D1994S (a KD mutation) and A53T α-syn showed greater neuropathology than A53T α-syn single transgenics13. However, since the same outcome was achieved co-overexpressing human LRRK2 G2019S and LRRK2 WT13, the Authors concluded that the expression levels of LRRK2 rather than its kinase activity were responsible for accelerating α-syn pathology. In fact, contrast to the present study, the kinase-enhancing G2019S mutation amplifies α-syn neuropathology in AAV-h-A53Tα-syn-injected12,16 or PFF α-syn-injected17,30 rodents. This paradox might be explained suggesting that a LRRK2 loss-of-function amplifies α-syn neuropathology, as previously shown for other LRRK2-mediated cell functions. Indeed, LRRK-deficient mice were shown to develop greater α-syn pathology accompanied with an age-dependent autophagic vacuoles accumulation before the onset of nigral degeneration31. Another possible explanation for the paradoxical finding that both a kinase-silencing and a kinase-enhancing LRRK2 mutation facilitate α-syn pathology relies in transgene levels achieved after AAV-h-A53Tα-syn injection. In fact, G2019S KI mice injected with AAV-h-A53Tα-syn showed greater nigrostriatal degeneration and pSer129 α-syn accumulation than WT mice, but transgene levels were similar between genotypes12. Conversely, in the present study, transgene levels were higher in AAV-h-A53Tα-syn-injected LRRK2 KD than WT mice. Therefore, the higher levels of h-A53Tα-syn in LRRK2 KD mice might translate into a greater α-syn pathology and pSer129 α-syn phosphorylation than in WT counterparts. The possibility that such a greater transgene accumulation in LRRK2 KD mice compared to WT mice is due to systematic delivery of higher amounts of AAV-h-A53Tα-syn in the SN of LRRK2 KD mice is quite remote since both genotypes were injected in the same surgical sessions. Moreover, the inconsistencies in SN targeting and AAV vector delivery in SN are minimized by the current bilateral injection design that allowed us to average in the same tissue section the signals of area of interests in the right and left striatum or SN. This supports the view that greater h-A53Tα-syn load is truly dependent on the kinase-silencing phenotype, perhaps be due to a greater viral transduction and/or a defective clearance of the human protein. To the best of our knowledge there is no evidence that silencing of LRRK2 kinase activity favors AAV penetration and/or viral expression in neurons. Nonetheless, it is well known that LRRK2 regulates immune response and host defense. In fact, in a model of reovirus-induced encephalitis, LRRK2 KO mice were found to have a higher brain load of viral proteins, with KD mice showing a greater number of colony-forming-units, although in lungs but not brain32. Although it is believed that LRRK2 and, to a greater degree, G2019S LRRK2 confer protection against microbial infection via an increase of the inflammatory response32,33, the mechanisms regulating neuronal defense are far to be clear and the possibility that LRRK2 might modulate additional steps of viral infections, e.g. viral entry and/or replication, deserves further investigations. A more plausible explanation of the higher transgene levels achieved in LRRK2 KD mice relates to ALP impairment associated with LRRK2 silencing in these mice21. In fact, the viral-mediated overload of h-A53Tα-syn might further compromise the already dysfunctional ALP of old LRRK2 KD mice, thus ultimately leading to greater human protein deposition and phosphorylation of endogenous α-syn. Therefore, although in the AAV-h-A53Tα-syn model in vivo either hyperactivation or silencing of LRRK2 kinase activity worsens α-syn pathology, this might be mediated by independent mechanisms. Interestingly, the increase of α-syn burden in the striatum and SNpc of KD mice was associated with a mild motor decline in the drag test in the absence of nigral degeneration. Mild behavioral alterations were observed also in the case of synucleinopathy not associated with nigrostriatal damage10,34, possibly indicating a role for compensatory non-dopaminergic mechanisms in motor dysfunction.
In conclusion, viral-mediated nigral overexpression of α-syn in mice carrying a KD mutation of LRRK2 results in greater transgene levels and α-syn pathology with respect to WT mice. The reasons for the higher viral transduction remain to be identified although it is possible that the ALP impairment observed in these mice might have some bearing. This study extends previous findings in G2019S KI mice in the same model12, indicating that both a kinase-silencing and a kinase-enhancing mutation leads to greater Ser129 α-syn phosphorylation and α-syn pathology, likely via different mechanisms.
Methods
Animals
All procedures involving animals complied with the ARRIVE guidelines35 and the EU Directive 2010/63/EU for animal experiments. They were approved by the Ethical Committee of the University of Ferrara and the Italian Ministry of Health (license 714/2017-PR). Male homozygous D1994S LRRK2 KI (LRRK2 KD) mice backcrossed for at least 9 generations on a C57BL/6J background, were used21,36,37,38. Founders were obtained from Novartis Institutes for BioMedical research (Novartis Pharma AG, Basel, Switzerland)23. A colony of non-transgenic WT mice was used as a control. Colonies were grown in the vivarium (LARP) of the University of Ferrara and kept under regular lighting conditions (12 h light/dark cycle), with free access to food (4RF21 standard diet; Mucedola, Settimo Milanese, Milan, Italy) and water. Animals were housed in groups of 5 for a 55 × 33 × 20 cm polycarbonate cage (Tecniplast, Buguggiate, Varese, Italy) with a Scobis Uno bedding (Mucedola, Settimo Milanese, Milan, Italy) and environmental enrichment.
Experimental design
Nineteen (19) 12-month-old LRRK2 KD mice and 19 age-matched wild-type controls were used. Mice were trained in the drag test for 4 days, until their motor performance became reproducible, then an experimental session was performed to set a baseline. A week later, mice were randomly divided into four balanced treatment groups and subjected to stereotactic injection of AAV-h-A53Tα-syn or AAV-GFP: LRRK2 KD AAV-h-A53Tα-syn (n = 10) and LRRK2 KD AAV-GFP (n = 9), WT AAV-h-A53Tα-syn (n = 9) and WT AAV-GFP (n = 10). Mice were challenged again in the drag test at 30, 60 and 90 days after surgery. The day after the last behavioral test, mice were euthanized with trancardial perfusion with paraformaldehyde (PFA) and brains processed for immunohistochemical analysis.
AAV-h-A53Tα-syn injection
Recombinant AAV vectors carrying h-A53Tα-syn (pAAV-CMVie/Syn-hA53T-α-syn-WPRE) were obtained from Vectorology Core Facility of the University of Bordeaux26. Mice were anaesthetized with isoflurane, placed in a stereotaxic apparatus and injected bilaterally in the SNpc (coordinates in mm from bregma: antero-posterior − 3.3, medio-lateral ± 1.25; dorso-ventral − 4.6) with 1 µl of AAV-h-A53Tα-syn (2.35 × 1013 genome containing particles/µl) or control vector (AAV-GFP)12. Viral vectors were injected at the speed of 0.5 µl/min via a 30 Ga Hamilton syringe left in place for 4 min after injection to prevent leakage during withdrawal12.
Behavioral task
The stepping test measures the ability of the animal to balance its body posture with the forelimbs in response to backwards dragging39,40,41. It gives information regarding the time to initiate (akinesia) and execute (bradykinesia) a movement. Animals were gently lifted from the tail, leaving the forepaws on the table, and then dragged backwards at a constant speed (about 20 cm/s) for a fixed distance (100 cm). The number of steps made by each paw was recorded by two blinded observers. Five determinations were collected for each animal. The test was performed 30, 60 and 90 days after AAV vector injection.
Tissue processing
Mice were euthanized by transcardial perfusion with phosphate buffer solution (PBS) followed by 4% paraformaldehyde (PFA) in PBS (pH 7.4) at 4 °C (Sigma Aldrich, Saint Louis, Missouri, USA), under terminal anaesthesia with isoflurane. Brains were removed, post-fixed in 4% PFA for 24 h and then transferred in 30% sucrose in 1×PBS at 4 °C to store them at − 80 °C. Coronal sections of PFA-fixed brains (50 μm thick) were cut with a cryostat (Leica, Buffalo Grove, Illinois, US) and stored in cryoprotective medium (0.2% sodium azide in 1×PBS) at 4 °C.
TH and α-syn immunohistochemistry
Coronal free-floating sections of striatum (AP from + 1.0 to − 1.25 mm from bregma; Paxinos and Franklin, 2001) and SN (AP from − 3.16 to − 3.52 mm from bregma) were rinsed three times in PBS, then incubated with 3% H2O2 in PBS for 10 min to inhibit endogenous peroxidases11,12,37,41. After 3 washes in PBS, sections were first blocked in 2% BSA solution in PBST, then incubated overnight with the following primary antibodies: rabbit anti-TH (ab112; 1:1000, Abcam, Cambridge, UK), rabbit anti-α-syn (ab52168; 1:200, Abcam), rabbit anti hα-syn (ab138501; 1:150, Abcam) and rabbit anti pSer129-α-syn (ab51253; 1:1000, Abcam). Sections were rinsed in PBS and incubated for 1 h with a rabbit HRP-conjugated secondary antibody (ab6721, 1:2000, Abcam). DAB substrate kit (ab64238, Abcam) was then used to reveal the staining. Sections were mounted onto gelatin coated slides, dehydrated and coverslipped. Only for human-α-syn staining, sections were pre-incubated with an antigen retrieval solution. Images were acquired by using a Leica DM600B motorized microscope and analyzed either through stereology (see below) or with Fiji ImageJ Software (NIH).
The signals of h-A53Tα-syn and total α-syn were quantified using a densitometric analysis, similar to that described for TH (see below). Regarding the quantification of pSer129 α-syn signal, instead, after being mounted, slides were scanned, each area of interest (striatum or SN) was delimited at 2.5× magnification and, moving to 10× magnification, each delimited area was subdivided into enough squares to represent 70% of the total area. For each section proposed by the software in a randomised manner, an image was acquired for subsequent analysis. The resulting images obtained were then analysed using Fiji software by converting colour images to 8-bit greyscale. The area of interest was delimited, and the threshold was applied to compare the signal emitted by the immunopositive surface given by pSer129 α-syn. The area occupied by pSer129 α-syn was expressed as a percentage of the total area.
Thanks to the bilateral injection design, for each tissue section, both hemispheres were analysed and values averaged to minimize inconsistencies in AAV vector delivery to the targeted SN. Moreover, two sections were analysed and averaged for each animal.
Stereology and neuronal counting
Stereological analysis was performed using an unbiased stereological sampling method based on optical fractionator stereological probe12,27. TH + neurons in SNpc were counted in 5 serial slices cut at 50 μm thickness and 200 μm apart and magnified at 63×. Stereo Investigator software (MBF Europe, Delft, The Netherlands) was used to estimate the total number of neurons calculated from the number of neurons counted within a Systematic Randomly Sampled set of unbiased virtual counting areas covering the entire region of interest with a uniform distance between unbiased virtual counting in spaces in directions X, Y, and Z. Neural counting was performed by investigators blinded to the experimental condition. Each value was obtained by averaging the number of neurons counted in the right and left SN of each tissue section.
TH, h-A53Tα-syn and total α-syn quantification
Images of striatum and SN were taken at 2.5× or 10× magnification with a Leica DM6B motorized microscope, and optical densitometry analyzed off-line as grey level with ImageJ using the corpus callosum as background12.
Data presentation and statistical analysis
Data are expressed as mean ± SEM (standard error of mean) of n mice. Statistical analysis was implemented on GraphPad Prism version 8.4.3. (GraphPad Software Inc., CA, USA). Motor performance in the drag test was presented as number of steps, the density of striatal TH+ terminals, h-A53α-syn and α-syn levels as mean absolute value of grey scale between the two striata, and pSer129 α-syn positive staining as percentage surface area. Data presented in Figs. 1, 3, 4 and 5 passed the Shapiro–Wilk normality test and were then handled using parametric statistical procedures, i.e. three-way ANOVA (Fig. 1) or factorial two-way ANOVA (Figs. 3B, 4B and 5) followed by the Tukey’s test to compare treatment groups. Data presented in Figs. 3A and 4A were analysed using the Student’s t-test, two-tailed for unpaired data. Values presented in Fig. 2, instead, did not have Gaussian distribution and similar variances. For this reason, we adopted a nonparametric equivalent of factorial ANOVA, namely the aligned rank transform (ART) ANOVA followed by the Bonferroni’s test for multiple comparisons. Statistical significance was set at p < 0.05.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Paisan-Ruiz, C. et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 44, 595–600. https://doi.org/10.1016/j.neuron.2004.10.023 (2004).
Zimprich, A. et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44, 601–607. https://doi.org/10.1016/j.neuron.2004.11.005 (2004).
Nalls, M. A. et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet. 46, 989–993. https://doi.org/10.1038/ng.3043 (2014).
Cookson, M. R. The role of leucine-rich repeat kinase 2 (LRRK2) in Parkinson’s disease. Nat. Rev. Neurosci. 11, 791–797. https://doi.org/10.1038/nrn2935 (2010).
West, A. B. et al. Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc. Natl. Acad. Sci. U. S. A.. 102, 16842–16847. https://doi.org/10.1073/pnas.0507360102 (2005).
Greggio, E. et al. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol. Dis. 23, 329–341. https://doi.org/10.1016/j.nbd.2006.04.001 (2006).
Yao, C. et al. LRRK2-mediated neurodegeneration and dysfunction of dopaminergic neurons in a caenorhabditis elegans model of Parkinson’s disease. Neurobiol. Dis. 40, 73–81. https://doi.org/10.1016/j.nbd.2010.04.002 (2010).
Domenicale, C., Magnabosco, S. & Morari, M. Modeling Parkinson’s disease in LRRK2 rodents. Neuronal. Signal. 7, NS20220040. https://doi.org/10.1042/NS20220040 (2023).
Xiong, Y., Dawson, T. M. & Dawson, V. L. Models of LRRK2-associated Parkinson’s disease. Adv. Neurobiol. 14, 163–191. https://doi.org/10.1007/978-3-319-49969-7_9 (2017).
Xiong, Y. et al. Overexpression of Parkinson’s disease-associated mutation LRRK2 G2019S in mouse forebrain induces behavioral deficits and alpha-synuclein pathology. eNeuro. 4 https://doi.org/10.1523/ENEURO.0004-17.2017 (2017).
Domenicale, C. et al. Dopamine transporter, PhosphoSerine129 alpha-synuclein and alpha-synuclein levels in aged LRRK2 G2019S knock-in and knock-out mice. Biomedicines. 10 https://doi.org/10.3390/biomedicines10040881 (2022).
Novello, S. et al. G2019S LRRK2 mutation facilitates alpha-synuclein neuropathology in aged mice. Neurobiol. Dis. 120, 21–33. https://doi.org/10.1016/j.nbd.2018.08.018 (2018).
Lin, X. et al. Leucine-rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson’s-disease-related mutant alpha-synuclein. Neuron 64, 807–827. https://doi.org/10.1016/j.neuron.2009.11.006 (2009).
Daher, J. P. et al. Neurodegenerative phenotypes in an A53T alpha-synuclein Transgenic mouse model are independent of LRRK2. Hum. Mol. Genet. 21, 2420–2431. https://doi.org/10.1093/hmg/dds057 (2012).
Herzig, M. C. et al. High LRRK2 levels fail to induce or exacerbate neuronal alpha-synucleinopathy in mouse brain. PLoS One. 7, e36581. https://doi.org/10.1371/journal.pone.0036581PONE-D-12-03076 (2012). [pii].
Daher, J. P. et al. Leucine-rich repeat kinase 2 (LRRK2) Pharmacological Inhibition abates alpha-Synuclein Gene-induced neurodegeneration. J. Biol. Chem. 290, 19433–19444. https://doi.org/10.1074/jbc.M115.660001 (2015).
Bieri, G. et al. LRRK2 modifies alpha-syn pathology and spread in mouse models and human neurons. Acta Neuropathol. 137, 961–980. https://doi.org/10.1007/s00401-019-01995-0 (2019).
Cuervo, A. M., Stefanis, L., Fredenburg, R., Lansbury, P. T. & Sulzer, D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 305, 1292–1295. https://doi.org/10.1126/science.1101738 (2004).
Cuervo, A. M. Autophagy and aging: keeping that old broom working. Trends Genet. 24, 604–612. https://doi.org/10.1016/j.tig.2008.10.002 (2008).
Albanese, F., Domenicale, C., Volta, M. & Morari, M. Modeling Parkinson’s disease in LRRK2 mice: focus on synaptic dysfunction and the autophagy-lysosomal pathway. Biochem. Soc. Trans. 50, 621–632. https://doi.org/10.1042/BST20211288 (2022).
Albanese, F. et al. Constitutive Silencing of LRRK2 kinase activity leads to early glucocerebrosidase deregulation and late impairment of autophagy in vivo. Neurobiol. Dis. 159, 105487. https://doi.org/10.1016/j.nbd.2021.105487 (2021).
Albanese, F., Novello, S. & Morari, M. Autophagy and LRRK2 in the aging brain. Front. NeuroSci. 13 https://doi.org/10.3389/fnins.2019.01352 (2019).
Herzig, M. et al. (ed, C.) LRRK2 protein levels are determined by kinase function and are crucial for kidney and lung homeostasis in mice. Hum. Mol. Genet. 20 4209–4223 https://doi.org/10.1093/hmg/ddr348 (2011).
Schapansky, J. et al. Familial knockin mutation of LRRK2 causes lysosomal dysfunction and accumulation of endogenous insoluble alpha-synuclein in neurons. Neurobiol. Dis. 111, 26–35. https://doi.org/10.1016/j.nbd.2017.12.005 (2018).
Singh, F. et al. Pharmacological rescue of impaired mitophagy in Parkinson’s disease-related LRRK2 G2019S knock-in mice. eLife 10 https://doi.org/10.7554/eLife.67604 (2021).
Bourdenx, M. et al. Lack of additive role of ageing in nigrostriatal neurodegeneration triggered by alpha-synuclein overexpression. Acta Neuropathol. Commun. 3, 46. https://doi.org/10.1186/s40478-015-0222-2 (2015).
Arcuri, L. et al. Genetic and Pharmacological evidence that endogenous Nociceptin/orphanin FQ contributes to dopamine cell loss in Parkinson’s disease. Neurobiol. Dis. 89, 55–64. https://doi.org/10.1016/j.nbd.2016.01.016 (2016).
Sun, X. et al. Comparison of the expression and toxicity of AAV2/9 carrying the human A53T alpha-synuclein gene in presence or absence of WPRE. Heliyon 7, e06302. https://doi.org/10.1016/j.heliyon.2021.e06302 (2021).
Decressac, M., Mattsson, B., Lundblad, M., Weikop, P. & Bjorklund, A. Progressive neurodegenerative and behavioural changes induced by AAV-mediated overexpression of alpha-synuclein in midbrain dopamine neurons. Neurobiol. Dis. 45, 939–953. https://doi.org/10.1016/j.nbd.2011.12.013 (2012).
Volpicelli-Daley, L. A. et al. G2019S-LRRK2 expression augments alpha-Synuclein sequestration into inclusions in neurons. J. Neurosci. 36, 7415–7427. https://doi.org/10.1523/JNEUROSCI.3642-15.2016 (2016).
Giaime, E. et al. Age-dependent dopaminergic neurodegeneration and impairment of the autophagy-lysosomal pathway in LRRK-deficient mice. Neuron. 96, 796–807 e796. https://doi.org/10.1016/j.neuron.2017.09.036 (2017).
Shutinoski, B. et al. Lrrk2 alleles modulate inflammation during microbial infection of mice in a sex-dependent manner. Sci. Transl. Med. 11 https://doi.org/10.1126/scitranslmed.aas9292 (2019).
Rocha, J. D., Schlossmacher, M. G. & Philpott, D. J. LRRK2 and Nod2 promote lysozyme sorting in Paneth cells. Nat. Immunol. 16, 898–900. https://doi.org/10.1038/ni.3255 (2015).
Koprich, J. B. et al. Progressive neurodegeneration or endogenous compensation in an animal model of Parkinson’s disease produced by decreasing doses of alpha-synuclein. PLoS One. 6, e17698. https://doi.org/10.1371/journal.pone.0017698 (2011).
Kilkenny, C., Browne, W. J., Cuthill, I. C., Emerson, M. & Altman, D. G. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol. 8, e1000412. https://doi.org/10.1371/journal.pbio.1000412 (2010).
Longo, F., Russo, I., Shimshek, D. R., Greggio, E. & Morari, M. Genetic and pharmacological evidence that G2019S LRRK2 confers a hyperkinetic phenotype, resistant to motor decline associated with aging. Neurobiol. Dis. 71, 62–73. https://doi.org/10.1016/j.nbd.2014.07.013 (2014).
Novello, S. et al. In vivo susceptibility to energy failure parkinsonism and LRRK2 kinase activity. Neurobiol. Dis. 105579 https://doi.org/10.1016/j.nbd.2021.105579 (2021).
Mercatelli, D. et al. Leucine-rich repeat kinase 2 (LRRK2) inhibitors differentially modulate glutamate release and Serine935 LRRK2 phosphorylation in striatal and cerebrocortical synaptosomes. Pharmacol. Res. Perspect. 7, e00484. https://doi.org/10.1002/prp2.484 (2019).
Marti, M. et al. Blockade of Nociceptin/orphanin FQ transmission attenuates symptoms and neurodegeneration associated with Parkinson’s disease. J. Neurosci. 25, 9591–9601. https://doi.org/10.1523/JNEUROSCI.2546-05.2005 (2005). 25/42/9591 [pii].
Viaro, R. et al. Nociceptin/orphanin FQ receptor Blockade attenuates MPTP-induced parkinsonism. Neurobiol. Dis. 30, 430–438. https://doi.org/10.1016/j.nbd.2008.02.011 (2008).
Longo, F. et al. Age-dependent dopamine transporter dysfunction and Serine129 phospho-alpha-synuclein overload in G2019S LRRK2 mice. Acta Neuropathol. Commun.. https://doi.org/10.1186/s40478-017-0426-8 (2017).
Acknowledgements
This work was supported by a FAR 2022 grant from the University of Ferrara.
Author information
Authors and Affiliations
Contributions
F.A. Investigation, formal analysis, writing—original draft; C.D. Investigation, formal analysis; D.M. Investigation, formal analysis, A.B. Investigation, S.D. Investigation, E.B. Supervision, writing—review and editing, M.M. Conceptualization, formal analysis, funding acquisition, writing—review and editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Albanese, F., Domenicale, C., Mercatelli, D. et al. Viral mediated α-synuclein overexpression results in greater transgene levels and α-synuclein overload in mice bearing kinase dead mutation of LRRK2. Sci Rep 15, 9992 (2025). https://doi.org/10.1038/s41598-025-94165-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-94165-0
