
Abstract
The new era of messenger RNA (mRNA) vaccines has led to development of a novel, state-of-the-art characterization method for this class of molecules. Currently, flow cytometry-based assays with antigen-specific antibodies are utilized for monitoring in-vitro expression (IVE) of mRNA. Here we present development, optimization, and application of an in-vitro expression liquid chromatography tandem mass spectrometry (IVE-LC/MS/MS) assay as an orthogonal method to IVE-flow cytometry that can be used for in-depth characterization of the expressed protein antigens and monitoring their relative expression levels in the cell post-mRNA transfection. The IVE-LC/MS/MS assessment accomplished the detection of influenza hemagglutinin (HA) antigens of four distinct strains simultaneously. The workflow is presented here, highlighting the optimization of all necessary steps required for protein purification and mass spectrometry method setup. The IVE-LC/MS/MS assay is a robust and versatile technique that complements the IVE-flow cytometry method and offers several advantages, such as being antibody-free, capable of multiplexing, and highly sensitive and selective. The various studies in this work, including evaluating dose–response relationships, refining transfection protocols, and examining mRNA-LNP stability under various conditions showcase the significant benefits of applying IVE-LC/MS/MS across different experimental settings. IVE-LC/MS/MS is a powerful tool for understanding and improving the performance and quality of mRNA LNPs.
Similar content being viewed by others
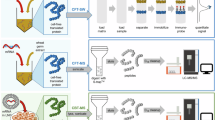


Introduction
Advancement of messenger RNA (mRNA) technologies has led to a new frontier in health care, especially in vaccine development1. Successful development and delivery of the world’s first mRNA-based vaccine for COVID-19 showed that mRNA could be an effective vaccine platform2,3, with the potential to prevent other infectious diseases including influenza4. The manufacturing processes for any mRNA-based influenza vaccine would be simpler and faster relative to the traditional technologies used to manufacture the flu protein vaccines that require the use of chicken eggs or mammalian cells. These traditional technologies, due to a long virus replication time and mutations while adapting to the environment, result in seasonal vaccines that prevent only 40 to 60% of the disease in the best-matched seasons. Because mRNA can be manufactured in a relatively short time frame5, the mRNA vaccine for flu could potentially allow manufacturers to better match the strains in circulation in any given season, leading to a greater efficacy. Additionally, mRNA-based vaccines can leverage a universal manufacturing process that can be adapted to new strains.
The mechanism of action for mRNA vaccines is to deliver the genetic code of the pathogen’s relevant antigen to the host (humans). In other words, mRNA is translated by the host to produce the relevant protein from the pathogen, allowing the host to develop an immune response against the foreign pathogen. The traditional flu vaccines primarily target highly immunogenic human influenza hemagglutinin (HA) proteins of representative strains from A(H1N1), A(H3N2), B-Victoria, and B-Yamagata lineages, the four influenza subtypes endemic in humans. Pfizer’s quadrivalent mRNA flu vaccine candidates similarly encode human influenza hemagglutinin (HA) viral proteins from four distinct strains, as nominated by the World Health Organization semi-annually6. Designs of mRNA constructs including 5′-cap, 3′-poly A tail, 5′ and 3′ untranslated regions (UTRs), incorporation of modified nucleosides as well as delivery vehicle including lipid nanoparticles affect the translational efficiency of mRNA into desired antigen protein. Additionally, the structure and stability of lipid nanoparticles (LNPs), including factors such as lipid composition and impurities7, formulation buffers8, and storage temperatures9, can affect mRNA delivery and translation, thereby influencing the efficacy of the vaccine.
Monitoring levels of expressed antigens in-vitro is an important step in vaccine development and manufacturing. Currently, flow cytometry-based assays with antibodies are utilized for monitoring in-vitro expression (IVE) of mRNA10 to support Comirnaty (SARS-Cov2) mRNA vaccine manufacturing. Each expressed antigen requires a sufficiently specific antibody that was raised in animals through time-consuming immunization, purification, and characterization. For seasonal mRNA vaccine development, if significant strain changes occur to the pathogens in circulation, there might not be sufficient time to raise new antibodies specific to the antigen(s) of interest for flow cytometry analysis. An alternative method is desired to meet this challenge.
Herein, we present the development, optimization, and application of an in-vitro expression liquid chromatography tandem mass spectrometry (IVE-LC/MS/MS) assay as an orthogonal method to IVE-flow cytometry that can be used for in-depth characterization of the expressed HA antigens as well as monitoring their relative expression levels in Human Embryonic Kidney (HEK) 293 T cells post mRNA transfection. Given the capability of LC/MS/MS to detect multiple peptides representing many proteins with high selectivity and sensitivity, coupling it as a detection method to monitor mRNA expression in-vitro has gained broader interests for supporting mRNA development11. There is precedent for IVE-LC/MS of mRNA. The application of mass spectrometry in combination with isotope dilution (IDMS)12 has been reported previously in literature to quantify the amount of spike protein after transfection of SARS-Cov2 mRNA vaccines in baby hamster kidney cells (BHK)13. In addition, a high-resolution targeted proteomics method by LC/MS/MS in parallel reaction monitoring (PRM) mode has been reported to quantitate low abundance proteins in whole cell lysates14 and showed enhanced sensitivity and selectivity over conventional PRM or selected reaction monitoring (SRM)15. We further this IVE-MS paradigm and show that a single IVE-LC/MS/MS assay enables the detection of multiple expressed antigens from multivalent influenza mRNA vaccines. We also show that multiplexing capability of IVE-LC/MS/MS method allows the addition of other antigens from a different pathogen such as SARS-Cov2 spike protein in support of development of combination mRNA vaccines.
The work highlights the optimization of all necessary steps required for protein purification and MS detection method PRM for quantitative HA protein analyses. Furthermore, the implementation of a Hamilton liquid handler for high-throughput sample preparation has enabled automation of the overall workflow. The data demonstrate that our IVE-LC/MS/MS assay can be successfully applied to characterization and monitoring of relative expression of multi-antigen mRNA-based influenza and other combination mRNA vaccines in a single analysis. The IVE-LC/MS/MS assay is selective and robust with a wide dynamic range and sensitivity. Specific applications of the IVE-LC/MS/MS assay are presented and indicate robust performance of the assay in a variety of samples with good alignment with IVE-flow cytometry results.
Results
IVE-LC/MS/MS synopsis
The in-vitro expression-liquid chromatography mass spectrometry (IVE-LC/MS/MS) method is fundamentally a PRM method, in which the analytes are select peptides unique to the expressed target protein antigens. The well characterized and readily available HEK293T cells were selected for their suitable growth characteristics and the ability to support robust antigen expression. Cells are expanded and transfected as is done for antigen expression assays using flow cytometry16. As few as 200,000 cells may be transfected with 3 to 500 ng mRNA in lipid nanoparticle (LNP) formulation. Usually multiple identical-cell-count plate wells are dosed with different levels of mRNA to probe dose response. After a period of culture time (usually 1 day), the transfected cells are harvested and may be split for IVE-flow cytometry and IVE-LC/MS/MS.
Robust, reproducible, and sensitive MS-based PRM necessitates mastering three method aspects: 1) Processing of the cell materials to yield the peptide analytes; 2) Selection of the peptide analytes; 3) Mass spectrometry analysis of the peptide analytes. Important considerations for the processing of the cell material are as follows (Fig. 1): 1) Incubation in sodium dodecyl sulfate (SDS) anionic surfactant for 15 min at 90 °C, followed by 3-cycle pulsed sonication, ensures a target-agnostic lysis that solubilizes all cellular components. This approach will work for any expressed protein no matter its mRNA-coded cellular routing. After this point liquid handling systems such as the Hamilton Vantage (Hamilton, Reno, NV) may be employed to streamline high throughput analysis and improve method precision. 2) Lysate proteins’ disulfide bonds are reduced, and cysteine thiols alkylated; this is especially important if the target has one or more disulfide bonds. Performing reduction and alkylation before the precipitation step, rather than after protein precipitation and reconstitution as described in many protocols, not only enables efficient purification by taking advantage of the disparity in physiochemical properties of large biomolecules compared to small molecule reactants but also simplifies the entire workflow. 3) Proteins, DNA and RNA are precipitated from the lysate/SDS/alkylation matrix by room temperature acetonitrile treatment17, which is safer than commonly used dichloromethane/ methanol-based protein extraction18 and simpler and less reagent dependent than more complicated protein purification approaches like S-Trap19. 4) The proteinaceous pellet is resolubilized with sonication and a universal nuclease is added to completely destroy DNA and RNA, which further improves chromatography robustness and performance. 5) Lys-C/Trypsin protease digestion then proceeds for 2 h to yield target peptides.

Overview of In-Vitro Expression (IVE)-Flow Cytometry and IVE-LC/MS/MS Workflow. IVE begins with the production of approximately 200—400 k viable HEK293T cells. These are transfected with mRNA encapsulated in LNPs such that the total dose of mRNA is controlled, usually from 3 to 500 ng. After 24 h the transfected cells are harvested and split for flow cytometry and LC/MS/MS PRM analysis. For LC/MS/MS PRM analysis, pelleted cells are fully solubilized in sodium dodecyl sulfate with 90 °C heating and sonication, then protein disulfides are reduced, and cysteine thiols are alkylated. Proteins and nucleic acids are acetonitrile-precipitated, and the pellet is resolubilized in a nuclease digestion reaction solution. Finally, proteins are Lys-C/Trypsin-proteolyzed in a 2 h incubation. No further sample handling is required prior to LC/MS/MS injection. Figure created with Biorender.com.
Selection of peptide analytes follows the common advisement for “quantotypic” PRM: peptides that are 8–25 amino acids, unique for the target, lack miscleavage sites, and lack problematic modification sites20. Empirical data for peptide selection is also required, and is best generated using null-transfected cells spiked with recombinant protein. In this way candidate target peptides’ retention times and best charge states can be cataloged, and candidate peptides can be stricken if there are conspicuous host cell proteome peptides that elute at the same time and have nearly the same m/z. The best 2–4 MS/MS fragment ions for each target peptide are also identified in this selection process.
The LC/MS/MS PRM method specifies each target peptide’s precursor m/z, retention time, and fragment ions’ m/z values. A high-resolution mass spectrometer (HRMS) such as an Orbitrap Fusion Lumos Tribrid HRMS is beneficial to limit interference from background proteome peptides by taking advantage of high resolving power (we use 240,000 RP at 200 m/z). The choice of flow rate and source ionization is important. Using an analytical flow rate (0.2 mL/min) and an ultra-high performance liquid chromatography (UHPLC) system for reversed phase 70 min separation of peptides, plumbed post-column to the mass spectrometer with an intervening divert valve, affords sufficient sensitivity while providing high reproducibility and robustness. The divert valve precludes the need for any post-digestion off-line desalting, which may add unknown bias by non-specific protein adsorption and may contribute to diminished precision. Of practical importance is that the UHPLC-HRMS configuration ensures the PRM UHPLC-HRMS system can be used for other kinds of experiments. Finally, by virtue of its robustness and reproducibility, this analytical flow format also allows for high-throughput analysis of IVE samples. The overall method throughput has been significantly improved with implementation of automated sample preparation using a Hamilton liquid handler, yet the overall scale-up is influenced by the constraints imposed by extended chromatographic separations.
Quantitation approaches
Various approaches for quantitating targeted proteins in a complex biological matrix have been employed by the MS community21,22. These include relative quantitation using either label-free23 or label-based24 methods, and absolute quantitation using targeted or non-targeted approaches25 along with reference standards26. In this work, we developed a label-free relative quantitation strategy. This strategy involves dividing the average signals from two representative peptides (Table S1) per antigen protein by the average signals from five representative HEK293T cell protein peptides (HEK peptides), enabled by targeted monitoring approaches such as parallel reaction monitoring (PRM) (see section "Key considerations for LC/MS/MS method development and method performance demonstration" for more discussions on the method). The two representative peptides for each antigen protein are selected based on general criteria such as appropriate amino acid length, minimal sequence liability, uniqueness to the antigen protein, and experimental observations including sufficient MS response, no carryover, and effective on-column separation. The five HEK protein peptides are selected from four highly abundant HEK proteins, serving as platform HEK protein surrogates. The robustness of the selected five HEK peptides is confirmed after meeting the theoretical peptide selection criteria. This straightforward relative quantitation strategy is our routine approach for the assay as it is easy to implement, eliminates the need for cell counting, and reduces method variation introduced from cell expression, sample preparation, and MS instrumentation. Additionally, the intensities of HEK peptides can be indicative of cell viability as modulated by treatment toxicity especially when IVEs are performed side-by-side (see an example in Fig. 4A, section "Dose–response and time-course of mRNA transfection by IVE-LC/MS/MS"). The relative quantitation approach enables direct comparison of the same antigen protein under various conditions or when testing batch-to-batch consistency.
This routine relative quantitation reporting strategy can be augmented with absolute quantitation using recombinant protein standards. The absolute quantitation strategy involves using a recombinant protein standard that is spiked into a negative control cell pellet at a known amount. The relative quantitation of the sample is then compared to the spiked control to calculate the absolute amount of protein antigens in the sample tested. This approach enables a direct comparison of the expression levels among different antigen proteins (see section "Dose–response of mRNA transfection measured by IVE-LC/MS/MS and IVE-flow cytometry" and "Dose–response and time-course of mRNA transfection by IVE-LC/MS/MS" for applications of absolute quantitation). Additionally, isotopically labeled peptides could be used as internal standards in addition to HEK293T protein peptides to further improve robustness of the assay. To minimize the use of reagents and cost, this approach was not employed. Absolute quantitation was not envisioned as the routine reportable because we sought a method that does not depend on maintaining critical reagents (such as recombinant proteins); moreover, relative quantitation is sufficiently informative for monitoring the activities (i.e., in-vitro expression) of mRNA LNPs of the same product under various conditions throughout product lifetime. This has been demonstrated with most of the IVE-LC/MS/MS studies we have completed, including the results presented here.
Key considerations for LC/MS/MS method development and method performance demonstration
The LC-parallel reaction monitoring (PRM) acquisition method selected for quantification of low abundance mRNA-expressed antigens in complex cellular lysates has been evaluated under different liquid chromatographic and mass spectrometric conditions to identify the optimal instrument parameters suitable for the highly selective, sensitive, and specific detection of peptides and their fragments. A medium length of LC gradient (70 min), in combination of careful selection of peptides to be monitored, enables sufficient spreading out between each peptide such that no more than three peptides are monitored at any given retention time window to accommodate the optimized MS/MS parameters discussed below. This ensures a minimum of 10 MS/MS scans per precursor ion to well define the chromatographic peak shape for accurate quantitation. An example of the high-quality LC-PRM profile is provided in Fig. 2A. Our final optimized key MS/MS parameters include narrow MS1 isolation window (0.4 m/z), high resolution MS/MS (240,000 or 240 K), high AGC target (1000%) and maximum injection time (502 ms), which are overall consistent with a previous publication14.

Spiking Study of Four Recombinant Flu Hemagglutinin and Covid Spike Antigens Using Various MS Parameters. (A) LC-PRM profile of four recombinant influenza hemagglutinin (HA) proteins (Wisconsin, Darwin, Austria, and Phuket) and recombinant SARS-CoV-2 S1 domain of spike protein at 2 pmol each spiked into negative control HEK293T cell pellets. (B) Quantitation results of spike study at high resolution (method 1). Antigen peptide signal (%) for each unique antigen relative to specific HEK293T peptide signal at each spike level were plotted and fit with linear curve. The fit slope and R2 value for each antigen are provided in the table below. Orange: Covid; Black: Austria; Blue: Wisconsin; Magenta: Phuket; Red: Darwin. (C) Representative PRM profile of Wisconsin HA peptide EQLSSVSSFER at 0.13 and 2 pmol spike-in levels using high (method 1), medium (method 2), and low (method 3) resolution settings. (D) Quantitation results of spike study at medium resolution (method 2). Antigen peptide signal (%) for each unique antigen relative to specific HEK293T peptide signal at each spike level were plotted and fit with linear curve. The fit slope and R2 value for each antigen are provided in the table below. Orange: Covid; Black: Austria; Blue: Wisconsin; Magenta: Phuket; Red: Darwin.
To demonstrate method robustness, linearity, and sensitivity, we evaluated a 6-point dilution series spanning 0.02 pmol to 2 pmol of five recombinant proteins spiked into six individually prepared side-by-side negative control HEK293T cells. Four of the recombinant proteins were hemagglutinin (HA) proteins expressed from four different influenza strains: A/Wisconsin/67/2022, A/Darwin/6/2021, B/Austria/1,359,417/2021, and B/Phuket/3073/2013. The fifth recombinant protein in this study was the S1 domain of the SARS-CoV-2 spike protein. Similar signal intensity, peak shape and retention time for HEK peptides are observed (one representative HEK peptide is shown in Figure S1A), demonstrating method robustness and consistent cell pellet size when the same cells were used for IVE preparation. No interference was observed in the negative control samples for any of the antigen peptides, and good signal-to-noise ratio and peak shapes were observed across all concentrations, demonstrating method selectivity and sensitivity (see Figure S1B for a representative antigen peptide). Importantly, the relative quantitation of all five recombinant antigens demonstrated linearity with R2 values greater than 0.99 (Fig. 2B).
In addition, we compared the optimized parameters to two other sets of parameters with lower resolution (60,000 and 30,000), wider isolation window (1 and 1.5 m/z) and lower maximum ion injection time (118 and 54 ms), using the same spiked-in serial dilution sample sets. The optimized MS/MS parameters (method 1) resulted in the highest signal-to-noise and smooth MS/MS peak profile whereas reduced qualities or partial interference are observed using method 2 and 3, as shown in Fig. 2C for recombinant Wisconsin HA peptide EQLSSVSSFER. High resolution is most responsible for eliminating interference, because non-target ions with m/z values that are very close to the target peptides are not resolved from the target m/z at lower resolution. This is consistent with observations reported by Nguyen et al14. This results in distortion of the mass spectra and quantitation of the target peptides. The isolation window width has much less impact on the mass accuracy of the precursor ions. The reduced qualities of the sub-optimal methods are more pronounced at lower concentrations. The combination of mass spectrometric parameters of 60K with 1 m/z and 118 ms (method 2 in Fig. 2C) was still suitable for peak integration and quantitation, despite the reduced qualities. Figure 2D showed that linear fit for the 100-fold dilution curve could be achieved for all five different antigen representing peptides for method 2 with very similar slopes to method 1. For method 3, the jagged LC-PRM profile can be mitigated by increasing the maximum injection time from 54 to 500 ms (method 4: 30K with 1.5 m/z and 500 ms) and a linear fit can be achieved after some significant interferences are removed for quantitation, albeit with slightly different slope compared to methods 1 and 2 (Figure S2).
Dose–response of mRNA transfection measured by IVE-LC/MS/MS and IVE-flow cytometry
A multivalent mRNA vaccine dose response study was conducted to compare the performance of antigen expression quantitation by IVE-LC/MS/MS assay vs. IVE-flow cytometry assay. HEK293T cells were transfected using a quadrivalent mRNA vaccine (1:1:1:1 ratio of mRNA constructs) with doses ranging from 16 to 250 ng total mRNA per well. The relative abundances of the expressed protein antigens (normalized to the level of selected HEK reference protein peptides) were measured by IVE-LC/MS/MS. All 4 protein antigens were detected within approximately a twofold relative abundance range (Fig. 3A). It cannot be inferred that the proteins differ in their concentrations by twofold, because peptides have differing mass spectrometry response factors due to their unique ionization and detection efficiencies. Consequently, each peptide has a unique absolute signal level even when they are present at equimolar concentrations. The relative abundance of each antigen correlates well with the administered mRNA dose from < 16 ng (the limit of quantitation, LOQ) to a dose of 250 ng total mRNA per well of cultured cells, as demonstrated by linear regression of these data (Fig. 3A). To overcome the issues of different antigen peptide responses in mass spectrometer, absolute quantitation using recombinant proteins was performed. All four antigens have consistent total levels of expression (Fig. 3B). In this way, the IVE-LC/MS/MS method provides a direct measurement for the level of each unique antigen expressed by the cells.

mRNA Vaccine Dose Response. The mRNA vaccine dose response, encapsulating the efficiency of mRNA transfection into HEK293T cells and subsequent expression of the encoded antigen proteins, was measured by IVE-LC/MS/MS and IVE-flow cytometry. HEK293T cells were dosed with a quadrivalent mRNA vaccine containing an equal theoretical load of each mRNA construct. The resulting data from the IVE-LC/MS/MS assay are graphed as % antigen peptide signal for each unique antigen relative to specific HEK293T peptide signal vs. total mRNA dosed (ng) per plate well (R2 for Wisconsin, Darwin, Austria and Phuket are: 0.963, 0.985, 0.99 and 0.980 respectively) (A). A linear regression of these same data in pmol antigen per pellet is plotted in B (R2 for Wisconsin, Darwin, Austria and Phuket are: 0.992, 0.970, 0.997 and 0.995 respectively). IVE-flow cytometry assay results are graphed as % cells positively expressing the antigen protein of interest (C) and mean fluorescence intensity (MFI) ratio (D) vs. the total mRNA (ng) per plate well.
Antigen expression was also assessed using IVE-flow cytometry assay for comparison (Fig. 3C). The flow cytometry primary reportable is the percentage of antigen expressing cells—it does not directly measure the level of the expressed antigen. Additionally, flow cytometry can assess level of expressed antigens using mean fluorescence intensity (MFI) ratio. As expected, the percentage of cells transfected with each antigen positively trends with the administered dose of the mRNA vaccine and is similar for all 4 constructs. Both IVE-LC/MS/MS and IVE-flow cytometry demonstrate a dose–response behavior. Notably, the IVE-flow cytometry assay reports that the percentage of cells expressing three of the four antigens are highly comparable, while Darwin is moderately higher at equivalent dosing (Fig. 3C) and the mean fluorescence intensity (MFI) ratio of Darwin-expressing cells is higher than those of the other antigen-expressing cells at doses > ~ 10 ng total mRNA per well (Fig. 3D). However, the apparent inter-antigen differences in percentage of antigen positive cells and MFI ratio may be confounded by antibody affinity and the intensity of the fluorophores chosen for detection. Without further information, this may lead one to conclude that transfection induces a higher level of Darwin expression and therefore a higher dose response. However, the IVE-LC/MS/MS results demonstrate that the absolute abundance of all 4 antigens (Fig. 3B), including Darwin, is similar and increases at the same rate, therefore all antigens have comparable levels of absolute expression (Fig. 3B). This may be expected due to the 1:1:1:1 ratio of their underlying antigen mRNA transcripts in the starting quadrivalent mRNA vaccine. Thus, IVE-LC/MS/MS with an absolute quantitation approach provides a clearer understanding of the relative level of each antigen expressed, and therefore the true dose response for all 4 antigens.
Dose–response and time-course of mRNA transfection by IVE-LC/MS/MS
We transfected LNPs containing multivalent mRNA at six dose levels from 31.25 to 500 ng/well of total mRNA per well for 24 and 48 h, without changing the cell medium after 24 h. Figure 4 (panel A) shows representative PRM profiles for the IVVDYMVQK peptide from Austria antigen at 48 h post-transfection, indicating increases in peak intensity with increasing dose, except for the highest dose of 500 ng/well. This may be due to cell toxicity induced by the highest dose27. We observed a similar dose-dependent increase in peptide peak intensity and antigen peak intensity normalized to the HEK peptide intensity (%Antigen/HEK) for all antigen strains. Figure 4, panels B and D, depict the representative %Antigen/HEK for Wisconsin and Darwin at 24- and 48-h post-transfection, respectively. The dose curves for both time points overlapped well, suggesting that the rate of antigen production was constant. However, the total amount of antigens per pellet was about three to fourfold higher at 48 h than at 24 h post-transfection (Fig. 4, panels C and E). One mole of mRNA yielded approximately five times more antigen proteins at 24 h and twenty times more by 48 h. This is due to cell growth and division, resulting in larger cell pellets with greater protein content as depicted in Fig. 4, panels D and E. The intensity of the average HEK peptides signals tightly matched protein concentration levels measured by UV absorbance post-sample clean-up (refer to Fig. 4, panels F and G) at both transfection time points. Consequently, the HEK peptides’ average peak area allows for standardizing varying cell pellet sizes seen in cell-based assays.

Dose–response and Time-course Study of mRNA Transfection. The mRNA vaccine in-vitro expression levels over different doses and time points were measured using IVE-LC/MS/MS. HEK293T cells were dosed with a quadrivalent mRNA vaccine. Panel A presents the raw PRM data for the IVVDYMVQK peptide in response to varying doses. Panels B and C show the antigen peptide signal overlays at 24 h (in red) and 48 h (in blue) relative to specific HEK peptide signals. This is plotted against the total mRNA (in pmol) dosed per well for two representative HA antigens—Wisconsin HA in Panel B and Darwin HA in Panel C. Panels D and E illustrate the absolute amounts of Wisconsin and Darwin HA antigens in cell pellets relative to the total mRNA (in pmol) dosed per well. Panels F (R2 = 0.77) and G (R2 = 0.936) display the correlation between total protein per sample, measured by UV280 absorbance, and the average HEK peptide peak area at 24 and 48 h, respectively.
LNP stability time-course
The development of thermally stable mRNA LNP vaccines has been a growing demand and remains a significant challenge as both mRNA and LNP are susceptible to degradation upon storage28. To evaluate the developmental stability of quadrivalent mRNA vaccine, we used the IVE-LC/MS/MS method to measure LNP transfection after storing the vaccine at -80 °C and 5 °C for up to 12 months. We also compared the IVE-LC/MS/MS results with the IVE-flow cytometry results, which included the percentage of antigen expressing cells and MFI ratio. For each time point, cells were transfected with LNPs for both LC/MS/MS and flow cytometry analyses using the same protocol with a total mRNA concentration at 250 ng/well, except that two wells were pooled for LC/MS/MS analysis. Figure 5 shows the time-course curves for expressed antigen by IVE-LC/MS/MS and the percentage of antigen positive cells and MFI ratio by flow cytometry for the four individual strains in the quadrivalent mRNA LNP. The panels A, B, C, and D show the percent change from time t = 0 for these three measurements at -80 °C storage condition, while the panels E, F, G, and H show the same measurements at 5 °C storage condition. The data indicate that the LNPs stored at -80 °C maintained a consistent expression level for 12 months and showed similar trends among the three measurements. However, the LNPs stored at 5 °C produced lower levels of expression over time as measured by the IVE-LC/MS/MS assay. The %Antigen/HEK data and flow cytometry data for 0-, 1-, 3-, and 6-month time points are also trending similarly (Fig. 5, panels E, F, G and H; the flow cytometry data for percentage of antigen positive cells and MFI ratio were not available for 9- and 12-month time points).

Stability Study of Quadrivalent Flu LNPs: Relationship between %Antigen/HEK, % Antigen Positive Cells and MFI. The mRNA vaccine stability was measured by IVE-LC/MS/MS and IVE-flow cytometry. HEK293T cells were dosed with a quadrivalent mRNA vaccine. Overlays of the change in expressed antigen by IVE-LC/MS/MS and the percentage of antigen positive cells and MFI ratio by flow cytometry for up to 12 months at -80 °C (Panels A-D) and at 5 °C (Panels E–H) for four HA antigens. Each data point measured by IVE-LC/MS/MS is representing the average biological replicates (n = 3, %RSD ≤ 6%) and each IVE-Flow cytometry is representing the average of 2 biological replicates.
Impact of formulation buffers on the mRNA transfection as measured by IVE-LC/MS/MS
Buffers play a crucial role in the stability and formulation of mRNA LNPs and are important to the proper delivery and transfection/translation of the mRNA therapeutic29. The flexibility and range of applications of the IVE-LC/MS/MS method were demonstrated by investigating the impact of LNP formulation buffers and final buffer composition on transfection. LNPs encapsulating monovalent influenza hemagglutinin Phuket mRNA were formulated with either acetate or acetate/NaCl, using the same mixing device and method as described in previous work30. Final buffer composition was either 10 mM Tris/300 mM Sucrose or 10 mM Tris/150 mM NaCl depending on the sample. An overview of the samples formulated and tested by IVE-LC/MS/MS is shown in Table 1. Each sample was tested at four different mRNA transfection doses (25, 50, 100 and 150 ng/well) in duplicate. Results are presented in Fig. 6 and reported as expressed %antigen/HEK. As shown in Fig. 6, buffer 3 containing 150 mM NaCl in both formulation and final buffer resulted in approximately tenfold higher expression relative to other buffers at all four tested doses of mRNA. Buffers 1 and 2 started with the same formulation buffer (50 mM acetate) and different final buffers (10 mM Tris with 300 mM sucrose and 10 mM Tris with 150 mM NaCl, respectively). These gave similar %Antigen/HEK dose–response curves. The similar dose response curves for buffer 1 and buffer 2 samples indicate that the final buffer composition has no impact on in-vitro expression measured by IVE-LC/MS/MS. Conversely, the tenfold in-vitro expression difference between buffer 1/2 and buffer 3 samples indicates the importance of the LNP formulation buffer and its potential impact on in-vitro expression. This was recapitulated by IVE-flow cytometry: the same samples were tested at a single 16 ng/well concentration of mRNA and showed a similar trend, with the highest percentage of antigen positive cells belonging to cells transfected with LNPs formulated in Buffer 3 (supplementary material Table S2). This demonstrates that formulation buffers can have a profound impact on in-vitro expression, and that this can be appropriately monitored by IVE-LC/MS/MS or flow cytometry.

Effect of Various Buffers on Phuket Monovalent Flu LNP Expression. Antigen expression in different buffers was measured by IVE-LC/MS/MS. HEK293T cells were dosed with a monovalent mRNA vaccine at four different doses. The figure depicts overlays of % antigen peptides signal to HEK293T peptides signal vs. total mRNA (ng) dosed per well for three buffer combinations. n = 2 for each transfection dose. R2 for Buffers 1, 2 and 3 are 0.993, 0.981 and 0.996 respectively.
Assessment of LNP shaking time on mRNA Transfection by IVE-LC/MS/MS and IVE-flow cytometry
Recent publications have shown that the impact of air entrainment, introducing liquid–air interfaces, caused by mixing and shaking can have profound impact on classical mRNA-LNP product quality attributes30. We used the IVE-LC/MS/MS and IVE-flow cytometry methods to evaluate the impact of vigorous shaking of LNPs on antigen expression. LNPs containing monovalent Austria mRNA underwent shaking at the maximum speed of 100 upward movements per minute at two different time points of 30 and 240 min at mRNA concentrations 0.12, 0.06, 0.01 mg/mL. All three shaking conditions, including non-shaken control samples (NS), 30- and 240-min shake samples, were tested by IVE-LC/MS/MS at four different mRNA transfection doses: 25, 50, 100, and 150 ng/well and by IVE-flow cytometry at 8, 16, 32, and 48 ng mRNA/well (Fig. 7). The non-shaken samples at 0.12 and 0.06 mg/mL of mRNA concentration resulted in very similar expression levels as measured by IVE-LC/MS/MS and IVE-flow cytometry (% antigen positive cells and MFI ratio). On the other hand, the non-shaken sample at the 0.01 mg/mL mRNA concentration produces higher antigen expression as compared to 0.12 and 0.06 mg/mL mRNA concentrations. After 30 min of shaking no drastic impact is observed on expression for all three mRNA concentrations which is consistent with the effect on other quality attributes30. Nevertheless, after 240 min of shaking, a significant impact on expression is observed. For all three mRNA concentrations (0.12, 0.06, and 0.01 mg/mL), LNPs show a decrease in antigen expression. This decrease is measured by IVE-flow cytometry (percentage of antigen-positive cells in Fig. 7, panels A, B, C; MFI ratio in Fig. 7, panels D, E, F) and by IVE-LC/MS/MS (Fig. 7, panels G, H, I). The 0.01 mg/mL mRNA concentration has a more profound decrease in expression than the two higher mRNA concentrations, as demonstrated by a comparison of 30 min shaking and 240 min shaking expression results for both IVE-LC/MS/MS and IVE-flow cytometry (Fig. 8). We note that IVE-LC/MS/MS was more sensitive to the change induced by 240 min shaking than the IVE-flow cytometry methods. These data show that the impact of air–liquid interfaces is more profound for lower concentration mRNA-LNP formulations, which is consistent with the previously published observations.30.

Antigen Expression is diminished after LNP shaking. Antigen expression of LNPs containing monovalent mRNA was measured after shaking for 30 and 240 min at mRNA concentrations 0.12, 0.06, and 0.01 mg/mL using IVE-flow cytometry (% HA positive cell, panels A, B, and C, and MFI ratio, panels D, E, and D) and IVE-LC/MS/MS (% Antigen/HEK, panels G, H, and I). Black squares depict non-shaken, green triangles and red diamonds depict 30- and 240-min shaking, respectively. R2 for NS, 30 min and 240 min conditions respectively are as follows: D, 0.98, 0.99, 0.99; E, 0,95, 0,95, 0,99; F, 0.98, 0.98, 0.98; G, 0.98, 0.99, 0.98; H, 0.99, 0.96, 0.99; I, 0.99, 0.98, 0.99. n = 2 for IVE-flow cytometry dose–response data.

Relative levels of antigen compared to non-shaking. Antigen expression of LNPs containing monovalent mRNA was measured after shaking for 30 and 240 min at mRNA concentrations 0.12, 0.06, and 0.01 mg/mL using IVE-flow cytometry (% HA positive cell, panel A, and MFI ratio, panel B) and IVE-LC/MS/MS (% Antigen/HEK, panel C). Data represented as % change from non-shaking condition (NS). Change from NS is calculated based on average of 3 doses (IVE-MS: 50,100,150 ng/well; Flow cytometry: 8, 16, 32 ng/well).
Discussion
A high-quality LC/MS/MS method was developed for quantitation of in-vitro expression of mRNA Influenza vaccines. The IVE-LC/MS/MS method’s capability to simultaneously quantify multiple antigenic targets, including the SARS-CoV-2 spike protein, represents a significant advancement in vaccine characterization. This method not only broadens the scope of detectable proteins but also enhances the reliability of the results. Furthermore, the automation of sample preparation is a pivotal development, ensuring greater robustness and reproducibility in the analytical process.
The optimal PRM performance is achieved with at least 10 MS/MS scans per precursor ion to define chromatographic peak shape, an optimized HCD collision energy, a narrow MS1 isolation window (0.4 m/z), a high-resolution MS/MS (240,000), a high AGC target (1000%), and a long maximum injection time (502 ms). The method robustness, selectivity, sensitivity, and linearity were demonstrated with four recombinant influenza hemagglutinin (HA) proteins (from Wisconsin, Darwin, Austria, and Phuket influenza strains) and recombinant S1 domain of the SARS-CoV-2 spike protein. Each protein was spiked into and diluted with negative control HEK293T cells to give a 6-point dilution series spanning 0.02 pmol to 2 pmol. Optimal method parameters were determined by varying the resolution (240,000, 60,000, and 30,000), peptide precursor ion isolation width (0.4, 1, and 1.5 Da), and maximum ion injection time (502, 118, and 56 ms) using the spiked-in dilution series. The optimized PRM method demonstrated excellent data quality, indicating high sensitivity and selectivity of the IVE-LC/MS/MS method. Though data quality is reduced when the system resolution, isolation width, and maximum injection time are concomitantly changed to less optimized settings (resolution lowered from 240,000 to 60,000, isolation width increased from 0.4 Da to 1 Da, and injection time shortened from 502 to 118 ms), a comparable linear fit of recombinant protein level to instrument response is observed. Thus, both conditions are suitable for the selective detection and quantitation of mRNA-translated antigens in complex cellular matrices. This suggests mid-range Orbitrap mass spectrometers may be sufficient for the characterization of mRNA vaccine expression.
The comparative study between the IVE-LC/MS/MS assay and IVE-flow cytometry assay has provided insights into the quantitation of antigen expression for mRNA vaccines from the two methods. The usage of HEK293T cells transfected with a quadrivalent mRNA vaccine across varying doses has allowed for a robust analysis of the relative abundances of expressed protein antigens. IVE-LC/MS/MS and IVE-flow cytometry both display dose-dependent antigen expression. The linear correlation of antigen levels with the administered mRNA dose, as demonstrated by IVE-LC/MS/MS, underscores its sensitivity and reliability. One of the pivotal findings from this study is the ability of the IVE-LC/MS/MS assay to offer a direct and accurate measurement of antigen levels. Unlike IVE-flow cytometry, which primarily reports the percentage of transfected cells, IVE-LC/MS/MS can quantify the absolute abundance of antigen expression using recombinant proteins and reveals that all four antigens encoded in a 1:1:1:1 ratio of mRNA constructs are expressed at comparable levels, providing a clearer picture of the true dose response for each antigen in the quadrivalent mRNA vaccine.
The dose–response and time-course of mRNA transfection study results underscore the efficiency and effectiveness of extending the transfection period to 48 h for optimizing HA antigen production. The linear increase in peak intensity with increasing doses highlights the dose-dependent nature of mRNA transfection. The decreased intensity at 500 ng/well suggests potential cell toxicity. The overlap in dose curves at 24 and 48 h indicates a consistent production rate of HA antigens across the time points, highlighting the advantage of relative quantitation approach for standardizing results with variations in cell pellet sizes and reaffirming the robustness of the transfection process. We commonly employed a 24 h transfection protocol for practical considerations and because the IVE-LC/MS/MS is sufficiently sensitive. If the mRNA transfection level is low, this study shows doubling the transfection protocol to 48 h will improve sensitivity and may rescue target observation from a below limit-of-detection result.
The results from the stability study of quadrivalent mRNA LNPs, as depicted in Fig. 5, revealed similar trending between IVE-LC/MS/MS and IVE-flow cytometry. The LNPs’ performance at different storage conditions (-80 °C and 5 °C) was assessed over a 12-month period. At -80 °C, the LNPs maintained consistent expression levels across all four antigens throughout the study duration, as observed among IVE-LC/MS/MS, the percentage of antigen positive cells and the mean fluorescent intensity (MFI) by IVE-flow cytometry. Conversely, the LNPs stored at 5 °C displayed a noticeable decline in expression levels over time, as indicated by the IVE-LC/MS/MS assay. Although the flow cytometry data for the 9- and 12-month time points were not available, similar trending of %Antigen/HEK data and the other measurements was confirmed for the initial 0-, 1-, 3-, and 6-month time points. The consistency between IVE-LC/MS/MS and IVE-flow cytometry results underscores the robustness of these techniques in providing reliable data.
IVE-LC/MS/MS was also used to evaluate the impact of buffer composition and shaking on LNP transfection. The results from buffer study highlight the significant value of the IVE-LC/MS/MS method in accurately assessing the impact of formulation buffers on mRNA transfection efficiency. By demonstrating a tenfold increase in in-vitro expression with the use of a 150 mM NaCl buffer, this method proves its sensitivity and precision. The shaking study’s findings indicate that shaking time significantly impacts mRNA transfection efficiency and the quality attributes of LNPs. While 30 min of shaking showed no significant effect on antigen expression, a notable decrease in expression was observed after 240 min, particularly at the lowest mRNA concentration (0.01 mg/mL) compared to the high concentrations (0.12 and 0.06 mg/mL). Both IVE-LC/MS/MS and IVE-flow cytometry methods indicated that the sensitivity to shaking-induced changes was more pronounced in lower concentration mRNA-LNP formulations. These results show that IVE-LC/MS/MS is an excellent tool to evaluate degradation of mRNA-LNPs structural and functional capabilities under various conditions.
In this manuscript we have presented the development of a high-quality, straightforward IVE-LC/MS/MS method for quantifying in-vitro mRNA vaccine expression, and demonstrated its versatility, accuracy, robustness, and sensitivity. Its superior performance makes it an invaluable asset for detailed antigen analysis, offering direct and precise measurements that complement IVE-flow cytometry. Our data showed that LC/MS/MS and flow cytometry results are trending similarly, underscoring the complementary nature of these techniques. While peptide-based workflows like IVE-LC/MS/MS are essential for certain aspects, flow cytometry is required when assessing the higher-order structural aspects vital to vaccine efficacy. Thus, the IVE-LC/MS/MS method is developed as an orthogonal characterization method to IVE-flow cytometry. The superior quantitation capabilities of IVE-LC/MS/MS are complemented by the single-cell resolution and population-level expression analysis of Flow Cytometry based expression methods.
The various studies in this work demonstrate the values of applying IVE-LC/MS/MS in different experimental settings, from assessing dose–response relationships, understanding transfection protocols to evaluating the stability of mRNA-LNP under varying stability and buffer conditions. The ability of IVE-LC/MS/MS to provide a deeper understanding of antigen expression and the impact of various factors on transfection efficiency under various experimental conditions, underscores its critical role in advancing mRNA vaccine research and development.
Methods
Materials
In this study, we utilized a range of high-quality reagents and antigens to ensure the accuracy and reliability of our results. Specifically, we employed Native Antigen Influenza A [A/Darwin/6/2021 (H3N2)-like virus] Hemagglutinin (REC32020), Influenza B [B/Austria/1,359,417/2021 (B/Victoria lineage)-like virus] Hemagglutinin (Product Code: REC32004), Influenza B [B/Phuket/3073/2013 (B/Yamagata lineage)-like virus] Hemagglutinin (Product Code: REC31954), and Influenza A [A/Wisconsin/588/2019 (H1N1)pdm09-like virus] Hemagglutinin (Product Code: REC32000) as well as recombinant SARS-CoV-2 B.1.1.7 spike protein S1 (Cat#797,004) from BioLegend. Additionally, Promega Trypsin Platinum, Mass Spectrometry Grade (Cat#VA9000) and FujiFilm Wako Pure Chemical Corporation Lysyl Endopeptidase, Mass Spectrometry Grade (Lys-C) 10AU/Vial were used for protein and Thermo Scientific Pierce Universal Nuclease for Cell Lysis (Cat# 88,702) for nucleic acid digestion.
mRNA-LNP drug product formulations were sourced from internal programs. mRNA-LNP material used for studies in this manuscript were in-house produced using a formulation process similar to the one described in reference30. All mRNA-LNP formulations contained PEG-lipids to avoid LNP aggregation and provided sufficient encapsulation efficiency to mitigate mRNA degradation by external factors. Additionally, degradation was prevented by minimizing sample manipulation during assay execution and sample shipment. Samples were stored under frozen or refrigerated conditions and liquid samples were shipped with minimal headspace to reduce potential sample degradation. In case of multivalent mRNA-LNP drug products correct mRNA ratios were assured at formulation level and verified by adequate release testing.
HEK293T cell transfection and harvest
The HEK293T adherent cells (obtained internally from Pfizer inc.) were seeded at 2E5 to 4E5 viable cells/mL based on the mean viable cell count. The cells were transfected by incubating with the equivalent of 250 ng or as specified of total mRNA per well or buffer for negative controls. After culturing for 16–24 h at 37 °C and 5% CO2, the cells were washed with DPBS and incubated with Accutase solution for 10 ± 5 min to release the adherent cells. Transfected and negative control cells from two wells were pooled into separate tubes and centrifuged at 130 rcf 5 min. The cells were washed with DPBS twice. The buffer was decanted, and the cell pellets were flash frozen with liquid N2 and stored at -80 °C for IVE-LC/MS/MS analysis. For IVE-LC/MS/MS sample tests conducted using a dose–response, a single measurement was performed, whereas for sample tests with a single mRNA concentration, measurements of three independent preparations were taken except for LNP stability at -80 °C. A similar set of wells were also harvested for determination of expression by Flow Cytometry. For both IVE Flow Cytometry and IVE-LC/MS/MS, the burden of cell propagation and manipulation is a major factor influencing method throughput.
Sample preparation for IVE-LC/MS/MS analysis
Transfected and negative control cell pellets were lysed with 5% SDS, 50 mM NaCl, 50 mM Tris–HCl (pH 8.4) buffer at 90 °C for 15 min with shaking at 750 rpm. Next, the samples were sonicated for 3 cycles of 5 s on/10 s off at 50% amplitude to help shear DNA and RNA using Qsonica Q800R3 sonicator. Post lysis, 4 µL of 250 mM TCEP was added and incubated at 55 °C for 45 min with shaking at 750 rpm to reduce disulfide bonds. An alkylation step with 6 µL of 500 mM IAA was performed for 30 min in the dark at 25 oC. ACN equivalent to 8X sample volume was added to precipitate protein at 25 oC for 30 min with gentle shaking of 300 rpm. Samples were then centrifuged at 600 rpm for 5 min at 4 oC to form a more compact pellet. Then the pellets were washed twice with 10X the volume of ACN, centrifuged at 600 rpm for 2 min at 4 °C. To resuspend the dry pellets, 100 µL of 50 mM Tris pH 8 was added, followed by sonication for 10 cycles of 15 s on/5 s off at 100% amplitude. To further mitigate DNA interference in LC/MS/MS analyses, 10 µL of Universal Nuclease (Pierce) was added followed by an incubation at 37 °C for 30 min. The samples were digested for 2 h at 37 oC with shaking at 450 rpm with a 1:5 total enzyme to protein ratio using equivalent levels of Trypsin Platinum (Promega) and Lysyl Endopeptidase (Wako). Post-digestion, the samples were spun down at 4000 rcf for 20 min and the supernatant was transferred into LC/MS/MS vials for analysis.
Liquid chromatography tandem mass spectrometry
The digested cellular and recombinant peptides were separated on an Thermo Scientific Vanquish HPLC system coupled to the Orbitrap Fusion Lumos Tribrid mass spectrometer. Peptides were separated on a reversed phase XSelect Premier CSH C18 column (130 Å, 2.5 µm, 2.1 × 150 mm) (Waters, Milford, MA, USA) using a binary gradient consisting of A: 0.1% formic acid (FA) in water and B: 100% acetonitrile, 0.1% FA at a flow rate of 0.250 mL/min. The gradient increased linearly from 1% B to 30% B over 60 min at column temperature 50 °C. Three washing steps at 99% of organic solvent B at the end of the gradient were used to prevent carry-over into the next run. For the mass spectrometry (MS) analysis, parallel reaction monitoring (PRM) was used in positive ion mode. The normalized collision energy was typically set at 21% (except for peptide FLPFQQFGR requiring higher energy at 29%) and normalized AGC target value at 1000%. The resolution was set at 240,000, isolation width m/z value was set to 0.4, and injection time was set at 502 ms for high-resolution mode. For the low-resolution mode acquisition, the resolution was set at 30,000, isolation width m/z value was set to 1, and injection time was set at 100 ms. The list of target peptide sequence, m/z, charge state, and RT window for each monitored antigen and HEK proteins that are used as cell endogenous controls is shown in Supplemental Table S1.
Data processing
The raw files were imported to Skyline31 and data was processed automatically with a minimum of two fragment ions per precursor peptide and 5 ppm mass tolerance for m/z of each fragment ion. Occasional manual reintegration of peak areas was performed if needed.
IVE-Flow Cytometry
Staining
Cells were harvested as described above except the 0.25 mL of quenched cell suspension was transferred to individual wells (n = 2 per sample) of a 96-well plate. The cells were permeabilized, fixed, and stained with a live/dead aqua dye (ThermoFisher), a specific primary antibody for each antigen and where appropriate, a secondary antibody. During each staining incubation, cells were pelleted, and the supernatant was removed, and the pelleted cells were washed 1X with 0.25 mL of 1X permeabilization wash buffer.
Flow-Cytometry
BD-FACSLyric (BD Biosciences) flow cytometers were used to analyze IVE of the antigen proteins. In the IVE assay, gates were set to assess cell viability (live/dead aqua stain), single vs. double events, and antigen expression (Ab signal). Analysis for each strain was achieved separately by use of a specific primary Ab, either directly conjugated to a fluorophore, or with a common secondary Ab. The multi-laser instrument excited the fluorophore, and the mean fluorescent intensity was recorded. The in-vitro expression of the antigen was determined from the average percent of viable single cells bound with fluorophore-conjugated antibody. This expression result was derived from averaging two independent preparations for each test sample and reported as the mean % antigen positive for each analyzed antigen. Mean fluorescence intensity (MFI) ratio of the sample relative to a matched negative control was additionally calculated as an adjunct measure of relative antigen expression. The gating strategy is provided in Figure S3 of the supplementary material.
Method validation
While Flow Cytometry results presented in the manuscript were generated with methods validated per ICH guidelines, the generated IVE-LC/MS/MS results were solely for characterization purposes. Implementation of the presented method for release and/or stability testing would require additional validation per relevant guidelines (e.g. ICH ).
As part of IVE-LC/MS/MS method development all typical performance characteristics such as specificity, assay precision (intra and inter-day), Lower and Upper Limits of Quantification (LOQ), linear response, accuracy (recovery) and matrix effects were evaluated during the development process. A summary on method performance characteristics is provided in Table S3 of the supplementary material. A graphical example of method precision is shown in supplementary Figure S4 for intra and inter-day testing as well as autosampler stability.
Statistical information
Statistical analysis was performed using GraphPad Prism (V10.2.1) software. Both simple linear and nonlinear regression fits were used to plot the data. The simple linear fit was calculated using the least squares method with 95% confidence interval. Additionally, the nonlinear regression was performed using the least squares method, adjusted for the specified weights of 1/X. The nonlinear model utilized a four-parameter logistic model with the curve representing the best fit with the 95% confidence interval. The resulting model parameters were estimated with high precision, and the goodness-of-fit was assessed using the coefficient of determination (R2) and residual analysis.
The method developed and presented in this manuscript was used for drug product characterization. From this perspective adding dose response experiments was often more valuable than increasing the number of replicates. In some cases, multiple replicate measurements were performed. From a characterization perspective, we emphasize that dose response curves provide additional relevant information while simultaneously confirming transfection differences for multiple instances. Replicate data is indicated in the respective figure captions.
Data availability
The data generated and evaluated in this manuscript are included in the published article and its supplementary materials. The mRNA-LNP formulations tested and discussed in the manuscript are sourced from various Pfizer internal programs, consequently it is not possible to make available any of the mRNA-LNP material presented here. Other non-proprietary reagents and antigens are described in the materials section.
References
Pardi, N., Hogan, M. J. & Weissman, D. Recent advances in mRNA vaccine technology. Curr. Opin. Immunol. 65, 14–20. https://doi.org/10.1016/j.coi.2020.01.008 (2020).
Park, J. W., Lagniton, P. N. P., Liu, Y. & Xu, R.-H. mRNA vaccines for COVID-19: what, why and how. Int. J. Biol. Sci. 17, 1446–1460. https://doi.org/10.7150/ijbs.59233 (2021).
Fang, E. et al. Advances in COVID-19 mRNA vaccine development. Signal Transduct. Target. Ther. 7, 94. https://doi.org/10.1038/s41392-022-00950-y (2022).
Deviatkin, A. A. et al. Universal Flu mRNA Vaccine: Promises, Prospects, and Problems. Vaccines (Basel) https://doi.org/10.3390/vaccines10050709 (2022).
Zhong, Z. et al. mRNA therapeutics deliver a hopeful message. Nano Today 23, 16–39. https://doi.org/10.1016/j.nantod.2018.10.005 (2018).
Hauguel, T. et al. Preclinical immunogenicity and safety of hemagglutinin-encoding modRNA influenza vaccines. npj Vaccines 9, 183. https://doi.org/10.1038/s41541-024-00980-3 (2024).
Packer, M., Gyawali, D., Yerabolu, R., Schariter, J. & White, P. A novel mechanism for the loss of mRNA activity in lipid nanoparticle delivery systems. Nat. Commun. 12, 6777. https://doi.org/10.1038/s41467-021-26926-0 (2021).
Chheda, U. et al. Factors Affecting Stability of RNA – Temperature, Length, Concentration, pH, and Buffering Species. J. Pharm. Sci. 113, 377–385. https://doi.org/10.1016/j.xphs.2023.11.023 (2024).
Le Vay, K., Salibi, E., Song, E. Y. & Mutschler, H. Nucleic Acid Catalysis under Potential Prebiotic Conditions. Chem. Asian J. 15, 214–230. https://doi.org/10.1002/asia.201901205 (2020).
Poveda, C., Biter, A. B., Bottazzi, M. E. & Strych, U. Establishing Preferred Product Characterization for the Evaluation of RNA Vaccine Antigens. Vaccines 7, 131 (2019).
Stiving, A. Q. et al. Enabling functionality and translation fidelity characterization of mRNA-based vaccines with a platform-based, antibody-free mass spectrometry detection approach. bioRxiv https://doi.org/10.1101/2024.05.14.594137 (2024).
Pierce, C. L. et al. Immunocapture isotope dilution mass spectrometry in response to a pandemic influenza threat. Vaccine 35, 5011–5018. https://doi.org/10.1016/j.vaccine.2017.07.049 (2017).
Sutton, W. et al. Quantification of Sars-CoV-2 Spike Protein Expression from mRNA Vaccines Using Isotope Dilution Mass Spectrometry. Vaccine https://doi.org/10.1016/j.vaccine.2023.04.044 (2023).
Nguyen, C. D. L. et al. A sensitive and simple targeted proteomics approach to quantify transcription factor and membrane proteins of the unfolded protein response pathway in glioblastoma cells. Sci. Rep. 9, 8836. https://doi.org/10.1038/s41598-019-45237-5 (2019).
Bourmaud, A., Gallien, S. & Domon, B. Parallel reaction monitoring using quadrupole-Orbitrap mass spectrometer: Principle and applications. Proteomics 16, 2146–2159. https://doi.org/10.1002/pmic.201500543 (2016).
Patel, H. K. et al. Characterization of BNT162b2 mRNA to Evaluate Risk of Off-Target Antigen Translation. J. Pharm. Sci. 112, 1364–1371. https://doi.org/10.1016/j.xphs.2023.01.007 (2023).
Neubert, H. et al. Protein biomarker quantification by immunoaffinity liquid chromatography-tandem mass spectrometry: current state and future vision. Clin. Chem. 66, 282–301. https://doi.org/10.1093/clinchem/hvz022 (2020).
Wessel, D. & Flügge, U. I. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal. Biochem. 138, 141–143. https://doi.org/10.1016/0003-2697(84)90782-6 (1984).
HaileMariam, M. et al. S-Trap, an Ultrafast Sample-Preparation Approach for Shotgun Proteomics. J. Proteome Res. 17, 2917–2924. https://doi.org/10.1021/acs.jproteome.8b00505 (2018).
Rauniyar, N. Parallel Reaction Monitoring: A Targeted Experiment Performed Using High Resolution and High Mass Accuracy Mass Spectrometry. Int. J. Mol. Sci. 16, 28566–28581 (2015).
Boja, E. S. & Rodriguez, H. Mass spectrometry-based targeted quantitative proteomics: achieving sensitive and reproducible detection of proteins. Proteomics 12, 1093–1110. https://doi.org/10.1002/pmic.201100387 (2012).
Pan, S. et al. Mass spectrometry based targeted protein quantification: methods and applications. J. Proteome Res. 8, 787–797 (2009).
Neilson, K. A. et al. Less label, more free: Approaches in label-free quantitative mass spectrometry. Proteomics 11, 535–553. https://doi.org/10.1002/pmic.201000553 (2011).
Sivanich, M. K., Gu, T. J., Tabang, D. N. & Li, L. Recent advances in isobaric labeling and applications in quantitative proteomics. Proteomics 22, 2100256. https://doi.org/10.1002/pmic.202100256 (2022).
Meyer, J. G. & Schilling, B. Clinical applications of quantitative proteomics using targeted and untargeted data-independent acquisition techniques. Expert review proteomics 14, 419–429. https://doi.org/10.1080/14789450.2017.1322904 (2017).
Calderón-Celis, F., Encinar, J. R. & Sanz-Medel, A. Standardization approaches in absolute quantitative proteomics with mass spectrometry. Mass Spectrum. Rev. 37, 715–737. https://doi.org/10.1002/mas.21542 (2018).
Haluck-Kangas, A., Fink, M., Bartom, E. T. & Peter, M. E. CD95/Fas ligand mRNA is toxic to cells through more than one mechanism. Mol. Biomed. 4, 11. https://doi.org/10.1186/s43556-023-00119-1 (2023).
Hashiba, K. et al. Overcoming thermostability challenges in mRNA–lipid nanoparticle systems with piperidine-based ionizable lipids. Commun. Biol. 7, 556. https://doi.org/10.1038/s42003-024-06235-0 (2024).
Chen, H., Ren, X., Xu, S., Zhang, D. & Han, T. Optimization of Lipid Nanoformulations for Effective mRNA Delivery. Int. J. Nanomed. 17, 2893–2905. https://doi.org/10.2147/ijn.S363990 (2022).
Matthessen, R., Van Pottelberge, R., Goffin, B. & De Winter, G. Impact of mixing and shaking on mRNA-LNP drug product quality characteristics. Sci. Rep. 14, 19590. https://doi.org/10.1038/s41598-024-70680-4 (2024).
Pino, L. K. et al. The Skyline ecosystem: Informatics for quantitative mass spectrometry proteomics. Mass Spectrom. Rev. 39, 229–244. https://doi.org/10.1002/mas.21540 (2020).
Acknowledgements
The authors thank Pfizer leadership Matthew Thompson, Margaret Ruesch, Tom Lerch, and Justin Sperry for their support and advice during the development and application of this method.
Funding
This study was funded by Pfizer Inc.
Author information
Authors and Affiliations
Contributions
IVE-LC/MS/MS method conceptualization and development – J.B, B.C.G, A.W.D; LNP material preparation – R.V.P, R.M, B.G; IVE-LC/MS/MS sample preparation, data collection and analysis – R.E, K.K, G.R; Hamilton method development and implementation for IVE-LC/MS/MS – B.B, K.K; IVE-flow cytometry sample preparation, data collection and analysis – A.C, J.E, C.E, M.W; the manuscript drafting, revising and editing – L.W, B.C.G, A.W.D, O.V.F; the final manuscript reviewing and editing – R.V.P, E.B.B, J.C.R; the overall study support – L.W, O.V.F, E.B.B, J.C.R, D.C. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
All authors are employees of Pfizer Inc. Some authors may be shareholders of Pfizer Inc. The authors declare no other competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, H.L., Kajbaf, K., Gau, B.C. et al. A novel in-vitro expression assay by LC/MS/MS enables multi-antigen mRNA vaccine characterization. Sci Rep 15, 10336 (2025). https://doi.org/10.1038/s41598-025-94616-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-94616-8
This article is cited by
-
Elucidating the underlying mechanism of mechanical stress-induced impact on mRNA-LNP structure
Scientific Reports (2025)
