
Abstract
DNA damage exhibits a strong correlation with gastric cancer (GC). However, there is still a paucity of comprehensive, in-depth investigations into this relationship. We aimed to explore the association between DNA damage-related genes and GC to provide insights into its molecular mechanisms and potential biomarkers. For this study, Bulk RNA sequencing data of GC were obtained from The Cancer Genome Atlas (TCGA), single-cell RNA sequencing datasets were retrieved from the Gene Expression Omnibus (GEO), and a DNA damage-associated gene set was sourced from the GeneCards database. Through the application of survival analysis, differential expression gene analysis, and weighted gene co-expression network analysis, we identified DNA damage-related genes potentially linked to GC. Subsequently, multiple machine learning approaches were employed to screen out hub genes with considerable diagnostic potential. Analysis of bulk RNA sequencing data verified gene expression patterns in GC. Single-cell analysis further demonstrated cell-type-specific gene expression, and immunohistochemical experiments were conducted to validate the potential biomarker utility of key genes. Our findings revealed that thirteen DNA damage-related genes that may be linked to GC were identified. Subsequently, CLSPN and SALL4 were screened out as hub genes possessing considerable diagnostic potential. Analysis of bulk RNA sequencing data verified the upregulated expression of these two genes in GC, thereby underscoring their predictive significance. Across multiple machine learning methods, CLSPN was consistently ranked as the gene with the highest importance. Single-cell analysis further demonstrated that CLSPN is predominantly highly expressed in tumor cells, which emphasizes its cell-type-specific function in GC. To validate CLSPN’s potential as a biomarker, we conducted immunohistochemical experiments; these experiments showed high CLSPN expression in GC tissues, and the expression levels were significantly correlated with age, tumor size, pT stage and lymph node metastasis. This study reinforces the link between DNA damage and GC and offers fresh perspectives on its underlying molecular mechanisms. Nonetheless, further validation in clinical evaluation is essential to confirm its practical value for GC management strategies.
Similar content being viewed by others


Introduction
GC is one of the most common cancers, with more than 600,000 deaths every year. Asia, South America and Eastern Europe have a high incidence and are rapidly younger (age < 50 years)1. In China, because the national GC screening has not yet been carried out, it is only targeted at the population in high-risk areas, resulting in GC at the time of diagnosis is mostly advanced, and the prognosis is poor2. Histological classifications alone are not enough to adequately categorize patients for personalized treatment and enhance clinical outcomes, therefore, advanced diagnostic tools and treatments are essential for discovering new therapeutic targets in GC patients3,4.
Maintaining the integrity of DNA structure and function is crucial to supporting regular life processes. Exposure to environmental threats and endogenous toxicants can disrupt DNA integrity, destabilize the genome, and play a key role in the onset or progression of various diseases, especially cancer5. DNA damage arises through two pathways, namely direct effects and indirect effects. In the first, insults from inside or outside the cell strike DNA itself, scrambling both structure and activity. In the second, the inside or outside insults spark a burst of reactive materials—such as free radicals—that go on to attack the genome6. The DNA damage response (DDR) pathway serves as the central hub that identifies, signals and repairs DNA damage. By imposing tight restraint on cell cycle progression and promoting DNA repair, it minimizes the transmission of damage to daughter cells7. DNA damage encompasses multiple types, among which the most harmful lesions and the gravest threat to cells are double-strand breaks (DSBs) formation. Nevertheless, radiation therapy and many chemotherapeutic agents through DSBs to eliminate tumor cells across numerous cell division cycles, a mechanism especially deadly to cancer cells6,8,9. Although existing studies have emphasized the critical role of DNA damage in the development and progression of GC, it remains a difficult undertaking to identify and confirm the key genes that play a role in regulating it.
In the present research, we utilized bioinformatic methodologies to carry out a systematic analysis of DNA damage-associated genes and their biological roles in GC. To start with, we extracted datasets related to GC and DNA damage from the TCGA database and GeneCards. We performed survival analysis, differential expression analysis, and weighted gene co-expression network analysis (WGCNA) with the aim of pinpointing candidate genes that are closely linked to GC. Following this, a variety of machine learning algorithms were employed to further filter for hub genes. Leveraging single-cell RNA sequencing (scRNA-seq) and spatial transcriptomics (ST) data, we examined the expression patterns of hub genes across diverse cell types and explored differences in intercellular communication. Lastly, we collected GC tissues from our own institution to verify the differential expression of hub genes between normal and tumor samples through immunohistochemistry. Additionally, we analyzed the clinicopathological features of patients with varying expression levels of hub genes, aiming to explore their potential as diagnostic and therapeutic targets.
Methods
Data acquisition and preprocessing
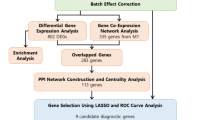
The original gene expression matrix and relative clinical datasets for STAD (stomach adenocarcinoma) were downloaded from the The Cancer Genome Atlas database using the TCGAbiolinks package (2.32.0) in R (version 4.3.3). The gene expression matrix was filtered to retain only the “protein_coding” genes, which were then subjected to preprocessing. For the clinical data, cases lacking survival prognosis information were excluded. Ultimately, the final dataset was obtained. The entire research workflow is illustrated in Fig. 1. Datasets GSE84433 and GSE15459 were downloaded from the GEO database and used for validation after the preprocessing procedures.

Study workflow.
Kaplan-Meier survival analysis
The TPM-formatted gene expression matrix was matched with the corresponding clinical data. Kaplan-Meier (KM) survival analysis was performed using the survival package (3.8.3) and survminer package (0.4.9) in R. Based on the median grouping of gene expression levels, the association between gene expression and patients’ survival time was evaluated, and only those genes with P < 0.01 were retained for further analysis.
Univariate Cox regression analysis
Univariate Cox regression analysis was performed for each gene using the coxph function to calculate the impact of each gene on patients’ survival. The summary(cox) function was used to extract the regression results, and only genes with P < 0.01 were retained. The hazard ratio (HR) and 95% confidence interval (CI) were calculated based on the regression coefficients of these retained genes.
Identification of differentially expressed genes
For differential expression gene analysis of TCGA-STAD gene expression matrix, genes with no expression across all samples were first removed. Then, differential gene expression analysis was performed using ‘limma-voom’ with the limma package (3.58.1), using a Counts-formatted gene expression matrix, with screening criteria set as a P < 0.05 and |logFC| > [mean(|logFC|) + 2sd(|logFC|)]10.
Analysis of the weighted gene co-expression network
The TPM-formatted gene expression matrix of TCGA-STAD was processed using the WGCNA package (1.72.5) to evaluate trait-related modules. Gene filtering was performed with a mean filtering threshold of 0 and selection of 6,000 genes based on median absolute deviation (MAD). A soft threshold of 7 was established to identify the core modules, with minModuleSize set to 80.
DNA damage gene set
The DNA damage gene set was sourced from GeneCards (https://www.genecards.org/)11.
Candidate genes discovery and machine learning model Building
UpSet analysis was conducted to identify the intersecting feature genes screened out by all five methods, with the consistently identified genes selected as candidate genes. To rank the importance of these intersecting genes and uncover hub genes, machine learning analyses were performed with C5.0, Neural Network (NNET), DecisionTree, Gradient Boosting Machine (GBM), K-Nearest Neighbors (KNN), Random Forest (RF), and Extreme Gradient Boosting (XGBoost) algorithms, all of which were implemented based on the caret package (7.0.1). A random seed of 400 was set before dataset splitting. The merged dataset was randomly split into a training set (70%) and a test set (30%), where the training set was used for model training and parameter optimization, and the test set for evaluating generalization performance. During model training, 10-fold cross-validation with 1 repetition was implemented to optimize hyperparameters. Gene importance was quantified using the varImp function from the caret package (the specific content can be found in the tutorial authored by Dr. Max Kuhn: https://topepo.github.io/caret/index.html). Notably, all preprocessing parameters were estimated solely from the training set and then applied to the test set to avoid data leakage.
Functional enrichment analysis
Using ClusterProfiler package (4.10.1), we performed Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses12,13 on differentially expressed genes (DEGs) and conducted Gene Set Enrichment Analysis (GSEA) to explore potential biological differences.
Nomogram construction
Nomogram analysis was conducted via the rms package (6.8.2) to forecast 1-, 3-, and 5-year survival as well as recurrence rates in GC patients. This analysis comprised an upper scoring component and a lower prediction component. Total scores—derived by summing the points assigned to each factor—enabled accurate prediction of the outcomes. Calibration curves were applied to evaluate the accuracy of survival forecasts.
Assessment of hub genes
Based on the identified hub genes, pan-cancer analyses were conducted using the Gene_DE Module in the TIMER3 portal (https://compbio.cn/timer3/)14 (http://timer.comp-genomics.org/). Gene_DE Module instantly contrasts tumor-normal gene expression across TCGA cancer types. Using the Correlation Analysis Module of the GEPIA2 (http://gepia2.cancer-pku.cn/#index)15, the correlation of candidate genes in ‘STAD Tumor’ was explored. To assess the diagnostic efficacy of the selected signature genes, GraphPad (Version 8.0) generated Receiver Operating Characteristic (ROC) curves and calculated the Area Under Curve (AUC), which evaluated their classification performance across varying sensitivity and specificity.
Transcription factor analysis
Transcription factor (TF) prediction was performed using the TFTF package (0.1.0)16 and various transcription factor databases to identify the transcription factors associated with the hub genes.
Single-cell RNA sequencing analysis
A public scRNA-seq dataset of GC, GSE264203, was downloaded from the GEO database (https://www.ncbi.nlm.nih.gov/geo/). The data first underwent initial filtering, and the raw dataset was preprocessed using the Seurat package (5.2.1). The minimum RNA molecule count was set to 1000, and cells expressing at least 200 genes were retained to remove empty droplets or vesicles. To reduce the risk of doublets, the maximum RNA molecule count was set to 50,000 and the maximum number of detected genes was set to 6000. The maximum percentage of mitochondrial genes was set to 15% to remove dead cells. A total of 2000 highly variable genes were selected using the FindVariableFeatures function. Regarding batch effects, no obvious batch effects were observed among the three samples after visualization. Subsequently, dimensionality reduction was performed via the Seurat::RunPCA function. For cell clustering, the FindNeighbors function was executed with the first 8 principal components as input features, and the FindClusters function was implemented with a resolution parameter set to 1 to assign cluster identities to cells. Additionally, the clustering results were further visualized using the Seurat::RunUMAP function, with the UMAP projection constructed based on the aforementioned principal components.
GO and KEGG enrichment analyses in scRNA-seq
Subpopulations of tumor cells from the above-mentioned scRNA-seq were extracted. Based on the median expression value of the hub gene, tumor cells were divided into high-expression and low-expression groups. Differential analysis was then conducted using the FindAllMarkers function in the Seurat package with its default parameters. Subsequently, to determine the biological processes and metabolic pathways associated with the candidate genes, GO and KEGG pathway enrichment analyses were performed using the enrichGO and enrichKEGG functions in the clusterProfiler package (4.10.1).
Pseudotime analysis
Pseudotime analysis aligns cells along a trajectory, simulating a temporal sequence and inferring their positions in distinct differentiation states based on gene expression profiles. For this trajectory analysis, Monocle3 package (1.3.7) was employed to infer tumor cell developmental trajectories. Selection began with the hub genes’ expression variation across cells as a starting point, given the hub genes may drive changes in cell states. Cells were pseudotime-ordered by the hub genes’ expression dynamics to uncover potential lineage relationships.
Cell-cell communication analysis
For the analysis of communication among various cell types, the CellChat package (2.1.2) was employed to systematically explore and visualize the detailed communication networks between these cell types, and infer cell type-specific interaction relationships, thereby gaining an understanding of the intercellular communication during the pathogenesis of GC.
Spatial transcriptomic analysis
The GSE251950 dataset was downloaded from the GEO, and the ST data of three patients (GSM7990473, GSM7990474, GSM7990475) were selected for analysis. The Seurat package was used for ST analysis. Data normalization was performed using NormalizeData, dimensionality reduction was conducted via RunPCA, clustering of ST spots was performed using FindNeighbors and FindClusters, and data visualization was achieved using RunUMAP.
Immune infiltration analysis
Based on the preprocessed TCGA-STAD gene expression matrix (tumor tissues only, after log2 transformation, and MAD filtering with retention of the top 10,000 genes), patients were divided into high hub gene expression and low candidate gene expression groups using the median candidate gene expression value. The IOBR package (0.99.8) was used to calculate immune scores via the CIBERSORT, CIBERSORT-ABS, quanTIseq algorithm.
Drug sensitivity analysis
Drug sensitivity analysis was performed using the pRRophetic package (0.5), which predicts drug sensitivity based on gene expression data. The Wilcoxon rank-sum test was employed to assess the differences in drug sensitivity between distinct gene expression groups. To visualize the results and further analyze the relationship between gene expression and drug sensitivity (as measured by IC50 values), boxplots and correlation scatter plots were generated using the ggplot2 package.
Immunohistochemistry
A total of 70 paired samples of GC tissue and adjacent normal tissue were gathered at the Sunshine Union Hospital, with formal patient authorization secured. The research protocol was endorsed by the Ethics Committee of Sunshine Union Hospital (Approval No.: 2025032123) and adhered to the principles of the Helsinki Declaration. Surgically resected tissue specimens were locally sampled after fixation in 10% neutral formalin fixative for 48 h, followed by routine procedures including dehydration, paraffin embedding, sectioning, and staining before diagnosis under a light microscope. Immunohistochemical detection was performed using the Dako EnVision FLEX+ system on 3-µm-thick tissue sections: after dewaxing and rehydration, antigen retrieval was conducted with EDTA buffer (pH = 9.0) under high temperature; the tissue sections were then rinsed with Tris-buffered saline, incubated with 10% goat serum blocking solution (diluted in PBS) for 25 min at room temperature, and after discarding the blocking solution, primary antibody against CLSPN (1:600 dilution) was added for 25 min of room temperature incubation. Following rinsing to remove the primary antibody, the sections were treated with hydrogen peroxide for 5 min, rinsed again, and incubated with secondary antibody for 20 min at room temperature in the dark; after another rinse, DAB chromogenic reaction was performed with the reaction time controlled under a microscope, and finally the sections were counterstained with hematoxylin, followed by dehydration, clearing, and mounting. The primary antibody against CLSPN (ab3720) was purchased from Abcam plc, while the immunohistochemistry kit and EDTA antigen retrieval buffer were obtained from Agilent Technologies, Inc. IHC assays were executed by seasoned pathologists, with candidate gene protein levels assessed via the immunoreactive score (IRS). Calculation of the IRS involved multiplying the staining intensity (SI) by the proportion of positive cells (PP)17. SI ratings were assigned as 0 (no staining), 1 (light yellow, weak), 2 (brown, moderate), or 3 (dark brown, strong). PP values were categorized as 0 (0%–5%), 1 (6%–25%), 2 (26%–50%), 3 (51%–75%), and 4 (> 75%).
Statistical analysis
Data processing and statistical analyses were performed using R 4.3.3 and GraphPad Prism 8.0. The disparity in continuous variables between the two groups was assessed using the Wilcoxon, differences between three or more groups were evaluated using one-way analysis of variance (ANOVA). Correlations between variables were evaluated using Pearson correlation coefficient analysis. The expression difference of hub genes between GC tissues and normal tissues was analyzed using the independent samples t-test. The correlation between hub genes expression and clinicopathological characteristics of GC was analyzed using the chi-square test (χ2 test) or Fisher’s exact test. The Log-rank test was applied to analyze the relationship between hub genes expression and postoperative overall survival (OS) of GC patients, and Kaplan-Meier survival curves were plotted. P < 0.05 was considered statistically significant.
Results
Survival analysis of gene expression and screening of significant genes
Regarding the expression matrix, a total of 448 patient tissues were finally obtained (412 tumor tissues and 36 normal tissues), and regarding the clinical data, survival and prognostic information for 340 patients was ultimately acquired. The gene expression matrix in TPM format was integrated with the corresponding clinical information for subsequent analyses. Survival analyses using the KM method were implemented with the survival and survminer packages in R. Patients were divided into two groups according to the median value of each gene’s expression level. The relationship between gene expression and patient survival duration was then assessed, and only genes showing a statistically significant association (P < 0.01) were selected for further investigations. A total of 1,653 genes were ultimately identified (Supplementary Table 1).
Univariate Cox regression-based screening of survival-associated genes
Univariate Cox regression analyses were conducted for each gene through the coxph function to assess the influence of individual genes on patient survival outcomes. The regression results were retrieved using the summary(cox) function, and based on the derived regression coefficients, the HR along with their corresponding 95% CI were computed to quantify the survival impact of each gene. A total of 1,628 genes with a P < 0.01 were ultimately identified (Supplementary Table 2).
Identification of differential genes
For the TCGA-STAD gene expression matrix, differential expression analysis using the limma package identified 1,043 DEGs after applying the thresholds of P < 0.05 and |logFC| > 1.98052 (Supplementary Table 3, Supplementary Fig. 1).
WGCNA-based module identification and candidate gene screening
Ranking all genes by MAD, we retained the top 6000 most variable genes from the expression matrix before entering WGCNA. The optimal soft-threshold β = 7 was selected via pickSoftThreshold function to attain scale-free topology (R2 ≥ 0.85) (Fig. 2A), and a dendrogram was cut at height h = 0.25 with minimum module size 80, yielding 9 colour-coded modules (Fig. 2B); the module turquoise most strongly correlated with tumor (r = 0.58, P = 4 × 10-41) contained 1573 genes (Fig. 2C, D; Supplementary Table 4). Further analysis revealed that the strongest correlation between module membership (MM) and gene significance (GS) exists within the turquoise module (Fig. 2E).

Establishment of a weighted gene co-expression network (WGCNA). (A) Soft thresholds selection. (B) Distinct colored branches correlate to 9 distinct modules. (C) Module-trait correlation heatmap. (D) Cluster dendrogram and heatmap of adjacency eigenvalue in the network. (E) The relationship between gene significance (GS) for tumor and module membership (MM) in turquoise module.
DNA damage gene set in genecards
To construct the DNA damage-related gene set, we retrieved candidate genes from the GeneCards database using the key term ‘DNA damage’ for targeted screening. We selected the top 3000 genes ranked by the Relevance score to ensure biological specificity and reduce data noise (Supplementary Table 5).
13 genes as candidate genes and their enrichment analysis.
Through the five aforementioned methods, we conducted UpSet plot analysis on the screened feature genes, using the UpSet function to identify genes consistently selected by all five methods; this analysis yielded 13 intersecting genes, which were designated as important candidate genes (Fig. 3A; Supplementary Table 6). We performed GO and KEGG pathway enrichment analyses on these 13 candidate genes (Fig. 3B). Within the biological process (BP) of GO analysis, these genes showed significant enrichment in processes associated with cell division, including cell cycle checkpoint signaling, regulation of cell cycle phase transition, negative regulation of cell cycle phase transition, and negative regulation of cell cycle process. In terms of cellular components (CC), the genes were primarily concentrated in condensed chromosome and chromosomal centromeric regions. Molecular function (MF) analysis revealed that the genes exhibited notable functional activities, such as DNA-binding transcriptional activator activity (RNA polymerase II-specific), single-stranded DNA binding, and DNA secondary structure binding. KEGG pathway analysis demonstrated that these candidate genes were enriched in pathways such as Cell cycle, Homologous recombination, and Fanconi anemia pathway. These findings imply that the 13 candidate genes may exert their biological roles through the regulation of signaling pathways involved in cell cycle progression.

Candidate Gene Screening and Its Enrichment Analysis. (A) 13 candidate genes were obtained by taking the intersection via UpSet. (B) GO and KEGG enrichment analyses of the 13 candidate genes (In this study, all raw KEGG data used can be accessed via www.kegg.jp/kegg/kegg1.html.).
Machine learning-based identification of CLSPN and SALL4 as hub genes
To further prioritize the most critical genes among these 13 candidates, we employed 7 widely used machine learning algorithms for feature importance ranking—C5.0, NNET, DecisionTree, GBM, KNN, RF, and XGBoost—and evaluated their performance using three key metrics: AUC (measures the model’s ability to distinguish between classes), Accuracy (measures the proportion of correctly classified samples), and F1-score (measures the balanced performance of precision and recall). The dataset used for machine learning comprised a total of 448 cases, including 412 gastric cancer tissues and 36 normal tissues; it was split into a training set (70%, 315 cases) and a test set (30%, 133 cases). The performance of each algorithm was as follows: C5.0 with AUC = 0.987, Accuracy = 0.977, and F1-score = 0.988; NNET with AUC = 0.998, Accuracy = 0.977, and F1-score = 0.988; DecisionTree with AUC = 0.946, Accuracy = 0.985, and F1-score = 0.992; GBM with AUC = 0.994, Accuracy = 0.977, and F1-score = 0.988; KNN with AUC = 0.993, Accuracy = 0.992, and F1-score = 0.996; RF with AUC = 0.998, Accuracy = 0.985, and F1-score = 0.992; and XGBoost with AUC = 0.997, Accuracy = 0.985, and F1-score = 0.992. These models are designed to output a ranking of gene importance, all models exhibited high performance, confirming the robustness of the feature importance ranking, and ultimately, Claspin (CLSPN) and Spalt-like transcription factor 4 (SALL4) appeared most frequently among the top 2 in the importance scores across all 7 algorithms, indicating they are the hub genes among the candidate genes (Fig. 4).

Seven methods including C5.0, NNET, DecisionTree, GBM, KNN, RF, XGBoost all indicate that CLSPN and SALL4 are the hub genes. The corresponding ROC curves on the right demonstrate that all these models achieve high AUC values, suggesting excellent predictive performance in identifying these hub genes.
Hub genes exhibit excellent evaluative value
Pan-cancer analysis using TIMER3 revealed that CLSPN and SALL4 are significantly altered in multiple common types of tumors (Fig. 5A). This widespread alteration pattern suggests that the hub genes may play a crucial role in the pathological processes of various cancer types, including STAD. Subsequently, ROC curves were used to further assess the expression of the 2 hub genes and their diagnostic utility. Results indicated that the AUC values distinguishing tumor samples from controls were 0.9819 (95% CI: 0.9706–0.9903) for CLSPN, and 0.9621 (95% CI: 0.9403–0.9839) for SALL4 (Fig. 5B). These findings imply the two hub genes have strong predictive potential for GC, laying a key foundation for their use in clinical diagnosis. The GEPIA2 database shows that these two genes exhibit significant correlations (r = 0.21) in STAD tumors (Fig. 5C). Based on the hub genes, a nomogram was developed for predicting the prognosis of GC patients. Total prognostic scores were computed by summing the individual points of CLSPN and SALL4, with the cumulative score mapping to the predicted probability of 1-, 3-, and 5-year OS. Calibration plots for the training dataset were generated to assess the concordance between the nomogram’s predicted 1-, 3-, and 5-year OS probabilities and the actual observed outcomes in the training population. The GSE15459 dataset was used as the validation cohort for generating calibration plots. After undergoing a series of preprocessing steps including duplicate row removal, extraction of protein-coding genes, data normalization, and exclusion of patients without survival prognosis data, 192 patients remained for subsequent validation analysis. Calibration plots for 1-year, 3-year, and 5-year OS demonstrated reasonable concordance between predicted and observed values (Fig. 5D).

Profiling pivotal gene expression and establishing nomogram model for GC diagnosis. (A) TIMER3-based pan-cancer assessment shows hub genes frequently altered across major tumor types. (B) Diagnostic values of hub genes. (C) Correlations between hub genes. (D) Nomogram for predicting gastric cancer prognosis and training dataset (left) as well as validation dataset (right) calibration plots for evaluating the predictive performance of the nomogram.
GSEA further reveals hub genes associated with DNA damage
GSEA results showed that hub genes are involved in multiple DNA damage-related pathways, such as Cell cycle, DNA replication, TGF-beta signaling pathway and Wnt signaling pathway, further supporting their critical role in the pathogenesis of DNA damage in GC (Fig. 6A). The GSE84433 dataset was subjected to validation after undergoing duplicate row removal, protein-coding gene extraction, and data normalization, and similar results were obtained (Fig. 6B).

GSEA of hub genes in two gastric cancer datasets. (A) Results from the TCGA-STAD dataset. (B) Validation in the independent GSE84433 dataset.
HNF4A is the common transcription factor of the hub genes.
After defining CLSPN, SALL4 as the hub genes, we sought to identify theirs potential upstream transcription factors. Nine TF-centric databases—CHEA, PWMEnrich_JASPAR, ENCODE, hTFtarget, FIMO_JASPAR, GTRD, ChIP-Atlas, KnockTF and TRRUST—were interrogated for hub genes regulators. Subsequently, UpSet analysis revealed that HNF4A is the common transcription factor for CLSPN and SALL4, and it is the one predicted by the largest number of the aforementioned databases (Supplementary Fig. 2, Supplementary Table 7), thereby laying a foundation for in-depth clarification of the genetic regulatory network.
Dimensionality-reduction clustering annotation and single-cell landscape
After stringent quality control (Supplementary Fig. 3A), the cleaned matrix retained 31,520 cells and 19,580 genes. The leading 8 PCs (Supplementary Fig. 3B) guided graph clustering, yielding 22 robust clusters (Supplementary Fig. 3C). Manually curated canonical markers were used to assign cell identities to each cluster. These markers guided the annotation of 6 distinct cell types: immune cells, fibroblasts, epithelial cells, tumor cells, neuroendocrine cells, and endothelial cells (Fig. 7A). UMAP visualized global cell topography (Fig. 7B).

Cell type annotation of scRNA-seq. (A) UMAP plot demonstrating the expression patterns of marker genes for each cell type. (B) Distribution of cell types in scRNA-seq.
CLSPN is highly expressed in tumor cells
In machine learning analyses (Fig. 4), CLSPN was consistently ranked as the most important feature across all algorithms, and thus we focused on its expression characteristics. ScRNA-seq results showed that CLSPN was mainly expressed in tumor cells and epithelial cells, with a higher expression level in tumor cells than in epithelial cells (Fig. 8A). To further evaluate the spatial distribution characteristics of CLSPN, we analyzed ST data from 3 GC patients, with the number of principal components selected for each patient’s dataset set to 4, 8, and 8, respectively. Annotation was performed by integrating the original literature and canonical genes, yielding the spatial distribution of tumor cells and epithelial cells. We found that the spatial distribution of CLSPN exhibited similar characteristics to those in the scRNA-seq data (Fig. 8B).

Expression and spatial distribution of CLSPN. (A) ScRNA-seq data showed that CLSPN was highly expressed mainly in tumor cells. (B) Spatial transcriptomics data showed that CLSPN was highly expressed mainly in tumor areas.
CLSPN-linked GO and KEGG pathways by scRNA-seq
Using scRNA-seq, we isolated the tumor cells subpopulation and dichotomized it into CLSPN-high and CLSPN-low groups based on the median CLSPN expression level. Then differential gene expression between these two groups was then systematically evaluated. DEGs expression patterns were visualized via volcano plots (Supplementary Fig. 4). GO enrichment analysis indicated that the DEGs were primarily involved in DNA replication, chromosome segregation, mitotic nuclear division and sister chromatid segregation in BP; in CC, they were mainly enriched in condensed chromosome, chromosomal region, and replication fork; and in MF, they were associated with ATP hydrolysis activity, single−stranded DNA binding, helicase activity, and damaged DNA binding. Regarding KEGG enrichment, it demonstrated significant enrichment in pathways such as Cell cycle, DNA replication, Homologous recombination and Cellular senescence (Fig. 9). These results further support the critical role of CLSPN in the pathogenesis of GC.

Enrichment analysis of CLSPN-high/low tumor cell subpopulations via scRNA-seq.
Cell-cell communication and Pseudotime analysis in CLSPN+ tumor cells
To clarify the biological functions of CLSPN in tumor cells, we divided tumor cells into CLSPN-positive (CLSPN+) and CLSPN-negative (CLSPN-) groups by CLSPN expression in scRNA-seq, and assessed the number and interaction of their cell-cell communication with other cell types. CLSPN+ tumor cells exhibited more interactions with other cells compared with CLSPN- tumor cells, with particularly closer synergistic interactions with fibroblasts (Fig. 10A). Current studies have found that CLSPN can promote tumor proliferation, migration, and invasion via the Wnt/β-catenin signaling pathway, and also revealed that the Wnt signaling pathway, DNA damage, and cell cycle are closely related through multiple mechanisms18,19,20,21,22. Therefore, we focused on WNT signaling pathway in cell-cell communication. The results showed that under the WNT signaling pathway, CLSPN+/- tumor cells exhibited a primary role as signal-receiving cells, with the incoming signal intensity in CLSPN+ tumor cells being higher than that in CLSPN- tumor cells (Fig. 10B, C). Subsequently, Monocle 3 was employed to perform pseudotime analysis on tumor cells and normal epithelial cells. Using the site with the lowest CLSPN expression in Fig. 9 as the origin for this analysis, we observed that with pseudotime progression, tumor cells gradually transitioned from low to high CLSPN expression along three trajectories (Fig. 10D), consistent with our abovementioned findings in this study, suggesting that this process may drive cell fate transitions.

(A) The number and strength of cell-cell communications between CLSPN+/- tumor cells and other cell types. (B) Dynamic changes in intercellular interactions among different cell types under the WNT signaling pathway. (C) WNT signaling pathway network. (D) UMAP plot showing the pseudotime distribution of cells, illustrating their developmental trajectory.
CLSPN exhibits links to macrophages and NK/T cells in the tumor microenvironment
To explore the relationship between the tumor microenvironment and CLSPN, we analyzed the immune cell enrichment scores using three methods, namely CIBERSORT, CIBERSORT-ABS, and quanTIseq, in groups stratified by CLSPN expression levels. The macrophage and NK/T cell scores exhibit significant differences between the high CLSPN expression group and the low CLSPN expression group (Fig. 11, Supplementary Fig. 5), suggesting that CLSPN may influence the tumor microenvironment in GC.

Evaluate the differences in immune cell infiltration between the CLSPN high-expression group and the CLSPN low-expression group.
CLSPN expression is associated with sensitivity to multiple drugs
Based on the median CLSPN expression level, the patients were divided into high-expression and low-expression groups. Drug sensitivity was predicted using the pRRophetic package. Results of correlation analysis showed that the expression level of CLSPN was significantly negatively correlated with the half-maximal inhibitory concentration (IC50) of BI-2536, Methotrexate, Ruxolitinib, Epothilone B, and Tipifarnib (P < 0.01), as assessed by Pearson correlation analysis (Fig. 12). Such observations could present a novel potential avenue for clinical chemotherapy via targeting CLSPN.

CLSPN expression and drug sensitivity analysis.
CLSPN was correlated with multiple clinicopathological factors of gastric cancer
Protein expression levels of CLSPN were assessed in 70 pairs of normal gastric tissues and GC tissues via immunohistochemical (IHC) staining (Fig. 13A). It was observed that CLSPN exhibited a significant upregulation in GC tissues compared with their normal counterparts, implying that CLSPN contributes to the initiation and progression of GC. To further investigate the association between CLSPN in GC and clinicopathological features, we analyzed the correlation of CLSPN expression with clinicopathological parameters of GC patients, along with its prognostic significance. Specifically, CLSPN expression showed no significant correlation with patient sex and neural/vascular invasion (all P > 0.05). However, it was closely associated with age (P = 0.016), maximum tumor diameter (P = 0.017), pT stage (P = 0.001) and lymph node metastasis (P = 0.003, Table 1). Additionally, Log-rank test results revealed that GC patients with high CLSPN protein expression had significantly shorter OS compared to those with low CLSPN expression (P = 0.024, Fig. 13B). The GSE15459 dataset was utilized to validate the association between CLSPN expression and prognosis in GC patients. The findings further confirmed that GC patients with elevated CLSPN expression exhibited notably reduced OS relative to those with low CLSPN expression (P = 0.035, Fig. 13C).

The differential expression of CLSPN between gastric cancer tissues and normal tissues. (A) Immunohistochemical analysis revealed that CLSPN exhibits distinct expression patterns across different tissues. (B,C) High CLSPN expression is associated with poor prognosis.
SALL4 Spatial pattern, immune infiltration and drug sensitivity
To investigate the spatial expression pattern of SALL4 in GC, we analyzed the ST data of the aforementioned 3 GC patients (GSM7990473, GSM7990474, GSM7990475). The ST analysis results showed distinct differences in SALL4 spatial expression among the three patients: For GSM7990474 and GSM7990475, SALL4 expression signals were significantly enriched in the tumor areas, while in the normal areas, SALL4 expression was relatively low. This spatial expression pattern further confirms the tumor-specific expression characteristics of SALL4, which is consistent with its potential role in GC progression. However, the difference in SALL4 expression between tumor areas and normal areas was not obvious in GSM7990473 (Supplementary Fig. 6). We also employed three immune infiltration analysis methods—CIBERSORT, CIBERSORT-ABS, and quanTIseq—to compare patients stratified into high and low SALL4 expression groups based on the median value of SALL4 expression versus low SALL4 expression. This analysis revealed significant differences in T cells, particularly CD8 T cells, between the two groups, suggesting that SALL4 may also influence the tumor microenvironment in GC (Supplementary Fig. 7). Leveraging the above-defined groups stratified by SALL4 expression median, we further conducted drug sensitivity analysis to explore the association between SALL4 expression and the efficacy of potential therapeutic agents. The results demonstrated that SALL4 expression levels were significantly negatively correlated with the half-maximal inhibitory concentration (IC50) of TAE684 and GNF-2 (Supplementary Fig. 8).
Discussion
GC is associated with a poor prognosis due to late detection and limited treatment options. It ranks among the major causes of cancer-related deaths and has a more significant impact on men’s health. Therapeutic approaches that target specific molecular targets have led to improved survival outcomes23. However, many biomarker-targeted drugs have achieved only limited clinical success, highlighting an urgent need to develop more targets to address the higher health demands of patients. Comprehensive multi-omics analyses of tumors can identify new biomarkers, which may help predict the response to targeted therapy.
In this study, TCGA-STAD gene and clinical data to screen candidate genes associated with GC. We first performed Kaplan-Meier survival analysis, univariate Cox regression, WGCNA, DEG analysis and collected DNA damage-related gene sets from GeneCards. Then UpSet analysis found 13 candidate genes, and 7 machine learning algorithms ranked CLSPN and SALL4 as the hub genes. The nomogram constructed using hub genes showed robust predictive performance, and ROC curves indicated that hub genes exhibited promising potential in the diagnostic prediction of GC, while GSEA further supported their key roles in the pathogenesis of DNA damage. Subsequently, we set out to investigate upstream TFs of the hub genes. Through the analysis of 9 TF databases, we found that HNF4A was consistently identified as a TF of the hub genes across all these databases. It plays a crucial role in disease pathogenesis through multiple mechanisms and has been primarily studied in the context of regulating hepatic metabolic pathways24,25. Numerous studies have demonstrated a strong association between HNF4A and GC. Cao et al. demonstrated that HNF4A plays a key role in suppressing GC by regulating the MIR194-2HG/miR-194/192/BTF3L4 axis. HNF4A directly activates the transcription of the miRNA host gene lncRNA MIR194-2HG and its derived tumor-suppressive miRNAs (miR-194 and miR-192)26. Zhou et al. reported in their study that HNF4A drives gastric cancer progression via the HNF4A-BAP31-VDAC1 axis: HNF4A upregulates BAP31 by binding its promoter, and BAP31 then promotes cancer cell proliferation and inhibits ferroptosis through VDAC1 polyubiquitination and degradation; BAP31 knockdown can reverse these effects and boost cell sensitivity to 5-FU and erastin, making the axis a promising therapeutic target27. Zhang et al. further demonstrated that HNF4A is key to gastric cancer chemosensitivity: chidamide inhibits the HDAC3/HNF4A/TYMS axis by enhancing HNF4A acetylation and reducing its phosphorylation, thereby downregulating TYMS and boosting gastric cancer cells’ sensitivity to 5-FU chemotherapy28. In our study, multiple databases consistently identify HNF4A as a transcription factor of hub genes, underscoring its reliability. This not only provides concrete molecular interaction clues for deciphering gene regulatory networks, clarifying previously obscure regulatory relationships, but also establishes a solid foundation for subsequent in vitro and in vivo validation experiments and for elucidating the dynamic regulatory mechanisms of these networks.
Notably, in machine learning analysis, we found that CLSPN consistently ranked first in importance among the five methods. CLSPN was initially extracted and isolated from the Xenopus egg extracts29, and it is crucial for cell cycle checkpoints and supporting DNA damage repair30. In-depth investigation of the role of CLSPN in tumors is crucial, as the CLSPN gene is closely associated with tumor development31. Chen et al.32 employed multi-omics analysis across 33 cancer types, revealing its upregulation in most tumors and strong associations with poor prognosis, immunosuppressive microenvironment, microsatellite instability, DNA repair, tumor mutational burden, and stemness scores. Functional assays further demonstrated that CLSPN promotes cell proliferation and cell cycle progression in lung adenocarcinoma, while its knockdown significantly inhibits tumor growth. Benevolo et al.33 investigated the role of CLSPN in high-grade cervical lesions associated with human papillomavirus (HPV), finding that CLSPN expression significantly correlates with both HPV infection and the severity of cervical lesions. In normal cervical tissues, CLSPN expression is low. However, as lesions progress, CLSPN levels rise, showing high expression in advanced lesions and cancerous tissues. These findings suggest that CLSPN can serve as an auxiliary marker for diagnosing HPV-related cervical lesions. Xie et al.30 demonstrated that the expression levels of CLSPN are often elevated in melanoma patients, and that these higher levels are associated with poorer prognoses. The researchers uncovered that CLSPN, by activating the IFI44L/JAK/STAT1 signaling pathway, promotes both the growth and progression of melanoma. Moreover, the study suggests that CLSPN is linked to an immunosuppressive tumor microenvironment, specifically through the polarization of tumor-associated macrophages (TAMs) towards the M2-type, which is known for its immunosuppressive characteristics. These findings position CLSPN as a potential therapeutic target, with the implication that targeting CLSPN could enhance the efficacy of immunotherapy and improve the prognosis for melanoma patients. Including but not limited to the aforementioned studies, CLSPN has been shown to exert strong effects in various types of tumors. However, to the best of our knowledge, limited studies have explored the relationship between CLSPN and GC, and the evidence regarding their association remains insufficient. Therefore, investigating the role of CLSPN in the occurrence and development of GC, as well as its underlying molecular mechanisms, holds significant scientific and clinical value.
Previous studies have reported that CLSPN can regulate DNA damage, or closely related processes such as cell cycle and DNA replication, through multiple mechanisms34,35,36. Based on this, we utilized single-cell data and grouped by taking the median of CLSPN expression levels as the cutoff, then performed GO and KEGG analyses. The results showed that differentially expressed genes were also significantly enriched in the cell cycle and DNA replication, which was consistent with previous findings. Subsequently, cell-cell communication analysis revealed that CLSPN+ cells engage in significantly more interactions, exhibiting particularly tight synergistic relationships with fibroblasts. Given prior evidence that CLSPN drives tumor progression via the Wnt/β-catenin signaling, we focused on this pathway and found that both CLSPN + and CLSPN- cells operate primarily as receivers, and CLSPN+ cells displayed higher incoming signal intensities. These data indicate that CLSPN augments the capacity of tumor cells to capture Wnt ligands. Pseudotime analysis, initiated from the CLSPN-low state, showed that tumor cells progressively transition from low to high CLSPN expression along three distinct trajectories, revealing heterogeneous regulatory routes of CLSPN activation and indicating that CLSPN upregulation is a dynamic driver of cell fate switching.
Results of immune infiltration and drug sensitivity analyses suggest that CLSPN possesses both immunoregulatory and clinical therapeutic guiding value in GC. Analyzed by CIBERSORT, CIBERSORT-ABS, and quanTIseq, CLSPN expression correlated significantly with enrichment scores of macrophages and NK/T cells, suggesting it may remodel the tumor microenvironment by modulating these key immune populations. Meanwhile, drug sensitivity prediction showed that high CLSPN expression was significantly negatively correlated with the IC50 values of multiple drugs, indicating that patients with high CLSPN expression may be more sensitive to these drugs. This finding provides a new direction for enhancing chemotherapy efficacy and optimizing individualized treatment of GC by targeting CLSPN.
Importantly, we anticipate that CLSPN could serve as a valuable immunohistochemical antibody for pathologists. Immunohistochemical results showed that CLSPN expression in GC tissues was significantly higher than that in normal gastric tissues; furthermore, compared with GC patients with low CLSPN expression, high CLSPN expression was significantly associated with advanced age, larger maximum tumor diameter, higher tumor pT stage, lymph node metastasis, and poor prognosis. These results are consistent with previous studies18,37,38,39, indicating that CLSPN is a potential biomarker for GC; however, further in-depth research is still required. As a classic cell proliferation marker in clinical pathological diagnosis, the Ki-67 protein (also known as MKI67) is widely used in tumor diagnosis. A study by Stamatiou et al.40 in 2024 revealed that Ki-67 is necessary during DNA replication for fork protection and genome stability, confirming its function in maintaining genome stability. The significant correlation (r = 0.72) between CLSPN and MKI67 in gastric cancer tissues, as indicated by the GEPIA2 database, provides clues for their functional linkage (Supplementary Fig. 9). On one hand, the co-expression of the two may serve as a molecular signature of high proliferative activity in gastric cancer cells, reflecting the regulatory synergy of tumor cells. On the other hand, it is speculated that CLSPN and Ki-67 may have direct or indirect interactions, and the regulatory network formed by them jointly safeguards genome integrity. This association also holds potential clinical translational value. As a routine pathological detection indicator, if Ki-67 can be combined with the expression level of CLSPN, it may more accurately evaluate the proliferative activity and genome instability of tumor cells, providing a novel combined biomarker for prognostic stratification and therapeutic target selection of tumors such as gastric cancer. Meanwhile, interventions targeting the synergistic pathway of Ki-67 and CLSPN are expected to offer new ideas for therapeutic strategies that target the DNA replication process of tumor cells.
Machine learning analysis also indicated that SALL4 is the second most important gene in terms of feature importance ranking. SALL4, part of the SALL family, is a regulator of embryonic stem cells and is vital for cell renewal and proliferation; its reactivation in cancer cells drives tumor development and imparts malignant traits41. Previous studies on SALL4 have primarily focused on germ cell tumors42,43,44, however, an increasing number of recent studies indicate that SALL4 is closely associated with various other malignancies, including gastric cancer45,46,47. Han et al. reported that serum SALL4 levels are significantly associated with tumor recurrence and survival outcomes in patients with hepatocellular carcinoma48. Building on Han’s findings, a prospective investigation could be conducted to examine perioperative changes in serum SALL4 levels across different stages of gastric cancer, assessing its potential as an early predictor of recurrence, while also exploring differences in SALL4 expression among various gastric cancer subtypes. Such a study could also synchronously detect SALL4 alongside traditional gastric cancer biomarkers like CEA and CA19-9, to verify whether SALL4 can complement existing indicators and improve the accuracy of prognostic stratification. These insights would further clarify the tissue-specific role of SALL4 in digestive system malignancies and offer new tools for optimizing gastric cancer patient follow-up and risk assessment.
Conclusion
By integrating a DNA damage-associated gene set with bulk RNA-seq data derived from TCGA-STAD and applying machine learning algorithms, we identified two hub genes including CLSPN and SALL4 that shed new light on GC pathogenesis. Notably, our findings further underscore the key effector role of CLSPN in GC progression and uncover striking heterogeneity across distinct cell populations. Beyond advancing our knowledge of GC’s biological mechanisms, these genes also identify promising therapeutic targets for subsequent intervention research.
Data availability
The datasets generated and/or analysed during the current study are available in the TCGA database (https://portal.gdc.cancer.gov, TCGA-STAD expression and clinical data), the GEO database (https://www.ncbi.nlm.nih.gov/geo/, GSE84433, GSE15459, GSE264203, and GSM7990473, GSM7990474, GSM7990475 from GSE251950), the GEPIA2 platform (http://gepia2.cancer-pku.cn/#index), and the TIMER3.0 website (https://compbio.cn/timer3/).
Abbreviations
- GC:
-
Gastric Cancer
- TCGA:
-
The cancer genome atlas
- STAD:
-
Stomach adenocarcinoma
- GEO:
-
Gene Expression Omnibus
- WGCNA:
-
Weighted gene co-expression network analysis
- DEGs:
-
Differentially expressed genes
- scRNA-seq:
-
Single-cell RNA sequencing
- ST:
-
Spatial transcriptomics
- TF:
-
Transcription factor
- KM:
-
Kaplan-Meier
- HR:
-
Hazard Ratio
- CI:
-
Confidence Interval
- AUC:
-
Area Under Curve
- ROC:
-
Receiver Operating Characteristic
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- GSEA:
-
Gene Set Enrichment Analysis
- IC50:
-
Half-maximal inhibitory concentration
- IHC:
-
Immunohistochemistry
- IRS:
-
Immunoreactive Score
- SI:
-
Staining intensity
- PP:
-
Proportion of positive cells
- OS:
-
Overall survival
- DDR:
-
DNA damage response
- DSBs:
-
Double-Strand Breaks
- MAD:
-
Median absolute deviation
- MM:
-
Module membership
- GS:
-
Gene significance
- ANOVA:
-
Analysis of variance
References
Sundar, R. et al. Gastric cancer. Lancet 405(10494), 2087–2102 (2025).
Qiu, H. et al. Cancer incidence, mortality, and burden in china: a time-trend analysis and comparison with the united States and united Kingdom based on the global epidemiological data released in 2020. Cancer Commun. (Lond). 41(10), 1037–1048 (2021).
Guan, W. L. et al. Gastric cancer treatment: recent progress and future perspectives. J. Hematol. Oncol. 16(1), 57 (2023).
Körfer, J. et al. Molecular targets for gastric cancer treatment and future perspectives from a clinical and translational point of view. Cancers (Basel). 13(20), 5216 (2021).
Tubbs, A. et al. Endogenous DNA damage as a source of genomic instability in cancer. Cell 168(4), 644–656 (2017).
Huang, R. et al. DNA damage repair: historical perspectives, mechanistic pathways and clinical translation for targeted cancer therapy. Signal. Transduct. Target. Ther. 6(1), 254 (2021).
Federica, G. et al. Targeting the DNA damage response in cancer. MedComm 5(11), e788 (2020). (2024).
Reisländer, T. et al. DNA damage and cancer immunotherapy: A STING in the Tale. Mol. Cell. 80(1), 21–28 (2020).
Harding, S. M. et al. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548(7668), 466–470 (2017).
Jia, R. et al. Identification of key genes unique to the luminal a and basal-like breast cancer subtypes via bioinformatic analysis. World J. Surg. Oncol. 18(1), 268 (2020).
Stelzer, G. et al. The genecards suite: from gene data mining to disease genome sequence analyses. Curr. Protoc. Bioinf. 54, 1301–13033 (2016).
Kanehisa, M. et al. KEGG: biological systems database as a model of the real world. Nucleic Acids Res. 53(D1), D672–D677 (2025).
Kanehisa, M. et al. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28(1), 27–30 (2000).
Li, T. et al. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 48(W1), W509–W514 (2020).
Li, C. et al. GEPIA2021: integrating multiple deconvolution-based analysis into GEPIA. Nucleic Acids Res. 49(W1), W242–W246 (2021).
Wang, J. T. F. T. F. An R-Based integrative tool for decoding human transcription Factor-Target interactions. Biomolecules 14(7), 749 (2024).
Yang, S. et al. Single-Cell and Spatial transcriptome profiling identifies the transcription factor BHLHE40 as a driver of EMT in metastatic colorectal cancer. Cancer Res. 84(13), 2202–2217 (2024).
Shi, Y. et al. How CLSPN could demystify its prognostic value and potential molecular mechanism for hepatocellular carcinoma: A crosstalk study. Comput. Biol. Med. 172, 108260 (2024).
Karimaian, A. et al. The crosstalk between Wnt/β-catenin signaling pathway with DNA damage response and oxidative stress: implications in cancer therapy. DNA Repair. (Amst). 51, 14–19 (2017).
Jun, S. et al. LIG4 mediates Wnt signalling-induced radioresistance. Nat. Commun. 7, 10994 (2016).
Moreno-Londoño, A. P. et al. Canonical Wnt pathway is involved in chemoresistance and cell cycle arrest induction in colon cancer cell line spheroids. Int. J. Mol. Sci. 24(6), 5252 (2023).
Wang, L. et al. Negative regulator of E2F transcription factors links cell cycle checkpoint and DNA damage repair. Proc. Natl. Acad. Sci. U. S. A. 115(16), E3837–E3845 (2018).
Albrecht, P. et al. Gastric cancer: from biomarkers to functional precision medicine. Trends Mol. Med. S1471-4914(25), 00118–00112 (2025).
Wang, Y. et al. TIFAB modulates metabolic pathways in KMT2A::MLLT3-induced AML through HNF4A. Blood Adv. 9(4), 844–855 (2025).
Yang, Y. H. et al. HNF4A mitigates sepsis-associated lung injury by upregulating NCOR2/GR/STAB1 axis and promoting macrophage polarization towards M2 phenotype. Cell. Death Dis. 16(1), 120 (2025).
Cao, H. et al. MIR194-2HG, a MiRNA host gene activated by HNF4A, inhibits gastric cancer by regulating MicroRNA biogenesis. Biol. Direct. 19(1), 95 (2024).
Zhou, Q. et al. HNF4A-BAP31-VDAC1 axis synchronously regulates cell proliferation and ferroptosis in gastric cancer. Cell. Death Dis. 14(6), 356 (2023).
Zhang, X. et al. Chidamide enhances the sensitivity of gastric cancer to 5-fluorouracil chemotherapy by suppressing the HDAC3/HNF4A/TYMS axis. Cell. Death Dis. Online (2025).
Kumagai, A. et al. Claspin, a novel protein required for the activation of Chk1 during a DNA replication checkpoint response in xenopus egg extracts. Mol. Cell. 6(4), 839–849 (2000).
Xie, Y. et al. Potential of CLSPN as a therapeutic target in melanoma: a key player in melanoma progression and tumor microenvironment. J. Transl Med. 23(1), 470 (2025).
Azenha, D. et al. Claspin functions in cell homeostasis-A link to cancer? DNA Repair. (Amst). 59, 27–33 (2017).
Chen, Y. et al. A multi-omics analysis reveals CLSPN is associated with prognosis, immune microenvironment and drug resistance in cancers. Biol. Proced. Online. 25(1), 16 (2023).
Benevolo, M. et al. Claspin as a biomarker of human papillomavirus-related high grade lesions of uterine cervix. J. Transl Med. 10, 132 (2012).
Bennett, L. N. et al. Regulation of Claspin degradation by the ubiquitin-proteosome pathway during the cell cycle and in response to ATR-dependent checkpoint activation. FEBS Lett. 580(17), 4176–4181 (2006).
Gao, D. et al. Cdh1 regulates cell cycle through modulating the claspin/Chk1 and the Rb/E2F1 pathways. Mol. Biol. Cell. 20(14), 3305–3316 (2009).
Smits, V. A. J. et al. Claspin-checkpoint adaptor and DNA replication factor. FEBS J. 286(3), 441–455 (2019).
Jia, Y. et al. CLSPN is a potential biomarker associated with poor prognosis in low-grade gliomas based on a multi-database analysis. Curr. Res. Transl Med. 70(4), 103345 (2022).
Hou, Z. et al. CLSPN actives Wnt/β-catenin signaling to facilitate Glycolysis and cell proliferation in oral squamous cell carcinoma. Exp. Cell. Res. 435(2), 113935 (2024).
Kobayashi, G. et al. Clinicopathological significance of Claspin overexpression and its association with spheroid formation in gastric cancer. Hum. Pathol. 84, 8–17 (2019).
Stamatiou, K. et al. Ki-67 is necessary during DNA replication for fork protection and genome stability. Genome Biol. 25(1), 105 (2024).
Moein, S. et al. SALL4: an intriguing therapeutic target in cancer treatment. Cells 11(16), 2601 (2022).
Cao, D. et al. SALL4 is a novel sensitive and specific marker of ovarian primitive germ cell tumors and is particularly useful in distinguishing yolk sac tumor from clear cell carcinoma. Am. J. Surg. Pathol. 33(6), 894–904 (2009).
Cao, D. et al. SALL4 is a novel sensitive and specific marker for metastatic germ cell tumors, with particular utility in detection of metastatic yolk sac tumors. Cancer 115(12), 2640–2651 (2009).
Trecourt, A. et al. SALL4 as a useful marker for the distinction of various gestational trophoblastic disease subtypes: choriocarcinoma from other trophoblastic lesions and early complete hydatidiform mole from partial mole and nonmolar villi. Am. J. Surg. Pathol. 49(5), 417–428 (2025).
Huang, J. et al. SALL4 promotes cancer stem-like cell phenotype and radioresistance in oral squamous cell carcinomas via methyltransferase-like 3-mediated m6A modification. Cell. Death Dis. 15(2), 139 (2024).
Lospinoso Severini, L. et al. SALL4 is a CRL3 substrate that drives Sonic Hedgehog-dependent Medulloblastoma. Cell. Death Differ. 31(2), 170–187 (2024).
Abouelnazar, F. A. et al. SALL4 promotes angiogenesis in gastric cancer by regulating VEGF expression and targeting SALL4/VEGF pathway inhibits cancer progression. Cancer Cell. Int. 23(1), 149 (2023).
Han, S. X. et al. Serum SALL4 is a novel prognosis biomarker with tumor recurrence and poor survival of patients in hepatocellular carcinoma. J. Immunol. Res. 2014, 262385 (2014).
Acknowledgements
We acknowledge all participants who have provided assistance for this study.
Funding
This work was supported by Weifang Municipal Health Commission Research Project (WFWSJK-2025-322).
Author information
Authors and Affiliations
Contributions
QM: Conceptualization, Formal analysis, Writing – original draft, Writing – review & editing, Validation, Final approval. LL: Conceptualization, Writing – original draft, Writing – review & editing, Validation, Final approval. XJY: Data curation, Investigation, Writing – review & editing, Validation, Final approval. NYS: Data curation, Investigation, Writing – review & editing, Final approval. LML: Writing – review & editing, Validation, Final approval. JB: Writing – review & editing, Validation, Final approval.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
The research protocol was endorsed by the Ethics Committee ofSunshine Union Hospital (Approval No.: 2025032123) and adheredto the principles of the Helsinki Declaration.
Informed consent
Informed consent was obtained from all subjects and/or their legal guardian(s).
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ma, Q., Yang, X., Sun, N. et al. Integrating multi-omics analysis identifies DNA damage-related gene CLSPN as a biomarker in gastric cancer. Sci Rep 16, 7789 (2026). https://doi.org/10.1038/s41598-026-39387-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-026-39387-6
