
Abstract
Clonal hematopoiesis (CH) is an age-related expansion of white blood cell (WBC) progenitors linked to risk of hematological malignancy. Patients with cancer have increased CH prevalence compared to healthy populations, but the characteristics and relevance of CH in advanced urological cancers are unknown. We interrogated CH and circulating tumor DNA (ctDNA) in 299 patients with metastatic urothelial or renal cell carcinoma using error-corrected targeted sequencing of matched WBC DNA and plasma cell-free DNA (cfDNA). 73% of patients carried CH variants at ≥0.25% allele frequency, with 13% exhibiting large CH populations marked by variants ≥10%. CH presence, clone size, and genotype did not impact patient survival. However, CH variants frequently affected solid cancer driver genes and were not individually discriminable from ctDNA variants based on cfDNA features including fragment length. In contrast, matched WBC DNA sequencing to ≥25% of cfDNA depth sufficiently resolved CH from ctDNA variants. Serial profiling revealed ctDNA and CH temporal dynamics including treatment-related expansion of PPM1D-mutated CH clones following platinum chemotherapy. Our data reveal the molecular landscape of CH in urological cancers and suggest that CH interferes in clinical ctDNA genotyping. We urge test providers to comprehensively filter CH from ctDNA results using matched WBC sequencing and propose a cost-effective framework for its integration into existing plasma-only assays.
Similar content being viewed by others

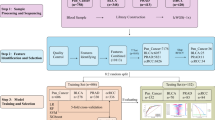

Introduction
Genomic mosaicism is a hallmark of aging cells. As tissues age, cells can acquire mutations that are propagated through subsequent divisions, forming distinct somatic clones1,2,3. Although most mosaicism is associated with propagation of functionally inert mutations through neutral genetic drift, a subset of mutations can confer a fitness advantage and enable rapid expansion4. Clonal hematopoiesis (CH) refers to expanded somatic mosaicism in the hematopoietic system without overt hematological malignancy, and is most commonly associated with mutations in epigenetic regulators DNMT3A and TET2. Over the age of 60, at least one (albeit typically small) clonally-expanded hematopoietic population is nearly universal in otherwise healthy individuals5. CH is an emerging risk factor for aging-associated pathologies including cardiovascular and neurodegenerative diseases6,7.
In patients with cancer, the prevalence of CH is higher than age-matched cancer-free individuals, partly due to cancer treatment exposures8,9,10,11. In patients receiving anticancer drugs, CH has been associated with an elevated risk of treatment-induced adverse events including myeloid neoplasms12, cytokine release syndrome13 and cardiovascular toxicity14, and in rare scenarios can progress to secondary hematologic malignancy (e.g. treatment-related acute myeloid leukemia [t-AML]) under the selective pressure of chemo- or radio-therapy15,16. However, prior studies investigating CH in solid tumors focused on heterogeneous pan-cancer and pan-stage populations using low-resolution techniques not designed to characterize CH8,11,17. The diversity of treatments applied across these cancer types may differentially modify the CH landscape, while correlative insights derived from cohorts enriched for primary malignancies may not generalize to metastatic disease. Consequently, the characteristics and clinical relevance of CH in specific malignancies with distinct age distributions and molecular etiologies remain unclear. Previous work in various mostly localized solid tumor types are conflicting regarding the precise features and prognostic relevance of CH14,18,19,20,21,22.
Plasma circulating tumor DNA (ctDNA) profiling is increasingly incorporated into routine clinical management for advanced malignancies. However, most commercial ctDNA genotyping platforms do not sequence synchronous white blood cell (WBC) DNA, meaning that CH is liable to contaminate tumor variant reporting since the majority of cell-free DNA (cfDNA) originates from hematopoietic populations23,24. Resolving CH mutations from true ctDNA variants poses a potential challenge for precision oncology. While more than half of CH mutations fall in DNMT3A and TET2 and could be automatically excluded during ctDNA mutation analysis based on gene identity, CH mutations in oncology genes with predictive and/or prognostic utility including BRCA1/2, TP53, ATM, and CHEK2 are ostensibly less easily resolved and may confound tumor genotyping. There is concern whether liquid biopsy platforms that sequence cfDNA without patient-matched WBC DNA are able to distinguish CH mutations from tumor-derived variants, although the extent of this potential problem has not been characterized8,25,26.
Here, we investigated synchronous CH and ctDNA features in two aggressive immune active urologic malignancies with distinct treatment landscapes: metastatic renal cell carcinoma (mRCC) and metastatic urothelial carcinoma (mUC). In both cancers, ctDNA profiling is emerging as a key tool for translational research and patient management, particularly for minimal residual disease detection in the perioperative setting27 but also for identification of genomic alterations associated with response to approved systemic therapies (e.g. FGFR3 alterations and FGFR inhibitors in mUC)27,28. Despite this emerging role for plasma testing, the frequency and characteristics of CH in mRCC and mUC are understudied. We demonstrate that CH with variant allele frequency (VAF) ≥ 0.25% is present in the majority of patients with mRCC or mUC, and the precise spectrum of affected genes depends on prior treatment exposures and is subject to evolution. In these metastatic tumor types with short life expectancy, CH had no impact on survival, although CH-related mutations in pan-cancer relevant genes showed high potential to confound ctDNA-based cancer genotyping. Our data provide a new blueprint for assessing CH features and suggest that matched WBC DNA sequencing is a practical and comprehensive approach to accurately resolve CH from ctDNA.
Results
Cohort composition and sequencing strategy
We analyzed blood from 299 patients with metastatic urologic cancer from a research biobank: 184 mRCC and 115 mUC (Fig. 1a). All patients provided a blood sample at enrollment (termed as “baseline” sample), predominantly prior to commencing a line of systemic therapy for metastatic disease. Follow-up sample(s) were available from a subset of patients (Supplementary Fig. 1).

a Study design. b Bar chart showing CH prevalence in mUC and mRCC at different detection thresholds. Patient numbers are indicated above bars. c Comparison of matched white blood cell (WBC) DNA and cell-free DNA (cfDNA) variant allele frequencies (VAFs). Inset plot shows VAF < 2%. d [Left] Bar chart showing the percentage of CH+ patients with mutations in the 15 most commonly mutated genes. Differences in per gene frequency between cancer types is indicated with unadjusted and Benjamini-Hochberg corrected p-values (denoted by asterisks). Patient numbers are provided to the left of each bar. [Right] Dot plot showing the WBC DNA VAFs for each gene in log10 scale. e Bar chart showing the distribution of the number of CH mutations detected in each patient (patient numbers are provided at the top of the chart). The superimposed dotplot shows the WBC DNA VAFs in log10 scale. f Box plot comparing the age at baseline blood draw for patients with CH detected at different WBC VAF thresholds. g Dot plot showing the CH prevalence across different age groups. The human figure in Fig. 1a was obtained and modified from Wikimedia Commons (author: Cancer Research UK) available under the Creative Commons Attribution-Share Alike 4.0 International license: https://commons.wikimedia.org/wiki/File:Diagram_showing_where_in_the_body_cancer_tends_to_spread_to_CRUK_381.svg.
Among patients with mRCC, 80% (148/184) had baseline blood drawn before systemic treatment for metastatic disease (Table 1). For patients whose first blood sample was collected during/after ≥1 line of therapy (n = 36), prior treatment included immunotherapy (IO) and/or receptor tyrosine kinase inhibitors (TKIs). Baseline characteristics of the patient population were reflective of real world cohorts. 71.3% of patients with mUC (82/115) provided blood prior to first- or second-line therapy for metastatic disease (mainly constituting platinum chemotherapy and IO, respectively) (Table 1). Prior to first blood collection, 50% (58/115) of mUC patients had received systemic platinum chemotherapy (for metastatic disease or as (neo)adjuvant therapy), and 21% (24/115) received single-agent IO. Median age at baseline blood draw in the mUC cohort was higher than that of mRCC (71 vs 65, Mann-Whitney U [MWU] P = 0.00047).
We interrogated both plasma cell-free DNA (cfDNA) and patient- and time-matched white blood cell (WBC) DNA to accurately discriminate CH from germline single nucleotide polymorphisms (SNPs) and tumor-derived variants (Fig. 1a). Deep targeted cfDNA and WBC DNA sequencing was performed using a custom panel where CH variants were required to be present in both analytes with a WBC VAF ≥ 0.25% (Methods). Median deduplicated read-family depth of 2290× in cfDNA and 2060× in WBC DNA was achieved (Supplementary Data 2). Aligning with prior landmark studies28,29, WBC VAF is used to quantify CH clonal prevalence unless otherwise stated.
CH is pervasive in mRCC and mUC
Incorporating all patients with mRCC or mUC, we detected 604 CH mutations across 45 genes in 73% (219/299) of baseline samples (Supplementary Data 3). 71% (131/184) and 76% (88/115) of mRCC and mUC were CH positive (CH+), respectively at 0.25% VAF detection threshold (Fig. 1b), without difference in prevalence by cancer type (Fisher’s exact P = 0.75) or biological sex (Supplementary Fig. 2). Overall population prevalence was 34% (103/299) and 13% (38/299) for CH+ clones above 2% and 10% VAF, respectively (Fig. 1b; Supplementary Fig. 3a)—accepted thresholds for clonal hematopoiesis of indeterminate potential (CHIP) and for larger CH clones8,30. 14 patients had exceptionally high CH VAFs (above 30%), potentially interfering with germline mutation calling. For example, one patient with mRCC (and no overt hematological malignancy) exhibited four CH mutations with VAFs between 38% and 41%. Median plasma cfDNA and WBC VAF of CH mutations was 0.88% and 0.90%, respectively (Supplementary Data 3) (cfDNA interquartile range [IQR]: 0.45–2.07%; WBC IQR: 0.47–1.94%), and expectedly, cfDNA and WBC VAF were closely correlated (Spearman’s correlation = 0.82, P = 4.86 × 10−148) (Fig. 1c).
Frequently mutated genes were consistent with the known landscape of CH in unselected populations28,31,32, and included DTA genes (DNMT3A, TET2, ASXL1), splicing factors (e.g. U2AF1, SF3B1, SRSF2), and genes involved in DNA damage response (DDR; e.g. ATM, CHEK2, TP53) (Fig. 1d). PPM1D, ATM and CHEK2 were more commonly mutated in mUC than mRCC among CH+ patients (corrected Z-test P < 0.05). Truncating CH mutations in PPM1D are known to be positively selected during platinum chemotherapy (received by 50% (58/115) of patients with mUC in our study) and are enriched in patients with therapy-related myeloid neoplasms16,17,33,34. Accordingly, in our study prior platinum chemotherapy exposure was associated with PPM1D CH in patients with mUC (OR = 3.41, P = 0.041) (Supplementary Data 5). CH mutations in ATM, ASXL1, and CHEK2 have also been observed in post-platinum settings17,35. At 0.25% VAF threshold, PPM1D mutations were mostly exon 6 truncation events associated with PPM1D gain-of-function and downstream TP53 suppression36,37 (Supplementary Fig. 4). There were no per-gene differences in median mutation VAF between mRCC and mUC (adjusted for multiple testing). 30% (67/219) of CH+ patients carried a single CH mutation with VAF ≥ 0.25% (Fig. 1e); conversely, 70% of patients harbored multiple CH mutations (range: 2–17) at the same detection threshold, with no apparent relationship between mutation multiplicity and VAF (ANOVA P = 0.436). The highest number of CH mutations detected in a single patient (88 years old, mUC, no prior systemic therapy) was 17 across 9 genes including multiple ASXL1, TET2, and TP53 mutations all with a VAF below 3%.
In addition to SNVs and indels, larger structural variation such as chromosomal aneuploidy may contribute to CH and increase risk of hematological malignancy38,39. Using our targeted sequencing data, we identified one patient with a putative germline duplication involving the JAK2 locus but did not identify any examples of CH-derived CNVs (Supplementary Fig. 5). Therefore, we re-analyzed WBC DNA from the entire mUC cohort (n = 115) plus 17 select patients with mRCC exhibiting CH mutations above 10% VAF (representing scenarios where >20% of WBCs originate from a single precursor population) using a genome-wide probe grid designed to identify large CNVs in liquid biopsy40. Leveraging this data, we identified a patient with putative trisomy 8 mosaicism (Warkany Syndrome 2) and one presumptively CH-derived ~7.6 Mb deletion of chromosome 5 (Supplementary Fig. 5).
Expectedly, CH+ (VAF ≥ 0.25%) patients were older than those without detected CH mutations (70 vs 60, MWU P = 5.5 × 10−12) (Fig. 1f)31,41. Risk of carrying a CH mutation with VAF > 10% increased by 1.6-fold (P = 0.0073) per additional decade across both cohorts (Fig. 1g). Previous work in a non-cancer population indicated that CH mutations in RNA spliceosome genes are acquired late in life but undergo rapid exponential growth, in contrast to DNMT3A CH mutations which can be acquired early but expand slowly42. Consistent with this study, we found that carriers of CH mutations in spliceosome-associated genes were older than those carrying mutations in DNMT3A without concurrent CH mutations in spliceosome genes (74 vs 70, MWU P = 0.0065). Overall, age and number of CH variants were positively correlated (Spearman’s ρ = 0.36, P = 4.29 × 10−8).
CH confounds accurate cfDNA-only tumor testing
To explore differences or overlap in the somatic landscapes of ctDNA and CH in the two urologic malignancies, we next analyzed mutations exclusive to plasma cfDNA and not independently detected in the WBC compartment, representing tumor-originating mutations. 58% of patients at baseline had cfDNA-only mutations indicative of ctDNA (mRCC: n = 89/184, 48%; mUC: n = 85/115, 75%) (Supplementary Fig. 6a). Median ctDNA mutation VAF in patients with mRCC was 1.56% and significantly higher at 4.49% in mUC (MWU P = 4.31 × 10−12) (Supplementary Fig. 6a)—consistent with the established ctDNA abundance of both cancer types43,44. For both, median ctDNA VAF exceeded the median CH plasma VAF (mUC: 4.49% vs 0.74%, MWU P = 1.55 × 10−39, mRCC: 1.57% vs 1.01%, MWU P = 8.78 × 10−8) indicating a higher burden of ctDNA versus CH populations represented in blood. The distribution of ctDNA-mutated genes in our cohort aligned with the established (and distinct) mRCC and mUC mutational landscapes: the most commonly mutated genes in ctDNA+ patients with mRCC were VHL (37%), ARID1A (27%), PBRM1 (21%), and SETD2 (19%), while ctDNA+ patients with mUC most commonly carried mutations in TP53 (47%), TERT promoter (42%), ARID1A (28%), and KMT2D (28%) (Fig. 2b, Supplementary Fig. 6)45,46,47. Overall, we did not find an association between CH presence and ctDNA presence (Supplementary Fig. 7a), consistent with their distinct somatic origins. However, we observed that the correlation between cfDNA-derived VAFs and WBC-derived VAFs for CH mutations was less strongly correlated in patients carrying ctDNA mutations with VAF ≥ 10% compared to those carrying ctDNA mutations with VAF < 10% (Supplementary Fig. 7b) In samples with large ctDNA populations, proportionally there are fewer reads originating from a CH compartment, resulting in a skewed VAF relative to the WBC population. Similarly there was a positive correlation between the highest ctDNA VAF and the mean cfDNA-WBC VAF deviation among CH mutations per sample (Supplementary Fig. 7c).

a Bar chart [top] showing the percentage of all somatic mutations detected in cfDNA that are CH-derived, on a per gene basis. Bar chart [bottom] indicates the total number mutations detected in each gene across all patients. Genes with notable relevance in solid tumors are indicated in bold typeface. b Oncoprint showing the breakdown of somatic mutations among patients with CH and/or ctDNA mutations. For each gene, the upper tracks in lighter gray background indicate CH mutations and the darker gray track indicates ctDNA mutations. The top three bar charts indicate the highest per patient variant allele frequency (VAF) for a ctDNA mutation, the highest VAF for a CH mutation, and the total mutation count per patient, respectively. The bar chart to the right of the oncoprint shows the number of mutations detected per gene with the mutation VAFs shown in the superimposed dot plot.
CH is thought to confound commercial tumor cfDNA biomarker tests that do not profile synchronous WBC DNA, potentially leading to inaccurate clinical test results48,49. However the extent of this problem in specific cancers and the likelihood of CH-ctDNA misattribution on a per-gene level is not known. When evaluating genes linked with blood cancers (e.g. MYD88, U2AF1, DNMT3A, TET2), over 90% of the somatic mutations detected in cfDNA had a CH origin (i.e. were also detected in WBC DNA). Conversely, 100% of cfDNA somatic mutations identified in the solid cancer driver genes VHL, FGFR3, RB1 and PTEN had presumed ctDNA origin due to lack of detection in WBC DNA (Fig. 2a)—highlighting these genes as relatively specific markers of plasma ctDNA presence. However, multiple genes with established biological and/or clinical relevance in urologic malignancies harbored CH and ctDNA mutations at a similar prevalence. For example, 91%, 70%, and 27% of all detected cfDNA mutations in CHEK2, ATM, and BRCA2 respectively originated from CH—genes associated with PARP inhibitor candidacy in solid cancers50—as well as 33% of mutations in ERBB2 (n = 5), a candidate biomarker for anti-HER2 antibody-drug conjugate therapy in mUC51. 32% of plasma cfDNA mutations in TP53 were CH. Similar per-gene overlap in CH and ctDNA mutation prevalence persisted when only considering CH mutations with VAF ≥ 1%, above the approximate SNV limit of detection utilized by commercial cfDNA tumor tests (Supplementary Fig. 8a)48,49. 18 patients carried both CH and ctDNA mutations in the same gene, including genes such as TP53, CHEK2, and ATM (Supplementary Fig. 8b, Supplementary Data 3-4)—complicating the distinction between mutations that arise through CH and those of tumor origin when not profiling matched WBC DNA. Although rare, we observed 20 ctDNA-originating mutations in 20 patients across DNMT3A, TET2, and ASXL1 with a cfDNA VAF above 0.50% and without independent WBC detection (WBC read support ≤2 reads) (Supplementary Data 4, Supplementary Fig. 9). Importantly, 22% (25/115) of the mUC cohort and 37% (68/184) of the mRCC cohort was CH+ but had no detected ctDNA mutations (Supplementary Fig. 7a). In these patients, the CH affected genes had a wide distribution of cfDNA VAFs and included solid cancer driver genes such as TP53, CHEK2, and STAG2 (Supplementary Fig. 7d) which, failing WBC comparison, could be misattributed as tumor-derived.
Synchronous WBC sequencing accurately discerns cfDNA variant etiology
Given that CH and tumor mutations are not completely discriminable based on gene identity, we sought to clarify whether other somatic features can delineate CH and ctDNA in the absence of synchronous WBC profiling. We first dichotomized samples into two groups based on whether their maximum ctDNA mutation VAF (as a surrogate for overall ctDNA fraction) was ≥10% or <10%, representing scenarios with high and low tumor burden, respectively. In both cases, we observed substantial overlap in the distribution of ctDNA VAFs and cfDNA-derived CH VAFs, indicating that distinguishing mutation origin based solely on cfDNA VAF is not reliable (Fig. 3a), especially for low ctDNA-shedding cancers like mRCC44,52 or in clinical scenarios where ctDNA abundance is suppressed by effective therapy. A similar degree of overlap remained even after excluding the most commonly CH-mutated genes DNMT3A and TET2 (Supplementary Fig. 8c).

a Box plot showing that the variant allele frequency (VAF) distribution of ctDNA and CH variants exhibits notable overlap in context of both high and low ctDNA levels (indicated by max ctDNA VAFs above and below 10% respectively). Number of patients represented in each box plot is indicated at the top. P values are derived from the Mann-Whitney U test. b cfDNA fragment length distribution for wildtype (WT) reads and variant-carrying reads across genomic positions in the context of CH or ctDNA variants. c The left schematic provides a summary of the computational approach used to differentiate the fragment length distribution of variant versus WT fragments. The median fragment lengths for mutated and wildtype fragments were compared at each genomic position with a somatic mutation using the Mann-Whitney U test. CH mutations [middle scatter plot] and ctDNA mutations [right] with significantly different median fragment lengths compared to WT fragments are highlighted in light blue. Pie charts show the percentage of mutations with significant differences. In (b) and (c) only variants with >15 supporting reads are shown. d Random in silico downsampling of the WBC DNA sequencing data illustrating the sequencing depth needed to detect all CH variants [left] and CH variants with VAF > 1% [right] by different numbers of supporting reads. e [Left] Application of common heuristic filters (aimed at removing CH-derived variants from plasma-only sequencing data) to all detected somatic mutations, showing the number of CH and ctDNA variants that would be excluded after applying each criterion (in a theoretical scenario where matched WBC DNA was not profiled). Pie charts indicate the sensitivity and specificity of each filtering approach. [Right] Fraction of discarded and retained somatic mutations in select genes after applying the filtering criteria.
Building on previous studies suggesting wild-type and tumor-derived cfDNA may be distinguished based on their fragmentomic profiles26,53,54, we next compared the fragment size distribution of somatically mutated and wild-type fragments. All cfDNA fragments followed a bimodal size distribution with characteristic mono- and di-nucleosomal peaks at ~167 bp and ~320 bp, respectively (Fig. 3b)26,54. cfDNA fragments originating from cancer were shorter than those originating from CH (average difference of 10 bp)-driven primarily by an enrichment for subnucleosomal (<167 bp) ctDNA fragments (Kruskal-Wallis P = 0.0003). Conversely, the size distribution of CH-mutated and wildtype cfDNA fragments was not significantly different (Kruskal-Wallis P = 0.218), consistent with a shared hematopoietic origin. In examining individual mutated loci, we found that only 29% (129/442) of ctDNA mutations had a median fragment length (across all mutant-supporting fragments) significantly different from that of reference allele fragments, although this was in contrast to only 4% (16/391) of CH mutations (Fig. 3c).
While WBC DNA sequencing can aid in distinguishing CH from ctDNA, there is no established consensus on the minimum technical standards (e.g. sequencing depth/coverage) needed to accurately differentiate most CH and ctDNA variants. To address this, we performed random in silico downsampling of WBC sample coverage at fixed increments while keeping the depth of cfDNA samples constant, and iteratively reperformed CH mutation calling using the same methodology. Encouragingly, sequencing WBC samples to ~25% the depth of matched cfDNA (i.e. median 2290× cfDNA and 600× WBC) enabled detection of 90% of CH mutations with VAF > 1%, with the remaining 10% liable to be miscalled as cancer-derived ctDNA variants. Although sequencing WBC at a higher depth recovered progressively larger proportions of our ground-truth denominator of all CH variants, we observed diminishing returns for downsampled WBC depth above ~600× (1:4 ratio of WBC/cfDNA depth). Requiring fewer minimum mutant-supporting reads for CH detection boosted recovery of CH variants across the range of surveyed WBC depths. Collectively, this in silico experiment proposes a minimum recommended WBC DNA sequencing depth for cost-effective discrimination of CH from ctDNA variants (versus the more expensive sequencing of both compartments to equivalent depths) while minimally compromising specificity (Fig. 3d).
Finally, we empirically tested the accuracy of several common rules-based heuristics aimed at removing CH interference that are utilized by plasma-only ctDNA genotyping platforms48,49. We tested three strategies and their impact on sensitivity and specificity for ctDNA-variant detection, including i) discarding all mutations in DTA genes, ii) excluding mutations with VAF < 1%, and iii) excluding putatively subclonal mutations (defined as a mutation with VAF less than 25% the sample’s maximum somatic VAF [applied only to samples with >1 mutation]). Leveraging our curated set of 1166 somatic cfDNA mutations (562 ctDNA and 604 CH), we applied these filtering strategies individually and calculated the sensitivity (i.e. proportion of true ctDNA variants recovered) and specificity (i.e. proportion of all recovered variants that are genuinely from ctDNA) of each approach to be between 75–96% (sensitivity) and 56–66% (specificity) (Fig. 3e). However, the filtered set of mutations from each approach included 34–44% of CH variants as false positives. Applying these criteria in series led to a sensitivity/specificity of 62%/30% and a final set of mutations that still contained 100 CH mutations with a median VAF of 2.91% (Fig. 3e). Importantly, this approach erroneously discarded genuine ctDNA mutations in clinically-relevant genes such as FGFR3 (erdafitinib eligibility for mUC55; 3/11 discarded), TP53 (16/63 discarded) and VHL (key gene for clear cell RCC pathogenesis56; 10/34 discarded).
CH does not strongly influence survival in mUC or mRCC
Detection of ctDNA in baseline blood was associated with poor overall survival (OS) in multivariable models adjusting for known cancer-specific clinical prognostic factors for mUC (HR = 2.35 [95% CI: 1.29–4.29], P = 0.0052) and mRCC (HR = 2.33 [95% CI: 3.43–1.58], P = 2 × 10-4) (Fig. 4a, b), consistent with the established prognostic effect of ctDNA in solid cancers43,44,57,58. In the subgroup of patients starting first-line treatment for metastatic disease, ctDNA detection was similarly prognostic for inferior OS in mRCC (HR = 2.96 [95% CI: 1.65–5.33], P = 2.89 × 10−4) but did not reach significance in mUC (HR = 1.7 [95% CI: 0.71–4.06], P = 0.235) (Supplementary Fig. 10a, b). In contrast, CH detection had no impact on OS in either cohort (mUC: HR = 1.1 [95% CI: 0.64-1.87], P = 0.735; mRCC: HR = 0.89 [95% CI: 0.59–1.34], P = 0.579) (Fig. 4c, d), even when dichotomizing patients by presence of larger CH events exceeding 2% VAF (Supplementary Fig. 11a, b), restricting analysis to patients starting first-line treatment for their metastatic disease (Supplementary Fig. 11c–f), or in any subgroup analysis across various gene groupings (Fig. 4e). CH status did not stratify OS among ctDNA+ patients (Supplementary Fig. 10c, d).

a, b Overall survival (OS) for patients with mUC or mRCC, stratified by ctDNA detection. c, d OS stratified by CH detection. e Forest plot from multivariable Cox Proportional Hazards model. f CH prevalence in patients with mRCC stratified by occurrence of immune-related adverse events. P values are derived from Fisher’s Exact Test.
We hypothesized that hematopoietic dysregulation via CH may modulate frequency of immune-related adverse events (irAEs) in patients receiving treatment59. Therefore, we evaluated the prevalence of grade 3–4 irAEs that occurred during systemic therapy administration and its relationship to baseline CH detection among 161 patients with mRCC (Supplementary Data 6). 30% (49/161) experienced any grade 3–4 irAEs. The two most common grade 3–4 irAEs were adrenal insufficiency and colitis, occurring in 7% (12/161) and 9% (14/161) of patients, with two experiencing both. There was no association between CH detection and occurrence of any grade 3–4 irAEs (i.e. as a single binary variable) (Fig. 4f). Interestingly, grade 3–4 adrenal insufficiency was less prevalent in patients with CH ≥ 0.25% or CH ≥ 2% (Fig. 4f); median age did not differ between patients with and without adrenal insufficiency (63.5 vs 64.5, MWU P = 0.65). No association was observed between CH and colitis. No patient developed treatment-related hematologic malignancy.
Evolutionary dynamics of CH during therapy
36% (n = 41) of patients with mUC had serial cfDNA (median collection interval: 17 weeks, IQR: 13-25 weeks), enabling us to track the evolutionary dynamics of CH mutations in DTA, DDR and splicing genes during systemic therapy. Mutations previously undetected in baseline samples due to insufficient read support (which may represent treatment-emergent mutations) were predominantly identified in PPM1D and DNMT3A (six and four, respectively) (Fig. 5a, Supplementary Data 7), although overall there were no per-gene differences in CH detection rate between the baseline timepoint and next subsequent blood collection among patients providing >1 sample (two proportion Z-test). CH mutation VAF was generally stable over time (median absolute VAF change: 0.37%), although CH mutations in TP53, PPM1D, and CHEK2 had a modestly higher absolute VAF difference between timepoints compared to DTA genes (ΔVAF 0.47% vs 0.20%, MWU P = 0.0075), and more commonly increased in VAF over time compared to DTA genes where VAF flux was less directional (Fig. 5b, Supplementary Fig. 12). Overall DDR mutations had significantly higher growth rates than DTA mutations, and platinum chemotherapy as first-line treatment for metastatic disease was also associated with significantly higher growth rates in DDR mutations (Fig. 5c, d). Interestingly, the largest changes in VAF all occurred in patients (n = 3) with PPM1D mutations who received intervening platinum-containing chemotherapy between their serial cfDNA collections (Fig. 5e). Expectedly, ctDNA and CH mutations showed divergent VAF dynamics during systemic treatment, indicating their distinct origin. Interestingly, in patient P123, one PPM1D frameshift mutation increased to 46% VAF while a second PPM1D stopgain mutation remained stable at less than 1% VAF, suggesting that different PPM1D mutations may have varying fitness under systemic therapy.

a Number of mutations detected per gene in blood samples collected at baseline and follow-up during treatment for mUC. b Temporal changes in variant allele frequency (VAF) for mutations detected in DTA genes (DNMT3A, TET2, ASXL1) or DNA damage response (DDR) genes (TP53, PPM1D, CHEK2). c Comparing growth rates in CH mutations in DTA genes versus DDR genes and d between treatment groups within each gene category. e VAF changes for PPM1D mutations across the course of systemic treatment in three patients with mUC. Carbo/Gem Carboplatin/Gemcitabine.
Discussion
CH is critically understudied in advanced solid cancers. We provide the first characterization of synchronous CH and ctDNA in patients with mUC or mRCC. Our results establish the prevalence and precise landscape of genes harboring CH variants in these malignancies, including distinct differences between cancers likely due to divergent prior treatment. Methodologically, our study is unique through independent error-corrected detection of low VAF somatic variants in both WBC DNA and cfDNA compartments, and this approach revealed notable overlap between genes that are mutated in CH and ctDNA. We posit that CH is highly clinically relevant due to the potential for CH-related variants to interfere with emerging liquid biopsy-guided patient management paradigms.
The high frequency of CH in our study (especially above the established 2% definition of CHIP) is consistent with elderly noncancer populations and pan-cancer studies that predominantly surveyed patients with localized malignancy. There were no previous reliable estimates of CH prevalence in mUC and mRCC, although two mRCC ctDNA studies had identified incidental CH in a small number of patients44,60. In a published pan-cancer pan-stage dataset including RCC (n = 183, stage unspecified), less than 25% of RCC patients had CH, but it is difficult to compare directly to the current work since this prior study applied much lower sequencing coverage than our evaluation (419× versus 2290×)8. Another pan-cancer pan-stage CH study including UC and RCC (stage unspecified) demonstrated an association between cytotoxic therapy and CH, but provided no insight into UC- or RCC-specific CH topology17. Ultimately our data suggest that increasingly sensitive clinical liquid biopsy approaches will render all patients CH+ at some level, crucially necessitating a shift from binary CH detection (current conceptual paradigm) to a more nuanced assessment of clone size, diversity, and genotype.
cfDNA-only tumor biomarker testing (i.e. not incorporating matched WBCs) represents the current liquid biopsy paradigm among widely-available companion diagnostic assays. Our data reaffirm that cfDNA-only sequencing harbors significant risk of misattribution of CH as tumor-derived variants, corroborating a prior suggestion that 8% of reported ctDNA mutations in CH-associated genes identified by a commercial ctDNA test had hematopoietic origin61. Similarly, early reports of the genomic landscapes in RCC and other malignancies using cfDNA-only assays were also likely contaminated by CH62,63,64,65. Discerning the etiology of mutations detected in blood is important for all tumor types where liquid biopsy may have clinical utility, especially in tumors where CH and ctDNA mutational landscapes have a high degree of overlap. For example, treatment regimens for lung and gynecological cancers often involve platinum-chemotherapy with known risk of t-AML. Frequent occurrence of TP53 mutations in gynecologic malignancies66 and RNA splicing alterations in lung adenocarcinoma67 underscores the potential added value of WBC sequencing in liquid biopsy workflows beyond urological malignancies. Our data suggest that heuristics for discriminating ctDNA and CH predicated on fragmentomic features, gene identity, and/or mutation VAF do not sufficiently rectify otherwise suboptimal sensitivity/specificity for clinical tumor genotyping. Similar individual molecular features between CH and ctDNA further imply that machine learning models for discriminating the two are unlikely to substantially improve classification—evidenced in the performance of prior multi-feature models being predominantly driven by categorical DTA gene identity and (low) VAF, with negligible additional discriminatory power afforded by other input features (e.g. mutation signatures)68. Reliance on low VAF to distinguish ctDNA and CH risk failure especially in low ctDNA shedding cancers like RCC, and/or in other malignancies during therapy response which is often accompanied by decreased ctDNA abundance.
Ultimately, failure to accurately resolve ctDNA- and CH-derived variants in cfDNA-only assays is liable to interfere with precision oncology workflows in five ways:
-
1.
False-positive CH variants in treatment-predictive genes like BRCA1/2 in prostate and breast cancers50,69 may lead to inappropriate targeted treatment allocation.
-
2.
Imprecise computational strategies for classifying tumor- versus CH-derived variants (in the absence of matched WBC sequencing) may erroneously discard genuine ctDNA mutations relevant for clinical management and precision oncology trial eligibility (e.g. in ERBB2, FGFR3), leading to false negative detection and withholding of potentially effective targeted therapy.
-
3.
False-positive CH mutations without any other detected tumor-derived variants may falsely imply nonzero sample ctDNA fraction, leading to the erroneous interpretation of negative tumor mutation status for all other reported genes—potentially precluding follow-up tissue genotyping and access to genome-aware targeted therapies70.
-
4.
False-positive CH variants may inflate higher-order metrics including tumor mutation burden which is relevant for immunotherapy allocation in solid tumors.
-
5.
In patients with large ctDNA populations and CH clones, the VAF of mutations in either compartment can be skewed due to competition for sequencing reads, resulting in less reliable estimates of overall population size (and consequently, inferences around variant clonality).
CH can also interfere with unpaired tumor tissue genotyping due to inadvertent capture of vasculature (containing WBCs) and/or tumor infiltration of CH-expanded leukocytes71,72, but is less confounding versus liquid biopsy where hematological admixture is guaranteed. Consequently, tumor-tissue informed ctDNA assays for detecting minimal residual disease are unlikely to be affected by CH, especially following appropriate selection of mutations in solid cancer-specific genes and/or clonal mutations (as is typically performed)73.
Our data corroborate prior suggestions that sequencing matched WBCs is a reliable strategy to remove CH interference in cfDNA testing, while additionally proposing a new minimum depth requirement for achieving cost-efficiency. Moreover, parallel WBC sequencing enables improved suppression of sequencing artefacts while simultaneously permitting interrogation of clinically relevant germline alterations (even at a lower depth). Currently, equivocal germline findings from commercial plasma-only ctDNA tests necessitate a subsequent blood draw for follow-up dedicated germline testing—which is ultimately both more resource-intensive and inefficient versus parallel WBC and cfDNA profiling. We anticipate that our findings are generalizable to other cancers with similar age distributions (e.g. advanced prostate cancer) where commercial ctDNA profiling is widely performed and CH interference has been reported14,25.
In contrast with prior studies in noncancer and heterogenous pan-cancer populations, CH detection in our cohort was not associated with survival in either mRCC or mUC8,17,28. Furthermore, CH did not correlate with grade 3–4 irAEs during systemic mRCC therapy, excepting a minor association between CH > 0.25% and a lower prevalence of grade 3–4 adrenal insufficiency (notwithstanding a lack of formal mechanistic explanation). We speculate that CH may have reduced clinical relevance in lethal metastatic cancers including mRCC and mUC, owing to the short life expectancy of late-stage disease curtailing cumulative risk of CH-associated comorbidities that could otherwise threaten survival (and/or erode quality of life); this aligns with a prior report showing that the 18-month cumulative incidence of developing t-AML in CH-positive patients with solid tumors is only 1%8. Consequently, monitoring for CH may be more beneficial in less aggressive and/or earlier settings—likely especially true in patients being managed with long-term continuous therapy (e.g. androgen deprivation therapy in castration-sensitive prostate cancer), given findings from our study and others that specific anticancer agents can strongly positively select for CH. In general, as systemic therapies are shifted earlier in the disease course, we anticipate that the precise oncologic impact of CH prevalence and genotype will become increasingly salient and may inform whether incidental CH findings should be incorporated into clinical decision-making. Future longitudinal studies exploring temporal clonal dynamics of low VAF CH (<2%) are especially warranted, since preexisting small clones that expand over time were found to be associated with adverse outcomes in other settings61,74.
Our study has the following limitations. First, our results are centered on coding SNVs and small indels, given our application of a 0.248 Mb exon-focused targeted sequencing panel that could not resolve intronic variants, or chromosomal copy-number changes and structural variants emblematic of rare large-scale mosaicism75. The small panel size may also have resulted in underestimation of sample ctDNA fraction (and/or frequency of ctDNA-positivity). Secondly, owing to the real-world aspect of our hypothesis-generating retrospective study, the patient cohort was not standardized with respect to timing of the baseline blood collection, prior treatment exposures and availability of clinical data (including grade 1–2 irAEs or any AEs in muC). It is plausible that our study may be underpowered to detect potentially modest associations between CH and survival, since prior studies reporting an association between CH and survival in cancer and non-cancer patients examined substantially larger cohorts (e.g. 5000 to 10,000+ patients)28. Evaluating CH in large prospective studies involving patients from various lines of therapy are needed to clarify the impact of CH on cancer outcomes and treatment-related adverse events, as well as to resolve the longitudinal clonal dynamics of CH during treatment.
Methods
We queried our British Columbia-based genitourinary cancer research biobank for patients with mUC or mRCC with ≥1 blood samples collected between 01/2017 and 05/2023. All histological subtypes were eligible. Clinical data were retrospectively collected for all 299 patients with mUC or mRCC, including baseline clinical characteristics and prior therapy exposure. Disease volume and sites of metastases were exclusively available for the mRCC cohort. Treatment information post first-line was unavailable in the mRCC cohort. Each patient contributed a baseline blood sample collected before initiating a new line of systemic anti-cancer therapy. 41 patients with mUC provided additional follow-up sample(s) (n = 50) at ~4-months post-treatment initiation and/or at disease progression. Overall survival was defined as the interval from the date of baseline blood collection to date of death from any cause, or date of last follow-up. Biological sex data were collected for all patients and incorporated into correlative analyses as appropriate; gender identity was not collected. All samples were de-identified at time of collection, and researchers were blind to patient gender identity and gender presentation. For all patients in the mRCC cohort that received systemic treatment, information regarding the most commonly observed grade 3 or 4 irAEs was collected. For the adverse event to be attributable to treatment, it was required to have occurred while the patient was receiving treatment. The correlative research study on banked blood samples was conducted in compliance with the Declaration of Helsinki, with ethics approval obtained from the University of British Columbia Clinical Research Ethics Board (certificate number H23-01547). All participants provided written informed consent before enrolling to our biobank and providing blood samples. The biobank was approved by the BC Cancer Clinical Research Ethics Board (certificate number H23-01402). Participation in the biobank was voluntary and patients were not financially compensated for their involvement.
cfDNA and WBC DNA was extracted from plasma and buffy coat using the QIAamp Circulating Nucleic Acid Kit (catalog number: 55114) and Promega Maxwell RSC Blood DNA Kit (catalog number: AS1400), respectively. Library pools for WBC DNA were generated from 25 to 250 ng of DNA using the KAPA HyperPlus Kit (catalog number: KK8512-07962401001), following the protocol in the technical manual (version KR1145 - v9.23), with an initial enzymatic fragmentation step to ~200 bp. In contrast, cfDNA libraries employed 10–50 ng inputs using the KAPA HyperPrep Kit (catalog number: KK8502-07962347001) following the procedures outlined in the technical manual (version KR0961 - v7.19). Subsequent steps included end-repair and A-tailing, followed by adaptor ligation that incorporated IDT 3-bp xGen CS Unique Molecular Identifiers (UMIs), with PCR amplification performed using IDT Unique Dual Index (UDI) primers to yield 1 µg libraries. Each resulting library was quantified using a NanoDrop spectrophotometer and underwent quality control on a 1.3% SYBR-Safe agarose gel. Libraries were then multiplexed to create single pools and hybridized to a custom KAPA HyperChoice target enrichment panel. This probe set captures the coding sequences of 14 genes commonly altered in genitourinary malignancies including FGFR3, ARID1A, BAP1, and VHL, as well as 42 genes associated with CH mutations or linked to hematologic malignancies such as DNMT3A, ASXL1, TET2, and TP53 (Supplementary Data 1). Additionally, probes targeting the TERT promoter region were included. Final libraries were purified according to the KAPA HyperCap Workflow and sequenced on the Illumina NovaSeq6000. Samples that did not achieve deduplicated 1000X sequencing coverage or exhibited signs of oxidative damage (e.g. excessive G>T mutations) were excluded from the study.
All mUC WBC DNA libraries were re-sequenced on another custom KAPA HyperChoice target enrichment panel containing regularly tiled probes across the whole genome, capturing ~9000 SNPs at high heterozygous frequency in the human population. Select mRCC WBC DNA libraries with mutational evidence of large CH clones (VAF greater than 10%) were re-sequenced using this panel. These sequencing data were used for genome-wide copy number modeling to search for copy number variations with somatic origin in the WBC populations.
FASTQ files from deep targeted sequencing were processed with fastp (0.23.2)76 to remove adapter sequences and trim low quality read tails using a 4 bp sliding window and a minimum base quality score of 20. Processed FASTQs were used to generate unmapped BAM files using the fgbio FastqToBam (2.0.2) tool. Unmapped BAMs were aligned to the hg38 reference genome using BWA mem (0.7.17)77. Aligned BAM files were subject to paired cfDNA-WBC DNA indel realignment using ABRA2 (2.24)78 with the additional “--no-edge-ci –mad 20000” arguments. Mate-pair information was verified and fixed using picard FixMateInformation (2.27.4). The resulting BAM files had their base quality scores recalibrated using gatk BaseRecalibrator and gatk ApplyBQSR (4.2.0.0). To enable sensitive resolution of true somatic variants from sequencing artifacts, dual UMI barcoded reads were collapsed into single-strand consensus families where each family was required to contain ≥2 unique reads. Aligned and recalibrated reads were then grouped by 5′ mapping position and UMI sequence using fgbio GroupReadsByUmi (2.0.2). One mismatch was permitted in each UMI and only reads with mapping quality greater than 20 were considered for grouping. The resulting groups were collapsed into single-strand consensus sequences (SSCS) using fgbio CallMolecularConsensusReads (2.0.2). The parameter --min-input-base-quality was set to 20. The resulting SSCS were first filtered using sambamba view -F “proper_pair” (0.6.8)79 to exclude improperly paired reads. Subsequently fgbio FilterConsensusReads (2.0.2) tool was used to correct the orientation of base-specific tags, --min_base_quality and --max_no_call_fraction parameters were set to 30 and 0.15, respectively. Resulting alignments were coordinate sorted and indexed using samtools sort and samtools index modules (1.13).
Freebayes (1.3.6)80, Mutect2 (4.2.0.0)81, and VarDictJava (1.8.3)82 were used for calling SNVs and indels in the cfDNA and WBC DNA, independently. We constructed a per-base error rate profile (“background error rate”) using DNA from ten healthy individuals under age 30 by performing a pileup of base and InDel counts across all genomic positions using pysamstats (1.12) and computing the overall error rate at each locus. All variant calls were normalized using bcftools norm (1.17)83 followed by vt decompose_blocksub to decompose biallelic block substitutions (2015.11.10)84. Variants were annotated with ANNOVAR85 using the refGene, gnomad211_exome, cosmic97_coding, avsnp150, and clinvar_20221231 databases.
The resulting variants from each variant caller were filtered using the following criteria applied to both the cfDNA and WBC DNA libraries from each patient blood collection:
-
≥200× coverage at position
-
≥5 variant supporting reads (unique SSCS)
-
Identified by 2 out of 3 variant callers
-
VAF ≥10× higher than the background error rate at that position
-
VAF ≥ 0.25%
-
GNOMAD database frequency <0.05%
-
Protein-coding changes or splice site variants only
-
TERT promoter mutations were also retained
We also performed testing for strand bias in each of the called variants using the Fisher’s Exact Test from scipy (1.10.0)86. Variants with p-value < 0.05 and odds ratio >4 or <0.25 in both the cfDNA and WBC DNA samples were discarded. Variants with zero variant read support in a given direction from both the cfDNA and WBC DNA were also discarded.
Filtered calls from each of the three variant callers were intersected and merged using bcftools (1.15) to produce a callset containing only variants detected by at least two out of three callers. The final call set was produced by merging the WBC DNA calls with the cfDNA calls and retaining only those present in both (for each sample). All variants were further manually curated by visualization in the Integrated Genomics Viewer (IGV; 2.11.9)87 to eliminate any remaining false positives.
To distinguish high VAF CH-related variants from genuine germline events, we performed the Tukey’s Fences test for statistical outliers on all mutations with WBC DNA VAF between 40% and 60%. Coding variants falling between the fences were extracted and filtered by removing duplicate variants and variants with a population allelic frequency >0.05% as reported in gnomAD (4.0). The resulting variant list was manually reviewed to assess the likelihood of CH origin by evaluating the gene, protein change, and sequence context in IGV. Finally the VAF of variants in the X chromosome was halved in male patients.
The GRCh38 reference build contains an incorrect duplication of the U2AF1 locus in the p-arm of chromosome 21 which results in poor mapping quality and failure to detect somatic variants, as described by ref. 88. To recover mutations in U2AF1 we used bedtools maskfasta to mask the duplicated region using a bed file of the offending regions provided by the Genome Research Consortium.
To identify tumor-originating mutations, we ran Mutect2 and VardictJava in tumor-normal paired mode with default parameters. VardictJava outputs were filtered using the testsomatic.R script provided with the software distribution. Resulting variants were normalized using bcftools norm and biallelic block substitutions were decomposed using vt decompose_blocksub. ctDNA mutation background error rate file was generated using the entire set of WBC samples in the study. The following criteria were applied to each candidate ctDNA mutation:
-
≥200× cfDNA coverage at position
-
≥25× WBC coverage at position
-
≥5 variant-supporting reads in the cfDNA sample
-
≤2 variant-supporting reads in the WBC sample
-
ctDNA VAF ≥ 0.5%
-
Identified by two out of two variant callers
-
The cfDNA VAF must be more than five times higher than the WBC VAF
-
Protein-coding changes or splice site variants only
-
TERT promoter mutations were also retained
Copy number variants (CNVs) were detected using CNVkit (0.9.10)89 where biases stemming from GC content and regional coverage were normalized using the cnvkit.py fix module. Since cfDNA samples are likely to harbor somatic CNVs derived from ctDNA, only WBC DNA samples were assessed for potential CH-related CNVs. Pooled reference normals for each of the two target enrichment panels were constructed from the WBC DNA samples of all patients that did not harbor any detectable CH mutations. Samples were otherwise processed according to default settings and copy number plots were produced using CNVkit’s cnvlib.do_scatter() or seaborn (0.12.2)90 in Python (3.10.0).
Fragmentomics analyses were performed on a subset of CH and ctDNA variants with at least 15 supporting reads. fetch() command from Pysam (0.22.1)91 was used to iterate through reads and determine those carrying alterations based on CIGAR strings. Then the template_length() command was used to calculate the fragment length associated with the read. Figure 3b was generated using the kdeplot() function from seaborn using fragment length as inputs. The bw_adjust parameter was set to 0.05. In Fig. 3c for each individual CH and ctDNA mutation, the median fragment length of WT and mutated fragments was compared using the Mann Whitney U test. The P values were corrected for multiple testing using the Benjamini-Hochberg procedure.
For each mutation we detected in the subset of the cohort with additional on treatment blood samples, we calculated the growth rate using the following formula:
where VAFOnTreatment and VAFBaseline represent the WBC VAF of the mutation detected at the two time points, and TOnTreatment−TBaseline represent the time difference between baseline and on treatment blood draws in weeks. For VAFs at each time point we also calculated confidence intervals using the binomial test. The MWU test was used to test for significant differences in growth rates between DTA and DDR gene mutations. Within each gene group, we further compared growth rates between patients treated with first-line platinum-based chemotherapy for metastatic disease and those who received other systemic treatments.
Statistical tests and data analysis was performed in Python 3.9.12 (using pandas v.1.4.2, numpy v.1.23.5, scipy v.1.10.1, statsmodels v.0.13.5). Visualizations were generated using matplotlib v.3.7.1 and seaborn v.0.13.0. The following bioinformatics/genomic analysis software was used: BWA mem (0.7.17), bcftools (1.15), ABRA2 (2.24), fastp (0.23.2), fgbio FastqToBam (2.0.2), gatk ApplyBQSR (4.2.0.0), picard (2.27.4), sambamba (0.6.8), Mutect2 (4.2.0.0), VardictJava (1.8.3), Freebayes (1.3.6), GRIPSS (2.3.2), bedtools v.2.26, samtools v.1.8 (htslib v.1.8), and ANNOVAR (v.20191024). All box-plots are centered at the median unless otherwise specified and show IQR. Whiskers extend 1.5 × IQR past the quartiles. All raw data are shown where possible. Survival curves for time-to-event (OS) outcomes were estimated using the Kaplan-Meier method. Univariable and multivariable Cox proportional hazards models were used to calculate hazard ratios and 95% confidence intervals. All hypothesis tests were two-tailed and used a 5% significance threshold. Benjamini-Hochberg method was used for multiple testing corrections. The following cohort-specific covariates were included in multivariable Cox models: mRCC, age at blood draw, biological sex, IMDC risk stratification, pathological subtype, presence of visceral metastases; mUC, Age at blood draw, biological sex, smoking status. The retrospective and hypothesis-generating nature of this correlative study meant that sample size and a statistical analysis plan (including power calculations) were not formally prespecified. Statistical analyses are reported without correction for multiple testing unless otherwise stated. Rationale for patient/sample exclusion from specific sub-analyses is clearly indicated in the manuscript text and/or figure legend. We did not perform any analyses requiring randomization.
Data availability
Sequencing data generated in this study have been submitted to the European Genome Archive under the accession code EGAD50000001267. Sequencing data are available indefinitely for research use only under standard EGA controlled access: data access inquiries should be directed to A.W.W. (alexander.wyatt@ubc.ca). Timeframe for data access will be subject to EGA policy and process. All other supporting data are available within the supplementary data files. Custom code for carrying out consensus BAM generation and variant calling are available in https://github.com/amunzur/CH_in_metastatic_urologic_malignancies.
References
Martincorena, I. et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 348, 880–886 (2015).
Lawson, A. R. J. et al. Extensive heterogeneity in somatic mutation and selection in the human bladder. Science 370, 75–82 (2020).
Martincorena, I. et al. Somatic mutant clones colonize the human esophagus with age. Science 362, 911–917 (2018).
Nik-Zainal, S. et al. The life history of 21 breast cancers. Cell 149, 994–1007 (2012).
Young, A. L., Challen, G. A., Birmann, B. M. & Druley, T. E. Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat. Commun. 7, 12484 (2016).
Zekavat, S. M. et al. TP53-mediated clonal hematopoiesis confers increased risk for incident atherosclerotic disease. Nat. Cardiovasc. Res. 2, 144–158 (2023).
Arends, C. M. et al. Associations of clonal hematopoiesis with recurrent vascular events and death in patients with incident ischemic stroke. Blood 141, 787–799 (2023).
Coombs, C. C. et al. Therapy-related clonal hematopoiesis in patients with non-hematologic cancers is common and associated with adverse clinical outcomes. Cell Stem Cell 21, 374–382.e4 (2017).
Teipel, R. et al. Prevalence and variation of CHIP in patients with aggressive lymphomas undergoing CD19-directed CAR T-cell treatment. Blood Adv. 6, 1941–1946 (2022).
Mouhieddine, T. H. et al. Clonal hematopoiesis is associated with adverse outcomes in multiple myeloma patients undergoing transplant. Nat. Commun. 11, 2996 (2020).
Zhang, Y. et al. Pan-cancer circulating tumor DNA detection in over 10,000 Chinese patients. Nat. Commun. 12, 11 (2021).
Gibson, C. J. et al. Clonal hematopoiesis associated with adverse outcomes after autologous stem-cell transplantation for lymphoma. J. Clin. Oncol. 35, 1598–1605 (2017).
Miller, P. G. et al. Clonal hematopoiesis in patients receiving chimeric antigen receptor T-cell therapy. Blood Adv. 5, 2982–2986 (2021).
Jensen, J. L. et al. Clonal hematopoiesis and clinical outcomes in metastatic castration-resistant prostate cancer patients given androgen receptor pathway inhibitors (Alliance A031201). Clin. Cancer Res. 30, 4910–4919 (2024).
Wong, T. N. et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature 518, 552–555 (2015).
Hsu, J. I. et al. PPM1D mutations drive clonal hematopoiesis in response to cytotoxic chemotherapy. Cell Stem Cell 23, 700–713.e6 (2018).
Bolton, K. L. et al. Cancer therapy shapes the fitness landscape of clonal hematopoiesis. Nat. Genet. 52, 1219–1226 (2020).
Lee, K. S. et al. Clinical relevance of clonal hematopoiesis and its interference in cell-free DNA profiling of patients with gastric cancer. Clin. Chem. Lab. Med. 62, 178–186 (2024).
Diplas, B. H. et al. Clinical importance of clonal hematopoiesis in metastatic gastrointestinal tract cancers. JAMA Netw. Open 6, e2254221 (2023).
Arends, C. M. et al. Clonal hematopoiesis is associated with improved survival in patients with metastatic colorectal cancer from the FIRE-3 trial. Blood 139, 1593–1597 (2022).
Hong, W. et al. Clonal hematopoiesis mutations in patients with lung cancer are associated with lung cancer risk factors. Cancer Res. 82, 199–209 (2022).
Nead, K. T. et al. Impact of cancer therapy on clonal hematopoiesis mutations and subsequent clinical outcomes. Blood Adv. 8, 5215–5224 (2024).
Hallermayr, A. et al. The utility of liquid biopsy in clinical genetic diagnosis of cancer and monogenic mosaic disorders. Med. Genet. 35, 275–284 (2023).
Lui, Y. Y. N. et al. Predominant hematopoietic origin of cell-free DNA in plasma and serum after sex-mismatched bone marrow transplantation. Clin. Chem. 48, 421–427 (2002).
Jensen, K. et al. Association of clonal hematopoiesis in dna repair genes with prostate cancer plasma cell-free DNA testing interference. JAMA Oncol. 7, 107–110 (2021).
Chabon, J. J. et al. Integrating genomic features for non-invasive early lung cancer detection. Nature 580, 245–251 (2020).
Bjerggaard Jensen, J. et al. 1960O Identification of bladder cancer patients that could benefit from early post-cystectomy immunotherapy based on serial circulating tumour DNA (ctDNA) testing: Preliminary results from the TOMBOLA trial. Ann. Oncol. 35, S1133 (2024).
Jaiswal, S. et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 371, 2488–2498 (2014).
Genovese, G. et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 371, 2477–2487 (2014).
Marston, N. A. et al. Clonal hematopoiesis, cardiovascular events and treatment benefit in 63,700 individuals from five TIMI randomized trials. Nat. Med. 30, 2641–2647 (2024).
Bick, A. G. et al. Inherited causes of clonal haematopoiesis in 97,691 whole genomes. Nature 586, 763–768 (2020).
Kar, S. P. et al. Genome-wide analyses of 200,453 individuals yield new insights into the causes and consequences of clonal hematopoiesis. Nat. Genet. 54, 1155–1166 (2022).
Kahn, J. D. et al. truncating mutations confer resistance to chemotherapy and sensitivity to PPM1D inhibition in hematopoietic cells. Blood 132, 1095–1105 (2018).
Ali, A. Y., Abedini, M. R. & Tsang, B. K. The oncogenic phosphatase PPM1D confers cisplatin resistance in ovarian carcinoma cells by attenuating checkpoint kinase 1 and p53 activation. Oncogene 31, 2175–2186 (2012).
Bertrums, E. J. M. et al. Selective pressures of platinum compounds shape the evolution of therapy-related myeloid neoplasms. Nat. Commun. 15, 6025 (2024).
Burocziova, M. et al. Truncated PPM1D impairs stem cell response to genotoxic stress and promotes growth of APC-deficient tumors in the mouse colon. Cell Death Dis. 10, 818 (2019).
Milosevic, J. et al. High expression of induces tumors phenotypically similar to loss-of-function mutations in mice. Cancers 13, 5493 (2021).
Loh, P.-R. et al. Insights into clonal haematopoiesis from 8342 mosaic chromosomal alterations. Nature 559, 350–355 (2018).
Niroula, A. et al. Distinction of lymphoid and myeloid clonal hematopoiesis. Nat. Med. 27, 1921–1927 (2021).
Tolmeijer, S. H. et al. Early on-treatment changes in circulating tumor DNA fraction and response to enzalutamide or abiraterone in metastatic castration-resistant prostate cancer. Clin. Cancer Res. 29, 2835–2844 (2023).
Mitchell, E. et al. Clonal dynamics of haematopoiesis across the human lifespan. Nature 606, 343–350 (2022).
Fabre, M. A. et al. The longitudinal dynamics and natural history of clonal haematopoiesis. Nature 606, 335–342 (2022).
Vandekerkhove, G. et al. Plasma ctDNA is a tumor tissue surrogate and enables clinical-genomic stratification of metastatic bladder cancer. Nat. Commun. 12, 184 (2021).
Bacon, J. V. W. et al. Plasma circulating tumor DNA and clonal hematopoiesis in metastatic renal cell carcinoma. Clin. Genitourin. Cancer 18, 322–331.e2 (2020).
Culliford, R. et al. Whole genome sequencing refines stratification and therapy of patients with clear cell renal cell carcinoma. Nat. Commun. 15, 5935 (2024).
Robertson, A. G. et al. Comprehensive molecular characterization of muscle-invasive bladder cancer. Cell 171, 540–556.e25 (2017).
Gulati, S. et al. Molecular analysis of primary and metastatic sites in patients with renal cell carcinoma. J. Clin. Investig. 134, e176230 (2024).
Vandekerkhove, G. et al. Toward informed selection and interpretation of clinical genomic tests in prostate cancer. JCO Precis Oncol. 8, e2300654 (2024).
Tukachinsky, H. et al. Genomic analysis of circulating tumor DNA in 3,334 patients with advanced prostate cancer identifies targetable BRCA alterations and AR resistance mechanisms. Clin. Cancer Res. 27, 3094–3105 (2021).
de Bono, J. et al. Olaparib for metastatic castration-resistant prostate cancer. N. Engl. J. Med. 382, 2091–2102 (2020).
Patelli, G. et al. The evolving panorama of HER2-targeted treatments in metastatic urothelial cancer: a systematic review and future perspectives. Cancer Treat. Rev. 104, 102351 (2022).
Kotecha, R. R. et al. Matched molecular profiling of cell-free DNA and tumor tissue in patients with advanced clear cell renal cell carcinoma. JCO Precis. Oncol. 6, e2200012 (2022).
Rose et al. Enhanced specificity of clinical high-sensitivity tumor mutation profiling in cell-free DNA via paired normal sequencing using MSK-ACCESS. Nat. Commun. 12, 3770 (2021).
Mouliere, F. et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci. Transl. Med. 10, eaat4921 (2018).
Loriot, Y. et al. Erdafitinib or chemotherapy in advanced or metastatic urothelial carcinoma. N. Engl. J. Med. 389, 1961–1971 (2023).
Sato, Y. et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat. Genet. 45, 860–867 (2013).
Fonseca, N. M. et al. Prediction of plasma ctDNA fraction and prognostic implications of liquid biopsy in advanced prostate cancer. Nat. Commun. 15, 1828 (2024).
Reichert, Z. R. et al. Prognostic value of plasma circulating tumor DNA fraction across four common cancer types: a real-world outcomes study. Ann. Oncol. 34, 111–120 (2023).
Hsiehchen, D., Sfreddo, H. J., Zhao, K., Han, C. Y. & Morris, L. G. T. Clonal hematopoiesis and differential outcomes after immune checkpoint blockade. Cancer Cell 40, 1071–1072 (2022).
Lasseter, K. et al. Plasma cell-free DNA variant analysis compared with methylated DNA analysis in renal cell carcinoma. Genet. Med. 22, 1366–1373 (2020).
Coombs, C. C. et al. Identification of clonal hematopoiesis mutations in solid tumor patients undergoing unpaired next-generation sequencing assays. Clin. Cancer Res. 24, 5918–5924 (2018).
Pal, S. K. et al. Evolution of circulating tumor DNA profile from first-line to subsequent therapy in metastatic renal cell carcinoma. Eur. Urol. 72, 557–564 (2017).
Kurokawa, K. et al. Genomic characterization of thymic epithelial tumors in a real-world dataset. ESMO Open 8, 101627 (2023).
Astier, C. et al. Molecular profiling of biliary tract cancers reveals distinct genomic landscapes between circulating and tissue tumor DNA. Exp. Hematol. Oncol. 13, 2 (2024).
Zengin, Z. B. et al. Complementary role of circulating tumor DNA assessment and tissue genomic profiling in metastatic renal cell carcinoma. Clin. Cancer Res. 27, 4807–4813 (2021).
Vang, R. et al. Molecular alterations of TP53 are a defining feature of ovarian high-grade serous carcinoma: a rereview of cases lacking TP53 mutations in The Cancer Genome Atlas Ovarian study. Int. J. Gynecol. Pathol. 35, 48–55 (2016).
Imielinski, M. et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 150, 1107–1120 (2012).
Fairchild, L. et al. Clonal hematopoiesis detection in patients with cancer using cell-free DNA sequencing. Sci. Transl. Med. 15, eabm8729 (2023).
Moore, K. et al. Maintenance olaparib in patients with newly diagnosed advanced ovarian cancer. N. Engl. J. Med. 379, 2495–2505 (2018).
Herberts, C. & Wyatt, A. W. Technical and biological constraints on ctDNA-based genotyping. Trends Cancer Res. 7, 995–1009 (2021).
Kleppe, M. et al. Somatic mutations in leukocytes infiltrating primary breast cancers. NPJ Breast Cancer 1, 15005 (2015).
Comen, E. A. et al. Evaluating clonal hematopoiesis in tumor-infiltrating leukocytes in breast cancer and secondary hematologic malignancies. J. Natl. Cancer Inst. 112, 107–110 (2020).
Abbosh, C. et al. Tracking early lung cancer metastatic dissemination in TRACERx using ctDNA. Nature 616, 553–562 (2023).
Abelson, S. et al. Prediction of acute myeloid leukaemia risk in healthy individuals. Nature 559, 400–404 (2018).
Saiki, R. et al. Combined landscape of single-nucleotide variants and copy number alterations in clonal hematopoiesis. Nat. Med. 27, 1239–1249 (2021).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Mose, L. E., Perou, C. M. & Parker, J. S. Improved indel detection in DNA and RNA via realignment with ABRA2. Bioinformatics 35, 2966–2973 (2019).
Tarasov, A., Vilella, A. J., Cuppen, E., Nijman, I. J. & Prins, P. Sambamba: fast processing of NGS alignment formats. Bioinformatics 31, 2032–2034 (2015).
Garrison, E. & Marth, G. Haplotype-based variant detection from short-read sequencing. https://doi.org/10.48550/ARXIV.1207.3907 (2012).
Benjamin, D. et al. Calling somatic SNVs and indels with Mutect2. bioRxiv https://doi.org/10.1101/861054 (2019).
Lai, Z. et al. VarDict: a novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res. 44, e108 (2016).
Goraichuk, I., Leyson, C. & Alkhamis, M. A. Sequencing and Phylogenetic Analysis as a Tool in Molecular Epidemiology of Veterinary Infectious Diseases (Frontiers Media SA, 2023).
Tan, A., Abecasis, G. R. & Kang, H. M. Unified representation of genetic variants. Bioinformatics 31, 2202–2204 (2015).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010).
Virtanen, P. et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods 17, 261–272 (2020).
Thorvaldsdóttir, H., Robinson, J. T. & Mesirov, J. P. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 14, 178–192 (2013).
Miller, C. A. et al. Failure to detect mutations in U2AF1 due to changes in the GRCh38 reference sequence. J. Mol. Diagn. 24, 219–223 (2022).
Talevich, E., Shain, A. H., Botton, T. & Bastian, B. C. CNVkit: Genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput. Biol. 12, e1004873 (2016).
Waskom, M. et al. Mwaskom/seaborn: v0.8.1 (September 2017) (Zenodo, https://doi.org/10.5281/ZENODO.883859).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Acknowledgements
This work was supported by a CIHR Project Grant (PJT 180417), the Canadian Cancer Society (via a Challenge Grant #707339), the Prostate Cancer Foundation, a Terry Fox New Frontiers Program Project Grant, and the BC Cancer Foundation. A.D.M. was supported by a BC Cancer Rising Star Award and the Four Year Doctoral Fellowship program provided by the University of British Columbia. No funding sources were involved in the design or execution of the study. The authors are grateful to all participating patients and their families.
Author information
Authors and Affiliations
Contributions
A.D.M.: Conceptualization, data curation, software construction, formal analysis, visualization, methodology, writing-original draft, project administration, writing-review and editing; J.V.W.B.: Conceptualization, data curation, software construction, formal analysis, visualization, methodology, writing-original draft, project administration, writing-review and editing. F.F.: Conceptualization, data curation, methodology, writing-original draft, project administration, writing-review and editing. C.H.: Writing-original draft, project administration, formal analysis, writing-review and editing. G.D.: Data curation, validation, methodology, writing-review and editing. C.Q.B.: Data curation, validation, methodology, writing-review and editing. K.P.: Data curation, visualization, validation, methodology, writing-review and editing. G.V.: Data curation, validation, methodology, writing-review and editing. D.C.M.: Data curation, validation, methodology, writing-review and editing. Maria. S. Data curation, writing-review and editing. Y.L.: Data curation, writing-review and editing. L.N.: Resources, writing-review and editing. D.K. Resources, writing-review and editing. C.M.D.: Resources, writing-review and editing. K.N.C. Resources, writing-review and editing. B.J.E.: Resources, writing-review and editing. C.K.: Resources, writing-review and editing. Maryam. S.: Conceptualization, resources, data curation, project administration, supervision, writing-review and editing; A.W.W.: Conceptualization, resources, project administration, supervision, funding acquisition, writing-original draft, writing-review and editing. All authors reviewed and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
G.V. reports research funding from Gilead Sciences, and travel expenses, honoraria and/or advisory board roles from Gilead Sciences, Janssen, and EMD Serono. L.N. has served on advisory boards and/or received honoraria from AstraZeneca, Bayer, Eisai, EMD-Serono, Ipsen, Janssen, Merck, Novartis, Pfizer, TerSera. C.M.D. reports Honoria from MSD, Bristol-Myers Squibb, Medison and Pfizer and consulting fees from Biomica LTD. K.N.C. reports grants from Janssen, Astellas, and Sanofi during the conduct of the study. K.N.C. also reports grants and personal fees from Janssen, Astellas, AstraZeneca, and Sanofi, as well as personal fees from Constellation Pharmaceuticals, Daiichi Sankyo, Merck, Novartis, Pfizer, Point Biopharma, and Roche outside the submitted work. B.J.E. reports honoraria from Seagen, EMD Serono, Pfizer, Janssen, BMS, Gilead, Merck, LOXO-Lilly and AstraZeneca. Maryam.S. has served on advisory boards and/or received honoraria from Pfizer, Ipsen, Novartis, Bayer, AstraZeneca, Alpha-9 Oncology, consulting fees from Bayer, Ipsen, Novartis and Eisai Limited, and travel support from Ipsen and Pfizer. A.W.W. has served on advisory boards and/or received honoraria from AstraZeneca, Astellas, Bayer, EMD Serono, Janssen, Merck, and Pfizer. A.W.W.’s laboratory has a contract research agreement with ESSA Pharma, Tyra Biosciences, and Promontory Therapeutics. The remaining authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Munzur, A.D., Bacon, J.V.W., Fishbein, F. et al. Clonal hematopoiesis in metastatic urothelial and renal cell carcinoma. npj Precis. Onc. 9, 177 (2025). https://doi.org/10.1038/s41698-025-00965-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41698-025-00965-y

{kind=link}