
Abstract
N6-methyladenosine (m6A) methylation modification can influence lung adenocarcinoma (LUAD) progression by regulating gene expression. Ferroptosis, a novel regulated cell death, is involved in various malignant progression. This study aimed to elucidate the role of ferroptosis-related long non-coding RNA (lncRNA) with m6A modification in LUAD. By integrating MeRIP-seq and RNA-seq data from in-house samples and public databases, we identified that lncRNA RBMS3-AS3 was abnormally downregulated in LUAD. The low expression of RBMS3-AS3 predicted poor prognosis in LUAD patients. RBMS3-AS3 was downregulated by m6A “eraser” ALKBH5 in an m6A-dependent manner. Reduced RBMS3-AS3 expression promoted LUAD progression in vitro and in vivo through inhibiting ferroptosis. Mechanistically, RBMS3-AS3 directly bound to HNRNPDL, recruiting it from the nucleus to the cytoplasm to stabilize ZEB1 mRNA. ZEB1 interacted with the E-box motif in the GPX4 promoter, suppressing GPX4 transcription and thereby promoting ferroptosis. In conclusion, RBMS3-AS3, epigenetically downregulated by ALKBH5, facilitates LUAD progression by inhibiting ferroptosis via the HNRNPDL/ZEB1/GPX4 axis, and may serve as a novel therapeutic target for LUAD.
Similar content being viewed by others


Introduction
Lung cancer accounts for approximately one-fifth of all deaths caused by malignant tumors around the world1,2. Lung adenocarcinoma (LUAD), the predominant pathological subtype of non-small cell lung cancer (NSCLC), causes a serious challenge due to its high incidence and poor prognosis3. Despite significant advances in medical strategies over the past few decades, the mortality rate of LUAD constantly increases, and the 5-year overall survival rate remains below 20%4,5. Therefore, it is necessary to explore the key molecules involved in the tumorigenesis and progression of LUAD from novel perspectives, and to reveal the underlying molecular mechanisms.
N6-methyladenosine (m6A) is the most abundant RNA modification in eukaryotic cells and can influence malignant progression by regulating biological processes of RNA, such as transcription, splicing, and translation6,7. Emerging evidence showed that m6A modification can not only affect protein-coding genes, but also non-coding genes, including lncRNAs8. For instance, METTL3-mediated m6A modification upregulated the expression of lncRNA SNHG17 by stabilizing its RNA transcript in LUAD9. The expression of lncRNA MSTRG.292666.16 was enhanced by METTL14 in an m6A-dependent manner10. Previous studies have depicted the transcriptome-wide m6A methylome in LUAD11. However, the detailed molecular mechanism of m6A-modified lncRNA in LUAD remains to be further explored.
Recently, a novel form of programmed cell death known as ferroptosis has gained significant attention12,13,14. During ferroptosis, intracellular iron acts as a pro-oxidant, reacting with hydrogen peroxide to produce lipid reactive oxygen species (ROS). These ROS can be converted into non-toxic lipid alcohols by the antioxidant enzyme glutathione peroxidase 4 (GPX4) and glutathione (GSH)15. Previous studies have elucidated the crucial role of lncRNAs in influencing malignancy progression by modulating ferroptosis. For instance, LINC00336 facilitated lung cancer progression and inhibited ferroptosis via sponging microRNA-685216. LncRNA RGMB-AS1 prevented the ubiquitination of heme oxygenase 1 and enhanced N-alpha-acetyltransferase 10 activity to induce ferroptosis and suppress NSCLC progression17. The high dependence of cancer cells on iron prompts researchers to develop new therapeutic strategies targeting the ferroptosis process18. Nowadays, the role of ferroptosis in LUAD treatment has been investigated19. For example, miR-6077-induced suppression of ferroptosis was associated with resistance of LUAD cells to cisplatin or pemetrexed20. This indicated that combining ferroptosis inducers with other chemotherapeutic agents might improve therapeutic outcomes for patients with cisplatin or pemetrexed resistance. Furthermore, sorafenib and Chinese medicines have been reported to influence the ferroptosis process in LUAD cells21,22,23. However, these previous studies did not systematically analyze the role of ferroptosis-related lncRNAs with m6A modification in LUAD progression, highlighting the need to further identify functional ferroptosis-related lncRNAs and explore their underlying molecular mechanisms.
In this study, we systematically screened m6A-modified lncRNAs by conjoint analyses of MeRIP-seq and RNA-seq data from Jiangsu Province Hospital (JSPH). Meanwhile, we identified ferroptosis-related lncRNAs through integrated analyses using multiple public databases. Here, we uncovered the role of RBMS3-AS3 with m6A modification in modulating tumor growth and ferroptosis in LUAD. Our findings revealed that the decreased expression of RBMS3-AS3 was mediated by m6A regulation, which facilitated cancer progression and suppressed ferroptosis through the HNRNPDL/ZEB1/GPX4 axis in LUAD.
Results
Conjoint analyses of MeRIP-Seq and RNA-Seq data in LUAD
We performed MeRIP-seq analysis of LUAD tissues and tumor-adjacent normal lung tissues from 8 LUAD patients. A total of 1031 significantly upregulated and 821 downregulated m6A peaks were found in LUAD tissues compared with normal lung tissues (| log2(fold change)| ≥ 1 and p-value < 0.05) (Fig. 1A)24. The top 20 altered m6A peaks are listed in Table S1. Furthermore, m6A peaks in LUAD tissues showed a different pattern compared with normal lung tissues (Supplementary Fig. S1A). Analysis of the distribution of m6A peaks revealed that most genes had one m6A peak (Supplementary Fig. S1B). All the modified m6A peaks were mapped to human chromosomes, and the dysregulated m6A peaks were observed in all chromosomes with significant presence in chr1 (Supplementary Fig. S1C). The significantly enriched biological process, cellular component, and molecular function of genes with modified m6A peaks were shown in Supplementary Fig. S1D. KEGG pathway analysis showed that genes with differential m6A peaks were significantly associated with the pathway in TGF−β, Prion diseases, Phospholipase D, and cancer (Supplementary Fig. S1E).

A The significantly differential m6A peaks were shown in volcano plots. B Volcano plots showing the differentially expressed genes in LUAD tissues compared with normal lung tissues. C The distribution of genes with significant alterations in both the m6A modification and RNA levels was shown in four-quadrant plots. D The differentially expressed lncRNAs in normal lung tissues vs LUAD tissues were shown in volcano plots. E The screening of ferroptosis-related lncRNAs with m6A modification in TCGA, JSPH, and GSE130779 data sets. F–H The expression of RBMS3-AS3 was detected in JSPH, TCGA, and GSE130779 cohorts. I The expression of RBMS3-AS3 in 32 paired LUAD from JSPH was determined by RT-qPCR. J, K The low expression of RBMS3-AS3 predicted poor LUAD patient OS in GSE31210 and TCGA cohorts using the Kaplan–Meier method. L The relative m6A level of RBMS3-AS3 in LUAD was detected by MeRIP-seq. M MeRIP combined with specific RT-qPCR was conducted to assess the m6A level of RBMS3-AS3 in LUAD cell lines and 16HBE cells. N Several m6A sequence motifs with high confidence in RBMS3-AS3 were predicted by SRAMP. O The RIP assay was performed to detect the interaction between RBMS3-AS3 and the proteins ALKBH5 and FTO in LUAD cells. P ALKBH5 overexpression shrank the expression of RBMS3-AS3 in LUAD cells while ALKBH5 knockdown enhanced RBMS3-AS3 expression. Q The expression of RBMS3-AS3 was neither affected by FTO overexpression nor by FTO knockdown via RT-qPCR. R MeRIP-qPCR assay uncovered that the m6A methylation level of RBMS3-AS3 was decreased in LUAD cells overexpressing ALKBH5, and the knockdown of ALKBH5 had the opposite effect. S The expression of RBMS3-AS3 was examined by RT-qPCR in LUAD cells treated with actinomycin D for the indicated times after modulating ALKBH5 expression. Data were representative images or were expressed as the mean ± standard deviation. ns: no significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Then, we performed RNA-seq (MeRIP-seq input library) to explore the transcriptome profiles of LUAD tissues versus normal lung tissues from eight LUAD patients. We found 2220 upregulated genes and 1901 downregulated genes in LUAD tissues compared with normal lung tissues (| log2(fold change)| ≥ 1 and p-value < 0.05) (Fig. 1B). The top 20 differentially expressed genes are listed in Table S2. The GO and KEGG pathway analyses of differentially expressed genes were conducted and the results were shown in Supplementary Fig. S1F and Fig. S1G. By integrating MeRIP-seq and RNA-seq data, the differential m6A peaks with differential gene expression were divided into four groups. We found 277 hypermethylated m6A peaks in genes that were significantly upregulated (178; hyper-up) or downregulated (99; hyper-down), while 244 hypomethylated m6A peaks in genes that were significantly upregulated (144; hypo-up) or downregulated (100; hypo-down) (Fig. 1C). According to the conjoint analyses of MeRIP-seq and RNA-seq data, we found 112 upregulated and 123 downregulated lncRNAs (Fig. 1D). Among them, we screened 88 differentially expressed lncRNAs with m6A regulation (|log2(fold change)| ≥ 1 and p-value < 0.05).
Ferroptosis-related lncRNA RBMS3-AS3 is downregulated by ALKBH5 through m6A modification in LUAD
We downloaded ferroptosis-related genes from FerrDb databases and conducted Pearson’s correlation analysis (|r| ≥ 0.4) to identify 1069 ferroptosis-related lncRNAs with differential expression (|log2(fold change)| ≥ 1 and p-value < 0.05). After analyzing the intersection of m6A-modified lncRNAs and ferroptosis-related lncRNAs, we identified a total of 10 ferroptosis-related lncRNAs regulated by m6A modification. Further analysis of DEGs in the TCGA and GSE130779 data sets revealed one ferroptosis-related lncRNA with differential expression and m6A regulation: RBMS3-AS3 (Fig. 1E). Compared with para-cancerous tissue, RBMS3-AS3 was significantly downregulated in LUAD tissues in JSPH (p < 0.0001), TCGA (p < 0.0001), and GSE130779 data set (p < 0.001) (Fig. 1F–H). Total RNA extracted from 32 paired LUAD clinical samples from JSPH was measured to further confirm the above results via RT-qPCR (p < 0.001) (Fig. 1I). The low expression of RBMS3-AS3 in LUAD patients was highly correlated with poor prognosis in both GSE31210 (p = 0.0098) and TCGA data sets (p = 0.0055) (Fig. 1J, K). Additionally, RBMS3-AS3 expression was associated with patient EGFR mutation status in the GSE31210 cohort (Table S3). In the TCGA-LUAD cohort, RBMS3-AS3 expression was associated with patient gender, TNM stage, EGFR mutation status, and smoking history (Table S4). Next, we examined RBMS3-AS3 expression in the human bronchial epithelial cell line (16HBE) and in LUAD cell lines. The results showed that RBMS3-AS3 expression in LUAD cell lines was lower than in 16HBE cells through RT-qPCR (Supplementary Fig. S2A). In this study, among the LUAD cell lines tested, A549 and PC9 cell lines exhibited moderate RBMS3-AS3 expression levels and were thus selected for further investigation. All these findings revealed that RBMS3-AS3 was downregulated in LUAD, and its low expression was correlated with poor prognosis in LUAD patients.
According to our MeRIP-seq, the relative m6A level of RBMS3-AS3 in LUAD and normal tissues was shown in Fig. 1L. The global m6A methylation level in LUAD cell lines and 16HBE cells was detected by conducting the MeRIP-qPCR assay. The results showed that compared with 16HBE cells, the m6A methylation level of RBMS3-AS3 was lower in LUAD cell lines, including H1299, A549, PC9, H358, and H1975 (Fig. 1M). Additionally, we found several m6A sequence motifs in RBMS3-AS3 through the online bioinformatics database SRAMP (http://www.cuilab.cn/sramp/) (Fig. 1N). Because of the decreased m6A methylation level of RBMS3-AS3, we speculated whether m6A “eraser” enzymes, including FTO alpha-ketoglutarate dependent dioxygenase (FTO) and Alk B homolog 5 (ALKBH5), regulated RBMS3-AS3 expression.
Therefore, we conducted RIP assays and the results showed that both FTO and ALKBH5 could bind to RBMS3-AS3 (Fig. 1O). Then, siRNAs and plasmids were designed to silence and overexpress FTO (si-FTO, OE-FTO) and ALKBH5 (si-ALKBH5, OE-ALKBH5) (Supplementary Fig. S2B–E). However, perturbing both genes revealed that only ALKBH5 knockdown or overexpression altered RBMS3-AS3 expression (Fig. 1P, Q). We conducted RT-qPCR to detect the expression of ALKBH5 in 32 paired LUAD clinical samples from JSPH. The results showed that ALKBH5 was significantly upregulated in LUAD tissues compared with para-cancerous tissue (p < 0.001) (Supplementary Fig. S2F). We then performed survival analysis of the TCGA-LUAD cohort and several GEO-LUAD cohorts on the Kaplan–Meier plotter website, which confirmed that high ALKBH5 expression correlated with significantly worse overall survival (Supplementary Fig. S2G). These findings support ALKBH5’s role as a key tumor-promoting regulator in LUAD. In addition, the results of correlation analysis in 32 paired LUAD samples showed a strong inverse correlation between ALKBH5 and RBMS3-AS3 expression (r = –0.4989, p = 0.0037) (Supplementary Fig. S2H).
The results of the MeRIP assays showed that compared with the control group, overexpressing ALKBH5 decreased the m6A methylation level of RBMS3-AS3, whereas knocking down ALKBH5 had the opposite effect in LUAD cells (Fig. 1R). Given that ALKBH5 had been reported to regulate target genes by modulating their RNA stability25, ALKBH5 silenced or overexpressed cells were treated with actinomycin D to measure RBMS3-AS3 stability. The results revealed that silencing ALKBH5 led to a significant elevation of RBMS3-AS3 stability while overexpressing ALKBH5 decreased RBMS3-AS3 stability in LUAD cells (Fig. 1S). These data indicated that the downregulation of RBMS3-AS3 in LUAD cells was related to ALKBH5-mediated m6A modification by regulating RBMS3-AS3 transcript stability.
The downregulation of RBMS3-AS3 facilitates tumor progression and suppresses ferroptosis in LUAD
We used siRNAs to knock down RBMS3-AS3 (si-RBMS3-AS3) and the lentivirus for stably knocking down (sh-RBMS3-AS3) or overexpressing RBMS3-AS3 (OE-RBMS3-AS3) in A549 and PC9 cell lines (Supplementary Fig. S3A). Subsequently, CCK-8, EdU, colony formation, and transwell assays indicated that the knockdown of RBMS3-AS3 promoted cell proliferation and migration, while RBMS3-AS3 overexpression resulted in the opposite effect (Fig. 2A–E). These results demonstrate that the low expression of RBMS3-AS3 facilitates LUAD progression in vitro.

A–E The cell proliferation and migration of LUAD cells with RBMS3-AS3 knockdown and overexpression were evaluated by CCK-8 assay (A), colony formation assay (B), EdU staining assay (C), and transwell assay (D). F CCK-8 assay was carried out to determine the cell viability of LUAD cells treated with RSL3 or Fer-1 after modulating RBMS3-AS3 expression. G, H The knockdown and overexpression of RBMS3-AS3 influenced the intracellular lipid ROS of A549 and PC9 cells treated with RSL3, which was detected by C11-BODIPY581/591. Representative images of lipid ROS in each group (G). The relative lipid ROS levels were examined using ImageJ software (H). I–N With the treatment of RSL3, the level of intracellular ferroptosis was detected by measuring the GSH/GSSG ratio (I, J), Fe2+ level (K, L), and MDA level (M, N) in A549 and PC9 cells subjected to the knockdown or overexpression of RBMS3-AS3. Data were representative images or were expressed as the mean ± standard deviation. ns: no significant, *p < 0.05, **p < 0.01, ***p < 0.001.
To investigate the role of RBMS3-AS3 in the ferroptosis process in LUAD cells, we treated LUAD cells with RSL3, a known ferroptosis inducer. Then, we assessed the role of RBMS3-AS3 in the regulation of ferroptosis. The CCK-8 assays indicated that knockdown of RBMS3-AS3 resisted the RSL3-induced ferroptosis while RBMS3-AS3 overexpression had the opposite effects (Fig. 2F), which were dose- and time-dependent (Supplementary Fig. S3B, C). Moreover, we detected several ferroptosis indicators to determine intracellular ferroptosis levels, such as intracellular concentrations of Fe2+, the end-product of lipid peroxides malondialdehyde (MDA), the key ferroptosis inhibitor GSH, and lipid ROS. The results showed that RBMS3-AS3 overexpression increased intracellular concentrations of lipid ROS, Fe2+, and MDA while reducing the GSH to glutathione disulfide (GSSG) ratio in LUAD cells. Conversely, knocking down RBMS3-AS3 resulted in opposite effects (Fig. 2G–N). These findings show that the low expression of RBMS3-AS3 induces resistance to ferroptosis in LUAD cells.
Low expression of RBMS3-AS3 promotes LUAD progression via inhibiting ferroptosis in vitro and in vivo
Since RBMS3-AS3 simultaneously affected tumor progression and intracellular ferroptosis levels, we wondered whether the oncogenic effect of RBMS3-AS3 downregulation in LUAD was mediated by ferroptosis. The colony formation and EdU assays showed that the enhanced cell proliferation caused by RBMS3-AS3 knockdown was rescued by the treatment of RSL3, while Fer-1 restored the tumor suppressive effect mediated by RBMS3-AS3 overexpression (Fig. 3A, B). Transwell assays suggested that RSL3 could suppress LUAD cell migration and reverse the enhanced migratory capacity resulting from RBMS3-AS3 knockdown (Fig. 3C). Conversely, Fer-1 could promote cell migration and rescue the reduced migratory ability induced by RBMS3-AS3 overexpression (Fig. 3C).

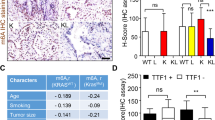
A–D The effects of RBMS3-AS3 expression modulation on LUAD cell proliferation and migration were assessed using colony formation assay (A), EdU staining assay (B), and transwell assay (C) following treatment with RSL3 or Fer-1. E The in vivo function of RBMS3-AS3 was evaluated through subcutaneous tumor formation assays in the presence of RSL3 or Fer-1 treatment. F The volume of tumors was measured every 4 days, and the curve was plotted. G The tumors were weighed. H–J Ferroptosis levels in extracted tumor tissues were detected by measuring the GSH/GSSG ratio (H), Fe2+ level (I), and MDA level (J). K–M Tumor sections were stained with H&E, Ki67 antibody, and GPX4 antibody. The number of Ki-67 or GPX4 positive cells was measured using ImageJ software. Data were representative images or were expressed as the mean ± standard deviation. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Furthermore, xenograft tumors in the sh-RBMS3-AS3 group exhibited a prominent increase in size, volume, and weight compared to the control group. This effect could be reversed with RSL3 treatment. In contrast, Fer-1 rescued the abrogated tumor growth caused by RBMS3-AS3 overexpression (Fig. 3E–G). Subsequently, we assessed the ferroptosis levels in the extracted tumor tissues. The results showed that the increased GSH to GSSG ratio and decreased Fe2+ and MDA levels induced by RBMS3-AS3 knockdown were restored by RSL3. In contrast, Fer-1 abrogated the enhanced ferroptosis levels caused by RBMS3-AS3 overexpression (Fig. 3H–J). Then, immunohistochemical analysis indicated that tumors in the sh-RBMS3-AS3 group exhibited increased expression of Ki-67 and GPX4, whereas the OE-RBMS3-AS3 group had opposite effects. These changes could be reversed by treatment with RSL3 or Fer-1 (Fig. 3K–M).
RBMS3-AS3 upregulates ZEB1 expression and ZEB1 promotes ferroptosis and suppresses LUAD progression both in vitro and in vivo
Through the previous Pearson’s correlation analysis, 29 ferroptosis-related genes correlated with RBMS3-AS3 (|r| ≥ 0.3) were identified. Among them, we selected the top 3 ferroptosis-related genes based on their correlation coefficient, namely zinc finger E-box binding homeobox 1 (ZEB1), ATM serine/threonine kinase (ATM), and phosphatase and tensin homolog (PTEN) (Fig. 4A, Supplementary Fig. S4A). In addition, the results of RT-qPCR and WB suggested that only ZEB1 RNA and protein levels were prominently downregulated by knocking down RBMS3-AS3 and upregulated by overexpressing RBMS3-AS3 in A549 and PC9 cells (Fig. 4B, C, Supplementary Fig. S4B).

A 29 ferroptosis-related genes related to RBMS3-AS3 expression were screened in the heatmap of co-expression analysis. B, C The expression of ZEB1, PTEN, and ATM was examined by RT-qPCR and WB in A549 and PC9 cells knocking down or overexpressing RBMS3-AS3. D, E ZEB1 was downregulated in LUAD tissues from TCGA and GTEx cohorts (D) and the JSPH cohort (E). F The RNA levels of ZEB1 in 32 paired LUAD samples from JSPH were measured by RT-qPCR. G RBMS3-AS3 expression showed a positive correlation with ZEB1 RNA levels in 32 paired LUAD samples from JSPH. H, I The decreased expression of ZEB1 predicted an unfavorable patient OS in the GSE31210 and GSE3141 cohorts. J, K The intracellular lipid ROS of A549 and PC9 cells treated with RSL3 were examined by C11-BODIPY581/591 following the modulation of ZEB1 expression. L–Q The role of ZEB1 in modulating the intracellular ferroptosis level in LUAD cells was detected by measuring the GSH/GSSG ratio (L, M), Fe2+ level (N, O), and MDA level (P, Q). Data were representative images or were expressed as the mean ± standard deviation. ns: no significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
We then explored the ZEB1 expression in LUAD. The results from TCGA and GTEx databases (p < 0.05) and JSPH (p < 0.0001) showed that the expression of ZEB1 in LUAD tissues was significantly lower than in para-cancerous tissue (Fig. 4D, E). The RT-qPCR analysis in 32 paired LUAD samples obtained from JSPH revealed that the expression of ZEB1 in LUAD tissues was notably decreased compared to para-cancerous tissues (p < 0.0001) and showed a positive correlation with RBMS3-AS3 expression at the RNA level (r = 0.5477, p = 0.0012) (Fig. 4F, G). We then evaluated ZEB1 protein levels in the same 32 paired LUAD samples using WB and performed correlation analysis with RBMS3-AS3 expression, and these results further confirmed our previous findings (r = 0.4657, p = 0.0072) (Supplementary Fig. S4C–E). Although ZEB1 expression showed no prognostic value in the TCGA-LUAD cohort (Supplementary Fig. S4F), low ZEB1 expression predicted poor prognosis in LUAD patients in both the GSE31210 (p = 0.0021) and GSE3141 (p = 0.0019) cohorts (Fig. 4H, I). Additionally, no association between ZEB1 expression and patient clinical characteristics was observed in the TCGA-LUAD and GSE31210 cohorts (Tables S5-6). Together, ZEB1 expression was positively correlated with RBMS3-AS3 expression. ZEB1 was downregulated in LUAD, and its low expression predicted a worse prognosis.
Therefore, the siRNA and overexpression lentivirus vectors were designed to knock down ZEB1 (si-ZEB1) or overexpress ZEB1 (OE-ZEB1) in A549 and PC9 cells (Supplementary Fig. S4G-H). The CCK-8, colony formation, EdU, and transwell assays showed that ZEB1 knockdown promoted LUAD cell progression in vitro while its overexpression resulted in opposite effects (Supplementary Fig. S5A–D). We then stably knocked down ZEB1 by the lentivirus (sh-ZEB1) to perform the xenograft tumor assay (Supplementary Fig. S5E). The results suggested that ZEB1 overexpression suppressed LUAD progression in vivo and showed decreased Ki-67 expression (Supplementary Fig. S5F–K).
Moreover, we assessed the intracellular ferroptosis levels in LUAD cells with the treatment of RSL3. The results suggested that the knockdown of ZEB1 increased the GSH to GSSG ratio and decreased lipid ROS, Fe2+, and MDA levels, verified by detecting the impact of ZEB1 overexpression in LUAD cells (Fig. 4J–Q). Additionally, we examined the role of ZEB1 overexpression in RBMS3-AS3 knockdown LUAD cells. The results of CCK-8, colony formation, EdU, and transwell assays indicated that the overexpression of ZEB1 restored the tumor progression induced by RBMS3-AS3 knockdown in vitro (Fig. 5A–D). Xenograft tumor assays revealed that the significant increases in the size, weight, and volume of subcutaneous xenograft tumors resulting from RBMS3-AS3 knockdown were effectively reversed by ZEB1 overexpression (Fig. 5E–G). Furthermore, the decreased intracellular ferroptosis levels caused by RBMS3-AS3 knockdown were rescued by ZEB1 overexpression (Fig. 5H–L). These data suggest that ZEB1 inhibits tumor growth in LUAD cells by enhancing intracellular ferroptosis, and the effect of RBMS3-AS3 on LUAD is mediated by ZEB1.

A–D The rescue CCK-8 assay (A), colony formation assay (B), EdU staining assay (C), and transwell assay (D) showed that the enhanced cell proliferation and migration of LUAD cells induced by knocking down RBMS3-AS3 were partly restored by ZEB1 overexpression. E The xenograft tumor assay with A549 cells was carried out to determine the effect of ZEB1 on tumorigenesis induced by stably knocking down RBMS3-AS3 in vivo. F Tumor volume was measured and recorded every 4 days and the curve was plotted. G The tumors were weighed. H, I The reduced lipid ROS level caused by silencing RBMS3-AS3 was rescued by overexpressing ZEB1 in A549 and PC9 cells with the treatment of RSL3. J–L ZEB1 overexpression partially restored the decreased intracellular ferroptosis level induced by RBMS3-AS3 knockdown via detecting the GSH/GSSG ratio (J), Fe2+ level (K), and MDA level (L). Data were representative images or were expressed as the mean ± standard deviation. *p < 0.05, **p < 0.01, ***p < 0.001.
ZEB1 induces ferroptosis in LUAD cells by inhibition of GPX4 transcription
Given that GPX4 is the crucial protein in the process of ferroptosis and ZEB1 has been reported to promote ferroptosis via regulating GPX4 expression in cancer26,27, we speculated whether ZEB1 affected GPX4 expression in LUAD cells. Through Pearson’s correlation analysis, we found a significant negative correlation between the expression of GPX4 and ZEB1 in the TCGA-LUAD cohort (r = –0.431, p < 3.06e–25) and 32 paired LUAD samples (r = –0.5092, p = 0.0029) (Fig. 6A, Supplementary Fig. S6A). Furthermore, GPX4 expression was significantly upregulated in tumor tissues compared to adjacent normal lung tissues in 32 paired LUAD samples (p < 0.001) (Supplementary Fig. S6B). However, no significant difference in GPX4 expression was observed between tumor and normal tissues in the TCGA-LUAD and GTEx cohorts (Supplementary Fig. S6C). Survival analysis showed that GPX4 expression was not associated with the prognosis of LUAD patients in the TCGA-LUAD cohort (Supplementary Fig. S6D). In addition, GPX4 expression was associated with patient smoking history in the TCGA-LUAD cohort (Table S7).

A The correlation coefficient of ZEB1 and GPX4 in LUAD tumors from the TCGA cohort. B, C The expression of GPX4 was examined by RT-qPCR (B) and WB (C) in A549 and PC9 cells subjected to the knockdown or overexpression of ZEB1. D The ZEB1 binding sites in the GPX4 promoter were predicted by JASPAR. E, F The products of the ChIP assay in LUAD cells were checked by agarose gel electrophoresis (E) and RT-qPCR (F). G, H Knocking down RBMS3-AS3 in A549 cells led to a reduction in GPX4 enrichment by ZEB1. I, J The effect of RBMS3-AS3 on GPX4 expression was detected by RT-qPCR (I) and WB (J) in LUAD cells. K, L ZEB1 overexpression restored the increased GPX4 expression induced by silencing RBMS3-AS3. M The schematic of the GPX4 promoter with luciferase reporter vectors. N The Dual-luciferase assay was conducted to assess the relative luciferase activity of reporter plasmids containing the GPX4 promoter and its mutant type in ZEB1 overexpression LUAD cells. Data were representative images or were expressed as the mean ± standard deviation. ns: no significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
The results of RT-qPCR and WB showed that the expression of GPX4 was inversely associated with ZEB1 (Fig. 6B, C, Supplementary Fig. S6E). Considering that ZEB1 has been demonstrated to bind to the E-box regions in the promoter of target genes28,29, we inferred that ZEB1 could bind to the E-box motif in the GPX4 promoter and subsequently suppress its transcription. The results found in the JASPAR (https://jaspar.elixir.no/) indicated that ZEB1 could bind to four ‘CACCTG’ motifs in the GPX4 promoter with high confidence (Fig. 6D). The results of ChIP assays and RT-qPCR demonstrated that two pairs of primers designed to the ‘CACCTG’ motif were immunoprecipitated by anti-ZEB1 both in A549 and PC9 cells (Fig. 6E, F). Moreover, silencing RBMS3-AS3 significantly suppressed the interaction between ZEB1 and GPX4 promoter in A549 cells (Fig. 6G, H). The results of RT-qPCR and WB revealed that the RBMS3-AS3 also downregulated GPX4 expression (Fig. 6I, J). The increased GPX4 level caused by RBMS3-AS3 silencing was restored by ZEB1 overexpression through RT-qPCR and WB (Fig. 6K, L, Supplementary Fig. S6F). Finally, luciferase reporter vectors were constructed with GPX4 promoter regions or their mutants and transfected into A549 and PC9 cells (Fig. 6M). The results demonstrated that ZEB1 overexpression significantly decreased GPX4 luciferase activity in the wild type compared to the control group, while the GPX4 luciferase activity remained unchanged in the mutant type (Fig. 6N), which suggested that ZEB1 could directly bind to the ‘CACCTG’ motif in the GPX4 promoter in LUAD. Taken together, ZEB1 directly binds to the promoter of GPX4 to suppress GPX4 transcription, leading to enhanced intracellular ferroptosis in LUAD cells.
RBMS3-AS3 directly interacts with HNRNPDL and promotes its cytoplasmic translocation
We further determined the underlying mechanism by which RBMS3-AS3 enhanced ZEB1 expression. The results of RIP assays demonstrated that RBMS3-AS3 could not directly interact with ZEB1 (Supplementary Fig. S6G). Considering that RBMS3-AS3 could regulate ZEB1 at the transcriptional level, we constructed the luciferase reporter plasmid containing the ZEB1 promoter. The results elucidated that the luciferase activity of the ZEB1 promoter was not affected by RBMS3-AS3 knockdown or overexpression (Supplementary Fig. S6H). Furthermore, actinomycin D (5 μg/ml) was used to treat LUAD cells to suppress their transcription and we found that RBMS3-AS3 silencing prominently reduced the half-life of ZEB1 mRNA while RBMS3-AS3 overexpression dramatically enhanced the half-life of ZEB1 mRNA (Supplementary Fig. S6I), which suggested that RBMS3-AS3 regulates ZEB1 expression through enhancing ZEB1 mRNA stability.
Considering that lncRNAs could exert biological functions by interacting with RNA-binding proteins (RBPs)30,31, we reasoned that a certain RBP interacted with both RBMS3-AS3 and ZEB1 mRNA and mediated this process. The results of FISH and nucleocytoplasmic separation assays showed that RBMS3-AS3 was primarily located in the cytoplasm (Fig. 7A, B). Therefore, specific RBMS3-AS3 probes were designed to perform RNA pull-down assays, silver staining assays, and mass spectrometry analysis. We found that a differential protein band located around 38 kDa was present in the RBMS3-AS3 group but not in the control group (Fig. 7C). And according to the mass spectrometry analysis results, we identified 20 RBPs that were only pulled down by RBMS3-AS3 probes both in A549 and PC9 cells (Fig. 7D). Finally, heterogeneous nuclear ribonucleoprotein D like (HNRNPDL) was chosen for further study because of its molecular weight and its ability to regulate the stability of target mRNA. WB assays suggested that HNRNPDL was pulled down by RBMS3-AS3 probe both in A549 and PC9 cells (Fig. 7E). The results of RIP assays indicated that RBMS3-AS3 enrichment in the anti-HNRNPDL group was significantly higher than that in the anti-IgG group in A549 and PC9 cells (Fig. 7E). These results suggest that RBMS3-AS3 can directly bind to HNRNPDL.

A, B The subcellular localization of RBMS3-AS3 was determined by FISH assay (A) and nucleocytoplasmic separation assay (B). C The RNA pull-down assay was conducted in LUAD cells with the biotinylated-RBMS3-AS3 and antisense probe, and subsequently subjected to SDS-PAGE with silver staining. D The results of mass spectrometry identified 20 RBPs that were only pulled down by RBMS3-AS3 probes in LUAD cells. E The binding of RBMS3-AS3 and HNRNPDL was validated by WB using RNA pull-down samples and RIP assay. F, G The nuclear and cytoplasmic proteins (F) and RNAs (G) were extracted to examine the subcellular localization of HNRNPDL by WB and RT-qPCR. H The co-localization of RBMS3-AS3 and HNRNPDL in A549 cells subjected to RBMS3-AS3 knockdown or overexpression. I In silico protein-RNA molecular docking simulations. J Illustration of full-length Flag-labeled HNRNPDL and other mutants. K RIP assays quantified RBMS3-AS3 enrichment bound to Flag-tagged HNRNPDL (full-length and truncated variants). L Secondary structure of RBMS3-AS3 predicted by RNAfold Website. M The RNA pull-down experiments using three truncated fragments of RBMS3-AS3 were performed. N The half-life of ZEB1 mRNA was measured by RT-qPCR in HNRNPDL knockdown or overexpression LUAD cells and this effect could be rescued by overexpressing or knocking down RBMS3-AS3, respectively. O The schematic of luciferase reporter vectors containing the ZEB1 mRNA 3′-UTR or its mutant variant. P The Dual-luciferase assay was conducted to assess the relative luciferase activity of reporter plasmids containing ZEB1 mRNA 3′-UTR or its mutant type in HNRNPDL knockdown LUAD cells. Data were representative images or were expressed as the mean ± standard deviation. ns: no significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Furthermore, we found that RBMS3-AS3 expression was not correlated with HNRNPDL expression in the TCGA-LUAD and JSPH cohorts (Supplementary Fig. S7A-B). No significant differential expression of HNRNPDL was observed in either the JSPH or TCGA databases (Supplementary Fig. S7C-D). HNRNPDL expression showed no significant association with LUAD prognosis in the TCGA-LUAD cohort (Supplementary Fig. S7E). However, in the GSE31210 cohort, low expression of HNRNPDL was significantly associated with poor prognosis for LUAD patients (Supplementary Fig. S7F). In addition, HNRNPDL expression correlated with patient EGFR mutation status in the TCGA-LUAD cohort (Table S8).
RT-qPCR and WB assays showed that neither silencing nor overexpressing RBMS3-AS3 altered the expression of HNRNPDL (Supplementary Fig. S7G-H). Given that HNRNPDL was located in both the nucleus and the cytoplasm, we speculated that RBMS3-AS3 recruited it from the nucleus to the cytoplasm. The nuclear and cytoplasmic RNAs and proteins were extracted from RBMS3-AS3 knockdown or overexpression A549 cells. The results suggested that RBMS3-AS3 overexpression increased the enrichment of HNRNPDL protein in the cytoplasm while silencing RBMS3-AS3 had the opposite effect (Fig. 7F, Supplementary Fig. S7I). Nevertheless, the RNA distribution of HNRNPDL was not altered by RBMS3-AS3 (Fig. 7G). We then conducted immunofluorescence via confocal laser scanning microscope and the results further validated the above results (Fig. 7H, Supplementary Fig. S7J-K).
Next, we elucidated the precise molecular determinants of the interaction of RBMS3-AS3 and HNRNPDL. We performed in silico protein-RNA molecular docking simulations combined with targeted mutagenesis experiments (Fig. 7I). This integrated approach has now identified that both the RRM1 (amino acids 148–230) and RRM2 (amino acids 233–312) domains of HNRNPDL mediate stronger interaction with RBMS3-AS3 (Fig. 7J, K). Next, we conducted in silico analysis of RBMS3-AS3 using RNAfold WebServer to predict conserved secondary structures and then constructed various truncations of RBMS3-AS3 based on their predicted binding sites (Fig. 7L). RNA pull-down experiments identified the 113–339 nt fragment of RBMS3-AS3 as the predominant interaction region with HNRNPDL (Fig. 7M). These data indicate that RBMS3-AS3 facilitates the cytoplasmic translocation of HNRNPDL by directly binding to it.
HNRNPDL binds to ZEB1 mRNA and enhances its stability
Since RBMS3-AS3 could regulate the stability of ZEB1 mRNA, we wondered whether HNRNPDL mediated this process. The RIP assays showed that compared with the anti-IgG group, a significant ZEB1 mRNA enrichment was observed in the anti-HNRNPDL group both in A549 and PC9 cells while silencing RBMS3-AS3 significantly abrogated this effect (Supplementary Fig. S8A-B). We silenced and overexpressed HNRNPDL via siRNA (si-HNRNPDL) and plasmid vector (OE-HNRNPDL) (Supplementary Fig. S8C-D). Pearson’s correlation analysis revealed no significant correlation between ZEB1 and HNRNPDL expression in the TCGA-LUAD cohort (r = 0.151, p = 5.06e–04) (Supplementary Fig. S8E). By contrast, in 32 paired LUAD samples, ZEB1 expression was significantly positively correlated with HNRNPDL expression (r = 0.4541, p = 0.0090) (Supplementary Fig. S8F). RT-qPCR and WB assays suggested that both RNA and protein levels of ZEB1 were regulated by HNRNPDL knockdown or overexpression (Supplementary Fig. S8G-H). Furthermore, the decreased ZEB1 expression induced by RBMS3-AS3 knockdown was rescued by HNRNPDL overexpression (Supplementary Fig. S8I-K). Then, the results of the mRNA stability assay indicated that with the treatment of actinomycin D, HNRNPDL overexpression reduced the degradation rate of ZEB1 mRNA while HNRNPDL silencing shortened the half-life of ZEB1 mRNA. This effect could be rescued by knocking down or overexpressing RBMS3-AS3 (Fig. 7N).
HNRNPDL has been reported to interact with poly(A)+ RNAs to regulate mRNA stability via directly binding to the 5′-ACUAGC-3′ RNA sequence in the 3′-UTR32. Consequently, we constructed luciferase reporter vectors containing the ZEB1 mRNA 3′-UTR or its mutant and transfected them into LUAD cells (Fig. 7O). The results showed that si-HNRNPDL repressed luciferase activity in the wild type whereas this effect was almost totally abrogated in the mutant type, suggesting that HNRNPDL could directly bind to the 3′-UTR of ZEB1 mRNA (Fig. 7P). These data indicate that HNRNPDL could stabilize ZEB1 mRNA by binding to its specific RNA motif in the 3′-UTR.
Moreover, we performed functional experiments to determine whether HNRNPDL mediated the effect of ZEB1 on LUAD cells. CCK-8, colony formation, and EdU assays demonstrated that HNRNPDL knockdown rescued the suppressed cell proliferation resulting from ZEB1 overexpression in LUAD (Fig. 8A–D). Additionally, transwell assays revealed that the impaired cell migration induced by ZEB1 overexpression was reversed by HNRNPDL knockdown in LUAD (Fig. 8E, F). Further WB assays indicated that the decreased GPX4 expression induced by ZEB1 was restored by HNRNPDL knockdown (Fig. 8G–I). We then detected the ferroptosis levels and found that HNRNPDL knockdown abrogated the enhanced intracellular ferroptosis levels caused by ZEB1 (Fig. 8J–N). Therefore, we conclude that HNRNPDL mediates ZEB1-induced tumor suppression and ferroptosis induction via stabilizing its mRNA. Altogether, as pictorially modeled in Fig. 9, our findings revealed that ALKBH5 mediated the low expression of RBMS3-AS3 via m6A modification and elucidated the role of RBMS3-AS3 downregulation in triggering tumor progression and suppressing ferroptosis in LUAD via HNRNPDL/ZEB1/GPX4 axis.

A–F CCK-8 (A), colony formation (B, C), EdU staining (D, F), and transwell assay (E, F) were carried out to assess the cell functions of ZEB1 overexpression in LUAD cells with HNRNPDL1 silencing. G, H HNRNPDL knockdown in LUAD cells rescued the ZEB1 overexpression-induced decrease in GPX4 expression, as measured by RT-qPCR and WB. I The GPX4 protein levels shown in Fig.8H were quantified using ImageJ software. J–N The effect of silencing HNRNPDL on the enhanced ferroptosis level caused by overexpressing ZEB1 in LUAD cells treated with RSL3 was examined by detecting the lipid ROS level (J, K), Fe2+ level (L), GSH/GSSG ratio (M), and MDA level (N). Data were representative images or were expressed as the mean ± standard deviation. ns: no significant, *p < 0.05, **p < 0.01, ***p < 0.001.

ALKBH5 downregulated RBMS3-AS3 expression in LUAD by the m6A modification. The decreased expression of RBMS3-AS3 promoted tumor progression and suppressed intracellular ferroptosis levels in LUAD via the HNRNPDL/ZEB1/GPX4 axis.
Discussion
Despite the indispensable role of ferroptosis-related lncRNAs with m6A modification in LUAD progression, there are limited therapeutic strategies targeting lncRNAs to modulate ferroptosis in patients with LUAD. Therefore, we systematically identified ferroptosis-related lncRNAs associated with m6A regulation by integrating MeRIP-seq and RNA-seq data from JSPH and multiple public databases. In this study, we prioritized RBMS3-AS3 as a ferroptosis-related lncRNA with m6A modification.
Recent studies have elucidated that RNA m6A modification exists in most eukaryotes and plays a significant role in regulating target RNA splicing, translation, and stability. M6A modification was also indispensable for the regulation of lncRNAs in various cancers33,34. A previous study has discovered the transcriptome-wide m6A methylome in LUAD11. In this study, we focused on m6A-modified lncRNAs in LUAD by conjoint analyses of MeRIP-seq and RNA-seq data from 8 LUAD patients. As a result, 88 differentially expressed lncRNAs with m6A modification were screened. Subsequent Pearson’s correlation analysis and differential expression analysis revealed that RBMS3-AS3 was a ferroptosis-related lncRNA with m6A modification.
Our investigations revealed that the m6A “eraser” ALKBH5 downregulated RBMS3-AS3 expression in LUAD via destabilizing it. Existing literature provides insights into potential regulators of ALKBH5 in cancer. For instance, exposure of breast cancer cells to hypoxia caused HIF-dependent upregulation of ALKBH535. Endogenous ROS was reported to inhibit ALKBH5 demethylase activity via ERK/JNK-mediated ALKBH5 phosphorylation, leading to increased global mRNA m6A levels36. Additionally, DNA methylation may contribute to the regulation of ALKBH5 expression. In KRAS-mutated lung cancer, LKB1 loss upregulated ALKBH5 expression by DNA hypermethylation of the CTCF-binding motif on the ALKBH5 promoter. ALKBH5 demethylation of m6A stabilized oncogenic drivers to aggravate the oncological behavior of KRAS-mutated lung cancer37. Consequently, multiple factors may contribute to the upregulation of ALKBH5 in LUAD. More potential triggers that may drive ALKBH5-mediated downregulation of RBMS3-AS3 require further investigation. The m6A modification is a complex epigenetic modification process, and whether other crucial m6A enzymes or molecules are involved in regulating RBMS3-AS3 expression requires further investigation.
RBMS3-AS3 is an antisense lncRNA transcribed from the antisense strand of the RNA binding motif single stranded interacting protein 3 (RBMS3)38. The RBMS3 protein belongs to the c-Myc single-strand binding protein family and participates in regulating various biological processes. RBMS3 has been identified as a tumor suppressor gene in multiple cancers, including gallbladder carcinoma, gastric cancer, esophageal squamous cell carcinoma, and nasopharyngeal carcinoma39,40,41,42. Notably, a recent study reported that metformin could induce ferroptosis and suppress ovarian cancer progression by upregulating RBMS343. Previously, RBMS3-AS3 has only been reported in several bioinformatics articles and its specific biological functions remained uninvestigated. This study uncovered that RBMS3-AS3 was downregulated in LUAD, and its low expression was significantly associated with poorer prognosis in LUAD patients. Functional assays demonstrated that the downregulation of RBMS3-AS3 promoted LUAD progression via restraining ferroptosis in vitro and in vivo.
We subsequently explored the mechanism by which RBMS3-AS3 influenced LUAD progression through regulating the ferroptosis process. As a novel non-apoptotic cell death, ferroptosis could effectively inhibit tumor progression44,45. Previous studies have identified dozens of lncRNAs involved in the regulation of ferroptosis across various cancers46,47,48. For example, LINC00336 could inhibit ferroptosis in lung cancer by competitively binding to miR-685222. LncRNA-PMAN suppressed ferroptosis by inducing the cytoplasmic translocation of ELAVL1 in gastric cancer49. In this study, we highlighted the ferroptosis-related lncRNA with m6A modification by integrating MeRIP-seq and RNA-seq data from JSPH and other online databases.
Through co-expression analysis and our experiments, we found that the expression of ZEB1 was positively associated with RBMS3-AS3. ZEB1, a transcription factor, plays a key role in epithelial-mesenchymal transition (EMT) and promotes tumor invasion and metastasis50. Previous study suggested that ZEB1 accelerated the malignancy in LUAD by activating LINC01123 via NOTCH signaling pathway51. In this study, we found that ZEB1 inhibited LUAD progression and enhanced ferroptosis both in vitro and in vivo. Mechanistically, ZEB1 interacted with the E-box motif in the GPX4 promoter, abrogating GPX4 transcription. This reduction in GPX4 expression led to excessive ferroptosis induction in LUAD cells. Similarly, recent studies have shown that ZEB1 can promote ferroptosis and inhibit tumor progression in various cancers26,52,53,54. Furthermore, one study has reported that ZEB1 regulates the differential expression of various key enzymes, such as stearoyl-CoA desaturase 1 (SCD) and long-chain acyl-CoA synthetase 4 (ACSL4), thereby reshaping the composition of phospholipid fatty acids and enhancing ferroptosis susceptibility in cancer cells55. These studies indicate that ZEB1 plays a dual role in tumors, and its ultimate effect is context-dependent.
To explore how RBMS3-AS3 regulated the expression of ZEB1, we performed the RNA pull-down assay and mass spectrometry analysis to identify the RBPs involved in this process. Through systematic screening, in silico protein-RNA molecular docking simulations, and targeted mutagenesis experiments, we found that both the RRM1 and RRM2 domains of HNRNPDL mediate a stronger interaction with the 113–339 nt fragment of RBMS3-AS3. HNRNPDL is a poorly studied RNA-binding protein belonging to the heterogeneous nuclear ribonucleoproteins (hnRNPs) family56. HNRNPDL is present in both the nucleus and cytoplasm and can shuttle between the nucleus and cytoplasm via its C-terminus57,58. It has also been reported to interact with a unique RNA motif ACUAGC33. Specifically, HNRNPDL could bind to the ACUAGC RNA sequence in the 3′-untranslated region (3′-UTR) of pre-B-cell leukemia homeobox 1 (PBX1) mRNA and stabilize its mRNA59. Our study revealed that HNRNPDL could bind to the ACUAGC RNA motif in the 3′-UTR of ZEB1 mRNA, resulting in increased ZEB1 expression through mRNA stabilization. More studies are warranted to further explore additional mechanisms by which RBMS3-AS3 regulates ZEB1.
In summary, our study demonstrated that RBMS3-AS3 was downregulated by ALKBH5 in an m6A-dependent manner in LUAD. The downregulation of RBMS3-AS3 inhibited ferroptosis and facilitated LUAD progression through the HNRNPDL/ZEB1/GPX4 axis. These findings provided novel potential therapeutic targets for LUAD.
Methods
Patients and tissue specimens
The clinical specimens used in this study were collected from Jiangsu Province Hospital (Nanjing, China). Eight paired LUAD and adjacent normal tissue specimens were used for MeRIP-seq and RNA-seq to detect m6A-regulated genes in LUAD. Total RNA and protein extracted from these tissues were used to measure the expression of the target gene in LUAD and adjacent normal tissues. The clinical characteristics of these 8 LUAD patients are listed in Table S9. Additionally, total RNA extracted from 32 additional LUAD patients was used for validation (Table S10). This study was approved by the Ethics Committee of Jiangsu Province Hospital (2019-SR-123), and written informed consent was obtained from all patients. The study was conducted in compliance with the Declaration of Helsinki.
MeRIP sequencing and RNA sequencing
Total RNA from each sample was isolated and purified using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and quantified using NanoDrop ND-1000 (NanoDrop, Wilmington, DE, USA). The RNA integrity was evaluated using the Bioanalyzer 2100 (Agilent, CA, USA) and confirmed by electrophoresis with the denaturing agarose gel. The Epicentre Ribo-Zero Gold Kit (Illumina, San Diego, USA) was utilized to deplete ribosomal RNA, following the manufacturer’s protocol. Subsequently, the ribosomal-depleted RNA was fragmented into small pieces using Magnesium RNA Fragmentation Module (NEB, e6150, USA) at 86 °C for 7 min. Then the cleaved RNA fragments were incubated for 2 h at 4 °C with m6A-specific antibody (No. 202003, Synaptic Systems, Germany) in IP buffer (50 mM Tris-HCl, 750 mM NaCl, and 0.5% Igepal CA-630). Furthermore, the IP RNA was reverse transcribed to generate complementary DNA (cDNA) using SuperScript™ II Reverse Transcriptase (Invitrogen, 1896649, USA). This cDNA was then utilized to synthesize U-labeled second-stranded DNAs with E. coli DNA polymerase I (NEB, m0209, USA), RNase H (NEB, m0297, USA), and dUTP Solution (Thermo Fisher, R0133, USA). An adenine base (A) was added to the blunt ends of each strand to prepare them for ligation with indexed adapters. Each adapter features a thymine base (T) overhang, facilitating the ligation to the A-tailed fragmented DNA. Single- or dual-index adapters were ligated to fragments, and size selection was carried out using AMPureXP beads. Following treatment of the U-labeled second-stranded DNAs with the heat-labile UDG enzyme (NEB, m0280, USA), the ligated products were amplified using PCR under the following conditions: an initial denaturation at 95 °C for 3 min; 8 cycles of denaturation at 98 °C for 15 s, annealing at 60 °C for 15 s, and extension at 72 °C for 30 s; and a final extension at 72 °C for 5 min. The average insert size for the final cDNA library was 300 ± 50 bp. Finally, we performed 2 × 150 bp paired-end sequencing (PE150) on an Illumina NovaSeq™ 6000 (LC-Bio Technology CO., Ltd, Hangzhou, China) following the vendor’s recommended protocol.
The fastp software (https://github.com/OpenGene/fastp) was utilized to remove reads containing adaptor contamination, low-quality bases, and undetermined bases using the default parameters. The sequence quality of both the IP and Input samples was assessed with FastQC and RseQC. Next, the HISAT2 software (v2.0.4) was used to map the reads to the reference genome Homo sapiens (Version: v101). Peak calling and differential peak analysis were conducted using the R package exomePeak2 (v1.5.0), with peak annotation performed through intersection with gene architecture via the R package ANNOVAR. The StringTie software (v2.1.2) was then used to calculate expression levels for all transcripts and genes from the input libraries, determining FPKM (total exon fragments / mapped reads (millions) × exon length (kB)). Differentially expressed transcripts and genes were identified with log2(fold change) ≥1 or log2(fold change) ≤ –1 and p-value < 0.05 using the R package edgeR (v4.1).
Bioinformatic analysis
We downloaded the transcriptome data of LUAD from the Cancer Genome Atlas (TCGA) database, as well as transcriptional data of ferroptosis-related genes from the FerrDb database (http://www.zhounan.org/ferrdb/current/). The GSE130779, GSE31210, and GSE3141 datasets were downloaded from the public database Gene Expression Omnibus (GEO). Using R software (version 4.4.1, USA), we systematically screened for ferroptosis-related lncRNAs based on the correlation coefficient between the TCGA and FerrDb databases, applying a threshold of |R| ≥ 0.3. The lncRNAs identified as ferroptosis-related met the criteria of |log2(fold change)| ≥ 1 and p-value < 0.05. Then, the significantly differentially expressed genes (DEGs) of GSE130779 were identified. We also downloaded transcriptome data of 57 pairs of LUAD tissue specimens from the TCGA database and filtered for significantly differentially expressed lncRNAs (|log2(fold change)| ≥ 1 and p-value < 0.05). Survival analysis of the GSE31210 and GSE3141 datasets was performed using the Kaplan–Meier method.
Cell lines and reagents
The human LUAD cell lines (H1299, PC9, A549, H358, H1975) and human bronchial epithelial cell line (16HBE) were purchased from the American Type Culture Collection (ATCC). All human cell lines were routinely tested for mycoplasma contamination using N-GRADE Mycoplasma PCR Reagent set (Euroclone S.p.A., Pero, Italy) and authenticated by STR profiling (Eurofins MWG-Biotech, Ebersberg, Germany). All human LUAD cell lines were cultured in DMEM (Gibco, NY, USA) with 10% fetal bovine serum (Gibco, NY, USA) and 1% penicillin and streptomycin (Beyotime, Shanghai, China). 16HBE cells were cultured in RPMI-1640 (Gibco, NY, USA) with the same ingredients. All cell lines were cultured at 37 °C with 5% CO2.
Methylated RNA immunoprecipitation (MeRIP) and MeRIP-qPCR
We performed the MeRIP assay using the Magna MeRIP m6A Kit (Millipore, USA) to determine the m6A level of RNAs. Briefly, total RNA was treated with gDNAwiper mix (Vazyme, Nanjing, China) to remove genomic DNA. Magnetic beads were incubated with an anti-m6A antibody (ab284130, Abcam, Cambridge, UK) or normal rabbit IgG (30000-0-AP, Proteintech) to immunoprecipitate RNA. Then, the m6A-enriched fragmented RNAs were extracted by N6-methyladenosine 5′-monophosphate salt and subsequently reverse transcribed. Afterward, RT-qPCR analysis was used to quantify the relative m6A enrichment normalized to the input.
Plasmids and siRNAs transfection
In this study, all plasmids were synthesized by Genechem (Shanghai, China), and siRNAs were generated in Proteinbio (Nanjing, China). The sequences of used siRNAs are summarized in Table S11. Cell transfection was conducted using Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol.
Lentivirus construction and infection
Lentivirus for knocking down or overexpressing RBMS3-AS3 and ZEB1. The corresponding control viruses (Control) were purchased from GeneChem (Shanghai, China). The efficiency of cell infection was enhanced using HiTransG A or HiTransG P solution (GeneChem, China). The sequences of used shRNAs are summarized in Table S11.
RNA extraction and quantitative real-time polymerase chain reaction (RT–qPCR)
Total RNAs from cell lines or tissues were extracted using the Trizol (Invitrogen, USA) reagent, and the quantification of RNA was measured via Nanodrop (Thermo Scientific, USA). 1 μg of total RNA was reverse transcribed into cDNA, which was subsequently subjected to RT-PCR by using SYBR Green Mix (Vazyme, Nanjing, China). GAPDH was used to analyze relative gene expression using the 2(–ΔΔCT) method. The RT-qPCR primer sequences involved in this study are listed in Table S12.
Cell viability, colony formation, EdU, and cell migration assay
The cell viability assay was performed by CCK-8 Cell Counting Kit (A311-01, Vazyme). Cells were planted into 96-well plates at a density of 2000 cells per well with 200 μl of medium. Afterward, CCK-8 solution (10 μl) was added to each well and then incubated for 2 h at 37 °C. The absorbance at 450 nm of each well was measured and calculated.
For colony formation assay, 1 × 103 NSCLC cells were planted into 6-well plates per well and incubated in a complete medium for 2 weeks. Next, the cells were washed with normal saline and fixed with 4% paraformaldehyde for 15 min at room temperature, followed by staining with 0.5% crystal violet solution (Beyotime, Shanghai, China) for 15 min. The number of colonies was measured using ImageJ software.
Cells were cultured in a 96-well plate until 50–80% confluent. EdU assays were performed by using YF®555 Click-iT EdU Imaging Kits (PC5516, Proteinbio) following the manufacturer’s instructions. The images were captured by an inverted fluorescence Microscope, and the percentage of EdU-staining cells was quantified.
In the cell migration assay, 4 × 104 starved LUAD cells, incubated in 200 μl serum-free medium, were seeded into the upper chamber by using a transwell apical chamber (Corning, NY, USA) while the lower chambers were filled with 700 µl complete medium. After 36 h, cells passing through the membrane were fixed with 4% paraformaldehyde. Subsequently, the fixed cells were stained with 0.5% crystal violet solution (Beyotime, Shanghai, China), while cells attached to the upper compartment were removed by cotton swabs. Then the migratory cells were pictured under the inverted microscope in five random fields of view and measured using ImageJ software.
Assessment of ferroptosis
Malondialdehyde (MDA), iron, lipid ROS, and Glutathione (GSH) assays were performed to examine the level of ferroptosis in LUAD cells. RSL3 (HY-100218A) and Ferrostatin-1 (Fer-1) (HY-100579) were purchased from MedChemExpress (MCE, Shanghai, China). A549 and PC9 cells were treated with RSL3 (1.0 μM) or Fer-1 (1.0 μM) for 24 h before conducting experiments. For MDA assay, the MDA Assay Kit (M496, DOJINDO) was used to detect the relative cellular MDA contents by assessing the absorbance at Ex: 540 nm, Em: 590 nm with a fluorescence microplate reader following the manufacturer’s recommendations. We assessed intracellular ferrous iron (Fe2+) by using FerroOrange (F374, DOJINDO, Japan) according to the manufacturer’s protocol. The fluorescence microplate reader was used to detect the absorbance at Ex: 543 nm/Em: 580 nm for each well to calculate the relative Fe2+ level. Furthermore, the Ferrous Iron Colorimetric Assay Kit (E-BC-K773-M, Elabscience, China) was used to measure the Fe2+ level in LUAD and normal tissues following the manufacturer’s protocol. In lipid ROS assays, the C11-BODIPY581/591 (D3861, Thermo Scientific, USA) was used to measure lipid peroxidation levels following the manufacturer’s instructions. In brief, after incubation with 5 μM C11-BODIPY581/591 at 37 °C for 30 min away from light, cells were washed with PBS and then photographed using a fluorescence microscope. For the Glutathione (GSH) assay, the total Glutathione (T-GSH) / Oxidized Glutathione (GSSG) Colorimetric Assay Kit (Elabscience, Wuhan, China) was used to detect the intracellular GSH / GSSG ratio by measuring the absorbance at 412 nm.
Xenograft mouse models
Four-week-old female BALB/c nude mice were purchased from Gempharmatech (Nanjing, China). All animal experiments were performed according to NIH animal care guidelines and the Experimental Animals Ethics Committee of Nanjing Medical University (IACUC-2501012). In brief, A549 cells with indicated treatments were collected in PBS, and then 1 × 106 cells were inoculated subcutaneously into the back of the mice. The volume of the tumor was measured and recorded every 4 days and determined as: larger diameter × (smaller diameter)2/2. The nude mice were euthanized via cervical dislocation at the indicated time. And the tumors were dissected, weighed, photographed, fixed with 4% formaldehyde, and embedded in paraffin for further analysis.
Immunohistochemistry (IHC) assay
Tumor xenograft sections were fixed with 4% paraformaldehyde, embedded in paraffin, sectioned to 6-μm thickness, and immunostained with specific antibodies, including ZEB1 (1:200; 21544-1-AP, Proteintech) and Ki67 (1:200; 12202S, CST, Boston, USA). Images were captured with a light microscope (Leica, Germany). The percentage of positive cells was measured by ImageJ software.
Western blotting (WB)
Cells or tissues were lysed in RIPA buffer (P0013B, Beyotime, Shanghai, China) containing the cocktail (MCE, Shanghai, China). The extracted protein was separated by SDS-PAGE and then transferred to PVDF membranes (Millipore, USA). After blocking with 5% non-fat dry milk in TBST for 2 h, the membranes were subsequently incubated in the primary antibody at 4 °C for at least 12 h. Subsequently, the membranes were incubated with the corresponding secondary antibodies at room temperature for 1 h and visualized with ECL substrate (RM00021, ABclonal, China) by using an imaging analysis system (BioRad, USA). The primary antibodies in this study are listed in Table S13.
mRNA stability assay
Actinomycin D (HY-17559) was purchased from MedChemExpress (MCE, Shanghai, China). Briefly, cells were incubated with actinomycin D (Act-D, 5 μg/ml) for 0, 2, 4, 6, and 8 h. RNA was collected by Trizol reagent and reverse transcribed to perform RT-qPCR. The remaining RNA level was normalized to the values detected at 0 h.
Dual-luciferase reporter assay
For the dual-luciferase reporter assay, dual-luciferase reporters, including ZEB1 mRNA 3′UTR WT, ZEB1 mRNA 3′UTR Mut, GPX4 promoter WT, and GPX4 promoter Mut, were designed and synthesized by Vazyme (Nanjing, China) using pGL3 vector. In brief, cells were seeded in 6-well plates and subsequently co-transfected with corresponding plasmid using Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA). The luciferase activity was detected by using the Dual-Luciferase Reporter Assay Kit (Vazyme, Nanjing, China) according to the manufacturer’s protocol.
Nuclear and cytoplasmic extraction
The PARIS™ Kit (AM1921, Thermo Scientific, USA) was used to separate nuclear and cytoplasmic RNAs or proteins from total RNAs or proteins according to the manufacturer’s protocol. Briefly, add 500 µl Cell Fractionation Buffer to 5 million cells and gently mix well. After incubating on ice for 10 min, the suspension was centrifuged (500 × g, 5 min). The supernatant (cytoplasmic fraction) was carefully collected, and the precipitate (nuclear fraction) was added to the Cell Disruption Buffer. The cytoplasmic and nuclear lysates were used to perform the WB assay to detect protein levels. For RNA isolation, the above lysate was mixed with 2X Lysis/Binding Solution and 100% ethanol, followed by centrifugation (15,000 × g, 1 min) in a Filter Cartridge. After washing, the RNA was eluted with the Elution Solution.
RNA pull-down assay and mass spectrometry analysis
5’Biotin-labeled RBMS3-AS3 full-length sense and antisense ChIRP probes were designed by Ribobio (Guangzhou, China). Then, we performed the RNA pull-down assay using the RNA-Protein pull-down Kit (P0202, Geneseed, Guangzhou, China) following the manufacturer’s recommendations. The cell lysates were incubated with magnetic bead-conjugated probes, and the co-precipitated proteins were eluted from the beads. Next, the proteins interacting with the probe were separated by SDS-PAGE and subsequently silver-stained by using a Fast Silver Stain Kit (P0017S, Beyotime) according to its instructions. Furthermore, GeneChem (Shanghai, China) subjected this target protein eluent to mass spectrometry analysis.
RNA immunoprecipitation (RIP) assay
The Magna RIP RBP Immunoprecipitation Kit (Millipore, USA) was used to perform RIP assay according to the manufacturer’s instructions. In brief, 2 × 107 cells were collected and homogenized in RIP lysis buffer on ice. The specific antibody and control rabbit IgG were incubated with the magnetic beads at room temperature. After 30 min, the complex was added to the cell lysates at 4 °C overnight. The 1 × PBS was used to rinse the cell lysates, and subsequently the RNA samples were purified and subjected to RT-qPCR analysis.
Chromatin immunoprecipitation (ChIP) assay
The ChIP assay was performed using the Enzymatic ChIP Assay Kit (P2083S, Beyotime) following the manufacturer’s instructions. 1 × 107 cells were collected and fixed with 1% formaldehyde for 10 min at 37 °C. Subsequently, the chromatin in cell lysates was fragmented by micrococcal nuclease and ultrasound. The DNA fragments were subjected to immunoprecipitation with the primary antibody against ZEB1 (21544-1-AP, Proteintech, Wuhan, China) and normal rabbit IgG (30000-0-AP, Proteintech, Wuhan, China). The purified DNA samples were tested through RT-qPCR, which was visualized by agarose gel electrophoresis. Primers involved in the ChIP assay are listed in Table S12.
Fluorescent in situ hybridization (FISH) and Immunofluorescence
The Ribo fluorescence in situ hybridization kit (C10910, RiboBio) was used to conduct the RNA-FISH assay according to the manufacturer’s protocol. Briefly, 3 × 105 cells were fixed with 4% paraformaldehyde, which was subsequently permeabilized with 0.5% Triton in PBS and blocked with 5% BSA. These fixed cells were incubated overnight with FITC-labeled RBMS3-AS3 probes from RiboBio or primary antibodies at 4 °C, followed by corresponding Fluor-labeled secondary antibodies (1:200, Thermo Scientific). Fluorescence imaging was taken by Stellaris STED laser scanning confocal microscopy (LSCM) (Leica, Germany).
Statistical analysis
Statistical analyses were performed using GraphPad Prism 9.5. The statistical tests applied included Student’s t-test, one-way ANOVA, two-way ANOVA, and chi-square tests. All continuous variables underwent normality and log-normality tests. Student’s t-test was used for data following a normal distribution. For data that did not follow a normal distribution or had an ambiguous distribution, the nonparametric Wilcoxon rank-sum test was employed. The data were represented as mean ± standard deviation. Survival analysis was assessed using the Kaplan–Meier log-rank test. A two-tailed p-value of less than 0.05 was considered statistically significant. All experiments were independently repeated at least three times.
Data availability
The TCGA database for the lung adenocarcinoma cohort can be accessed and analyzed by Gene Expression Profiling Interactive Analysis (GEPIA, http://gepia.cancer-pku.cn/). The GSE31210 and GSE3141 datasets can be accessed from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/gds/). The remaining data used and/or analyzed in the present study are available from the corresponding author upon reasonable request.
Code availability
The underlying code for this study is available from the corresponding author upon reasonable request.
References
Mao, Y., Yang, D., He, J. & Krasna, M. J. Epidemiology of lung cancer. Surg. Oncol. Clin. N. Am. 25, 439–445 (2016).
Sung, H. et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71, 209–249 (2021).
Thai, A. A., Solomon, B. J., Sequist, L. V., Gainor, J. F. & Heist, R. S. Lung cancer. Lancet 398, 535–554 (2021).
Wang, B. Y. et al. The comparison between adenocarcinoma and squamous cell carcinoma in lung cancer patients. J. Cancer Res Clin. Oncol. 146, 43–52 (2020).
Duma, N., Santana-Davila, R. & Molina, J. R. Non-small cell lung cancer: epidemiology, screening, diagnosis, and treatment. Mayo Clin. Proc. 94, 1623–1640 (2019).
Chen, Y., Lin, Y., Shu, Y., He, J. & Gao, W. Interaction between N(6)-methyladenosine (m(6)A) modification and noncoding RNAs in cancer. Mol. Cancer 19, 94 (2020).
Zaccara, S., Ries, R. J. & Jaffrey, S. R. Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol. 20, 608–624 (2019).
Yang, X. et al. Epigenetic modulations of noncoding RNA: a novel dimension of cancer biology. Mol. Cancer 19, 64 (2020).
Zhang, H. et al. m6A methyltransferase METTL3-induced lncRNA SNHG17 promotes lung adenocarcinoma gefitinib resistance by epigenetically repressing LATS2 expression. Cell Death Dis. 13, 657 (2022).
Ji, X. et al. METTL14 enhances the m6A modification level of lncRNA MSTRG.292666.16 to promote the progression of non-small cell lung cancer. Cancer Cell Int. 24, 61 (2024).
Mao, W. et al. Comprehensive analysis of the transcriptome-wide m6A methylome in lung adenocarcinoma by MeRIP sequencing. Front. Oncol. 12, 791332 (2022).
Dixon, S. J. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072 (2012).
Dixon, S. J. & Olzmann, J. A. The cell biology of ferroptosis. Nat. Rev. Mol. Cell Biol. 25, 424–442 (2024).
Jiang, X., Stockwell, B. R. & Conrad, M. Ferroptosis: mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 22, 266–282 (2021).
Yang, W. S. et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331 (2014).
Wang, M. et al. Long noncoding RNA LINC00336 inhibits ferroptosis in lung cancer by functioning as a competing endogenous RNA. Cell Death Differ. 26, 2329–2343 (2019).
Gao, G. B. et al. LncRNA RGMB-AS1 inhibits HMOX1 ubiquitination and NAA10 activation to induce ferroptosis in non-small cell lung cancer. Cancer Lett. 590, 216826 (2024).
Xu, T. et al. Molecular mechanisms of ferroptosis and its role in cancer therapy. J. Cell Mol. Med. 23, 4900–4912 (2019).
Chen, X., Kang, R., Kroemer, G. & Tang, D. Broadening horizons: the role of ferroptosis in cancer. Nat. Rev. Clin. Oncol. 18, 280–296 (2021).
Bi, G. et al. miR-6077 promotes cisplatin/pemetrexed resistance in lung adenocarcinoma via CDKN1A/cell cycle arrest and KEAP1/ferroptosis pathways. Mol. Ther. Nucleic Acids 28, 366–386 (2022).
Lachaier, E. et al. Sorafenib induces ferroptosis in human cancer cell lines originating from different solid tumors. Anticancer Res. 34, 6417–6422 (2014).
Mo, X. et al. Tetrandrine citrate suppresses lung adenocarcinoma growth via SLC7A11/GPX4-mediated ferroptosis. Discov. Oncol. 14, 85 (2023).
Huang, F. et al. Hedyotis diffusa injection induces ferroptosis via the Bax/Bcl2/VDAC2/3 axis in lung adenocarcinoma. Phytomedicine 104, 154319 (2022).
Bardou, P., Mariette, J., Escudié, F., Djemiel, C. & Klopp, C. jvenn: an interactive Venn diagram viewer. BMC Bioinform. 15, 293 (2014).
Liu, Z. et al. The roles of N6-methyladenosine and its target regulatory noncoding RNAs in tumors: classification, mechanisms, and potential therapeutic implications. Exp. Mol. Med. 55, 487–501 (2023).
Viswanathan, V. S. et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547, 453–457 (2017).
Han, X. et al. ZEB1 directly inhibits GPX4 transcription contributing to ROS accumulation in breast cancer cells. Breast Cancer Res. Treat. 188, 329–342 (2021).
Zhang, J. et al. ZEB1 induces ER-α promoter hypermethylation and confers antiestrogen resistance in breast cancer. Cell Death Dis. 8, e2732 (2017).
Krebs, A. M. et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell Biol. 19, 518–529 (2017).
Bian, Y. et al. m(6)A modification of lncRNA ABHD11-AS1 promotes colorectal cancer progression and inhibits ferroptosis through TRIM21/IGF2BP2/ FOXM1 positive feedback loop. Cancer Lett. 596, 217004 (2024).
Mattick, J. S. et al. Long non-coding RNAs: definitions, functions, challenges and recommendations. Nat. Rev. Mol. Cell Biol. 24, 430–447 (2023).
Kamei, D. & Yamada, M. Interactions of heterogeneous nuclear ribonucleoprotein D-like protein JKTBP and its domains with high-affinity binding sites. Gene 298, 49–57 (2002).
Zheng, J. et al. Epigenetically silenced lncRNA SNAI3-AS1 promotes ferroptosis in glioma via perturbing the m(6)A-dependent recognition of Nrf2 mRNA mediated by SND1. J. Exp. Clin. Cancer Res. 42, 127 (2023).
Xie, J. et al. M6A-mediated-upregulation of lncRNA BLACAT3 promotes bladder cancer angiogenesis and hematogenous metastasis through YBX3 nuclear shuttling and enhancing NCF2 transcription. Oncogene 42, 2956–2970 (2023).
Zhang, C. et al. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m⁶A-demethylation of NANOG mRNA. Proc. Natl Acad. Sci. USA 113, E2047–E2056 (2016).
Yu, F. et al. Post-translational modification of RNA m6A demethylase ALKBH5 regulates ROS-induced DNA damage response. Nucleic Acids Res. 49, 5779–5797 (2021).
Zhang, D. et al. Suppression of m6A mRNA modification by DNA hypermethylated ALKBH5 aggravates the oncological behavior of KRAS mutation/LKB1 loss lung cancer. Cell Death Dis. 12, 518 (2021).
Yang, Y., Quan, L. & Ling, Y. RBMS3 inhibits the proliferation and metastasis of breast cancer cells. Oncol. Res. 26, 9–15 (2018).
Zhang, T. et al. Low expression of RBMS3 and SFRP1 are associated with poor prognosis in patients with gastric cancer. Am. J. Cancer Res. 6, 2679–2689 (2016).
Li, Y. et al. Downregulation of RBMS3 is associated with poor prognosis in esophageal squamous cell carcinoma. Cancer Res. 71, 6106–6115 (2011).
Chen, J. et al. RBMS3 at 3p24 inhibits nasopharyngeal carcinoma development via inhibiting cell proliferation, angiogenesis, and inducing apoptosis. PLoS ONE 7, e44636 (2012).
Wu, Y. et al. Increased expression of RBMS3 predicts a favorable prognosis in human gallbladder carcinoma. Oncol. Rep. 44, 55–68 (2020).
Zhao, Y., Wang, Y., Zhang, X., Han, S. & Yang, B. Metformin-induced RBMS3 expression enhances ferroptosis and suppresses ovarian cancer progression. Reprod. Biol. 25, 100968 (2024).
Zuo, Y. B. et al. Ferroptosis in cancer progression: role of noncoding RNAs. Int. J. Biol. Sci. 18, 1829–1843 (2022).
Zhang, R., Kang, R. & Tang, D. Ferroptosis in gastrointestinal cancer: from mechanisms to implications. Cancer Lett. 561, 216147 (2023).
Guo, Y. et al. Identification of a prognostic ferroptosis-related lncRNA signature in the tumor microenvironment of lung adenocarcinoma. Cell Death Discov. 7, 190 (2021).
Lu, L., Liu, L. P., Zhao, Q. Q., Gui, R. & Zhao, Q. Y. Identification of a ferroptosis-related LncRNA signature as a novel prognosis model for lung adenocarcinoma. Front. Oncol. 11, 675545 (2021).
Yao, J. et al. Characterization of a ferroptosis and iron-metabolism related lncRNA signature in lung adenocarcinoma. Cancer Cell Int. 21, 340 (2021).
Lin, Z. et al. Hypoxia-induced HIF-1α/lncRNA-PMAN inhibits ferroptosis by promoting the cytoplasmic translocation of ELAVL1 in peritoneal dissemination from gastric cancer. Redox Biol. 52, 102312 (2022).
Caramel, J., Ligier, M. & Puisieux, A. Pleiotropic roles for ZEB1 in cancer. Cancer Res. 78, 30–35 (2018).
Zhang, M. et al. ZEB1-activated LINC01123 accelerates the malignancy in lung adenocarcinoma through NOTCH signaling pathway. Cell Death Dis. 11, 981 (2020).
Zhang, Z. et al. Inhibition of KLF5 promotes ferroptosis via the ZEB1/HMOX1 axis to enhance sensitivity to oxaliplatin in cancer cells. Cell Death Dis. 16, 28 (2025).
Lee, J., You, J. H., Kim, M. S. & Roh, J. L. Epigenetic reprogramming of epithelial-mesenchymal transition promotes ferroptosis of head and neck cancer. Redox Biol. 37, 101697 (2020).
Wang, X. et al. O-GlcNAcylation of ZEB1 facilitated mesenchymal pancreatic cancer cell ferroptosis. Int. J. Biol. Sci. 18, 4135–4150 (2022).
Schwab, A. et al. Zeb1 mediates EMT/plasticity-associated ferroptosis sensitivity in cancer cells by regulating lipogenic enzyme expression and phospholipid composition. Nat. Cell Biol. 26, 1470–1481 (2024).
Dreyfuss, G., Matunis, M. J., Piñol-Roma, S. & Burd, C. G. hnRNP proteins and the biogenesis of mRNA. Annu. Rev. Biochem. 62, 289–321 (1993).
Imasaki, T. et al. Structural basis for substrate recognition and dissociation by human transportin 1. Mol. Cell 28, 57–67 (2007).
Kawamura, H. et al. Identification of the nucleocytoplasmic shuttling sequence of heterogeneous nuclear ribonucleoprotein D-like protein JKTBP and its interaction with mRNA. J. Biol. Chem. 277, 2732–2739 (2002).
Ji, D. et al. Oncogenic heterogeneous nuclear ribonucleoprotein D-like modulates the growth and imatinib response of human chronic myeloid leukemia CD34(+) cells via pre-B-cell leukemia homeobox 1. Oncogene 39, 443–453 (2020).
Acknowledgements
This study was supported by the National Natural Science Foundation of China (82203296), the Major Program of Science and Technology Foundation of Jiangsu Province (BE2023832), the Key Project of Jiangsu Commission of Health (ZD2022055), and the specific cohort for early-staged lung cancer of the First Affiliated Hospital of Nanjing Medical University (DL202402).
Author information
Authors and Affiliations
Contributions
Weibing Wu, Zhihua Li, and Wenke Ge conceived and designed this study. Wenke Ge, Qi Wang, and Hongshun Wang performed the experiments and wrote the manuscript. Weiran Zhang and Liang Chen analyzed the initial data. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ge, W., Wang, Q., Wang, H. et al. M6A-ALKBH5-dependent RBMS3-AS3 down-regulation suppresses ferroptosis to promote lung adenocarcinoma progression through HNRNPDL/ZEB1/GPX4 axis. npj Precis. Onc. 9, 356 (2025). https://doi.org/10.1038/s41698-025-01141-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41698-025-01141-y
