
Abstract
Metabolic syndrome (MetS)—encompassing obesity, insulin resistance, dyslipidemia, and hypertension—is prevalent in both humans and horses, offering a unique opportunity to explore shared pathophysiological mechanisms across species in a controlled model organism. In this first report from the Pioneer 100 Horse Health Project (P100HHP), we conducted a longitudinal, multi-omic analysis of 108 deeply phenotyped horses to interrogate individual health trajectories for precision insights into MetS. We identified two primary metabotypes: one characterized by elevated unsaturated triglycerides (TGs) and the other by increased levels of primary bile acids (BAs), notably taurocholic acid and taurochenodeoxycholic acid. Horses with higher circulating levels of taurocholic acid had significantly higher plasma insulin concentrations, especially after an oral sugar challenge (P = 0.01), indicating that specific BAs are associated with hyperinsulinemia—a key phenotype of MetS. Metabolomic signatures predicted body condition score (relative adiposity) with high performance, underscoring their potential for precision diagnostics. Seasonal variations influenced BA levels and were associated with shifts in the fecal microbiota, particularly in Clostridium and Proteobacteria populations. Additionally, we observed an inverse relationship between genetic diversity—measured by runs of homozygosity—and insulin levels, suggesting a genetic component to MetS susceptibility. Our findings demonstrate the power of deep phenotyping and multi-omic approaches to effectively delineate MetS subtypes in horses, highlighting the pivotal roles of bile acids and the microbiome in MetS pathogenesis. These insights not only advance the understanding of equine MetS but also establish the horse as a valuable translational model for human MetS, with potential implications for targeted diagnostics and therapeutics in both veterinary and human medicine
Similar content being viewed by others

Introduction
Metabolic syndrome (MetS) is common to both humans and horses and shares many features, including obesity, insulin resistance, dyslipidemia, and hypertension1,2. Despite differences in diet types, horses encounter MetS at a similar rate to that of the human population, with up to 40% of horses considered obese or overweight3,4. This shared health crisis makes horses a powerful naturally occurring disease analog to determine intrinsically shared mechanisms driving MetS.
MetS demonstrates the end effect of the complex relationship between inherent genetic risks, current physiological state(s) and environmental exposomes5. The clinical phenotypes concurrent within an individual affected by MetS strongly suggest shared or convergent underlying pathophysiologic mechanisms6. Despite this, MetS in both humans and horses remains a complex and syndromic condition, with a heterogeneous distribution of contributing phenotypes6.
Many studies have indicated that integration of metabolome and microbiome data enables a more complete picture for drivers of MetS7,8,9. The gastrointestinal (GI) microbiome is increasingly recognized as an active participant in the development of obesity and MetS10,11. Much of this effect in humans has been attributed to the modernization of food production systems in the post-industrialized world12. Individuals in Western societies possess a distinct, yet less diverse, GI microbial population, often referred to as the ‘industrial microbiome’. Principally, people of Western societies have a distinct, yet less complex, GI microbial population, regarded as the “industrial” microbiome. This stands in contrast to non-Western and indigenous GI microbiomes, where richness and diversity are maintained, correlating with a reduced risk of metabolic-related diseases12,13,14. Rodents have been most commonly used in experimental studies of the “industrial” microbiome. The premise being that, with successively more restrictive microbial colonization of rodents housed in modern vivaria, this situation approximates the shift seen with industrialization15,16. While valuable, rodent studies cannot fully recapitulate the occurrence of both the industrial/domestic GI microbiome and spontaneous MetS.
Domestication of horses delineates one of the initial steps towards an industrialized society17. Similar to the industrialization of human society, the domestication of horses had a comparable effect on their microbiota. Non-domesticated horses maintain a higher microbial diversity than their domestic counterparts18. In addition to industrialized dietary habits of domestic horses, they now also share modifying exposures like those of humans, namely the use of antimicrobial medications18.
One advantage of studying metabolic syndrome in horses is the ability to control environmental and physiological parameters in ways that are difficult to achieve in human studies. As a result, horses offer a unique opportunity to disentangle the complex interactions between genetics, environment, and microbiome contributions to MetS.
Concurrent evaluation of the metabolome and microbiome brings together genetic, physiologic and environmental contributions to MetS. Integration of multiple high-dimensional datasets provides valuable context, and indeed enables a deeper dive into each individual that we refer to as a N-of-1 aggregates analysis19. The effectiveness of this approach in resolving microbiome-metabolome signatures in health and disease has been well validated with the human Arivale cohort19,20,21. The Arivale cohort is a continuation of the human pilot study. In the pilot study, the Pioneer 100, 108 individuals were sufficient to define important correlational families in cross-omic and phenotypic datasets19. In a similar manner, our study has generated high-density datasets for over 100 individual horses, with the intent of defining molecular signatures for MetS and, through this, improving the molecular classification of MetS sub/endotypes.
Using data generated by the P100HHP, we aimed to explore host and microbiota-directed dysregulation of metabolic homeostasis to improve clinical designation and management of MetS. We hypothesized that MetS will segregate into sub/endotypes, termed metabotypes, that are defined by specific metabolomic signatures. We further hypothesized that metabolome-microbiome integration would allow for the detection of host, microbiome, and environmental directionality underlying MetS in horses.
Results
Study design and cohort description
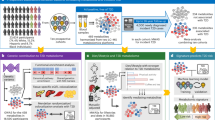
This study used data generated from the P100HHP cohort, a longitudinal and deeply phenotyped population of horses (n = 108) living at a single research facility (Fig. 1). This population included horses of both sexes and seven major breed groups (Supplementary Data 1). Individuals were prospectively phenotyped by body system, with a detailed sample repository generated.

Animals were actively selected into the cohort from the resident population of horses at the UC Davis Center for Equine Health. Animals were required to meet age, breed, and sex ratio selection prior to entry into the study. The minimum amount of data collection for animals included in the study requires a high coverage whole-genome sequence (WGS) and an accessible medical record. Animals underwent extensive prospective phenotyping by body system and sample collection for biorepository creation. Disease phenotypes that occur spontaneously or were known at entry in the study are also documented. Created in BioRender. Donnelly, C. (2025) https://BioRender.com/s82ldgd.
Individuals were repeatedly evaluated for the cluster of phenotypic traits associated with equine MetS: insulin dysregulation (ID), pituitary pars intermedia dysfunction (PPID), and obesity. Briefly, individuals were longitudinally evaluated for insulin regulation by oral sugar test (OST), baseline adrenocorticotropin hormone (ACTH), and thyrotropin-releasing hormone stimulation test (TRH-stim), body condition scoring and body weight measurement (see Methods). ID was defined as a fasting insulin of >20 μIU/mL or a post sugar administration plasma insulin of >45 μIU/mL at 60- or 90-min. Individuals were considered MetS if they were ID in 2/3 OST-matched metabolomic datasets. If an individual was ID periodically (Spring or Fall) in <2 OST, they were designated as seasonally MetS (SMetS). This more typically occurred in the Fall than Spring. Animals that remained insulin-regulated at all time points were considered metabolically healthy. Animals were considered affected by equine PPID based on results of TRH-stimulation testing and suggestive physical phenotype (hypertrichosis, retrobulbar fat deposition, and muscle atrophy), in accordance with current diagnostic guidelines22. An animal was regarded as obese if the body condition score (BCS) was >7/9 and overweight if >6/923.
Individuals were also phenotyped for other, non-MetS health-related phenotypes. In concert with phenotype assessment, contemporaneous samples were collected for evaluation of plasma metabolome and fecal microbiota (Methods). As part of the entry requirement for individuals into the study, whole-genome sequence data and electronic medical record data had already been collected (Supplementary Data 2).
Fecal microbiota is stable in horses irrespective of insulin regulation
Horses were sampled over a period of 2 years (2020–2021) to determine fecal microbiota by 16S rRNA sequencing of the V3–V4 region. This included 418 individual samples across the participating horses in the study. Fecal samples were not used if horses had received antimicrobials within 30 days of the sampling window. Metabolic phenotypes did not influence differences in the microbiota composition of horses in this study. Grouping by combined ID phenotypes (SMetS and MetS; “EMS”) compared to regulated individuals demonstrated stable α- and β-diversity across the study period (Fig. 2). In horses managed in a single environment with a consistent diet, fecal microbiota based on 16S rRNA sequencing remained largely stable over time.

A Violin plot of mean plasma deoxycholic acid (DCA) concentrations across the three sampling times grouped by regulated or insulin dysregulated (SMetS and MetS combined), showing median and range (P = 0.038). B Violin plot of mean plasma taurochendeoxycholic acid (TCDCA) concentrations across the three sampling times grouped by regulated or insulin dysregulated (SMetS and MetS separate), showing median and range (P = 0.039). C Dendrogram of hierarchical clustering based on outlier analysis of metabolites. More insulin dysregulated individuals segregated in clade 1, and more insulin-regulated individuals segregated in clade 2. Animals were classed as MetS (ID; red), SMetS (Seasonal; blue) or Regulated (green). D Heatmap of the top 20 metabolites used to cluster individuals by phenotype. E Box and whisker plots of Shannon diversity index across all sample time points, grouping by insulin-regulated and dysregulated, demonstrating at a community level, no differences in alpha diversity were detected by metabolic phenotype. F Box and whisker plots of Chao 1 diversity index across all sample time points, grouping by insulin-regulated and dysregulated demonstrating at a community level no differences in sample richness were detected by metabolic phenotype.
Metabolites segregate with MetS
To characterize the metabolite communities of individuals, the mean metabolite concentrations across all time points were evaluated by outlier analysis (see Methods). Twenty-four metabolites, with five individuals/metabolite outliers, were selected for further analysis. The mean number of outliers per individual was 22 ± 47. One individual had considerably more metabolite outliers (354 metabolites) as compared to the rest of the cohort. After initially segregating alone, this individual was not included in further analysis. Unsupervised hierarchical clustering of outliers by Euclidean distance identified two major clades in the data (Fig. 2). These major clades matched insulin regulation states, with a more insulin-regulated clade and a more ID clade. In the MetS group, 85% of individuals segregated to one clade and 82% of regulated individuals clustered to the opposing major clade. SMetS individuals were intermediary between the extreme ends, with half occurring in each of the major clades. Segregating major clades was not driven by breed (P = 0.3), age (P = 0.2) or sex (P = 0.4). Mean body condition scores (relative adiposity) were significantly higher for the more ID clade (mean ± SD; 6.7 ± 0.9), as compared to the more insulin-regulated clade (5.9 ± 0.8; P = 0.0003). Similarly, MetS individuals had a higher mean BCS (7.1 ± 0.9) as compared to regulated individuals (5.8 ± 0.7; P = 0.0007), regardless of segregating clade. SMetS individuals did not separate from MetS (P = 0.4) nor from regulated (P = 0.05) individuals based on BCS. Metabolite outliers were closely associated with the overlapping phenotypes of obesity and/or ID for MetS and regulated individuals. The data from the outlier analysis demonstrated unique metabotypes in conjunction with ID that are independent of population substructure, including breed, age, and sex.
We next evaluated the data for metabolite classes that were driving subclusters to define metabotypes. Two major subclusters emerged, one driven primarily by MetS individuals and the other by SMetS individuals. The MetS predominant subcluster was dominated by monoarachidonic triglyceride (TG) species, ranging in length from 14 to 18 carbons, and could be summarized as an overabundance of unsaturated TGs. The second major subcluster was defined by the increased abundance of primary bile acid and bile acid conjugates (Supplementary Data 3, 4).
MetS are defined by triglyceride metabolite classes
To identify differences between regulated and MetS individuals, we applied generalized linear models to each of the three collected time points. Similar to the previous outlier analysis, triglycerides defined individuals with MetS. This was most prominent in Fall 2020, with 49 triglyceride species enriched in individuals with MetS. Monoarachidonic triglyceride species, ranging in size from 15 to 18 carbons, predominate and summarily represent an overabundance of unsaturated triglycerides.
Bile acids segregate with a seasonal effect on MetS
We also explored the association between SMetS and metabolites using a similar approach. Here, we found a larger number of changes associated with the SMetS phenotype. For Fall 2020, we found a larger number of changes in bile acids and conjugates (Fig. 2). The Spring 2021 time point also showed changes in bile acids, as well as a large number of triglycerides. The Fall 2021 time point did not have any significant changes in metabolites associated with the SMets phenotype.
Seasonal association of fecal microbiota and bile acids with MetS
The overall abundance and diversity of microbiota was stable across the sampling period. (Supplementary Data 5–8). Two important signals were detected when the microbiota was evaluated at both the seasonal and phyla level. A significant enrichment for Clostridium Sensu Stricto 1 (log2 FC = 2.19, Padj = 0.02) was detected for Spring 2021. Further, a positive correlation was detected for taurochenodeoxycholic acid (TCDCA) and the proportion of Proteobacter (Fig. 3).

A Plasma concentration of TCDCA positively correlates with increased abundance of bacteria from the Proteobacter phyla (Pearsons r = 0.253. P = 0.035). B Enrichment analysis of metabolite pathways for significantly differentially abundant metabolites in MetS individuals. C Network analysis for the top differentially abundant metabolites in SMetS individuals. D Box and whisker plots of delta BCS prediction for individuals by group. TCDCA taurochenodeoxycholic acid, BCS body condition score.
Biologic body condition score models predict adiposity
Metabolic body mass index (BMI) has been shown to distinguish phenotypic states of host metabolism in human studies24. Using the metabolic data, we trained machine learning models to predict BCS, an equine equivalent to BMI. Due to the small sample size and BCS group size, we applied leave-one-out cross-validation using the least absolute shrinkage and selection operator (LASSO) algorithm to the mean metabolite data and seasonal data (Supplementary Data 8). The mean metabolite model reached an r2 of 0.21. The prediction of the seasonal models varied due to inconsistent BCS group sizes (Supplementary Data 9). Interestingly, the delta prediction (BCS measured – BCS predicted) showed differences between SMetS and MestS with regulated individuals (Fig. 3). Increasing deviation from predicted BCS may be a strategy for identification of individuals in transition to SmetS or MetS and warrants further investigation.
Genetic diversity correlates with insulin regulation phenotype
The three largest breed groups within the cohort (American Quarter Horse, Thoroughbred and Warmblood) were evaluated for genetic diversity by determining runs of homozygosity (ROH) from whole-genome sequence data. Thoroughbreds had the longest ROH (mean \(\pm \,\)SD; 148, 464\(\pm\)24,497 base pairs), with Quarter Horses having the shortest (81,141\(\pm\)11,509 base pairs; Fig. 4A). Correlation analysis of ROH with the mean insulin response across all phenotyping events showed a striking inverse relationship between length of ROH and plasma insulin concentration. Lower ROH length strongly correlated with increased plasma insulin concentrations at baseline and following oral sugar challenge (r = −0.283-[−0.402], P < 0.0001; Fig. 4B). Notably, the correlation coefficient for ROH and plasma insulin paralleled the correlation between body condition score (BCS) and insulin (r = 0.46–0.53, P < 0.0001).

A Box and Whisker plot of runs of homozygosity (ROH). Horses from the thoroughbred breed group had the highest across-genome ROH (mean ± SD; 148,464 ± 24,497 bp). The other two major groups, Quarter Horse (81,141 ± 11,509 bp) and Warmblood (85,629 ± 15,418 bp), had approximately half the ROH length as Thoroughbreds. The small number of Thoroughbred x Quarter Horse crossed horses demonstrated an intermediary ROH. B Heatmap of Pearson correlation coefficients for ROH, insulin and BCS/ All correlations were significant (P < 0.0001). QH quarter horse, WBxQH warmblood cross quarter horse, TB thoroughbred, TBxQH thoroughbred cross quarter horse, WB warmblood, ROH runs of homozygosity, bp base pairs, BCS body condition score.
Discussion
For the first time, we have demonstrated the use of an N-of-1 aggregates approach to defining metabotypes for MetS in horses. Particularly, we demonstrated that MetS in horses segregate into two predominant metabotypes; a triglyceride (TG) dominant and a bile acid (BA) dominant metabotype. Further, we demonstrated that metabolomics could predict relative adiposity in horses. Our data show that deep phenotyping, in concert with multi-omic analysis, can be as effective in nonhuman species as they are in humans. Lastly, our data suggest that horses are a useful model of spontaneous MetS that recapitulates the phenotypes and underlying molecular drivers of the analogous human syndrome.
Defining MetS in horses by distinct subtypes is an important step toward improving the diagnosis and treatment of the syndrome. This has not been a focus of previous metabolomic studies, where the predominant aim has been a single metabolite biomarker or metabogenomic marker discovery25,26,27,28,29. Previous studies have been hampered by one or more limitations, including small sample size, single breed investigation and single time point sampling26,27,28,29. The current study was specifically designed to address these limitations and to detect metabolomic signatures that are biologically reflective of MetS and MetS subtypes.
Dyslipidemia, particularly hypertriglyceridemia, is strongly associated with insulin resistance and type 2 diabetes in humans30. We demonstrated that enrichment for unsaturated TGs defines a subcluster that is dominated by MetS individuals and, to a lesser degree, SMetS or regulated individuals. Various unsaturated TGs distinguished MetS from SMetS and regulated horses. Outlier analysis indicated that specific dysregulated TGs represent MetS better in some individuals than total TGs. Additionally, this relationship is not linear, with the severity of hypertriglyceridemia not associated with the severity of ID, merely that the two are likely to exist together. In support of this finding, the individual removed from the group analysis had a total unsaturated TGs 20 times higher than that of the next highest horse, yet had a normal fasted insulin and mildly unhealthy postprandial response. In a previous study of obese horses not phenotyped for ID, untargeted lipidomic profiles identified highly complex obesity associated signatures.29 However, due to individuals coming from multiple farms (environments), clustering in that particular study was highly biased. In individuals phenotyped for MetS from a single environment, untargeted lipidomics identified many dysregulated lipid species, with a predominance of TGs. Collectively, the current study confirms that TGs are associated non-linearly with the MetS phenotype. We add to the existing known metabolites and lipids associated with MetS, and we suggest that TG identifies a metabotype within the MetS spectrum. Further, measurement of total TGs may not be sufficient to capture the nature of dysregulation in MetS individuals. Therefore, the development of assays for specific TGs may improve diagnosis and targeted therapy for equine MetS.
Bile acid (BA) mediated pathways are highly associated with the development of MetS in humans31. To date, the effect of BAs, as they relate to equine MetS, has not been demonstrated. Through their interaction with the Farnesoid X receptor (FXR) and Takeda G protein receptor 5 (TGR5), BAs have direct and indirect roles on insulin secretion, gluconeogenesis, glycogen synthesis, energetic regulation, and inflammation in multiple organs31. Although total bile acids are routinely measured in horses, compositional analysis has rarely been reported. From a limited sample number, Washizu et al previously determined that taurochenodeoxycholic acid (TCDCA) and taurocholic acid (TCA) are the major components (>80%) of bile acid in horses32. Our data confirms that these tauro-cojugated species are the primary components of equine plasma bile acids. Additionally, we also identified the secondary bile acids taurolithocholic acid (TLCA) and deoxycholic acid (DCA). This finding may be due to improved analytic sensitivity to detect lower abundance BAs. Importantly, the relative and absolute abundance of primary BAs segregated with MetS and SMetS. We directly show that reduced postprandial tolerance of glucose is associated with elevated primary BAs in both MetS and SMetS animals. Mirroring that of human MetS, higher relative abundance of primary versus secondary BAs promotes insulin resistance33. Our data suggest that BA signaling may be a permissive factor in the development of MetS in horses and that alterations in the BA profile may, in part, explain horse’s periodicity of ID in SMetS. Although further study is required, compositional BA analysis may indicate animals at risk of developing ID and subsequently MetS. In the current study, TCDCA concentrations were more closely correlated with insulin than that of TGs. This relationship is expected, as BAs have a direct effect on glucose homeostasis, whereas TGs are both a consequence and indirect reason forID33. Primary BAs through interaction with their canonical receptors, including Farnesoid X receptor, may directly impact insulin secretion and hepatic gluconeogenesis33. Importantly, we present a new avenue for MetS diagnostic and therapeutic research in the horse with specific reference to BA dysregulation. Additionally, this feature elevates the horses as a naturally occurring model of MetS for translational studies, as it recapitulates the human syndrome with an additional layer of fidelity33.
Bile acid regulation is emblematic of the interaction between the host and microbiota. Microbial-derived secondary bile acids are well characterized in humans34 but have yet to be evaluated in horses. While lower in overall abundance, the secondary BA, DCA, tended to segregate with more metabolically normal individuals. DCA is the product of cholic acid metabolism, primarily from bacteria of the Clostridium genus35,36. DCA directly promotes insulin sensitivity and is under investigation as a supplement for humans with MetS37. Importantly, the relationship between BAs and the microbiota is bidirectional, with one affecting the other. To date, defining the microbiota population in obese and MetS horses has been challenging. Again, efforts have been hampered by a small sample size and single sampling opportunities. On a microbiota population level, the current study does not differ greatly from previous studies, with no differences in diversity indices between MetS and regulated individuals26. The current study does have the advantage of being able to specifically detect individual shifts in microbiota over time. In conjunction with BA dysregulation, we show that there is a relationship with Clostridiales, insulin regulation and seasonality, at least at the family level. Clostridial bacteria are responsible for the conversion of primary to secondary BAs, with higher primary BAs potentially explained by the lower abundance of Clostridium35,36. Additionally, we demonstrate that, with increased levels of primary BA, the microbiota becomes richer in Proteobacter. Proteobacter enterotypes have been associated with antibiotic use and diarrhea in horses and are thought to represent a dysbiosis shift38,39. Indeed, with antibiotic use, there is widespread reduction of Firmicutes, which include the family Clostridiales38. Conversely, not all Firmicutes promote glucose homeostasis. Rumminococcaceae have been highly and consistently associated as a pathobiont in the development of type 2 diabetes in humans40. Ruminococcaceae are a major constituent of the equine microbiota due to the high cellulose content of their diets18. Typically, this species is associated with a healthy microbiota41. Within a certain range, this is likely still the case, but, in the current study, we suggest that when Rumminococcaceae predominates and falls out of regulation with other Firmicutes, such as Lachnospiraceae, it is associated with reduced glucose tolerance.
The inverse relationship between heterozygosity and insulin regulation was an unexpected finding of this study. The impact of genetics on MetS is complex, with individual genes contributing variably to the development of the syndrome. It would therefore be expected that increased and not decreased homozygosity would concentrate deleterious variants that could drive the development of features of the syndrome. Although little is known about the effects of inbreeding in horses on body composition, humans with higher levels of inbreeding are leaner42,43. As such the relationship between ROH and insulin may a surrogate to the potential effects that inbreeding has on body condition score. This study includes a snapshot of horse breeds, with homozygosity only evaluated in the largest breed subsets. As such, the complexity of genetic variation and susceptibility will require evaluation of a more diverse array of precisely phenotyped horses.
Collectively, we demonstrate a relationship between the microbiota, BA regulation and the development of phenotypes associated with equine MetS. Although this finding is promising, it must be tempered with the limitations of the techniques used. Without metagenomic and/or metatranscriptomic approaches, species identity and the necessary array of genes responsible for BA metabolism cannot be fully determined. Follow-up evaluation using techniques that allow more precise designation of microbiota constituents at the genus or species-level is required to validate the current findings. Similarly, while it is a strength of this study that all individuals reside at one institution, it also limits external validity. Evaluation of additional individuals from different environments will be needed to verify the generatability of these findings amongst horses with and without MetS. Bile acid regulation represents the convergence of host and microbial metabolism. As such, it will be key to establish if BA associations with MetS are intrinsic to host metabolism, if they are a product of the microbial community or are a synergism between both mechanisms in a wider cohort of horses.
The complex relationship between the host, microbiota and environment have been difficult to capture in the context of equine MetS. These data are the first to show that there is a potential microbiota-metabolome crosstalk that promotes a tendency to MetS. With our systems biology approach, we have been able to leverage a smaller sample size through deep phenotype and multi-omic evaluation. Our data suggest that microbial interventions, such as probiotics or dietary modifications that promote beneficial bacterial populations, could influence bile acid profiles and mitigate the progression of MetS in both horses and humans. Future studies establishing the direct mechanisms of bile acid signaling are needed to develop new therapies around this proposed pathophysiologic mechanism.
Methods
Animals
All procedures in this experiment were performed in accordance with the institutional care and use committee guidelines for the University of California, Davis (Protocol #21700). Animals used in this experiment are a part of the Pioneer 100 Horse Health Project (P100HHP). Age was used as the primary selection criteria for entering the study, with a median age of 12 years (yrs) for females (range 2–18 yrs), and a median age of 9 yrs for males (range 2–18 yrs). Horses included in the cohort have been resident at UC Davis Center for Equine Health (UCD-CEH) for a median of four years (range 0–17 yrs) at the time of inclusion in the study. Age and time in residence at UCD-CEH were positively correlated (R = 0.60, P = 6.08 × 10−12). A maximum age of 15 years, unless the animal had a unique or important phenotypic trait that was of interest to the wider goals of the P100 project were enrolled. Included in this cohort are 60 females (entire N = 52, ovariectomized (OVX) N = 8) and 48 males (entire N = 3, orchidectomized = 45). Breed was a secondary selection criterion, with seven breed groups represented in the P100HHP cohort and encompassing 20 recognized breeds (Supplementary Data 1). Body condition scoring (BCS) was performed as part of the mandatory deep phenotype protocol according to Henneke during the Spring of 2020 and 202123. All animals were scored by two veterinary experts, with the consensus score recorded as the adiposity phenotype. Animals additionally had contemporaneous still photographs and videos at walking and trotting gaits recorded for verification of BCS and lameness phenotypes.
Endocrine phenotypes
Animals were phenotyped in Spring (May) and Fall (Oct-Nov) of 2020 and 2021. During spring phenotyping, insulin regulation and pituitary pars intermedia dysfunction (PPID) phenotypes were determined at the same sampling event according to ref. 44. One horse included in the cohort had laboratory and clinical exam findings consistent with PPID. Insulin regulation was evaluated by oral sugar test (OST) as previously described44. Horses were acclimated to stalls and fasted overnight. Baseline serum and plasma samples were obtained between 6 a.m. and 7 a.m. for all horses in both Spring and Fall. In the Spring sampling, horses were administered 1 mg of thyrotropin-releasing hormone (TRH) intravenously, with plasma sampled ten minutes following administration. Immediately following the TRH-stimulation test, 0.15 mL/kg Lite Karo syrup was orally administered. Plasma and serum samples were obtained 60 and 90 min following oral sugar administration. In the Fall sampling, only OST was performed. All blood samples were immediately placed on ice and processed into plasma or serum (by centrifugation) within 30–60 min of sampling. Samples were stored at −80 °C until analysis and analyzed within 6 months of collection.
Plasma samples at baseline and at 10-min post-TRH stimulation were analyzed for adrenocorticotropin releasing hormone (ACTH) using a chemiluminescent immunometric assay validated for use in horses (Diagnostic Products Cor, Los Angeles, CA, USA)45. Plasma insulin concentration was measured at baseline, 60- and 90-minutes post OST. Insulin concentrations were determined by an equine validated immunoradiometric assay (Immuno-Biological Laboratories, Minneapolis, MN, USA)46.
Targeted metabolomics
Metabolomic analysis was performed on heparinized plasma samples obtained at the same time as endocrine phenotype samples for a subset of individuals (n = 72). Three time points were evaluated for each individual, spanning 1 year, with samples taken in November 2020, May 2021, and October-November 2021. Samples were analyzed using the Biocrates Quant500 (Biocrates Life Sciences AG, Innsbruck, Austria) quantitative targeted metabolomic panel. This panel includes 630 directly measured metabolites/lipids and 234 calculated values and indices. All samples were analyzed at the Center for Metabolomics, Institute of Metabolic Diseases, Baylor Scott & White Research Institute.
Fecal microbiota
Fecal samples were collected at five time points; May 2020 (n = 68), November 2020 (n = 82), March 2021(n = 93), May 2021 (n = 89), and October/November 2021(n = 86). Free catch samples were used. Only samples where passage was observed were retained. Samples were immediately placed on ice and stored for transport and were transferred to −80 °C within 3 h of collection. Sample genomic DNA extraction and 16S rRNA sequencing was performed by Laragen Inc (Culver City, CA, USA). Paired-end sequencing of the V3–V4 region of the 16S domain was performed, with a target read length of 250 base pairs and a targeted depth of 100,000 reads per sample. All samples were sequenced on an Illumina MiSeq platform.
16S data preprocessing
Sample fastqs were processed using a custom pipeline previously described (https://doi.org/10.1038/s42255-021-00348-0). In short, samples were processed using DADA2 to generate amplicon sequence variants (ASV) for each sample. Forward and reverse ASVs were merged, followed by chimaera removal using DADA2 (14.67% of reads). Taxonomic assignment of ASVs was done using the RDP method in DADA2 with the SILVA database (version 138.2). The taxonomic classification for Family was 96.5%, Genus = 64% and Species = 0%. Considering the low species-level classification, it was not considered for further analysis. Finally, a phyloseq object was generated from the resulting ASV abundance tables and taxonomy classifications47. Log abundance values were used for correlation with metabolites.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Statistics and reproducibility
Metabolic statistical analysis
Prior to analysis, we performed QC on the metabolomics data using standard principal component analysis (PCA). One sample was dropped from the analysis, as it provided 95% percent of the variance in the first principal component. All other samples were kept in the analysis, which were preprocessed first by dropping metabolites with greater than 15% NA values. The remaining metabolites were then imputed using the median value of the non-missing samples. For each metabolite, linear regressions were run using the LIMMA R package48. P values were adjusted using the Benjamini–Hochberg correction to control for the false-discovery rates. Each regression used either the binary MetS or SMetS response variable and included age, sex, and breed as covariates.
For biological body condition score prediction, metabolic data were further standardized with z-score using the mean and standardization of each metabolite. Predicted body condition scores were generated using LASSO regression from the glmnet R package and leave-one-out cross-validation. Values were predicted for each of the available seasons and the MetS or SMETs response variable. The difference between the predicted value and the measured value was calculated and compared using a Student’s t-test. The same analysis was also performed using age as a response variable, but did not result in a significant difference.
Whole-genome sequencing
Venous blood was acquired by jugular venipuncture into EDTA vacutainers. Whole blood was used to isolate DNA using the Promega Wizard Genomic DNA Kit. Concentration and ds260/280 absorbance were measured using a Qiagen Qiexpert. A minimum concentration of 50 ng/μL of DNA with a 260/280 ratio of 1.8–1.9 was submitted for whole-genome sequencing. DNA fragmentation was evaluated by gel electrophoresis using 1.5% agarose, with an adjusted concentration of 50 ng/μL for each animal. Libraries were prepared by Novogene. Libraries were sequenced on an Illumina NovaSeq with 150 bp pair-end reads with a target depth of 20X.
Whole-genome data processing
Raw sequences were trimmed to remove adapters and low-quality bases using TrimGalore49 and Cutadapt50 and subsequently aligned to the reference genome, EquCab3.051, using BWA MEM52 with default parameters. Aligned reads were filtered using SAMTools53 to remove duplicates and multimapping reads.
Whole-genome population structure
Variants were called using GATK (v4.1.9.0) HaplotypeCaller and GenotypeGVCFs following best practice for germline variant calling54. Called variants were filtered using VCFtools and GATK VariantFiltration (v4.1.9.0) to retain only single-nucleotide variants with depth-normalized quality scores over 2 (QD >2) and minor allele frequencies over 0.1 (MAF >0.1)55. The final variant set was then used to calculate eigenvectors and eigenvalues for all samples using PLINK(v1.90)56. The same variant set was also used to estimate runs of homozygosity (ROH) using BCFtools57. Results of PCA and ROH were visualized using matplotlib and seaborn56,57.
Change history
23 February 2026
In this article the grant number “D21EQ-050” relating to Morris Animal Foundation was omitted. The original article has been corrected.
References
Durham, A. E. et al. ECEIM consensus statement on equine metabolic syndrome. J. Vet. Intern. Med. 33, 335–349 (2019).
Ahmed, M. et al. Metabolic syndrome; definition, pathogenesis, elements, and the effects of medicinal plants on it’s elements. J. Diabetes Metab. Disord. 21, 1011–1022 (2022).
Thatcher, C. D., Pleasant, R. S., Geor, R. J. & Elvinger, F. Prevalence of overconditioning in mature horses in southwest Virginia during the summer. J. Vet. Intern. Med. 26, 1413–1418 (2012).
Flegal, K. M., Kruszon-Moran, D., Carroll, M. D., Fryar, C. D. & Ogden, C. L. Trends in obesity among adults in the United States, 2005 to 2014. JAMA 315, 2284–2291 (2016).
Ojanen, X. et al. Towards early risk biomarkers: serum metabolic signature in childhood predicts cardio-metabolic risk in adulthood. EBioMedicine 72, 103611 (2021).
Hyötyläinen, T. in: A Systems Biology Approach to Study Metabolic Syndrome (eds Orešič, M. & Vidal-Puig, A.) (Springer, 2014).
Fromentin, S. et al. Microbiome and metabolome features of the cardiometabolic disease spectrum. Nat. Med. 28, 303–314 (2022).
Hu, X. et al. Alterations of gut microbiome and serum metabolome in coronary artery disease patients complicated with non-alcoholic fatty liver disease are associated with adverse cardiovascular outcomes. Front. Cardiovasc. Med. 8, 805812 (2022).
Vallianou, N. et al. Understanding the role of the gut microbiome and microbial metabolites in non-alcoholic fatty liver disease: current evidence and perspectives. Biomolecules 12, 56 (2021).
Hild, B. et al. Neonatal exposure to a wild-derived microbiome protects mice against diet-induced obesity. Nat. Metab. 3, 1042–1057 (2021).
Diener, C. et al. Baseline gut metagenomic functional gene signature associated with variable weight loss responses following a healthy lifestyle intervention in humans. mSystems 6, e0096421 (2021).
Sonnenburg, J. L. & Sonnenburg, E. D. Vulnerability of the industrialized microbiota. Science 366, eaaw9255 (2019).
Clemente, J. C. et al. The microbiome of uncontacted Amerindians. Sci. Adv. 1, e1500183 (2015).
Pasolli, E. et al. Extensive unexplored human microbiome diversity revealed by over 150,000 genomes from metagenomes spanning age, geography, and lifestyle. Cell 176, 649–662.e20 (2019).
Rosshart, S. P. et al. Wild mouse gut microbiota promotes host fitness and improves disease resistance. Cell 171, 1015–1028.e13 (2017).
Rosshart, S. P. et al. Laboratory mice born to wild mice have natural microbiota and model human immune responses. Science 365, eaaw4361 (2019).
Orlando, L. Ancient genomes reveal unexpected horse domestication and management dynamics. Bioessays 42, 1900164 (2020).
Kauter, A. et al. The gut microbiome of horses: current research on equine enteral microbiota and future perspectives. Anim. Microbiome 1, 14 (2019).
Price, N. D. et al. A wellness study of 108 individuals using personal, dense, dynamic data clouds. Nat. Biotechnol. 35, 747–756 (2017).
Wilmanski, T. et al. Gut microbiome pattern reflects healthy ageing and predicts survival in humans. Nat. Metab. 3, 274–286 (2021).
Wilmanski, T. et al. Blood metabolome predicts gut microbiome α-diversity in humans. Nat. Biotechnol. 37, 1217–1228 (2019).
Equine Endocrinology Group. https://sites.tufts.edu/equineendogroup/ (2022).
Henneke, D. R., Potter, G. D., Kreider, J. L. & Yeates, B. F. Relationship between condition score, physical measurements and body fat percentage in mares. Equine Vet. J. 15, 371–372 (1983).
Watanabe, K. et al. Multiomic signatures of body mass index identify heterogeneous health phenotypes and responses to a lifestyle intervention. Nat. Med. 29, 996–1008 (2023).
Delarocque, J. et al. Metabolic changes induced by oral glucose tests in horses and their diagnostic use. J. Vet. Intern. Med. 35, 597–605 (2021).
Coleman, M. C., Whitfield-Cargile, C. M., Madrigal, R. G. & Cohen, N. D. Comparison of the microbiome, metabolome, and lipidome of obese and non-obese horses. PloS one 14, e0215918 (2019).
Delarocque, J. et al. Metabolic profile distinguishes laminitis-susceptible and -resistant ponies before and after feeding a high sugar diet. BMC Vet. Res. 17, 56 (2021).
Patterson Rosa, L., Mallicote, M. F., Long, M. T. & Brooks, S. A. Metabogenomics reveals four candidate regions involved in the pathophysiology of equine metabolic syndrome. Mol. Cell. Probes 53, 101620 (2020).
Elzinga, S., Wood, P. & Adams, A. A. Plasma lipidomic and inflammatory cytokine profiles of horses with equine metabolic syndrome. J. Equine Vet. Sci. 40, 49–55 (2016).
Grundy, S. M. Hypertriglyceridemia, insulin resistance, and the metabolic syndrome. Am. J. Cardiol. 83, 25F–29F (1999).
Shapiro, H., Kolodziejczyk, A. A., Halstuch, D. & Elinav, E. Bile acids in glucose metabolism in health and disease. J. Exp. Med. 215, 383–396 (2018).
Washizu, T., Tomoda, I. & Kaneko, J. J. Serum bile acid composition of the dog, cow, horse and human. J. Vet. Med. Sci. 53, 81–86 (1991).
Wu, T. et al. Effects of taurocholic acid on glycemic, glucagon-like peptide-1, and insulin responses to small intestinal glucose infusion in healthy humans. J. Clin. Endocrinol. Metab. 98, E718–E722 (2013).
Ramírez-Pérez, O., Cruz-Ramón, V., Chinchilla-López, P. & Méndez-Sánchez, N. The role of the gut microbiota in bile acid metabolism. Ann. Hepatol. 16, s15–s20 (2017).
Figueiredo, G., Gomes, M., Covas, C., Mendo, S. & Caetano, T. The unexplored wealth of microbial secondary metabolites: the Sphingobacteriaceae case study. Microb. Ecol. 83, 470–481 (2022.
Doden, H. L. et al. Completion of the gut microbial epi-bile acid pathway. Gut Microbes 13, 1–20 (2021).
Stacpoole, P. W. The pharmacology of dichloroacetate. Metabolism 38, 1124–1144 (1989).
Shin, N. R., Whon, T. W. & Bae, J. W. Proteobacteria: microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 33, 496–503 (2015).
Costa, M. C. et al. Comparison of the fecal microbiota of healthy horses and horses with colitis by high throughput sequencing of the V3-V5 region of the 16S rRNA gene. PLoS ONE 7, e41484 (2012).
Gurung, M. et al. Role of gut microbiota in type 2 diabetes pathophysiology. EBioMedicine 51, 102590 (2020).
Julliand, V. & Grimm, P. HORSE SPECIES SYMPOSIUM: the microbiome of the horse hindgut: history and current knowledge. J. Anim. Sci. 94, 2262–2274 (2016).
Fareed, M. & Afzal, M. Evidence of inbreeding depression on height, weight, and body mass index: a population-based child cohort study. Am. J. Hum. Biol.26, 784–795 (2014).
Fatma, R., Chauhan, W. & Afzal, M. Assessment of inbreeding depression on morphometric traits among North Indian population cohorts. Discov. Public Health 21, 14 (2024).
Hodge, E. et al. Effect of thyrotropin-releasing hormone stimulation testing on the oral sugar test in horses when performed as a combined protocol. J. Vet. Intern. Med. 33, 2272–2279 (2019).
Beech, J., McFarlane, D., Lindborg, S., Sojka, J. E. & Boston, R. C. α-Melanocyte-stimulating hormone and adrenocorticotropin concentrations in response to thyrotropin-releasing hormone and comparison with adrenocorticotropin concentration after domperidone administration in healthy horses and horses with pituitary pars intermedia dysfunction. J. Am. Vet. Med. Assoc. 238, 1305–1315 (2011).
Kerrigan, L. E., Thompson, D. L. Jr, Chapman, A. M. & Oberhaus, E. L. Effects of epinephrine, detomidine, and butorphanol on assessments of insulin sensitivity in mares. J. Equine Vet. Sci. 85, 102842 (2020).
McMurdie, P. J. & Holmes, S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8, e61217 (2013).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Krueger, F. (2015). Trim galore. A wrapper tool around Cutadapt and FastQC to consistently apply quality and adapter trimming to FastQ files (2015).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 10–12 (2011).
Kalbfleisch, T. S. et al. Improved reference genome for the domestic horse increases assembly contiguity and composition. Commun. Biol. 1, 197 (2018).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Van der Auwera, G. A. & O’Connor, B. D. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra 1st edn (O’Reilly Media, 2020).
Danecek, P. etr et al. 1000 Genomes Project Analysis Group, the variant call format and VCFtools. Bioinformatics ume 27, 2156–2158 (2011).
Purcell, S. et al (2007) PLINK: a toolset for whole-genome association and population-based linkage analysis. Am. J. Hum. Genet. 81, 559–575 (2007).
Narasimhan, V. et al. BCFtools/RoH: a hidden Markov model approach for detecting autozygosity from next-generation sequencing data. Bioinformatics 32, 1749–1751 (2016).
Acknowledgements
Funding for this study provided by Morris Animal Foundation (D21EQ-050).
Author information
Authors and Affiliations
Contributions
C.G.D., C.J.F. and N.D.P. designed and supervised experiments. J.M. and M.C. contributed to phenotype analysis. S.P., L.P., N.R. and C.G.D. performed all data analysis and interpretation. C.G.D. wrote the manuscript. All authors discussed the results, revised the draft manuscript, and read the approved final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Tanja Hess and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Tobias Goris. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Donnelly, C.G., Peng, S., Pflieger, L. et al. Bile acids segregate metabolic syndrome in a cohort of 100 deeply phenotyped horses. Commun Biol 8, 1711 (2025). https://doi.org/10.1038/s42003-025-09111-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-09111-7
