
Abstract
Small cell lung cancer (SCLC) is highly lethal, often developing rapid chemo-resistance and progression after initial response to platinum-based chemotherapy. The role of DNA methylation in driving this progression remains poorly understood. Here, genome-wide methylation profiling via methylated DNA immuno-precipitation sequencing (MeDIP-seq) reveals hypermethylated and hypomethylated regions in both SCLC tissue DNA and plasma cell free DNA (cfDNA). A hypermethylated region in the promoter of RASA4 gene, common to SCLC tumor DNA and progression-associated cfDNA, is identified. Pharmacologic and, more specifically, targeted demethylation reactivate RASA4 expression in SCLC. The down-regulation of RASA4, a negative regulator of Ras signal, leads to the activation of Ras-MAPK pathway in SCLC. RASA4 overexpression inhibits SCLC growth, invasion and chemo-resistance, whereas its knockdown promotes these malignant phenotypes by enhancing epithelial-mesenchymal transition (EMT) and stemness. We further demonstrate that RASA4 negatively regulates the EMT-related protein SERPINE2, and directly interacts with it. Immunohistochemistry analysis of clinical specimens validates that RASA4 is significantly down-regulated in SCLC tumors, and its low expression correlates with SCLC patients’ poor survival. Taken together, the epigenetic silencing of RASA4 drives SCLC progression through the induction of EMT and stemness. Our findings underscore the potential of RASA4 as a diagnostic biomarker and therapeutic target.
Similar content being viewed by others
Introduction
Globally, lung cancer is the most prevalent malignant tumor and the leading cause of cancer-related mortality1. Small cell lung cancer (SCLC) constitutes approximately 10–15% of all lung cancer cases, and is the most lethal subtype with a very low 5-years survival rate of <7%2. SCLC is characterized by neuroendocrine features, high invasiveness, and rapid doubling time, which contribute to its strong tendency for early and widespread metastasis3. Platinum-based chemotherapy is the first-line treatment for SCLC. However, despite initially dramatic responses, most of the SCLC patients develop drug resistance and recurrence quickly within 3–6 months4. Therefore, there is an unmet need for SCLC to discover new molecular targets.
Epigenetic modifications, particularly DNA methylation, play a crucial role in regulating transcriptional programs of cancer cells, thereby promoting an aggressive phenotype5. However, it is poorly understood how DNA methylation alteration promotes the progression and chemo-resistance in SCLC. Using SCLC primary tissues and cell lines, the first genome-wide DNA methylation analysis by CpG islands microarray revealed a large number of methylated genes, some of which were functionally enriched in neuronal differentiation6. By analyzing drugs responses and DNA methylations in 66 SCLC cell lines, a study has identified multiple significant correlations between DNA methylation changes and chemo-sensitivity in SCLC7. Since SCLC patients rarely undergo surgery, adequate tissue samples are seldom obtained for DNA methylation sequencing8. Recent studies utilized circulating free DNA (cfDNA) from SCLC patients’ plasma to profile the methylome. Using an enrichment-based methylation sequencing approach (T7-MBD-seq), the cfDNA methylome of SCLC patients was profiled. By integrating tumor-specific methylome from SCLC patients-derived mice models, the study further constructed an accurate model for classifying SCLC and its subtypes by cfDNA methylome9. Using reduced-representation bisulfite sequencing (RRBS) of tumor tissue DNA, a study developed a highly accurate DNA methylation-based classifier for SCLC subtypes. When applied to cfDNA, this classifier not only provided a non-invasive alternative to tissue biopsy but also revealed dynamic shifts in subtypes throughout disease progression10. Another study demonstrated that subtracting peripheral blood leukocyte methylation signatures improved tumor specificity of cfDNA methylome in SCLC11. These findings indicate that the dynamic changes of the DNA methylome with SCLC progression could be reflected by the plasma cfDNA methylome. Therefore, to identify progression-associated DNA methylation alterations, we compared the cfDNA methylomes from serial samples taken before treatment and at progression in SCLC.
Bisulfite sequencing is widely regarded as the gold standard for studying genome-wide DNA methylation. However, bisulfite modification results in considerable DNA degradation, potentially leading to the loss of critical methylation signals, particularly for the low-abundance cfDNA12. In the study, a bisulfite-free technique, methylated DNA immunoprecipitation and high-throughput sequencing (MeDIP-seq) was employed to enrich methylated DNA fragments by anti-5-methylcytidine antibody11,13. To find SCLC-specific methylated DNA sites, differentially methylated regions (DMRs) in tissue genomic DNA between SCLC tumor and adjacent normal tissues were identified. To find tumor progression-associated DNA methylated sites, DMRs in plasma cfDNA of SCLC patients between pre-treatment and progression were identified. Integration of DMRs from the two comparisons revealed frequent promoter methylation of the RASA4 gene, which encodes a Ca2+-dependent Ras GTPase-activating protein. In SCLC, the promoter methylation of RASA4 inhibited its expression, and conversely activated the Ras-MAPK pathway, thereby promoting cellular proliferation, invasion, and chemo-resistance by the enhanced epithelial-mesenchymal transition (EMT) and stemness. Using clinical specimens, RASA4 was validated to be down-regulated in SCLC tumors, which correlated with poor survival of SCLC patients. In summary, the study suggests that RASA4 could be a potential therapeutic target and prognostic biomarker for SCLC.
Results
The genome-wide DNA methylation profiling in plasma cfDNA and tissue DNA of SCLC
To investigate the global change of DNA methylation associated with chemotherapy resistance and disease progression in SCLC, we collected plasma cfDNA samples at pre-treatment and progression from two extensive stage (ES) SCLC patients, and conducted MeDIP-seq on these cfDNA samples. After removing the PCR duplication, average sequence reads of 26.1 million (approximately 4G data/sample) were obtained from all samples, 83.6% of which was mapped to the human reference genome (GRCh37/hg19). To identify DNA methylation regions associated with SCLC, we collected formalin-fixed and paraffin-embedded (FFPE) samples from six patients with limited-stage (LS) SCLC who underwent surgery. Genomic DNA was extracted from both tumor and adjacent normal tissues for MeDIP-seq. MeDIP-seq generated average sequence reads of 17.8 million (approximately 2G data/sample) in all FFPE samples, 87.3% of which was mapped to the human reference genome (GRCh37/hg19).
To characterize the progression-associated DNA methylation profile, we compared the DMRs of cfDNA between pre-treatment and progression. The screening criteria were set at P < 0.05 and |LogFC | ≥1 to generate volcano plots. In the whole genome level, we identified 13,619 DMRs with increased methylation levels in the cfDNA of SCLC with progression, and 11,462 DMRs whose methylation levels were decreased (Fig. 1A). By a same threshold (P < 0.05 and |LogFC | ≥1), an analysis of DMRs between tumors and adjacent normal tissues identified 29,641 hypermethylated DMRs and 66,421 hypomethylated DMRs in SCLC tumors (Fig. 1A). The visual DMR signals of hypomethylation and hypermethylation mapped to whole genome were presented in Fig. 1B. We examined the genomic distribution of both hypomethylated and hypermethylated DMRs, and found a considerable fraction of DMRs located in intergenic regions and introns. The SCLC tumor tissues exhibited a dramatic hypomethylation (Fig. 1C).

A Volcano plot of DMRs across the whole genome in plasma cfDNA between pre-treatment and progression from ES-SCLC patients (left panel). Volcano plot of the DMRs across the whole genome in tissue DNA between tumors and adjacent normal tissues from LS-SCLC patients (right panel). B Distribution of the DMRs on chromosomes. The red areas represent the hypermethylated regions, and the blue areas represent the hypomethylated regions. C The distribution of genomic locations for the DMRs. D Volcano plot of DMRs within the promoter regions in plasma cfDNA between pre-treatment and progression from ES-SCLC patients (left panel). Volcano plot of the DMRs within the promoter regions in tissue DNA between tumors and adjacent normal tissues from LS-SCLC patients (right pannel). E GO and KEGG analysis of genes associated with the DMRs within the promoter regions in tissue DNA. F GO and KEGG analysis of genes associated with the DMRs within the promoter regions in cfDNA.
Aberrant DNA methylation in the promoter of a gene results in abnormal gene expression. In promoter regions, volcano plots showed that 1635 DMRs were hypermethylated in the cfDNA of SCLC with progression, and 1600 DMRs were hypomethylated (Fig. 1D). In SCLC tumor tissues, 1632 hypermethylated DMRs and 16,836 hypomethylated DMRs located within promoter regions (Fig. 1D). To understand the biological functions of the genes associated with the DMRs in promoter regions, we performed Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis. In SCLC tissue DNA, differentially methylated genes were enriched in GTPase activity, Ras protein signal transduction and so on (Fig. 1E). In plasma cfDNA with SCLC progression, differentially methylated genes were enriched in embryonic development, channel activity, axon guidance, tight junction and so on (Fig. 1F). Therefore, these results reveal the global change of DNA methylation in plasma cfDNA and tissue DNA, which may be associated with SCLC pathogenesis and progression.
Frequent promoter methylation of RASA4 in SCLC
To identify epigenetically regulated genes that associated with chemo-resistance and disease progression in SCLC, we intersected the DMRs among the three comparisons (tumors VS adjacent normal tissues, cfDNA at pre-treatment VS cfDNA at progression in patient #1, cfDNA at pre-treatment VS cfDNA at progression in patient #2) (Fig. 2A). 154 DMRs were shared among the three comparisons. The 154 DMRs were associated with 142 genes (Supplementary Data 1). KEGG enrichment analysis of the 142 genes revealed significant enrichment in the MAPK and Ras signaling pathways as well as SCLC (Fig. 2B). Among the 154 DMRs, 31 DMRs located within the promoter region, and 2 DMRs of which exhibited consistent methylation changes in cfDNA and tissue DNA. The 2 DMRs were associated with 2 genes, RASA4 and CTRB2. RASA4 is particularly notable for its function in regulating MAPK and Ras signaling.

A The workflow of the integrated analysis of MeDIP-seq data of plasma cfDNA and tissue DNA. B KEGG analysis of genes associated with the 154 shared DMRs. C Methylation peaks of the DMR (Chr7: 102254100-102244400) within the RASA4 promoter in plasma cfDNA and tissue DNA, as determined by MeDIP-seq. D The DMR (Chr7: 102,254,100-102,244,400) contains binding sites for transcription factors TFAP2A and RUNX1, predicted in the JASPAR database. E BSP analysis of the CpG sites in the DMR (Chr7: 102254100-102244400) within the RASA4 promoter in 10 paired SCLC tumor and adjacent normal tissues. The data are shown as mean ± SD from 10 SCLC patients (n = 10, paired Student’s t-test).
In plasma cfDNA of SCLC patients with progression, a genome browser snapshot of MeDIP-seq signals across the RASA4 gene indicated the increased methylation levels in the identified DMR (chr7:102254100-102254400) within the RASA4 promoter. In the genomic DNA of SCLC tumors, a genome browser snapshot of MeDIP-seq signals indicated that the methylation levels of the DMR (chr7:102254100-102254400) were higher in SCLC tumor tissues, compared with adjacent normal tissues (Fig. 2C). By aligning with the JASPAR database14, the DMR (chr7:102254100-102254400) was predicted to contain binding sites for the transcription factors TFAP2A and RUNX1 (Fig. 2D).
To validate the MeDIP-seq data, we performed high-resolution analysis of the methylation status of 6 CpG sites within the DMR (chr7:102254100-102254400) by bisulfite sequencing PCR (BSP). The results indicated that the methylation frequency of 6 CpG sites within the DMR (chr7:102254100-102254400) was significantly higher in FFPE tumor tissues from 10 SCLC patients, compared to adjacent normal tissues (Fig. 2E). These results demonstrate that the promoter methylation of RASA4 frequently occurs in SCLC.
RASA4 expression is repressed by promoter CpG methylation in SCLC
To further demonstrate whether promoter CpG methylation in the RASA4 gene silences its expression, we performed pharmacological demethylation in SCLC cells. Firstly, we detected RASA4 expression in eight SCLC cell lines (H82, H69, H526, H446, SBC-2, DMS114, SHP77, and DMS273) and in a normal human bronchial epithelial cell line, BEAS-2B. RASA4 expression was found to be lower in all SCLC cell lines compared to BEAS-2B cells. In SCLC cell lines, RASA4 expression was lower in H82, H526, DMS114, SHP77 and DMS273 cells (Fig. 3A). Then, we treated SCLC cells (H82, H526, DMS114 and SHP77) with the demethylating agent 5-aza-2′-deoxycytidine (5-Aza) at doses of 5 µM and 10 µM for 72 h. The quantitative reverse transcription-PCR (qRT-PCR) and western blot analyses demonstrated a significant increase in both mRNA and protein levels of RASA4 in SCLC cells treated with 5-Aza (Fig. 3B, C). To further evaluate the methylation-dependent regulation of RASA4 expression, we performed targeted demethylation of the DMR (Chr7: 102254100-102254400) within the RASA4 promoter using the dCas9-Tet1 system. The results indicated that co-transfection with dCas9-Tet1/sgRNA complex targeting the specific DMR (Chr7: 102254100-102254400) significantly reduced methylation levels at the target CpG sites in both DMS114 and SHP77 cells, compared to control cells expressing dCas9-Tet1 alone. Concordantly, RASA4 mRNA expression was significantly increased in DMS114 and SHP77 cells following targeted demethylation of the DMR (Chr7: 102254100-102254400) by dCas9-Tet1/sgRNA (Fig. 3D). These results indicate that the methylation of the RASA4 promoter silences its expression in SCLC.

A Western blot analysis of RASA4 expression in SCLC cell lines (H82, H69, H526, H446, SBC-2, DMS114, SHP77, and DMS273) and the normal bronchial alveolar epithelial cell line (BEAS-2B). B qRT-PCR shows that the treatment of 5-Aza increases the transcription of RASA4 mRNA in H82, H526, SHP77, and DMS114 SCLC cells. Data are shown as mean ± SD from three independent biological replicates (n = 3, unpaired Student’s t-test). C Western blot shows that the treatment of 5-Aza increases the expression of RASA4 protein in H82, H526, SHP77, and DMS114 SCLC cells. D Targeted demethylation of the DMR (chr7:102254100-102254400) within the RASA4 promoter by dCas9-Tet1 and sgRNA. BSP analysis indicates a significant decrease in methylation levels at the target CpG sites in both DMS114 and SHP77 cells following co-transfection with vectors expressing dCas9-Tet1 and the sgRNA targeting the specific DMR (Chr7: 102254100-102254400). qRT-PCR indicates that RASA4 mRNA expression is significantly increased in DMS114 and SHP77 cells following targeted demethylation of the DMR (Chr7: 102254100-102254400) by dCas9-Tet1 and sgRNA. For the BSP analysis, data are shown as mean ± SD from six independent DNA clones (n = 6, unpaired Student’s t-test). For the qRT-PCR analysis, data are shown as mean ± SD from three independent biological replicates (n = 3, unpaired Student’s t-test). E Western blot shows that the treatment of 5-Aza inhibits the phosphorylation of MEK and ERK in H526 SCLC cells. F Ras activation assay shows that knockdown of RASA4 increases Ras activity in SBC-2 SCLC cells. The overexpression of RASA4 decreases Ras activity in SHP77 SCLC cells. G Western blot shows that knockdown of RASA4 increases the phosphorylation of MEK and ERK in SBC-2 SCLC cells. The overexpression of RASA4 decreases the phosphorylation of MEK and ERK in SHP77 SCLC cells.
The silence of RASA4 activates Ras-MAPK signal in SCLC
Previous studies have indicated that RASA4 functions as a negative regulator of Ras signal15. After treatment of H526 cells with 5-Aza, Ras downstream signals (p-MEK and p-ERK) were inhibited (Fig. 3E). Next, we knocked down and overexpressed RASA4 in SCLC cells. In SBC-2 cells with stable RASA4 knockdown, we observed increased Ras activity and elevated phosphorylation levels of MEK and ERK (Fig. 3F, G). Conversely, stable RASA4 overexpression in SHP77 cells decreased Ras activity and the expression of phosphorylated MEK and ERK (Fig. 3F, G). These results indicate that RASA4 is a negative regulator of the Ras-MAPK signaling pathway in SCLC.
RASA4 inhibits the malignant phenotype in SCLC
Next, to indicate whether RASA4 inhibits the malignant phenotype, RASA4 was overexpressed or knocked down in SCLC cells. When RASA4 were stably over-expressed in DMS114 and SHP77 cells, the ability of cellular proliferation and colony formation was significantly reduced (Fig. 4A, B). Matri-gel invasion assay indicated that RASA4 overexpression significantly inhibited the cellular invasion in DMS114 cells. In SBC-2 cells with stable RASA4 knock-down, the ability of cellular invasion was significantly increased when compared to the scramble shRNA-transfected SBC-2 cells (Fig. 4C). Wound healing assay revealed that RASA4 over-expression in DMS114 cells reduced the migratory ability, while stable RASA4 knock-down in SBC-2 cells increased the migratory ability (Fig. 4D). Furthermore, to indicate the effect of RASA4 on chemo-treatment, we examined the IC50 values of carboplatin and etoposide in SCLC cells with stable RASA4 knock-down and over-expression. RASA4 knockdown in SBC-2 cells resulted in an increase of the IC50 values for carboplatin and etoposide, compared to the scramble shRNA-transfected SBC-2 cells (Fig. 4E). Overexpression of RASA4 in SHP77 cells resulted in a decrease of the IC50 values for carboplatin and etoposide, compared to the vector-transfected SHP77 cells (Fig. 4E).

A Cell viability assay shows that the stable over-expression of RASA4 inhibits the proliferation in SHP77 and DMS114 cells. Data are shown as mean ± SD from three independent biological replicates (n = 3, unpaired Student’s t-test). B Colony formation assay shows that the ability of colony formation of SHP77 and DMS114 cells with stable over-expression of RASA4 is reduced. The colonies were quantified and shown by mean ± SD from three biologically independent experiments (n = 3, unpaired Student’s t-test). C Matri-gel invasion assay shows that the stable over-expression of RASA4 inhibits the invasion in DMS114 cells. The stable knockdown of RASA4 increases the invasion in SBC-2 cells. The invaded SCLC cells were quantified and shown by mean ± SD from three biologically independent experiments (n = 3, unpaired Student’s t-test). (scale bar = 200 μm) (D) Wound healing assay shows that the stable over-expression of RASA4 inhibits the migration in DMS114 cells. The stable knockdown of RASA4 increases the migration in SBC-2 cells. Wound closure was quantified as a percentage of the initial wound area. Data are shown as mean ± SD from three biologically independent experiments (n = 3, unpaired Student’s t-test). (scale bar = 200 μm) (E) The growth inhibition curves demonstrate that stable RASA4 over-expression sensitizes SHP77 cells to etoposide and carboplatin, respectively, as evidenced by decreased IC50 values, whereas RASA4 knock-down conversely increases the IC50 values in SBC-2 cells. Data are shown as mean ± SD from three independent biological replicates. F Subcutaneous tumorigenesis assay in nude mice. SHP77 cells with stable transfection of the empty vector and the RASA4-expressing vector were subcutaneously injected into nude mice. The growth curves show the tumor volume over a period of 6 weeks. Upon euthanasia at the end of the experiment, tumors were dissected and weighed. Images of tumors from each group were taken. Data are shown as mean ± SD from 5 mice for each group (n = 5, unpaired Student’s t-test). G H&E and Ki67 IHC staining in SHP77 xenografts stably transfected with empty vector or RASA4-expressing vector. (scale bar = 40 μm for 20×, scale bar = 200 μm for 4×) (H) The knockdown of RASA4 promotes the metastasis of SCLC cells. SBC-2 cells stably expressing scramble and RASA4 shRNA were intravenously injected into nude mice. After 6 weeks, the lungs of mice were dissected and photographed using an animal imaging system. Green fluorescence-expressing metastatic nodules were indicated. Each group comprised five mice injected with SBC-2 cells stably expressing scramble or RASA4 shRNA. Three mice in each group developed metastatic lung nodules, as observed during dissection. Metastatic lung nodules are quantified as mean ± SD from three mice (n = 3, unpaired Student’s t-test).
To further indicate the tumor-inhibitory effect of RASA4 in SCLC in vivo, we established subcutaneous xenografts using SHP77 cells with stable RASA4 overexpression. As shown Fig. 4F, over-expression of RASA4 led to a significant reduction in tumor size and weight, compared to the group inoculated by vector-transfected SHP77 cells. H&E and immunohistochemistry (IHC) staining indicated that the expression of proliferation marker Ki-67 was lower in SCLC tumors with stable RASA4 overexpression (Fig. 4G). To demonstrate the effect of RASA4 on SCLC metastasis, we further injected SBC-2 cells with stable RASA4 knockdown through the tail vein in nude mice. After 2 months of the injection, the mice were dissected. In the nude mice injected with SBC-2 cells with stable RASA4 knockdown, we observed more metastatic foci (as shown in the foci number and fluorescence intensity) in the lungs, compared to the mice injected with scramble shRNA-transfected SBC-2 cells (Fig. 4H). In summary, all these results indicate that RASA4 inhibits the malignant phenotype of SCLC in vitro and in vivo.
RASA4 inhibits EMT and stemness of SCLC cells by the inhibition of SERPINE2
To investigate the signaling pathways and biological processes regulated by RASA4 on the malignant phenotype of SCLC cells, we performed RNA sequencing (RNA-seq) of SBC-2 cells with RASA4 knockdown. The differential expression genes of SBC-2 cells with RASA4 shRNA and scramble shRNA transfection were shown in Fig. 5A and Supplementary Data 2. Pathway enrichment analysis of GO and KEGG indicated that the differential expression genes were primarily enriched in biological processes related to the “response to mechanical stimulus” (Supplementary Data 3). A major consequence of persistent activation of Ras signaling is the activation of downstream factors that drive EMT, which in turn promotes tumor metastasis16. EMT can be triggered by various mechanical stimuli, including shear stress and alterations in basement membrane stiffness17. A gene in the gene set of “response to mechanical stimulus” is SERPINE2 (Serpin Family E Member 2), which is a crucial regulator in the EMT signal pathway. SERPINE2 regulates extracellular matrix remodeling and promotes cell migration and invasion by inhibiting plasminogen activator18. RNA-seq data indicated that SERPINE2 was significantly up-regulated in SBC-2 cells with RASA4 knock-down (Fig. 5A). We further validated that mRNA expression of SERPINE2 was increased when RASA4 was knocked down in SBC2 and H446 cells, while mRNA expression of SERPINE2 was decreased when RASA4 was over-expressed in SHP77 and DMS114 cells (Fig. 5B). Enzyme-linked immunosorbent assay (ELISA) indicated that SBC2 and H446 cells with RASA4 knock-down secreted more SERPINE2 protein into extracellular culture medium (Fig. 5C). Intriguingly, immunofluorescence assay revealed that SERPINE2 protein was primarily expressed in the nucleus of SBC-2 cells. However, SERPINE2 protein was released to the cytoplasm and even the extracellular region when RASA4 was knocked down in SBC-2 cells. In H446 cells with RASA4 knock-down, immunofluorescence assay indicated that the expression of SERPINE2 protein was dramatically increased, compared to scramble-shRNA transfected H446 cells (Fig. 5D). Co-immunoprecipitation (Co-IP) showed that there was an interaction between RASA4 and SERPINE2 protein (Fig. 5E). By western blot, we observed a significant increase in SERPINE2 protein and mesenchymal markers (N-cadherin, Vimentin and Snail proteins) in SBC-2 cells with RASA4 knock-down. However, in DMS114 cells with RASA4 overexpression, the protein expression of SERPINE2 and mesenchymal markers (N-cadherin, Vimentin, and Snail proteins) were reduced (Fig. 5F). These findings suggest that the downregulation of RASA4 promotes the expression of SERPINE2 and its secretion to the extracellular region, thereby leading to the degradation of the extracellular matrix and cellular invasion.

A Volcano plot of differentially expressed genes in SBC-2 cells with stable RASA4 knockdown, relative to those transfected with scramble shRNA. B qRT-PCR shows that SERPINE2 mRNA levels were significantly lower in DMS114 and SHP77 cells with stable over-expression of RASA4, compared to cells stably transfected with an empty vector. SERPINE2 mRNA levels were significantly higher in H446 and SBC-2 cells with stable knockdown of RASA4, compared to cells stably transfected with scramble shRNA. Data are shown as mean ± SD from three independent biological replicates (n = 3, unpaired Student’s t-test). C ELISA analysis shows that H446 and SBC-2 cells with RASA4 knockdown secreted more SERPINE2 protein to cell culture medium, compared to cells stably transfected with scramble shRNA. Data are shown as mean ± SD from three independent biological replicates (n = 3, unpaired Student’s t-test). D Immunofluorescence staining of SERPINE2 protein in SBC-2 and H446 cells with stable transfections of scramble shRNA and RASA4 shRNA. (scale bar = 10 μm for SBC-2 cells, scale bar = 20 μm for H446 cells). E Co-IP analysis of protein interaction between RASA4 and SERPINE2 in DMS114 cells with RASA4 stable over-expression. Cell lysates from DMS114 cells with stable transfection of FLAG-tagged RASA4 were immunoprecipitated with anti-FLAG antibody, followed by western blot with anti-SERPINE2 antibody. IgG was used as a negative control. F Western blot shows that the protein expression of SERPINE2 and EMT markers (N-cadherin, E-cadherin, Vinmintin, and snail) following RASA4 stable knockdown and overexpression in SBC-2 and DMS114 cells. G qRT-PCR analysis of stemness markers NANOG, OCT4, SOX2, and MYC in SBC-2 and DMS114 cells with RASA4 stable knockdown and overexpression. Data are shown as mean ± SD from three independent biological replicates (n = 3, unpaired Student’s t-test). H Sphere formation assay in SBC-2 and H446 cells with RASA4 stable knockdown, and in DMS114 and SHP77 cells with RASA4 stable overexpression. (scale bar = 50 μm).
Previous studies have indicated that EMT is not only associated with the metastatic potential of SCLC, but also contributes to its resistance to treatment19. As EMT promotes invasiveness and stem cell-like features (stemness) of cancer cells, we further investigated whether RASA4 affects the stemness of SCLC cells. qRT-PCR indicated that the expression of classical stem cell markers, including SOX2, NANOG, OCT4, and MYC, were up-regulated in SBC-2 and H446 cells with stable RASA4 knock-down, compared to cells with stable scramble shRNA transfection. However, in DMS114 and SHP-77 cells with stable over-expression of RASA4, the mRNA levels of SOX2, OCT4, NANOG, and MYC were significantly down-regulated (Fig. 5G).
Furthermore, using the sphere formation assay, we assessed the stemness of SCLC cells with RASA4 knockdown and overexpression. The stable knockdown of RASA4 significantly augmented both the size and quantity of cell spheres formed in SBC-2 and H446 cells (Fig. 5H). However, the number and size of cell spheres formed in the SHP77 and DMS114 cells with stable over-expression of RASA4 were significantly diminished, compared to the control group (Fig. 5H). In summary, these data indicate that the downregulation of RASA4 promotes EMT and stemness properties of SCLC.
RASA4 is down-regulated in SCLC tumor tissues and correlates with poor clinical prognosis
To further investigate the clinical significance of RASA4 in SCLC, we conducted IHC staining using SCLC clinical samples. We collected FFPE specimens from 29 SCLC patients, including both SCLC tumor tissues and adjacent normal tissues (Supplementary Data 4). IHC staining showed that RASA4 protein expression was down-regulated in SCLC tumor tissues, compared with adjacent normal tissues. The IHC scores for RASA4 staining were evaluated in tumor tissues and adjacent normal tissues. The results indicated that the scores in tumor tissues were significantly lower than the scores in the corresponding normal tissues (Fig. 6A, B, E). We also performed IHC staining for SERPINE2 on FFPE specimens from the 29 SCLC cases. The results indicated that SERPINE2 expression levels in SCLC tumor tissues were significantly higher, compared with adjacent normal tissues (Fig. 6A, C, E). Interestingly, we found that SERPINE2 expression was elevated in invasive front of SCLC tumor, suggesting the positive association between SERPINE2 expression and SCLC invasion (Fig. 6D). IHC staining indicated that RASA4 and SERPINE2 exhibited significantly opposing expression patterns in tumor and adjacent normal tissues, with SERPINE2 being upregulated in tumor tissues and RASA4 predominantly upregulated in adjacent normal tissues (Fig. 6F). Based on RASA4 staining scores, SCLC patients were classified into weak and moderate groups for survival analysis. SCLC patients in the moderate group demonstrated improved overall survival after surgical treatment (Fig. 6G). Finally, we performed Cox regression analysis to assess the prognostic value of RASA4 staining scores along with clinical characteristics, including gender, age, smoking history, and pathological T and N stages. In both univariate and multivariate analyses, T stage and RASA4 staining scores were statistically significant, indicating them as independent risk factors for SCLC patient prognosis (Table 1). In summary, these clinical data demonstrates that low RASA4 expression is significantly associated with poor survival of SCLC patients, suggesting its potential as a SCLC prognostic biomarker.

A Representative images of H&E staining in tumor and adjacent normal tissues from FFPE specimens of three surgical SCLC patients. B IHC staining of RASA4 in tumor and adjacent normal tissues from FFPE specimens of the same three SCLC patients. The results show significantly reduced RASA4 expression in the tumor tissues compared to the adjacent normal tissues. C IHC staining of SERPINE2 in tumor and adjacent normal tissues from FFPE specimens of the same three SCLC patient samples. The results show significantly increased SERPINE2 expression in the tumor tissues compared to the adjacent normal tissues. (scale bar = 20 μm for 40×, scale bar = 100 μm for 8×) (D) IHC staining shows that SERPINE2 expression is elevated in the invasive front of the SCLC tumor. (scale bar = 40 μm for 20×, scale bar = 200 μm for 5×) (E) IHC scores of RASA4 and SERPINE2 in tumor and adjacent normal tissues from 29 SCLC patients who underwent surgery (n = 29, Mann–Whitney test). F Statistical analysis indicates the negative association between RASA4 and SERPINE2 expression in SCLC tumor and adjacent normal tissues (n = 29, Chi-square test for R × C contingency tables). G Kaplan–Meier survival analysis of 29 SCLC patients who underwent surgery, stratified by RASA4 IHC scores in tumor tissues. The results show that SCLC patients with moderate scores of RASA4 expression have significantly better survival compared to those with weak scores. The statistical significance of the difference in survival was determined by the log-rank test.
Discussion
In this study, through an integrated genome-wide methylation analysis of tissue DNA and blood cfDNA, we identified a promoter-methylated gene, RASA4, in SCLC. As a member of Ras GTPase activating proteins (RasGAP), epigenetic silencing of RASA4 leads to the abnormal activation of Ras signaling in SCLC. The down-regulation of RASA4 enhanced SCLC proliferation, invasion, and chemoresistance through the increased stemness and EMT. Furthermore, the study found a RASA4 negatively-regulated protein SERPINE2, which interacted with RASA4 in SCLC. Finally, the down-regulation of RASA4 was found to be associated with poor clinical prognosis in SCLC. Therefore, the study demonstrates that RASA4 is a potential target and biomarker in SCLC.
RAS mutations are frequently found in human cancers, especially for pancreatic cancer, colorectal cancer, non-small cell lung cancer, and melanoma etc20,21,22,23. Point mutations in the KRAS gene commonly result in its constitutive activation in a GTP-bound state, which in turn initiates downstream oncogenic pathways16,24. However, in human cancers with infrequent RAS mutations, alternative mechanisms are responsible for the aberrant activation of Ras signaling. Such mechanisms include the over-activation of upstream kinases, inactivation of GTPase-activating proteins, alterations in downstream Ras effectors, and regulation by non-coding RNAs25,26,27,28,29. Previous genomic analyses of SCLC revealed a near absence of RAS mutations30. Rubio et al. have reported that ITGB2 integrin receptor signaling activated Ras signaling in SCLC31. Zhang et al. have demonstrated that lncRNA ITPR1-AS1 was up-regulated in SCLC, and it promoted malignant phenotype through the activation of cRaf-MEK-ERK by the alternative transcript splicing of HRAS gene32. Wang et al have found a RhoGEFs family gene ARHGEF19, which was overexpressed in SCLC and promoted SCLC growth. ARHGEF19 interacted with HRAS and activated the MAPK/ERK pathway in SCLC33. Our study identified significant hypermethylation of the RASA4 promoter in SCLC. Therefore, the epigenetic inactivation of RASA4, a negative regulator of Ras, is a novel alternative mechanism of Ras signal activation in SCLC.
RasGAPs subfamily includes five members (RASA1, RASA2, RASA3, RASA4, and RASA5) and their loss often occurs in various cancers26. Kwon et al. have indicated that RASA1 mutations were found in 7% of the patients with gastric cancer, and correlated with distant metastasis. RASA1 loss enhanced Wnt signaling and stemness in gastric cancer34. The whole-exome sequencing identified the RASA2 gene, which was mutated in 5% of melanomas. Loss-of-function mutations in the RASA2 gene promoted melanoma cell growth and migration by activating RAS35. The study of RASA3 on cancer is limited. A previous study identified RASA3 as an oncogene that promoted migration and invasion by recruiting to focal adhesions and inactivating R-RAS, thereby increasing focal adhesion turnover36. RASA4 and RASA5 mutations were rare. Li et al. have found that promoter CpG methylation of RASA5 was frequently observed in various cancers, including esophageal cancer, nasopharyngeal cancer, and breast cancer. The epigenetic silence of RASA5 activated Ras signaling, thereby promoting malignant features37. Previous studies have found that RASA4 was down-regulated in some cancer types, such as cervical cancer and rectal cancers38,39. RASA4 hypermethylation correlated with adverse clinical outcomes in juvenile myelomonocytic leukemia40. Our results demonstrate that RASA4 is frequently silenced by promoter methylation in SCLC. It is plausible that RASA4 hypermethylation also occurs in other cancers, which warrants further investigation.
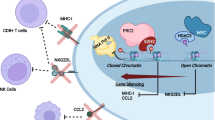
RNA-seq revealed that SERPINE2 was a downstream target of RASA4 in SCLC. Previous studies have indicated that SERPINE2 plays a crucial role in modulating extracellular matrix (ECM) dynamics by interacting with proteases that degrade ECM components41. Buchholz et al. have found that SERPINE2 overexpression enhanced the invasion of pancreatic cancer cells by altering ECM production and organization within the tumor42. Previous studies have established the promotive role of SERPINE2 in metastasis across various cancers. In liver cancer, SERPINE2 promoted metastasis by interacting and stabilizing EGFR, thus enhancing cell adhesion and ECM remodeling43. In esophageal cancer, SERPINE2 enhanced metastatic potential via BMP4-induced EMT44. Previous studies have indicated that elevated SERPINE2 was associated with tumor invasion and metastasis, underscoring its value as a poor prognostic biomarker in various cancer types, including urothelial carcinoma, oral squamous cell carcinoma, endometrial cancer, and osteosarcoma45,46,47,48. Our study identified an interaction between RASA4 and SERPINE2. Similarly, a recent study has also reported an interaction of KRAS and SERPINE149. Our findings suggest that RASA4 down-regulation enhances the secretion of SERPINE2 into the ECM, where it may contribute to the metastasis and invasion of SCLC. We propose that in RASA4-expressing cells, RASA4 interacts with SERPINE2, preventing its extracellular secretion, a model that requires further investigation.
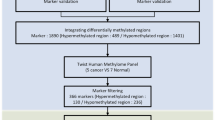
Due to the challenge of recruiting a sufficient number of SCLC patients for long-term monitoring, the inclusion of only two SCLC patients in the analysis of progression-associated cfDNA methylation represents a limitation for robust statistical evaluation. In order to minimize potential false discoveries and select reliable DMRs associated with SCLC progression, we analyzed the MeDIP-seq data through a series of filtering steps. (1) For plasma cfDNA from two SCLC patients (each with pre-treatment and progression samples), we conducted a paired comparison (progression vs pre-treatment) for each patient. This approach reduces the variability introduced by inter-patient differences. (2) To select the DMRs derived from tumor tissue, DMRs were screened by identifying the overlapping DMRs between tissue samples and plasma samples. (3) DMRs within promoter regions, which have the potential to regulate gene expression, were selected for further analysis. (4) DMRs-associated genes were functionally characterized through GO and KEGG enrichment analyses, which revealed significant enrichment in pathways including MAPK and Ras signaling. Furthermore, we validated the identified DMR using bisulfite sequencing PCR in 10 pairs of SCLC FFPE specimens. In addition, low RASA4 expression was also validated by IHC in 29 pairs of SCLC FFPE specimens. Taken together, these results indicate that RASA4 is identified through systematic enrichment rather than by random selection.
However, dynamic changes in RASA4 methylation in cfDNA throughout disease progression were not validated. In the future study, we propose to initiate a multi-center trial to recruit a sufficient cohort of SCLC patients. A methylation-specific PCR assay will be developed to enable sensitive quantification of RASA4 methylation in cfDNA. Using serial cfDNA samples collected throughout treatment, we will monitor dynamic changes in RASA4 methylation level. We will evaluate whether RASA4 methylation in cfDNA is associated with SCLC progression to assess its potential as a clinical biomarker.
In summary, our study reveals a novel epigenetic mechanism of RAS activation in SCLC. RASA4 is frequently silenced by promoter methylation in SCLC, thereby activating Ras signaling. The silence of RASA promotes SCLC proliferation and invasion through enhanced EMT and stemness. Using clinical specimens, our study indicates that low expression of RASA4 is a poor prognostic marker in SCLC, suggesting its potential as a diagnostic biomarker and therapeutic target.
Methods
Human specimens
FFPE specimens and blood samples of SCLC patients were collected under a protocol (2022-BE(H)-130) approved by the Ethics Committee of the First Affiliated Hospital of USTC (University of Science and Technology of China). Written informed consent to participate in the study was obtained from patients. All ethical regulations relevant to human research participants were followed.
FFPE specimens of paired SCLC tumors and adjacent normal tissues were collected from 35 LS-SCLC patients who underwent surgery at the First Affiliated Hospital of USTC between January 2011 and April 2022. All SCLC tumor and non-tumor tissue samples were confirmed by at least two experienced pathologists. Whole blood samples at pre-treatment and progression were collected from two ES-SCLC patients receiving chemotherapy at the First Affiliated Hospital of USTC from January 2021 to December 2022. Clinical data of the SCLC patients, including age, gender, staging, smoking, and follow-up information, were collected (Supplementary Data 4).
DNA extraction
For blood samples, plasma cfDNA was extracted. Briefly, 10 mL of peripheral blood was collected in EDTA anticoagulant tubes and processed within 4 h of collection. After the separation of plasma, cfDNA was extracted using the Circulating Nucleic Acids Kit (#55114, Qiagen, Hilden, Germany) according to the manufacturer’s instructions.
For FFPE samples, tissue genomic DNA was extracted. Briefly, 5–10 μm thick sections were used to extract tissue DNA by the GeneRead DNA FFPE Kit (#180134, Qiagen, Hilden, Germany) according to the manufacturer’s instructions.
For cell line samples, genomic DNA was extracted using the TIANamp Genomic DNA Kit (#DP304, Tiangen, Beijing, China) following the manufacturer’s instructions.
MeDIP-seq and DMRs analysis
The MeDIP-seq libraries were constructed according to previous studies50,51. Specifically, cfDNA samples were first subjected to end repair, A-tailing, and adapter ligation using the KAPA HyperPrep Kit (#KK8504, KAPA Biosystems, Boston, MA, USA) and NEBNext Multiplex Oligos (#E7335L, New England BioLabs, Ipswich, MA, USA). Methylated DNA immunoprecipitation was then performed with the MagMeDIP kit (#C02010021, Diagenode, Seraing, Belgium) to enrich methylated fragments. Finally, the immunoprecipitated libraries were amplified by PCR using KAPA HiFi Hotstart ReadyMix (#KK2602, KAPA Biosystems, Boston, MA, USA), and then sequenced to generate 150-bp paired-end reads on an Illumina Nova 6000 system (Illumina, San Diego, CA, USA). The quality and quantity of raw reads were examined using FastQC. Raw reads were trimmed using Trim Galore with default settings in paired-end mode. The trimmed reads were then aligned to the human reference genome hg19 using Bowtie 2. The R package MEDIP was used to quantify the counts within 300-bp sliding windows across the genome. The sequencing data for all 16 samples (6 FFPE tissue pairs and 2 plasma cfDNA pairs) were summarized in Supplementary Data 5.
To identify the DMRs, differential expression analysis of the counts was performed using the DESeq2 or edgeR package. DMR regions enriched in more than 45% of samples with counts >0 were included in the differential expression analysis for 6 paired FFPE samples. The coverage threshold for DMR calling was not set for 2 paired cfDNA samples. The DESeq2 package was used to analyze DMRs between SCLC tumor and adjacent normal tissues (n = 6 for each group), and DMRs of cfDNA from SCLC patients with progression versus pre-treatment (n = 2 for each group). The edgeR package was used for paired comparison (progression vs pre-treatment) for each patient. The exactTest function in edgeR was applied for the comparison between two groups without replicates, under the assumption of a negative binomial distribution with the dispersion estimation manually set to 0.05. For all differential expression analyses using DESeq2 and edgeR packages, all P values were adjusted using Benjamini–Hochberg FDR correction with a threshold set at FDR < 0.05.
Volcano plots were generated to visualize the DMRs. Reads per kilobase per million mapped reads (RPKM) was used to normalize the number of reads by deeptools and visualized using Integrative Genomics Viewer (IGV) tools.
BSP
Bisulfite conversion of DNA (1–2 µg) was performed with the EZ DNA Methylation Kit (#D5001, Zymo Research, Irvine, CA, USA), according to the manufacturer’s instructions. The bisulfite-converted DNA was purified using a kit (#DP214, Tiangen, Beijing, China). Nested PCR was performed to amply the methylation region of the RASA4 promoter using the primers listed in Supplementary Data 6. The PCR products were cloned into the T-vector (#TSV-007S, Tsingke, Shijiazhuang, China), and the colonies were randomly chosen for Sanger sequencing.
Cell lines
Human SCLC cell lines, H82, H69, H526, H446, DMS-114, SHP-77, and DMS-273 were obtained from ATCC (Manassas, VA, USA). SBC-2 cell line was purchased from HyCyte (#TCH-C432, Suzhou, China). BEAS-2B cells were obtained from Anhui Medical University. All cell lines were cultured in RPMI-1640 (Gibco, Carlsbad, CA, USA) containing 10% fetal bovine serum (FBS) (ExCell Bio, Shanghai, China) and 1× penicillin/streptomycin (Hyclone, Logan, UT, USA). All cells were maintained in a humidified incubator with 5% CO2 at 37 °C.
The generation of SCLC cells with stable RASA4 knockdown or overexpression
For SCLC cells with stable RASA4 knockdown, SBC-2 and H446 cells were used. The lentiviral particles containing scramble and RASA4 shRNA were obtained from GENERAL BIOL (Chuzhou, China). SBC-2 and H446 cells were infected by the lentiviral particles with 5 µg/ml polybrene overnight, followed to grow in fresh medium for 24 h. Then, puromycin (2 µg/mL for SBC-2 cells, 1 µg/mL for H446 cells) was added for selection. After 7–10 days of selection, puromycin-resistant cells were collected for subsequent validation of RASA4 knockdown and further experiments.
For SCLC cells with stable RASA4 overexpression, SHP77 and DMS114 cells were used. The RASA4 cDNA was cloned into the pCMV-3Tag expression vector. SHP77 and DMS114 cells were transfected with pCMV-3Tag empty vector and RASA4-expressing vector using Effectene Transfection Reagent (#301425, QIAGEN, Hilden, Germany). After 48 h of transfection, a fresh medium containing G418 (#345810, Sigma, St. Louis, MO, USA) at 800 µg/mL was used for selection. Individual G418-resistant colonies were selected and expanded, followed by validation of RASA4 overexpression and subsequent experiments.
Cell viability and colony formation assay
To detect the growth of SCLC cells, SCLC cells (1000 cells/well) were plated onto 96-well plates. After 1, 2, and 3 days, cell viability was measured by CellTiter-Glo Luminescent assay (#G7572, Promega, Madison, WI, USA).
To detect the growth inhibition of SCLC cells by drugs, SCLC cells (3000 cells/well) were seeded in 96-well plates. After 24 h, cells were treated with different concentrations of drugs for 72 h. After drug treatment, cell viability was evaluated by CellTiter-Glo Luminescent assay (#G7572, Promega, Madison, WI, USA).
For colony formation, SCLC cells were seeded in 6-well plates at a density of 500 cells per well and cultured for 12 days. Colonies were fixed with 4% paraformaldehyde and stained with 0.5% crystal violet.
Cell invasion and migration assay
To detect the ability of cell invasion, a Matrigel invasion assay was conducted. 1 × 10⁵ SCLC cells in serum-free medium were seeded onto the upper chamber of a Transwell insert (8 μm pore size, Corning, USA) pre-coated with Matrigel (#356231, BD Biosciences, Sparks, NJ, USA). The lower chamber was filled with complete medium. After 24 h of incubation, the invaded cells in the lower surface of the insert were fixed with 4% paraformaldehyde and then stained with 0.1% crystal violet. The invaded cells were observed using a microscope (Leica Microsystems, Wetzlar, Germany).
To detect the ability of cell migration, a wound healing assay was conducted. SCLC cells (5 × 10⁵) were seeded in 6-well plates, scratched with a pipette tip, and rinsed to remove detached cells. After incubation in serum-free medium, migration in the scratched area was imaged at 0 and 24 h. Migration distance was measured at five fixed points, and wound closure was quantified by comparing the initial and final images.
Ras activity
Ras activity was examined using a Ras Activation Assay Kit (#BK008-S, Cytoskeleton, Denver, CO, USA) according to the manufacturer’s protocol. Briefly, SCLC cells were lysed in ice-cold lysis buffer containing protease inhibitors. For each sample, 500 µg of total protein was incubated with the GST-tagged Ras-binding domain of Raf1, which specifically binds to GTP-bound active Ras. After incubation, active Ras was precipitated using glutathione-agarose beads, washed, and eluted in SDS sample buffer. The levels of active Ras were analyzed by Western blot with an anti-Ras antibody.
Immunofluorescence assay
SCLC cells were fixed with 4% paraformaldehyde for 20 min and then washed three times with PBS. Permeabilization was carried out with 0.5% Triton X-100 for 10 min, followed by blocking with 5% FBS and 2% bovine serum albumin for 1 h. The cells were incubated overnight at 4°C with the SERPINE2 polyclonal antibody (1:100 dilution, #11303-1-AP, Proteintech, Wuhan, China). After washing with TBST, the cells were incubated with Alexa Fluor 568 goat anti-mouse secondary antibody (1:1000 dilution, #A11004, Thermo Fisher Scientific, Waltham, MA, USA) for 40 min, followed by DAPI (#C0065, Solarbio, Beijing, China) staining for 5 min. Finally, the cells were mounted and imaged using a digital slide scanner (3DHISTECH, Budapest, Hungary).
Co-IP
Co-IP was performed to investigate RASA4 and SERPINE2 interaction. DMS114 cells with stable transfection of pCMV-3Tag-RASA4 were lysed using IP lysis buffer. 500 μg of protein per sample was used for immunoprecipitation. One aliquot was incubated with anti-Flag antibody (1:1000 dilution, #SLCR2991, Sigma, St. Louis, MO, USA), while the other one was incubated with IgG (1:1000 dilution, #A7016, Beyotime, Beijing, China). After incubation overnight at 4 °C, the protein was incubated with magnetic beads for 2 h at room temperature. Following washing, the beads were incubated with Flag peptide solution (#P9801, Beyotime, Beijing, China) at a concentration of 150 µg/mL for 1 h at room temperature. The eluted samples were mixed with SDS sample buffer for a western blot to detect SERPINE2 and Flag-RASA4 using anti-SERPINE2 antibody (1:1000 dilution, #ab154591, Abcam, Cambridge, MA, USA) and anti-Flag antibody (1:3000 dilution, #SLCR2991, Sigma, St. Louis, MO, USA).
ELISA
SERPINE2 concentration in cell culture medium was quantified using the Human Glia-Derived Nexin (SERPINE2) ELISA Kit (#CSB-EL021082HU, Cusabio, Wuhan, China) following the manufacturer’s protocol. Standards and samples were added to a microplate pre-coated with anti-SERPINE2 antibodies. After incubation, a biotin-conjugated secondary antibody and HRP-avidin were sequentially introduced, followed by the addition of TMB substrate for color development. After termination, absorbance was measured at 450 nm. SERPINE2 concentrations were calculated using a standard curve and normalized to the total protein content.
qRT-PCR
Total cellular RNA was extracted using the RNA extraction kit (#DP451, Tiangen, Beijing, China) following the manufacturer’s protocol. One-step reverse transcription was performed using the Reverse Transcription Kit (#AT301, TransGen, Beijing, China) according to the manufacturer’s protocol. The real-time PCR system (HealForce, Shanghai, China) was utilized to perform quantitative PCR. Relative gene expression was determined using the 2−∆∆Ct method, with β-actin as the internal control, and presented as mean ± SD relative to the control group. The specific primer sequences are listed in Supplementary Data 6.
Sphere formation assay
SCLC cells (1 × 105 cell/well) were seeded in 6-well ultra-low attachment plates with a stem cell-specific TSCM pellet medium (#P2401, QIDABIO, Shanghai, China). The cells were incubated for 10 days to allow spheroid formation. Spheroids were imaged using a microscope (Leica, Wetzlar, Germany). ImageJ (NIH, Bethesda, MD, USA) was employed to count spheroids with diameters ≥100 μm.
Western blot analysis
Cellular protein was extracted using cell lysis buffer (150 mM NaCl, 50 mM Tris-HCl (pH 8.0), 1% Triton X-100, 2 mM EDTA, 1 × Proteinase Inhibitor), and then quantified by a BCA assay (#P0009, Beyotime, Shanghai, China). 20 μg of protein sample was run for SDS-PAGE and then transferred to a PVDF membrane. Membranes were blocked in 5% nonfat milk for 2 h and probed with primary antibodies at 4 °C overnight, following by horseradish peroxidase-conjugated secondary antibodies at room temperature for 1 h. Blots were developed by using the chemiluminescence system (NcmECL Ultra, Nanjing, China).
The primary RASA4 and SERPINE2 antibodies were purchased from Abcam (#ab157190 and #ab154591, Cambridge, MA, USA) and used at dilutions of 1:1000 and 1:3000, respectively. Other primary antibodies were obtained from Cell Signaling Technology (Danvers, MA, USA), including Phospho-MEK1/2 (1:1000 dilution, #9154), MEK1/2 (1:1000 dilution, #8727), Phospho-p44/42 MAPK (Erk1/2) (1:2000 dilution, #4370), p44/42 MAPK (Erk1/2) (1:1000 dilution, #4695), N-Cadherin (1:1000 dilution, #13116), E-Cadherin (1:1000 dilution, #3195), Vimentin (1:1000 dilution, #5741), and Snail (1:1000 dilution, #3879). The β-actin antibody (1:10000 dilution, #HC201, TransGen Biotech, Beijing, China) was used as a loading control.
H&E and IHC staining
For H&E staining, paraffin-embedded tissue sections (4–5 µm) were deparaffinized, rehydrated, and stained with Mayer’s hematoxylin, followed by eosin Y counterstaining. Then, the sections were dehydrated, cleared in xylene, mounted, and imaged using a digital slide scanner (3DHISTECH, Budapest, Hungary).
IHC staining was carried out according to a previous study52. Specifically, paraffin-embedded tissue sections were baked at 60 °C for 3 h, deparaffinized in xylene, and rehydrated through a graded ethanol series (100%, 95%, 85%, and 75%; 5 min each). Antigen retrieval was then performed by heating the sections in citric acid buffer. After blocking endogenous peroxidases with a blocking solution (#A600332-0100, Sangon Biotech, Shanghai, China) for 30 min, the sections were incubated overnight at 4 °C in a humidified chamber with primary antibodies against RASA4 (1:100 dilution, #ab157190, Abcam, Cambridge, MA, USA) and SERPINE2 (1:100 dilution, #ab154591, Abcam, Cambridge, MA, USA). Followed by the incubation with a secondary antibody (1:100 dilution, #KIT-5020, MaxVision-HRP mouse/rabbit, Maxim, Fuzhou, China) for 30 min, signal detection was carried out using a DAB kit (ZLI-9018, Zhongshan Golden Bridge, Beijing, China), The sections were counterstained with hematoxylin, dehydrated, mounted, and imaged using a digital slide scanner (3DHISTECH, Budapest, Hungary). The results were evaluated based on staining intensity (0, 1+, 2+, and 3+) and the percentage of positive cells, categorized as follows: 0 (0%), 1 (1–25%), 2 (26–50%), 3 (51–75%), and 4 (76–100%). The final assessment score, based on a combination of the staining intensity with the percentage of staining cells, was categorized as follows: negative (0 score), weak (1–4 score), moderate (5–8 score), and strong (9–12 score).
RNA-seq analysis
Total cellular RNA was extracted using Trizol (#15596018CN, Thermo Fisher, Waltham, MA, USA). Quality of RNA was assessed by Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA) and NanoDrop (Thermo Fisher, Waltham, MA, USA). mRNA was enriched using the VAHTS mRNA Capture Beads (#N403, Vazyme, Nanjing, China). RNA-seq libraries were constructed with the VAHTS Universal V8 RNA-Seq Library Prep Kit (#NR605-01, Vazyme, Nanjing, China), and then sequenced on Illumina NovaSeq 6000 machines (Illumina, San Diego, CA, USA) to generate 150 bp paired-end reads. Raw reads were trimmed and aligned to the GRCh37 genome. Quantification was performed with RSEM (v1.3.1). Normalization and differential expression analysis were conducted using R package edgeR (v3.38.4) with gene count matrices.
In vivo xenograft models
The animal study was approved by the Institutional Animal Care and Use Committee of Hefei Cancer Hospital of the Chinese Academy of Sciences (DWLLPF-2024031001). We have complied with all relevant ethical regulations for animal use.
5-week-old female nude mice (Vital River Laboratory Animal Technology, Beijing, China) were inoculated subcutaneously with SCLC cells (2 × 106/100 μL medium) mixed with 100 μL of Matrigel (BD Biosciences, Franklin, NJ, USA). The tumor volume was measured twice weekly using calipers, with the following formula: V = (L×W×W)/2, where L is the longest diameter, and W is the shortest diameter. At the end of the experiment, the mice were euthanized by CO2 asphyxiation. The tumors were dissected and fixed in 4% paraformaldehyde for 24 h, subsequently subjected to H&E and IHC staining.
SBC-2 cells (1 × 106) with stable scramble-shRNA and RASA4-shRNA transfection were injected into the tail veins of nude mice. shRNA vector contains green fluorescent protein ZsGreen. After 60 days of injection, the mice were euthanized. The lungs were dissected, and metastatic nodules in both lungs were imaged using an in vivo imaging system (Biolight Biotechnology, Guangzhou, China).
Targeted demethylation by dCas9-Tet1 system
The small guide RNA (sgRNA: TGCCAAGGCCCTAAATATAG) was designed to target the DMR (chr7:102254100-102254400) within the promoter of the RASA4 gene using the CRISPOR online tool. The sgRNA was cloned to the LentiGuide-Puro vector. The plasmid (Fuw-dCas9-Tet1CD-P2A-BFP) expressing dCas9-Tet1 was obtained from Saicheng Biotechnology (Guangzhou, China). SHP77 and DMS114 cells were co-transfected with the dCas9-Tet1 expression plasmid with or without the LentiGuide-Puro vector expressing sgRNA using the Effectene Transfection Reagent (#301425, QIAGEN, Hilden, Germany). Cells were harvested 72 h post-transfection for BSP and qRT-PCR analysis.
Statistical analysis and reproducibility
R (v4.1.1) and GraphPad Prism9 (v9.0.0) were used for statistical analysis. Tests for differences between two groups were performed using a two-tailed Student t test or Mann-Whitney test. KEGG and GO enrichment analyses were used to identify the significantly enriched pathways of differentially expressed genes. Kaplan–Meier analysis was used to compare the difference in overall survival between the two groups.
The sample size (n) for each experiment was defined in the corresponding figure legend and represented the number of biologically independent samples, or biologically independent experimental replicates. Details regarding the number of replicates, error bars, and the statistical tests were described in the corresponding figure legends. P <0.05 was considered as statistical significance (P <0.05*, P <0.01**, P <0.001***, P <0.0001****). All numerical source data and exact P values are provided in the Supplementary Data 7.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data avaliability
All the data can be found in either the main text or the supplementary materials. MeDIP-seq raw data of SCLC samples and RNA-seq raw data of SBC-2 cells upon RASA4 knockdown have been deposited in the Genome Sequence Archive (GSA) in National Genomics Data Center, China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (GSA-Human: HRA014533, HRA014880) that are publicly accessible at https://ngdc.cncb.ac.cn/gsa-human. The source data behind the graphs in the paper can be found in Supplementary Data 7. Uncropped western blot images are provided in the Supplementary Information as Supplementary Fig. S1.
References
Sung, H. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA cancer J. clin. 71, 209–249 (2021).
Rudin, C. M., Brambilla, E., Faivre-Finn, C. & Sage, J. Small-cell lung cancer. Nat. Rev. Dis. Prim. 7, 3 (2021).
Megyesfalvi, Z. et al. Clinical insights into small cell lung cancer: tumor heterogeneity, diagnosis, therapy, and future directions. CA cancer J. clin. 73, 620–652 (2023).
Iams, W. T., Porter, J. & Horn, L. Immunotherapeutic approaches for small-cell lung cancer. Nat. Rev. Clin. Oncol. 17, 300–312 (2020).
Nishiyama, A. & Nakanishi, M. Navigating the DNA methylation landscape of cancer. Trends Genet. TIG 37, 1012–1027 (2021).
Kalari, S., Jung, M., Kernstine, K. H., Takahashi, T. & Pfeifer, G. P. The DNA methylation landscape of small cell lung cancer suggests a differentiation defect of neuroendocrine cells. Oncogene 32, 3559–3568 (2013).
Krushkal, J. et al. Epigenome-wide DNA methylation analysis of small cell lung cancer cell lines suggests potential chemotherapy targets. Clin. epigenetics 12, 93 (2020).
Meijer, J. J. et al. Small cell lung cancer: novel treatments beyond immunotherapy. Semin. cancer Biol. 86, 376–385 (2022).
Chemi, F. et al. cfDNA methylome profiling for detection and subtyping of small cell lung cancers. Nat. cancer 3, 1260–1270 (2022).
Heeke, S. et al. Tumor- and circulating-free DNA methylation identifies clinically relevant small cell lung cancer subtypes. Cancer cell 42, 225–237.e225 (2024).
Ul Haq, S. et al. Cell-free DNA methylation-defined prognostic subgroups in small-cell lung cancer identified by leukocyte methylation subtraction. iScience 25, 105487 (2022).
Gouil, Q. & Keniry, A. Latest techniques to study DNA methylation. Essays Biochem. 63, 639–648 (2019).
Corcoran, R. B. & Chabner, B. A. Application of cell-free DNA analysis to cancer treatment. N. Engl. J. Med. 379, 1754–1765 (2018).
Rauluseviciute, I. et al. JASPAR 2024: 20th anniversary of the open-access database of transcription factor binding profiles. Nucleic acids Res. 52, D174–d182 (2024).
Wang, Y., Qi, YX., Qi, Z. & Tsang, SY. TRPC3 regulates the proliferation and apoptosis resistance of triple negative breast cancer cells through the TRPC3/RASA4/MAPK pathway. Cancers 11, 558 (2019).
Shin, S., Dimitri, C. A., Yoon, S. O., Dowdle, W. & Blenis, J. ERK2 but not ERK1 induces epithelial-to-mesenchymal transformation via DEF motif-dependent signaling events. Mol. cell 38, 114–127 (2010).
Dong, Y. et al. Higher matrix stiffness as an independent initiator triggers epithelial-mesenchymal transition and facilitates HCC metastasis. J. Hematol. Oncol. 12, 112 (2019).
Monard, D. SERPINE2/Protease Nexin-1 in vivo multiple functions: does the puzzle make sense?. Semin. cell dev. Biol. 62, 160–169 (2017).
Yang, C. et al. EHMT2-mediated transcriptional reprogramming drives neuroendocrine transformation in non-small cell lung cancer. Proc. Natl. Acad. Sci. USA 121, e2317790121 (2024).
Waters, AM. & Der, CJ. KRAS: the critical driver and therapeutic target for pancreatic cancer. Cold Spring Harb. perspect. med. 8, a031435 (2018).
Tan, E. S. et al. Prognostic and Predictive Value of PIK3CA Mutations in Metastatic Colorectal Cancer. Target. Oncol. 17, 483–492 (2022).
Meng, D. et al. Prognostic value of K-RAS mutations in patients with non-small cell lung cancer: a systematic review with meta-analysis. Lung cancer 81, 1–10 (2013).
Bravo-Soto, G. A., Brañes, R., Peña, J. & Nervi, B. Palliative chemotherapy for advanced gallbladder cancer. Medwave 21, e8045 (2021).
Chen, Y., Liu, Q. P., Xie, H. & Ding, J. From bench to bedside: current development and emerging trend of KRAS-targeted therapy. Acta pharmacol. Sin. 45, 686–703 (2024).
Yarden, Y. & Pines, G. The ERBB network: at last, cancer therapy meets systems biology. Nat. Rev. Cancer 12, 553–563 (2012).
Maertens, O. & Cichowski, K. An expanding role for RAS GTPase activating proteins (RAS GAPs) in cancer. Adv. Biol. Regul. 55, 1–14 (2014).
Slack, F. J. & Chinnaiyan, A. M. The Role of Non-coding RNAs in Oncology. Cell 179, 1033–1055 (2019).
Stephen, A. G., Esposito, D., Bagni, R. K. & McCormick, F. Dragging Ras back in the ring. Cancer cell 25, 272–281 (2014).
Network. CGA Genomic classification of cutaneous melanoma. Cell 161, 1681–1696 (2015).
George, J. et al. Comprehensive genomic profiles of small cell lung cancer. Nature 524, 47–53 (2015).
Rubio, K. et al. Non-canonical integrin signaling activates EGFR and RAS-MAPK-ERK signaling in small cell lung cancer. Theranostics 13, 2384–2407 (2023).
Zhang, Q. et al. ITPR1-AS1 promotes small cell lung cancer metastasis by facilitating P21(HRAS) splicing and stabilizing DDX3X to activate the cRaf-MEK-ERK cascade. Cancer Lett. 577, 216426 (2023).
Wang, Z. et al. ARHGEF19 regulates MAPK/ERK signaling and promotes the progression of small cell lung cancer. Biochem. biophys. Res. Commun. 533, 792–799 (2020).
Kwon, J. W. et al. Combined inhibition of Bcl-2 family members and YAP induces synthetic lethality in metastatic gastric cancer with RASA1 and NF2 deficiency. Mol. cancer 22, 156 (2023).
Arafeh, R. et al. Recurrent inactivating RASA2 mutations in melanoma. Nat. Genet. 47, 1408–1410 (2015).
Li, H. et al. Phosphoinositide conversion inactivates R-RAS and drives metastases in breast cancer. Adv. Sci. 9, e2103249 (2022).
Li, L. et al. Tumor suppression of Ras GTPase-activating protein rasa5 through antagonizing ras signaling perturbation in carcinomas. iScience 21, 1–18 (2019).
Chen, J. et al. RASA4 inhibits the HIFα signaling pathway to suppress proliferation of cervical cancer cells. Bioengineered 12, 10723–10733 (2021).
Xiao, Y. et al. Overexpression of SYNGAP1 suppresses the proliferation of rectal adenocarcinoma via Wnt/β-Catenin signaling pathway. Discov. Oncol. 15, 135 (2024).
Poetsch, A. R. et al. RASA4 undergoes DNA hypermethylation in resistant juvenile myelomonocytic leukemia. Epigenetics 9, 1252–1260 (2014).
Pera, EM., Nilsson-De Moura, J., Pomeshchik, Y., Roybon, L. & Milas, I. Inhibition of the serine protease HtrA1 by SerpinE2 suggests an extracellular proteolytic pathway in the control of neural crest migration. Stem Cell Rep. 15, 256–273 (2024).
Buchholz, M. et al. SERPINE2 (protease nexin I) promotes extracellular matrix production and local invasion of pancreatic tumors in vivo. Cancer Res. 63, 4945–4951 (2003).
Zhang, S. et al. SERPINE2 promotes liver cancer metastasis by inhibiting c-Cbl-mediated EGFR ubiquitination and degradation. Cancer Commun. 44, 384–407 (2024).
Zhang, J., Luo, A., Huang, F., Gong, T. & Liu, Z. SERPINE2 promotes esophageal squamous cell carcinoma metastasis by activating BMP4. Cancer Lett. 469, 390–398 (2020).
Chuang HW. et al. SERPINE2 overexpression is associated with poor prognosis of urothelial carcinoma. Diagnostics 11, 1928 (2021).
Sasahira, T., Kurihara-Shimomura, M., Shimomura, H. & Kirita, T. SERPINE2 is an oral cancer-promoting factor that induces angiogenesis and lymphangiogenesis. Int. J. Clin. Oncol. 26, 1831–1839 (2021).
Shen, Y., Wang, X., Xu, J. & Lu, L. SerpinE2, a poor biomarker of endometrial cancer, promotes the proliferation and mobility of EC cells. Cancer Biomark. Sect. A Dis. markers 19, 271–278 (2017).
Mao, M. & Wang, W. SerpinE2 promotes multiple cell proliferation and drug resistance in osteosarcoma. Mol. Med. Rep. 14, 881–887 (2016).
Huang, Y. K. et al. Inhibition of ADAM9 promotes the selective degradation of KRAS and sensitizes pancreatic cancers to chemotherapy. Nat. cancer 5, 400–419 (2024).
Shen, S. Y. et al. Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature 563, 579–583 (2018).
Qi, J. et al. Prediction model for malignant pulmonary nodules based on cfMeDIP-seq and machine learning. Cancer Sci. 112, 3918–3923 (2021).
Fu, M. et al. CD3, CD8, IFN-γ, tumor and stroma inflammatory cells as prognostic indicators for surgically resected SCLC: evidences from a 10-year retrospective study and immunohistochemical analysis. Clin. Exp. Med. 24, 99 (2024).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant Number: 81872438), the Collaborative Innovation Program of Hefei Science Center, CAS (Grant Number: 2022HSC-CIP015), the Program of Clinical Medical Translational Research in Anhui Province (Grant Number: 202304295107020092), the USTC Research Funds of the Double First-Class Initiative (Grant Number: YD9110002062), and the Research Funds of Centre for Leading Medicine and Advanced Technologies of IHM (Grant Number: 2025IHM01130). We would like to thank the patients who gracefully provided their samples and time for this study.
Author information
Authors and Affiliations
Contributions
B.H., H.W., and M.F. designed the study. M.F., Z.W., and J.W. performed the experiments. Q.Z., J.Q., and X.Z. performed the bioinformatic analysis. M.F., X.H., Z.H., W.W., and H.W. collected the SCLC clinical samples. M.F. and B.H. wrote the manuscript. M.F., W.W., B.H., and W.H. edited the manuscript. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Mario Perez-Medina and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Kaliya Georgieva.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Fu, M., Zhu, Q., Qi, J. et al. Integrated methylome analysis identifies an epigenetically silenced tumor suppressor RASA4 in small cell lung cancer. Commun Biol 9, 161 (2026). https://doi.org/10.1038/s42003-025-09440-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-09440-7
