
Abstract
Differences in the ϒ-aminobutyric acid (GABA) system contribute to an excitatory-inhibitory imbalance in autism, particularly affecting sensory processing. However, the brain’s broader response to interventions targeting GABA pathways in individuals with autism remains poorly understood. This study tested the hypothesis that GABAergic control of information transfer across large-scale brain functional networks is altered in autism. We conducted a phase-amplitude coupling (PAC) analysis of resting-state EEG signals within and between these networks. Responses were compared after double-blind, randomized oral administration of either a placebo or 15/30 mg of arbaclofen, a GABAB receptor agonist. Twenty-four non-autistic (9 males; 19-53 years) and 15 autistic participants (9 males; 20-51 years) completed 93 study visits. Autistic participants exhibited significantly higher theta-beta PAC, especially within the limbic network. High-dose arbaclofen shifted PAC metrics in visual and somatomotor networks towards non-autistic levels, but had minimal effects on networks related to higher cognitive functions. Interestingly, altered PAC within and between networks in the limbic system of autistic participants was normalized by low-dose arbaclofen, yet reemerged after high-dose administration. These findings provide compelling evidence for altered GABAergic responsivity in autism, helping explain some of the challenges in prescribing medications for autistic individuals, such as paradoxical reactions and dose sensitivity.
Similar content being viewed by others


Introduction
Imbalance between excitatory glutamate and inhibitory ϒ-aminobutyric acid (GABA) neural activity in the brain, which alters information processing, is a unifying hypothesis for diverse streams of neurobiological and genetic differences that may contribute to autism1,2. The evidence especially converges on a disruption within inhibitory GABA pathways, as demonstrated in rodent genetic models3,4, postmortem analyses5,6, bulk tissue concentrations as measured using in vivo magnetic resonance spectroscopy7, and GABA-dependent sensory functions associated with autism8. However, to date, studies supporting the hypothesis of GABAergic dysfunction in autism have been largely indirect, for example, based on ‘static’ baseline comparisons and correlational approaches. There has been limited investigation into the key question of how the GABA system in autistic brains dynamically responds to challenge. Addressing this gap not only holds the promise of establishing a causal link between GABA dysfunction and autism, but it also carries profound implications for the use of pharmacotherapies in autism9,10.
In our prior ‘shiftability’ studies involving both autistic and non-autistic adults11, we reported that autistic individuals have significant differences in GABAergic responsivity. For example, we demonstrated that the autistic brain does not suppress brain and retinal responses to visual flickering stimuli12,13, nor brain responses to repeated auditory stimuli14; but suppression was established by a single 30-mg dose of the GABA type B (GABAB) receptor agonist arbaclofen. In contrast, arbaclofen had a diametrically opposite effect in non-autistic participants, abolishing their usual suppression of neural responses to repeated visual and auditory stimuli.
These first steps provided evidence for autistic differences in GABAergic responsivity of the visual or auditory domains. However, autism is a complex condition that involves more than just sensory systems15. The autistic differences in social communication and interaction, repetitive behaviors16, and associated cognitive17,18 and/or neurological and mental health difficulties such as epilepsy, attention deficit hyperactivity disorder (ADHD), and obsessive compulsive disorder (OCD)19, suggest the involvement of complex mechanisms of information transfer requiring recruitment of functionally connected large-scale brain networks, extending far beyond the sensory processing regions of the brain20,21,22,23. However, it is unknown if altered GABA responsivity in the autistic brain extends beyond sensory networks.
Therefore, in this study, we extended our previous work to test the hypothesis that GABAergic control of large-scale brain systems is also altered in autism. To achieve this, we implemented a measure of information transfer using electroencephalography (EEG), namely, cross-frequency analysis of the phase-amplitude coupling (PAC) between different frequencies of neural activity. PAC refers to a phenomenon where the amplitude envelope of a high-frequency oscillation is modulated by the phase of a slower oscillation. A classic example of PAC is the interaction between theta (4-8 Hz) and gamma (>30 Hz) oscillatory activity observed in the hippocampus24,25. PAC has been suggested to support the transfer of information from slow, large-scale brain networks to the fast, local cortical processing necessary for effective computation and synaptic plasticity26. Evidence from preclinical studies and computational modeling has demonstrated that PAC can arise from various configurations of nested excitatory-inhibitory networks27,28,29 or from interactions within purely inhibitory populations of GABAergic interneurons30.
Previous studies report PAC alterations in individuals with autism. Higher posterior-midline alpha-gamma PAC during resting state has been observed in children with autism compared to their typically developing (TD) counterparts31. A follow-up study determined that children with autism demonstrated regionally specific abnormalities in PAC, with higher alpha-gamma PAC for a central midline source and lower PAC at lateral sources32. In this study, we wished to establish what role the GABA system may have in the regulation of PAC in autistic individuals compared to controls. We hypothesized that atypical PAC indices may persist in adult individuals, may be more widespread than previously reported, and may also be differently modulated by the GABA system. Hence, we used PAC to investigate differences in GABAergic responses across a broad range of brain regions in autistic adults.
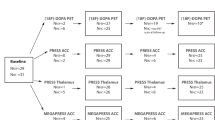
To understand the spatial as well as temporal landscape of GABAergic responses, we localized resting-state EEG signals to 400 regions of interest (ROIs) in the cortical source space. These 400 ROIs were further allocated to Yeo 7 functional subnetworks according to their spatial distributions to investigate cross-frequency coupling effects within and between these networks [respectively, the frontoparietal network (FPN), default mode network (DMN), limbic network (LN), ventral attention network (VAN), somatomotor network (SMN), dorsal attention network (DAN) and visual network (VN)33]. To test our hypothesis, PAC indices between different spectral components of EEG were measured and compared between autistic and non-autistic adults at placebo and following a single low (15 mg) or high (30 mg) oral dose of arbaclofen (see Fig. 1).

A Preprocessed EEG signals collected at placebo or drug administration are source-localized and projected onto 400 regions of interest (ROIs) on the cortex, resulting in a matrix of source space time courses. For the time course of each ROI, the phase-slope index (PSI) is calculated, which estimates the slope of the phase difference as a function of frequency within a specified frequency band. The phase-amplitude coupling (PAC) value is obtained based on the maximum PSI value between two frequency bands. B The 400 ROIs are divided into 7 brain functional networks, including the Frontoparietal Network (FPN), Default Mode Network (DMN), Dorsal Attention Network (DAN), Ventral Attention Network (VAN), Sensorimotor Network (SMN), Visual Network (VN), and Limbic Network (LN). Quantitative analysis is then conducted on the cross-frequency coupling effects within and between these networks using PAC as the metric.
Results
Clinical cohort
Twenty-four non-autistic (controls) and 15 autistic individuals were included, and 93 study visits were completed: 32 placebo (P) visits (20 controls and 12 autistic; Table 1), 36 low dose (L) visits (21 controls and 15 autistic), and 25 high dose (H) visits (13 controls and 12 autistic). Thus, we considered six cells determined by clinical status and drug dose: Control_P, Control_L, Control_H, Autism_P, Autism_L, and Autism_H (Table 2). Twenty-two participants (12 controls and 10 autistic) completed all three visits, 10 participants (6 controls and 4 autistic) completed two visits, and 7 participants (6 controls and 1 autistic) completed only one visits. The most common reason for dropout was (expected) adverse side effects, affecting 6 controls and 3 autistic participants. Other reasons included study cancellation due to expired drugs (3 controls) and personal preference to discontinue participation (3 controls and 2 autistic). Visits were at least one week apart to ensure complete drug washout.
All participants were adults. Demographic characteristics, including biological sex and IQ intelligence quotient (IQ; on the Wechsler Abbreviated Scale of Intelligence; WASI-II34), were matched between the control and autism groups (Table 1). The age range of the autism and control groups was also similar (20-51 years and 19–53 years, respectively), although the mean age of the autism group (37.7 ± 9.7 years) was higher than that of the control group (29.5 ± 8.3 years; t(37) = 2.7, p = 0.01). However, there was no significant relationship between any electrophysiological measures and age in either group, indicating that age differences were unlikely to account for any reported group differences. As expected, there was a highly significant difference between the two groups in AQ (t(36) = 7.9, p < 0.001). Statistical results reported were corrected for multiple comparisons.
Placebo differences
Four conventional PAC metrics were compared between autistic individuals and controls; theta-beta35, theta-gamma36, alpha-beta37, and alpha-gamma38 PAC. Fig. 2A depicts a heatmap of results comparing placebo PAC measurements within each network between autistic and non-autistic participants. All significant group differences were observed during the eyes-closed state. Specifically, autistic participants exhibited higher theta-beta PAC across nearly the entire brain during the eyes-closed state (FPN: t(30) = 3, p = 0.007; DMN: t(30) = 3.7, p = 0.003; LN: t(30) = 4, p = 0.001; VAN: t(30) = 2.7, p = 0.01; SMN: t(30) = 3.2, p = 0.005; VN: t(30) = 4.5, p = 0.001; Fig. 2B). The only exception was in the DAN, where theta-beta PAC was comparable between the two groups (t(30) = 1.1, p = 0.3). As shown in Fig. 2C, the remaining three PAC parameters derived from the limbic network during the eyes-closed state were significantly higher in autistic participants compared to non-autistic individuals (alpha-beta PAC: t(30) = 2.9, p = 0.0075; theta-gamma PAC: t(30) = 3.3, p = 0.004; alpha-gamma PAC: t(30) = 3.3, p = 0.004). Finally, theta-gamma PAC in the SMN in autistic participants was significantly higher than non-autistic participants (t(30) = 2.7, p = 0.01). No discernible differences in other networks (Table 3).

A Heatmap plotted using t-values for group comparisons. The four different PAC values under eyes-open and eyes-closed conditions (column) are compared between the autism group and the control group across seven networks (row). Colored rectangular boxes represent conditions with significant group differences, with darker colors indicating higher t values. B Scatter plots of theta-beta PAC values for group comparisons within six networks (excluding the limbic network, LN) under the eye-closed condition. Red represents the autism group, and blue indicates the control group. C Scatter plots of the four PAC values for group comparisons within the LN under the eye-closed condition. All t values were achieved through independent t tests and the corresponding p-values reported were corrected for multiple comparisons using the FDR-BH method. FPN: Frontoparietal Network; DMN: Default Mode Network; DAN: Dorsal Attention Network; VAN: Ventral Attention Network; SMN: Sensorimotor Network; VN: Visual Network; LN: Limbic Network. * the difference is statistically significant with corrected p < 0.05; ** corrected p < 0.01; and *** corrected p < 0.001. Error bars show the standard deviation (SD).
Effects of arbaclofen on theta-beta PAC in the SMN and VN
Low dose (15 mg) arbaclofen
There was no statistically significant effect of low-dose arbaclofen on theta-beta PAC in the SMN or VN—higher levels in the autistic participants persisted. The regulatory effect of arbaclofen on theta-beta PAC of six networks (except LN) is illustrated in Fig. 3A.

A Scatter plots depict the theta-beta PAC responses of the autism group (red) and the control group (blue) under placebo/drug administrations. theta-beta PAC was chosen due to its sensitivity to group differences across almost the entire brain under placebo conditions. The six functional networks included in the plots are FPN, DMN, DAN, VAN SMN, and VN. B The four PAC values of the LN network under placebo/drug administrations are specifically plotted, with theta-beta, alpha-beta, theta-gamma, and alpha-gamma PAC displayed from left to right. Significant differences between the autism group under placebo or drug administrations and the control group under placebo conditions are indicated by asterisks. FPN: Frontoparietal Network; DMN: Default Mode Network; DAN: Dorsal Attention Network; VAN: Ventral Attention Network; SMN: Sensorimotor Network; VN: Visual Network; LN: Limbic Network. * the difference is statistically significant with corrected p < 0.05; ** corrected p < 0.01; and *** corrected p < 0.001. Error bars show the standard deviation (SD).
High dose (30 mg) arbaclofen
reduced (higher) theta-beta PAC in the SMN (t(30) = 3.2, p = 0.005) and VN (t(30) = 4.4, p = 0.001) of autistic participants to a level comparable to that of placebo controls (Table 4, the upper part). LMM results confirmed a significant group-drug interaction in theta-beta PAC of both the SMN (t89 = −2.1, p = 0.04) and VN (t89 = −2.6, p = 0.009); the effect of drug in the SMN (t(37) = −2.3, p = 0.02) and the VN (t(37) = −2.9, p = 0.006) was also significant within the autistic group, but not within the control group.
Effects of arbaclofen on PAC in the LN
Low dose (15 mg) arbaclofen
reduced (higher) theta-beta PAC (t(30) = 4, p = 0.0015), theta-gamma PAC (t(30) = 3.3, p = 0.004), alpha-beta PAC (t(30) = 2.9, p = 0.0075), and alpha-gamma PAC (t(30) = 3.3, p = 0.004) in the autistic group (Table 4, lower part). No drug effects were observed in controls in the low-dose condition.
High dose (30 mg) arbaclofen
did not shift limbic PAC values which remained atypically high in the autistic group compared to the control baseline (beta-theta PAC: t(30) = 3.1, p = 0.01; gamma-theta PAC: t(30) = 2.9, p = 0.01, gamma-alpha PAC: t(30) = 3.1, p = 0.01). The only exception was the higher alpha-beta PAC in autistic individuals, which was rescued by 30-mg arbaclofen (t(30) = 1.7, p = 0.12). No drug effects were observed in controls in the high-dose condition. The regulatory effect of arbaclofen on PAC values of LN is shown in Fig. 3B.
The LMM results confirmed a significant drug-group interaction in autistic individuals on theta-beta PAC (t(86) = -2, p = 0.04), alpha-beta PAC (t(34) = -2.5, p = 0.01) and alpha-gamma PAC (t(86) = −2.3, p = 0.02) in the LN.
The LMM returned a significant drug effect on alpha-beta PAC in controls (t(52) = −2.6, p = 0.01); 30 mg of arbalcofen reduced alpha-beta PAC in the control group, but this result did not reach statistical significance (Control_P vs. Control_H: t(31) = 2.2, p = 0.08).
Effects of arbaclofen on between-network connections in autistic individuals
We examined whether there were alterations in between-network PAC connections in autistic individuals, and explored the impact of arbaclofen. We first assessed the theta-beta PAC connection between any two ROIs from different networks (see Methods for details). A set of discriminative point-to-point connections associated with the autistic alterations was identified following placebo or drug administrations (Fig. 4A–C, top). To examine the spatial distribution of these connections, we counted the number of occurrences where each brain region was selected as a connection terminus, and then divided these counts by the total number of ROIs in each network to obtain the participation rates (Fig. 4A, B and C bottom). A higher rate value indicates a greater involvement of the network in the aberrant connectivity in the autism group relative to non-autistics. At placebo, the LN (participation rate = 0.19), SMN (participation rate = 0.16), VN (participation rate = 0.13), and FPN (participation rate = 0.12) were notably involved, with their participation rates over 0.1 (Fig. 4A). Low dose arbaclofen reduced the participation rates of these networks, leaving only the LN remaining above 0.1 (Fig. 4B). After high dose administration, the participation rates were further decreased globally, with none of the networks exceeding 0.08 (Fig. 4C).

A Top: The spatial distribution of discriminative hyper- (red edge) and hypo-connections (blue edge) for placebo comparison between the Autism_P group and the Control_P group. Bottom: The histogram illustrating the extent to which various networks are affected as quantified by the participation rate. The node or bar color represents the corresponding functional network. B The spatial distribution of discriminative connections (top) and the histogram of affected networks (bottom) for comparison between Autism_L and Control_P. C The spatial distribution of discriminative connections (top) and the histogram of affected networks (bottom) for comparison between Autism_H and Control_P. The overall between-network connectivity based on network characterizations is also plotted for comparisons among Autism_P vs. Control_P (D), Autism_L vs. Control_P (E), and Autism_P vs. Control_P (F). FPN: Frontoparietal Network; DMN: Default Mode Network; DAN: Dorsal Attention Network; VAN: Ventral Attention Network; SMN: Sensorimotor Network; VN: Visual Network; LN: Limbic Network.
Next, for each participant, we averaged all theta-beta PAC connections between the same pair of networks to assess the overall between-network connectivity between these two networks. As shown in Fig. 4D, under the placebo condition, several average between-network connections had significant differences between the autism and control groups, primarily involving the LN, DMN, and SMN. After a low dose of arbaclofen in the autism group, there were no longer between-network differences compared to non-autistic placebo condition (Fig. 4E). However, after high dose administration in autistics, atypical connections reemerged, and all of them were related to the LN (Fig. 4F).
Effects of arbaclofen on LN-related connections in autistic individuals
To elucidate how the limbic network (LN) is specifically modulated by arbaclofen, we quantified all aberrant between-network point-to-point connections and average connectivity related to the LN in autistic individuals under both placebo and drug conditions. This process encompassed all four types of PAC connections (see Methods for details). At placebo, the LN-related between-network point-to-point connections associated with the autism group involved networks including the DMN (participation rate = 0.077), FPN (participation rate = 0.04), VN (participation rate = 0.026), and SMN (participation rate = 0.02) (Fig. 5A). A low dose of arbaclofen in autistics reduced the participation rates in most of the affected networks mentioned above (DMN: 0.033; FPN: 0.019; VN: 0.013), but had no effect on the SMN (Fig. 5B). Following high-dose arbaclofen, the participation rates of most networks remained at low levels, but the anomalous connections between the LN and the SMN/DMN notably increased (Fig. 5C).

A Top: The spatial distribution of LN-related discriminative connections for placebo comparison between Autism_P and Control_P. Bottom: The histogram illustrating the extent to which various networks are affected as quantified by the participation rate. B The spatial distribution of LN-related discriminative connections (top) and the histogram of affected networks (bottom) for comparison between Autism_L and Control_P. C The spatial distribution of LN-related discriminative connections (top) and the histogram of affected networks (bottom) for comparison between Autism_H and Control_P. LN-related between-network connectivity based on network characterizations, as measured by different PAC values, is also plotted for comparisons among Autism_P vs. Control_P (D), Autism_L vs. Control_P (E), and Autism_P vs. Control_P (F). FPN: Frontoparietal Network; DMN: Default Mode Network; DAN: Dorsal Attention Network; VAN: Ventral Attention Network; SMN: Sensorimotor Network; VN: Visual Network; LN: Limbic Network.
Similar results were also observed in the between-network average connectivity. As shown in Fig. 5D, at placebo, the aberrant average connectivity in the autistic group relative to non-autistics was primarily located between the LN and the DMN/FPN, also involving the SMN/VAN/VN. In terms of PAC frequency bands, they primarily involved theta-beta and theta-gamma PACs. A low dose administration of arbaclofen rescued connectivity in autistic individuals, resulting in no significant difference compared to the control group in the placebo condition (Fig. 5E). However, in the high-dose condition, the between-network average connectivity in the autism group once again showed abnormalities, primarily located between the LN and the SMN/VAN (Fig. 5F). Hence, low dose arbaclofen removes group differences by its action in the autism group, whereas high-dose arbaclofen causes the re-emergence of aberrant cross-network PAC connections between the LN and SMN in the autism group. To further elaborate on this point, exemplary spaghetti plots are provided to illustrate the modulatory effects of the drug on the LN-SMN between-network connectivity at the individual level (Fig. 6). Low dose arbaclofen tended to reduce the connectivity measured by theta-beta PAC in autistic individuals, while the high dose of drug significantly increased theta-beta PAC compared to the low dose (t(8) = 2.5, p = 0.04; Fig. 6A). A similar result was also observed for the LN-SMN between-network connectivity as measured by theta-gamma PAC (t(10) = 2.5, p = 0.03; Fig. 6B).

Spaghetti plots are provided for autistic participants who completed all three visits (placebo, low dose, and high dose). The between-network connectivity was measured using either theta-beta PAC (A) or theta-gamma PAC (B). * the difference is statistically significant with corrected p < 0.05.
Discussion
Our results provide the first human in vivo insights into the differential responsivity of distinct brain networks in autism to the targeted modulation of GABA.
Phase-amplitude coupling (PAC) captures an organizational principle of brain function where the amplitude envelope of a high-frequency oscillation is modulated by the phase of a slower oscillation. Also, PAC arises from configurations of nested excitatory-inhibitory networks and/or from interactions within inhibitory intraneuronal populations27,28,29,30. GABAergic signaling is key to this dynamic process, which is essential to the transfer of information from slow, large-scale brain networks to the fast, local cortical processing necessary for effective computation. Here, we confirmed that PAC is altered in individuals with autism and differentially responds to GABAergic stimulation. In autistic people, altered cross-frequency couplings were evident across large-scale brain functional networks at placebo. Specifically, autistic participants have higher theta-beta PAC than non-autistic participants across most brain regions during the eyes-closed state. Moreover, we discovered that autistic participants have higher limbic alpha-beta, theta-gamma, and alpha-gamma PAC; and theta-gamma PAC in the SMN. Activating GABAB receptor function through a single oral dose of arbaclofen had varying dose-dependent effects on atypical coupling metrics across different functional networks in autism.
The details of microcircuitry underlying PAC are still uncertain, but there is consensus that a prominent PAC indicates high-frequency dynamics, or excitability of specific neuronal populations, that are phase-locked to slow rhythms39,40. These high-frequency rhythms are thought to reflect localized cortical processing domains, whereas low-frequency brain activity is synchronized across extensive brain areas through long-range connectivity conveyed by white matter networks26,41. Thus, PAC may serve as a mechanism to synchronize local and global neuronal populations. This is in turn considered fundamental for maintaining E-I balance within and between brain functional networks42,43. There are a number of proposed explanations for PAC. Computational modeling demonstrates that PAC can be generated from E-I interactions mediated by an inter-laminar coupling circuit distributed between supragranular layers and superficial layers44. Others emphasize GABA. For example, it has been shown that synchronized cross-frequency information originates from inhibitory postsynaptic potentials generated during GABAergic synaptic transmission45,46,47 and concurrent generation of low- and high-frequency rhythms and their coupling can be achieved by a purely GABAergic network incorporating both slow and fast feedback30. These findings support the notion that PAC may reflect neuronal dynamics closely tied to E-I interactions, particularly those involving GABA-related mechanisms48. Therefore, the presence of PAC alterations in autistic participants extends an E-I theory of autism based largely on indirect evidence from preclinical studies and genetic, postmortem, and correlational studies in humans2,49 to confirm differences in in-vivo E-I dynamics in the human brain.
Specifically, we reportthat the atypically high PAC in autistic individuals was in functional networks closely associated with sensory processing, including the VN and SMN. In contrast, other networks, such as the FPN, DMN, and VAN, showed minimal to no response to either dose of arbaclofen. The LN response to arbaclofen was quite distinctive. At low dose arbaclofen, LN PAC in autistic participants was down regulated; after high dose administration, LN PAC was elevated. The reason(s) for this regulatory disparity across functional networks are likely complicated. Nonetheless, our observed effects may be attributed to regional variations in the distribution of GABAegic receptors across different brain areas50,51. If GABA concentrations are low, as has been reported in autism52, networks with a higher density of GABAB receptors may require a higher dose of arbaclofen to achieve optimal modulation (as observed in sensory processing networks), whereas networks with fewer GABAB receptors may reach saturation and exhibit overmodulation under high-dose conditions (as seen in the LN). In cases where the concentration of GABA is adequate to occupy available receptors, arbaclofen would have limited or no targets to act upon, resulting in no response to the challenge. However, this explanation conflicts with studies that have shown no difference in GABA concentrations within the visual network between individuals with and without autism7,12 and reports of lower GABAergic receptor density6,53,54.
A more plausible explanation, therefore, is that there is a difference in the functioning of GABAB receptor circuits within networks in the autistic brain. GABAB receptors are conventionally thought to provide tonic inhibition; however, they can also exert both pre- and postsynaptic cellular effects and cause regional excitation through disinhibition55. They also exhibit crosstalk both with excitatory glutamate receptors56 and inhibitory GABAA receptors57. Supporting evidence comes from recent advances in animal models of autism, which show enhanced, rather than decreased, GABAergic function in the anterior cingulate cortex (ACC) of male mice58. This enhancement is closely linked to stress-induced social impairments. Thus, activating GABAB through oral medication alone may trigger a range of complex, even counteracting mechanisms that regulate E-I function. It could ultimately result in altered or no observable modulation within these networks responsible for higher-order cognitive functions (FPN, DMN, VAN, and DAN). In contrast, sensory processing networks, such as the VN and SMN, involve relatively concise hierarchical neural pathways that enable rapid and efficient processing of sensory inputs59. Consequently, abnormalities in GABAergic function within sensory networks may manifest as a more consistent reduction in inhibitory tone60,61, leading to a monotonic dose-dependent modulation effect that strengthens with increasing activation of GABAB receptors.
Our results identify the limbic network (LN) as a region of particular interest. The limbic system is comprised of the orbital frontal cortex (OFC) and the medial temporal lobe, which includes the hippocampus and the amygdala. This region has long been implicated in the neuropathology of autism due to its strong association with social and emotional functioning62,63,64. Previous studies have shown significant differences in structural circuit connectivity within the limbic system between autistic and non-autistic individuals65,66. In our results, all four PAC metrics within LN showed significant placebo differences between participants with and without autism. We further demonstrated that a low dose of arbaclofen modulated these aberrant within-network PAC values and between-network connectivity in the autism group to levels comparable to the placebo state of the control group. When GABAB activation was excessive, as in the 30-mg condition, no modulation effect was observed on either the LN-related atypical within-network PAC or between-network connectivity. These results extend previously reported structural abnormalities in the limbic system of individuals with autism to dysfunctions in oscillatory synchronizations and couplings measured by PAC, particularly linking them to E-I imbalance. Importantly, we demonstrate that the observed LN-related dysfunctions can be modulated through targeted activation of GABAB receptor, which is highly sensitive to drug dosage, with excessive activation leading to a ‘re-emergence’ of E-I imbalance within the LN. These results offer possible explanations for the clinically mixed reports of paradoxical reactions and dose sensitivity of arbalcofen.
In our previous work, we have shown that the steady-state visual evoked potentials (SSVEPs) elicited by external inhibitory visual stimuli were resistant to interference12, and event-related potentials (EPRs) induced by repetitive auditory stimuli did not suppress in the autistic brain14; but sensory dynamics similar to non-autistics were established by a single 30-mg dose of arbaclofen. Our findings from this study of a monotonic dose-dependent modulation effect on resting-state PAC in sensory networks are consistent with our previous task-based observations. The elevated PAC in autistic individuals at placebo may indicate a tight coupling between the brain’s local plasticity mechanisms and the endogenous low-frequency landscape. This, in turn, may weaken the brain’s flexibility and adaptability to respond to changes in external sensory stimuli, leading to the observed decrease in task-based sensory sensitivity. Arbaclofen, by activating GABAB receptors, may help “loosen” the endogenous coupling and allow the autistic brain to regain plasticity across sensory information processing networks.
The study has some limitations: We acknowledge the modest sample size in this study. In mitigation, we used a within-subject design with the same participants returning for repeat visits, thereby reducing heterogeneity and increasing power. A critical consideration in both basic research involving acute dose study designs and clinical trial designs aimed at assessing potential therapeutics for autism and related disorders is the selection of the dosing regimen. In this study, we only had two single arbaclofen dose conditions, 15 mg and 30 mg. Consequently, it is possible that we may have overlooked the impact on functional networks that could require different doses to manifest effects. Furthermore, the potential impact of arbaclofen on other neurotransmitter systems and receptors may have influenced the observed results. For example, activation of GABAB receptors can exhibit crosstalk with both excitatory glutamate receptors56 and inhibitory GABAA receptors57. This complex effect might allow for redundancy and/or secondary neurosignalling processes which ‘mask’ GABAB effects, particularly in networks associated with higher-order cognitive functions, where we did not observe significant effects of arbaclofen. However, our study in humans cannot address this level of detail and we will need further investigation in preclinical models to elucidate these mechanisms.
Although our study demonstrates differential effects of GABAB receptor agonism in autism, it also demonstrates that findings are heterogeneous in terms of dose and network effects, especially within the LN. However, the LN-related between-network connectivity results exhibited a dose-dependent response, with the LN-related connectivity patterns reverting to a more typical pattern under the low-dose condition in the autism group. This between-network response to arbaclofen in LN was more consistent across autistic participants (Fig. 6). It also echoes a similar pattern in an fMRI study of the effect of arbaclofen on between-network connectivity of resting-state networks11, suggesting these findings are not spurious.
Although our findings do not speak to the clinical efficacy of arbaclofen, they may nevertheless have important implications for the development and interpretation of interventions targeting autistic symptomatology. Arbaclofen is the biologically active R-enantiomer of baclofen, exhibiting over 100-fold higher specificity for the GABAB receptor compared to the S-enantiomer and approximately five times greater specificity than racemic baclofen67. It has been shown to be safe and well-tolerated in individuals with autism68. Inspired by the success in preclinical animal models69,70,71, several clinical trials have been conducted to investigate the use of arbaclofen for targeting core symptoms of autism; however, the results have been mixed68,72. Translation from preclinical research to clinical application is always challenging, and this may be particularly pertinent in the context of autism. Besides the inherent heterogeneity in genetic and environmental aetiological influences73, and the ongoing debates regarding diagnostic criteria74, we propose that the complex dynamic response to a single oral dose of arbaclofen observed here may reflect complex E-I responses across diverse brain regions in autistic individuals; some beneficial, some not. An important next step therefore is to adapt clinical trial design and establish if drug response dynamics predict clinical response at an individual level.
Materials and methods
Study design
Thirty-nine participants completed a total of 93 study visits. Of these, 24 were non-autistic individuals and 15 were autistic. In this placebo-controlled, cross-over and double-blind study, participants were administered either a placebo or a single oral dose of 15 or 30 mg of arbaclofen (STX209) on the designated study day, with the order of administration randomized across visits. The compound was donated by Clinical Research Associates (CRA), a non-profit subsidiary of the Simons Foundation. Resting-state EEG data were collected three hours after placebo/drug administration, within the active physiological window of arbaclofen [69]. To ensure drug wash-out, visits for each participant were scheduled at least one week apart. This study was approved by King’s College Research Ethics Committee (RESCM-17/18-4081 and LRS-15/16-3582) and the Medicines and Health Regulatory Authority (MHRA) in the UK confirmed the design was not a clinical trial as arbaclofen was used as a pharmacological probe to explore potential GABAergic functional differences in brains of autistic and non-autistic people. Informed consent was obtained from all participants. All ethical regulations relevant to human research participants were followed.
Medical or nursing cover was nevertheless provided throughout each study visit, and participants were asked to remain at our unit for at least four hours after drug/placebo administration. The clinician and the researchers were ‘blind’ to the order of administration but had access to the visit-specific placebo/drug allocation information for unblinding if necessary. No emergency code break was required, but in cases where a participant experienced side effects that were deemed more than ‘moderate’ by the study medic, and after consultation with the chief investigator, unblinding occurred to avoid exposing the participant to a higher dose of arbaclofen during a subsequent visit.
Participants
Diagnoses of autism were confirmed by experienced clinicians and supported by the Autism Diagnostic Interview-Revised (ADI-R)75 where an informant was available, and/or the Autism Diagnostic Observation Schedule (ADOS-2)76. Participants with autism caused by a known genetic syndrome, such as fragile X syndrome or 22q11 deletion syndrome, were excluded from the study. Autism traits were also assessed in both the control group and the autism group using the autism quotient [AQ70;]; see Table 1.
Other inclusion criteria were as follows: an IQ score above 70, the ability to provide informed consent, no comorbid psychiatric illness (e.g., psychotic illness and major mood disorder), no history of seizures or a diagnosis of epilepsy, and no significant physical illness, such as heart disease, high blood pressure, or renal insufficiency. In the month preceding participation, ten participants (2 controls and 8 autism) were taking regular medication, including drugs like citalopram and sertraline, which did not directly affect glutamate or GABA. All other participants were medication free.
Resting-state EEG acquisition
Resting-state EEG data were collected three hours after placebo/drug administration within arbaclofen half-life77. Testing was conducted in a dimly lit room at the Institute for Translational Neurodevelopment, King’s College London. The participant was seated comfortably and instructed to remain relaxed and avoid making large movements during the experiment. Participant behavior was carefully monitored by the experimenter throughout the test and the procedures were well tolerated. The test consisted of six resting-state trials, with three trials conducted with eyes open and three with eyes closed. Each trial lasted for one minute with a 10-second interval between trials, and a beep sound was used to signal the end of each trial. The order of the eyes-open and eyes-closed trials was alternated and counterbalanced among the participants. Continuous scalp EEG signals were recorded using a 64-channel standard actiCAP (EASYCAP GmbH) with a sampling rate of 5 kHz and amplified by a BrainAmp amplifier (Brain Products GmbH). The default reference was FCz, and electrode placements adhered to the international 10–20 system78. Impedances between the scalp and electrodes were maintained below 15 kΩ.
EEG preprocessing
The recorded EEG data were preprocessed in MATLAB 9.2.0 (MathWorks Inc.) using in-house scripts. The steps of our preprocessing follow a standard pipeline79 and are described as follows1: The raw EEG data from all recording channels were resampled to \({Fs}=250{{\rm{Hz}}}\);(2) nonphysiological slow drifts were removed using a 0.1-Hz high-pass filter;(3) line noise and artifacts with high-frequency components, such as the electromyography, were further removed using a 45-Hz low-pass filter;(4) significant muscle artifacts were corrected by ‘BurstCriterion’ in EEGLAB with interpolation;(5) bad channels were identified and rejected based on low correlation with other channels by “ChannelCriterion” in EEGLAB. Then, they were interpolated using the spherical interpolation method, with the “MaxBadChannels” parameter set to 20%, which corresponds to an upper limit of 12 interpolated channels for our 64-channel montage;(6) data from 1 to 59 s of each resting trial was considered as one data segment, with the first and last second of data discarded to eliminate edge effects;(7) remaining artifacts were removed using independent component analysis (ICA) by invoking the ‘pop_runica’ function provided by EEGLAB80, which facilitated automatic removal of independent components associated with muscle, ocular, and electrocardiogram artifacts;(8) any segments exceeding 200 microvolts would be rejected;(9) these preprocessed data segments were saved separately for eyes-open and eyes-closed trials for further analysis.
Source-space time course extraction
We conducted an EEG source localization to identify specific brain regions that generate scalp physiological activities and to reconstruct source-space time courses on the cortical layer81. This process was performed using the BrainStorm tool82 in MATLAB by adopting standardized low-resolution electromagnetic tomography (sLORETA83;). Specifically, the mapping between scalp electrode space and source space at a certain sampling point can be expressed by the Maxwell’s equation as follows:
where \(\varPhi \in {{\mathbb{R}}}^{C\times 1}\) and \(X\in {{\mathbb{R}}}^{V\times 1}\) represent the scalp vector recorded from the C electrodes (channels) and the source vector consisting of V regions of interest (ROIs), respectively; \(M\in {{\mathbb{R}}}^{C\times V}\) is the lead field matrix and \(\varepsilon \in {{\mathbb{R}}}^{C\times 1}\) represents noise vector. The core idea of LORETA is to solve the following inverse problem:
With a given M, \(\varPhi\) and a regularization parameter α ≥ 0, the explicit solution for the above optimization problem is
where superscript ‘+’ indicates the Moore-Penrose pseudoinverse and \(H\in {{\mathbb{R}}}^{C\times C}\) represents the centering matrix. Next, \(\hat{X}\) is standardized to get the sLORETA estimate of each ROI. In this study, we divided the cortex into 400 ROIs in source space according to the Schaefer parcellation atlas83. Time courses of all ROIs were extracted by repeating the above procedure, forming the reconstructed source matrix \({JJ}\in {{\mathbb{R}}}^{400\times N}\), where N is the number of sample points.
PAC calculation for each ROI
We then adopted a state-of-the-art method to assess the coupling between the phase of slower oscillations and the power envelope of faster oscillations by employing the phase-slope index (PSI)84. The time course of one of the source voxels extracted from the reconstructed source matrix \({JJ}\in {{\mathbb{R}}}^{400\times N}\) is denoted as \(X=({x}_{1},{x}_{2},...,{x}_{N})\). In our PAC measurements, the time series \(X\) was compared to the temporal evolution of the power of the activity at frequency v, which was termed \({Y}^{v}=({y}_{1}^{v},{y}_{2}^{v},...,{y}_{N}^{v})\). This \({Y}^{v}\) presented power as a function of time and was estimated by applying a discrete Fourier transform to successive segments extracted from \(X\) using a sliding window with M samples:
where \({y}_{n}^{v}\) is the power of the activity at frequency v and sample point n, \(h\in {{\mathbb{R}}}^{1\times M}\) represents a Hanning taper to reduce spectral leakage. The length of M was chosen to be five cycles long with respect to the frequency v, that is \(M=5\cdot {{\rm{Fs}}}/{{\rm{\nu }}}\), as such the length of the window decreases with increasing frequency.
Next, \(X\) and \({Y}^{v}\) were segmented into a set of S successive segments with l overlapping time points. The time course and the temporal evolution of power of segment s are referred to as \({X}^{s}\) and \({Y}^{v,s}\), respectively. Their spectral representations are calculated using a standard Fast Fourier Transform (FFT):
where \({{{\rm{X}}}}_{{{\rm{FFT}}}}^{{{\rm{s}}}}\) denotes Fourier transforms centered at the frequencies \(f\in \left\{0,\frac{{Fs}}{{n}_{{FFT}}},...,\frac{{Fs}}{2}\left(1-\frac{1}{{n}_{{FFT}}}\right)\right\}\), \({n}_{{FFT}}=256\) defines the frequency resolution of FFT with \(\triangle f={{\rm{Fs}}}/({{{\rm{n}}}}_{{{\rm{FFT}}}}* 2)=0.5{{\rm{Hz}}}\). The complex coherence between the signal \(X\) and the power envelope of the signal \({Y}^{v}\) was obtained by the following formula:
with frequencies \(f\in \left\{0,\frac{{Fs}}{{n}_{{FFT}}},...,\frac{{Fs}}{2}\left(1-\frac{1}{{n}_{{FFT}}}\right)\right\}\), and ‘*’ denotes the complex conjugate.
The quantification of the PAC was based on the estimation of the phase-slope index (PSI) following a framework proposed by Jiang et al.84. The PSI estimates the slope of the phase difference as a function of frequency within a specified frequency band, where the sign of the slope indicates the direction of interaction. We applied the PSI to the signal \(X\) and the power envelope of the signal \({Y}^{v}\) at frequency tile (\(v\), \({f}_{j}\)):
where \({f}_{j}\) denotes the low frequency of the signal \(X\), \(\Delta f\) is frequency resolution and \({Im}\) denotes the imaginary part. We used \(\beta =2\) Hz to denote the bandwidth for which the phase slope was calculated. The PAC value between the phase of a low frequency band \({B}_{p}\) (theta or alpha) and the amplitude of a high frequency band \({B}_{a}\) (beta or gamma) is defined as the maximum PSI value among all pairwise combinations (\({f}_{k},{f}_{l}\)) in the two bands:
This PAC calculation method based on PSI is not only capable of calculating the coupling relationship between low- and high-frequency components of the acquisition EEG signal within a single channel, but also can be easily extended to compute the signal coupling relationship across different channels.
Within-network PAC evaluation
The 400 ROIs in the source space were assigned to Yeo’s 7 functional networks based on their spatial distributions33. These networks include the visual network (VN), somatomotor network (SMN), dorsal attention network (DAN), ventral attention network (VAN), limbic network (LN), frontoparietal network (FPN), and default mode network (DMN). The within-network PAC is defined as the average PAC value across all ROIs within a specific network:
where PAC is calculated between the phase of a low frequency band \({B}_{p}\) and the amplitude of a high frequency band \({B}_{a}\), and \(T{C}_{{{net}}_{i}}\left(h\right)\in {{\mathbb{R}}}^{1\times N}\) denotes the time course of the h-th ROI within a specific network \({{net}}_{i}\), and \({{\rm{H}}}\) represents the number of ROIs contained in the network.
Identification of discriminative between-network PAC connections
First, we evaluated the point-to-point connection between two ROIs with terminations originating from any pair of functional networks. The connection strength was quantified as the theta-beta PAC value, which was calculated between the phase of the low-frequency components of an ROI in one network and the amplitude of the high-frequency components of an ROI in the other network.
Discriminative point-to-point connections were identified using a rigorous threshold of statistical significance (p < 1\(\times\)10-4). We then defined a participation rate to quantify the degree to which each of the seven functional networks is involved in the spatial distribution of these connections. Specifically, we counted the number of instances where each functional network served as one of the endpoints of a connection, and subsequently divided these counts by the total number of ROIs within each network to obtain the participation rate.
Next, all point-to-point connections between two networks with the same phase-amplitude direction were averaged to represent between-network connectivity based on network characterizations. The procedure can be described by the following formula:
where \({{{\rm{H}}}}_{1}\) and \({{{\rm{H}}}}_{2}\) represents the number of ROIs contained in network \({{{\rm{net}}}}_{{{\rm{i}}}}\) and \({{{\rm{net}}}}_{{{\rm{j}}}}\), respectively. Given that the number of average connectivity is significantly lower than the number of point-to-point connections, we applied another rigorous threshold (p < 1 \(\times\) 10−2) for the identification of discriminative average connectivity.
Statistics and reproducibility
A two-sided independent-sample t test was used to assess differences in within-network PAC values between the autism and control groups at placebo. Prior to the analysis, the Shapiro-Wilk test was conducted to verify the normality of the PAC responses. Extreme points (one case in the autism group) located outside two standard deviations were excluded. The results were corrected for multiple comparisons using the Benjamini-Hochberg method, implemented through the FDR-BH function in Matlab.
Repeated measures analysis of the differential drug effects on the within-network PAC values of the autism group and the control group was conducted using a linear mixed-effects model (LMM). For each of the seven functional networks, we built an LMM model for the theta-beta PAC value, with group (autism/control) and drug dose (placebo/low/high) as fixed effect factors, and subject as the random effect factor. This allowed us to measure the differential modulations by arbaclofen in autistic and non-autistic participants as a group-drug interaction. When a significant group-drug interaction or an overall group difference was observed (p < 0.05), the LMM model shrank to a simple linear model with drug dose as the fixed-effect variable and subject as the random-effect variable, which was then separately applied to the control and autism groups. Additionally, a two-sided independent-sample t test was also used to assess the changes in PAC values in the autism group under placebo and different drug conditions, with the control group under placebo serving as the baseline. The results were corrected for multiple comparisons using the Benjamini-Hochberg method. Lastly, age was added as a fixed variable to the LMM model, and the analysis was repeated to control for any potential age effect on the main outcomes.
To identify discriminative between-network connections, we first conducted a two-sided independent-sample t-test to compare the PAC strength of each connection between the control group and the autism group. Given the large number of point-to-point connections, we applied a rigorous threshold (p < 1 \(\times\) 10−4) to determine statistical significance and control for multiple comparisons.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Data have been deposited in PAC_across_networks and are accessible in the data section of the repository at (https://github.com/didi226/PAC_across_networks). Source data are hosted in this repository. All other data are available from the corresponding author upon reasonable request.
Code availability
Custom code used for data processing, analysis, and figure generation is available in the code section of PAC_across_networks repository (https://github.com/didi226/PAC_across_networks). All scripts, parameters, and settings required to reproduce the analyses are provided within the repository.
References
Lord, C. et al. Autism spectrum disorder. Nat. Rev. Dis. Prim. 6, 1–23 (2020).
Rubenstein, J. & Merzenich, M. M. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes, Brain Behav. 2, 255–267 (2003).
Peñagarikano, O. et al. Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell 147, 235–246 (2011).
Paulsen, B. et al. Autism genes converge on asynchronous development of shared neuron classes. Nature 602, 268–273 (2022).
Fatemi, S. H., Reutiman, T. J., Folsom, T. D. & Thuras, P. D. GABA A receptor downregulation in brains of subjects with autism. J. Autism Dev. Disord. 39, 223 (2009).
Oblak, A. L., Gibbs, T. T. & Blatt, G. J. Decreased GABAB receptors in the cingulate cortex and fusiform gyrus in autism. J. Neurochem. 114, 1414–1423 (2010).
Gaetz, W. et al. GABA estimation in the brains of children on the autism spectrum: measurement precision and regional cortical variation. Neuroimage 86, 1–9 (2014).
Robertson, C. E., Ratai, E.-M. & Kanwisher, N. Reduced GABAergic action in the autistic brain. Curr. Biol. 26, 80–85 (2016).
Krystal, J. et al. Glutamate and GABA systems as targets for novel antidepressant and mood-stabilizing treatments. Mol. Psychiatry 7, S71–S80 (2002).
Möhler, H. The GABA system in anxiety and depression and its therapeutic potential. Neuropharmacology 62, 42–53 (2012).
Whelan, T. P. et al. Editorial Perspective: Bridging the translational neuroscience gap in autism—development of the ‘shiftability’ paradigm. J. Child Psychol. Psychiatry 65, 862–865 (2024).
Huang, Q. et al. GABAB receptor modulation of visual sensory processing in adults with and without autism spectrum disorder. Sci. Transl. Med. 14, eabg7859 (2022).
Huang, Q. et al. Retinal GABAergic alterations in adults with autism spectrum disorder. J. Neurosci. 44, e1218232024 (2024).
Huang, Q. et al. Exploratory evidence for differences in GABAergic regulation of auditory processing in autism spectrum disorder. Transl. Psychiatry 13, 320 (2023).
Lord, C., Elsabbagh, M., Baird, G. & Veenstra-Vanderweele, J. Autism spectrum disorder. Lancet 392, 508–520 (2018).
D. American Psychiatric Association, A. P. Association, Diagnostic and Statistical Manual of Mental Disorders: DSM-5 Vol. 5 (American psychiatric association, 2013)
Desaunay, P. et al. Memory in autism spectrum disorder: A meta-analysis of experimental studies. Psychol. Bull. 146, 377 (2020).
Dawson, M., Mottron, L. & Gernsbacher, M. A. Learning in autism. Learn. Mem.: A Compr. Ref. 2, 759–772 (2008).
Lai, M.-C. et al. Prevalence of co-occurring mental health diagnoses in the autism population: a systematic review and meta-analysis. Lancet Psychiatry 6, 819–829 (2019).
Kitzbichler, M. G. et al. Altered development and multifaceted band-specific abnormalities of resting state networks in autism. Biol. psychiatry 77, 794–804 (2015).
Gutierrez, R. et al. Altered synchrony and connectivity in neuronal networks expressing an autism-related mutation of neuroligin 3. Neuroscience 162, 208–221 (2009).
Belmonte, M. K. et al. Autism and abnormal development of brain connectivity. J. Neurosci. 24, 9228–9231 (2004).
Holiga, Š et al. Patients with autism spectrum disorders display reproducible functional connectivity alterations. Sci. Transl. Med. 11, eaat9223 (2019).
Lisman, J. E. & Jensen, O. The theta-gamma neural code. Neuron 77, 1002–1016 (2013).
Canolty, R. T. et al. High gamma power is phase-locked to theta oscillations in human neocortex. Science 313, 1626–1628 (2006).
Canolty, R. T. & Knight, R. T. The functional role of cross-frequency coupling. Trends Cogn. Sci. 14, 506–515 (2010).
Hyafil, A., Giraud, A.-L., Fontolan, L. & Gutkin, B. Neural cross-frequency coupling: connecting architectures, mechanisms, and functions. Trends Neurosci. 38, 725–740 (2015).
Onslow, A. C., Jones, M. W. & Bogacz, R. A canonical circuit for generating phase-amplitude coupling. PLoS ONE 9, e102591 (2014).
Zhang, X., Kendrick, K. M., Zhou, H., Zhan, Y. & Feng, J. A computational study on altered theta-gamma coupling during learning and phase coding. PLoS ONE 7, e36472 (2012).
White, J. A., Banks, M. I., Pearce, R. A. & Kopell, N. J. Networks of interneurons with fast and slow γ-aminobutyric acid type A (GABAA) kinetics provide substrate for mixed gamma-theta rhythm. Proc. Natl. Acad. Sci. 97, 8128–8133 (2000).
Berman, J. I. et al. Alpha-to-gamma phase-amplitude coupling methods and application to autism spectrum disorder. Brain Connectivity 5, 80–90 (2015).
Port, R. G. et al. Children with autism spectrum disorder demonstrate regionally specific altered resting-state phase–amplitude coupling. Brain Connectivity 9, 425–436 (2019).
Yeo, B. T. et al. The organization of the human cerebral cortex estimated by intrinsic functional connectivity. J. Neurophysiol. 106, 1125–1165 (2011).
Wechsler, D. Wechsler Abbreviated Scale of Intelligence Second Edition (WASI-II) (Pearson, 2011).
Sacks, D. D. et al. Longitudinal associations between resting-state, interregional theta-beta phase-amplitude coupling, psychological distress, and wellbeing in 12–15-year-old adolescents. Cereb. Cortex 33, 8066–8074 (2023).
Chaieb, L. et al. Theta-gamma phase-phase coupling during working memory maintenance in the human hippocampus. Cogn. Neurosci. 6, 149–157 (2015).
Grabot, L. et al. The strength of alpha–beta oscillatory coupling predicts motor timing precision. J. Neurosci. 39, 3277–3291 (2019).
Port, R. G. et al., Children with autism spectrum disorder demonstrate regionally specific altered resting-state phase–amplitude coupling. Brain Connect. 9, 425–436 (2019).
Young, C. K. & Eggermont, J. J. Coupling of mesoscopic brain oscillations: recent advances in analytical and theoretical perspectives. Prog. Neurobiol. 89, 61–78 (2009).
Jensen, O. & Colgin, L. L. Cross-frequency coupling between neuronal oscillations. Trends Cogn. Sci. 11, 267–269 (2007).
Kopell, N. J., Gritton, H. J., Whittington, M. A. & Kramer, M. A. Beyond the connectome: the dynome. Neuron 83, 1319–1328 (2014).
Poil, S.-S., Hardstone, R., Mansvelder, H. D. & Linkenkaer-Hansen, K. Critical-state dynamics of avalanches and oscillations jointly emerge from balanced excitation/inhibition in neuronal networks. J. Neurosci. 32, 9817–9823 (2012).
Buzsaki, G. & Draguhn, A. Neuronal oscillations in cortical networks. Science 304, 1926–1929 (2004).
Mejias, J. F., Murray, J. D., Kennedy, H. & Wang, X.-J. Feedforward and feedback frequency-dependent interactions in a large-scale laminar network of the primate cortex. Sci. Adv. 2, e1601335 (2016).
Hasenstaub, A. et al. Inhibitory postsynaptic potentials carry synchronized frequency information in active cortical networks. Neuron 47, 423–435 (2005).
Wulff, P. et al. Hippocampal theta rhythm and its coupling with gamma oscillations require fast inhibition onto parvalbumin-positive interneurons. Proc. Natl. Acad. Sci. 106, 3561–3566 (2009).
Tort, A. B., Rotstein, H. G., Dugladze, T., Gloveli, T. & Kopell, N. J. On the formation of gamma-coherent cell assemblies by oriens lacunosum-moleculare interneurons in the hippocampus. Proc. Natl. Acad. Sci. 104, 13490–13495 (2007).
Peterson, E. J. & Voytek, B. Balanced oscillatory coupling improves information flow. BioRxiv 030304 (2015).
Sohal, V. S. & Rubenstein, J. L. Excitation-inhibition balance as a framework for investigating mechanisms in neuropsychiatric disorders. Mol. Psychiatry 24, 1248–1257 (2019).
Hansen, J. Y. et al. Mapping neurotransmitter systems to the structural and functional organization of the human neocortex. Nat. Neurosci. 25, 1569–1581 (2022).
Zilles, K. & Palomero-Gallagher, N. Multiple transmitter receptors in regions and layers of the human cerebral cortex. Front. Neuroanat. 11, 78 (2017).
Puts, N. A. et al. Reduced GABA and altered somatosensory function in children with autism spectrum disorder. Autism Res. 10, 608–619 (2017).
Oblak, A., Gibbs, T. & Blatt, G. Decreased GABAA receptors and benzodiazepine binding sites in the anterior cingulate cortex in autism. Autism Res. 2, 205–219 (2009).
Horder, J. et al. GABAA receptor availability is not altered in adults with autism spectrum disorder or in mouse models. Sci. Translational Med. 10 (2018).
Marshall, F.H. in Inhibitory Regulation of Excitatory Neurotransmission. (ed. Darlison, M.G.) 87–98 (Springer-Verlag Berlin Heidelberg, 2008).
Kantamneni, S. Cross-talk and regulation between glutamate and GABAB receptors. Front. Cell. Neurosci. 9, 135 (2015).
Connelly, W. M. et al. GABAB receptors regulate extrasynaptic GABAA receptors. J. Neurosci. 33, 3780–3785 (2013).
Guo, B. et al. CB1R dysfunction of inhibitory synapses in the ACC drives chronic social isolation stress-induced social impairments in male mice. Neuron 112, 441–457. e446 (2024).
Felleman, D. J. & Van Essen, D. C. Distributed hierarchical processing in the primate cerebral cortex. Cereb. Cortex) 1, 1–47 (1991).
Chen, Q. et al. Dysfunction of cortical GABAergic neurons leads to sensory hyper-reactivity in a Shank3 mouse model of ASD. Nat. Neurosci. 23, 520–532 (2020).
Robertson, C. E. & Baron-Cohen, S. Sensory perception in autism. Nat. Rev. Neurosci. 18, 671–684 (2017).
Catani, M., Dell’Acqua, F. & De Schotten, M. T. A revised limbic system model for memory, emotion and behaviour. Neurosci. Biobehav. Rev. 37, 1724–1737 (2013).
Dawson, G., Meltzoff, A. N., Osterling, J. & Rinaldi, J. Neuropsychological correlates of early symptoms of autism. Child Dev. 69, 1276–1285 (1998).
Bachevalier, J. Medial temporal lobe structures and autism: a review of clinical and experimental findings. Neuropsychologia 32, 627–648 (1994).
Stephens, K. et al. Associations between limbic system white matter structure and socio-emotional functioning in children with ADHD+ ASD. J. Autism Dev. Disord. 51, 2663–2672 (2021).
Haznedar, M. M. et al. Limbic circuitry in patients with autism spectrum disorders studied with positron emission tomography and magnetic resonance imaging. Am. J. Psychiatry 157, 1994–2001 (2000).
Froestl, W. Chemistry and pharmacology of GABAB receptor ligands[M]. in Advances in Pharmacology (eds Blackburn, T. P. & Enna, S. J.) 58, 19–62 (Academic Press, 2010).
Erickson, C. A. et al. STX209 (arbaclofen) for autism spectrum disorders: an 8-week open-label study. J. Autism Dev. Disord. 44, 958–964 (2014).
Silverman, J. L. et al. GABA B receptor agonist R-baclofen reverses social deficits and reduces repetitive behavior in Two mouse models of autism. Neuropsychopharmacology 40, 2228–2239 (2015).
Han, S. et al. Autistic-like behaviour in Scn1a+/− mice and rescue by enhanced GABA-mediated neurotransmission. Nature 489, 385–390 (2012).
Gandal, M. et al. GABAB-mediated rescue of altered excitatory–inhibitory balance, gamma synchrony and behavioral deficits following constitutive NMDAR-hypofunction. Transl. Psychiatry 2, e142–e142 (2012).
Veenstra-VanderWeele, J. et al. Arbaclofen in children and adolescents with autism spectrum disorder: a randomized, controlled, phase 2 trial. Neuropsychopharmacology 42, 1390–1398 (2017).
Chaste, P. & Leboyer, M. Autism risk factors: genes, environment, and gene-environment interactions. Dialogues Clin. Neurosci. 14, 281–292 (2012).
Mottron, L. & Bzdok, D. Autism spectrum heterogeneity: fact or artifact?. Mol. Psychiatry 25, 3178–3185 (2020).
Lord, C., Rutter, M. & Le Couteur, A. Autism Diagnostic Interview-Revised: a revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J. Autism Dev. Disord. 24, 659–685 (1994).
Lord, C. et al. The Autism Diagnostic Observation Schedule—Generic: a standard measure of social and communication deficits associated with the spectrum of autism. J. Autism Dev. Disord. 30, 205–223 (2000).
Sanchez-Ponce, R. et al. Metabolic and pharmacokinetic differentiation of STX209 and racemic baclofen in humans. Metabolites 2, 596–613 (2012).
Jasper, H. H. The ten-twenty electrode system of the International Federation. Electroencephalogr. Clin. Neurophysiol. 10, 370–375 (1958).
Wu, W. et al. An electroencephalographic signature predicts antidepressant response in major depression. Nat. Biotechnol. 38, 439–447 (2020).
Delorme, A. & Makeig, S. EEGLAB: an open source toolbox for analysis of single-trial EEG dynamics including independent component analysis. J. Neurosci. Methods 134, 9–21 (2004).
Jiang, L. et al. Spatial–rhythmic network as a biomarker of familial risk for psychotic bipolar disorder. Nat. Ment. Health 1, 887–899 (2023).
Tadel, F., Baillet, S., Mosher, J. C., Pantazis, D. & Leahy, R. M. Brainstorm: a user-friendly application for MEG/EEG analysis. Comput. Intell. Neurosci. 2011, 879716 (2011).
Pascual-Marqui, R. D. Standardized low-resolution brain electromagnetic tomography (sLORETA): technical details. Methods Find. Exp. Clin. Pharm. 24, 5–12 (2002).
Jiang, H., Bahramisharif, A., van Gerven, M. A. & Jensen, O. Measuring directionality between neuronal oscillations of different frequencies. Neuroimage 118, 359–367 (2015).
Acknowledgements
This project was funded by an Independent Investigator Award (to G.M.M.) from the Brain & Behavior Research Foundation and by Clinical Research Associates, L.L.C., an affiliate of the Simons Foundation. Support is also acknowledged from Autistica (to A.C.P.) and the Institute for Translational Neurodevelopment at King’s College London and EU-AIMS (European Autism Interventions)/EU AIMS-2-TRIALS, an Innovative Medicines Initiative Joint Undertaking under Grant Agreement No. 777394. Q.H. is funded by the National Natural Science Foundation under Grant No. 62406173 and No. 2024A1515011690, also partially supported by the Young Talent Support Project under Grant No. 2023QN100110 and No. 2024D01J0075. In addition, this paper represents independent research part funded by the National Institute for Health and Care Research (NIHR) Maudsley Biomedical Research Centre (BRC) at South London and Maudsley NHS Foundation Trust and King’s College London. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR, or the Department of Health and Social Care.
Author information
Authors and Affiliations
Contributions
G.M.M. conceived the study. E.D., D.G.M.M., and G.M.M.designed the study. Q.H., D.C., A.C.P., A.L., C.L.E, H.V., C.M.P., and G.M.M. advised on the methods. A.C.P., A.L., H.V., M.D., C.L.E, F.M.P., N.M.L.W., E.D., and L.K. collected the data. Q.H. and D.C. preprocessed the data. Q.H. and D.C. analyzed the data. Q.H., D.C., and G.M.M. drafted the manuscript. All authors edited and approved the final draft.
Corresponding authors
Ethics declarations
Competing interests
The authors acknowledge that this investigator-initiated study was part-funded by Clinical Research Associates, a subsidiary of the Simons Foundation. This included the donation of arbaclofen. The funders did not influence the study design, its conduct or decision to publish outcomes. No other competing interests are declared.
Peer review
Peer review information
Communications Biology thanks Stavros Trakoshis, Talitha C. Ford and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Benjamin Bessieres. [A peer review file is available].
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Huang, Q., Chen, D., Pereira, A.C. et al. Differential GABA dynamics across brain functional networks in autism. Commun Biol 9, 283 (2026). https://doi.org/10.1038/s42003-026-09563-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-026-09563-5
