
Abstract
Citrin deficiency (CD) is caused by the inactivation of SLC25A13, a mitochondrial membrane protein required to move electrons from cytosolic NADH to the mitochondrial matrix in hepatocytes. People with CD do not like sweets. Here we show that SLC25A13 loss causes the accumulation of glycerol-3-phosphate (G3P), which activates the carbohydrate response element-binding protein (ChREBP) to transcribe FGF21, which acts in the brain to restrain intake of sweets and alcohol and to transcribe key genes driving lipogenesis. Mouse and human data suggest that G3P–ChREBP is a mechanistic component of the Randle Cycle that contributes to metabolic-dysfunction-associated steatotic liver disease and forms part of a system that communicates metabolic states from the liver to the brain in a manner that alters food and alcohol choices. The data provide a framework for understanding FGF21 induction in varied conditions, suggest ways to develop FGF21-inducing drugs and suggest potential drug candidates for lean metabolic-dysfunction-associated steatotic liver disease and support of urea cycle function in CD.
Similar content being viewed by others


Main
Citrin deficiency (CD) is an autosomal recessive disease caused by mutation of the citrin gene, SLC25A13, which encodes a mitochondrial membrane protein highly expressed in hepatocytes with a key role in moving high energy electrons from the cytosol to the mitochondrial matrix1,2. Infants with CD are diagnosed in their first month with jaundice and elevated circulation of ammonia, citrulline and arginine, resembling a urea cycle disorder3, coincident with elevated lactate, resembling a mitochondrial disease4. Though CD is panethnic5, it is most frequently diagnosed in the Far East. Data indicate a pathological allele frequency of up to 1 in 28 in southern China, 1 in 45 across other parts of China6 and 1 in 50–100 elsewhere in the Far East7. CD is underdiagnosed outside of the Far East8 with a global disease burden that remains not well calculated and with unknown effects for SLC25A13-mutation carriers.
By weight, carbohydrates constitute the largest class of macronutrient in human breast milk9 such that infant livers are primed to use carbohydrates as fuel, which requires nicotinamide adenine dinucleotide (NAD) coenzymes in the hydride-accepting NAD+ form in both the cytosol and the mitochondrial matrix10. As shown in Fig. 1a, glycolysis yields pyruvate and NADH in the cytosol. Whereas pyruvate can be transported to the mitochondrial matrix for further oxidation11, cytosolic NADH does not cross the mitochondrial membrane12. Rather, the high energy electrons—termed reducing equivalents—picked up by NAD+ are moved to mitochondrial electron transport by two major NADH shuttle systems, the malate–aspartate (Asp) shuttle (MAS)13 and the glycerol-3-phosphate (G3P) dehydrogenase shuttle (GPDS)14. In hepatocytes, SLC25A13 is the component of the MAS that mediates Asp entry into cytosol in exchange for glutamate (Glu) transport into the mitochondrial matrix15. Another Glu/Asp antiporter encoded by SLC25A12 has higher expression in brain and muscle—its expression in hepatocytes is considered a primary mechanism of disease modification and/or escape in CD16.

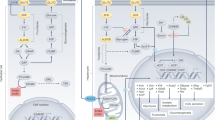
FGF21 is induced in a wide variety of conditions that have eluded a unified theory of induction. The following diagrams of metabolite flow in hepatocytes as affected by conditions of metabolic stress led us to propose G3P as the activator of ChREBP that can resolve the paradoxes of FGF21. a, The metabolic flow in unperturbed hepatocytes is facilitated by two NADH shuttles without the induction of FGF21. The MAS1 facilitates the transfer of reducing equivalents from cytosolic NADH to oxaloacetate, transiently forming malate, which is reoxidized at complex I in the mitochondrial matrix. The GPDS13 facilitates the transfer of reducing equivalents from cytosolic NADH to DHAP, transiently forming G3P with reducing equivalents transferred to FAD, forming FADH2, which is reoxidized with electron transfer to coenzyme Q in the mitochondrial electron transfer chain (METC). b, With either mitochondrial insufficiency, disruption of the MAS in CD, disruption of the GPDS, ethanol metabolism or elevated glucose, cytosolic NADH is expected to rise, which would be expected to cause the accumulation of G3P, which we propose to be the activator of ChREBP, driving the FGF21 expression. c, Fructolysis is predicted to elevate G3P, with one G3P equivalent formed from DHAP and another formed from glyceraldehyde (GA) via the function of alcohol dehydrogenase (ADH) and glycerol kinase107. The resulting G3P is proposed as the activating ligand for ChREBP activation and FGF21 expression. d, Triglyceride lipolysis is expected to produce glycerol and free fatty acids. Conversion of glycerol to G3P is proposed to activate ChREBP, which would co-operate with free fatty-acid-activated PPARα to drive transcription of FGF21. OAA, oxaloacetate; α-KG, α-ketoglutarate; 1,3-BPG, 1-3-bisphosphoglycerate; FFA, free fatty acid. Figure created with BioRender.com.
The resemblance of CD to a mitochondrial disease can be explained by failure of CD hepatocytes to obtain mitochondrial energy from complete oxidation of carbohydrates, with elevated lactate being an expected outcome of elevated cytosolic NADH. The resemblance of CD to a urea cycle disorder can be explained by a deficiency in hepatocytosolic Asp, which is required for the citrulline-consuming step of the urea cycle5. After diagnosis, children with CD, classified as cases of neonatal intrahepatic cholestasis caused by CD2,3, are managed nutritionally with a diet in which carbohydrates are largely replaced by medium chain triglycerides (MCTs)5. In most cases, neonatal intrahepatic cholestasis caused by CD goes into remission, and people with CD are able to grow and lead relatively normal lives, though they do not like sweets3,17 and are prone to metabolic-dysfunction-associated steatotic liver disease (MASLD) despite a lean body mass18. In other cases, there is a failure to thrive with dyslipidemia caused by CD. In adulthood, CD symptomology can reactivate with diagnoses of adult-onset type II citrullinemia or adult-onset CD, which are characterized by hyperammonaemia, MASLD, pancreatitis and neuropsychiatric complications2,3. The conventional nutritional management of hyperammonaemia, that is, a lower protein, higher carbohydrate diet, does not benefit patients with CD19.
When Slc25a13 was knocked out in mice, there was no clear physiological phenotype20. Reasoning that the GPDS is more highly expressed in mouse liver than in human liver, researchers proceeded to inactivate the GPDS component Gpd2. Mice homozygous for the disruption of Slc25a13 and Gpd2 formed an excellent CD model, exhibiting hyperlactataemia, hyperammonaemia, hyperargininemia, hypercitrullinemia and MASLD14. When provided a choice between water and saccharine, Slc25a13−/−Gpd2−/− mice behaved similarly to wild-type mice, choosing saccharine by a wide margin, indicating that there is no defect in detection of or desire for sweet taste in naive mice with an inactivation of the two NADH shuttle systems21. However, when provided with a choice between sucrose and water, Slc25a13−/−Gpd2−/− double mutants fail to prefer sucrose, suggesting that incomplete carbohydrate oxidation is required to produce the carbohydrate-aversive phenotype. Though wild-type mice consumed greater than 15 g of sucrose or 3 g of ethanol or 4 g of glycerol per 25-g mouse per day, Slc25a13−/−Gpd2−/− double mutants had impaired preferences for sucrose, ethanol and glycerol when given a choice between water and these energy-containing liquids21. Single homozygous mutants in the MAS component Slc25a13 and the GPDS component Gpd2 generally had intermediate phenotypes. Metabolomic analysis showed the accumulation of glycerol and G3P in Slc25a13−/−Gpd2−/− double mutants21. These data suggested that loss of the MAS and GPDS coupled with the provision of specific macronutrients result in a type of metabolic stress that leads to a behavioural change to avoid these compounds.
Fibroblast growth factor 21 (FGF21) is a secreted polypeptide made in the liver, adipose and muscle in response to a wide variety of stress conditions22. FGF21 has multiple sites of action including specific β-klotho-expressing regions of the brain, where FGF21 functions to restrain the intake of sweets23 and ethanol24, and the periphery, where it increases energy expenditure25 and body temperature26. FGF21 was first termed a starvation hormone because it is released into circulation by the liver in response to fasting27,28 and ketogenic diet (KD)29 in mice. Though these conditions result in the release of free fatty acids, which activate PPARα function at peroxisome proliferator response elements30 in the FGF21 promoter, the deletion of PPARα from mouse liver did not completely eliminate the induction of FGF21 by KD29.
Literature on FGF21 induction is considered paradoxical31 because FGF21 is not only induced by fasting and the near absence of dietary carbohydrates but also induced by the provision of simple carbohydrates32,33,34, particularly fructose35,36. Moreover, in addition to being induced by fasting27,28,37 and exercise38,39, FGF21 is induced by refeeding40, obesity41, type 2 diabetes42 and mitochondrial disease43. It has also been shown that FGF21 is induced by a loss-of-function variant of glucokinase regulator GCKR44 and ethanol45 via reductive stress, that is, conditions that increase the NADH/NAD+ ratio in hepatocytes46, and by protein restriction47.
We considered whether mitochondrial disease, NADH shuttle disruption, ethanol, fructolysis and the conditions involving lipolysis, that is, fasting and exercise, might produce a common metabolite that would activate FGF21 transcription. As shown in Fig. 1b, we reasoned that mitochondrial disease, NADH shuttle disruption, elevated glycaemia and/or ethanol metabolism would elevate cytosolic NADH, leading to a buildup of G3P at the expense of dihydroxyacetone phosphate (DHAP). As shown in Fig. 1c, we reasoned that fructolysis could also lead to a buildup of G3P with G3P formed from DHAP and from glyceraldehyde. Finally, as shown in Fig. 1d, we reasoned that triglyceride lipolysis would produce glycerol and, consequently, G3P due to the activity of glycerol kinase.
Two of the key transcription factors acting within the FGF21 promoter are PPARα and carbohydrate response element-binding protein (ChREBP)31,34. Although it is clear that PPARα strongly contributes to turning on FGF21 in conditions of fasting27,28,29 and that FGF21 is both upstream and downstream of PPARα in the adaptive response to starvation28, the PPARα-independent component of FGF21 induction in mice fed a KD29 suggested the action of a different metabolite-sensing factor.
ChREBP, encoded by the MLXIPL gene, is a member of the MYC and MAX superfamily of heterodimerizing helix–loop–helix transcription factors that is abundantly expressed in the liver48,49,50. As a heterodimer with MAX-like protein X (MLX) and at elevated levels of glucose metabolites, ChREBP activates the transcription of target promoters containing carbohydrate response elements (ChoREs)51. Well-characterized ChoREs drive the ChREBP-dependent transcription of ChREBPβ—a shorter, carbohydrate-induced form of ChREBP52, liver pyruvate kinase (PKLR)53, FGF2131 and other genes that are important for carbohydrate adaptations including those for de novo lipogenesis51 such as Ac-coA lysase (ACLY), Ac-coA carboxylase (ACACA) and fatty acid synthetase (FASN).
In the extensive literature on ChREBP48,49,50, researchers have reported that ChREBP is activated by a glucose metabolite that engages the N-terminal glucose-sensing module (GSM)54, which is conserved between ChREBP and the related MondoA transcription factor55. The specific identity of this metabolite remains less clear, however, as evidence has been presented for glucose-6-phosphate (G6P)55,56, fructose-2-6-bisphosphate (F2,6BP)57 and xylulose-5-phosphate (X5P)58. It is difficult to distinguish between these proposed mechanisms because typical ChREBP activation conditions involve a shift from low glucose to high glucose that would simultaneously elevate all proposed ChREBP GSM-activating ligands, and none of these metabolites have been shown directly to bind the GSM. Moreover, it is challenging to reconcile the previously proposed ChREBP ligands with data showing that glycerol treatment strongly activates hepatic ChREBP in vivo59.
Recent work has shown that the key transcription factor for the ethanol induction of FGF21 is ChREBP and that the ChREBP transcription programme is downstream of an increase in the NADH/NAD+ ratio that occurred with a rise in a select group of metabolites that include G3P but not G6P or X5P60. We hypothesize that patients with CD and mouse models have a sweet- and ethanol-aversive phenotype because their livers activate a G3P–ChREBP–FGF21 transcription programme (Fig. 1b). Moreover, we suggest that this mechanism resolves what have been considered paradoxical aspects of FGF21 induction because fasting and refeeding, KD and simple carbohydrates, mitochondrial disease, ethanol and fructose all have direct paths to producing G3P (Fig. 1b–d).
Here, we show that G3P accumulates in the liver of the mouse model of CD and that ChREBP is activated in this model with the transcription of FGF21 and the activation of a lipogenic transcriptional programme. Our data further show that G3P is a specific ligand of the ChREBP GSM and suggest that features of the G3P–ChREBP activation mechanism can account for why fructose is more lipogenic than glucose, provide a unifying mechanism for non-alcoholic and alcoholic hepatic steatogenesis, resolve paradoxes of FGF21 expression, and explain key aspects of CD pathogenesis including lean MASLD, the favourable effects of MCTs and severe urea cycle dysfunction. This work suggests drug targets for treatment of CD, lean MASLD and common MASLD and also implicates CD mutation carriers, who number in the millions, as people with a distinct metabolic profile consistent with elevated circulation of FGF21.
Results
Deletion of NADH shuttle systems and provision of glycerol cause increased FGF21 circulation in mice
Sperm from C57BL/6J mice of genotype Slc25a13−/−Gpd2−/−(ref. 21) were used for the in vitro fertilization of C57BL/6J females. Subsequent crosses generated male and female Slc25a13−/− mice, Gpd2−/− mice and, at much lower than Mendelian ratios, Slc25a13−/−Gpd2−/− mice. Consistent with our first prediction, as shown in Fig. 2a, mice with deletion of either Slc25a13 or Gpd2 tended to have elevated circulating FGF21, whereas mice inactivated for both NADH shuttle systems had around threefold-elevated FGF21. This result was significant for mice of both sexes (Fig. 2a), for male mice (Fig. 2b) and for females (Fig. 2c). Mice of each genotype with 5% (w/v) glycerol in their drinking water for 2 days had significantly elevated circulating FGF21 by virtue of glycerol exposure, and the effects of genotype and glycerol were additive. The effect of glycerol on the induction of FGF21 was highly significant in each genotype irrespective of sex (Fig. 2a) and in males (Fig. 2b). Further, Slc25a13−/−Gpd2−/− females have significantly higher circulating FGF21 than wild-types or either single mutant upon exposure to glycerol (Fig. 2c). Thus, consistent with the hypothesis that sweet aversion in CD is due to a G3P–ChREBP–FGF21 induction programme, the mouse model of CD overproduces FGF21 and superinduces FGF21 when exposed to glycerol.

a, The serum FGF21 from the mice of the indicated genotypes was measured after 2 days with ad libitim access to food and water. (Wild type is represented by circles, n = 17; Slc25a13−/− is represented by up triangles, n = 20; Gpd2−/− is represented by the down triangles, n = 14; Slc25a−/−Gpd2−/− is represented by squares, n = 5). In parallel, additional mice were provided access to food and 5% (w/v) glycerol (wild type is represented by circles, n = 21; Slc25a13−/− is represented by up triangles, n = 18; Gpd2−/− is represented by down triangles, n = 13; Slc25a−/−Gpd2−/− is represented by squares, n = 7). The data show that across both sexes, Slc25a−/−Gpd2−/− mice have elevated FGF21 and that each genotype has its FGF21 circulation elevated by glycerol. b, Male subgroup analysis of food and water (wild type is represented by circles, n = 9; Slc25a13−/− is represented by up triangles, n = 14; Gpd2−/− is represented by the down triangles, n = 5; Slc25a−/−Gpd2−/− is represented by squares, n = 3) or food and 5% (w/v) glycerol (wild type is represented by circles, n = 10; Slc25a13−/−is represented by up triangles, n = 10; Gpd2−/− is represented by down triangles, n = 5; Slc25a−/−Gpd2−/− is represented by squares, n = 5). The data show that male Slc25a−/−Gpd2−/−mice have elevated FGF21 and that each genotype has its FGF21 circulation elevated by glycerol. c, Female subgroup analysis of food and water (wild type is represented by circles, n = 8; Slc25a13−/− is represented by up triangles, n = 6; Gpd2−/− is represented by down triangles, n = 9; Slc25a13−/−Gpd2−/− is represented by squares, n = 2) or food and 5% (w/v) glycerol (wild type is represented by circles, n = 11; Slc25a13−/− is represented by up triangles, n = 8; Gpd2−/− is represented by down triangles, n = 8; Slc25a13−/−Gpd2−/− is represented by the squares, n = 2). The data show that female Slc25a−/−Gpd2−/− mice have elevated FGF21 with respect to the wild type at baseline. In addition, the data show that female Slc25a−/−Gpd2−/− mice have elevated FGF21 on glycerol with respect to each of the other genotypes on glycerol. Significant differences were calculated by applying two-way analysis of variance and Tukey’s multiple comparisons test in which *P < 0.05, **P < 0.005, ***P < 0.0005 and ****P < 0.0001. The error bars represent means ± s.e.m. WT, wild type.
Deletion of NADH shuttle systems and provision of glycerol activate hepatic ChREBP, FGF21 and lipogenic transcription
To determine whether the deletion of NADH shuttle systems and/or provision of glycerol result in a ChREBP transcriptional programme, we collected livers from 40 mice representing water- and glycerol-exposed males of the four genotypes, prepared complementary DNA (cDNA) and performed bulk paired-end 150 base-pair RNA sequencing (RNA-seq)61 using an Illumina NovaSeq X Plus sequencer at >20 million paired reads per sample. As shown in Fig. 3, the ChREBP transcriptional programme is evident as judged by induction of well-characterized ChREBP target genes including ChREBPβ; Fgf21; Pklr; Khk, Aldob and Tkfc (the three key enzymes of fructolysis); Gpi1 and Pgd (phosphoglucose isomerase and 6-phosphogluconate dehydrogenase); Fasn, Elovl6 and Agpat2 (key enzymes for triglyceride synthesis); and Tm6sf2 (very low-density lipoprotein synthesis factor).

Hierarchical clustering of mean liver gene expression levels (fragments per kilobase of transcript per million mapped reads) of select ChREBP target genes across four genotypes of male mice exposed to either water (Wat) or glycerol (Gly). Experimental sample n numbers are provided in Table 1.
Many of the genes of de novo lipogenesis are dually activated by ChREBP and sterol response element-binding protein 1c (SREBP-1c)62, encoded by the Srebf1 gene. To test whether the mouse model of CD dysregulates either of these transcription factors and to examine the relationship with Fgf21 expression, we calculated the levels of specific transcripts using RSEM63. As presented in Extended Data Table 1, Srebf1 expression was unaffected by deletion of Slc25a13 and/or Gpd2 and was unaffected by addition of glycerol. The ChREBPβ transcript was induced by about 2.4-fold by glycerol in the wild-type strain, about 4.5-fold by loss of either of the NADH shuttle genes and nearly 13-fold by the inactivation of both genes. The ChREBPα transcript was largely unaffected by genotype but was induced approximately two- to threefold by glycerol in wild-type and single mutant strains. In the double mutant, which has the highest level of circulating FGF21 and the highest level of ChREBPβ transcript, there was no further induction of of ChREBP transcripts by glycerol. Consistent with the effect of FGF21 on feeding behaviours, at the mRNA level, glycerol induced Fgf21 to a greater degree in the wild-type and Slc25a13−/− strains in which basal Fgf21 expression was lowest and induced Fgf21 the least in the Gpd2−/− and Slc25a13−/−Gpd2−/− strains in which basal Fgf21 expression was highest. As the experiment was performed with 2 days of ad libitim access to 5% glycerol as the water source, and it has been shown that the CD mouse model has a sucrose, ethanol and glycerol-aversive phenotype21, one would expect that the high basal levels of FGF21 in the Slc25a13−/−Gpd2−/− strain significantly limit their glycerol intake and further increases in Fgf21 mRNA expression.
Deletion of NADH shuttle systems result in accumulation of hepatic G3P
We performed targeted a metabolomic analysis of liver extracts from male mice of the four genotypes to determine whether G3P or compounds previously termed ChREBP activators accumulate as a function of NADH shuttle disruption. As shown in Fig. 4a, G3P was quantified at ~1 mM in livers from wild-type mice. The deletion of Slc25a13, Gpd2 or both NADH shuttle genes resulted in levels of hepatic G3P that tended to be higher than levels of G3P in wild-type mouse livers. Similarly, the glycerol-exposed mouse livers of each of the four genotypes had levels of G3P that tended to be higher. On the basis that the ChREBPβ transcript is induced in Slc25a13−/−, Gpd2−/− and Slc25a13−/−Gpd2−/− mouse livers, we compared G3P levels in wild-type versus all mutant livers and observed a significant increase in G3P from 1.07 ± 0.20 mM to 1.95 ± 0.60 mM. As G6P55,56, F2,6BP57 and X5P58 have been previously proposed to be activating ligands of ChREBP, we tested whether hexose phosphates or pentose phosphates were altered by conditions that activate ChREBP, namely glycerol and the deletion of NADH shuttle genes. As shown in Fig. 4c,d, the hepatic concentrations of hexose phosphates were not altered by genotype and/or addition of glycerol. As shown in Fig. 4e,f, relative levels of pentose phosphates were also unaffected. Thus, consistent with the proposed G3P–ChREBP–FGF21 activation mechanism in Fig. 1b and the observation that Slc25a13−/−Gpd2−/− mice experiencing sweet- and ethanol-aversive behaviours have elevated hepatic G3P21, we show that deletion of the NADH shuttle systems is sufficient specifically to elevate the proposed ChREBP-activating ligand.

a–f, We performed quantitative metabolomics for hepatic G3P (a and b) and hexose phosphates (c and d) and qualitative metabolomics for hepatic levels of pentose phosphates (e and f). The numbers of mice analysed were wild type (represented by circles, n = 3), Slc25a13−/− (represented by up triangles, n = 5), Gpd2 (represented by down triangles, n = 5) and Slc25a13−/−Gpd2−/− (represented by squares, n = 2) in mice with 2 days of ad libitim access to food and water. The numbers of mice analysed were wild type (represented by circles, n = 5), Slc25a13−/− (represented by triangles, n = 5), Gpd2 (represented by down triangles, n = 5) and Slc25a13−/−Gpd2 −/− (represented by squares, n = 5) with 2 days of ad libitim access to food and 5% w/v glycerol. In a, c and f, all mouse genotypes were analysed separately. In b, d and f, the three mutant genotypes were pooled to compare wild type (represented by circles, n = 3) with the three genotypes containing one or both NADH shuttle systems disrupted (represented by filled squares, n = 12) with exposure to water. In a, the data show that each mutant genotype tends to have higher G3P than wild type and that glycerol tends to elevate G3P with respect to water. In b, the data show that the deletion of one or both NADH shuttle systems significantly elevates G3P (P = 0.03 by unpaired t-test, t = 2.43, degrees of freedom: 13). In c and d, no differences were observed in the hexose phosphate levels across the four genotypes and two conditions. In e and f, no differences were observed in the hexose phosphate levels across the four genotypes and two conditions. *P < 0.05. The error bars represent means ± s.e.m. n.s., not significant; WT, wild type.
Genetic manipulation of G3P drives ChREBP activation in a reconstituted system
HEK293T cells do not express ChREBP and show poor expression of MLX, thereby allowing the reconstitution of condition-dependent, ChREBP- and MLX-dependent, ChoRE-dependent transcription60. The robust ChoRE-luciferase activity in HEK293T cells depends on the introduction of both ChREBPα and MLX. The reconstituted transcriptional activity is depressed by the expression of L. brevis NADH oxidase (LbNOX), which lowers the NADH/NAD+ ratio, and increased by expression of Escherichia coli soluble transhydrogenase (EcSTH), an enzyme that uses reducing equivalents from NADPH to elevate the cytosolic NADH/NAD+ ratio60. In prior work, this system was used to show that three metabolites, namely G3P, glyceraldehyde-3-phosphate (GA3P) and fructose-1,6-bisphosphate (F1,6BP), were highly correlated with ChREBPα activation but that levels of G6P and X5P were uncorrelated with ChREBP transcriptional activity60. To distinguish between potential ChREBPα-activating ligands, we introduced glycerol kinase (GK), GPD1 and GAPDH genes into the reconstituted ChREBPα-MLX HEK293T system alongside green fluorescent protein (GFP), LbNOX and EcSTH as inactive, ChoRE-luciferase-depressing and ChoRE-luciferase-activating controls, respectively. Though GAPDH had a minor ChoRE-luciferase depressing effect, and GK was without effect potentially due to the absence of a supply of glycerol, GPD1 strongly increased ChoRE-luciferase activity (Fig. 5a). Relative quantification of 137 metabolites showed that in these cells, GPD1 strongly depressed levels of GA3P and DHAP, while elevating G3P. The correlation coefficient (CC) for luciferase activity with relative G3P in the six resulting HEK293T transfected cell lines was 0.96 (Fig. 5b), exceeding all the other compounds tested (Supplementary Data). G6P (Fig. 5c), which has been considered a candidate ChREBP-activating ligand55,56, was uncorrelated with ChoRE-luciferase activity (CC of −0.11). GA3P (Fig. 5d), which had appeared to be correlated to ChoRE-luciferase activity in prior work60, became uncorrelated with inclusion of the effects of GK, GPD1 and GAPDH (CC of 0.18).

a, HEK293T cells cotransfected with ChREBPα, MLX, ChoRE-luciferase and the indicated genetic constructs were assessed for ChoRE-luciferase activity. The data show that LbNOX depresses, EcSTH increases and GPD1 greatly increases ChoRE-luciferase activity. Significant differences between were calculated by applying one-way analysis of variance and Dunnett’s multiple comparisons test in which ***P < 0.0005 and ****P < 0.0001. Numbers of biological replicates were: eGFP (n = 11); GK (n = 4); GPD1 (n = 3); GAPDH (n = 5); LbNOX (n = 3); and EcSTH (n = 3). b–d, Relative levels of 137 metabolites were determined by LC–MS; by plotting ChoRE-luciferase activity against each metabolite, we show that G3P is highly correlated (CC of 0.96) with ChREBPα-dependent ChoRE-luciferase activity (b), whereas G6P (c) and GA3P (d) are not. P values tested the hypothesis of a non-zero slope. e, HEK293T cells cotransfected with ChREBPβ or eGFP plus ChoRE-luciferase were assessed for ChoRE-luciferase activity. Numbers of biological replicates were: eGFP (n = 3); ChREBPβ (n = 3); unpaired t-test (two-tailed), P = 0.0248. f, The HEK293T cells cotransfected with ChREBPβ with MLX or eGFP plus ChoRE-luciferase were assessed for ChoRE-luciferase activity. The numbers of biological replicates were: eGFP (n = 3); MLX (n = 3); unpaired t-test (two-tailed), P = 0.0009. g, HEK293T cells cotransfected with MLX plus ChoRE-luciferase plus either ChREBPα or ChREBPβ were assessed for ChoRE-luciferase activity. Numbers of biological replicates were: ChREBPα (n = 3): ChREBP β (n = 3); unpaired t-test (two-tailed), P = 0.0178. h,i, Finally, HEK293T cells cotransfected with ChREBPβ, MLX and ChoRE-luciferase plus were assayed for ChoRE-luciferase activity with either eGFP or LbNOX (h) or EcSTH (i). There were three biological replicates for each sample. For h, the unpaired t-test (two-tailed): P = 0.7680. For i, the unpaired t-test (two-tailed): P = 0.5023. The data in e and i show that ChREBPβ has significant ChoRE-luciferase activity in HEK293T cells, e, that is, further boosted by MLX transfection, f. g, The data show that the ChoRE-luciferase activity of ChREBPβ-MLX exceeds that of ChREBPα-MLX. However, in contrast to the ability of LbNOX to depress and EcSTH to increase ChoRE-luciferase activity of ChREBPα-MLX, a, ChREBPβ-MLX was not regulated by either LbNOX, h, or EcSTH, i, thereby mapping the modulation of ChREBPα to the N-terminal GSM domain. Significant differences were calculated with unpaired t-tests in which *P < 0.05 and ***P < 0.0005. The error bars represent means ± s.e.m. n.s., not significant.
To further map the site of G3P activity, we used the HEK293T system to characterize the sensitivity of ChREBPβ-MLX to altered levels of metabolites. As shown in Fig. 5e–g, ChREBPβ—the form of ChREBP without the N-terminal GSM52—was introduced into HEK293T cells and shown to induce ChoRE-luciferase in a manner that depends on MLX cotransfection. Moreover, ChoRE-luciferase activation from ChREBPβ-MLX was about threefold more potent than that from ChREBPα-MLX (Fig. 5h). However, when ChREBPβ-MLX-dependent ChoRE-luciferase was challenged by LbNOX (Fig. 5i) and EcSTH (Fig. 5j) expression, there was no modulation of ChREBP transcriptional activity60. These data indicate that the metabolite that responds to an elevated NADH/NAD+ ratio thereby driving ChREBP-MLX transcription60 acts on the ChREBPα-specific N-terminus and is fully correlated with accumulation of G3P.
The GSM domain of ChREBP is a G3P-sensing module
Various constructs have been used to obtain structural or biophysical data on the GSM of ChREBP. Notably, a construct from residue 1 to 250 of mouse ChREBP was purified as a His-tagged protein in E. coli for structural characterization. However, this molecule was found to have been proteolyzed to a fragment of the GSM from residue 81 to 196 and, when mixed with the 14-3-3β protein for structural characterization, the full length of 14-3-3β and only residues 117–137 of ChREBP were structured64. Based on knowledge that both ChREBP and the homologous MondoA are responsive to glucose metabolites54,65, we performed a careful multiple sequence alignment of ChREBP and MondoA and chose to define residues 43–307 of mouse ChREBP (ChREBP43–307) as a candidate stable, globular GSM.
After expression of His-tagged ChREBP43-307 in E. coli and purification by nickel nitrilotriacetic acid affinity chromatography, we characterized ligand binding by isothermal titration calorimetry66. As presented in Table 1, we tested glucose, G6P, F6P, F1,6BP, GA3P, DHAP, G3P and X5P for binding and were able to detect saturable binding with each ligand. However, the equilibrium dissociation constant (Kd) values for all but two ligands were greater than 130 μM. G6P, which has been considered a candidate GSM ligand55,56 but is uncorrelated with ChREBP transcriptional activation60 (Fig. 5c), showed half-saturated binding at 64.3 ± 13.5 μM, suggesting an association that could be displaced by a higher-affinity ligand whose abundance is sensitive to conditions that activate ChREBP. Indeed, G3P, which is greatly increased by the deletion of the NADH shuttle systems (Fig. 4) and GPD1 overexpression (Fig. 5a) and which correlates with ChREBP activation (Fig. 5B), binds the GSM with a Kd value of 17.0 ± 1.1 μM. Thus, biophysical, genetic, metabolomic and cellular reconstitution data indicate that the GSM of ChREBP should be termed a G3P-sensing module that drives the transcription of ChREBPβ, FGF21 and other ChREBP target genes. Notably, this model can account for the PPARα-independent induction of FGF21 in conditions of lipolysis29; for fructose and glycerol as activators of ChREBP59; and for ethanol45, reductive stress46, hyperglycaemia42 and mitochondrial dysfunction43 as drivers of FGF21 transcription.
After this work was available in preprint form67, support for G3P as the activator of ChREBP was provided in an experiment in which reporter plasmid transcription from ChREBP and MLX was reconstituted in HEK293T cells and shown to increase in response to GPD1 overexpression68 as we showed in Fig. 5a. Moreover, these investigators added G3P to HEK293T cells expressing ChREBP and MLX to show that the G3P addition protected ChREBP from thermal precipitation68. As all models of ChREBP activation depend on new protein interactions upon small molecule engagement48,49, the cellular thermal denaturation assay does not constitute evidence for the direct binding of G3P. However, the work supports our physiological, metabolomic and biophysical evidence for G3P as the activator of ChREBP.
The G3P–ChREBP activation system suggests new mechanistic components of the Randle cycle and cooperation between ChREBP and other transcription factors
Philip Randle’s medical school lectures, published in 1963, contained two simple sketches that depicted what he termed the glucose fatty-acid cycle69. The first sketch schematized glucose as the source of G3P, which is the backbone for triglyceride synthesis through the Kennedy pathway70. The second sketch illustrated Randle’s observation that, in conditions of high fatty acid availability, there are mechanisms to block glycolysis and that, in conditions of high glucose availability, there are mechanisms to block fatty acid oxidation. In subsequent years, citrate production and inhibition of phosphofructokinase 1 were identified as mechanisms for fatty acid oxidation blocking glycolysis71. Regulation in what is termed the sweet side of the Randle cycle has been explained by the function of malonyl-coA as an essential substrate of fatty acid synthase and as an inhibitor of carnitine palmitoyltransferase 1, which mediates long-chain fatty acid (LCFA) entry into mitochondria72. Thus, when acetyl coA carboxylase converts cytosolic Ac-coA to malonyl-coA, it is committing carbon flow not only to synthesize LCFA but also to block LCFA oxidation.
Although Randle’s principles of fuel utilization have been important in guiding research and medicine, many problems in metabolism remain elusive. For example, though fructose is known to be more lipogenic than glucose, it is not clear that this is fully explained on the basis of the higher affinity of fructokinase for fructose versus glucokinase for glucose, and the bypass of phosphofructokinase regulation73. According to our model, ChREBP evolved specifically to respond to the formation of G3P by stimulating transcription of enzymes that convert carbohydrates to LCFAs and transcription of Kennedy pathway enzymes that link LCFAs to G3P in triglyceride synthesis. Fructose would thus be more lipogenic than glucose because it results in greater ChREBP activation59 because fructolysis (Fig. 1c) would tend to produce more G3P than glycolysis. Notably, because G3P is not a direct glycolytic intermediate such as DHAP or GA3P but rather an electron carrier in the GPDS, the production of G3P from carbohydrates is a signal of carbohydrate overload from diet, diabetes, GCKR variants or a signal that would be generated at normoglycaemia by fructose, ethanol, anoxia or mitochondrial insufficiency. Reductive stress was previously noted as a shared mechanism underlying metabolic features of both alcoholic and non-alcoholic hepatic steatosis via ChREBP activation60. Identification of G3P as the activator of ChREBP further unites the mechanisms of hepatic steatogenesis downstream of ethanol, fructose, hyperglycaemia and mitochondrial insufficiency.
We do not suggest that G3P-driven ChREBP-activated transcription is fully responsible for complex metabolic switches. Lipogenesis requires the activation of both ChREBP and SREBP-1c62, which occurs with depression of the carnitine palmitoyltransferase and beta oxidation systems. By contrast, fasting-induced lipolysis, which is expected to produce G3P and activate ChREBP, also activates PPARα and the beta oxidation programme27,28,29. It is thus to be expected that complex interactions between fatty acid ligands, PPAR isoforms, SREBP and other transcription factors modulate the metabolic outputs of ChREBP in changing conditions. Two expected differences between G3P formation from glucose, ethanol and mitochondrial insufficiency (Fig. 1b) and G3P formation from lipolysis is that lipolytic G3P formation is expected to require GK activity and to produce PPARα-activating fatty acids (Fig. 1d). Thus, it is interesting to note that GK expression has been shown to drive lipolytic gene expression in the mouse liver, though this was attributed to the transcriptional activation of SREBP-1c rather than enzymatic activity74.
The mouse CD model shows an ISR that may exacerbate urea cycle dysfunction
The integrated stress response (ISR) is an adaptive response to amino acid deprivation that is mediated by phosphorylation of eukaryotic translational initiation factor eIF2α, the production of specific transcription factors ATF4 and/or ATF5 and the resulting gene expression to restore protein homoeostasis75. Under conditions of protein limitation, new protein synthesis is focused on resolving amino acid deficiency. For example, FGF21 is an ISR-activated hormone that drives protein ingestion76, whereas SLC3A2 is an ISR-activated transporter that increases cellular amino acid import77. The classical ISR-transcribed enzymes asparagine (Asn) synthetase (ASNS)78 and cystathionine γ-lyase (CTH)79 are induced to produce Asn and cysteine (Cys), respectively.
Mitochondrial defects are known to induce the ISR80 via mechanisms that are not completely understood. However, it was shown that complex I inhibition of C2C12 mouse myoblasts induces the ISR by virtue of depressing Asp and Asn synthesis81. When amino acids are in limited supply, uncharged transfer RNAs (tRNAs) activate GCN2 protein kinase to phosphorylate eIF2α82. In myoblasts, the complex I inhibition-mediated induction of the ISR can be relieved by lowering the NADH/NAD+ ratio and by provision of Asp or Ans81.
RNA-seq data from the mouse model of CD supports Asp limitation as a mechanism by which the ISR can be activated by mitochondrial dysfunction in the liver. Further, the data suggest that the ISR may aggravate urea cycle dysfunction in CD. As shown in Fig. 6a, we noted that Asns78 is strongly upregulated with deletion of Slc25a13. Moreover, with the inactivation of both NADH shuttle systems (Fig. 6b), additional components of an ISR were observed including elevation of the Atf5 transcription factor83. Exposure of Slc25a13−/−Gpd2−/− mice to glycerol (Fig. 6c) increased expression of Slc3a277 and Cth79, which are recognized as part of the ISR84.

a–c, Differentially expressed hepatic genes shown in volcano plots for slc25a13−/− mice versus wild type (a), slc25a13−/−gpd2−/− mice versus wild type (b) and slc25a13−/−gpd2−/− glycerol-exposed mice versus wild-type water-exposed (c). d, Mitochondrial generation of Asp and the role of Slc25a13 in providing Asp to hepatocyte cytosol are depicted. Asp is consumed by Asn synthetase (Asns) to make Asn, is used in protein synthesis and is ligated to citrulline by argininosuccinate synthetase 1 (Ass1) in the urea cycle. e, An ISR is proposed to be initiated by shortage of cytosolic Asp and the consequent accumulation of uncharged tRNA Asp and/or tRNA Asn, which activate Gcn2 kinase activity on eIF2α. Subsequent induction of Atf5 and Atf5 target genes could aggravate the urea cycle dysfunction because Fgf21 drives protein ingestion, Slc3a2 increases amino acid uptake, Asns consumes Asp, further limiting the availability of Asp for nitrogen disposal, and Cth produces ammonia which may exacerbate the nitrogen disposal problem. FC, fold change; n.s., not significant; OAA, oxaloacetic acid. Panels d and e created with BioRender.com.
As depicted in Fig. 6d, Asp is produced in mitochondria in a process requiring the activities of MDH2, GOT2 and the mitochondrial electron transfer chain85. Asp enters the cytosol through SLC25A13, where Asp is used for protein synthesis, ASNS-dependent conversion to Asn and nitrogen disposal through the urea cycle. As in the case of myoblasts treated with piericidin to inhibit complex I, which produced an ISR that was reversed by the provision of Asp or Asn81, our data show that the loss of SLC25A13 is sufficient to induce an ISR. However, whereas the ISR evolved to be an adaptive system to resupply amino acids and restore protein homoeostasis75, it is potentially maladaptive with respect to the ammonia disposal problem in CD because FGF21 may cause people to eat more protein76, SLC3A2 will drive cellular amino acid import into hepatocytes already challenged by ammonia disposal77, ASNS expression would tend to commit urea-cycle-limiting levels of Asp for Asn synthesis78, and CTH enzyme activity directly produces ammonia79. Thus, we suggest that the partial inhibition of ATF5 and/or target enzymes such as ASNS and CTH could be considered as pharmacological approaches to improve urea cycle function in CD.
SLC25A13-mutation carriers have distinct metabolic traits and signs of potential FGF21 overexpression
Given the links between the loss of both copies of SLC25A13 with liver disease and dietary preference in humans, we considered whether heterozygosity for SLC25A13 disease mutations might be associated with anthropometrics, dietary patterns or unusual biomarkers. We used gnomAD86 to assemble a list of the most commonly occurring SLC25A13 alterations that are scored as pathological or likely pathological (Extended Data Table 2) and subjected them to several tests of genetic association. In deep phenotype genome-wide association studies, rs80338722 was moderately associated with body weight and body mass index in BioBank Japan (P = 8.3 × 10−6 and P = 4.1 × 10−4, respectively)87 and associated with triglyceride levels in non-diabetic individuals88, among other traits. Three additional alterations of SLC25A13 variants were associated with total cholesterol88, leptin88 and autoimmune hepatitis87. Our interpretation of these data is that, at a population level, SLC25A13 heterozygosity may bias the liver metabolism to produce greater G3P–ChREBP-dependent transcriptional output, which could either predispose to lipogenesis and/or produce a signal for elevated FGF21 as reported89 for the GCKR polymorphism44,46. Indeed, based on known biology90 and Mendelian randomization91, one might expect higher FGF21 to be associated with higher sodium clearance from the kidney and with dietary preference for fatty fish versus sweets76. As shown in Fig. 7a, a phenome-wide association study for rs80338722 within BioBank Japan showed an association with low body weight. When we aggregated rare variant gene-based burden tests between SLC25A13 alterations and 189 complex traits from the Common Metabolic Disease Knowledge Portal, the data suggested a distinct metabolic profile associated with low C-peptide and low incidence of type 1 diabetes but elevated levels of bilirubin and apolipoprotein B and a strong signal for high urinary sodium excretion (Fig. 7b). Consistent with the hypothesis that the inactivation of one copy of the SLC25A13 gene elevates FGF21, common variant gene-based tests for SLC25A13 and 189 complex traits from the Common Metabolic Disease Knowledge Portal88 identified a strong association with oily fish consumption (Fig. 7c). This result is complementary to reports that a loss-of-function variant in FGF21 is associated with higher sugar and alcohol consumption92 and less desire for fish93.

a, Phenome-wide association study for rs80338722 within BioBank Japan identified an association with low body weight. The x axis represents phenotypes within Biobank Japan, whereas the y axis is the −log10(P value) for the association between rs80338722 and each trait. The dashed grey line represents the moderate significance threshold (P < 1 × 10−3). b, The aggregated rare variant gene-based burden tests between SLC25A13 and 189 complex traits from the Common Metabolic Disease Knowledge Portal identified phenotypic associations, including high urinary sodium excretion and elevated bilirubin. The traits above the orange dashed line (significance threshold P < 5 × 10−2) are statistically significant. Tiangles pointing up indicate associations with increased trait levels or disease risk, whereas the triangles pointing down indicate associations with decreased traits levels or disease risk. c, Common variant gene-based tests for SLC25A13 and 189 complex traits from the Common Metabolic Disease Knowledge Portal identified a strong association with oily fish consumption. Traits above the orange dashed line (threshold P < 2.5 × 10−6) are nominally significant. ApoA, apolipoprotein A; ApoB, apolipoprotein B; BMI, body mass index; adj, adjusted; HDL, high density lipoprotein; ASCVD, atherosclerotic cardiovascular disease; NAFLD, nonalcoholic fatty liver disease; HOMA-IR, homeostatic model assessment of insulin resistance; CCS, coronary calcium score.
Data on human variation suggest that pathological SLC25A13 mutations appeared multiple times in the Far East15. Our data suggest that loss-of-function allele persistence may be mediated by beneficial effects of FGF21 expression on renal function, insulin sensitivity and food choices. Indeed, if it is true that SLC25A13-mutation carriers circulate higher levels of FGF21, one might expect that lower ethanol and/or fructose consumption would protect their livers from the potentially lipogenic combination of these energy inputs with diminished flux through the MAS. Specifically, one might predict that SLC25A13-mutation carriers drink less alcohol than the general population but that those mutation carriers who drink have a greater hazard of hepatic steatosis.
Discussion
Just as rare mutations in model organisms have revealed the complexities of morphogenesis and gene regulation, many fundamental biological insights have been revealed from rare human diseases. In the case of CD, it became apparent that without a key component of the MAS, people and mice are sensitive to the development of MASLD despite being lean and do not enjoy sweets despite an intact initial preference for sweets. We propose that a key to both of these CD presentations is the accumulation of hepatic G3P. The G3P-ChREBP programme drives a lipogenic transcriptional programme that induces Pklr, Acly, Acaca, Fasn, Elovl6 and other genes to synthesize LCFAs and Agpat2 and other genes to link newly synthesized LCFAs to the G3P backbone for triglyceride synthesis. Notably, the product of Acaca, malonyl-coA, is not only the key substrate for LCFA synthesis but also the key inhibitor of LCFA entry into mitochondria for β-oxidation72. Thus, G3P activation of ChREBP has the potential to transcriptionally direct triglyceride synthesis causing hepatic steatogenesis while also promoting resistance to hepatic lipolysis. Indeed, based on the low ATP state of the liver in CD models21, one might have expected brisk usage of stored hepatic triglycerides during the fasting daytime of CD mice or the fasting nighttime of patients with CD. However, the fact that lean MASLD is common in CD18 and the clinical observation that MCTs are preferable to common fats5 suggest that CPT1A may be inhibited in CD, thereby rendering stored triglycerides to be resistant to oxidation. In addition, at the RNA level, our data show that the Slc25a13−/−Gpd2−/−model significantly depresses expression of Cpt1a (Fig. 6b). Thus, patients with CD and others with MASLD driven by the proposed G3P–ChREBP lipogenesis programme may benefit from small molecule activators of CPT1A94. Additionally, it may be worth testing activators of AMP kinase95, which would be expected to turn off Ac-coA carboxylase and thereby relieve CPT1A inhibition.
It is notable that the FGF21 induction system responds to a number of conditions of metabolic stress including fasting and the ingestion of fructose and ethanol. Although FGF21 signals to the brain to limit sweets and alcohol and to eat protein76, it also makes complex signals to the periphery, which have the potential to treat common MASLD96. Discovery of the G3P–ChREBP induction system suggests strategies to develop lipidated prodrugs of G3P that would induce FGF21 expression, potentially in combination with fibrates to activate PPARα-dependent lipolysis and synergistically superinduce FGF2127,29. Although chronic FGF21-elevated conditions such as mitochondrial disease are potentially FGF21-resistant43, G3P-releasing FGF21-inducing prodrugs that increase energy expenditure and alter food choices could be valuable to address overweight and MASLD, particularly if combined with glucagon-like peptide-1 receptor agonists97.
Though ChREBP has long been known to connect carbohydrate oxidation to lipogenesis48,49,50, and G3P has long been known to serve as the backbone for triglyceride synthesis70 with a unique location in the glucose fatty-acid cycle69, the accumulation of G3P and induction of a G3P–ChREBP transcriptional programme in the mouse model of CD allowed us to propose that the most distinctive presentations of CD, namely sweet aversion, lean MASLD and the beneficial effects of MCTs in CD are due to G3P–ChREBP signalling. Ongoing work will further probe the components of FGF21 transcriptional induction mechanisms and determine how the ISR and ChREBP induction systems interact in health and disease.
Methods
Generation of mice and mouse samples
All mouse breeding and experiments were performed with protocols approved by the City of Hope Institutional Animal Care and Use Committee and Institutional Biosafety Committee. Sperm from a male Slc25a13−/−Gpd2−/− C57BL6/J mouse14 were a kind gift of Dr Saheki and the Citrin Foundation. In vitro fertilization was performed with C57BL6/J eggs and a pseudopregnant C57BL6/J female recipient by Walter Tsark in the City of Hope Transgenic Mouse Core. The resulting diheterozygous offspring were intercrossed to generate wild-type, Slc25a13−/−, Gpd2−/− and Slc25a13−/−Gpd2−/− mice of both sexes. At weaning, genotypes were determined by the polymerase chain reaction of tail tissues (Transnetyx), and mice were single-sex group-housed until experimental use. The mice were maintained at 21 °C under a standard 12–12-h light–dark cycle and provided with ad libitim access to food and water. For analysis of FGF21 expression, hepatic gene expression and metabolite accumulation, 8 to 16-week-old mice were single-housed with chow (LabDiet irradiated PicoLab Rodent Diet 20, 5053) and a single bottle of either water or 5% (w/v) glycerol for 2 days. Mice were then killed by decapitation via guillotine without sedation and exsanguinated with a funnel for blood collection into 1.5-ml microcentrifuge tubes on ice, and livers were freeze-clamped in liquid nitrogen immediately after collection98,99. Sera were obtained by centrifuging blood at 8,000g for 10 min at 4 °C. Liver samples were pulverized with a mortar and pestle cooled by liquid nitrogen. Data for all mouse samples are provided in Source Data Figs. 2–4.
FGF21 quantification
The Mouse/Rat FGF21 Quantikine ELISA Kit from R&D Systems (MF2100) was used for FGF21 quantification of mouse sera. A total of 50 μl of assay diluent RD1-41 was added to each well followed by equal volumes of standard, control and experimental samples. After incubation for 2 h at room temperature, wells were aspirated and washed five times with 400 μl of wash buffer. A total of 100 μl of horseradish-peroxidase-conjugated FGF21 antibody solution was then added to each well and incubated at room temperature for 2 h followed by five washes with 400 μl of wash buffer. After washing, 100 μl of freshly prepared substrate was added to each well and incubated for 30 min at room temperature. Assays were terminated with the addition of 100 μl of stop solution followed by reading at 450 nm with wavelength correction at 570 nm.
RNA-seq
The 20-mg liver aliquots were used for RNA isolation (Qiagen, RNeasy Mini Kit, 74104) and DNase I treatment (Qiagen, RNase-Free DNase Set, 79254). RNA integrity was assessed using an Agilent 4200 Tapestation, ensuring an RNA integrity number >5.0 for all samples. Library preparation and sequencing was performed by 7 Traits Genomics. Libraries were prepared using the NEBNext UltraExpress RNA Library Prep Kit (New England Biolabs), following the manufacturer’s protocol using 150 ng of RNA per sample. Messenger RNA (mRNA) was enriched using poly(A) selection. Enriched mRNA was fragmented, reverse-transcribed and converted to cDNA. The cDNA underwent end repair, adaptor ligation and size selection. Polymerase chain reaction amplification was performed (11 cycles) to ensure a sufficient library yield while minimizing amplification bias. Library quality and size distribution were assessed using a Tapestation and quantified by Qubit (Thermo Fisher Scientific). The libraries were pooled on an equimolar basis and sequenced on an Illumina NovaSeq X Plus platform using a 25B lane configuration with paired-end 150-bp reads with a minimum of 20 million paired reads per sample. Base calling and demultiplexing were performed using Illumina’s DRAGEN Bio-IT platform. Raw sequencing reads were processed for quality control using FastQC and trimmed to remove adaptors and low-quality bases using Trimmomatic100 and poly(A) tails using FASTP101. Processed reads were mapped back to the mouse genome (mm10) using STAR software (v. 2.6.0.a)102. HTSeq software (v.0.11.1) was used to generate the count matrix with default parameters103. Differential expression analysis was performed by normalizing read counts to expression values using the trimmed mean of M values normalization method in edgeR104,105. Generalized linear models were applied to identify differentially expressed genes between glycerol and water-exposed liver samples of the same genotype or between liver samples of different genotypes. Normalized expression levels from the trimmed mean of M values were used as the dependent variable, whereas sequencing batches were included as an independent variable to account for batch effects. Genes with a false-discovery-rate-adjusted Pvalue below 0.05 and a fold change greater than 2 or less than 0.5 were classified as significantly upregulated or downregulated, respectively.
Metabolomic analysis of mouse livers
For G3P quantification, 2.0-mg samples of pulverized frozen liver were spiked with 13C3-G3P (2.28 µM final concentration, Sigma-Aldrich). Frozen samples were rapidly processed with a boiling buffered ethanol extraction (600 µl of 25% 10 mM HEPES buffer and 75% ethanol) followed by vigorous vortexing. Samples were placed on a thermomixer for 5 min at 55 °C with shaking at 1,200 rpm then cooled on ice for 30 s. Samples were then placed in a water bath sonicator for 1 min followed by an additional 30 s on ice. Samples were clarified by centrifugation at 16,100g in a prechilled centrifuge at 4 °C; the supernatants were transferred to clean tubes followed by a second round of centrifugation. Clarified supernatants were transferred to new tubes and dried for 5 h in a vacuum centrifuge at 4 °C. Dried samples were resuspended in 80 µl of liquid chromatography–mass spectrometry (LC–MS) water. In total, 20-fold-diluted samples were transferred to mass spectrometry vials for analysis by liquid chromatography–tandem mass spectrometry. For the analysis of hexose phosphates and pentose phosphates, 2.0-mg samples of pulverized frozen liver were spiked with a 13C glucose-grown yeast extract (ISO101, Cambridge Isotope Laboratories) as an internal standard106, and the same workup was performed. The samples were analysed on a Vanquish Horizon UHPLC with a tandem Thermo Scientific Orbitrap Fusion mass spectrometer. The vials were maintained in autosampler at 4 °C. The instrument source parameters were held at 3 kV negative ion spray voltage, 300 °C ion transfer tube temperature, 250 °C vaporizer temperature, sheath gas of 20, auxiliary gas of 10 and sweep gas of 3. The liquid chromatography separation was carried out using an Acquity Premier HSS T3 column with VanGuard FIT, 1.8 µm, 2.1 mm × 150 mm with mobile phases A (5 mM tributylamine and 10 mM acetic acid with 5% v/v methanol in LC–MS-grade water) and B (LC–MS-grade methanol) at a constant flow rate of 0.5 ml min−1. The separation was carried out with a starting condition of 0% B; 0–10 min, 10.5% B; 10–18 min, 52.6% B; 18–19 min, 52.6% B; 19–20 min, 0% B; and 20–26 min, 0% B. Spectra for G3P were acquired using a targeted MS2 scan with parent ion of m/z 171.0058 with collision energy 25. Spectra for 13C3-G3P were acquired with parent ion m/z of 174.0165 with collision energy 25 with the primary fragment ion of m/z 78.9588. The hexose phosphate spectra were acquired using a targeted MS2 scan of parent ion m/z 259.0198. The 13C6-hexose phosphate spectra were acquired with a parent ion of m/z 265.0426 with collision energy of 20 and primary fragment ion m/z96.9690. The hexose phosphate spectra signals were predominantly from G6P, fructose-6-phosphate and glucose-1-phosphate, determined with pure synthetic standards. The pentose phosphate spectra were acquired using the targeted MS2 scan for the parent ion m/z 229.0124 at collision energy 30 with a primary fragment ion m/z of 78.9588. The pentose phosphate spectra were predominantly composed of ribose-5-phosphate, ribose-1-phosphate and X5P. The mass spectra data were analysed using the open source-software, Skyline v25.1.
Cellular reconstitution of ChREBP-MLX-dependent ChORE-luciferase activity
Human ChREBPα (accession number NM_032951.3), human ChREBPβ (accession number XM_047420437.1), human MLX (accession number NM_170607.3) and eGFP coding sequences were synthesized by Genewiz and cloned into pcDNA3.1 vectors (Invitrogen). GK was subcloned into pcDNA3.1 (Invitrogen) from pWZL-Neo-Myr-Flag-GK (Addgene plasmid 20493). pcDNA3.1-GPD1 and pcDNA3.1-GAPDH were purchased from GenScript (clone ID: OHu20325 and OHu20566). Plasmids pcDNA3.1-LbNOX, pcDNA3.1-EcSTH, pGL4.14 [luc2/Hygro]-ChoRE and pGL4.75[hRluc/CMV] were as described60. HEK293T cells (ATCC catalogue CRL-3216) were seeded into 24-well plates and maintained in Dulbecco’s modified Eagle medium (Gibco) with 10% foetal bovine serum (Gibco) and 1% Pen-Strep (Gibco) at 37 °C and 5% CO2 overnight. On the following day, media were replaced by OPTI-MEM (Gibco) 1 h before plasmid cotransfection. Lipofectamine 3000 reagent (Invitrogen) was used for cotransfections for 5 h, after which media were replaced with Dulbecco’s modified Eagle medium containing 10% foetal bovine serum for 48 h. The cells were then collected, and luciferase assays were performed using the Firefly and Renilla Single Tube Luciferase Assay Kit (Biotium) following the manufacturer’s protocol. A Tecan Infinite M Plex plate reader was used to measure Firefly and Renilla luminescence. Firefly luciferase values were normalized to corresponding Renillaluciferase measurements to correct for cell quantity and transfection efficiency. The underlying data are provided in Source Data Fig. 5.
Metabolomic analysis of transfected HEK293T cells
HEK293T cells were seeded into six-well plates overnight and then underwent plasmid cotransfection as described above for 48 h. Cells were then washed with phosphate-buffered saline, quenched with dry ice-cold 80% methanol and transferred to conical tubes. Relative quantification of 137 metabolites was performed as described60.
Biophysical characterization of the ChREBP GSM
The coding sequence for mouse ChREBPα (amino acids 43 to 307) was converted to E. coli-optimized codons and inserted into a pET vector carboxyl to MetHis6. The plasmid, pVB240306, was transformed into in E. coli Arctic Express, and expression was induced at 12.5 °C with 1 mM IPTG for 18 h. Total protein from the 1-litre culture was solubilized in 15 ml of denaturing lysis buffer (8 M urea, 100 mM NaH2PO4, 10 mM Tris Cl, pH 8.0, 0.05% Tween 20). The recombinant protein was captured by overnight incubation with 3 ml of Ni-NTA resin (Qiagen) followed by washing with 30 ml of denaturing buffer (8 M urea, 100 mM NaH2PO4, 10 mM Tris Cl, pH 6.3, 0.05% Tween 20). On-column protein refolding was performed by washing with 30 ml of 50 mM NaH2PO4, pH 8.0, 300 mM NaCl, 0.1% Triton X-100, followed by 30 ml of 50 mM NaH2PO4, pH 8.0, 300 mM NaCl, 5 mM β-cyclodextrin, 0.05% Tween 20 and 30 ml of 50 mM NaH2PO4, pH 8.0, 500 mM NaCl, 0.05% Tween 20. His-tagged ChREBP43–307 was recovered in 50 mM NaH2PO4, pH 8.0, 300 mM NaCl, 0.05% Tween 20, 350 mM imidazole at a yield of 1–2 mg l−1 of culture. All ligands were prepared in the same buffer. Ligand binding was determined by isothermal titration calorimetry using a Nano ITC (TA Instruments) to measure ligand concentration-dependent heat changes. Ligands were delivered in a 50-µl syringe to 185 µl of His-tagged ChREBP43–307 in the sample cell. After an initial 0.8-µl injection, 19 additional 2.5-µl injections were made with a time interval of 200 s between each injection. Measurements were made at 20 °C with stirring at 250 rpm. Informative ligand concentrations were determined empirically to observe saturable binding. Binding profiles were fitted to a blank and independent model with single-site binding using analysis software supplied with the instrument. Each experiment was performed in triplicate.
Statistics and reproducibility
Because the effect sizes and the variability of gene expression, FGF21 expression and metabolite levels were unknown until these experiments were performed, it was not possible to predetermine animal sample sizes. The size of mouse groups was limited by the availability of male and female Slc25a13−/−Gpd2−/− mice, which were obtained at lower than Mendelian ratios. Mice of each of the four genotypes were randomly assigned to water or glycerol exposure. Sera and frozen liver powders were stored in numbered tubes and were analysed by investigators who were blinded to genotype and treatment. Sera that were haemolysed were excluded from FGF21 quantification. Data distributions were assumed to be normal, but this was not formally tested. Statistical analysis was performed with GraphPad Prism software. Details of the statistical tests and the numbers of replicates are provided in the figure legends.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The mouse data, protein purification data and isothermal titration calorimetry data are available via OSF at https://osf.io/j5veb. The RNA-seq data are available via the Gene Expression Omnibus with accession no. GSE304958. The metabolomic data for Figs. 4 and 5 are available via the Metabolomics Workbench with accession nos. ST004126 and ST004125, respectively. The plasmids are available via Addgene. Source data are provided with this paper.
References
Palmieri, L. et al. Citrin and aralar1 are Ca2+-stimulated aspartate/glutamate transporters in mitochondria. EMBO J. 20, 5060–5069 (2001).
Saheki, T. & Kobayashi, K. Mitochondrial aspartate glutamate carrier (citrin) deficiency as the cause of adult-onset type II citrullinemia (CTLN2) and idiopathic neonatal hepatitis (NICCD). J. Hum. Genet. 47, 333–341 (2002).
Saheki, T. et al. Metabolic derangements in deficiency of citrin, a liver-type mitochondrial aspartate-glutamate carrier. Hepatol. Res. 33, 181–184 (2005).
Grunert, S. C. et al. Citrin deficiency mimicking mitochondrial depletion syndrome. BMC Pediatr. 20, 518 (2020).
Saheki, T., Moriyama, M., Funahashi, A. & Kuroda, E. AGC2 (citrin) deficiency—from recognition of the disease till construction of therapeutic procedures. Biomolecules https://doi.org/10.3390/biom10081100 (2020).
Lin, Y. et al. Combining newborn metabolic and genetic screening for neonatal intrahepatic cholestasis caused by citrin deficiency. J. Inherit. Metab. Dis. 43, 467–477 (2020).
Kikuchi, A. et al. Simple and rapid genetic testing for citrin deficiency by screening 11 prevalent mutations in SLC25A13. Mol. Genet. Metab. 105, 553–558 (2012).
Kido, J., Makris, G., Santra, S. & Haberle, J. Clinical landscape of citrin deficiency: a global perspective on a multifaceted condition. J. Inherit. Metab. Dis. 47, 1144–1156 (2024).
Kim, S. Y. & Yi, D. Y. Components of human breast milk: from macronutrient to microbiome and microRNA. Clin. Exp. Pediatr. 63, 301–309 (2020).
Belenky, P., Bogan, K. L. & Brenner, C. NAD+ metabolism in health and disease. Trends Biochem. Sci. 32, 12–19 (2007).
Bricker, D. K. et al. A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science 337, 96–100 (2012).
Goyal, S. et al. Dynamics of SLC25A51 reveal preference for oxidized NAD+ and substrate led transport. EMBO Rep. 24, e56596 (2023).
Dawson, A. G. Oxidation of cytosolic NADH formed during aerobic metabolism in mammalian cells. Trends Biochem. Sci. 4, 171–176 (1979).
Saheki, T. et al. Citrin/mitochondrial glycerol-3-phosphate dehydrogenase double knock-out mice recapitulate features of human citrin deficiency. J. Biol. Chem. 282, 25041–25052 (2007).
Tavoulari, S., Lacabanne, D., Thangaratnarajah, C. & Kunji, E. R. S. Pathogenic variants of the mitochondrial aspartate/glutamate carrier causing citrin deficiency. Trends Endocrinol. Metab. 33, 539–553 (2022).
Gonzalez-Moreno, L. et al. Exogenous aralar/slc25a12 can replace citrin/slc25a13 as malate aspartate shuttle component in liver. Mol. Genet. Metab. Rep. 35, 100967 (2023).
Saheki, T. et al. Reduced carbohydrate intake in citrin-deficient subjects. J. Inherit. Metab. Dis. 31, 386–394 (2008).
Komatsu, M. et al. Citrin deficiency as a cause of chronic liver disorder mimicking non-alcoholic fatty liver disease. J. Hepatol. 49, 810–820 (2008).
Fukushima, K. et al. Conventional diet therapy for hyperammonemia is risky in the treatment of hepatic encephalopathy associated with citrin deficiency. Intern. Med. 49, 243–247 (2010).
Sinasac, D. S. et al. Slc25a13-knockout mice harbor metabolic deficits but fail to display hallmarks of adult-onset type II citrullinemia. Mol. Cell. Biol. 24, 527–536 (2004).
Saheki, T. et al. Oral aversion to dietary sugar, ethanol and glycerol correlates with alterations in specific hepatic metabolites in a mouse model of human citrin deficiency. Mol. Genet. Metab. 120, 306–316 (2017).
Flippo, K. H. & Potthoff, M. J. Metabolic messengers: FGF21. Nat. Metab. 3, 309–317 (2021).
Jensen-Cody, S. O. et al. FGF21 signals to glutamatergic neurons in the ventromedial hypothalamus to suppress carbohydrate intake. Cell Metab. 32, 273–286 e276 (2020).
Flippo, K. H. et al. FGF21 suppresses alcohol consumption through an amygdalo-striatal circuit. Cell Metab. 34, 317–328 e316 (2022).
Xu, J. et al. Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet-induced obese mice. Diabetes 58, 250–259 (2009).
Zouhar, P. et al. A pyrexic effect of FGF21 independent of energy expenditure and UCP1. Mol. Metab. 53, 101324 (2021).
Inagaki, T. et al. Endocrine regulation of the fasting response by PPARα-mediated induction of fibroblast growth factor 21. Cell Metab. 5, 415–425 (2007).
Potthoff, M. J. et al. FGF21 induces PGC-1α and regulates carbohydrate and fatty acid metabolism during the adaptive starvation response. Proc. Natl Acad. Sci. USA 106, 10853–10858 (2009).
Badman, M. K. et al. Hepatic fibroblast growth factor 21 is regulated by PPARα and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 5, 426–437 (2007).
Forman, B. M., Chen, J. & Evans, R. M. Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors α and δ. Proc. Natl Acad. Sci. USA 94, 4312–4317 (1997).
Uebanso, T. et al. Paradoxical regulation of human FGF21 by both fasting and feeding signals: is FGF21 a nutritional adaptation factor? PLoS ONE 6, e22976 (2011).
Soberg, S. et al. FGF21 is a sugar-induced hormone associated with sweet intake and preference in humans. Cell Metab. 25, 1045–1053 e1046 (2017).
von Holstein-Rathlou, S. et al. FGF21 mediates endocrine control of simple sugar intake and sweet taste preference by the liver. Cell Metab. 23, 335–343 (2016).
Iroz, A. et al. A specific ChREBP and PPARα cross-talk is required for the glucose-mediated FGF21 response. Cell Rep. 21, 403–416 (2017).
Dushay, J. R. et al. Fructose ingestion acutely stimulates circulating FGF21 levels in humans. Mol. Metab. 4, 51–57 (2015).
Fisher, F. M. et al. A critical role for ChREBP-mediated FGF21 secretion in hepatic fructose metabolism. Mol. Metab. 6, 14–21 (2017).
Galman, C. et al. The circulating metabolic regulator FGF21 is induced by prolonged fasting and PPARα activation in man. Cell Metab. 8, 169–174 (2008).
Kim, K. H. et al. Acute exercise induces FGF21 expression in mice and in healthy humans. PLoS ONE 8, e63517 (2013).
Cuevas-Ramos, D. et al. Exercise increases serum fibroblast growth factor 21 (FGF21) levels. PLoS ONE 7, e38022 (2012).
Markan, K. R. et al. Circulating FGF21 is liver derived and enhances glucose uptake during refeeding and overfeeding. Diabetes 63, 4057–4063 (2014).
Zhang, X. et al. Serum FGF21 levels are increased in obesity and are independently associated with the metabolic syndrome in humans. Diabetes 57, 1246–1253 (2008).
Chen, W. W. et al. Circulating FGF-21 levels in normal subjects and in newly diagnose patients with type 2 diabetes mellitus. Exp. Clin. Endocrinol. Diabetes 116, 65–68 (2008).
Tyynismaa, H. et al. Mitochondrial myopathy induces a starvation-like response. Hum. Mol. Genet. 19, 3948–3958 (2010).
Cheung, C. Y. Y. et al. An exome-chip association analysis in Chinese subjects reveals a functional missense variant of GCKR that regulates FGF21 levels. Diabetes 66, 1723–1728 (2017).
Desai, B. N. et al. Fibroblast growth factor 21 (FGF21) is robustly induced by ethanol and has a protective role in ethanol associated liver injury. Mol. Metab. 6, 1395–1406 (2017).
Goodman, R. P. et al. Hepatic NADH reductive stress underlies common variation in metabolic traits. Nature 583, 122–126 (2020).
Laeger, T. et al. FGF21 is an endocrine signal of protein restriction. J. Clin. Invest. 124, 3913–3922 (2014).
Katz, L. S., Baumel-Alterzon, S., Scott, D. K. & Herman, M. A. Adaptive and maladaptive roles for ChREBP in the liver and pancreatic islets. J. Biol. Chem. 296, 100623 (2021).
Abdul-Wahed, A., Guilmeau, S. & Postic, C. Sweet sixteenth for ChREBP: established roles and future goals. Cell Metab. 26, 324–341 (2017).
Kawaguchi, T., Osatomi, K., Yamashita, H., Kabashima, T. & Uyeda, K. Mechanism for fatty acid “sparing” effect on glucose-induced transcription: regulation of carbohydrate-responsive element-binding protein by AMP-activated protein kinase. J. Biol. Chem. 277, 3829–3835 (2002).
Ma, L., Robinson, L. N. & Towle, H. C. ChREBP•Mlx is the principal mediator of glucose-induced gene expression in the liver. J. Biol. Chem. 281, 28721–28730 (2006).
Herman, M. A. et al. A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature 484, 333–338 (2012).
Shih, H. M., Liu, Z. & Towle, H. C. Two CACGTG motifs with proper spacing dictate the carbohydrate regulation of hepatic gene transcription. J. Biol. Chem. 270, 21991–21997 (1995).
Li, M. V., Chang, B., Imamura, M., Poungvarin, N. & Chan, L. Glucose-dependent transcriptional regulation by an evolutionarily conserved glucose-sensing module. Diabetes 55, 1179–1189 (2006).
McFerrin, L. G. & Atchley, W. R. A novel N-terminal domain may dictate the glucose response of Mondo proteins. PLoS ONE 7, e34803 (2012).
Li, M. V. et al. Glucose-6-phosphate mediates activation of the carbohydrate responsive binding protein (ChREBP). Biochem. Biophys. Res. Commun. 395, 395–400 (2010).
Arden, C. et al. Fructose 2,6-bisphosphate is essential for glucose-regulated gene transcription of glucose-6-phosphatase and other ChREBP target genes in hepatocytes. Biochem. J. 443, 111–123 (2012).
Kabashima, T., Kawaguchi, T., Wadzinski, B. E. & Uyeda, K. Xylulose 5-phosphate mediates glucose-induced lipogenesis by xylulose 5-phosphate-activated protein phosphatase in rat liver. Proc. Natl Acad. Sci. USA 100, 5107–5112 (2003).
Kim, M. S. et al. ChREBP regulates fructose-induced glucose production independently of insulin signaling. J. Clin. Invest. 126, 4372–4386 (2016).
Singh, C. et al. ChREBP is activated by reductive stress and mediates GCKR-associated metabolic traits. Cell Metab. 36, 144–158 (2024).
Kukurba, K. R. & Montgomery, S. B. RNA sequencing and analysis. Cold Spring Harb. Protoc. 2015, 951–969 (2015).
Linden, A. G. et al. Interplay between ChREBP and SREBP-1c coordinates postprandial glycolysis and lipogenesis in livers of mice. J. Lipid Res. 59, 475–487 (2018).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf. 12, 323 (2011).
Ge, Q. et al. Structural characterization of a unique interface between carbohydrate response element-binding protein (ChREBP) and 14-3-3beta protein. J. Biol. Chem. 287, 41914–41921 (2012).
Stoltzman, C. A. et al. Glucose sensing by MondoA:Mlx complexes: a role for hexokinases and direct regulation of thioredoxin-interacting protein expression. Proc. Natl Acad. Sci. USA 105, 6912–6917 (2008).
Velazquez-Campoy, A., Ohtaka, H., Nezami, A., Muzammil, S. & Freire, E. Isothermal titration calorimetry. Curr. Protoc. Cell Biol. https://doi.org/10.1002/0471143030.cb1708s23 (2004).
Tiwari, V. et al. Glycerol-3-phosphate activates ChREBP, FGF21 transcription and lipogenesis in citrin deficiency. Preprint at bioRxiv https://doi.org/10.1101/2024.12.27.630525 (2025).
Cheong, M. C. et al. Ethanol induction of FGF21 in the liver is dependent on histone acetylation and ligand activation of ChREBP by glycerol-3-phosphate. Proc. Natl Acad. Sci. USA 122, e2505263122 (2025).
Randle, P. J., Garland, P. B., Hales, C. N. & Newsholme, E. A. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1, 785–789 (1963).
Kennedy, E. P. Biosynthesis of complex lipids. Fed. Proc. 20, 934–940 (1961).
Hue, L. & Taegtmeyer, H. The Randle cycle revisited: a new head for an old hat. Am. J. Physiol. Endocrinol. Metab. 297, E578–E591 (2009).
McGarry, J. D., Leatherman, G. F. & Foster, D. W. Carnitine palmitoyltransferase I. The site of inhibition of hepatic fatty acid oxidation by malonyl-CoA. J. Biol. Chem. 253, 4128–4136 (1978).
Softic, S., Cohen, D. E. & Kahn, C. R. Role of dietary fructose and hepatic de novo lipogenesis in fatty liver disease. Dig. Dis. Sci. 61, 1282–1293 (2016).
Ouyang, S. et al. Glycerol kinase drives hepatic de novo lipogenesis and triglyceride synthesis in nonalcoholic fatty liver by activating SREBP-1c transcription, upregulating DGAT1/2 expression, and promoting glycerol metabolism. Adv. Sci.https://doi.org/10.1002/advs.202401311 (2024).
Costa-Mattioli, M. & Walter, P. The integrated stress response: from mechanism to disease. Science https://doi.org/10.1126/science.aat5314 (2020).
Khan, M. S. H. et al. FGF21 acts in the brain to drive macronutrient-specific changes in behavioral motivation and brain reward signaling. Mol. Metab. 91, 102068 (2024).
de la Ballina, L. R. et al. Amino acid transport associated to cluster of differentiation 98 heavy chain (CD98hc) is at the cross-road of oxidative stress and amino acid availability. J. Biol. Chem. 291, 9700–9711 (2016).
Lomelino, C. L., Andring, J. T., McKenna, R. & Kilberg, M. S. Asparagine synthetase: function, structure, and role in disease. J. Biol. Chem. 292, 19952–19958 (2017).
Kraus, J. P. et al. Cystathionine γ-lyase: clinical, metabolic, genetic, and structural studies. Mol. Genet. Metab. 97, 250–259 (2009).
Nikkanen, J. et al. Mitochondrial DNA replication defects disturb cellular dNTP pools and remodel one-carbon metabolism. Cell Metab. 23, 635–648 (2016).
Mick, E. et al. Distinct mitochondrial defects trigger the integrated stress response depending on the metabolic state of the cell. Elife https://doi.org/10.7554/eLife.49178 (2020).
Dong, J., Qiu, H., Garcia-Barrio, M., Anderson, J. & Hinnebusch, A. G. Uncharged tRNA activates GCN2 by displacing the protein kinase moiety from a bipartite tRNA-binding domain. Mol. Cell 6, 269–279 (2000).
Watatani, Y. et al. Stress-induced translation of ATF5 mRNA is regulated by the 5′-untranslated region. J. Biol. Chem. 283, 2543–2553 (2008).
Lee, J. I. et al. HepG2/C3A cells respond to cysteine deprivation by induction of the amino acid deprivation/integrated stress response pathway. Physiol. Genom. 33, 218–229 (2008).
Birsoy, K. et al. An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell 162, 540–551 (2015).
Gudmundsson, S. et al. Variant interpretation using population databases: lessons from gnomAD. Hum. Mutat. 43, 1012–1030 (2022).
Sakaue, S. et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat. Genet. 53, 1415–1424 (2021).
Costanzo, M. C. et al. The Type 2 Diabetes Knowledge Portal: an open access genetic resource dedicated to type 2 diabetes and related traits. Cell Metab. 35, 695–710 e696 (2023).
Larsson, S. C., Michaelsson, K., Mola-Caminal, M., Hoijer, J. & Mantzoros, C. S. Genome-wide association and Mendelian randomization study of fibroblast growth factor 21 reveals causal associations with hyperlipidemia and possibly NASH. Metabolism 137, 155329 (2022).
Turner, T. et al. FGF21 increases water intake, urine output and blood pressure in rats. PLoS ONE 13, e0202182 (2018).
Giontella, A. et al. Renoprotective effects of genetically proxied fibroblast growth factor 21: Mendelian randomization, proteome-wide and metabolome-wide association study. Metabolism 145, 155616 (2023).
Frayling, T. M. et al. A common allele in FGF21 associated with sugar intake is associated with body shape, lower total body-fat percentage, and higher blood pressure. Cell Rep. 23, 327–336 (2018).
May-Wilson, S. et al. Large-scale GWAS of food liking reveals genetic determinants and genetic correlations with distinct neurophysiological traits. Nat. Commun. 13, 2743 (2022).
Liang, K. Mitochondrial CPT1A: insights into structure, function, and basis for drug development. Front Pharm. 14, 1160440 (2023).
Srivastava, R. A. et al. AMP-activated protein kinase: an emerging drug target to regulate imbalances in lipid and carbohydrate metabolism to treat cardio-metabolic diseases. J. Lipid Res. 53, 2490–2514 (2012).
Yang, R., Xu, A. & Kharitonenkov, A. Another kid on the block: long-acting FGF21 analogue to treat dyslipidemia and fatty liver. J. Clin. Endocrinol. Metab. 107, e417–e419 (2022).
Gilroy, C. A. et al. Sustained release of a GLP-1 and FGF21 dual agonist from an injectable depot protects mice from obesity and hyperglycemia. Sci. Adv. 6, eaaz9890 (2020).
Trammell, S. A. et al. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat. Commun. 7, 12948 (2016).
Trammell, S. A. et al. Nicotinamide riboside opposes type 2 diabetes and neuropathy in mice. Sci. Rep. 6, 26933 (2016).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Anders, S. & Huber, W. Differential expression analysis for sequence count data. Genome Biol. 11, R106 (2010).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
McCarthy, D. J., Chen, Y. & Smyth, G. K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 40, 4288–4297 (2012).
Trammell, S. A. & Brenner, C. Targeted, LCMS-based metabolomics for quantitative measurement of NAD(+) metabolites. Comput. Struct. Biotechnol. J. https://doi.org/10.5936/csbj.201301012 (2013).
Sillero, M. A., Sillero, A. & Sols, A. Enzymes involved in fructose metabolism in liver and the glyceraldehyde metabolic crossroads. Eur. J. Biochem. 10, 345–350 (1969).
Acknowledgements
We thank T. Saheki (Kumamoto University) and the Citrin Foundation for introduction to the problem of CD and for donation of Slc25a13−/−Gpd2−/− mouse sperm, W. Tsark at City of Hope for in vitro fertilization and T.-F. Chou at the Proteome Exploration Laboratory of the California Institute of Technology for assistance with mass spectrometry. We thank P. Fueger, C. Mallet, J. Williams, Z. Wang, Y. Deng, S. Shuck and R. Natarajan for helpful consultations. This work was supported by National Institutes of Health grant nos. R01DK012170 and R01DK100425 (M.A.H.); National Institutes of Health grant nos. R01DK118011 and R01DK136671 (C.N.S.); American Diabetes Association grant no. 11-22-JDFPM-06 (C.N.S.); National Institutes of Health grant no. R01DK134675 (R.P.G.); Burroughs Wellcome Career Award for Medical Scientists (R.P.G.); National Institutes of Health grant no. R01HL147545 (C.B.); National Institutes of Health grant no. P30CA33572 (City of Hope shared resources); Alfred E. Mann Family Foundation and the Arthur Riggs Diabetes and Metabolism Research Institute (C.B.).
Author information
Authors and Affiliations
Contributions
C.B. conceived of the project with M.A.H., C.N.S. and R.P.G., all of whom also provided funding. V.T., B.J., O.S., E.D.J.L.G., H.S., A.Z., C.N.S., R.P.G. and C.B. performed experiments and experimental analysis. M.-H.C. and X.W. performed data analysis. C.B. wrote and edited the manuscript with the assistance of all authors.
Corresponding author
Ethics declarations
Competing interests
C.B. is the coinventor of US provisional patent application 63/739,033 for G3P-releasing prodrugs as FGF21-inducing compounds. The other authors declare no competing interests.
Peer review
Peer review information
Nature Metabolism thanks Araceli del Arco, Eleftheria Maratos-Flier and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Yanina-Yasmin Pesch, in collaboration with the Nature Metabolism team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Supplementary information
Source data
Source Data Figs. 2–4
Mouse data. The mice used in Fig. 2 are identified in column F. The FGF21 circulation data for Fig. 3 are in column G. The values in red were from haemolysed samples, excluded from analysis. The metabolite concentrations in mouse liver for Fig. 4 are in columns H, I and J.
Source Data Fig. 5
Luciferase data and metabolomic analysis from HEK293T cells.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tiwari, V., Jin, B., Sun, O. et al. Glycerol-3-phosphate activates ChREBP, FGF21 transcription and lipogenesis in citrin deficiency. Nat Metab 7, 2284–2299 (2025). https://doi.org/10.1038/s42255-025-01399-3
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s42255-025-01399-3
