
Abstract
Background
Normal cognitive aging is accompanied by molecular changes in the brain and periphery, but the specific proteomic and peptide-level alterations remain poorly defined. This study aimed to characterize age-related protein and peptide modifications by analyzing matched cerebrospinal fluid (CSF) and plasma samples from cognitively normal individuals.
Methods
Mass spectrometry was used to profile the proteome and peptide-level data of CSF and plasma samples from young (n = 52; mean age 29 ± 5.9 years; 4% male) and older (n = 40; mean age 69 ± 6.3 years; 48% male) adults. Differential abundance analysis, gene set enrichment, and weighted correlation network analyses were performed to identify age-associated pathways. Protein cleavage, alternative splicing, and phosphorylation events were examined to capture post-translational modifications linked to aging.
Results
In CSF, aging is associated with significant upregulation of extracellular matrix (ECM) components, coagulation, and inflammatory pathways. In plasma, the insulin-like growth factor-1 (IGF-1) signaling pathway is notably downregulated. Peptide-level analysis reveals novel alternative cleavage and phosphorylation patterns in key proteins involved in lipid metabolism, ECM structure, axonogenesis, and synaptic activity, including APP, APOE, COL4A2, NRXN1, and NRCAM. These findings highlight distinct and compartment-specific molecular changes that occur with aging.
Conclusions
This study provides a comprehensive proteomic and peptide-level landscape of normal cognitive aging, identifying both protein-level shifts and novel age-associated cleavage and phosphorylation events.
Plain language summary
As people age, their bodies and brains undergo gradual molecular changes, even in the absence of disease. In this study, we analyzed blood and cerebrospinal fluid (the fluid surrounding the brain and spinal cord) from healthy younger and older adults to understand how the proteins in these fluids change with normal aging. To do this, we used advanced mass spectrometry (MS), a powerful analytical method that measures the mass of molecules with extremely high precision. Our study found age-related changes in proteins involved in brain structure, inflammation, and metabolism. Notably, we also discovered subtle modifications in key proteins that may influence how cells communicate and maintain brain health over time. These findings provide new insights into the biology of healthy aging and may help researchers distinguish normal aging from the early stages of neurodegenerative diseases in the future.
Similar content being viewed by others


Introduction
Over the past half-century, advancements in healthcare and living conditions have led to a significant increase in life expectancy globally, with a similar but smaller increase in human healthspan1. Aging is characterized by a gradual decline in physiological functions, resulting from cumulative damage and a loss of cellular and tissue homeostasis1,2. The aging process is highly variable among individuals, with some aging faster or slower than others, affecting their lifespan, health span, and quality of life3,4.
Proteomics has emerged as a powerful tool for studying aging, given its ability to provide a comprehensive overview of the proteins and post-translational modifications (PTMs) involved in biological processes3,4,5,6. Johnson et al. systematically reviewed age-related proteins and pathways in plasma in 36 analyses and found extracellular matrix (ECM), inflammatory pathways, and protein regulatory mechanisms to be among the most replicated5. While there is a multitude of papers exploring the proteomics of plasma in the aging population, studies that delve into cerebrospinal fluid (CSF) changes in healthy cognitive aging are sparse2,6,7,8,9,10. A recent study investigated the interaction between APOE, sex, amyloid status, and age in three large CSF proteomics studies and reported an increase in inflammatory and complement-related proteins and a decrease in ECM-enriched components with age independently of amyloid status11. However, these studies primarily investigated individuals aged 60 to 80 and did not include CSF samples from young adults11. Baird et al. also measured approximately 800 proteins in CSF of young adults compared to octogenarians and reported similar biological pathway upregulation with age10,12.
Despite these advances in broader protein-level analyses, a more detailed look at the peptide and PTM levels, which are not fully captured by antibody-based methods, is missing. This study aims to fill this gap by leveraging mass spectrometry data to capture a more nuanced state of each protein with aging in addition to expression levels in matched CSF and plasma of cognitively normal younger and older adults. The matched sample approach allows us to minimize inter-individual variability and accurately investigate the interplay between the proteomics of CSF and plasma across different age groups. This approach allows for identifying novel alternative cleavage and phosphorylation events, which have not been studied before.
In this study, we show that aging is associated with widespread proteomic and peptide-level changes in both CSF and plasma. We identify an upregulation of extracellular matrix components, inflammatory mediators, and coagulation-related proteins in the CSF of older adults, along with a downregulation of IGF-1 signaling components in plasma. Importantly, we uncover novel age-associated alternative cleavage and phosphorylation patterns in key proteins such as APP, APOE, and NRXN1, alterations that are not detectable at the total protein level.
Methods
Participants
The participants included in this study were enrolled in the Johns Hopkins Alzheimer’s Disease Research Center and the Center for CSF Disorders in the Department of Neurology, who were cognitively normal. The clinical diagnostic classification followed the recommendations of the National Institute on Aging/Alzheimer’s Association workgroups13.
Moreover, young cognitively normal participants were recruited from the Johns Hopkins Center for CSF Disorders who were evaluated for CSF dynamic disorders and underwent a lumbar puncture as part of standard of care but were found to have normal opening pressures, no pleocytosis, normal glucose and protein and normal structural MRI with no evidence of stroke, tumor, demyelination, small vessel disease or ventriculomegaly.
All participants selected for this study were required to be: (1) cognitively normal based on Clinical Dementia Rating Scale (CDR) and (2) negative CSF AD biomarkers based on the cut-offs established by Greenberg et al.14. All participants signed institutional review board–approved consent forms before participating in the study.
Ethical compliance
This study was approved by the Institutional Review Board of Johns Hopkins University (Approval Number NA_00045104). All participants provided written informed consent prior to their participation in the study.
CSF and blood collection and CSF biomarker assays
CSF was collected from 40 cognitively normal older adults and 52 cognitively normal young adult participants (Fig. 1 and Table 1). Participants underwent a lumbar puncture and blood collection in the fasted state in the same visit. Twenty ml of CSF was collected in polypropylene tubes along with 20 ml of whole blood in EDTA tubes and underwent centrifugation at 2500×g for 15 min. Samples were then frozen at −80 °C within 2 h of collection. CSF Aβ1–42, Aβ1–40, total tau, and p-tau181 were measured using the Lumipulse G1200 assay (Fujirebio, Malvern, PA, USA). The intra-assay coefficient of variation for this assay was 3.4% for Aβ1–42, 2.7% for Aβ1–40, and 1.8% for p-tau181.

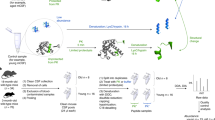
Cerebrospinal fluid (CSF) and blood samples were collected from cognitively normal young and old participants, and proteomics profiling was performed using mass spectrometry. Differential expression analysis, gene set enrichment analysis (GSEA), and weighted gene co-expression network analysis (WGCNA) were conducted to identify significant pathways and co-expression modules, highlighting age-related differences in the CSF and plasma proteome (Created with BioRender.com).
Sample preparation
Plasma samples were first filtered using hydrophilic PVDF membrane filter plates (0.22 µm; Millipore Cat. No. MAGVS2210) to remove larger particles and then depleted of the 14 most abundant proteins using a mouse-3 multi-affinity removal column (Agilent) connected to a Dionex UltiMate 3000 RS pump. Both plasma and cerebrospinal fluid (CSF) samples were further processed on a KingFisher™ Flex system (Thermo Scientific™) following Biognosys’ standard operating procedure. This protocol included reduction, alkylation, and overnight digestion at 37 °C with trypsin (Promega, 1:100 protease-to-total protein ratio) and Lys-C (Fujifilm Wako Chemicals, 1:200 ratio). Peptide clean-up was performed using an Oasis HLB µElution Plate (30 µm; Waters) according to the manufacturer’s instructions. The resulting peptides were dried in a SpeedVac and reconstituted in LC solvent A (1% acetonitrile/0.1% formic acid in water) containing Biognosys’ iRT-peptide mix for retention time calibration. Peptide concentrations were then measured using an mBCA assay (Pierce, Thermo Scientific™).
Library generation
To create a study-specific spectral library, 600 µg of pooled, depleted, and digested plasma samples were injected onto an Acquity UPLC CSH C18 column (1.7 µm, 2.1 × 150 mm; Waters) connected to a Dionex UltiMate 3000 RS pump (Thermo Fisher Scientific™). The separation was performed using LC solvents A (20 mM ammonium formate in water) and B (100% acetonitrile) with a nonlinear high-pH reverse-phase (HPRP) gradient from 1 to 40% B over 30 min, with fractions collected every 30 s. These fractions were sequentially pooled to yield 20 distinct fractions, which were then dried and resuspended in 0.1% formic acid and 1% acetonitrile with Biognosys’ iRT peptides spiked in.
Liquid chromatography mass spectrometry (LC-MS/MS)
For DIA LC-MS/MS acquisitions, 3.5 µg of peptides per sample were injected onto an in-house packed reversed-phase column on a Thermo Scientific™ EASY-nLC™ 1200 nano-liquid chromatography system. The LC separation was performed using solvents A (water with 0.1% formic acid) and B (80% acetonitrile with 0.1% formic acid) with a nonlinear gradient from 1 to 50% B over 210 minutes, followed by a 10-min column washing step at 90% B and an 8-min equilibration at 1% B, all at 60 °C and a flow rate of 250 nl/min. Data were acquired on a Thermo Scientific™ Orbitrap™ Exploris 480 mass spectrometer equipped with a Nanospray Flex™ ion source and a FAIMS Pro™ ion mobility device. The FAIMS-DIA method involved one full-range MS1 scan and 34 DIA segments per applied compensation voltage, as described in Bruderer et al. and Tognetti et al. For DDA LC-MS/MS measurements, peptides were separated under identical LC conditions with MS1 precursor scans acquired from 330–1650 m/z at 60,000 resolution and data-dependent MS2 scans at 15,000 resolution, using a cycle time of 1.8–2 s per compensation voltage; only precursors with charge states 2–6 were isolated. Instrument performance was monitored using quality control injections, and all samples were acquired in randomized order.
Mass spectrometry data analysis
The resulting DIA maps were analyzed using the Spectronaut software suite (version 18.4.231017.55695, Biognosys) with modified default settings using a hybrid library comprising all DIA and DDA runs obtained from previous studies15,16.
The resulting search archives were combined into a master library, and all DIA maps were subsequently searched against this library using modified default settings. These modifications included: (i) setting the digestion type to “Semi-specific,” (ii) specifying fixed modifications as “Carbamidomethyl (C),” (iii) including variable modifications “Acetyl (Protein N-term),” “Deamidation (NQ),” “Oxidation (M),” and “Phospho (STY),” and (iv) configuring quantification so that the minor (peptide) group was determined “by Modified Sequence.” Protein and peptide-level false discovery rates were maintained at 1%, with cross-run normalization performed using local normalization. To facilitate the investigation of post-translational modifications (PTMs), PTM localization was enabled with a probability cutoff of 0.75, and imputation was disabled for all searches.
Peptide outliers ±1.5 interquartile range (IQR) were removed, and peptide identification was done in Spectronaut with 1% false discovery rate (FDR) at the peptide level, and protein quantification was done in Spectronaut® with the MaxLFQ approach with 1% FDR at the protein level17.
Pre-processing steps
Initially, local normalization was conducted using the Spectronaut® software, specifically focusing on precursors that were identified across all runs. Subsequently, the data underwent a log2 transformation to stabilize variance and normalize distribution. Pre-filtering criteria were applied differently based on the type of biological specimen: for CSF, only peptides consistently measured across all liquid chromatography (LC) column batches were retained; for plasma, retention was limited to peptides consistently measured across all Field Asymmetric Ion Mobility Spectrometry (FAIMS) batches. Finally, to correct for batch effects associated with plate variations, we utilized the HarmonizR R package, employing the ComBat algorithm with the parameter “ComBat_mode” set to 3, which corresponds to “par.prior = FALSE” and “mean.only = FALSE”18.
Protein and peptide differential expression analysis
Differential abundance analysis was performed on the normalized plate-batch corrected data using the R package proDA with default settings19. Significantly differentially expressed peptides were identified by the p value of 0.001 and BH-adjusted p value of 0.05; and further used to search for alternative splicing, cleavages, protein domains regulation and differential phosphorylation.
Search for alternative splicing (AS), cleavages and protein domains regulation
In the exploration of alternative splicing (AS), proteolytic cleavages, and the regulation of protein domains, our analysis began with an initial filtering stage. First, significantly differentially expressed peptides were identified by the threshold of 0.58 log2 fold changes (logFC), p value of 0.001 and BH-adjusted p value of 0.05. Then, we designated cases as candidates for AS, cleavage, or differentially regulated protein domains if they involved more than one DA peptide per protein and exhibited significant logFC in both the up and down directions. To refine this list, we excluded AS candidates potentially influenced by technical artifacts and miscleavages. This exclusion involved removing peptides where lysine (K) or arginine (R) did not appear as the last amino acids or were not followed by proline (P), except when the peptide was positioned at the very beginning or end of the protein. Additionally, peptides that mapped to multiple positions within the protein sequences were disregarded.
Further filtration was required for differentiating post-translational modifications (PTMs). We eliminated peptides stemming from likely technical effects at the PTM level by retaining peptides that: (1) had only one modified (or unmodified) sequence per stripped sequence; (2) included phosphorylated peptides, which are more likely indicative of biological effects; and (3) from the remaining peptides, selected per stripped sequence the peptide with the highest abundance for those peptides with missing values in less than 75% of samples and an absolute difference in the number of missing values under 75% between younger and older groups.
Additionally, we extended the start and end of each peptide by ten amino acids to assess overlapping regions, retaining proteins that exhibited at least three overlapping extended peptides and proteins that displayed a biologically relevant pattern in the locations of DA peptides. Peptides were classified as tryptic or semi-tryptic based on the last amino acid and the one before the first amino acid.
Finally, while not a filtering criterion, we visually assessed AS and cleavage candidate proteins by evaluating the coverage of the protein sequence by DA peptides and examining their distribution in volcano plots.
Differential phosphorylation analysis
In our differential phosphorylation analysis, we employed a systematic approach to evaluate the regulatory roles of phosphorylation at the peptide level. Initially, for each stripped peptide, we evaluated the sign of log2 fold changes (logFC) of its phosphorylated and non-phosphorylated modified peptides separately. The cases with several modified peptides per stripped phosphorylated or non-phosphorylated peptide were retained due to difficulty to set apart which of these are potentially due to technical effects. We then narrowed our focus to those stripped peptides that exhibited both phosphorylated and non-phosphorylated forms and demonstrated logFC in opposite directions, specifically, scenarios where the phosphorylated form was upregulated while the non-phosphorylated form was downregulated, or vice versa. The cases with several modified peptides per stripped phosphorylated or non-phosphorylated peptide were again retained.
Subsequently, from this refined list, we selected peptides that were significantly differentially abundant (DA). The criteria for significance included a logFC threshold of 0.58 and an unadjusted p value threshold of 0.01. This selection process ensured that our analysis highlighted peptides where phosphorylation status correlated strongly with changes in abundance, suggesting potential biological significance.
Plasma-CSF protein levels correlation analysis
In our approach to analyzing the correlation between plasma and CSF protein levels, we adopted an individual-centric methodology. Specifically, we calculated the correlation of plasma-CSF protein levels for each individual separately. This method allowed us to capture the unique biological variance and interactions specific to each individual.
We handled missing values by removing them prior to analysis. The correlation between protein abundances in plasma and CSF for each individual was quantified using the Pearson correlation coefficient.
Furthermore, to estimate the effect size, we used a method where the distance between the medians of plasma and CSF protein levels was divided by the overall visible spread (DBM/OVS) of the data.
Gene set enrichment analysis (GSEA)
Gene set enrichment analysis (GSEA) was performed using GSEA 4.3.2 software. The GSEA calculates the signal-to-noise ratio for all proteins and orders gene sets by normalized enrichment scores (NES). We performed the GSEA with the default settings of the software, which included 1000 permutations, phenotype permutation type, exclusion of gene sets larger than 500 and smaller than 15 and using weighted enrichment statistics. FDR cut-off of 0.25 was adopted in this analysis as recommended by GSEA developers to avoid overlooking potentially significant pathways in the context of a relatively small number of gene sets being analyzed.
Subsequently, to investigate potential confounding of age and sex because of differences in age group sample size by sex, we performed single-sample gene set enrichment analysis (ssGSEA) to calculate gene set enrichment scores for each biological pathway. ssGSEA calculates an enrichment score for each sample in the dataset for each biological pathway or molecular signature, allowing further statistical analysis by covariates and factors. The gene set enrichment scores were used as the dependent variable (Y) in a linear statistical model that included age group (young or old), sex, and sex by age group interaction:
Y ~ b1 + b2*age_group + b3*sex + b4*age_group*sex + e
Only pathways that were identified as statistically significantly different by age group were included in this analysis.
Weighted correlation network analysis (WGCNA)
Weighted gene co-expression network analysis (WGCNA) was performed on the proteomics data. The raw protein expression data were imputed using the Random Forest (RF) method utilizing the MissForest package in R20. No outliers were detected using hierarchical clustering and principal component analysis (PCA). A variance stabilizing transformation (VST) was applied to normalize the data. The network was constructed with a soft-thresholding power of 5, employing a blockwise module detection approach. Modules were identified and their eigengenes were calculated. These module eigengenes were then correlated with clinical traits of interest, such as age, sex, CSF Aβ1–42/Aβ1–40, CSF p-tau181, and MoCA, using Pearson correlation. Module-trait relationships were visualized through heatmaps, and statistical significance was determined using p values adjusted for multiple testing using an FDR cut-off of 0.05.
After identifying modules with WGCNA, we conducted overrepresentation analysis (ORA) to identify biological processes or cellular component gene ontological terms corresponding to each module using the gene ontology (GO) biological process and cellular component database. ORA was performed using the WEB-based GEne SeT AnaLysis Toolkit (WebGestalt)21. The default settings of WebGestalt were used with the FDR cut-off of 0.05.
Statistics and reproducibility
All statistical analyses were performed using R (version 4.3.1) and the Spectronaut software suite (version 18.4.231017.55695, Biognosys). Differential abundance analysis for protein and peptide data was conducted using the proDA package, which accounts for missing values in label-free proteomics data using a probabilistic dropout model. A FDR-adjusted p value of <0.05 and an absolute log2 fold change of ≥0.58 were considered statistically significant. GSEA was performed with GSEA software (version 4.3.2) using 1000 phenotype permutations and standard filtering criteria. WGCNA was used to identify protein modules associated with clinical traits; correlations were assessed using Pearson’s r and adjusted for multiple testing using the Benjamini–Hochberg method.
The study included 92 participants: 52 cognitively normal young adults and 40 cognitively normal older adults. Each participant provided a single matched CSF and plasma sample. All proteomic and peptide-level measurements were acquired in technical duplicates using DIA mass spectrometry and processed in randomized batches to reduce bias. Biological replicates were defined as individual participants. No data were excluded unless specified in the Methods.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Results
Figure 1 illustrates our comprehensive workflow. Matched blood and cerebrospinal fluid samples were collected from cognitively normal young and older adults, with CSF biomarkers (Aβ1–42, Aβ1–40, and p-tau181) confirming normal status. Deep proteomic and peptidomic profiling was then performed using liquid chromatography coupled with FAIMS. Differential abundance analyses, including assessments of alternative splicing, proteolytic cleavage, and phosphorylation, were conducted to identify age-related molecular changes. Finally, correlation studies between plasma and CSF proteins, along with GSEA and WGCNA, revealed key pathways such as extracellular matrix remodeling, coagulation, inflammation, axonogenesis, and IGF-1 signaling.
Peptide and protein differential expression analysis
We identified 127,560 and 94,415 peptides in matched CSF and plasma samples from 40 cognitively normal older adults (ON) and 52 cognitively normal young adults (YN) participants. Comparative analysis revealed 7730 and 1379 peptides exhibiting differential expression in CSF and plasma between younger and older cognitively healthy participants, as determined by FDR-adjusted p values <0.05, detailed in Supplementary Data s1, s2. The proteomic analysis of CSF and plasma was conducted, resulting in the quantification of 5599 and 3112 proteins, respectively. Comparative analysis revealed 787 and 812 proteins exhibiting differential expression in CSF and plasma between younger and older cognitively healthy participants, as determined by FDR-adjusted p values <0.05, detailed in Supplementary Data s3, s4 and Fig. 2a, c. About 131 proteins were found to be overlapping between the CSF and plasma, significantly differentially abundant (DA) proteins, out of which 85 were differentially expressed in the same direction in both biofluids (Fig. 2b and Table s5). Moreover, 3368 and 690 peptides were significantly downregulated in CSF and plasma with age, respectively, as depicted in Fig. 2d, e.

a Volcano plot showing proteins differentially expressed in plasma with age; b Venn diagram illustrating the overlap of differentially expressed proteins between plasma and CSF; c Volcano plot showing proteins differentially expressed in CSF with age; d Volcano plot showing peptides differentially expressed in plasma with age; e Volcano plot showing peptides differentially expressed in CSF with age. Proteins and peptides highlighted in the volcano plots meet the criteria of p value <0.05 and log2 fold change (LogFC) <–0.58 or >0.58. The source data for Fig. 2 is in Supplementary Data s1–s4. (Created with BioRender.com).
Many of the proteins that were differentially expressed with age in a similar direction in plasma and CSF belonged to several major biological processes. These processes included matrisome core proteoglycans and glycoproteins (e.g., LTBP2, NPNT, MFAP4, VWCE, COMP, LUM, FBLN2, PXDN, and SVEP1), matrisome-associated proteins (e.g., CSPG4, REG1A, HRG, TIMP1, and SERPINI1), coagulation-associated proteins (e.g., FGA, FGB, and FGG), proinflammatory and complement activation cascade members (e.g., C7, C4BPB, C4BPA, CFD, and CHI3L1), and insulin-like growth factor-1 (IGF-1) signaling pathway (e.g., IGF2, IGFALS, IGFBP6, and IGFBP5).
Alternative splicing (AS) and alternative cleavage analysis in cerebrospinal fluid
Alternative splicing and/or proteolytic changes that alter protein function are well-known to occur in aging22,23. In our investigation, we identified 27 proteins as candidates for alternative splicing, proteolytic cleavages, and protein domain regulation in CSF based on the criteria set in the Methods section (Fig. s1 and Table s6). Notably, these proteins did not appear among those significantly differentially abundant at the protein level, following our set criteria. Alternative splicing leading to various protein isoforms that are reported in the Uniprot database were used to identify whether differentially expressed peptides identified in this analysis corroborate with previously established isoforms of the proteins or are a result of post-translational cleavage/proteolysis.
Of these 27 proteins, 13 have previously been reported to be associated with aging and longevity24. This subset includes A2M, ALB, APOE, APP, C4A, CADM1, CLU, COL4A2, DKK3, NRCAM, NRXN1, PAM, and PRNP. Furthermore, the same proportion (13 of 27 proteins, or 48%) was found to express more than one transcript according to the APPRIS database, which includes proteins such as APLP1, CADM1, CHL1, CLU, DAG1, DKK3, F5, NRXN1, NUCB1, PAM, PRNP, SERPINA1, and SPARCL125. An overlap in proteins associated with aging/longevity and alternative splicing was identified, comprising CADM1, CLU, DKK3, NRXN1, PAM, and PRNP. This overlap suggests a potential biological link between age-related gene expression modulation and the regulation through alternative splicing and protein processing.
An interesting example is provided by amyloid-beta precursor protein (APP), where a partially matching isoform has been described that lacks a part of the sequence between amino acids 637–65426 (Fig. 3a, b). We observed non-specific cleavage at the N-terminus around amino acids 631 and 634, which occurred more frequently in young individuals compared to older normal adults. Whether this alternative cleavage finding points to this alternative splicing or is an alternative proteolytic cleavage event needs to be further studied and validated.

a, b Volcano plot of differentially abundant peptides (A) and peptide mapping to the full-length protein sequence (b) for APP; c, d Volcano plot (c) and peptide mapping (d) for APOE; e, f Volcano plot (e) and peptide mapping (f) for NRXN1. The source data for Fig. 3 is in Supplementary Table s6.
Another notable finding pertains to APOE, where increased non-specific cleavage of the LDL receptor binding site around amino acid 144 in older individuals likely decreases the protein’s ability to bind to its LDL receptors compared to younger individuals (Fig. 3c, d). This alteration in cleavage pattern could have significant implications for the functional efficacy of APOE in aging populations.
Furthermore, NRXN1, a presynaptic hub protein that actively modulates synaptic transmission, is found to demonstrate an increase in cleavage at the 730 amino acid residue, which is located at the beginning of the Laminin G-like 4 domains (LG4) (Fig. 3e, f).
No proteins were identified in plasma that passed all the criteria placed in the Methods section.
Differential phosphorylation analysis
We selected peptides with opposing phosphorylation trends and filtered for those with significant differential abundance. Notably, our differential phosphorylation analysis uncovered several differentially abundant phospho-peptides (44 differentially abundant phospho-peptides with 54 phospho-sites on 22 proteins). Although these numbers are modest, the fact that these sites were identified without any phosphopeptide enrichment underscores the sensitivity of our approach and highlights the significance of these findings.
A total of 49 proteins were identified with significantly DA peptides, including
A2M, ACTA2, AHSG, ALB, APLP1, APOE, C3, C4A, C7, CFB, CFH, CHGA, CHGB, CHL1, CLU, COL4A2, CP, CSPG4, CST3, DAG1, ENPP2, F2, FGG, GSN, IGHA1, IGKV1D-16, IGKV1D-42, IGKV1D-43, IGKV3D-15, IGKV3D-7, MEGF8, NRCAM, NRXN1, PLTP, PRNP, PRRT2, PTGDS, SCG2, SELENOP, SERPINA1, SERPINA3, SERPINC1, SERPINF1, SERPING1, SPARCL1, SPP1, TF, UniProt:P0DOX5, and UniProt:P0DOX7 which do not have specific protein names but belong to immunoglobulin protein family (Table s7).
Among these, 22 proteins had significantly DA phosphorylated peptides
A2M, ALB, APOE, C4A, C7, CHGA, CHL1, CLU, COL4A2, CP, GSN, NRCAM, NRXN1, PRNP, PTGDS, SCG2, SERPINA1, SERPING1, SPARCL1, SPP1, TF, and UniProt:P0DOX5. It’s noteworthy that the remaining proteins exhibited significantly regulated non-phosphorylated peptides, suggesting diverse regulatory mechanisms affecting the expression of these proteins and their phosphorylation patterns (Fig. 4a–f and Table s7).

a Peptides volcano plot for the COL4A2 protein, which is an example of potentially differential phosphorylation even in aging; b Mapping of peptides to the protein sequence for (a); c Log2 abundances of phosphorylated and non-phosphorylated DA-peptides of COL4A2 (highlighted in (b); d Peptides volcano plot for the global analysis of differential phosphorylation in aging; e, f Examples of genes with aging associated differential peptide phosphorylation. The source data for Fig. 4 is in Supplementary Tables s6, s7.
Furthermore, 18 of the 49 proteins are recognized as associated with aging and longevity in a recent publication24. These proteins are
A2M, ALB, APOE, C3, C4A, C7, CFB, CFH, CLU, COL4A2, GSN, MEGF8, NRCAM, NRXN1, PRNP, SELENOP, SERPINF1, and SPP1.
This highlights a broader context of protein regulation in aging, encompassing both well-documented and newly identified pathways.
Plasma-CSF protein levels correlation analysis
In our study, we evaluated the correlation between protein levels in CSF and plasma among the two age groups. The results revealed a moderate correlation across all ages, with a slightly stronger correlation observed in older individuals compared to younger ones (Correlation coefficient were 0.5 in young adults and 0.53 in older adults, P value <0.001). This analysis was conducted using samples from 52 young and 40 older individuals.
For the analysis, proteins were grouped based on various criteria: all proteins, proteins measured in both age groups, and subsets excluding low-abundant proteins. Median numbers of protein groups (PGs) per sample were: 1757 for all proteins and those measured in both age groups, 894 for high-abundance proteins, 41 for low molecular weight proteins, and 62 for brain-specific proteins. These findings are displayed in Fig. 5a, b, providing a detailed overview of protein correlations in CSF and plasma within this cohort.

a Log2 abundances of proteins measured in both young and old samples, excluding low-abundant proteins (log2abundance below 10th percentile of correspondingly plasma/CSF protein abundances data); b Plasma-CSF Pearson correlation coefficient per sample for different groups of proteins: (1) all proteins without missing values, (2) proteins measured in both young and old samples, (3) proteins measured in both young and old samples, excluding low-abundant proteins (i.e., proteins shown in a), (4) low molecular weight (MW below 10th percentile) proteins measured in both young and old samples, excluding low-abundant proteins, (5) brain-specific proteins measured in both young and old samples, excluding low-abundant proteins. DBM/OVS: distance between medians divided by overall visible spread as a measure of effect size. The source data for Fig. 5 is deposited with the ProteomeXchange Consortium under accession number MSV000097888.
Gene set enrichment analysis (GSEA)
The results for pathways found to be significant in CSF are provided in Supplemental Table s8 and Fig. s3. “Regulation of coagulation”, “regulation of fibrinolysis”, “fibrinolysis”, and “positive regulation of coagulation” are among the most significantly upregulated pathways in older adults. Several GO terms related to ECM are also upregulated with age, including: “Proteoglycan binding”, “positive regulation of response to wounding”, and “ECM structural constituent conferring compression resistance”. Moreover, several neuroinflammatory biological pathways (BP) such as “type II interferon production”, “detection of biotic stimuli”, “phagocytosis recognition”, and “complement activation classical pathway” are found to be upregulated with age. No pathways were found to be significantly differentiated with age in plasma.
While the number of female and male samples was similar for the older age group (21 female and 19 male) there was a substantial imbalance in the younger age group (28 females and four males). To address the potential confounding of age with sex for the younger group and assess the sensitivity of the results, we conducted single-sample gene set enrichment analysis (ssGSEA) to determine if there were sex-specific effects and sex by group (ON and YN) interactions that potentially could confound the results of the age group GSEA comparisons.
In summary, the main effect of sex was not significant (p value >0.05) for all the pathways identified as significantly different by initial GSEA analysis comparing age groups. Moreover, the interaction between sex and group was also not significant (p value >0.05) for any of these biological pathways. Overall, this data indicates that sex does not appear to be a confounder with age group for the pathways identified as significantly different by initial GSEA between age groups (Table s9).
Weighted gene correlation network analysis (WGCNA)
The WGCNA demonstrates a significant negative correlation of ME10, which signifies the IGF-1 receptor signaling pathway with aging in plasma (Fig. 6a and Table s10).

a Plasma protein co-expression modules demonstrated in a heatmap with correlation coefficients and adjusted p values for associations between plasma protein modules and clinical traits (age, amyloid-β1–42 to amyloid-β1–40 ratio (Aβ1–42/Aβ1–40), phosphorylated tau181 (p-tau181), total tau, and sex); b Cerebrospinal fluid (CSF) protein co-expression modules demonstrated with a heatmap of correlations between CSF modules and clinical traits, highlighting modules involved in extracellular matrix organization, coagulation, inflammation, and synaptic function, which significantly associate with age and CSF p-tau181 levels. The source data for Fig. 4 is in Supplementary Tables s11, s12. (Created with BioRender.com).
The proteomics co-expression modules in CSF are more complex (Fig. 6b and Tables s10–12). The highest correlation between modules and age belongs to modules with roles in extracellular matrix (ECM) structure (ME9, 10, 17, and 19). One module containing collagen chains is found to be inversely correlated with age (ME6). There is a similar upregulation with age observed in modules related to coagulation (ME4) and multiple modules with a role in humoral inflammation (ME9, 29, and 4). Low-density lipoprotein receptor activity (ME27) is also demonstrated to be positively correlated with age.
While no modules were found to be correlated with Aβ1–42/Aβ1–40, there are multiple modules that are correlated with p-tau181 and total tau concentrations. Several modules related to ECM are negatively correlated (ME6, 16, 17, 22, and 24), while modules related to axonogenesis, and synapse organization (ME7 and 3) are positively correlated with p-tau181 and total tau concentrations (Fig. 6b and Tables s10–12).
Since the young adult participants were predominantly female, many of the modules have a significant relationship with age and sex (ME8, 28, 14, 21, 25, 4, 9, 19, 10, 12, and 13). Whether these modules are associated with age or sex can be deciphered based on the GSEA results and the p value and correlation coefficients of the modules with sex or age groups (Tables s10–12).
Discussion
This study provides a detailed proteomic analysis of CSF and plasma from cognitively normal younger and older adults, using mass spectrometry to focus on peptide-level data and post-translational modifications. This is the first study to develop a method to examine alternative cleavage and/or alternative splicing in the setting of normal aging. By examining alternative cleavage and phosphorylation events, we have uncovered intricate molecular changes associated with normal cognitive aging, which may influence key functional and structural aspects of proteins, beyond the differential analysis of proteins inferred from peptide-level data. Notably, our findings reveal significant alterations in several biological processes, including the upregulation of extracellular matrix components, modifications in lipoprotein metabolism, and enhanced inflammatory pathways.
Cells and tissues are surrounded by ECM, which provides structural support and influences tissue geometry, integrity, and function27. The ECM regulates intercellular communication by storing or transporting signaling molecules, such as growth hormones, with cell surface receptors like integrins linking the ECM to cell signaling27,28. A growing body of evidence suggests ECM deterioration as a hallmark of aging, during which the integrity of ECM declines due to collagen fragmentation, oxidation, glycation, and crosslinking, resulting in reduced ECM dynamics and loss of organ support and function28. The impact of the ECM modulation reflected as an increase in ECM components in plasma and CSF has been demonstrated in aging in previous publications1,5,29. This is in line with our differential expression analysis, and both the GSEA and WGCNA results, which demonstrate substantial upregulation of multiple matrisome-related protein pathways with age. Some of the most significantly upregulated proteins in CSF were core matrisome glycoproteins and proteoglycans (e.g., COMP, ACAN, LTBP2, FN1, HSPG2, and LUM) and matrisome-associated proteins (e.g., LOXL1, CSPG4, REG1A, HRG, TIMP1, and SERPINI1). While several collagens (e.g., COL15A1, COL21A1, COL18A1, COL6A2, and COL6A3), proteoglycans, and matrisome-regulating enzymes (LOXL1) were found to be upregulated in CSF with age in the present study, many collagens were in fact downregulated (e.g., COL1A1, COL1A2, COL4A2, COL3A1, COL26A1, COL19A1, COL12A1, and COL14A1). The loss of collagen mass with age and its consequential effect on various cellular functions (e.g., IGF-1 signaling and a proinflammatory state) has been demonstrated previously and links the various biological processes that we found to be dysregulated during aging28,30. The upregulation of a portion of matrisome-related proteins and downregulation of many collagens indicate an intricate and complex process of ECM remodeling that significantly affects many aspects of cellular function as we age.
As discussed above, there are multiple ECM-related modules that are downregulated with age (ME6) and more notably with p-tau181 and total tau concentrations (ME6, 22, and 24)31. Based on previous studies, ECM is a dynamic structural milieu of glycoproteins, proteoglycans, and enzymes that actively modulate and regulate the aggregation of p-tau18131. For instance, aggrecan perineuronal nets have been shown to decrease tau propagation and, therefore, inhibit p-tau aggregation31. Therefore, we speculate that the ECM components found in our study might act in a similar net-like fashion to inhibit tau propagation and therefore have an inverse correlation with p-tau concentrations; however, further studies are needed to investigate this hypothesis.
It is especially important to note that modulation of ECM components goes beyond proteomics changes and starts at the peptide and post-translational modification (PTM) level (Fig. 4a–d). For instance, we found that there is significant upregulation of phosphorylated Ser1475 residue of COL4A2 with age (Figs. Fig. 4c, D and s2). COL4A2 participates in the creation of trimeric Collagen IV, which constitutes one of the most abundant components of nearly all basement membranes and blood–brain barrier (BBB)32. While this is the first report of Ser1475 phosphorylation site and its change with age, its location near non-collagenous domain 1 (NC1) can potentially alter the interaction of COL4A2 with other collagen fibrils and collagen IV structure and cause vascular abnormalities33. Therefore, differential phosphorylation of this component of Collagen IV might have direct implications in the integrity of the BBB and intracranial vessels during normal cognitive aging34.
While we did not find differential expression of the lipoprotein pathway in plasma with aging, there is significant upregulation of APOA4, APOC1, APOC2, and APOC3 in the CSF, which points to an increase in high-density lipoprotein (HDL) or very low-density lipoprotein (VLDL) abnormalities in CSF with aging. The GSEA analysis results demonstrate an upregulation of HDL in the CSF with aging. Previous studies indicate that higher CSF APOC-III levels are associated with slower cognitive decline over a 12-year period in individuals with mild cognitive impairment (MCI)35. Additionally, it has been shown that CSF APOC-III levels are significantly lower in MCI compared to controls and are positively associated with CSF Aβ, suggesting a potential neuroprotective role36. Therefore, we speculate that the higher levels of APOC proteins in the cognitively normal older adult group might indicate the neuroprotective effect of these apolipoproteins in healthy cognitive aging. However, the role and mechanism of these lipoproteins in aging has not previously been explored and needs to be further investigated.
Moreover, even though the concentrations of APOE was not different with age based on our proteomics analysis, we found that semi-tryptic peptides (140/143 to 152 amino acid residue) located immediately before the LDL receptor binding region (located at 158 to 168) go up with age which demonstrates a proteolytic process that is lysing the APOE protein immediately before the LDL receptor binding region. This is the first time the upregulation of this semi-tryptic peptide upregulation is described with aging and there are no mechanistic descriptions of this peptide. However, multiple reported mutations located within this hinge region interfere with LDL receptor binding, heparin sulfate proteoglycan binding, and lead to hyperlipidemia and dysbetalipoproteinemia37,38,39. Moreover, there is significant downregulation of semi-tryptic peptides (282/283 to 292 amino acid residues) located at the VLDL and lipid binding region40,41. Therefore, we speculate that these proteolytic changes with age also significantly alter the function of APOE in relation to lipid and LDL metabolism.
A WGCNA proteomics module (ME3) in CSF was positively correlated with CSF p-tau181 and total tau concentrations without concurrently being correlated with age. These modules are involved in axonogenesis, synapse organization, and neuron projection biological processes (Fig. 6b). It is essential to note that the CSF p-tau181 concentrations included in this study are within the normal range (below 50.6 pg/mL) even though we found significantly higher concentrations of p-tau181 in the older adult group (Table 1)14. In addition, all participants in this study are cognitively normal. Therefore, in the absence of neurodegeneration, we found that CSF p-tau181 and total tau concentrations are positively correlated with axonogenesis, neuron projection, and synaptic proteins regardless of age. The translation of tau, an axonal cytoskeleton-stabilizing protein that conforms tubular junctions, impacts synaptic function and plasticity by modulating the organization of synapses and influencing the phosphorylation patterns of tau protein within dendritic spines, which may contribute to the regulation of dendritic signal transduction and synaptic stability42,43. Therefore, such dynamics contribute to the regulation of neural signal transduction and synaptic stability, revealing a critical role of tau beyond its traditionally understood pathological contributions in neurodegenerative diseases42,43,44.
In addition to pathway analysis results, we demonstrated that multiple proteins belonging to neuron assembly and axonogenesis modules go through alternative cleavages with age. Firstly, APP, known to play an important role in neural growth and maturation during brain development, was not found to be differentially expressed with age at the proteomic level. However, we found significant downregulation of several semi-tryptic peptides located immediately before beta secretase (BACE) cleavage sites (Fig. 3a) in the older cohort. The mentioned peptides do not overlap with previously well-established intramembranous sections of APP protein that give rise to Aβ1–42 and Aβ1–40 fibrils, which are traditionally associated with cognitive impairment45. This is in line with group comparison of CSF Aβ1–42/Aβ1–40 that also demonstrated no significant difference between older and younger adult groups (Table 1). The biologic cleavage sites of the mentioned downregulated peptides (N-terminus side) align with the N-terminus of several endogenous Aβ/APP fragments that were previously reported in human CSF45. While these cleavage sites were downregulated with aging, an upregulation of another semi-tryptic peptide located at the highly preserved copper binding site (CuBD) of APP was shown46. The cleavage of APP at this site (Asp142 residue), interferes with Cu-binding domain, which is known to modulate neuronal copper homeostasis, APP transport from endoplasmic reticulum to Golgi membrane, and homodimerization of the protein46,47. Therefore, major changes in APP function and transport with normal cognitive aging outside the framework of Aβ aggregation are suggested based on our findings.
Moreover, NRCAM and NRXN1, participating in ME3, both were noted to have age-associated alternative cleavage and phosphorylation changes. NRXN1 comprises multiple splicing variants that act as presynaptic adhesion hub proteins that regulate synaptic structure and signaling48. Peptide-level analysis showed an increase in cleavage of the NRXN1 protein at 730 amino acid residues, which is located at the beginning of the Laminin G-like 4 domain (LG4). The LG4 domain of NRXN1 participates in creating an L-shaped extracellular region that mediates protein adhesion to postsynaptic proteins49. Hence, disruption of this domain can potentially disrupt crucial protein-protein interactions at the synapse49. Similarly, two age-associated phosphorylation events were identified at the fibronectin III (FNIII) domain of NRCAM (Ser666 and Thr668) that can potentially alter its interaction with other proteins, especially gliomedin in Schwann cells50.
Coagulation pathways are among the most differentially expressed pathways in CSF of older cognitively healthy adults based on the GSEA results (Fig. s3). Three fibrinogen domains (FGA, FGB, and FGG) are among the core proteins of these upregulated pathways in both plasma and CSF of older adults. All three fibrinogen components are found to be increased by 12% in plasma (log2 fold change or LogFC = 0.16) and by 40% in CSF (LogFC = 0.50). While the rise of fibrinogen concentration in both plasma and CSF is a well-replicated phenomenon with age, the increase of fibrinogen, a plasma protein, in CSF of older adults has been classically interpreted as a sign and driver of blood–brain barrier (BBB) dysfunction with age51. Additionally, we found that the correlation between CSF and plasma protein concentrations increases with age, which could potentially indicate a disrupted BBB allowing facilitated diffusion of proteins across the BBB52,53. Moreover, fibrinogen is demonstrated to drive a neuroinflammatory response when present in CSF and brain tissue, which is in line with our findings of increased humoral and complement-related neuroinflammatory markers in CSF of older adults (C4BPA, C4BPB, C4A, C3, and C8B). Increased inflammatory processes with age, termed Inflammaging, has been extensively reported and proposed to be a driver of many age-related pathologies54.
Our pathway analysis identified a significant downregulation of the IGF-1 signaling pathway with age in plasma. Moreover, several proteins that were markedly downregulated in both plasma and CSF belonged to the IGF signaling pathway (e.g., IGF2, IGFALS, IGFBP6, and IGFBP5). Confirming previous literature, it has been shown that the IGF-1 pathway regulates organismal aging and longevity in many species, including humans and is a relatively preserved biological process with a potential for being a therapeutic target to increase and improve life span55,56.
Despite the significant findings and comprehensive analysis presented in this study, several limitations need to be acknowledged. First, the study included a relatively small sample size (40 older adults and 52 young adults), which might limit the generalizability of the findings. Additionally, the younger group had a notable imbalance in gender distribution, with significantly more females (28 females and four males), while the older group had a more balanced gender distribution (21 females and 19 males). This gender imbalance in the younger cohort could introduce bias and affect the interpretation of age-related differences. Although attempts were made to control for sex in downstream analysis, other potential confounding factors, such as lifestyle, genetic background, and comorbidities, were not accounted for, which could influence the results. Specifically, we used ssGSEA pathway analysis to account for sex distribution differences. This analysis allowed us to calculate gene set enrichment scores for each biological pathway and further analyze the impact of sex as a covariate. Moreover, although our study employs established methods for protein and peptide differential expression, alternative cleavage, and phosphorylation analysis, our findings have not been validated in an independent cohort due to the scarcity of CSF samples from healthy young individuals, and the unprecedented depth of our peptide-level data has not been reached before; nonetheless, our methodological framework provides a robust blueprint for future studies to replicate and expand upon these observations and elucidate their functional implications. Finally, the functional implications of the novel alterations with aging identified in this study are hypothesis-generating and will need considerable additional work to determine their clinical relevance.
This study provides a comprehensive proteomic and peptide-level analysis of CSF and plasma in cognitively normal aging individuals, revealing significant age-associated molecular changes. Our data indicate that with advancing age, there is an upregulation of extracellular matrix components, alterations in lipoprotein metabolism, and increased activity of inflammatory pathways. By focusing on alternative cleavage and phosphorylation events, we have uncovered previously unreported post-translational modifications in proteins involved in critical biological processes such as axonogenesis, synaptic activity, extracellular matrix organization, and lipid metabolism. These peptide-level changes (detected without any phosphopeptide enrichment) offer a more granular perspective on the molecular modifications occurring during normal aging, which conventional protein-level quantification alone cannot capture.
Our integrated approach, combining traditional protein-level analysis with deep peptide-level investigation, reveals a multifaceted layer of proteoform diversity in the aging proteome. Notably, key proteins including APOE, APP, and NRXN1 exhibit unique post-translational modifications, such as differential phosphorylation and alternative cleavage/splicing events. Importantly, while some proteins (e.g., APP) did not show significant differences at the protein level, their peptide-level profiles revealed marked alterations, underscoring the added value of this in-depth analysis.
Future studies with larger, more diverse cohorts and longitudinal designs are essential to validate these results and further elucidate the molecular mechanisms underlying cognitive resilience and the role of post-translational modifications in aging populations.
Data availability
The raw data have been deposited with the ProteomeXchange Consortium via the MassIVE partner repository under accession number MSV000097888. The full Spectronaut project, including search archives, settings, and MaxLFQ matrices, is included in the same submission and can be inspected using the free Spectronaut Viewer (www.biognosys.com/spectronaut-viewer). The dataset is accessible at: https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?task=00eb30f68bb44f008abdc4f9de6be1dd. The source data underlying the graphs and charts in the main figures are provided as Supplementary Tables s1–s12. Any unique reagents, protocols, or materials generated during this study are available from the corresponding author upon reasonable request. There are no restrictions on the availability of these materials.
References
Moaddel, R. et al. Proteomics in aging research: a roadmap to clinical, translational research. Aging Cell 20, e13325 (2021).
Basisty, N. et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 18, e3000599 (2020).
Lehallier, B., Shokhirev, M. N., Wyss-Coray, T. & Johnson, A. A. Data mining of human plasma proteins generates a multitude of highly predictive aging clocks that reflect different aspects of aging. Aging Cell 19, e13256 (2020).
Rutledge, J., Oh, H. & Wyss-Coray, T. Measuring biological age using omics data. Nat. Rev. Genet. 23, 715 (2022).
Johnson, A. A., Shokhirev, M. N., Wyss-Coray, T. & Lehallier, B. Systematic review and analysis of human proteomics aging studies unveils a novel proteomic aging clock and identifies key processes that change with age. Ageing Res. Rev. https://doi.org/10.1016/j.arr.2020.101070 (2020).
Shuken, S. R. et al. Limited proteolysis-mass spectrometry reveals aging-associated changes in cerebrospinal fluid protein abundances and structures. Nat. Aging 2, 379 (2022).
Coenen, L., Lehallier, B., De Vries, H. E. & Middeldorp, J. Markers of aging: unsupervised integrated analyses of the human plasma proteome. Front Aging https://doi.org/10.3389/fragi.2023.1112109 (2023).
Lehallier, B. et al. Undulating changes in human plasma proteome profiles across the lifespan. Nat. Med. 25, 1843–1850 (2019).
Oh, H. S. H. et al. Organ aging signatures in the plasma proteome track health and disease. Nature 624, 164–172 (2023).
Baird, G. S. et al. Age-dependent changes in the cerebrospinal fluid proteome by slow off-rate modified aptamer array. Am. J. Pathol. 180, 446–456 (2012).
Wesenhagen, K. E. J. et al. Effects of age, amyloid, sex, and APOE ε4 on the CSF proteome in normal cognition. Alzheimer’s. Dement. 14, 19 (2022).
Zhang, J. et al. Quantitative proteomic analysis of age-related changes in human cerebrospinal fluid. Neurobiol. Aging 26, 207–227 (2005).
Jack, C. R. et al. Introduction to the recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s. Dement. 7, 257–262 (2011).
Greenberg, B. D. et al. CSF Alzheimer disease biomarkers: time-varying relationships with MCI symptom onset and associations with age, sex, and apoE4. Neurology 99, e1640 (2022).
Muntel, J. et al. Surpassing 10 000 identified and quantified proteins in a single run by optimizing current LC-MS instrumentation and data analysis strategy. Mol. Omics. 15, 348–360 (2019).
Tognetti, M. et al. Biomarker candidates for tumors identified from deep-profiled plasma stem predominantly from the low abundant area. J. Proteome Res. 21, 1718–1735 (2022).
Cox, J. et al. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteom. 13, 2513–2526 (2014).
Voß, H. et al. HarmonizR enables data harmonization across independent proteomic datasets with appropriate handling of missing values. Nat. Commun. 13, 1–15 (2022).
Ahlmann-Eltze, C. proDA: differential abundance analysis of label-free mass spectrometry data. Bioconductor R package version 1.0 (2022).
Stekhoven, D. J. & Bühlmann, P. Missforest-non-parametric missing value imputation for mixed-type data. Bioinformatics 28, 112–118 (2012).
Liao, Y., Wang, J., Jaehnig, E. J., Shi, Z. & Zhang, B. WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 47, W199–W205 (2019).
Wang, K. et al. Comprehensive map of age-associated splicing changes across human tissues and their contributions to age-associated diseases. Sci. Rep. 8, 10929 (2018).
Semba RD, et al. A targeted proteomic assay for the measurement of plasma proteoforms related to human aging phenotypes. Proteomics. 17, 1600232 (2017).
Rafikova, E. et al. Open genes—a new comprehensive database of human genes associated with aging and longevity. Nucleic Acids Res. 52, D950–D962 (2024).
Rodriguez, J. M. et al. APPRIS 2017: principal isoforms for multiple gene sets. Nucleic Acids Res. 46, D213–D217 (2018).
König, G. et al. Identification and differential expression of a novel alternative splice isoform of the beta A4 amyloid precursor protein (APP) mRNA in leukocytes and brain microglial cells. J. Biol. Chem. 267, 10804–10809 (1992).
Ewald, C. Y. The matrisome during aging and longevity: a systems-level approach towards defining matreotypes promoting healthy aging. Gerontology 66, 266 (2019).
Statzer, C., Park, J. Y. C. & Ewald, C. Y. Extracellular matrix dynamics as an emerging yet understudied hallmark of aging and longevity. Aging Dis. 14, 670 (2023).
Tsumagari, K., Sato, Y., Aoyagi, H., Okano, H. & Kuromitsu, J. Proteomic characterization of aging-driven changes in the mouse brain by co-expression network analysis. Sci. Rep. 13, 1–8 (2023).
Ewald, C. Y., Landis, J. N., Abate, J. P., Murphy, C. T. & Blackwell, T. K. Dauer-independent insulin/IGF-1-signalling implicates collagen remodelling in longevity. Nature 519, 97 (2015).
Moretto, E., Stuart, S., Surana, S., Vargas, J. N. S. & Schiavo, G. The role of extracellular matrix components in the spreading of pathological protein aggregates. Front. Cell Neurosci. 16, 844211 (2022).
Reed, M. J., Damodarasamy, M. & Banks, W. A. The extracellular matrix of the blood–brain barrier: structural and functional roles in health, aging, and Alzheimer’s disease. Tissue Barriers 7, 1651157 (2019).
Verbeek, E. et al. COL4A2 mutation associated with familial porencephaly and small-vessel disease. Eur. J. Hum. Genet. 20, 844 (2012).
Costea, L. et al. The blood–brain barrier and its intercellular junctions in age-related brain disorders. Int. J. Mol. Sci. 20, 5472 (2019).
Wang, Q., Zhou, W. & Zhang, J. Alzheimer, for & Neuroimaging Initiative, D. Higher apolipoprotein C-III levels in cerebrospinal fluid are associated with slower cognitive decline in mild cognitive impairment. J. Alzheimer’s. Dis. 67, 961–969 (2019).
Zhang, X. & Initiative, O. behalf of the A. D. N. Higher plasma APOC-III was associated with a slower reduction of β-amyloid levels in cerebrospinal fluid among older individuals without dementia. Neuropsychiatr. Dis. Treat. 16, 1139 (2020).
Lohse, P., Alexander Mann, W., Stein, E. A. & Bryan Brewer, H. Apolipoprotein E-4Philadelphia (Glu13—Lys,Arg145—Cys). Homozygosity for two rare point mutations in the apolipoprotein E gene combined with severe type III hyperlipoproteinemia. J. Biol. Chem. 266, 10479–10484 (1991).
De Knijff, P. et al. Familial dysbetalipoproteinemia associated with apolipoprotein E3-Leiden in an extended multigeneration pedigree. J. Clin. Invest. 88, 643–655 (1991).
Wintjens, R. et al. Global molecular analysis and APOE mutations in a cohort of autosomal dominant hypercholesterolemia patients in France. J. Lipid Res. 57, 482 (2016).
Dong, L. M. et al. Human apolipoprotein E. Role of arginine 61 in mediating the lipoprotein preferences of the E3 and E4 isoforms. J. Biol. Chem. 269, 22358–22365 (1994).
Weisgraber, K. H. Apolipoprotein E distribution among human plasma lipoproteins: role of the cysteine-arginine interchange at residue 112. J. Lipid Res. 31, 1503–1511 (1990).
Brown, J. et al. Tau in cerebrospinal fluid induces neuronal hyperexcitability and alters hippocampal theta oscillations. Acta Neuropathol. Commun. 11, 67 (2023).
Mandell, J. W. & Banker, G. A. A spatial gradient of tau protein phosphorylation in nascent axons. J. Neurosci. 16, 5727–5740 (1996).
Mietelska-Porowska, A., Wasik, U., Goras, M., Filipek, A. & Niewiadomska, G. Tau protein modifications and interactions: their role in function and dysfunction. Int. J. Mol. Sci. 15, 4671–4713 (2014).
Brinkmalm, G. et al. An online nano-LC-ESI-FTICR-MS method for comprehensive characterization of endogenous fragments from amyloid β and amyloid precursor protein in human and cat cerebrospinal fluid. J. Mass Spectrom. 47, 591–603 (2012).
Barnham, K. J. et al. Structure of the Alzheimer’s disease amyloid precursor protein copper binding domain: a regulator of neuronal copper homeostasis. J. Biol. Chem. 278, 17401–17407 (2003).
Spoerri, L., Vella, L. J., Pham, C. L. L., Barnham, K. J. & Cappai, R. The amyloid precursor protein copper binding domain histidine residues 149 and 151 mediate APP stability and metabolism. J. Biol. Chem. 287, 26840 (2012).
Ishizuka, K. et al. Functional characterization of rare NRXN1 variants identified in autism spectrum disorders and schizophrenia. J. Neurodev. Disord. 12, 1–16 (2020).
Miller, M. T. et al. The crystal structure of the α-neurexin-1 extracellular region reveals a hinge point for mediating synaptic adhesion and function. Structure 19, 767 (2011).
Labasque, M., Devaux, J. J., Lévêque, C. & Faivre-Sarrailh, C. Fibronectin type III-like domains of neurofascin-186 protein mediate gliomedin binding and its clustering at the developing nodes of Ranvier. J. Biol. Chem. 286, 42426 (2011).
McAleese, K. E. et al. Extravascular fibrinogen in the white matter of Alzheimer’s disease and normal aged brains: implications for fibrinogen as a biomarker for Alzheimer’s disease. Brain Pathol. 29, 414–424 (2019).
Banks, W. A., Reed, M. J., Logsdon, A. F., Rhea, E. M. & Erickson, M. A. Healthy aging and the blood–brain barrier. Nat. Aging 1, 243–254 (2021).
Musaeus, C. S. et al. Cerebrospinal fluid/plasma albumin ratio as a biomarker for blood-brain barrier impairment across neurodegenerative dementias. J. Alzheimer’s. Dis. 75, 429–436 (2020).
Cullen. et al. Accelerated inflammatory aging in Alzheimer’s disease and its relation to amyloid, tau, and cognition. Sci. Rep. 11, 1–9 (2021).
Mao, K. et al. Late-life targeting of the IGF-1 receptor improves healthspan and lifespan in female mice. Nat. Commun. 9, 1–12 (2018).
Anisimov, V. N. & Bartke, A. The key role of growth hormone–insulin–IGF-1 signaling in aging and cancer. Crit. Rev. Oncol. Hematol. 87, 201–223 (2013).
Acknowledgements
This work was supported by the National Institutes of Aging (Grant numbers: U19AG033655 and P30 AG066507).
Author information
Authors and Affiliations
Contributions
All authors have contributed significantly to the conception, design, execution, and analysis of this study and to the writing of the manuscript. Specifically, M.W.L. and A.M. led the conceptualization and study design of this project, formulating the research idea and experimental strategy, while P.S., M.T., C.B., S.G.H., R.B., L.R., and Y.F. conducted the data acquisition through experiments and sample collection. A.K., M.T., C.B., M.W.L., and P.S. then carried out the data analysis and interpretation to extract meaningful insights from the findings, and finally, A.K., P.S., A.M., and M.W.L. prepared the manuscript. All authors have reviewed and approved the final version of the manuscript, ensuring the integrity and accuracy of the work.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Medicine thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kamalian, A., Shichkova, P., Tognetti, M. et al. Unveiling proteomic and peptide-level modifications in cerebrospinal fluid and plasma in normal cognitive aging. Commun Med 5, 474 (2025). https://doi.org/10.1038/s43856-025-01183-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s43856-025-01183-0
