
Abstract
Immune checkpoint inhibitor (ICI) therapies can increase the risk of cardiovascular events in survivors of cancer by worsening atherosclerosis. Here we map the expression of immune checkpoints (ICs) within human carotid and coronary atherosclerotic plaques, revealing a network of immune cell interactions that ICI treatments can unintentionally target in arteries. We identify a population of mature, regulatory CCR7+FSCN1+ dendritic cells, similar to those described in tumors, as a hub of IC-mediated signaling within plaques. Additionally, we show that type 2 diabetes and lipid-lowering therapies alter immune cell interactions through PD-1, CTLA4, LAG3 and other IC targets in clinical development, impacting plaque inflammation. This comprehensive map of the IC interactome in healthy and cardiometabolic disease states provides a framework for understanding the potential adverse and beneficial impacts of approved and investigational ICIs on atherosclerosis, setting the stage for designing ICI strategies that minimize cardiovascular disease risk in cancer survivors.
Similar content being viewed by others
Main
ICIs have transformed cancer immunotherapy, improving the survival of patients with previously untreatable malignancies1. These therapeutic monoclonal antibodies target IC molecules, reinvigorating cytotoxic T cell responses against tumor cells by reducing negative regulatory signals and amplifying co-stimulatory signals. In the United States, 11 ICIs targeting programmed cell death protein 1 (PD-1), programmed cell death ligand 1 (PD-L1), cytotoxic T lymphocyte-associated protein 4 (CTLA4) and lymphocyte-activation gene 3 (LAG3) are approved for 19 cancer-related indications2,3,4,5. With more than 40% of patients with cancer eligible for ICI treatment4,6, and ongoing clinical trials investigating additional ICIs targeting Cluster of Differentiation 47 (CD47), T cell immunoglobulin and mucin-domain containing-3 (TIM3), T cell immunoglobulin and ITIM domain (TIGIT) and B and T lymphocyte attenuator (BTLA), this class of drugs is poised to rapidly expand.
While ICIs aim to enhance the immune response within the tumor microenvironment, their impact can extend to healthy tissues. Immune-related adverse events (irAEs), including those affecting the heart, occur frequently in patients treated with ICIs, requiring treatment discontinuation in 10–40% of cases7. Furthermore, associations between ICI therapy and atherosclerotic cardiovascular disease (ASCVD)-related adverse events have been reported, including accelerated progression of atherosclerotic plaques and increased risk for stroke and myocardial infarction7,8,9. These clinical findings align with studies in preclinical animal models showing that inhibition of PD-1/PD-L1 and CTLA4 expedite atherosclerosis and increase plaque T cell accumulation and activation10,11. Moreover, the identification of T cells expressing PD-1 and other markers of exhaustion within human atherosclerotic plaques12 suggests that ICIs may activate plaque T cells and their pro-atherogenic functions. Notably, not all ICIs affect atherosclerosis in a similar fashion: LAG3 inhibition in mice increased plaque T cell activation without affecting plaque size13; deletion of Lgals9 (encoding galectin 9, which interacts with TIM3) in mice increased plaque burden14; and ICIs targeting CD47 have anti-atherosclerotic effects in patients15 and mice16. These observations emphasize the need to consider the expression of immune checkpoint molecules beyond the tumor microenvironment in the design of ICIs, with the goal of anticipating and potentially mitigating the adverse effects of ICIs on ASCVD and related ischemic cardiovascular events (for example, stroke and myocardial infarction) in survivors of cancer.
Cancer and atherosclerosis also share common risk factors, including hyperlipidemia and diabetes17. Indeed, close to 20% of patients with cancer have diabetes, a condition characterized by low-grade chronic inflammation. Therefore, a comprehensive understanding of irAEs stemming from ICI therapy in the context of ASCVD also necessitates examining how common comorbidities such as dyslipidemia and diabetes, and their treatment, influence IC expression. To bridge this knowledge gap, we conducted an extensive single-cell transcriptional mapping of ICs in human and mouse atherosclerotic plaques and identified how disease-modifying factors such as type 2 diabetes and lipid-lowering treatments influence IC expression and cellular crosstalk in that milieu.
Results
Immune checkpoint landscape of human atherosclerosis
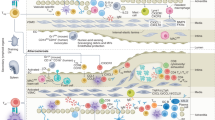
To investigate IC expression in human atherosclerotic plaque, we analyzed single-cell RNA sequencing (scRNA-seq) data from 69,442 immune cells isolated from 22 carotid plaques of patients undergoing carotid endarterectomy (CEA). We identified nine major identities and 69 sub-identities of plaque immune cells (Fig. 1a, Extended Data Fig. 1, Supplementary Fig. 1 and Supplementary Tables 1 and 2). As previously reported12,18,19,20, T cells accounted for ~70% of sequenced plaque immune cells (Fig. 1b,c), with CD8+ and CD4+ T cells comprising ~35%. A mixed T cell cluster, constituting ~20% of the dataset, matched CD8+ or CD4+ T counterparts despite lacking CD8A, CD8B1 or CD4 expression (Supplementary Fig. 2). Another cluster, identified as Tγδ cells, also lacked CD8A, CD8B1 or CD4 transcripts and comprised 10–20% of all immune cells, consistent with CD8–CD4– T cells identified by cytometry by time-of-flight (CyTOF) and corroborated by cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq) in an independent cohort (Supplementary Fig. 3 and Supplementary Table 3). CD8+CD4+ double-positive (DP) T cells, B cells, myeloid cells, natural killer (NK) cells and innate lymphoid cells (ILCs) accounted for the remaining ~25% of plaque immune cells (Fig. 1c).

a, Schematic depiction of experimental design. scRNA-seq was performed on CD45+ cells from atherosclerotic plaque tissue collected from patients undergoing CEA (n = 22). b, UMAP of scRNA-seq data clustered by cell type. c, Proportions of major immune cell identities per patient. Each dot represents one patient. Boxes represent interquartile ranges; center lines depict medians. Whiskers below and above boxes represent extent of lower and upper quartiles, respectively (n = 22). d, Dot plot of IC gene expression by immune cell subclusters in human atherosclerotic plaques. Dot color represents positive fold change (FC) values in each subcluster compared to the rest (false discovery rate (FDR) < 0.05). Among ‘FDA-approved targets’ are genes encoding direct targets of drugs approved by the US FDA to treat cancer-related indications as well as genes encoding their interaction partners. Among ‘Investigational targets’ are genes encoding ICs currently under investigation as potential cancer immunotherapy targets (ClinicalTrials.gov; 6 December 2023). ICs that do not fit either of these categories appear under ‘Other immune checkpoints’. Mye, myeloid cell; CTL, cytotoxic T lymphocyte; Reg, regulatory; Mono, monocyte; Mφ, macrophage; DC, dendritic cell.
To provide a comprehensive framework for investigating IC gene expression in human atherosclerotic tissues, we focused on a more granular characterization of major immune cells based on manually curated annotations aligned with the CellTypist consensus of immune cells across human tissues21 (Extended Data Fig. 1 and Supplementary Fig. 4). The most abundant plaque CD8+ T cell subsets corresponded to effector memory (EM) and terminally differentiated effector memory re-expressing CD45RA (EMRA) cells. An EM CD8+ T cell subcluster expressing GZMK accounted for ~18% of all CD8+ T cells, while an EMRA CD8+ T cell subcluster expressing CXC3R1, characterizing resident and highly cytotoxic CD8+ T cells22, accounted for ~16% (Extended Data Fig. 1a). Naive CD4+, mixed and DP T cell populations represented a substantial proportion (~15%, ~45% and ~22%) of their respective T cell subsets (Extended Data Fig. 1b–d and Supplementary Fig. 5). Mucosal-associated invariant T (MAIT) subclusters were also discernible within the CD4+, CD8+ and mixed T cell groups. Some immune cell subclusters, such as MZB1+ plasma B cells, CLEC4C+ plasmacytoid dendritic cells, ITGA6+IL17RA+ NK cells and IL1R1+ type 1 ILCs (ILC1; Extended Data Fig. 1e–g) remain poorly understood in the plaque; however, others, such as PLIN2+TREM1+ macrophages linked to plaque inflammation and TREM2+ macrophages associated with lipid processing and pro-resolving functions, have been previously characterized19,23.
Analysis of IC transcripts revealed heterogeneous expression levels of both co-inhibitory and co-stimulatory IC genes across this immunological map, highlighting the intricate equilibrium between immune activation and suppression within the atherosclerotic plaque microenvironment (Fig. 1d). Transcripts encoding the clinically approved targets PD-1 (PDCD1), CTLA4 (CTLA4) and LAG3 (LAG3) were primarily expressed in T cells, particularly in EM and EMRA. PDCD1 was expressed by most CD8+ and CD4+ EM and EMRA T cells, as well as by clusters of γδ T cells, but was absent in central memory (CM) and naive T cell clusters. CTLA4 was predominantly expressed in regulatory T cells, including LAIR2+ and CCR4+ CD4+ T cells, IL2RA+ mixed T cells and IL32+ DP T cells, but was absent in plaque CD8+ T cells. Subsets of EM CD4+ T cells, such ISG15+ and GZMK+, as well as IL9R+ and ITGA1+ resident memory (RM) CD4+ T cells, also expressed CTLA4. LAG3 was expressed by all EM, EMRA, RM and MAIT CD8+ T cell clusters; ISG15+ EM and LAIR+ regulatory CD4+ T cells; subclusters of γδ T cells (for example, TNF+, S1PR1+, GZMK+ and PTMS+); and NKG7+ EM and GIMAP4+ EMRA DP T cells. CD274 (which encodes PD-L1) was modestly expressed in some CD8+ (for example, IL7R+ naive, LGALS3+ EM, CCL42L+ EMRA and ISG20+ EMRA) and CD4+ (for example, ISG15+ EM, RGCC+ EMRA and ITGA1+ RM) T cell clusters.
Among plaque myeloid cells, CCR7+FSCN1+ dendritic cells (~0.7% of myeloid cells) exhibited elevated expression of CD274, PDCD1LG2 (encoding PD-L2) and the transcripts encoding the CTLA4 ligands CD86 and CD80. Except for CD274, plaque myeloid cells did not express genes encoding other US Food and Drug Administration (FDA)-approved ICI targets. PDCD1LG2 was expressed by CCR7+FSCN1+ dendritic cells and TREM2+ and HLA-DRB1+ macrophages, whereas the majority of monocyte, macrophage and dendritic cell clusters expressed CD86. CD86 can either activate T cells by interacting with CD28 (ref. 24) or inhibit them by binding to CTLA4 (ref. 25), suggesting that CTLA4 inhibitors may increase myeloid cell-mediated activation of T cells within plaques. CD80, was expressed in fewer myeloid clusters, indicating a less prominent role in the plaque.
Co-inhibitory signaling molecules under clinical investigation, including LGALS9 (encoding galectin 9), HAVCR2 (encoding T cell immunoglobulin and mucin domain 3, TIM3) and SIRPA (encoding signal regulatory protein α; SIRPα), showed elevated expression levels across various monocyte, macrophage and dendritic cell subpopulations. Notably, CCR7+FSCN1+ dendritic cells exhibited high expression of LGALS9, SIRPA and CD200 (encoding the co-inhibitory molecule CD200). A specific subcluster of TCL1A+ mature B cells also exhibited heightened expression of CD200, while its receptor, encoded by CD200R, was detected in several T cell subclusters and a few NK and ILC subclusters. Ongoing investigations are exploring the inhibition of the CD200–CD200R axis as a potential anticancer strategy26; however, CD200R expression on inflammatory monocytes in patients with coronary artery disease is associated with a more stable plaque phenotype and lack of Cd200 in mice exacerbates atherogenesis27, suggesting a possible pro-atherogenic effect of targeting CD200/CD200R. Another investigational IC target, CD47 (ref. 15), which encodes the ‘don’t-eat-me’ signal protein CD47, was lowly expressed in T cell subpopulations, such as RM CD8+ and CD4+ T cells, as well as NCAM1+ NK cells.
Various immune cells within human plaque expressed transcripts encoding co-stimulatory molecules targeted by investigational ICIs. CD40, the target of agonists designed to mimic the CD40 ligand (CD40L) to boost antitumor immune responses28, was expressed by plaque dendritic cells and macrophages, including CCR7+FSCN1+ dendritic cells, TREM2+ macrophages and most B cell clusters. NK cells and ILCs, which have cytotoxic and pro-inflammatory functions29, showed preferential expression of genes encoding TNF superfamily molecules with co-stimulatory functions. For example, KLRC1+NCAM1+ NK cells and IL1R1+ ILC1 expressed TNFRSF18, which encodes the co-stimulatory molecule GITR. TNFRSF18 was also highly expressed by CD4+, mixed and DP regulatory T cells. Notably, both CD40 and GITR aggravate pro-atherogenic processes30,31, indicating that investigational cancer immunotherapies enhancing their function may exacerbate atherosclerosis.
We also investigated IC expression differences between atherosclerotic and healthy vasculature, in both immune and nonimmune cells, using scRNA-seq of six human coronary artery specimens from five explanted hearts. These were classified by a blinded clinical pathologist (N.N.) as either atherosclerotic (n = 3) or showing adaptive intimal thickening (AIT; n = 3), a normal vascular adaptation to local mechanical forces evident from a young age32 (Extended Data Fig. 2a and Supplementary Table 4). Immune cell clusters within the coronary vasculature largely overlapped with those found in carotid plaques. The nonimmune compartment, comprising endothelial cells, fibroblasts, myofibroblasts and smooth muscle cells, accounted for ~20% of cells in atherosclerotic coronaries and ~34% of cells in AIT (Extended Data Fig. 2a–c). IC gene expression across the coronary cellular map largely confirmed the findings of the carotid plaque analysis, with some differences noted (Extended Data Fig. 2d). Most ICs were expressed by both AIT and atherosclerotic coronary artery immune cells. Among these, 21 IC genes were differentially expressed in specific subsets of immune and nonimmune cells with most, including LAG3, LGALS3, LGALS9, CD47 and CD40, being downregulated in atherosclerotic compared to AIT coronaries (Extended Data Fig. 2d). Analysis of ICs in nonimmune cells revealed that LGALS3 was significantly downregulated in endothelial cells and fibroblasts from atherosclerotic coronaries compared to AIT. Additionally, CD200 expression was significantly lower in myofibroblasts and NT5E expression was lower in fibroblasts, within atherosclerotic coronaries compared to AIT.
ICOS, CD40LG and VSIR were the only IC genes upregulated in atherosclerotic coronaries, specifically in T cell subsets such as GZMK+ EM CD8+ T cells, naive CD4+ and mixed T cells and GZMK+ γδ T cells (Extended Data Fig. 2d). ICOS was also highly expressed in most T cells within carotid plaques (Fig. 1d), suggesting that ICOS co-stimulation therapies under investigation for cancer33 may exacerbate atherosclerosis at different vascular sites, particularly when combined with CTLA4 or PD-1 blockade.
We also identified distinct IC expression patterns in immune and mesenchymal cells between healthy and atherosclerotic coronaries. For example, LGALS3 was expressed in CCR7+FSCN1+ dendritic cells only in AIT. In atherosclerotic coronaries, PDCD1 was exclusively found in LEF1+ naive CD4+ T cells and myofibroblasts, whereas in AIT it was expressed in CX3CR1+ EMRA CD8+ T cells, GZMB+ cytotoxic CD4+ T cells, monocytes, HLA-DRB1+ macrophages, CCL4+ NK cells, as well as endothelial cells, fibroblasts and smooth muscle cells. Other targets of US FDA-approved ICIs also showed exclusive expression in specific clusters in either AIT or atherosclerotic coronaries, indicating distinct susceptibility of normal and diseased vessels to ICIs.
Overall, our findings demonstrate that most T cells within human atherosclerotic plaques, whether carotid or coronary, can be directly influenced by US FDA-approved ICIs targeting PD-1, CTLA4 and LAG3. Among myeloid cells, CCR7+FSCN1+ dendritic cells predominantly expressed molecules targeted by US FDA-approved or investigational ICIs, indicating that these cells are key mediators of the immune response to ICIs within plaques.
IC-mediated interactions in human atherosclerotic plaques
To investigate IC interactions between immune cells, we conducted a cell–cell communication analysis using a manually curated list of established IC ligand–receptor pairs (Supplementary Table 5). CCR7+FSCN1+ dendritic cells mediated up to 15 co-stimulatory and co-inhibitory interactions (Extended Data Fig. 3), including pairs targeted by approved and investigational ICIs (Extended Data Fig. 4a). PD-L1–PD-1 and PD-L2–PD-1 signaling was predominantly identified between CCR7+FSCN1+ dendritic cells and various T cell populations, including EM, EMRA, RM, regulatory, γδ T cells, MAIT, NKT cells and cytotoxic T lymphocytes (CTLs) (Fig. 2a and Extended Data Fig. 4b). Notably, PDCD1 exhibited stronger signaling through CD274 engagement compared to PDCD1LG2 (Extended Data Fig. 4c). Multiplex immunofluorescence staining confirmed that PD-L1+CD11c+ dendritic cells and PD-1+ T cells were present in human carotid plaques and interacting based on neighborhood enrichment score (NES) analysis34 (Fig. 2b). Various myeloid populations, including CCR7+FSCN1+ dendritic cells, were predicted to interact with regulatory T cell subclusters through CD86–CTLA4 or CD80–CTLA4 signaling (Fig. 2c and Extended Data Fig. 4d), with CD86–CTLA4 identified as the stronger interaction within the plaque (Extended Data Fig. 4e). Immunofluorescent staining of carotid plaque sections confirmed that CTLA4+CD4+ T cells interacted with CD4–CD86+ cells in at least half of the specimens assayed (Fig. 2d). LAG3 interactions with LGALS3 (which encodes galectin 3) or SNCA (encoding the protein α-synuclein; αSyn) involved largely EM, EMRA and RM CD8+ T cell clusters and multiple myeloid subtypes (Fig. 2e and Extended Data Fig. 4f). CCR7+FSCN1+ dendritic cells were predicted to engage LAG3 via LGALS3 expression rather than SNCA (Extended Data Fig. 4g). Spatial aggregation analysis confirmed interactions between galectin 3+ cells and LAG3+CD8a+ T cells (Fig. 2f), suggesting that relatlimab-nivolumab ICI therapy, used in unresectable melanoma35, may aggravate atherosclerosis by increasing effector T cell responses within atherosclerotic lesions by targeting LAG3 and PD-1 in plaques. CD274, CD86 and LGALS3 were more robustly expressed in relevant myeloid cell populations when compared to PDCD1LG2, CD80, CLEC4G and FGL1, which were either absent or minimally expressed in our dataset (Extended Data Fig. 4h).

a,c,e, Chord plots illustrating the strongest interactions (P < 0.05) between: PD-L1 (encoded by CD274) and PD-1 (encoded by PDCD1) (a), CD86 and CTLA4 (c) and galectin 3 (encoded by LGALS3) and LAG3 (e). Direction of ligand → receptor interaction is denoted by chord arrows. b,d,f, Representative immunofluorescent staining of human carotid plaque specimens (left; n = 6–8) and corresponding cell segmentation of phenotypes of interest (top right) for spatial aggregation analysis by NES computation (bottom right). Scale bars, 10 μm. NES > 0 are depicted in red and <0 in blue.
Top predicted co-inhibitory interactions between human plaque immune cells included HLA-E and KLRC1 (encoding NKG2A), HMGB1 and HAVCR2 (encoding TIM3), and CD47 and SIRPA (encoding SIRPα) (Extended Data Fig. 4a). The HLA-E–KLRC1 axis, a promising cancer IC target36, was predicted to signal most strongly between T and NK cells (Extended Data Fig. 5a).
The HMGB1–HAVCR2 (HMGB1–TIM3) pair was predicted to signal between plaque T and NK cells (Extended Data Fig. 5b), while LGALS9–HAVCR2 (galectin 9–TIM3) interactions were strongest among myeloid cell subclusters, including CCR7+FSCN1+ dendritic cells (Extended Data Fig. 5c). Given the heightened plaque burden in mice treated with a Tim3 inhibitor14,37, our findings indicate that investigational ICIs38,39 targeting TIM3, may exacerbate innate immune responses within atherosclerotic tissue. CD47–SIRPA communication, which occurred mainly between many T cell subsets and LY6E+ monocytes and PLIN2+ macrophages (Extended Data Fig. 5d), acts as a ‘don’t-eat-me’ signal and its disruption can both remove cancer cells and ameliorate atherosclerosis15,16. We also predicted potential interactions between BTLA and TNFRSF14 (encoding HVEM) exclusively between B cells and EM, EMRA or regulatory T cells, especially CD4+ and DP T cells (Extended Data Fig. 5e), which would inhibit T cell activation and cytokine production40. Other IC interactions involved TIGIT, the target of a recently designated breakthrough therapy by the US FDA, and its four ligands41 NECTIN2–4 and PVR (encoding Poliovirus receptor) (Extended Data Fig. 5f).
Our model predicted weak interactions between TIGIT and NECTIN4, suggesting that inhibiting NECTIN 4 with enfortumab vedotin-ejfv, a conjugate of an anti-NECTIN 4 antibody and a microtubule inhibitor42, may not significantly impact atherosclerosis. Stronger interactions were predicted between TIGIT and NECTIN2 (Extended Data Fig. 5f), particularly in CCR7+FSCN1+ dendritic cells and various T and NK cell clusters (Extended Data Fig. 5g).
CCR7 + FSCN1 + dendritic cells coordinate IC-mediated interactions
Myeloid cells engaged in the largest number of significant IC interactions with effector and regulatory T cells within the CD8+, CD4+, mixed and DP compartments. Notably, CCR7+FSCN1+ dendritic cells were the main drivers of all the myeloid–T cell interactions (Fig. 3a), marking them as an immunoregulatory hub within human atherosclerotic plaques. CCR7 is crucial for dendritic cell maturation and chemotaxis43, while FSCN1 encodes fascin, an actin-bundling protein essential for dendritic cell activation, maturation and migration44. CCR7+FSCN1+ dendritic cells exhibited a distinctive transcriptional profile (for example, CCL19, CCL22, IL4I1, LAMP3, BIRC3, IDO1, RELB and SOCS2) (Fig. 3b and Supplementary Fig. 6a) similar to that of mature and migratory dendritic cells in tumors45,46,47. We detected CCR7+FSCN1+ dendritic cells in similar proportions in 13 of 22 carotid and five of six coronary artery samples (Supplementary Fig. 6b). Immunofluorescence staining and unbiased digital pathology analyses showed that CD11c+FSCN1+ cells constitute ~1% of human carotid plaque immune cells (Fig. 3c). As these cells expressed genes involved in the TH1 and TH2 pathways (Fig. 3d) and downregulated interleukin (IL)-10 signaling, they likely contribute to pro-atherogenic immune responses.

a, Heatmap showing the number of significant (P < 0.05) IC interactions in which ligand-expressing myeloid cells were predicted to engage with T cells within atherosclerotic plaques. P values were calculated by the CellChat package (v.1.1.3) in R (v.4.0.3). b, Violin plots showing normalized transcript abundance and distribution in myeloid cell subclusters engaging in IC interactions with T cells. NE, normalized expression. c, Representative immunofluorescent staining of human carotid plaque specimens (top; n = 3) and proportion of CD11c+ or CD11c+FSCN1+ cells as a percentage of total cells quantified per specimen (bottom). Scale bars, 10 μm. Each dot in the bar plot represents the average of three regions of interest. d, Canonical pathway analysis of differentially expressed transcripts in CCR7+FSCN1+ dendritic cells compared to other myeloid cell populations in human atherosclerotic tissue. Significantly upregulated or downregulated gene pathways terms appear as red or blue dots, respectively. Gray dots indicate pathways below cutoffs for significance (P value < 0.05 and |z-score| > 2), denoted by dotted lines. P values were calculated by the Ingenuity Pathway Analysis software. Some pathway names were edited for brevity. e, Chord plot illustrating the strongest significant IC interactions between ligand-expressing CCR7+FSCN1+ DCs (left) and receptor-expressing subclusters (right).
The most prominent IC-mediated interactions between CCR7+FSCN1+ dendritic cells and other plaque T cells involved HLA-DR, HLA-C (encoding MHC-I or MHC-II) and CD8 or CD4 (Supplementary Fig. 6c), consistent with their roles in antigen-presentation and T cell activation. These dendritic cells also engaged with other immune cells through interactions targeted by ICIs, such as CD86–CTLA4, NECTIN2–TIGIT, HMGB1–HAVCR2, LGALS3–LAG3 and LGALS9–HAVCR2 (Fig. 3e). Our data suggest that CCR7+FSCN1+ dendritic cells, which display the transcriptional signature of dendritic cells in tumors45,46, are deeply implicated in IC signaling in human atherosclerotic plaques.
Lipid lowering modulates IC profile of atherosclerotic plaques
Lipid-lowering treatment is crucial for preventing atherosclerotic cardiovascular disease7,8,10,48 and improving survival in patients with cancer49. Understanding its impact on IC expression and IC-mediated crosstalk in human atherosclerosis is essential, as it may affect the susceptibility of atherosclerotic plaques to ICIs. To investigate this, we fed Ldlr−/− mice a western diet for 16 weeks to induce atherosclerosis, then switched to standard chow diet for 4 weeks (Fig. 4a), reducing serum cholesterol levels without affecting plaque size (Fig. 4b and Supplementary Fig. 7). We performed scRNA-seq on CD45+ cells from the aortic arches before and after lipid-lowering, integrating the results with previous data by our group50,51,52,53. The combined dataset of 20,735 plaque CD45+ cells matched major immune cell types in human plaques and aligned with the Immunological Genome Project (Immgen) reference database54 (Supplementary Fig. 8). Lipid reduction did not substantially alter plaque immune cell composition (Fig. 4c) but induced marked changes in the IC landscape, with most IC genes downregulated after treatment (Extended Data Fig. 6a). Among the few upregulated IC genes, Lgals3 (encoding galectin 3) was markedly increased in H2-Eb1+, Spp1+ and Lyve+ macrophages. Given that loss of galectin 3 induces inflammation and promotes atherosclerosis in mice55, our data suggest that elevated Lgals3 expression following lipid-lowering confers an atheroprotective effect, potentially neutralized by ICI treatment.

a, Schematic depiction of experimental design. scRNA-seq was performed on CD45+ cells from the aortic arches of Ldlr−/− mice subjected to a western diet for 16 weeks with or without an additional 4 weeks of chow diet feeding as a lipid-lowering treatment (n = 10 per group). Raw counts were integrated with equivalent (control or lipid-lowered) specimens from Sharma et al.50, Afonso et al.52 and Schlegel et al.51 for analyses. b, Total cholesterol levels of atherosclerotic mice before and after lipid lowering. P values were determined by unpaired, two-tailed Student’s t-test. c, Proportion of major cell identities in mouse atherosclerotic plaques by scRNA-seq, stratified by treatment. d,f,h, Chord plots illustrating the strongest (P < 0.05) interactions between PD-L1 (encoded by Cd274) and PD-1 (encoded by Pdcd1) (d), CD86 and CTLA4 (f) and galectin 3 (encoded by Lgals3) and LAG3 (h) in mice that received no (left) or lipid-lowering treatment (right). The direction of the ligand → receptor interaction is denoted by chord arrows. Darker chords indicate selected interactions that change with lipid lowering. e,g,i, Heatmaps showing differential PD-L1–PD-1 (e), CD86–CTLA4 (g) and galectin 3–LAG3 (i) interaction probabilities predicted in untreated versus lipid-lowered mice (P < 0.05). P values were calculated by the CellChat package (v.1.1.3) in R (v.4.0.3). Nφ, neutrophil; Bφ, basophil; MoDC, monocyte/dendritic cell. CD8+ T cells are depicted in purple; CD4+ T cells are depicted in green; DP T cells are depicted in dark blue; Tγδ cells are depicted in cyan; myeloid cells are depicted in red; NK cells are depicted in orange; and ILCs are depicted in gray.
We mapped the IC ligand–receptor interactions mediated by orthologs of US FDA-approved ICIs, stratified by lipid-lowering treatment (Fig. 4d–i). Lgals3–Lag3 (galectin 3–Lag3) was identified as a top co-inhibitory interaction, along with Lgals9–Havcr2 (galectin 9–Tim3), CD86–Ctla4, CD274–Pdcd1 (Pd-l1–Pd-1) and CD200–CD200r1 (Extended Data Fig. 6b). Pd-l1, CD86 and galectin 3 were identified as primary ligands for Pd-1, Ctla4 and Lag3. Lipid lowering reduced the interaction strength of these ligand–receptor pairs (Extended Data Fig. 6c), highlighting the impact of cholesterol lowering on ICI targets in atherosclerotic plaques. Before lipid lowering, mouse atherosclerotic lesions showed findings consistent with human plaques. A cluster of mature, regulatory Ccr7+Fscn1+ dendritic cells with migratory capacity, played a pivotal role in signaling through the CD274–Pdcd1 (Pd-l1–Pd-1) axis with clusters of plaque CD8+, CD4+, DP T and γδ T cells (Fig. 4d). In hyperlipidemic mice, a subset of Gzmk+ EM CD8+ T cells engaged in CD274–Pdcd1 (Pd-l1–Pd-1)-mediated interactions with various other T cell populations, unlike in human plaques where GZMK+ EM CD8+ T cells communicated with CCR7+FSCN1+ dendritic cells (Fig. 2a).
Lipid lowering increased Pd-l1–Pd-1 interactions between Isg15+ EMRA CD4+ and other T cell clusters (Fig. 4e), indicating reshuffling of Pd-l1–Pd-1 dynamics. It also altered CD86–Ctla4 and Lgal3–Lag3 interactions between myeloid cells and T cells (Fig. 4f–i). For example, CD86–Ctla4 interactions between multiple myeloid subclusters (for example, H2-Eb1+ and Spp1+ macrophages and Ccr7+Fscn1+ dendritic cells) and Il2ra+ γδ CD8+ T cells increased following lipid lowering (Fig. 4f,g). Similarly, Lgal3–Lag3 interactions between various myeloid cells and Runx3+ cytotoxic DP T cells were upregulated (Fig. 4h,i). Changes in CD274–Pdcd1 communication strength were associated to PD-1 pathway downregulation in myeloid and CD4+ T cells (Extended Data Fig. 7a–c).
Overall, lipid lowering induced anti-inflammatory transcriptional reprogramming, upregulating alternative macrophage activation, TH1/TH2 pathways and phagosome formation in myeloid cells (Extended Data Fig. 7a). In CD4+ T cells, it upregulated TH1/TH2 and Gzmb pathways (Extended Data Fig. 7c), whereas in CD8+ T cells TCR, IL-2, IFN and IL-6 pathways were reduced (Extended Data Fig. 7b). We also observed downregulation of inflammatory cytokine genes (Il1b, Nfkb1, Ifnrgr1 and Ifnrgr2) in monocytes, macrophages and dendritic cells (Extended Data Fig. 7d) and Ifng, Ifngr1 and Il2rg in cytotoxic CD8+ and CD4+ T cells, with increased expression of Il10rb in these cells (Extended Data Fig. 7e,f). These findings underscore the significant impact of lipid lowering on the IC immune landscape of atherosclerotic plaques and its potential implications in ICI therapy.
IC inhibition enriched CCR7 + FSCN1 + dendritic cells in vitro
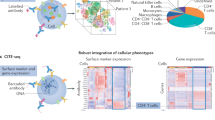
To investigate immune cell interactions in response to PD-1 and CTL4 inhibition, we performed CITE-seq on human peripheral blood mononuclear cells (PBMCs) treated with anti-PD-1 or anti-CTLA4 antibodies (Fig. 5a). After 24 h, CCR7+FSCN1+ dendritic cells were the only myeloid cells significantly enriched (Fig. 5b,c), expressing genes like CCL19, CCL22, IDO1, BIRC3 and LAMP3, indicating a mature, regulatory transcriptional program similar to those in human plaques and in solid tumors45,46 (Supplementary Fig. 9a,b). These cells upregulated pathways related to cell cycle, cholesterol biosynthesis and LXR/RXR activation, suggesting enhanced resistance to apoptosis and/or increased expansion (Fig. 5d). Anti-PD-1 and anti-CTLA4 treatments also upregulated PTEN signaling and expression of IL32 and CXCL9 in CCR7+FSCN1+ dendritic cells, which are involved in recruitment and activation of cytotoxic lymphocytes (Supplementary Fig. 9c), suggesting the reinvigoration of antitumoral CD8+ T cell function56.

a, Schematic depiction of experimental design. Cellular indexing of transcriptomes and epitopes followed by sequencing (CITE-seq) was performed on human PBMCs subjected to anti-CTLA4 or anti-PD-1 treatment for 24 h. b, Proportions of monocyte and dendritic cell subclusters from total myeloid cell compartment, with proportional changes in FSCN1+ dendritic cell abundance highlighted. c, Violin plots illustrating normalized transcript abundance and distribution of indicated genes in myeloid cell subclusters. d, Canonical pathway analysis of differentially expressed transcripts in FSCN1+ dendritic cells compared to other myeloid cell populations. e, Proportions of T cell subclusters from total CD8+ T cell compartment with proportional changes in NKG7+ EM and TFRC+ terminally differentiated EMRA CD8+ T cell abundances highlighted. f, Violin plots illustrating normalized transcript abundance and distribution of indicated genes in CD8+ T cell subclusters. g,h, Canonical pathway analysis of differentially expressed transcripts in NKG7+ EM CD8+ T cells (g) or TFRC+ EMRA CD8+ T cells (h) compared to other CD8+ T cell populations. i, Proportions of T cell subclusters from total CD4+ T cell compartment, with proportional changes in CCR4+ Reg and ENTPD1+ EM CD4+ T cell abundances highlighted. j, Violin plots illustrating normalized transcript abundance and distribution of indicated genes in CD4+ T cell subclusters. k,l, Canonical pathway analysis of differentially expressed transcripts in CCR4+ Reg CD4+ T cells (k) or ENTPD1+ EM CD4+ T cells (l) compared to other CD4+ T cell populations. P values < 0.05 and |z-score| > 2 (d,g,h,k,l). P values were calculated by the Ingenuity Pathway Analysis software. Some Gene Ontology terms were edited for brevity. NE, normalized expression; Tag, aging T cell; TFH, follicular helper T cell; ISG, interferon-stimulated gene; RAR, retinoic acid receptor. m, Schematic depiction of ex vivo anti-PD-1 experiment. CyTOF was performed on human carotid vascular explants subjected to α-PD-1 treatment ex vivo (n = 3). n, Heatmap depicting significant (P < 0.05) T cell marker protein expression by condition. z-score shows scaled average expression. o, Box plot depicting normalized expression of CD45RA in CD8+ or CD4+ T cells derived from control or anti-PD-1-treated explants. Boxes represent interquartile ranges; center lines depict medians. Whiskers below and above boxes represent extent of lower and upper quartiles, respectively. Each dot represents a cell. P values in n and o were determined by two-tailed Wilcoxon rank-sum test (n = 3 per group).
Analysis of the T cell compartment revealed that two CD8+ T subclusters, NKG7+ EM and TFRC+ EMRA CD8+ T cells, were expanded following CTLA4 or PD-1 blockade but nearly absent in untreated controls (Fig. 5e). NKG7+ EMRA CD8+ T cells upregulated IFNG and other genes involved in interferon signaling, hypercytokinemia/chemokinemia and cytokine storm pathways (Fig. 5f,g and Supplementary Fig. 9d). TFRC+ EMRA CD8+ T cells displayed increased expression of TNF and STAT2, and cell cycle markers, such as TOP2A, MKI67 and CCN1 (Fig. 5f and Supplementary Fig. 9d). Other upregulated genes were involved in cell cycle programs (for example, purine biosynthesis and chromosomal replication), whereas genes related to senescence, autophagy, alternative activation of macrophages, TGFβ signaling and the TH2 pathway were downregulated (Fig. 5h). This transcriptional profile indicated that TFRC+ EMRA CD8+ T cells were activated and proliferating following CTLA4 or PD-1 inhibition. Among CD4+ T cells, treatment with anti-PD-1 or anti-CTLA4-blocking antibodies enriched CCR4+ regulatory and ENTPD1+ EM CD4+ T cells (Fig. 5i). CITE-seq analysis showed that only CCR4+ regulatory CD4+ T cells expressed CTLA4 and PDCD1 (Fig. 5j and Supplementary Fig. 9e). Considering that intracellular CTLA4 protein is constitutively expressed and shuttles to the cell surface upon T cell activation57, we used CITE-seq protein data to confirm surface expression in CCR4+ regulatory CD4+ T cells (Supplementary Fig. 9f). Both CCR4+ regulatory and ENTPD1+ EM CD4+ T cells expressed IL2, IL2RA, TNFRSF4 (encoding OX40) and TNFRSF18 (encoding GITR), indicating T cell activation (Fig. 5j and Supplementary Fig. 9e). The CCR4+ regulatory CD4+ T cell subcluster also downregulated genes involved in inflammatory cytokine production (Fig. 5k), corroborating their regulatory phenotype. Conversely, the ENTPD1+ EM CD4+ T cell subcluster upregulated pro-inflammatory and type 1 immune response pathways (Fig. 5l), along with increased cell cycle gene expression (Supplementary Fig. 9g).
To validate our findings in human plaque tissue, we treated human carotid vascular explants from CEA patients with anti-PD-1 ex vivo and measured protein markers using CyTOF (Fig. 5m and Supplementary Table 6). Both CD8+ and CD4+ T cells in carotid explants treated with PD-1 blocking antibody displayed increased expression of activation, inflammation and cytotoxicity markers (Fig. 5n and Extended Data Fig. 8). Notably, CD45RA expression increased (Fig. 5o), indicating greater T cell activation and acquisition of TEM and TEMRA fates. CD161, associated with cytokine production58, was also elevated in both CD8+ and CD4+ T cells from atherosclerotic vascular explants treated with PD-1-blocking antibody (Extended Data Fig. 8b). CD8+ T cells displayed increased interferon-γ and decreased IL-2, while CD4+ T cells showed increased GZMB and decreased PRF1 (Extended Data Fig. 8c), indicating specific activation and cytotoxicity mechanisms downstream of PD-1 inhibition. Anti-PD-1 treatment also increased expression of CCR6 and CCR4 (Extended Data Fig. 8d), linked to pro-inflammatory TH17 migration59 and heightened expression of CXCR3 in CD4+ T cells, characteristic of TH1 cells60 (Extended Data Fig. 8e). CXCR5 expression remained unchanged (Extended Data Fig. 8f). These results show that PD-1 inhibition enhances the pro-inflammatory, effector and cytotoxic phenotype of T cells in human atherosclerotic plaques.
IC-mediated interactions are influenced by diabetes status
Given that individuals with type 2 diabetes (T2D) face elevated risks of both cancer and atherosclerosis, and recognizing the potential of shared immune mechanisms among the three conditions17,61, we next examined carotid plaques from patients with and without T2D (Fig. 6a and Supplementary Table 2). The proportions of major immune cell populations within the plaques from individuals with and without T2D were largely comparable, except for the B cell compartment, which showed greater abundance in patients with T2D (P = 0.036; Fig. 6b). Abundance of T and NK cell subclusters predicted to signal through PD-1, CTLA4 or LAG3 (Fig. 2a,c,e) remained largely unchanged by T2D (Supplementary Fig. 10a–g). In contrast, myeloid cells tended to decline (P = 0.29; Fig. 6b), with CCR7+FSCN1+ dendritic cell (P = 0.046; Fig. 6c), PLIN2+ macrophage (P = 0.057), TREM2+ macrophage (P = 0.044) and AXL+ dendritic cell (P = 0.021) subclusters reduced in plaques from patients with T2D compared to controls.

a, Schematic depiction of IC interaction analyses in atherosclerotic plaque immune cells from T2D versus nondiabetic patients (n = 22). b, Proportion of major cell identities found in human plaques by scRNA-seq, stratified by diabetes status. *P = 0.036. c, Proportions of CD274- or CD86-expressing myeloid cell subclusters engaging in the strongest significant interactions with PD-1 or CTLA4. Boxes represent interquartile ranges; center lines depict medians. Each dot represents a patient. Whiskers below and above boxes represent extent of lower and upper quartiles, respectively. P values in b and c were determined by unpaired, two-tailed Student’s t-tests (n = 9 nondiabetic and n = 13 T2D). d,e, Dot plot of differential PD-L1 (encoded by CD274)–PD-1 (encoded by PDCD1) (c) or CD86–CTLA4 (d) interaction probabilities in T2D versus nondiabetic patients (P < 0.05). P values were calculated by the CellChat package (v.1.1.3) in R (v.4.0.3).
To evaluate how T2D influences IC signaling within atherosclerotic tissue, we performed a ligand–receptor interaction analysis involving genes encoding PD-1, CTLA4 and their ligands. In patients with T2D, the interaction strength of both CD274–PDCD1 and CD86–CTLA4 across most immune subclusters was downregulated (Fig. 6d,e). Notably, the expression of PDCD1 transcript was largely similar between immune subclusters from T2D and control plaques, except for specific subclusters among those involved in CD274–PDCD1 interactions (Supplementary Fig. 10h). PDCD1 expression was increased in LGALS3+ EM CD8+ and S1PR1+ γδ T cells and decreased in CD69+ RM CD4+ T cells within T2D plaques (Supplementary Fig. 10h), though the frequency of these three populations remained unchanged (Supplementary Fig. 10a,b,e). In contrast, CCR7+FSCN1+ dendritic cells, which were less abundant in plaques of patients with T2D, showed no change in the expression of CD274 between T2D and control plaques (Supplementary Fig. 10i). These results suggest that the reduced CD274–PDCD1 interactions observed in T2D are largely related to the decrease of CCR7+FSCN1+ dendritic cells in T2D plaques. The reduced CD86–CTLA4 interaction seen in T2D plaques seemed to be driven by a significant downregulation of CTLA4 in IL32+ regulatory DP T cells (Supplementary Fig. 10j). CD86 expression was increased in plaque TREM2+ macrophages as well as TLR8+ and FCGR3A+ monocytes, but decreased in CD14+ monocytes of patients with T2D (Supplementary Fig. 10k). Of these, only FCGR3A+ monocytes showed a concomitant, albeit small, increase in the likelihood of communication across CD86–CTLA4 in T2D compared to nondiabetic plaques (Fig. 6e).
IC interactions in patients with T2D and the effect of lipid lowering
To establish the impact of lipid lowering on IC expression and IC-mediated cell–cell communication in patients with and without T2D, we analyzed a scRNA-seq dataset from PBMCs isolated from a subgroup of individuals with T2D and nondiabetic controls participating in the cholesterol lowering and residual risk in type 2 diabetes (CHORD) trial (NCT04369664; Supplementary Tables 7 and 8). Blood samples were collected at baseline and 4 weeks after starting a lipid-lowering treatment that included proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibition in combination with a statin or ezetimibe. PCSK9 inhibition dramatically lowers low-density lipoprotein (LDL) cholesterol62,63 and increases the efficacy of IC therapies in cancer64,65,66, without increasing the risk of developing T2D, unlike statins67,68. Analysis of 226,982 PBMCs from CHORD study participants (Fig. 7a) identified all major immune cell types, including CD8+, CD4+, unconventional and DP T cells; B cells; myeloid cells; NK cells; and ILCs (Fig. 7b and Supplementary Fig. 11). Principal-component analysis (PCA) of differential gene expression of IC genes segregated patients by diabetes status across PC2, accounting for 20% of the total variance (Fig. 7c), indicating a distinct IC transcriptional signature in patients with T2D.

a, Schematic depiction of scRNA-seq analyses of PBMCs from T2D (n = 8) or nondiabetic (n = 4) samples. b, Proportion of major cell identities found in human PBMCs by scRNA-seq. c, PCA of IC gene counts from PBMCs of T2D versus nondiabetic patients. d, Bar plot depicting the relative contribution of co-inhibitory IC interactions to overall cell–cell communication. e, Communication strengths of IC interactions targeted by cancer immunotherapies, ranked in order of probability, with the top 30 interactions depicted as rings. Colors indicate the receptor family, PD-1 (blue), CTLA4 (orange) or LAG3 (green). f–h, Chord plots illustrating the strongest (P < 0.05) predicted interactions between CD86 and CTLA4 (f), galectin 3 (encoded by LGALS3) and LAG (g) and αSyn (encoded by SNCA) and LAG3 (h). The direction of the ligand → receptor interaction is denoted by chord arrows. P values were calculated by the CellChat package (v.1.1.3) in R (v.4.0.3).
We noted elevated expression of several co-stimulatory ligand and receptor genes in specific subsets of immune cells from T2D versus controls (Extended Data Fig. 9a). For example, LGALS9 was downregulated in all myeloid populations of patients with T2D, suggesting reduced susceptibility to TIM3 inhibitors. CD28, encoding a co-stimulatory receptor that competes with CTLA4 for CD86 and CD80 binding69,70, was upregulated in naive, CM, EM and regulatory CD4+ T cells from patients with T2D. Additionally, TRDV2 and TRDC, encoding T cell receptors that can act as co-stimulatory molecules, were upregulated in KLRB1+ γδ T cells from patients with T2D compared to controls.
Modeling IC ligand–receptor pairs in circulating immune cells revealed the same top co-inhibitory interactions observed in human atherosclerotic plaques (for example, HLA-E–KLRC1, HMGB1–HAVCR2 and CD47–SIRPA) (Fig. 7d and Extended Data Fig. 4a). Among interactions targeted by US FDA-approved drugs, CD274–PDCD1, CD80–CTLA4 and CLEC4G–LAG3 were not active in PBMCs, whereas CD86–CTLA4 and LGALS3–LAG3 emerged as the strongest co-inhibitory interactions (Fig. 7e). CD86–CTLA4 interactions involved multiple circulating myeloid populations, including CCR7+FSCN1+ dendritic cells (Fig. 7f). These dendritic cells also engaged in LGALS9–HAVCR2 and BTLA–TNFRSF14 interactions (Extended Data Fig. 9b,c). As seen in atherosclerotic plaques, circulating regulatory T cells were primarily involved in CD86–CTLA4 interactions (Fig. 7f), whereas the LGALS3–LAG3 axis was mainly engaged by EM and EMRA CD8+ T cells (Fig. 7g). Notably, circulating ILC subclusters engaged in co-inhibitory CD86–CTLA4, LGALS3–LAG3 and LGALS9–HAVCR2 interactions (Fig. 7f,g and Extended Data Fig. 9b), which were not observed in atherosclerotic plaques (Fig. 2c,e and Extended Data Fig. 5c). SELP+ITGA2B+ cells and a population of GP9+ monocytes, possibly corresponding to leukocyte–platelet aggregates, showed strong signaling through SNCA–LAG3 with EM, EMRA and γδ T cells as well as CTLs (Fig. 7h).
Lipid-lowering treatment reduced circulating levels of total and LDL cholesterol in individuals with and without T2D (Fig. 8a,b). In T2D, CCR7+FSCN1+ dendritic cells engaged in strong CD86–CTLA4 interactions in patients with T2D before and after lipid lowering, but these cells were absent in individuals without diabetes (Fig. 8c). While the CD86–CTLA4 communication (Fig. 7f) was broadly increased in T2D, particularly between LAIR2+CD4+ and IL32+ mixed regulatory T cells, lipid lowering decreased these interactions between myeloid cells and regulatory T cells, often normalizing them to nondiabetic control levels (Fig. 8c,d). This suggests that lipid-lowering treatment may reduce the susceptibility to anti-CTLA4 therapies in patients with T2D.

a,b, Levels of total cholesterol (a) or LDL-c (b) in individuals with and without T2D, before and after lipid-lowering treatment (n = 12). c, Communication strengths of the strongest (P < 0.05) CD86–CTLA4 interactions before and after lipid-lowering treatment (gray lines; n = 44 interactions). Red lines depict interactions involving CD86-expressing FSCN1+ dendritic cells. Rings represent lack of interaction, hence interaction strength = 0. P values in a–c were determined by two-tailed Wilcoxon signed-rank tests. P values for interactions were calculated by the CellChat package (v.1.1.3) in R (v.4.0.3). d, Differential communication strengths of strongest (P < 0.05) CD86–CTLA4 interactions in CD86-expressing myeloid cell subclusters before (gray) and after (light blue) lipid-lowering treatment. Dots represent the mean FC between T2D and nondiabetic patients of the indicated interaction. Dotted line indicates log2(FC) = 0, which is the threshold at which interactions between T2D and nondiabetic patients were predicted to remain unchanged. P values were calculated by the CellChat package (v.1.1.3) in R (v.4.0.3). CM, classical monocyte; IM, intermediate monocyte; NCM, nonclassical monocyte.
LGALS3–LAG3 interactions, primarily involving effector and cytotoxic CD8+ T cells with monocytes, were as strong as CD86–CTLA4 (Fig. 7e,g). Lipid lowering increased LGALS3–LAG3 interactions between myeloid cells and CCL4+, RPSY4+ and CASP9AP2+ cytotoxic CD8+ T cells in patients with T2D (Extended Data Fig. 10) but decreased them between monocytes and ITGA1+ EM CD8+ T cells. Lipid lowering upregulated LAG3 and LGALS3 exclusively in male participants (Supplementary Fig. 12), indicating a potential sex bias. Female participants with T2D had elevated TIGIT, ENTPD1 and CD86, whereas LTB was the only IC upregulated in both sexes compared to nondiabetic controls. Lipid lowering also decreased LGALS3–LAG3 signaling between monocyte/dendritic cells and KLRB1+ γδ T cells in patients with T2D. These results indicate that lipid lowering increases the inhibition of cytotoxic CD8+ T cell function through LAG3, promoting anti-inflammatory effects in atherosclerosis.
Discussion
Clinical data on the effects of ICI therapies in patients with cancer indicate an increase in the incidence of pro-atherogenic irAEs that underlie cardiovascular events such as heart attacks, strokes and peripheral arterial disease7,8,9. Approximately 44% of patients with cancer in the United States qualify for US FDA-approved ICIs4,6 and this number is expected to increase with over 2,900 ongoing clinical trials (ClinicalTrials.gov; 2 December 2023). Our study addresses the critical need to understand how ICIs heighten cardiovascular risk by mapping IC expression across immune and nonimmune cells in human atherosclerotic tissue. We reconstructed the IC interactome in health and disease using single-cell transcriptomic assays, bioinformatic analyses and protein validation experiments and identified the key hazards of ICI treatment that drive atherosclerosis. Additionally, we show how diabetes, a risk factor for both cardiovascular events and cancer, influences the susceptibility of atherosclerotic plaques to both US FDA-approved and investigational ICIs. Our study also identifies how lipid lowering affects IC expression and cell communication within atherosclerotic plaques, potentially influencing their susceptibility to current and investigation ICI agents.
A major finding from our study is the identification of CCR7+FSCN1+ dendritic cells as a central hub for PD-1- and CTLA4-mediated communication within the atherosclerotic niche. These cells, previously observed in the tumor microenvironment45,46,47, were more prevalent following PD-1 or CTLA4 inhibition, which also induced a transcriptional reprogramming toward cell cycle activation and lipid synthesis, pathways that contribute to expansion, maturation and migration of dendritic cells to the lymph nodes in response to ICIs in cancer45,46,47. CCR7+FSCN1+ dendritic cell responses were associated with enhanced activation, enhanced cytotoxicity and proliferation of T cells. Our analysis predicts a pivotal role of plaque CCR7+FSCN1+ dendritic cells in executing IC-mediated crosstalk with myeloid cells through TIM3 and galectin 9, both targets of new investigational ICIs38,39,71,72. These results suggest that CCR7+FSCN1+ dendritic cells play a central role in the phenotype of exacerbated atherosclerosis observed with Lgals9 deletion or Tim3 inhibition14,37. CCR7+FSCN1+ dendritic cells within the plaque also engage in communication through LAG3 signaling with various myeloid cell subpopulations (for example, CD14+ monocytes and PLIN2+ macrophages) and effector T cell subsets (for example, EM, EMRA and RM CD8+ T cells). Collectively, our results emphasize the pivotal role of CCR7+FSCN1+ dendritic cells as a central hub for IC communication within plaques and as a target for both US FDA-approved and emerging investigational ICIs that may accelerate ASCVD.
This study also revealed the significant impact of lipid lowering, a cornerstone in the standard of care of ASCVD, on IC expression and IC-mediated crosstalk between immune cells in atherosclerotic plaques. Cholesterol lowering in atherosclerotic mice reduced the overall strength of cell–cell interactions targeted by US FDA-approved ICIs, (for example, Cd274–Pdcd1 and Cd86–Ctla4) and investigational drugs (for example, Lgals3–Lag3 and Cd200–Cd200r1); however, specific Cd274–Pdcd1-mediated interactions such as those between Isg15+ EMRA CD4+ and other T cell clusters were increased following lipid lowering. Similarly, Cd86–Ctla4 interactions between Il2ra+ γδ CD8+ T cells and multiple myeloid subclusters, including Ccr7+Fscn1+ dendritic cells were increased. Elevated Lgals3–Lag3 interactions between various myeloid cells and Runx3+ cytotoxic DP T cells were also identified following lipid lowering. Overall, these findings indicate that lipid lowering reshuffles immune cell cross-communication in the plaque. Recent studies suggest that lipid lowering strategies improve survival in patients with cancer49,64,65,66 and our study provides clues into how lipid lowering may impact IC expression and immune cell communication in that context.
Our study also reveals the substantial impact of cardiometabolic factors on CCR7+FSCN1+ dendritic cells and IC expression regulation, highlighting varying susceptibility between the bloodstream and atherosclerotic immune compartments to ICIs. In T2D plaques, the abundance of CCR7+FSCN1+ dendritic cells decreased, along with a downregulation of CD274–PDCD1 and CD86–CTLA4 interactions; however, PDCD1-mediated interactions remained stronger in atherosclerotic tissue than in blood, likely reflecting the higher expression of PD-1 in plaque immune cells12, suggesting increased susceptibility of atherosclerotic plaques to the effect of PD-1 inhibitors. We showed that IC expression in circulating immune cells of patients with T2D can be influenced by standard-of-care lipid-lowering treatments. CD86–CTLA4 interactions among circulating immune cells, initially elevated in T2D, decreased to nondiabetic levels upon lipid-lowering treatment, indicating reduced susceptibility to anti-CTLA4 ICI therapies. The lipid-lowering effects on LAG3-mediated signaling in circulating immune cells were more complex: LGALS3–LAG3 interactions between myeloid cells and specific cytotoxic CD8+ T cell subsets were increased, whereas signaling through LGALS3–LAG3 between monocytes and ITGA1+ EM CD8+ T cells were decreased in T2D compared to individuals without diabetes, as did monocyte and dendritic cell signaling to KLRB1+ γδ T cells through LGALS3–LAG3. These findings, coupled with unchanged LGALS3-expressing cell frequencies, suggest that lipid lowering enhances the inhibition of cytotoxic CD8+ T cell function through LAG3, potentially leading to anti-inflammatory effects, which could be neutralized by ICIs in the circulation.
Our study has some limitations. While data from the CHORD study were obtained from minimally processed PBMCs, carotid and coronary artery data required enzymatic tissue digestion which may have altered cell subtype frequencies. Additionally, our study design did not control for transient ischemic aneurysm/stroke or coronary artery bypass graft surgery in comparing carotid plaques from T2D and nondiabetic individuals. Despite these limitations, our findings provide valuable insights for designing ICI strategies that consider cardiovascular comorbidities and side effects.
In conclusion, our findings highlight the susceptibility of the atherosclerotic microenvironment to both US FDA-approved and investigational cancer immunotherapies and the influence of cardiometabolic comorbidities on the IC landscape and cardiovascular outcomes in patients with cancer receiving ICI treatment. The observation that lipid lowering reshapes the IC landscape in atherosclerosis, serves as a foundation to understand the cardiovascular impact of new and investigational ICIs and to identify therapeutic strategies to mitigate atherosclerosis and related cardiovascular outcomes in survivors of cancer. Similar mapping of other organs or disease states may aid in designing ICI treatments that consider tissue- and organ-specific immunological makeup, thereby reducing irAEs.
Methods
Sample collection and preparation for sequencing assays
Human carotid plaque samples for scRNA-seq
Carotid samples were obtained from 22 patients undergoing CEA enrolled in the ATHERO-IN (ATHEROsclerosis-INflammation study). This study was approved by the Institutional Review Board (IRB) of New York University (NYU) Langone Health (IRB no. 21-00429) and the Icahn School of Medicine at Mount Sinai (IRB no. 11-01427). All patients were over 18 years old, and those with current infection, autoimmune disease, active or recurrent cancer, severe renal failure requiring dialysis or peripheral arterial occlusive disease-causing pain at rest were excluded. Participants provided informed written consent. The cohort (Supplementary Tables 1 and 2) included 18 male and 4 female participants, with a mean age of 70.8 ± 6.4 years (median 71.5 years). Eleven patients with T2D and two patients with pre-diabetes were included in the T2D group for analyses. The remaining nine participants were nondiabetic. Carotid atherosclerotic tissue specimens (plaques) were placed in Dulbecco’s modified Eagle’s medium (DMEM; Corning, 10-013-CV) with 10% vol/vol fetal bovine serum (FBS; Gibco, 10082-147) or in HypoThermosol Storage Media (StemCell Technologies, 7935) on ice. Within 1 h, plaques were digested to obtain single-cell suspensions12. Digestion involved washing, mincing and treating plaques with DMEM with 10% vol/vol FBS, 1 mg ml−1 collagenase type IV (Sigma, C5138), 0.3 mg ml–1 DNase I (Sigma, DN25), hyaluronidase (Sigma, H3506), collagenase type XI (Sigma, C7657) and collagenase type II (Sigma, C6885) in a gentleMACS Octo Dissociator with Heaters (Miltenyi Biotec). Digested specimens were filtered through 70-μm and 40-μm cell strainers (Fisher Scientific, 22363547 and 22363548, respectively) and dead cells removed using an EasySep Dead Cell Removal (annexin V) kit (StemCell Technologies, 17899). CD45+ cells were isolated using EasySep Release Human CD45 positive selection kit (StemCell Technologies, 100-0105). Total live CD45+ cells were counted and loaded onto the Chromium Controller (10x Genomics) for library preparation using the Chromium Single Cell 3′ Library v3 reagent kit (10x Genomics; 1000268, 1000215 and 100127, respectively). DNA library quality was assessed using the TapeStation High Sensitivity D5000 ScreenTape system (Agilent) and the Bioanalyzer High Sensitivity DNA kit (Agilent, 5067-4626) and sequencing was performed on an Illumina NovaSeq 6000 at the Genomics Core Facility at the Icahn School of Medicine at Mount Sinai. Each sample was processed and sequenced separately.
Human coronary artery samples for scRNA-seq
Five patients with end-stage heart failure undergoing orthotopic heart transplantation were enrolled in the ATHERO-IN study approved by NYU Langone Health (IRB no. 21-00429). All patients were over 18 years old and undergoing heart transplantation for either ischemic or nonischemic heart failure. Exclusion criteria were current infection, autoimmune disease, active or recurrent cancer, severe renal failure requiring dialysis or peripheral arterial occlusive disease-causing pain at rest. Participants provided informed written consent. Left anterior descending (LAD) coronary arteries were extracted immediately following organ removal, cleaned of perivascular adipose tissue and then placed in HypoThermosol Storage Media (StemCell Technologies, 7935) on ice. Areas of AIT or plaque were identified by pathological examination of slides stained with hematoxylin and eosin by a clinical cardiovascular pathologist (N.N.). For one patient, both LAD and right coronary artery tissue were collected, characterized by different pathology (plaque and AIT, respectively) and processed independently. Within 1 h, specimens were processed as described for carotid specimens, without the CD45 enrichment step. Single-cell suspensions were counted and loaded onto a Chromium Controller (10x Genomics). Following library preparation and quality control, sequencing was carried out on an Illumina NovaSeq X Plus sequencer at the NYU Langone Genome Technology Center (GTC). Each sample was processed and sequenced separately.
Human peripheral blood samples for cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq)
PBMCs were isolated from 500 ml whole human blood from the New York Blood Center by density gradient centrifugation using Ficoll-Paque PLUS12. The PBMC layer was collected, washed, resuspended at 2–5 × 105 cells per ml and stored at −80 °C before been transferred to liquid N2 until use. PBMCs were thawed and acclimatized in RPMI-1640 supplemented with 10% vol/vol FBS and 100 μg ml−1 penicillin–streptomycin (Corning, MT30002CI) in a humidified incubator at 37 °C and 5% CO2 for 2 h. PBMCs were cultured at 5 × 105 cells per well of a 12-well plate coated with Dynabeads Human T-Activator CD3/CD28 (Gibco, 11161D). Next, 1 μg ml−1 anti-PD-1 (Bio X Cell, SIM0003), 20 μg ml−1 anti-CTLA4 (Bio X Cell, SIM0004) or vehicle were added to the corresponding wells for 24 h. Samples were then washed in PBS (Corning, 1-031-CV), centrifuged and placed in PBS supplemented with 1% vol/vol bovine serum albumin (BSA, Jackson ImmunoResearch, 149856) and 10 μg Anti-Fc Receptor Binding Inhibitor (Invitrogen, 14-9161-73) for 10 min at 4 °C to prevent nonspecific binding in later steps. Fc-blocked samples were incubated with 0.5 μg unique hashtag antibodies from the 3ʹ CellPlex Kit Set A (10x Genomics, PN-1000261), respectively, and stained with the TotalSeq-B Human Universal Cocktail, V1.0 (BioLegend, 399904) according to the manufacturer’s instructions. Stained cells were submitted to the NYU Langone GTC for CITE-seq analysis following the Single Cell 3′ v2 workflow and using a 3′ CellPlex Kit, Set A for multiplexing (10x Genomics, 1000261) through the complementary DNA amplification step. The transcriptome library was generated according to the protocol by Stoeckius et al.73 Separate PCRs were used to generate the CITE-seq antibody-derived tag (ADT) protein library (SI-PCR and RPI-x primers) and the hashtag library, respectively. Finally, the ADT and hashtag libraries were sequenced on an Illumina NovaSeq 6000 using the S4 200 cycle flow cell v.1.5, also at the NYU Langone GTC.
Human peripheral blood samples for scRNA-seq
Twelve patients with elevated plasma cholesterol levels (LDL-c > 100 mg dl–1 or Lp(a) > 50 mg dl–1) with or without T2D were enrolled for the CHORD (cholesterol lowering and residual risk in type 2 diabetes) study (NCT04369664) at NYU Langone Health (IRB no. 19-01964). Inclusion criteria for all participants regardless of diabetes status included age ≥18 and <90 years, LDL-c > 100 mg dl–1 and the ability and willingness to provide written informed consent for the study. Exclusion criteria for participants with T2D were established cardiovascular disease on antithrombotic therapy, triglycerides > 250 mg dl–1, use of a PCSK9 inhibitor, HbA1c > 10%, recent infection in the past 30 days, any hospitalization in the past 30 days, use of immunosuppressive therapy, use of any antithrombotic therapy, use of aspirin, use of NSAID within the past 72 h, pregnancy, anemia (hemoglobin < 9 g dl–1), thrombocytopenia (platelet count < 75 × 103 µl–1), thrombocytosis (platelet count > 600 × 103 µl–1) and a history of severe bleeding or bleeding disorders and chronic kidney disease (CrCl < 30 ml min–1). Exclusion criteria for participants without known diabetes included diabetes (type 1 or type 2) and all exclusions for participants with T2D. Supplementary Tables 7 and 8 summarize the demographic and clinical characteristics of the 12 participants (5 males and 7 females) with T2D (n = 8) or nondiabetic (n = 4), with a mean age of 52.6 ± 15.0 years (median 60.5 years) that were included in our analysis. Both T2D and nondiabetic patients were treated with either atorvastatin (up to 80 mg daily, orally; n = 6) or ezetimibe (10 mg daily, orally; n = 6) in combination with the PCSK9 inhibitor evolocumab (140 mg 15 days apart, intra-muscularly; n = 12) for 30 days. Before and after treatment fasting, peripheral venous blood collected into tubes containing anticoagulant citrate dextrose solution A (BD, 364606) was used to isolate PBMCs, as described above. Cryopreserved PBMCs were thawed, washed and their viability was assessed using a Cellometer Auto 2000 (Nexcellom). Single-cell partitioning, barcoding, loading as well as library preparation were carried out as described above for human plaque cells. DNA library quantity and quality were measured using the TapeStation High Sensitivity D5000 ScreenTape system (Agilent) and the Bioanalyzer High Sensitivity DNA kit, then sequenced on an Illumina NovaSeq 6000 sequencer at the NYU Langone GTC.
Mouse aortic arch samples for scRNA-seq
All experimental procedures were performed in accordance with the US Department of Agriculture Animal Welfare Act and the US Public Health Service Policy on Humane Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of the NYU Grossman School of Medicine (IA16-01079). Eight-week-old Ldlr−/− mice (The Jackson Laboratory, 002207) were placed on a western diet (21% wt/wt fat, 40% fat kcal and 0.3% cholesterol; Dyets, 101977GI) for 16 weeks and then switched to a chow diet (13% fat kcal and 0% cholesterol; LabDiet, 5053) for an additional 4 weeks as the lipid-lowering intervention (n = 5 male and 5 female mice per group). At the experimental end point, mice were killed with CO2, exsanguinated by cardiac puncture and perfused with PBS. Total plasma cholesterol was measured using a Wako Cholesterol E colorimetric assay (Wako Diagnostics, 99902601). We assessed the atherosclerotic status of mice as detailed elsewhere.53 In brief, the heart was separated from the aorta, embedded in optimal cutting temperature compound (Fisher, 23-730-571), snap-frozen, sectioned through the aortic root and stained with hematoxylin and eosin. All images were collected with a BZ-X800 microscope (Keyence) and analyzed using ImageJ software (https://fiji.sc/). For sequencing, whole aortic arches were collected following removal of perivascular adipose tissue, minced and incubated in a solution of 0.77 mg ml–1 liberase (Roche, 273582), 0.1 mg ml–1 hyaluronidase (Sigma, 3506), 0.06 mg ml–1 DNase I (Sigma, DN25) and 1 mol l–1 CaCl2 in a gentleMACS Octo Dissociator. Digested tissue was filtered through a 70-µm strainer, washed with cold PBS, centrifuged at 500g for 5 min at 4 °C and resuspended in PBS supplemented with 1% vol/vol BSA. Single-cell suspensions were placed on ice and stained with eFluor 780 Fixable Viability Dye (eBioscience, 65-0865-14) for 30 min, washed, blocked with TruStain FcX (anti-mouse CD16/32) antibody (BioLegend, 101319; 93; 1:100 dilution) for 5 min and then stained with PerCP/Cyanine5.5 anti-mouse CD45 antibody (BioLegend, 103132; 30-F11; 1:100 dilution) for 30 min. Live, CD45+ cells were purified by fluorescence-activated cell sorting using a BD FACSAria II instrument equipped with a 100-µm nozzle at the NYU Grossman School of Medicine Cytometry and Cell Sorting Laboratory. For sorting, specimens were pooled by experimental group. Single-cell partitioning, barcoding, loading as well as library preparation and quality control were carried out as described above for human carotid plaque immune cells at the NYU Langone GTC.
Single-cell sequencing data analyses
scRNA-seq of human carotid plaque immune cells
Data from ten patients had been previously published by our group (Fernandez et al.12 and Eberhardt et al.74) and can be accessed on the Gene Expression Omnibus (GEO) hosted by the National Center for Biotechnology Information under accession codes GSE224273 and GSE235437, respectively. Data for one patient of the Fernandez et al. cohort was obtained by CITE-seq. That sample was analyzed independently for Supplementary Fig. 3d–h, and only the RNA module was incorporated into the full human carotid plaque immune cell scRNA-seq dataset. Cell Ranger (10x Genomics, v.3.1.0) was used to perform alignment (GRCh38, Ensembl 93), filtering, barcode counting and unique molecular identifier (UMI) counting of sequencing reads from fastq files. The Seurat package (v.4.3.0)67,75 in R (v.4.0.3) was used to assess sample quality (quantify genes detected, total UMIs and fraction of UMIs aligning to mitochondrial genes) and filter out low-quality cells as well as multiplets. Data were then normalized using SCTransform as implemented in Seurat. To ease cell type identification, the influence of cell cycle phase on downstream clustering was mitigated by including scores for S and G2/M phases as regression coefficients in a second round of SCTransform. Cell cycle scores were computed using Seurat’s CellCycleScoring function on SCTransform-normalized data. For downstream differential expression and cell–cell communication inference, counts in each cell were also normalized using Seurat’s NormalizeData function. To align similar cell states across samples, data were integrated using Seurat’s reciprocal PCA integration workflow. PCA was then also run on the integrated data. A shared-nearest-neighbor (SNN) graph was computed on the first 30 principal components using Seurat’s FindNeighbors function. Twenty-one clusters were identified via the Louvain algorithm as implemented in Seurat’s FindClusters function with a resolution of 0.5. For visualization, uniform manifold approximation and projection (UMAP) was run on the first 30 principal components as well. Clusters were assigned to one of four groups: T cells, B cells, NK cells or myeloid cells. Unique expression of CD4 transcript and absence of both CD8A and CD8B was used to identify the CD4+ T cell compartment. Similarly, CD8+ T cells were identified by expression of CD8A or CD8B transcripts and absence of CD4. DP (CD8+CD4+) and double-negative (DN) (CD8–CD4–) T cells were identified from among the remaining T cells, respectively, by coexpression of either CD8A or CD8B and CD4 or by absence of all three transcripts. This approach yielded the following major cell groups: CD8+, CD4+, DN and DP T cells; B cells; myeloid cells; NK cells and ILCs. To ascertain cell identities more granularly, each of the major cell groups was subclustered by re-scaling the integrated data and implementing the Louvain algorithm as mentioned above. Canonical marker gene expression computed via the pseudobulk method developed by Squair et al.76 was used to identify and label the subclusters found within each major cell group. Cell annotations were validated using the automated cell annotation method CellTypist (v.1.5)21,77 against the ‘Immune_All_Low.pkl’ model with Python (v.3.9.7). Residual low-quality cells (for example, displaying low UMI counts or a large fraction of UMIs aligned to mitochondrial genes) and multiplets were filtered out from the dataset. In total, we sequenced 69,422 single human plaque leukocytes that passed quality controls and clustered into 69 identifiable subpopulations. Among DN T cells, six γδ T cell subclusters were identified by gene expression and corroborated by CyTOF and CITE-seq analyses (Supplementary Fig. 3). The remaining DN T cell clusters were relabeled mixed (CD8+ or CD4+) T cells based on neighborhood purity and pairwise cluster modularity analyses, which were carried out using the neighborPurity and pairwiseModularity functions, respectively, from the bluster package (v.1.11.4) in R. Differences in gene expression due to diabetes status were assessed within each cell type using the Wilcoxon rank-sum test as implemented in the Seurat package. Dot plots, box plots and stacked or grouped bar plots were generated using the ggplot2 package on R. Canonical pathway analyses were carried out using the Ingenuity Pathway Analysis platform (QIAGEN; v.01-22-01)78 with differential gene expression input generated using the pseudobulk method described above. Heatmaps were generated with the pheatmap package in R. Volcano and bar plots were generated using GraphPad Prism software (v.9.3.0). To infer the cell–cell communication network between immune cells, we ran the CellChat package (v.1.1.3)79 on the entire dataset. A comprehensive, manually curated human IC interaction module encompassing 27 unique receptors and 35 unique ligands engaging in 45 distinct interactions was added to the CellChat database (Supplementary Table 5) to enable analyses of all known IC interactions. Communication between plaque immune cell subclusters was quantified using the average expression values of a ligand by one immune cell population and that of a receptor by another and associating the resulting values with a probability value modeled by the law of mass action. Significant ligand–receptor interactions between any two subclusters were identified by randomly permutating cluster annotations and recalculating every interaction probability with its associated P value. Interactions with P value < 0.05 were considered significant. Among these, the 30 interactions with the lowest P values were considered strongest. To uncover the impact of diabetes status on the cell–cell communication network, data subsets corresponding to T2D or nondiabetic patients were run through the CellChat pipeline, respectively, and merged for condition-based analyses.
scRNA-seq of human coronary artery cells
Data from two patients had been previously published by our group (Cyr et al.53) and can be accessed on the GEO under accession code GSE252243. Cell Ranger (10x Genomics; v7.1.0) was used to perform alignment to a concatenated human and SARS-CoV-2 genome (GRCh38, Ensembl 98 and ASM993790v1, respectively) as well as filtering, barcode counting and UMI counting of sequencing reads from fastq files. Additionally, the remaining multiplets were detected and removed using the scDBLfinder package80 (v.1.8.0) in R. After an initial round of clustering, clusters were partitioned based on PTPRC expression before annotation based on canonical gene expression. PTPRC− (nonimmune) cluster annotation was based on Wirka et al.81.
CITE-seq of human PBMCs
CITE-seq-specific data processing was conducted as follows. Cell Ranger (10x Genomics, v.6.0.1) was used to perform alignment (GRCh38, Ensembl 98), filtering, barcode counting and UMI counting of sequencing reads from fastq files. To de-multiplex samples, hashtag-associated counts were extracted from the antibody capture matrix and normalized using centered log-ratio (CLR) transformations as implemented in Seurat’s NormalizeData function. To detect cells that could be confidently assigned to a single hashtag sequence, we used the HTOdemux function from Seurat. Predicted singlets were retained for further analysis. RNA and protein expression data were then normalized as well via SCTransform and CLR, respectively. RNA and protein data were integrated across samples independently following Seurat’s integration workflow82. Major cell types were identified using the integrated protein expression data matrix. In brief, we ran the PCA and then computed an SNN graph with Seurat’s FindNeighbors function using the first ten principal components. Next, we identified 16 clusters by using the Leiden algorithm via Seurat’s FindClusters function with a resolution of 0.75. Clusters were assigned to one of four groups: T cells, B cells, myeloid cells or NK cells. T cells were then split into CD8+, CD4+, DP or DN using a manual gating strategy based on the expression distribution of CD8 and CD4. To ascertain cell identities more granularly, each major immune cell group was processed as follows. First, RNA counts were re-normalized via SCTransform. To identify the most variable genes, Seurat’s SelectIntegrationFeatures function was used. PCA was then run on the integrated RNA data using only the most variable genes selected in the previous step. To incorporate both RNA and protein data, a weighted-nearest-neighbor graph was computed using Seurat’s FindMultimodalNeighbors function using the first ten principal components of the integrated RNA and protein data, respectively. The weighted-nearest-neighbor graph was then clustered using the Leiden algorithm via Seurat’s FindClusters function. Canonical marker expression computed via Seurat’s Wilcoxon rank-sum test as implemented by the FindAllMarkers function on both RNA and protein data, respectively, was used to identify and label the subclusters found within each major cell group. In total, 40 subclusters were identified and annotated. Canonical pathway analyses and data visualization techniques were carried out as described above for human carotid plaque immune cells.
scRNA-seq of human PBMCs
Cell Ranger (10x Genomics, v.6.0.1) was used to perform alignment (GRCh38, Ensembl 98), filtering, barcode counting and UMI counting of sequencing reads from fastq files. Additionally, the remaining multiplets were detected and removed using the DoubletFinder package (v.2.0.3) in R83. To further mitigate the influence of cell division and/or proliferation on cell type identification, a list of cell cycle genes published in Li et al.84 was removed from each sample before integration and clustering. We identified the same major cell populations also found in human plaques and two additional ones, hematopoietic stem and progenitor cells (HSPCs) and a cluster of SELP+ITGA2B+ cells. In total, we analyzed 226,982 single human PBMCs that passed quality controls and clustered into 81 identifiable subpopulations that were validated with a published PBMC CITE-seq dataset using Seurat’s query-reference mapping workflow82. DN T cell subclusters were relabeled γδ or unconventional T cells based on gene expression and support from the literature on circulating T cells85. CellChat was run as above to infer the cell–cell communication network between cells in PBMC samples. Data subsets corresponding to T2D or nondiabetic patients as well as pre-treatment or lipid lowering-treated patients were run through the CellChat pipeline, respectively, and merged for condition-based analyses. Data visualization techniques were carried out as described above for human carotid plaque immune cells.
scRNA-seq of mouse aortic arches
Data from four investigations has been previously published by our group (Sharma et al.50, Afonso et al.52, Schlegel et al.51 and Cyr et al.53) and can be accessed on GEO under accession codes GSE141038, GSE161494, GSE168389 and GSE252243, respectively. In these studies, atherosclerosis was induced in mice either by Ldlr knockout or adeno-associated viral delivery of Pcsk9 to the liver, respectively, followed by western diet feeding for 14–20 weeks. A cohort of mice was placed on a chow diet for an additional 4 weeks in two of the studies51,52. Cell Ranger (10x Genomics, v.6.0.1) was used to perform alignment (GRCm38, Ensembl 84), filtering, barcode counting and UMI counting of sequencing reads from fastq files. The Seurat package (v.4.3.0) in R (v.4.2.0) was used to assess sample quality (quantify genes detected, total UMIs and fraction of UMIs aligning to mitochondrial genes), filter out low-quality cells as well as multiplets and further downstream analyses. To mitigate the influence of cell division and/or proliferation on clustering, a list of cell-cycle-specific genes was generated following the method outlined in Li et al.84 and using Mki67, Pcna, Mcm3, Top2a and Ccnb2 as anchor genes with a correlation coefficient of 0.15 (Supplementary Table 9). These genes were removed from the integration workflow but maintained in the dataset for downstream analyses. Twenty-five clusters were identified via the Louvain algorithm as implemented in Seurat’s FindClusters function with a resolution of 0.55. For visualization, UMAP was run on the first 30 principal components as well. To aid in the identification of major cell groups, we performed reference-based annotation using the SingleR package (v.1.10.0) along with the complete Immunological Genome Project catalog as the reference dataset54,86 in R. Similarly to our analyses of human plaque immune cells, clusters were assigned to one of four groups: T cells, B cells, NK cells or myeloid cells. CD8+ T cells were identified by expression of CD8a or CD8b1 transcripts and the absence of CD4. Similarly, unique expression of CD4 transcript and absence of both CD8a and CD8b1 was used to identify the CD4+ T cell compartment. DP and DN T cells were identified from among the remaining T cells, respectively, by coexpression of either CD8a or CD8b1 and CD4 or by absence of all three transcripts. This approach yielded the following major cell groups: CD8+, CD4+, DN and DP T cells; B cells; myeloid cells; NK cells and ILCs. To ascertain cell identities more granularly, each of the major cell groups was subclustered by re-scaling the integrated data and implementing the Louvain algorithm as mentioned above. Canonical marker gene expression computed via Seurat’s Wilcoxon rank-sum test as implemented by Seurat’s FindAllMarkers function was used to identify and label the subclusters found within each major cell group. Cohorts of mice that received any treatment other than diet adjustments were removed from the analysis following integration and clustering. Residual low-quality cells (for example, displaying low UMI counts or a large fraction of UMIs aligned to mitochondrial genes) and multiplets were filtered out from the dataset. In total, we analyzed 20,735 single mouse plaque leukocytes that passed quality controls and clustered into 62 identifiable immune cell clusters. Consistently with the approach used for human plaque immune cells, murine DN T cell subclusters were relabeled γδ or mixed based on gene expression. Differences in gene expression due to lipid-lowering treatment were assessed within each cell type also using Seurat’s Wilcoxon rank-sum test. CellChat was run as above to infer the cell–cell communication network between murine plaque immune cells. A comprehensive mouse IC interaction module encompassing 32 unique receptors and 31 unique ligands engaging in 51 distinct interactions was added to the CellChat database (Supplementary Table 10) to enable analyses of all IC interactions cognate to their human counterparts. Data subsets corresponding to lipid-lowered and control mice were run through the CellChat pipeline, respectively, and merged for condition-based analyses. Canonical pathway analyses and data visualization techniques were carried out as described above for human carotid plaque immune cells.
Multiplex fluorescent immunohistochemistry
To validate PD-L1–PD-1 interactions, human carotid plaques corresponding to those from six patients from the scRNA-seq cohort (Supplementary Table 1) were formalin-fixed, paraffin-embedded, sectioned into 5-µm slices and submitted for MULTIPLEX staining using the PhenoCycler-Fusion platform (Akoya Biosciences) at the NYU Langone Experimental Pathology Core. In brief, samples were baked and deparaffinized before antigen retrieval and staining with the following antibodies, including anti-CD3e (Akoya Biosciences, 4550119; 1:200 dilution), anti-CD11c (Akoya Biosciences, 4550114; 1:200 dilution), anti-PD-L1(Akoya Biosciences, 4550072; 1:100 dilution) and anti-PD-1 (Akoya Biosciences, 4550038; 1:200 dilution) as well as and 4,6-diamidino-2-phenylindole (DAPI) (Akoya Biosciences, 7000003) for 3 h at room temperature (RT). Immunostained slides were then fixed and maintained in storage buffer (Akoya Biosciences, 240197) until ready to image. Whole slides were scanned at ×20 magnification using a Vectra Multispectral Imaging System (Akoya Biosciences/PerkinElmer), visualized using the Phenochart Whole Slide Viewer software (Akoya Biosciences; v.1.1.0) and acquired using the Phenochart Viewer software (v.2.0.1). Detection and quantification of PD-L1- and PD-1-expressing immune cell phenotypes was carried out using the HALO Image Analysis Platform (Indica Labs, v.3.6.4143). Cell segmentation was performed by training the nuclei segmentation neural network classifier on DAPI-stained nuclei from imaged carotid sections using the HALO AI nuclei identification software (v.3.6.4143). Probe quantification was based on color and constant image intensity thresholding. All parameters were kept constant across specimens. CD3e+PD-1+ and CD11c+PD-L1+ cells were identified and quantified using the HALO HighPlex FL algorithm (v.4.2). The likelihood of PD-L1–PD-1 interactions was determined by computing the NES as described elsewhere34. In brief, NES were implemented in the squidpy framework87 by counting the number of CD11c+PD-L1+ cells within 30 μm of CD3e+PD-1+ cells. Phenotypes were then permuted 1,000 times, recounting the number of CD11c+PD-L1+ cells within 30 μm of CD3e+PD-1+ cells. The NES was the resulting z-score of the original neighbor count compared to the permutations.
Single-plex fluorescent immunohistochemistry
To validate CD86–CTLA4 and galectin 3–LAG3 interactions as well as quantify the frequency of FSCN1+ dendritic cells, human carotid plaque tissue samples corresponding to six patients from the scRNA-seq cohort (Supplementary Table 1) were processed for single-plex staining. Plaque tissues were sectioned into 5-µm slices following deparaffinization and rehydration and before antigen retrieval using IHC Antigen Retrieval Solution – low pH (Invitrogen, 00-4955-58) in a water bath at 95 °C. Then, sections were blocked and permeabilized in PBS supplemented with 3% wt/vol BSA and 0.25% vol/vol Triton X 100 for 30 min in a humidified chamber at RT. Sections were next incubated with anti-CD86, (Abcam, ab220188; C86/1146; 1:100 dilution), anti-CD4 (Abcam, ab133616; EPR6855; 1:100 dilution) and anti-CTLA4 (Invitrogen, PA5-47547; polyclonal; 1:100 dilution); anti-galectin 3 (Invitrogen, 14-5301-82; eBioM3/38; 1:100 dilution), anti-CD8a (Abcam, ab17147; C8/144B; 1:100 dilution) and anti-LAG3 (Abcam, ab209236; EPR20261; 1:100 dilution); or anti-CD3e (Abcam, ab11089; CD3-12; 1:100 dilution), anti-CD11c (Invitrogen, MA1-46052; 3.9; 1:200 dilution) and anti-Fascin 1 (Invitrogen, MA5-32728; JM12-53; 1:100 dilution) overnight in a humidified chamber at 4 °C. Secondary antibody staining was carried out for 1 h and followed by nuclear counterstaining with DAPI (Thermo Scientific, 62248) for 10 min, both at RT. Autofluorescence was quenched using the TrueBlack Lipofuscin Autofluorescence Quencher (Biotium, 23007) following the manufacturer’s instructions. Stained samples were mounted with fluorescence mounting medium Fluoromount-G (Southern Biotech, 010001). All images were acquired at ×20 magnification using a BZ-X800 microscope. The quantification of immune cell phenotypes and NES computations were carried out as described above.
Cytometry by time-of-flight
Thirty-one patients undergoing CEA were enrolled in a clinical study approved by the IRB of NYU Langone Health (IRB no. 21-00429) and the Icahn School of Medicine at Mount Sinai (IRB no. 11-01427). Supplementary Tables 2 and 5 summarize the demographic and clinical characteristics of the patient cohort as employed, respectively, to validate the presence of CD8–CD4– T cells in carotid plaques at the protein level (n = 28) and to carry out the PD-1 inhibition experiment in vitro (n = 3). For the latter, carotid plaques (vascular explants) were stored in CryoStor CS10 (StemCell, 07959) as described in Arazi et al.88 and maintained in liquid nitrogen until experiment setup, when explants were thawed and cut into approximately 3-mm2 pieces and seeded onto wells of a 12-well plate in DMEM supplemented with 10% vol/vol FBS, 2 mM l-glutamine and 100 μg ml−1 penicillin–streptomycin. Minced explants were then treated with 150 μg anti-PD-1 antibody (Bio X Cell, SIM0003) in a humidified incubator at 37 °C and 5% CO2 for 24 h. Untreated explants were used as controls. All explants were stimulated with a Cell Activation Cocktail (BioLegend, 423303) containing phorbol 12-myristate-13-acetate, ionomycin and brefeldin A for the last 6 h of anti-PD-1 treatment, then washed, minced and digested as described above for human carotid plaque scRNA-seq. Next, single-cell suspensions were stained with Cell-ID Cisplatin-196Pt (Standard BioTools, 201196) as a viability dye. Each sample was then barcoded with a unique combination of three anti-human CD45 antibodies (Standard BioTools; 3111001B, 3113001B and 3114001B), washed, pooled and blocked using TrueStain FcX (BioLegend, 422301) for 10 min at RT. Cell staining was performed with the Maxpar Direct Immune Profiling Assay (Standard BioTools, 201325) according to the manufacturer’s instructions. Stained samples were washed, fixed and permeabilized with a BD Cytofix/Cytoperm Fixation/Permeabilization kit (BD Biosciences, 554714) also following the manufacturer’s instructions. Then, intracellular staining of permeabilized cells using Anti-Human IFNg-165Ho (Standard BioTools, 3165002B; 1:50 dilution), Anti-Human IL-2-112Cd (Standard BioTools, 3112002B; 1:50 dilution), anti-human Perforin1-175Lu (Standard BioTools, 3175004B; 1:50 dilution) and anti-human Granzyme B-198Pt (Standard BioTools, 3198002B; 1:50 dilution) was carried out for 1 h at RT. Cells were maintained in PBS containing 0.125 nM Cell-ID Intercalator-Iridium (Fluidigm, 201192) until acquisition using a CyTOF2 Helios mass cytometer (Standard BioTools). Data were normalized using the Helios normalizer software (Fluidigm, v.6.7.1014), concatenated and de-barcoded using the CyTOF Software (Fluidigm, v.7.1.16389.0) and further processed using the Cytobank Premium online platform (Beckman Coulter Life Sciences) to sequentially remove beads, debris and dead cells. T cells were gated on live CD45+CD19–CD66–CD56–CD3+ single cells and further gated on CD8a+ or CD4+ expression. Median intensities for gated T cells were exported for further quality control and analysis using R packages cytoqc (v.0.99.2), flowWorkspace (v.4.14.3), flowCore (v.2.14.2) and cytofCore (v.0.4). Tissue processing and data acquisition were carried out as detailed above (and without frozen storage, anti-PD-1 treatment, T cell activation, cell permeabilization or intracellular staining steps) on vascular explants for the CD8–CD4– T cell validation experiments. Supplementary Tables 11 and 12, respectively, contain a comprehensive list of antibodies used for this and the anti-PD-1 in vitro experiment.
Statistics and reproducibility
Comparisons were made by two-tailed Student’s t-tests, Wilcoxon rank-sum or signed-rank tests, Kolmogorov–Smirnov tests or two-way analysis of variance. Statistical tests were calculated using GraphPad Prism software (v.9.3.0); the Ingenuity Pathway Analysis platform (v.01-22-01); and the Seurat (v.4.3.0), CellChat (v.1.1.3), propeller (v.1.1.0) and ggpubr (v.0.6.0) packages as well as base functions in R (v.4.0.3). No statistical method was used to predetermine sample size. The experiments were not randomized and the investigators were not blinded to group allocation during experiments and outcome assessment. No data were excluded from the analyses.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
scRNA-seq data from ten human carotid artery plaques were previously published and available in the GEO repositories GSE224273 and GSE235437, hosted by the National Center for Biotechnology Information. Previously published human coronary artery scRNA-seq data can be accessed from GSE252243. Previously published murine plaque scRNA-seq data can be accessed from GSE141038, GSE161494, GSE168389 and GSE252243. Additional human scRNA-seq data from 12 carotid plaque specimens, 3 coronary artery specimens and 22 blood specimens as well as additional murine plaque scRNA-seq data can be accessed from GSE246318. CyTOF raw data are available at https://zenodo.org/records/12734792. The datasets generated and/or analyzed during the current study are provided. Source data are provided with the paper.
Code availability
Code used for data analyses is available on GitHub under https://github.com/giannarelli-lab.
References
Vuong, J. T. et al. Immune checkpoint therapies and atherosclerosis: mechanisms and clinical implications: JACC state-of-the-art review. J. Am. Coll. Cardiol. 79, 577–593 (2022).
Tawbi, H. A. et al. Relatlimab and nivolumab versus nivolumab in untreated advanced melanoma. N. Engl. J. Med. 386, 24–34 (2022).
FDA approves anti-LAG3 checkpoint. Nat. Biotechnol. https://doi.org/10.1038/s41587-022-01331-0 (2022).
Haslam, A. & Prasad, V. Estimation of the percentage of US patients with cancer who are eligible for and respond to checkpoint inhibitor immunotherapy drugs. JAMA Netw. Open 2, e192535 (2019).
US FDA. FDA Drug Database https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm (2023).
Zhang, L., Reynolds, K. L., Lyon, A. R., Palaskas, N. & Neilan, T. G. The evolving immunotherapy landscape and the epidemiology, diagnosis, and management of cardiotoxicity: JACC: cardiooncology primer. JACC CardioOncol. 3, 35–47 (2021).
Suero-Abreu, G. A., Zanni, M. V. & Neilan, T. G. Atherosclerosis with immune checkpoint inhibitor therapy: evidence, diagnosis, and management: JACC: cardiooncology state-of-the-art review. JACC CardioOncol. 4, 598–615 (2022).
Drobni, Z. D. et al. Association between immune checkpoint inhibitors with cardiovascular events and atherosclerotic plaque. Circulation 142, 2299–2311 (2020).
Herrmann, J. et al. Defining cardiovascular toxicities of cancer therapies: an International Cardio-Oncology Society (IC-OS) consensus statement. Eur. Heart J. 43, 280–299 (2022).
Poels, K. et al. Immune checkpoint inhibitor therapy aggravates T cell-driven plaque inflammation in atherosclerosis. JACC CardioOncol. 2, 599–610 (2020).
Bu, D. X. et al. Impairment of the programmed cell death-1 pathway increases atherosclerotic lesion development and inflammation. Arterioscler. Thromb. Vasc. Biol. 31, 1100–1107 (2011).
Fernandez, D. M. et al. Single-cell immune landscape of human atherosclerotic plaques. Nat. Med. 25, 1576–1588 (2019).
Mulholland, M. et al. LAG3 regulates T cell activation and plaque infiltration in atherosclerotic mice. JACC CardioOncol. 4, 635–645 (2022).
Krautter, F. et al. Galectin-9: a novel promoter of atherosclerosis progression. Atherosclerosis 363, 57–68 (2022).
Jarr, K. U. et al. Effect of CD47 blockade on vascular inflammation. N. Engl. J. Med. 384, 382–383 (2021).
Kojima, Y. et al. CD47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature 536, 86–90 (2016).
Gyldenkerne, C. et al. 10-year cardiovascular risk in patients with newly diagnosed type 2 diabetes mellitus. J. Am. Coll. Cardiol. 82, 1583–1594 (2023).
Wang, Z. et al. Pairing of single-cell RNA analysis and T cell antigen receptor profiling indicates breakdown of T cell tolerance checkpoints in atherosclerosis. Nat. Cardiovasc. Res. 2, 290–306 (2023).
Dib, L. et al. Lipid-associated macrophages transition to an inflammatory state in human atherosclerosis, increasing the risk of cerebrovascular complications. Nat. Cardiovas. Res. 2, 656–672 (2023).
Slysz, J. et al. Single-cell profiling reveals comparatively inflammatory polarization of human carotid versus femoral plaque leukocytes. JCI Insight https://doi.org/10.1172/jci.insight.171359 (2023).
Dominguez Conde, C. et al. Cross-tissue immune cell analysis reveals tissue-specific features in humans. Science 376, eabl5197 (2022).
Bottcher, J. P. et al. Functional classification of memory CD8(+) T cells by CX3CR1 expression. Nat. Commun. 6, 8306 (2015).
Depuydt, M. A. C. et al. Microanatomy of the human atherosclerotic plaque by single-cell transcriptomics. Circ. Res. 127, 1437–1455 (2020).
June, C. H., Ledbetter, J. A., Gillespie, M. M., Lindsten, T. & Thompson, C. B. T-cell proliferation involving the CD28 pathway is associated with cyclosporine-resistant interleukin 2 gene expression. Mol. Cell. Biol. 7, 4472–4481 (1987).
Linsley, P. S. et al. CTLA-4 is a second receptor for the B cell activation antigen B7. J. Exp. Med. 174, 561–569 (1991).
Liu, J. Q. et al. CD200-CD200R pathway in the regulation of tumor immune microenvironment and immunotherapy. Adv. Exp. Med. Biol. 1223, 155–165 (2020).
Kassiteridi, C. et al. CD200 limits monopoiesis and monocyte recruitment in atherosclerosis. Circ. Res. 129, 280–295 (2021).
Lutgens, E. et al. Immunotherapy for cardiovascular disease. Eur. Heart J. 40, 3937–3946 (2019).
Artis, D. & Spits, H. The biology of innate lymphoid cells. Nature 517, 293–301 (2015).
Shami, A. et al. Glucocorticoid-induced tumour necrosis factor receptor family-related protein (GITR) drives atherosclerosis in mice and is associated with an unstable plaque phenotype and cerebrovascular events in humans. Eur. Heart J. 41, 2938–2948 (2020).
Lacy, M. et al. Cell-specific and divergent roles of the CD40L–CD40 axis in atherosclerotic vascular disease. Nat. Commun. 12, 3754 (2021).
Stary, H. C. et al. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation 92, 1355–1374 (1995).
Sharma, N. et al. ICOS costimulation in combination with CTLA-4 blockade remodels tumor-associated macrophages toward an antitumor phenotype. J. Exp. Med. 221, e20231263 (2024).
Vanguri, R. S. et al. Integration of peripheral blood- and tissue-based biomarkers of response to immune checkpoint blockade in urothelial carcinoma. J. Pathol. 261, 349–360 (2023).
Schadendorf, D. et al. Health-related quality of life with nivolumab plus relatlimab versus nivolumab monotherapy in patients with previously untreated unresectable or metastatic melanoma: RELATIVITY-047 trial. Eur. J. Cancer 187, 164–173 (2023).
Borst, L., van der Burg, S. H. & van Hall, T. The NKG2A–HLA-E axis as a novel checkpoint in the tumor microenvironment. Clin. Cancer Res. 26, 5549–5556 (2020).
Foks, A. C. et al. T-cell immunoglobulin and mucin domain 3 acts as a negative regulator of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 33, 2558–2565 (2013).
Yasinska, I. M. et al. Ligand–receptor interactions of galectin-9 and VISTA suppress human T lymphocyte cytotoxic activity. Front. Immunol. 11, 580557 (2020).
Yang, R. et al. Galectin-9 interacts with PD-1 and TIM-3 to regulate T cell death and is a target for cancer immunotherapy. Nat. Commun. 12, 832 (2021).
Sedy, J. R. et al. B and T lymphocyte attenuator regulates T cell activation through interaction with herpesvirus entry mediator. Nat. Immunol. 6, 90–98 (2005).
Wu, B. et al. Poliovirus receptor (PVR)-like protein cosignaling network: new opportunities for cancer immunotherapy. J. Exp. Clin. Cancer Res. 40, 267 (2021).
O’Donnell, P. H. et al. Enfortumab vedotin with or without pembrolizumab in cisplatin-ineligible patients with previously untreated locally advanced or metastatic urothelial cancer. J. Clin. Oncol. 41, 4107–4117 (2023). JCO2202887.
Braun, A. et al. Afferent lymph-derived T cells and DCs use different chemokine receptor CCR7-dependent routes for entry into the lymph node and intranodal migration. Nat. Immunol. 12, 879–887 (2011).
Yamakita, Y. et al. Fascin1 promotes cell migration of mature dendritic cells. J. Immunol. 186, 2850–2859 (2011).
Maier, B. et al. A conserved dendritic-cell regulatory program limits antitumour immunity. Nature 580, 257–262 (2020).
Magen, A. et al. Intratumoral dendritic cell-CD4(+) T helper cell niches enable CD8(+) T cell differentiation following PD-1 blockade in hepatocellular carcinoma. Nat. Med. 29, 1389–1399 (2023).
Wu, S. Y. et al. CCL19(+) dendritic cells potentiate clinical benefit of anti-PD-(L)1 immunotherapy in triple-negative breast cancer. Medicine 4, 373–393 e378 (2023).
Libby, P. The changing landscape of atherosclerosis. Nature 592, 524–533 (2021).
Nielsen, S. F., Nordestgaard, B. G. & Bojesen, S. E. Statin use and reduced cancer-related mortality. N. Engl. J. Med. 367, 1792–1802 (2012).
Sharma, M. et al. Regulatory T cells license macrophage pro-resolving functions during atherosclerosis regression. Circ. Res. 127, 335–353 (2020).
Schlegel, M. et al. Silencing myeloid netrin-1 induces inflammation resolution and plaque regression. Circ. Res. 129, 530–546 (2021).
Afonso, M. S. et al. miR-33 silencing reprograms the immune cell landscape in atherosclerotic plaques. Circ. Res. 128, 1122–1138 (2021).
Cyr, Y. et al. The IRG1–itaconate axis protects from cholesterol-induced inflammation and atherosclerosis. Proc. Natl Acad. Sci. USA 121, e2400675121 (2024).
Heng, T. S., Painter, M. W. & Immunological Genome Project. The Immunological Genome Project: networks of gene expression in immune cells. Nat. Immunol. 9, 1091–1094 (2008).
Di Gregoli, K. et al. Galectin-3 identifies a subset of macrophages with a potential beneficial role in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 40, 1491–1509 (2020).
Chow, M. T. et al. Intratumoral activity of the CXCR3 chemokine system is required for the efficacy of anti-PD-1 therapy. Immunity 50, 1498–1512 e1495 (2019).
Alegre, M. L. et al. Regulation of surface and intracellular expression of CTLA4 on mouse T cells. J. Immunol. 157, 4762–4770 (1996).
Truong, K. L. et al. Killer-like receptors and GPR56 progressive expression defines cytokine production of human CD4(+) memory T cells. Nat. Commun. 10, 2263 (2019).
Annunziato, F. et al. Phenotypic and functional features of human Th17 cells. J. Exp. Med. 204, 1849–1861 (2007).
Groom, J. R. & Luster, A. D. CXCR3 in T cell function. Exp. Cell. Res. 317, 620–631 (2011).
Ling, S. et al. Inequalities in cancer mortality trends in people with type 2 diabetes: 20 year population-based study in England. Diabetologia 66, 657–673 (2023).
Sabatine, M. S. et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N. Engl. J. Med. 376, 1713–1722 (2017).
Shapiro, M. D., Tavori, H. & Fazio, S. PCSK9: from basic science discoveries to clinical trials. Circ. Res. 122, 1420–1438 (2018).
Liu, X. et al. Inhibition of PCSK9 potentiates immune checkpoint therapy for cancer. Nature 588, 693–698 (2020).
Wang, R. et al. Inhibition of PCSK9 enhances the antitumor effect of PD-1 inhibitor in colorectal cancer by promoting the infiltration of CD8(+) T cells and the exclusion of Treg cells. Front. Immunol. 13, 947756 (2022).
Quagliariello, V. et al. PCSK9 inhibitors in cancer patients treated with immune-checkpoint inhibitors to reduce cardiovascular events: new frontiers in cardioncology. Cancers 15, 1397 (2023).
Cohain, A. T. et al. An integrative multiomic network model links lipid metabolism to glucose regulation in coronary artery disease. Nat. Commun. 12, 547 (2021).
Sabatine, M. S. et al. Cardiovascular safety and efficacy of the PCSK9 inhibitor evolocumab in patients with and without diabetes and the effect of evolocumab on glycaemia and risk of new-onset diabetes: a prespecified analysis of the FOURIER randomised controlled trial. Lancet Diabetes Endocrinol. 5, 941–950 (2017).
Linsley, P. S., Clark, E. A. & Ledbetter, J. A. T-cell antigen CD28 mediates adhesion with B cells by interacting with activation antigen B7/BB-1. Proc. Natl Acad. Sci. USA 87, 5031–5035 (1990).
Azuma, M. et al. B70 antigen is a second ligand for CTLA-4 and CD28. Nature 366, 76–79 (1993).
Zhu, C. et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat. Immunol. 6, 1245–1252 (2005).
Li, H. et al. Tim-3/galectin-9 signaling pathway mediates T-cell dysfunction and predicts poor prognosis in patients with hepatitis B virus-associated hepatocellular carcinoma. Hepatology 56, 1342–1351 (2012).
Stoeckius, M. et al. Cell hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol. 19, 224 (2018).
Eberhardt, N. et al. SARS-CoV-2 infection triggers pro-atherogenic inflammatory responses in human coronary vessels. Nat. Cardiovasc. Res. 10, 899–916 (2023).
Hafemeister, C. & Satija, R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 20, 296 (2019).
Squair, J. W. et al. Confronting false discoveries in single-cell differential expression. Nat. Commun. 12, 5692 (2021).
Xu, C. et al. Automatic cell type harmonization and integration across Human Cell Atlas datasets. Cell 26, 5876–5891 (2023).
Krämer, A., Green, J., Pollard, J. Jr & Tugendreich, S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics 30, 523–530 (2013).
Jin, S. et al. Inference and analysis of cell–cell communication using CellChat. Nat. Commun. 12, 1088 (2021).
Germain, P. L., Lun, A., Garcia Meixide, C., Macnair, W. & Robinson, M. D. Doublet identification in single-cell sequencing data using scDblFinder. F1000Res 10, 979 (2021).
Wirka, R. C. et al. Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat. Med. 25, 1280–1289 (2019).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 e3529 (2021).
McGinnis, C. S., Murrow, L. M. & Gartner, Z. J. DoubletFinder: doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Cell Syst. 8, 329–337 e324 (2019).
Li, H. et al. Dysfunctional CD8 T cells form a proliferative, dynamically regulated compartment within human melanoma. Cell 176, 775–789 e718 (2019).
Pellicci, D. G., Koay, H. F. & Berzins, S. P. Thymic development of unconventional T cells: how NKT cells, MAIT cells and γδ T cells emerge. Nat. Rev. Immunol. 20, 756–770 (2020).
Aran, D. et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 20, 163–172 (2019).
Palla, G. et al. Squidpy: a scalable framework for spatial omics analysis. Nat. Methods 19, 171–178 (2022).
Arazi, A. et al. The immune cell landscape in kidneys of patients with lupus nephritis. Nat. Immunol. 20, 902–914 (2019).
Acknowledgements
We thank the Division of Advanced Research Technologies at NYU Langone Health, which supports the Histology and Immunohistochemistry Laboratory at the Center for Biospecimen Research and Development (RRID: SCR_018304), the GTC (RRID: SCR_017929) and the Cytometry and Cell Sorting Laboratory (RRID: SCR_019179). The Division of Advanced Research Technologies is supported by the National Institutes of Health (NIH)/National Cancer Institute grant P30CA016087. C.G. is supported by NIH/National Heart, Lung and Blood Institute grants R01HL153712 and R01HL165258 and Chan Zuckerberg Institute grant NFL-2020-218415. K.J.M. is supported by NIH/National Heart, Lung and Blood Institute grants R35HL135799 and R01HL084312. J.S.B., I.J.G., E.A.F. and C.G. are supported by the American Heart Association (AHA) Strategically Focused Research Network grant 20SFRN35210252. D.D. is supported by the AHA Strategically Focused Research Network grant (20SFRN35210252). N.E. is supported by the AHA Research Supplement to Promote Diversity in Science (965509) and M. Gourvest is supported by the AHA postdoctoral grant 23POST1029885.
Author information
Authors and Affiliations
Contributions
K.J.M. and C.G. designed the study. J.G.B.D., K.J.M. and C.G wrote the paper. S.S., K.R. and P.F. managed clinical data. N.N. assessed human plaque pathology. D.D., L.A., N.E., P.S., P.F. and B.C collected and processed human samples. D.D., L.A. and D.F. performed human in vitro experiments. M. Gourvest performed mouse experiments. J.G.B.D., M. Gildea and R.K. performed data analyses and visualization. D.D. and R.V. carried out the cell–cell interaction analysis in situ. J.B.S. and I.J.G. conducted the CHORD study and provided human PBMCs from the CHORD study. E.A.F. provided intellectual input. All authors edited and approved the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Cardiovascular Research thanks the anonymous reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Proportions and corresponding gene expression patterns of human atherosclerotic plaque immune cell subclusters by single-cell RNA sequencing.
a–g, Proportions of CD8+ T cell (a), CD4+ T cell (b), Mixed (CD8+ or CD4+) and γδ T (Tγδ) cell (c), double-positive (DP; CD8+ CD4+) T cell (d), B cell (e), myeloid (f), and natural killer (NK) cell and innate lymphoid cell (ILC) (g) subclusters (left) and dot plots showing scaled average expression of corresponding markers (right). EM: effector memory; EMRA: terminally differentiated effector memory re-expressing CD45RA; CM: central memory; RM: resident memory; CTL: cytotoxic T lymphocyte; Reg: regulatory; MAIT: mucosal-associated invariant; NKT: natural killer T cell; Mono: monocyte; Mφ: macrophage; DC: dendritic cell. NE: normalized expression.
Extended Data Fig. 2 Immune checkpoint (IC) gene expression pattern in human coronary artery.
a, Uniform Manifold Approximation and Projection (UMAP) of scRNA-seq data from human coronary arteries (left; n = 6 specimens from 5 subjects) and proportion of each cell type depicted (right), stratified by atherosclerotic disease status: adaptive intimal thickening (AIT, n = 3) or plaque (n = 3). b, Proportions of major immune cell identities sequenced per patient. Each dot represents one specimen. Boxes represent interquartile ranges; center lines depict medians. Whiskers below and above boxes represent extent of lower and upper quartiles, respectively (n = 3 per group). c, Heatmap depicting immune cell marker gene expression by cluster. z-score shows scaled average expression. d, Heatmap of scRNA-seq IC gene expression in AIT (pink), plaque (yellow), both (gray), or neither (white) specimen from human coronary arteries, shown by cell clusters and subclusters. For significantly differentially expressed genes in plaque vs. AIT (FDR < 0.05), fold change (FC) is shown instead. DP: double-positive (CD8+ CD4+); Tγδ: γδ T cell; Mye/M: myeloid cell; NK: natural killer; ILC: innate lymphoid cell; HSPC: hematopoietic stem and progenitor cell; Fibro: fibroblast; MyoF: myofibroblast; SMC: smooth muscle cell; EM: effector memory; EMRA: terminally differentiated effector memory re-expressing CD45RA; CM: central memory; RM: resident memory; CTL: cytotoxic T lymphocyte; Reg: regulatory; MAIT: mucosal-associated invariant; Mono: monocyte; Mφ: macrophage; DC: dendritic cell; FDA: [United States] Food and Drug Administration.
Extended Data Fig. 3 Distribution of significant immune checkpoint (IC) interactions across immune cell clusters and subclusters found in human atherosclerotic plaques by single-cell RNA sequencing.
Heatmap showing the number of significant (p < 0.05) IC interactions predicted between ligand-expressing (y-axis) and receptor-expressing (x-axis) immune cell clusters and subclusters within atherosclerotic plaques. p values were calculated by the CellChat package (v1.1.3) in R (v4.0.3). DP: double-positive (CD8+ CD4+); Tγδ: γδ T cell; NK: natural killer; ILC: innate lymphoid cell; EM: effector memory; EMRA: terminally differentiated effector memory re-expressing CD45RA; CM: central memory; RM: resident memory; CTL: cytotoxic T lymphocyte; Reg: regulatory; MAIT: mucosal-associated invariant; NKT: natural killer T cell; Mono: monocyte; Mφ: macrophage; DC: dendritic cell.
Extended Data Fig. 4 Strength and gene expression profile of clinically relevant immune checkpoint (IC) interactions.
a, Bar plot depicting the relative contribution of predicted IC interactions to overall cell-cell communication among human plaque immune cells. (*) indicates interactions targeted by immunotherapies approved by the U.S. Food and Drug Administration; (†) indicates interactions reported to increase risk of atherosclerotic cardiovascular events7,9,12. b,d,f, Chord plots illustrating the strongest interactions (p < 0.05) between: (b), PD-L2 (encoded by PDCD1LG2) and PD-1 (encoded by PDCD1); (d), CD80 and CTLA4; and (f), α-synuclein (αSyn, encoded by SNCA) and LAG3. Direction of ligand → receptor interaction is denoted by chord arrows. p values were calculated by the CellChat package (v1.1.3) in R (v4.0.3). c,e,g, Communication strengths of: (c), PD-L1– or PD-L2–PD-1 interactions; (e), CD86– or CD80–CTLA4 interactions; and (g), Galectin 3– or αSyn–LAG3 interactions, ranked in order of probability, with the top 30–40 interactions depicted as rings. p values were determined by two-tailed Kolmogorov–Smirnov tests. h, Violin plots illustrating normalized transcript abundance and distribution of IC ligands in myeloid cell subclusters engaging in significant IC interactions. NE: normalized expression. DP: double-positive (CD8+ CD4+); Tγδ: γδ T cell; NK: natural killer; EM: effector memory; EMRA: terminally differentiated effector memory re-expressing CD45RA; RM: resident memory; CTL: cytotoxic T lymphocyte; Reg: regulatory; MAIT: mucosal-associated invariant; NKT: natural killer T cell; Mono: monocyte; Mφ: macrophage; DC: dendritic cell.
Extended Data Fig. 5 Co-inhibitory immune checkpoint interactions targeted by experimental ICIs.
Chord plots illustrating the strongest interactions (p < 0.05) between: (a), HLA-E and and NKG2A (encoded by KLRC1); (b), HMGB1 and TIM3 (encoded by HAVCR2); (c), Galectin 9 (encoded by LGALS9) and TIM3; (d), CD47 and SIRPα (encoded by SIRPA); (e), BTLA and HVEM (encoded by TNFRSF14); and (g), NECTIN2 and TIGIT. Direction of ligand → receptor interaction is denoted by chord arrows. p values were calculated by the CellChat package (v1.1.3) in R (v4.0.3). DP: double-positive (CD8+ CD4+); NK: natural killer; ILC: innate lymphoid cell; EM: effector memory; EMRA: terminally differentiated effector memory re-expressing CD45RA; CM: central memory; RM: resident memory; CTL: cytotoxic T lymphocyte; Reg: regulatory; MAIT: mucosal-associated invariant; NKT: natural killer T cell; Tγδ: γδ T cell; Mono: monocyte; Mφ: macrophage; DC: dendritic cell. CD8+ T cells are depicted in purple; CD4+ T cells are depicted in green; Mixed (CD8+ or CD4+) T cells are depicted in light blue; DP T cells are depicted in dark blue; γδ T (Tγδ) cells are depicted in cyan; B cells are depicted in yellow; myeloid cells are depicted in red; NK cells are depicted in orange; ILC are depicted in gray. f, Communication strengths of NECTIN2, NECTIN3, PVR, or NECTIN4–TIGIT interactions, ranked in order of probability, with the top 30 interactions depicted as rings. p values were determined by two-tailed Kolmogorov–Smirnov tests. Range from lowest to highest p values computed is shown.
Extended Data Fig. 6 Immune checkpoint (IC) interactions in murine atherosclerotic plaque immune cells.
a, Dot plot of immune checkpoint (IC) gene expression by immune cell clusters and subclusters in murine atherosclerotic plaques. Dot color represents fold change (FC) values in each subcluster compared to the rest (FDR < 0.05). b, Bar plot depicting the relative contribution of predicted IC interactions to overall cell-cell communication among murine plaque immune cells. (*) indicates interactions targeted by immunotherapies approved by the U.S. Food and Drug Administration; (†) indicates interactions known to increase risk of cardiovascular events like myocardial infarction and stroke in humans7,9,12. c, Communication strengths of IC interactions targeted by cancer immunotherapies, ranked in order of probability and stratified by treatment, with the top 30 interactions depicted as rings. DP: double-positive (CD8+ CD4+); M: Mixed (CD8+ or CD4+) T cell; Tγδ: γδ T cell; NK: natural killer; ILC: innate lymphoid cell; EM: effector memory; RM: resident memory; Tc17: T cell 17; EMRA: terminally differentiated effector memory re-expressing CD45RA; Th17: T helper 17; Mono: monocyte; Mφ: macrophage; Nφ: neutrophil; Bφ: basophil; MoDC: monocyte/dendritic cell; DC: dendritic cell.
Extended Data Fig. 7 Characterization of anti-inflammatory program in murine atherosclerotic plaque immune cells that results from lipid-lowering treatment.
a–c, Canonical pathway analyses of differentially expressed transcripts in, respectively, myeloid, CD8+ T cells, or CD4+ T cells in lipid-lowered vs. untreated (control) mice. p value < 0.05 and |z-score| > 2. Some GO terms were edited for brevity. d–f, Heatmaps showing normalized transcript abundances of genes contributing to the respective GO terms for, respectively, myeloid, CD8+ T cells, or CD4+ T cells in lipid-lowered (light blue) vs. untreated (control; gray) mice. z-score shows scaled average expression.
Extended Data Fig. 8 Proteomic characterization by cytometry by time-of-flight (CyTOF) of human vascular explants subjected to PD-1 inhibition ex vivo.
a–f, Box plots depicting normalized expression of indicated markers in CD8+ or CD4+ T cells derived from control or anti (α)-PD-1-treated human carotid vascular explants. Boxes represent interquartile ranges; center lines depict medians. Whiskers below and above boxes represent extent of lower and upper quartiles, respectively. Each dot represents a cell. p values were determined by two-tailed Wilcoxon rank-sum tests (n = 3 per group).
Extended Data Fig. 9 Immune checkpoint (IC) gene expression and interactions in human peripheral blood mononuclear cells (PBMCs).
a, Dot plot of differential IC gene expression by immune cell cluster and subcluster found in human PBMCs from type 2 diabetic (T2D; n = 8) vs. non-diabetic (n = 4) patients. Dot color represents fold change (FC) values in each subcluster compared to the rest (FDR < 0.05). b,c, Chord plots illustrating the strongest (p < 0.05) interactions between: (c) Galectin 9 (encoded by LGALS9) and TIM3 (encoded by HAVCR2) and (d) BTLA and HVEM (encoded by TNFRSF14). Direction of ligand → receptor interaction is denoted by chord arrows. p values were calculated by the CellChat package (v1.1.3) in R (v4.0.3). Unconv: unconventional; NK: natural killer [cell]; ILC: innate lymphoid cell; HSPC: hematopoietic stem and progenitor cell; EM: effector memory; EMRA: terminally differentiated effector memory re-expressing CD45RA; CTL: cytotoxic T lymphocyte; CM: central memory; Reg: regulatory; ExReg: ex-regulatory; NKT: natural killer cell; MAIT: mucosal-associated invariant; Tγδ: γδ T cell; CMono: classical monocyte; IM: intermediate monocyte; NCM: non-classical monocyte; MoDC: monocyte/dendritic cell; DC: dendritic cell.
Extended Data Fig. 10 Comparative changes in significant Galectin 3–LAG3 interactions.
Differential communication strengths of strongest (p < 0.05) Galectin 3 (encoded by LGALS3)–LAG3 interactions in LAG3-expressing myeloid cell subclusters before (gray) and after (light blue) lipid-lowering treatment. Dots represent the mean fold change (FC) between T2D and non-diabetic patients of the indicated interaction. Dotted line indicates log2(FC) = 0, which is the threshold at which interactions between T2D and non-diabetic patients were predicted to remain unchanged. p values were calculated by the CellChat package (v1.1.3) in R (v4.0.3). Tγδ: γδ T cell; EM: effector memory; EMRA: terminally differentiated effector memory re-expressing CD45RA; CTL: cytotoxic T lymphocyte; Tγδ: γδ T cell; CM: classical monocyte; IM: intermediate monocyte; NCM: non-classical monocyte; MoDC: monocyte/dendritic cell; DC: dendritic cell.
Supplementary information
Supplementary Information
Supplementary Figs. 1–12.
Supplementary Table
Supplementary Tables 1–12
Source data
Source Data Fig. 1
Statistical Source Data.
Source Data Fig. 2
Statistical Source Data.
Source Data Fig. 3
Statistical Source Data.
Source Data Fig. 4
Statistical Source Data.
Source Data Fig. 5
Statistical Source Data.
Source Data Fig. 6
Statistical Source Data.
Source Data Fig. 7
Statistical Source Data.
Source Data Fig. 8
Statistical Source Data.
Source Data Extended Data Fig. 1
Statistical Source Data.
Source Data Extended Data Fig. 2
Statistical Source Data.
Source Data Extended Data Fig. 3
Statistical Source Data.
Source Data Extended Data Fig. 4
Statistical Source Data.
Source Data Extended Data Fig. 5
Statistical Source Data.
Source Data Extended Data Fig. 6
Statistical Source Data.
Source Data Extended Data Fig. 7
Statistical Source Data.
Source Data Extended Data Fig. 8
Statistical Source Data.
Source Data Extended Data Fig. 9
Statistical Source Data.
Source Data Extended Data Fig. 10
Statistical Source Data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Barcia Durán, J.G., Das, D., Gildea, M. et al. Immune checkpoint landscape of human atherosclerosis and influence of cardiometabolic factors. Nat Cardiovasc Res 3, 1482–1502 (2024). https://doi.org/10.1038/s44161-024-00563-4
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s44161-024-00563-4
This article is cited by
-
7-O-methylpunctatin is a potential inhibitor of human arachidonate 5-lipoxygenase: molecular and structural insights into anti-atherosclerosis therapeutics
Molecular Diversity (2026)
-
Cardiometabolic heart failure with preserved ejection fraction: from molecular signatures to personalized treatment
Cardiovascular Diabetology (2025)
-
Spatial transcriptomics elucidates localized immune responses in atherosclerotic coronary artery
EMBO Molecular Medicine (2025)
-
Immune checkpoint inhibitors and myocardial infarction
Journal of Thrombosis and Thrombolysis (2025)
-
Anti-atherogenic immune checkpoint TIM-3 as a promising pharmacologic target toward ischemic heart diseases: a prospective review
Molecular Biology Reports (2025)
