
Abstract
Type 2 diabetes (T2D) precipitates diabetic cardiomyopathy (dbCM), a condition characterized by chronic inflammation, metabolic dysregulation and impaired cardiac performance. Here we show that the glucokinase activator AZD1656, originally developed for glycemic control but later identified to have immunomodulatory effects, reverses cardiac dysfunction and metabolic remodeling in dbCM. In obese, hyperglycemic db/db mice with diastolic dysfunction, 6 weeks of AZD1656 treatment improved myocardial performance, reduced infarct size and enhanced post-ischaemic recovery. Integrated metabolic, functional and histological analyses revealed restoration of mitochondrial metabolism and attenuation of fibrosis. Mechanistically, AZD1656 remodeled the cardiac immune landscape by promoting infiltration of regulatory T cells. These findings demonstrate a link between cardiac inflammation and metabolic remodeling in dbCM and highlight that modulation of immune cells and metabolism can protect the diabetic heart. Targeting immunometabolic pathways may therefore offer a therapeutic strategy to alleviate cardiac dysfunction and reduce infarct vulnerability in T2D.
Similar content being viewed by others

Main
Diabetic cardiomyopathy (dbCM) is a complication of type II diabetes (T2D) characterized by impaired cardiac function, disrupted metabolism, systemic inflammation, an increased incidence of ischemic heart disease and heart failure1. The dbCM heart is metabolically inflexible, relying heavily on fatty acid (FA) oxidation, which leads to chronic energetic impairment2. Although metabolic derangements worsen dbCM prognosis, no current therapeutic agents specifically target cardiac metabolism.
Glucokinase (GCK) and hexokinase catalyze the first step of glycolysis, the conversion of glucose to glucose-6-phosphate. Hexokinase is widely expressed, whereas GCK is primarily found in pancreatic β-cells, liver and a subset of T cells, where it acts as a glucose sensor. Thus, GCK activation is an attractive approach for T2D treatment. AZD1656 was developed as a selective GCK activator with >100-fold specificity over hexokinase and was expected to improve glycemic control3. However, in 25 clinical trials involving 900 people with T2D, AZD1656 only transiently improved glucose levels for up to 4 months3.
Subsequent work revealed the immunoregulatory actions of AZD1656. Kishore et al.4 demonstrated that GCK activation enhances regulatory T cell (Treg) motility. In Treg cells, CD28 costimulation activates PI3K–mTORC2 signaling, linking metabolic reprogramming with cytoskeletal remodeling4 through Akt phosphorylation. This promotes GCK transcription and modulates its regulatory partner GCKR, releasing GCK to participate in localized glycolytic ATP production essential for actin remodeling and cell movement4. Pharmacological activation by AZD1656 therefore increases Treg glycolytic capacity and migration without affecting proliferation.
Clinical data support this mechanism. In the ARCADIA trial5, AZD1656 treatment improved outcomes in people with T2D and systemic inflammation, decreasing mortality and accelerating recovery, accompanied by improved immune homeostasis and reduced numbers of circulating cardiotropic Treg cells. Genetic evidence is consistent: individuals with the GCKR-P446L loss-of-function polymorphism, which diminishes GCK inhibition, display increased hepatic GCK activity, altered glucose and lipid metabolism and enhanced Treg motility4. Together, these findings identify GCK-dependent glycolysis as a key metabolic determinant of Treg trafficking. Building on this immunomodulatory foundation, the ADOPTION trial (NCT05216172), is a placebo-controlled, double-blind randomized study investigating AZD1656 as a strategy to modulate T cell immunometabolism in renal-transplant recipients with T2D.
Chronic inflammation is a hallmark of T2D, associated with increased numbers of circulating inflammatory T cells1. Hyperlipidemia promotes differentiation of pro-inflammatory effector memory T (TEM) cells and reduces that of Treg cells, disrupting immune balance6. High levels of circulating fatty acids drive TEM migration into non-lymphoid tissues, promoting cytokine release and systemic inflammasome activation6,7. However, whether T cell-mediated inflammation contributes to myocardial inflammation and metabolic dysfunction in dbCM remains unknown.
Given the immunomodulatory effects of AZD1656, we investigated its potential to regulate inflammation and improve cardiac metabolic function in dbCM. In a 6-week intervention in db/db mice, AZD1656 attenuated diastolic dysfunction, reversed metabolic remodeling, reduced infarct size, decreased cardiac inflammation and fibrosis. AZD1656 also normalized myocardial gene expression, including genes in pathways governing inflammation, metabolism and pro-survival signaling. These findings suggest that targeting immunometabolism with AZD1656 could be a therapeutic strategy for dbCM.
Results
AZD1656 treatment improves diastolic function in the diabetic heart and offers protection from IR injury
The impact of 6 weeks of AZD1656 treatment (Fig. 1a) on cardiac function in dbCM was assessed in vivo using CINE magnetic resonance imaging (CINE MRI) and echocardiography (Table 1). As has previously been noted8, no reduction in systolic function was observed in untreated db/db mice, with a small but significant alteration in CINE MRI-assessed ejection fraction observed alongside unaltered cardiac output (Table 1). There was no evidence of pulmonary congestion or any increase in overall heart weight in db/db animals (Table 1). Cine MRI assessment (Table 1) identified unaltered end diastolic volume and decreased end systolic volume. Untreated db/db mice exhibited the diastolic dysfunction that is typical of dbCM, evidenced by significant alterations in isovolumetric relaxation time, mitral valve deceleration time and the ratio of early to late diastolic mitral inflow velocities (Fig. 1b and Table 1). Ex vivo functional assessment by Langendorff perfusion (Fig. 1d) further confirmed diastolic dysfunction in db/db mice.

a, The AZD1656 treatment protocol. Mice were fed nutritionally standardized and matched Teklad Standard Base Diet (Envigo), with AZD1656 added to the diet used for the db/db treatment group (30 mg kg−1 body weight). b, In vivo echocardiography assessment of diastolic dysfunction: isovolumic relaxation time (IVRT) (control versus db/db P = 0.0001; control versus AZD P = 0.0002; db/db versus AZD P = 0.0015), mitral valve deceleration (MV decel) (control versus db/db P < 0.0001; control versus AZD P < 0.0036; db/db versus AZD P = 0.0397), myocardial performance index (MPI) (control versus db/db P = 0.0007; control versus AZD P = 0.811; db/db versus AZD P = 0.0521), mitral valve early versus late ratio (E/A) (control versus db/db P = 0.0003 by one-way ANOVA, AZD versus control P = 0.185 and AZD versus db/db P = 0.036 by Fischer’s exact test). The approach to statistical analysis for E/A is described in the Methods. Control n = 17, db/db n = 17, AZD n = 14. c, Representative Langendorff perfused heart. d, Langendorff perfused heart ex vivo function end diastolic pressure (control n = 9; db/db n = 9; AZD n = 6; control versus db/db P = 0.0001; control versus AZD P = 0.0001; db/db versus AZD P = 0.0344) by one-way ANOVA. e, Langendorff perfused heart ex vivo function systolic pressure (control n = 9; db/db n = 9; AZD n = 6; control versus db/db P = 0.0001; control versus AZD P = 0.0001; db/db versus AZD P = 0.0344) by one-way ANOVA. f, Representative TTC stained cardiac cross sections after 20 min of total global ischemia and 2 h reperfusion, used for quantification of the infarct size. g, Quantification of infarct size after ischemia–reperfusion injury (db/db, AZD n = 5 per group; control n = 3) by one-way ANOVA (control versus db/db P = 0.0005; control versus AZD P = 0.1372; db/db versus AZD P = 0.005). h, Improved left ventricular developed pressure (LVDP) recovery after ischemia in Langendorff perfused hearts (control n = 5; db/db n = 5; AZD n = 6; control versus db/db P = 0.0001; control versus AZD P = 0.0001; db/db versus AZD P = 0.034) by one-way ANOVA. n represents biological replicates (individual animals). Data are presented as mean ± s.e.m. NS, non-significant. Created with BioRender.com.
Treatment with AZD1656 enhanced in vivo cardiac function, reflected by improvements in diastolic function, myocardial performance index (Table 1 and Fig. 1b), the doppler-flow-assessed pulmonary artery velocity–time integral, pulmonary artery peak velocity (Table 1) and ascending aorta peak velocity (Table 1). This attenuation of myocardial dysfunction in db/db mice was accompanied by a protection from ischemia–reperfusion injury, evidenced by reduced infarct size (Fig. 1f,g) and significantly improved functional recovery after myocardial infarction versus untreated db/db mice (Fig. 1h).
AZD1656 treatment improves cardiac metabolism in dbCM
Untreated db/db mice were characterized by severe cardiac metabolic dysfunction (Fig. 2). Principal component analysis (PCA) of the metabolomic profile, assessed by high-resolution proton nuclear magnetic resonance (1H NMR) spectroscopy (Fig. 2a), confirmed that T2D leads to a distinct metabolomic profile of db/db hearts versus controls. Specifically, the metabolomic profile of T2D mice is characterized by a significant depletion of amino acids (glutamine, valine, glycine, taurine) and acetate, as well as altered levels of tricarboxylic acid cycle intermediates (depleted fumarate, elevated succinate, Fig. 2b).

a, PCA plot of 1H NMR metabolomic profiling. b, Fold change db/db versus control 1H NMR metabolomic spectroscopy profiling (n = 6 per group). Metabolite fold change versus control (control = 1) with propagated error of mean by t-test; two tailed P < 0.05. c, Fold change AZD-treated db/db versus control 1H NMR metabolomic spectroscopy profiling. n = 6 per group metabolite fold change versus control (control = 1) with propagated error of mean by t-test; two tailed P < 0.05. PCr, phosphocreatine. d, Cardionet metabolic flux balance analysis based on the 1H NMR metabolomic profiling and plasma metabolomic analysis (n = 6 per group). Statistical analysis for Cardionet is described in the Methods. e, Visualization of CardioNet metabolic flux predictions using Cytoscape. Metabolites and reactions are depicted as nodes and lines, respectively. The line thickness corresponds to the calculate flux rate. Statistical analysis for Cardionet is described in the Methods. f, Representative annotated spectrum from in vivo 13C dynamic nuclear polarization magnetic resonance spectroscopy cardiac metabolic flux assessment. g, Prediction of oxygen consumption rates based on CardioNet simulations (control n = 5; db/db n = 4; AZD n = 5) by one-way ANOVA (control versus db/db P = 0.0001, control versus AZD P = 0.174, db/db versus AZD P = 0.0001). h,i, In vivo 13C DNP MRS measurement during tricarboxylic acid cycle flux (control n = 12, db/db n = 16, AZD n = 10). Multiple group comparison by one-way ANOVA (lactate/pyruvate: control versus db/db P = 0.399; control versus AZD P = 0.99; db/db versus AZD P = 0.99 (h); bicarbonate/pyruvate control versus db/db P = 0.0001, control versus AZD P = 0.99, db/db versus AZD P = 0.0147 (i)). n represents biological replicates (individual animals). Data are presented as mean ± s.e.m.
Collectively, metabolomic changes in db/db hearts were associated with reduction in energy reserves (phosphocreatine, ATP and ADP, the ratio of phosphocreatine to ATP and ADP, Fig. 2b).
To further analyze the cardiac metabolic profile in db/db mice, we applied a systems-biology approach that combines metabolomics with constrained-based in silico modeling9,10. To infer flux distributions, we integrated targeted metabolomics data from control, db/db and AZD1656-treated groups into flux balance analysis (FBA). Metabolic flux distributions were calculated using the network of mammalian cardiac metabolism, CardioNet11, based on quantified fold changes of metabolite concentrations between control and experimental groups, as well as the metabolic profile of the circulation. CardioNet in silico flux balance analysis of db/db cardiac metabolism further confirmed that these mice had a markedly different metabolic profile than that of controls (Fig. 2d). Computational simulations of db/db mice revealed enhanced anaerobic glycolysis and glucose flux through the pentose phosphate pathway, whereas glucose oxidation in the Krebs cycle was decreased (Fig. 2e). The shift in extracellular nutrient supplies enhanced lipid oxidation (Fig. 2e), which increased generation of reactive oxygen species (ROS) (Fig. 2e) and markedly elevated oxygen consumption (Fig. 2g). Moreover, in vivo metabolic flux assessment using 13C hyperpolarized magnetic resonance spectroscopy (Fig. 2f) showed that in db/db mice, cardiac pyruvate-dehydrogenase flux was 95% lower in vivo, reflecting a reduction in cardiac glucose oxidation (Fig. 2i).
Remarkably, treatment with AZD1656 ameliorated cardiac metabolic dysfunction in db/db hearts. PCA analysis showed that the metabolomic profile after drug treatment was indistinguishable between AZD1656-treated db/db hearts and controls (Fig. 2a and Extended Data Fig. 2). The 1H NMR metabolomic profile in the heart was comparable between drug-treated db/db and control animals (Fig. 2c), with the exception of persistent supra-normal elevation of succinate, an increase in NADH and a reduction in glutamine concentration. In silico modeling also showed improvements in cardiac metabolic flux reactions (Fig. 2d), including markedly improved lipid oxidation, ROS production and hexosamine pathway utilization (Fig. 2e). Our CardioNet flux analysis predicts that AZD1656 treatment results in a marked reduction in anaerobic glycolysis leading to a reduction in lactate release compared with db/db hearts. One of the most notable adaptations following drug treatment is the striking twofold reduction in CardioNet-calculated oxygen consumption (Fig. 2g) returning it to near-control rate. Furthermore, 13C hyperpolarized magnetic resonance spectroscopy demonstrated that in vivo cardiac metabolic flux was improved (Fig. 2i), showing increased conversion of [1-13C]pyruvate to [1-13C]bicarbonate, indicative of increased PDH flux. Of note, in healthy hearts, acute AZD1656 treatment alone did not exert any metabolic or functional effects (Extended Data Fig. 2). There was no difference in lipoprotein lipase (LPL) activity between control and untreated db/db hearts (control = 0.84 ± 0.13; db/db = 0.66 ± 0.09 mUnits ml−1 mg−1), but AZD1656 treatment significantly increased LPL activity in db/db hearts (db/db = 0.66 ± 0.09 versus AZD1656 = 1.34 ± 0.13 mUnits ml−1 mg−1, P < 0.05). Furthermore, AZD1656 treatment had no impact on mitochondrial cristae density (Extended Data Fig. 2).
To further study the impact of AZD1656 on cardiac lipid metabolism, we comprehensively quantified the distribution and abundance of 13 major lipid classes in heart tissues by non-targeted MS/MS lipidomic analysis (Fig. 3 and Supplementary Tables 3 and 4). We analyzed lipidomics datasets by supervised multivariate classification using partial least square-discriminant analysis (PLS-DA).

a, Partial-least-square-discriminant analysis (PLS-DA) visualization of lipid abundances. Samples (n = 5 per group) are grouped according to biological replicates, and dimensions (Dim) 1 and 2 together captured 81.35% of variability. b, Contribution of significantly altered phospholipid (PL) classes across biological samples. MG, monoacylglycerols; PC, phosphatidylcholine (n = 5 per group). c, PCA of PC acyl-chain profiles. Total carbon lengths were compared across biological samples. Dimensions 1 and 2 captured 84.4% of variability. PC(40) and PC(26) were the strongest contributors to PC alterations in treatments. d,e, Average total chain length (d) and degree of saturation (e) varied between experimental groups (n = 5 per group). *P < 0.05 control versus db/db; #P < 0.05 control versus AZD1656 by one-way ANOVA. List of exact P values for comparison of carbon lengths and degree of saturation provided in Supplementary Table 3 (lipidomics comparison P values). n represents biological replicates (individual animals). Whisker bar plots were plotted with RStudio using the ggplot2 package. The minimum and maximum observation that is not an outlier are indicated by lines connected to the box. The first quartile (25th percentile of the data) and third quartile (75th percentile of the data) are represented by the lower end and the upper end of the box, respectively. The median (50th percentile of the data) is indicated by a line within the box. n represents biological replicates (individual animals). Source data provided in Supplementary Table 4.
Among treatment-dependent separation, 81% of the total variance was captured in the first two dimensions (Fig. 3a). Phosphatidylcholine (PC) and monoacyl-glycerols (MG) showed significant alterations across all three experimental groups (Fig. 3b). In the fatty acyl (FA) pools of PC, the most influential summed acyl chain lengths were identified by PCA (Fig. 3c), which showed that FA26, FA38 and FA40 were dominant. Additionally, FA34, FA35 and FA36 contributed to PC variability. Thus, we analyzed the PC fatty-acyl-chain composition (carbon length) and the degree of unsaturation (number of double bonds) across all experimental groups. We found that db/db mice had decreased FA26 and FA40 abundance, whereas FA38 was significantly upregulated compared with the level in control animals (Fig. 3d). By contrast, treatment with AZD1656 partially normalized FA profiles, reducing the overall contribution of FA38 and increasing the abundance of FA26 and FA40. Both the db/db genotype and AZD1656 treatment were associated with increased unsaturation of PC species from mono-unsaturated toward polyunsaturated fatty acyls with four to five double bonds. In untreated db/db mice, highly unsaturated PC acyl chains were generally longer (six or more double bonds) (Fig. 3d). By contrast, treatment with AZD1656 caused less saturated and shorter acyl chains. The di-unsaturated to mono-unsaturated ratio was typically lower in hearts from untreated db/db mice (0.07) than in those from AZD1656-treated animals (0.2) and controls (0.129) (Fig. 3e).
AZD1656 treatment effects are not mediated by changes in systemic metabolism
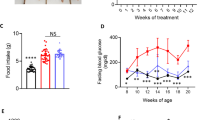
To examine whether the AZD1656-treatment-driven cardiometabolic improvements were a result of systemic metabolic alterations in db/db animals (Fig. 4a), extensive analyses of blood, liver, adipose tissue and skeletal muscle were performed. AZD1656 treatment did not improve obesity (body weight, Fig. 4a,b), hyperglycemia (Fig. 4c and Extended Data Fig. 3), hyperinsulinemia (Fig. 4d) or insulin resistance (adiponectin, Extended Data Fig. 3). Although AZD1656 treatment significantly reduced the concentration of free fatty acids in circulation (Fig. 4e), the remnant circulating metabolite profile did not improve (Fig. 4f,g and Extended Data Fig. 3). AZD1656 treatment did not improve the diabetic liver phenotype (Fig. 4h, alanine transferase, alkaline phosphatase, Extended Data Fig. 3) or metabolism (Liver 1H NMRs metabolomics Fig. 4i,j and Extended Data Fig. 3). AZD1656 treatment led to a minute increase in lean muscle mass compared with the db/db genotype (Fig. 4k) but had limited effect on the skeletal muscle metabolome (Fig. 4l,m and Extended Data Fig. 3). Furthermore, treatment with AZD1656 did not reduce whole body fat mass (Extended Data Fig. 4) or alter the lipid composition of the adipose tissue (Extended Data Fig. 4).

a, Representative images of control, db/db and AZD-treated db/db mice from the study. b, Body weight (control n = 23, db/db n = 17, AZD n = 20) multiple group comparison by one-way ANOVA (control versus db/db P = 0.0001, control versus AZD P = 0.0001, db/db versus AZD P = 0.623). c, Fasting plasma glucose (n = 9 per group). Control versus db/db P = 0.0001, control versus AZD P = 0.0001, db/db versus AZD P = 0.47. d, Plasma insulin (control n = 10, db/db n = 11, AZD n = 6; control versus db/db P = 0.0001, control versus AZD P = 0.0010; db/db versus AZD P = 0.8874). e–g, Circulating lipid metabolism constituents (free fatty acids (control n = 11, db/db n = 9, AZD n = 5) (e), cholesterol (control n = 7, db/db n = 10, AZD n = 5) (f), triacylglycerol (control n = 7, db/db n = 11, AZD n = 9) (g)). Multiple group comparison by one-way ANOVA (*P < 0.05, **P < 0.01, ***P < 0.001). Liver phenotype profiling. h, Liver weight (control n = 12, db/db n = 11, AZD n = 7). Multiple group comparison by one way ANOVA. i, Fold change db/db versus control liver 1H NMR metabolomic spectroscopy profiling. Two group comparison by two-tailed t-test. j, Fold change AZD versus control liver 1H NMR metabolomic spectroscopy profiling (control n = 6, db/db n = 7, AZD n = 7). Skeletal muscle phenotype (control n = 6, db/db n = 6, AZD n = 5), two group comparison by two-tailed t-test P < 0.05. k, Lean muscle mass by body composition analysis (n = 5 per group; control versus db/db P = 0.0001, control versus AZD P = 0.0001, db/db versus AZD P = 0.02). Multiple group comparison by one-way ANOVA. l, Fold change db/db versus control skeletal muscle 1H NMR metabolomic spectroscopy profiling. m, Fold change AZD versus control skeletal muscle 1H NMR metabolomic spectroscopy profiling (n = 5 per group). Two group comparison by two-tailed t-test. *P < 0.05, **P < 0.01, ***P < 0.001 multiple group comparison by one-way ANOVA, two-tail comparison by Student’s t-test. Of note, parameters measured over the course of the six-year study in multiple cohorts thus sample size varied. n represents biological replicates (individual animals). Data are presented as mean ± s.e.m. Metabolite fold change versus control (control = 1) with propagated error of mean.
AZD1656 treatment reduces T cell-mediated cardiac inflammation and fibrosis in dbCM
Untreated db/db mice were characterized as having low-grade systemic inflammation, observed from alterations to the circulating cytokine profile (Fig. 5a and Extended Data Fig. 5). Levels of IGF BP-1, LDL-R, PD-ECGF/thymidine phosphorylase, leptin and osteoprotegerin were increased.

a, Circulating XXL cytokine plasma panel heatmap summary (control n = 10, db/db n = 12, AZD n = 10). Multiple group comparison by one-way ANOVA. b, Representative FACS contour plots. c, Relative frequency of myocardial CD4+ T cells quantified by FACS (control n = 12, db/db n = 12, AZD n = 7). Multiple group comparison by one-way ANOVA (control versus db/db P = 0.0001, control versus AZD P = 0.163, db/db versus AZD P = 0.002). d, Masson trichrome stained representative cardiac cross sections. e, Quantification of Masson trichrome fibrosis staining intensity (control n = 5, db/db n = 4, AZD n = 4). Multiple group comparison by one-way ANOVA (control versus db/db P = 0.0001, control versus AZD P = 0.128, db/db versus AZD P = 0.0002). f, Relative frequency of myocardial Treg cells (CD4+CD25+FoxP3+) (n = 9 per group). Data are normalized to control (set to 1), and expressed as fold change. n, biological replicates (individual animals). Multiple group comparison by one-way ANOVA (control versus db/db P = 0.957, control versus AZD P = 0.02, db/db vereesus AZD P = 0.03). g, B cells, dendritic cells, neutrophils and macrophages (controls n = 8, db/db n = 8, AZD n = 5). Multiple group comparison by one-way ANOVA (B cells: control versus db/db P = 0.83, control versus AZD = 0.0082, db/db versus AZD P = 0.03; dendritic cells: control versus db/db P = 0.23, control versus AZD P = 0.70, db/db versus AZD P = 0.87; neutrophils: control versus db/db P = 0.89, control versus AZD P = 0.99, db/db versus AZD P = 0.99; macrophages: control versus db/db P = 0.77, control versus AZD P = 0.77, db/db versus AZD P = 0.44). *P < 0.05, **P < 0.01, ***P < 0.001. Multiple group comparison by ANOVA. n represents biological replicates (individual animals). Data are presented as mean ± s.e.m.
A significant decrease was observed in levels of circulating anti-inflammatory mediators interleukin-10 (IL-10) and IL-4, the growth repair factors EGF, PD-ECGF, WISP-1 and Serpin F, the matrix-remodeling enzymes MMP-3 and WISP-1, the chemokines CX3CL1, CXCL16 and CD40 and the immune cell regulators M-CSF, complement factor D, IL-12p40 and the adipokine and metabolic signal resistin (Fig. 5a and Extended Data Fig. 5).
Unlike resolving acute cardiac inflammation, in which numbers of infiltrating Treg cells are increased12, cardiometabolic and functional derangements in db/db mice were concomitant with unregulated T cell-mediated cardiac autoimmunity (Fig. 5b,c) and fibrosis (Fig. 5d,e) without Treg increase (CD4+CD25+FoxP3+, Fig. 5f). In terms of the infiltration of other inflammatory cell types, there was no evidence of increased presence of B cells (B220), CD11c+ dentritic cells, neutrophils (Ly6G+) or macrophages (F4/80+) (Fig. 5g). In pre-diabetic, high-fat–fed C57BL/6 mice displaying obesity, insulin resistance and early cardiac metabolic derangement preceding overt T2D, no myocardial infiltration by T cells or other inflammatory cells (macrophages, dendritic cells, neutrophils, B Cells) was observed (Supplementary Table 2 and Extended Data Fig. 6).
AZD1656 markedly attenuated cardiac inflammation in db/db mice. Treated hearts exhibited reduced interstitial fibrosis (Fig. 5d,e) and diminished T cell infiltration (Fig. 5c), alongside a pronounced increase in Foxp3⁺ regulatory T cell recruitment (Fig. 5f), evidence of enhanced Treg mobilization and a more favorable myocardial immune milieu13. AZD1656 treatment did not induce global CD4⁺ T cell expansion, as evidenced by unchanged splenic CD3⁺ T cell abundance, consistent with the absence of systemic lymphoproliferative effects (Extended Data Fig. 5). Although AZD1656 did not alter dendritic cell, neutrophil or macrophage trafficking (Fig. 5g), it significantly decreased B cell accumulation in the myocardium (Fig. 5g).
In terms of soluble cytokine profile, there was a marked change after AZD1656 treatment (Fig. 5a and Extended Data Fig. 5). Pharmacological intervention normalized plasma concentrations of IL-4, BAFF, CCL6, FGF-21, GDF-15, coagulation factor III, CXCL16, FGF acidic, Gas6, IL-12p40, M-CSF, CXC3CL1 and MMP3; decreased concentrations of IL-10, serpin F1, resistin, CD40, EGF and complement factor D; and increased concentrations of CCL20, CCL22, CXCL1, MMP-9, Serpin E1, P-selectin, myeloperoxidase, PDGF-BB, IFGBP-1, LDL-R, PD-ECGF, leptin and osteoprogenerin (Fig. 5a and Extended Data Fig. 5).
Treatment with AZD1656 normalizes the expression of genes regulating key intracellular pathways
Development of dbCM in db/db mice was characterized by significant alterations in cardiac gene expression (1,379 differentially expressed genes (DEGs) db/db versus control, 503 genes db/db versus AZD1656, 1,480 genes control versus AZD1656, Fig. 6a). G profiler gene enrichment analysis (Extended Data Fig. 7) shows that dbCM caused extensive alterations in the expression of genes governing key aspects of cardiac function: molecular pathways, biological processes, cellular components and protein complexes. Analysis of the 30 most upregulated genes in db/db versus control hearts shows that 19 of the top 30 overexpressed genes are proinflammatory mediators (Fig. 6b). Nlrp3 gene inflammasome levels were markedly elevated, showing a 2.5-fold increase in db/db animals compared with controls (RNA-sequencing data, Array Express accession E-MTAB-13849). This upregulation suggests that assembly of the inflammasome complex is enhanced, which likely drives increased caspase-1 activation and subsequent IL-1β and IL-18 release, promoting a pro-inflammatory myocardial environment.

a, Visualization of DEGs using hexbin plot summaries. The y axis vertical cut-off is log2(1.5) ± 0.585, and the x axis horizontal cut off is –log10(0.05) ≈ 1.301. Differential expression analysis was performed using GenXpro with a standard RNA-seq pipeline. Expression values were compared using statistical tests generating P values, followed by Benjamini–Hochberg false discovery rate (FDR) correction. b, The top 30 most upregulated DEG in db/db versus control, AZD versus db/db and AZD versus control groups. Red bars are proinflammatory regulators; blue bars are anti-inflammatory regulators. c, Venn diagram of overlapping homologous DEGs among the three data sets (control, db/db and AZD). The threshold was set to be FC > 1.5 (or <1/1.5) and P < 0.05. Sample genes (increased and decreased) from each of the subsets are given: abs_diff_log2(FC) = abs(log2(foldchange_AZD_vs_dbdb) log2(foldchange_dbdb_vs_CTRL). d–f, Heatmap plots of log(P) of DEGs resulting from hypergeometric test and Benjamini–Hochberg P correction to identify all ontology and pathway terms that contain a greater number of genes in common with an input list than expected by chance using Qiagen IPA software81: upstream regulators (d), biofunctions (e) and canonical pathways (f). This is expressed as log (P) in the heatmaps. All RNA-seq data presented in this figure are based on n = 6 hearts per group. n represents biological replicates (individual animals).
Further Venn diagram analysis (Fig. 6c) identified overlapping homologous DEGs in db/db, control and AZD1656-treated db/db hearts. The Venn diagram analysis of gene homology between the groups highlights the upregulation of inflammation-mediating genes (Frp2, Ifitm6, Camp, Ltf, Ngp, Saa3), including leukocyte migration mediator Selp in db/db animals versus lean controls (Fig. 6c). Pathway analysis further identified genetic hallmarks of dbCM and has shown the upregulation of canonical pathways, biofunction regulators and upstream regulators (Fig. 6d–f) of inflammation, apoptosis, T cell migration and infiltration, inflammatory signaling, metabolism of fats and carbohydrates (Extended Data Figs. 8–10).
Treatment with AZD1656 reversed pathologically elevated gene expression in the hearts of db/db T2D mice compared with their untreated counterparts (503 genes, AZD1656 versus db/db, Fig. 6a,b). Of the 30 most expressed DEGs in AZD1656 versus db/db hearts and AZD1656 versus control hearts, only 8 (AZD1656 versus db/db) and 6 (AZD1656 versus control) are proinflammatory, respectively. Of note, AZD1656 treatment also increased expression of Sfrp5 (frizzled-related sequence protein 5), which mediates protection against inflammation and apoptosis through the inhibition of the Wnt5a-JNK pathway14. AZD1656 treatment reduced the number of shared, upregulated inflammation mediators, compared with control (Fig. 6c). Pathway analysis reveals that drug treatment resulted in the opposite pattern of myocardial pathway gene expression: for instance, the allograft rejection pathway that showed the biggest increase in log fold change in expression in db/db versus control is the least expressed in db/db versus AZD1656 (Extended Data Fig. 7). These AZD1656-treatment-driven changes in gene expression also included a reduction in the expression of genes in ROS-production pathways, metabolism of lipids and multiple inflammation mechanisms and signals (Fig. 6d–f).
In addition, treatment with AZD1656 targeted and improved a series of pathways involved in biological functions (Fig. 6d–f). These included necrosis, cell movement, apoptosis, leukocyte migration and series of signaling pathways: sirtuin, Nrf2, hepatic signaling and leukocyte migration (Fig. 6d–f and Extended Data Figs. 8–10). Furthermore, AZD1656 treatment impacted a range of upstream regulators including improvement of proinflammatory signals (IFN-γ, IL-4, PPARα) (Extended Data Fig. 7).
Crucially, the mechanistic impact of the AZD1656 treatment on db/db hearts was shown by complete amelioration of the elevated Nlrp3 expression (RNA-sequencing data, Array Express accession E-MTAB-13849), as well as dysregulated leukocyte extravasation signaling governing T cell infiltration of the myocardium (change of signaling pathway intermediate components from red to green, detailed KEGG pathway in Extended Data Fig. 8) as well as of two key metabolic regulator pathways: HIF1α (Extended Data Fig. 9) and NRF2-mediated oxidative stress response (Extended Data Fig. 10).
Discussion
Given the reported immunomodulatory effects of AZD1656 (refs. 4,5), we investigated whether treatment could reduce cardiac inflammation and thereby attenuate cardiac remodeling in dbCM. Although systemic T cell-mediated inflammation is well-characterized in T2D and other chronic metabolic stress states, cardiac inflammation in T2D has not been thoroughly defined. Twenty-week-old db/db mice displayed hallmark features of human dbCM, including obesity, hyperglycemia and impaired diastolic function (in vivo and ex vivo). Diabetic hearts showed altered T cell dynamics with systemic proinflammatory cytokine changes, increased fibrosis and disrupted cardiac metabolism. Remarkably, six weeks of pharmacological treatment with AZD1656 attenuated the functional and metabolic remodeling observed in the untreated diabetic heart, resulting in improved functional recovery and reduced damage following ischemia.
One of the key challenges in dbCM is the loss of metabolic flexibility. However, AZD1656 drug treatment achieved significant improvements in the cardiac metabolomic profile and flux as observed from both in vivo and in silico metabolic assessment. These changes suggest that substrate utilization flexibility is improved. Furthermore, AZD1656 pharmacological intervention in db/db mice resulted in a 68% reduction in cardiac oxygen consumption. Increased myocardial oxygen consumption and O2 wasting due to uncoupled mitochondrial function have been shown to reduce cardiac efficiency in T2D15,16. Thus, the reduction in oxygen consumption after AZD1656 treatment is critical for the improvement in cardiac function and for the reduction in ischemia-reperfusion damage.
dbCM in db/db mice was also characterized by differences in the fatty acyl chain composition of glycerolipids, such as phosphatidylcholine as well as monoacyl-glycerols. AZD1656 treatment significantly improved the cardiac phospholipid composition, suggestive of improved lipid metabolism as well as cellular membrane and mitochondrial integrity17,18. Our studies demonstrated that pharmacologic interventions that attenuate obesity-induced alterations in phospholipid composition and homeostasis positively impact cardiac metabolism and function. Bulk cardiac tissue RNA-sequencing analysis showed that AZD1656 treatment significantly improved the gene expression profile, including genes governing key metabolic and inflammatory response pathways (that is, NLRP3, HIF1α, PGC1α, apoptosis, necrosis, leukocyte extravasation, fatty acid metabolism and oxidative phosphorylation).
Experimental AZD1656 treatment outcomes could have been achieved through two potential mechanisms of action: improved glycemic control (original drug indication) and/or modulation of the immune response5. Because no differences in glycemic control were detected between groups, it is unlikely that the differences in outcomes were a result of the glucose-lowering effect of AZD1656.
T cell inflammation has been previously linked with the development of heart failure in both animal disease models and humans19. T cell costimulatory blockade, using the rheumatoid arthritis drug abatacept, significantly reduced the severity of cardiac dysfunction in heart failure. This therapeutic effect occurred through the inhibition of activation and cardiac infiltration of T cells and macrophages, leading to reduced cardiomyocyte death19. The immunophenotyping data here suggest that AZD1656 treatment induced an immunomodulatory effect in db/db mice through improved Treg infiltration, a reduced number of proinflammatory CD4+ T cells and the resultant reduction in fibrosis. This is consistent with rebalancing of the immune response observed in the ARCADIA trial in COVID19 T2D patients5. Treg migration is metabolically demanding and fueled by glucokinase-regulated glycolysis4. This has been shown in human carriers, in whom a loss-of-function gene polymorphism in GCK regulatory protein (GKRP) causes Treg cells, rather than conventional T cells, to display altered motility ex vivo4. Although glycolysis fuels the migration of other T cell subtypes, including proinflammatory T effector cells, their migration is hexokinase I, rather than glucokinase IV dependent20. Thus, the GCK activator AZD1656 selectively enhanced Treg migration and resultant myocardial infiltration.
Increased infiltration of Treg cells into the db/db myocardium exerts multi-faceted cardioprotective effects by simultaneously modulating the immune landscape, metabolic efficiency and tissue remodeling. Treg cells secrete key anti-inflammatory cytokines, including IL-10 and TGF-β, which suppress proinflammatory macrophage activation and effector T cell infiltration, thereby attenuating the chronic low-grade inflammation characteristic of pathologies such as heart failure21,22. Treg-secreted anti-inflammatory cytokines limit NLRP3 inflammasome assembly, which could account for normalized Nlrp3 gene expression in AZD16556-treated db/db hearts23. The Treg-mediated immunosuppressive milieu directly translates into a reduction in maladaptive fibrotic remodeling through inhibition of cardiac fibroblast activation and downregulation of TGF-β-Smad signaling pathways24. Furthermore, this immunomodulation has the potential to restore insulin-signaling pathways, thereby improving cellular glucose uptake and mitochondrial function25. In organs with high metabolic demand, such as the heart, enhanced Treg presence also promotes expression of key metabolic regulators, such as PPARγ, further improving overall metabolic homeostasis26. Beyond immunoregulation, Treg cells influence vascular integrity by promoting endothelial quiescence and angiogenic support, improving myocardial perfusion in the context of microvascular dysfunction21. In models of cardiac ischemia-reperfusion injury, Treg-derived amphiregulin and IL-33 signaling not only promote tissue regeneration, but also limit infarct expansion27. This could account for the observed reduction in infarct size in AZD1656-treated db/db hearts. Treg cells further support cardiomyocyte survival by enhancing IL-10-mediated activation of STAT3 signaling28. Collectively, these findings position Treg cells as potent mediators of immunometabolic resilience in dbCM.
In addition to the improved T cell inflammatory profile, AZD1656 also caused a significant decrease in B cell infiltration in db/db animals versus their untreated counterparts. Increased B cell infiltration has been shown to increased antibody (IgG3) production that aids the recruitment and activation of a variety of innate and structural cell populations: neutrophils, fibroblasts, macrophages and T cells29.
Given that AZD1656 boosts Treg infiltration and suppresses CD4+ T cells, the myocardial environment shifts from proinflammatory to immunoregulatory. Treg cells secrete IL-10 and TGF-β, which downregulate key chemokines and endothelial adhesion molecules (for example, CXCL13, VCAM-1) that ordinarily recruit B cells to inflamed tissue. At the same time, the reduction in CD4+ deprives B cells of essential survival and activation signals, such as IL-21 and CD40L, impairing their retention and viability in cardiac tissue. Finally, by suppressing NLRP3 inflammasome activation and its downstream cytokines IL-1β and IL-18, AZD1656 would further diminish the pro-B cell milieu. Consistent with findings that BAFF levels rise after B cell depletion, and given the reduction of cardiac B cells observed after AZD1656 treatment, it is reasonable to expect a corresponding decrease in peripheral B cell numbers following treatment30. Together, these mechanisms lead to lower myocardial B cell numbers while restoring a balanced, protective immune state in db/db hearts. Given that B cells have been linked to the development of inflammation and maladaptive cardiac remodeling in heart failure29, immunomodulatory therapeutic strategies that target B cells and their products could be an effective treatment tool. Our study focused on heart-resident CD4+ T cells, so we did not assess peripheral blood or lymphoid tissues. Although sequencing the systemic TCR repertoire could provide complementary insights, it was beyond the scope of this cardiac-centric investigation.
AZD1656 delivers a finely tuned suppression of diabetogenic inflammation and remodeling through plasma cytokine rebalancing. Pharmacological intervention led to a coordinated increase in a suite of cytokines implicated in immune regulation, vascular remodeling and metabolic adaptation, collectively suggesting reversal of immune suppression and reactivation of cardiac repair pathways. Key chemokines, such as CCL20, CCL22 and CXCL1, promote the recruitment of Treg cells, fostering a reparative immune environment and reducing chronic inflammation31,32. CCL20 and CCL22 are ligands for CCR6 and CCR4, respectively, both expressed on Treg cells and implicated in their trafficking to inflamed tissue. Upregulation of BAFF and IL-12p40 supports maturation of tolerogenic dendritic cells and T cell differentiation, consistent with immune modulation rather than proinflammatory activation33. Alteration in circulating IL-12 levels is indicative of less T cell effector function and DC activation34. Increased FGF-21 and GDF-15 are markers of systemic metabolic rescue and mitochondrial stress tolerance, both shown to be cardioprotective under diabetic conditions35,36. Elevated MMP-9 and Serpin E1 levels reflect dynamic matrix turnover and remodeling, whereas enhanced levels of PD-ECGF, FGF acidic and Gas6 suggest restoration of angiogenic signaling and endothelial repair37,38. The AZD1656-triggered induction of P-selectin, CXCL16 and myeloperoxidase likely represents controlled inflammatory engagement, contributing to immune surveillance and clearance. Notably, levels of metabolic and vascular stress mediators, such as leptin, resistin, LDL-R and osteoprotegerin (OPG), were also elevated, which could reflect attempts at metabolic compensation and vascular remodeling. Increased IGF BP-1 might indicate improved IGF signaling balance or a response to restored metabolic demand39.
Metabolomic profiles of blood, liver, adipose tissue and skeletal muscle were largely unaffected, thus any beneficial effects observed in hearts of AZD1656-treated db/db animals were not driven by marked improvements in the metabolism of systemic peripheral tissue (liver, muscle). We have, however, observed a significant reduction in the circulating fatty acid concentration with an accompanying increase in body fat content after AZD1656 treatment. This could have contributed to the improvement of myocardial metabolism and inflammation. Reduced levels of fatty acids in circulation would have affected myocardial metabolism, specifically PDH flux through fatty-acid regulation of PDK4 (refs. 40,41) and reduced systemic proinflammatory T cell proliferation42.
The observed reduction in circulating free fatty acids could reflect the combined effects of AZD1656 on β-cell and hepatic glucokinase activity, leading to systemic reorganization of lipid metabolism43,44,45. In models with preserved β-cell function, glucokinase activation enhances glucose-stimulated insulin secretion and secondarily increases LPL activity through hyperinsulinemia46. However, db/db mice are already profoundly hyperinsulinemic and have limited β-cell capacity; therefore, the increase in cardiac LPL activity and altered lipid milieu following AZD1656 treatment most likely reflects hepatic substrate-driven signaling, rather than additional insulin release. Consistent with this interpretation, AZD1656 did not exacerbate hypertriglyceridemia, plasma triglycerides remained elevated but unchanged, while levels of circulating free fatty acids declined significantly, indicative of predominantly hepatic, insulin-independent effects. Hepatic glucokinase activation might enhance glycolytic flux and lipogenesis, maintaining output of triglycerides in very-low-density lipoproteins output without further elevating plasma triglyceride levels. The resulting lipid redistribution could stabilize myocardial LPL47. However, despite increased cardiac LPL activity, fatty-acid utilization was not augmented; rather, AZD1656 normalized PDH flux and restored a more balanced cardiac metabolic profile, suggesting improved glucose oxidation and metabolic flexibility through hepatic-cardiac metabolic cross-talk. Given negligible cardiac GCK expression and no direct metabolic effect in perfused hearts, enhanced myocardial LPL activity likely represents a secondary adaptive response, complementing the drug’s immunomodulatory actions. Supporting this, individuals carrying the GCKR P446L loss-of-function variant, which increases hepatic glucokinase activity, exhibit similar alterations in systemic lipid metabolism and enhanced Treg motility4.
If AZD1656 benefits truly depend on modulating Treg cells, why do we observe its anti-inflammatory effects exclusively in the heart? Ultimately, why T cells preferentially home to the diabetic myocardium despite systemic dysregulated metabolism remains an open, almost philosophical, question that transcends current mechanistic understanding. One of the key characteristics of dbCM is an increase in fibroblasts which have been shown to act as antigen presenting cells48 driving myocardial T cell infiltration and inflammation49.
In db/db mice, there are potentially four converging factors that render the myocardium uniquely immunogenic: (1) chronic hyperglycemia and lipotoxicity selectively upregulate CXCL10, CCL2, VCAM-1 and ICAM-1 in cardiomyocytes and the coronary endothelium, establishing ‘docking sites’ for lymphocytes50; (2) metabolic stress generates AGE- and oxidized-lipid neoantigens that are robustly presented on myocardial MHC-I/II, locally activating CD4⁺ T cell responses; (3) heightened TLR2 and TLR4 signaling in resident macrophages and cardiomyocytes fuels IL-1β, IL-6 and tumor necrosis factor release, amplifying antigen presentation and chemotaxis51; and (4) by contrast, skeletal muscle, liver and adipose tissue maintain lower basal levels of cardiac self-antigens and inducible adhesion molecules, failing to create the chemokine gradients or MHC upregulation needed for substantial T-cell homing. Together, these factors create a ‘perfect storm’ in diabetic hearts: enhanced antigen presentation, chemokine expression and endothelial adhesion molecule display converge to selectively recruit and retain T cells in the myocardium, driving inflammation and the dbCM phenotype. Together, these cardiac-restricted antigenic and adhesion cues explain the selective myocardial infiltration and thus the heart-specific anti-inflammatory effects of AZD1656-targeted Treg cells in diabetic cardiomyopathy. Future studies using CD4⁺ T-cell-deficient mice or antibody-mediated depletion would be valuable in delineating the mechanistic contribution of CD4⁺ T cells to dbCM. These studies are currently under consideration for follow-up work.
Despite the promising results presented in this work, questions remain regarding whether the changes in cardiac metabolism or immune response are the therapeutically exploitable culprits in triggering the dbCM pathology. Specifically, what is the temporal sequence of events: is mitochondrial dysfunction an early driver, or does inflammation occur first? To help to understand the sequence of events driving diabetic cardiomyopathy, we established a spectrum of cardiac dysfunction by inducing obesity and insulin resistance in mice via a high-fat diet, using age-, sex-, and strain-matched controls to db/db mice52. In this prediabetic model, we observed clear cardiac metabolic derangements before any evidence of myocardial immune cell infiltration. These findings indicate that systemic metabolic disturbances (hyperglycemia, glucose intolerance, obesity) and intrinsic mitochondrial dysfunction are primary events that precede, and likely trigger, subsequent cardiac inflammation in established T2D, such as that seen in db/db mice. This would be in agreement with a recently proposed model in which mitochondrial dysfunction itself is a potent pro-inflammatory trigger53. When oxidative phosphorylation falters, excess mitochondrial ROS and oxidized mtDNA leak into the cytosol, where they directly engage and activate the NLRP3 inflammasome. This drives caspase-1-dependent maturation of IL-1β and IL-18, which in turn amplify NF-κB signaling and downstream production of TNF and IL-6. Simultaneously, released mitochondrial lipids, such as cardiolipin, act as danger-associated molecular patterns, further escalating cytokine release53. Thus, broken mitochondria are not silent bystanders, but the very ‘damage sensors’ that convert metabolic stress into chronic, low-grade inflammation53.
A growing body of evidence demonstrates that pharmacological augmentation of Treg cells confers potent anti-inflammatory and plaque-stabilizing effects in models of cardiovascular disease. Agents as diverse as rapamycin and mycophenolate mofetil expand CD4⁺FoxP3⁺ Treg cells while concurrently depleting proinflammatory effector T cells and antigen-presenting cells, thereby reducing vascular inflammation and matrix metalloproteinase activity54. Statins, aspirin and PPAR-γ agonists (pioglitazone) similarly restore the balance between T helper 17 cells and Treg cells, elevate IL-10 and TGF-β levels and promote endothelial homeostasis. Even microbiome-derived metabolites (indole propionic acid) and micronutrients (vitamin D₃, amygdalin) have been shown to increase Treg frequencies in hypertensive and atherosclerotic models54. Collectively, these interventions validate Treg modulation as a unifying strategy to attenuate chronic vascular inflammation and improve metabolic resilience in cardiometabolic disease. However, the ARCADIA trial outcome has introduced immunometabolic therapeutic concept: pharmacological targeting of the endogenous immune cells to turn them into the therapeutic agents in the body5. Previously, this drug development approach, whether used in exogenously treated or engineered cells, has failed5,55,56,57.
Our study results therefore contribute to the growing field of cardio-immunology because they suggest that AZD1656 could be used as an immunomodulatory drug to attenuate cardiometabolic dysfunction in T2D as well as the whole host of other sterile-inflammation pathologies.
Methods
Animals
Commercially available 8-week-old T2D mice (db/db mouse, The Jackson Laboratory homozygous BKS.Cg-Dock7m +/+ Leprdb/J, male) were purchased, as were lean littermate controls (heterozygous Dock7m +/+ Leprdb). Animals were kept under pathogen-free conditions, 12 h light–dark cycle, controlled temperature (20–22 °C) and fed chow and water ad libitum. Circulating glucose (tail sample, Accu-Check, Roche) and body weight were monitored weekly. This investigation conformed to UK Home Office Guidance on the Operation of the Animals (Scientific Procedures) Act, 1986.
AZD1656 drug treatment protocol
AZD1656 (AZD) is a selective glucokinase activator. No significant clinical effects nor specific toxicology risks, other than hypoglycemia, were noted in clinical trials58. The db/db and lean controls were divided into 3 groups at 13 weeks of age: group 1, lean controls, Envigo diets control diet (2019 Teklad Global 19% Protein Rodent Diet, irradiated, Teklad Custom Diets, Envigo); group 2, db/db, Envigo diets control diet; group 3, db/db, AZD1656 diet (30 mg kg−1 body weight per day, Envigo Specialist Diets; drug dosing based on refs. 59,60) (Fig. 1a). Diets were fully matched in terms of nutritional standardization and food intake was not affected by dietary drug incorporation. Animals (study total: group 1, controls n = 64; group 2, db/db n = 63; group 3, AZD1656 n = 57) were kept on the feeding protocol from 14 until 20 weeks of age when the study reached endpoint. For the obesity-induction protocol, C57/BL6 8-week-old male mice (n = 18, Charles River) were fed a high-fat diet for 12 weeks ad libitum (HFD, cat. no. TD.200185; Envigo, 60.3% fat, 21.4% carbohydrate, and 18.3% protein; percentage of energy in kcal)52. In vivo physiology and heart function, as well as the ex vivo cardiac metabolome, were assessed as has been described52.
Body composition analysis
Body composition analysis was carried out by non-invasive magnetic resonance relaxometry using an EchoMRI Body Composition Analyser E26-348-MT. The accumulation factor was for extra-high precision (3×) resulting in a scan time of approximately 2.5 min.
In vivo assessment of left ventricular systolic and diastolic function
CINE magnetic resonance imaging
For the assessment of systolic function, mice were imaged in a 7T MRI instrument (Agilent) using CINE magnetic resonance imaging, as previously described61. Eight to 10 short-axis slices (slice thickness, 1.0 mm; matrix size, 256 × 256; field of view, 25.6 × 25.6 mm; echo time/repetition time, 0.3/4.6 ms; flip angle, 30°; and number of averages, 4) were acquired with a gradient echo, fast low-angle shot sequence62. Left ventricular volumes were derived using the freehand draw function in ImageJ (National Institutes of Health). For each heart, left ventricular mass, ejection fraction, stroke volume and cardiac output were calculated.
Echocardiography
M-mode and Doppler echocardiography was performed in 20-week lean controls, db/db and AZD1656-treated db/db mice at the end of the drug-treatment protocol as previously described52,63. Anesthesia was induced with 4% isoflurane and maintained at 1.5–2% for the duration of the procedure. Echocardiography images were recorded using a Vevo-3100 imaging system with a 40-MHz linear probe (VisualSonics). Morphological measurements were taken in the parasternal short axis view at the level of the papillary muscles, and the ejection fraction was calculated from M-mode images. Diastolic transmitral left ventricle (LV) inflow images were acquired from apical four-chamber views using pulsed-wave doppler to calculate early (E) and late (atrial, A) peak filling blood flow velocities and E-wave deceleration time. Analysis was performed using VevoLab 5.5.1 software. Researchers were blinded to phenotype when assessing cardiac function.
Hyperpolarized magnetic resonance spectroscopy
For the in vivo assessment of cardiac metabolism, hyperpolarized magnetic resonance spectroscopy was used to monitor the downstream fate of hyperpolarized [1-13C]pyruvate, as previously described64. Experiments were performed between 7 and 11 a.m., when mice were in the fed state. A total of 40 mg [1-13C]pyruvic acid (Sigma-Aldrich) doped with 15 mmol L−1 trityl radical (OXO63; GE Healthcare) and 3 ml Dotarem (1:50 dilution; Guerbet) was hyperpolarized in a prototype polarizer, with 30–60 min of microwave irradiation65. The sample was subsequently dissolved in a pressurized and heated alkaline solution, containing 2.4 g L−1 sodium hydroxide and 100 mg L−1 EDTA dipotassium salt (Sigma-Aldrich), to yield a solution of 80 mmol L−1 hyperpolarized sodium [1-13C]pyruvate with a polarization of ~30%. A total of 200 μl was injected over 10 s through the tail vein. 13C MR pulse–acquire cardiac spectra were acquired over 60 s following injection of hyperpolarized [1-13C]pyruvate (repetition time, 1 s; excitation flip angle, 15°; sweep width, 13,021 Hz; acquired points, 2,048; and frequency centred on the C1 pyruvate resonance)64. The 13C label from pyruvate and its metabolic products was summed over 60 s from the first appearance of pyruvate and fitted with the AMARES algorithm in jMRUI66. Data are reported as the ratio of metabolic product to the [1-13C]pyruvate signal to normalize for differences in polarization and delivery time.
Langendorff heart perfusions
Mice were terminally anesthetized. Their hearts were rapidly excised, cannulated and perfused, following standard Langendorff preparation as previously described67. The Krebs-Henseleit (KH) perfusion buffer was continuously gassed with 95% O2/5% CO2 (pH 7.4, 37 °C) and contained (in mM): NaCl (116), KCl (4.7), magnesium sulfate heptahydrate, KH2PO4 (1.2), NaHCO3 (25), CaCl2 (1.4) and enriched with metabolites (glucose (11), intralipid 0.4 mM, 1 sodium L-lactate; 0.1 sodium pyruvate; 0.5 L-glutamic acid monosodium salt monohydrate; 5 mU l−1 insulin (NovoRapid insulin, Novo Nordisk, Denmark)) and paced at 550 b.p.m. The impact of 1μM AZD4 on heart function and metabolism was assessed in unpaced hearts perfused using hyperglycaemic (11 mM) crystalloid KH buffer. At the end of each experiment, hearts, liver, adipose tissue and muscle (gastrocnemius and soleus) were immediately freeze-clamped using Wollenberger tongs for metabolic profiling.
Myocardial infarct size was quantified using triphenyltetrazolium chloride (TTC) staining68. In brief, after 20 min of equilibration, Langendorff perfused hearts were subject to 20 min of global normothermic ischemia (37 °C) and 2 h of reperfusion (37 °C). At the end of the protocol, hearts were perfused for 10 min with 3% TTC in KH Buffer followed by 10 min incubation in 3% TTC-KH. Tissue was sectioned (mouse heart gauge, Zivic instruments, USA) and infarct field was quantified using ImageJ Software (National Institute of Health).
Metabolomic profiling
Snap frozen and pulverized tissue (heart, liver, skeletal muscle(gastrocnemius+soleus), adipose tissue) was analyzed as previously described52,69. Non-targeted lipid profiling of the cardiac tissue was done using LC–MS/MS at the School of Chemical and Physical Sciences Mass Spectrometry Laboratory Services. Lipids were extracted from weighed heart tissue using a modified Folch protocol. Samples were homogenized in chloroform–methanol and washed with 0.9% NaCl to induce phase separation, and the organic layer was collected, dried under nitrogen and reconstituted in methanol for HPLC–MS analysis. Total lipids were separated using an ACQUITY UPLC system (Waters Corporation) equipped with a UPLC CSH C18 column (2.1 × 100 mm, 1.7 μm (Waters Corporation), maintained at 55 °C). The mobile phases consisted of (a) acetonitrile:water (60:40, vol/vol) containing 10 mM ammonium formate and 0.1% formic acid, and (B) isopropanol:acetonitrile (90:10, vol/vol) containing 10 mM ammonium formate and 0.1% formic acid. Lipid species were separated using a step gradient to 70% solvent B over 12 min at a flow rate of 0.45 ml min−1.
MS detection was performed on a Synapt G2-Si high-resolution mass spectrometer (Waters Corporation) operated in positive electrospray ionization (ESI) mode. Mass spectra were acquired over an m/z range of 50–1200. Nitrogen was used as both the cone and desolvation gas, with flow rates of 25 L h−1 and 900 L h−1, respectively. The source and desolvation temperatures were maintained at 125 °C and 350 °C. Leucine enkephalin (Leu-Enk) was continuously infused and used as a lock mass for real-time mass calibration. Data acquisition and instrument control were performed using MassLynx software (Waters Corporation). Cardiotonic lipids were analyzed in a non-targeted acquisition mode. Following mass spectrometry data collection, samples were processed using UNIFI software (Waters Corporation). The observed mass intensities of each identified lipid were matched against a curated cardiolipid library comprising approximately 270 lipid species. Lipid signal intensities were normalized to the weight of the corresponding heart tissue samples. Annotation of lipid species to lipid classes and categories was conducted using Lipidmaps70. MS intensities were then used to weight the molecular fatty acyl (FA) contributions and calculate class wise and total FA profiles. The carbon chain length distribution of phospholipid species was expressed as a percentage of the total signal within each class (carbon length (%)). For each phosphatidylcholine (PC) species, the total number of carbons across both fatty acyl chains was calculated, and the proportion of all PC species containing the total carbon number was determined. Similarly, the degree of unsaturation was assessed by calculating the total number of double bonds in the fatty acyl chains of each PC species and expressing the proportion of species carrying that total number of unsaturations as a percentage of all PC species (carbon bonds (%)).
Lipoprotein lipase assay
Lipoprotein lipase (LPL) activity was measured in cardiac tissue samples (control, db/db n = 10 per group, AZD n = 9) using a fluorometric assay (Cell Biolabs, STA-610, Lot:8724005). In brief, cardiac tissue samples were homogenized in 20 mM Tris (pH 7.5), 150 mM NaCl before centrifugation at 10,000g for 10 min at 4 °C. The supernatant was diluted 1:50 in LPL assay buffer and run alongside an LPL enzyme standard curve. Fluorescence intensities were measured at 485 excitation and 520 emission.
In silico analysis of cardiac metabolism
In silico simulations were performed using the metabolic network of the cardiomyocyte, CardioNet67. Mathematical modeling has previously been used to study the dynamics of cardiac metabolism in response to stress, and CardioNet has been successfully applied to identify limiting metabolic processes and estimate flux distributions10,67,71. Optimization problems were defined with the objective to minimize the total sum of fluxes through the CardioNet metabolic network72. Simulations for control, db/db or AZD1656 groups were based on the assumption that cardiomyocytes seek to maintain a certain ATP provision to sustain cardiac contractile function alongside the synthesis of macromolecules, including structural proteins and phospholipids for membranes72. We included the intracellular metabolite concentrations of 23 metabolites. Furthermore, we constrained extracellular metabolites that could be taken up from the blood, including glucose, lactate, cholesterol and free fatty acids. Linear programming was solved using the GUROBI solver (version 9.1.2 build v9.1.2rc0, Linux64)73. Details of all reactions and their metabolic subsystems used for CardioNet analysis were as classified in the Kyoto Encyclopedia of Genes and Genomes database74.
Tissue extraction and digestion for fluorescence-activated cell sorting
To generate leukocyte single cell suspensions for characterisation of immune cell populations, hearts were isolated from mice and digested. Mice were euthanised using an overdose of anaesthesia with 5 % isoflurane in 2 L min−1 O2. Cessation of pedal and corneal reflexes were checked, and death was confirmed by cervical dislocation.
Cardiac tissue suspensions were prepared by perfusing hearts with cold HBSS for 5 min before the atria was removed, followed by mincing of the ventricular tissue digested in collagenase I (Worthington Laboratories, C1639, 450 units ml−1), collagenase XI (Worthington Laboratories, C7657, 125 units ml−1), DNase 1 (Worthington Laboratories, D4527, 60 units ml−1) hyaluronidase (Sigma Aldrich, H3506, 60 unitsml−1) and 20 mM Hepes in PBS for 20 min at 37 ˚C with gentle agitation (Thermomixer, 750 r.p.m.). Spleens were mechanically dissociated by mashing through a cell strainer using the plunger end of a sterile syringe. Samples were passed through a 70-µm cell strainer, rinsed with cold 2% FBS/PBS and centrifuged at 400g for 10 min at 4˚C. The supernatant was removed, and pellets were resuspended in 5 ml red blood cell lysis buffer (BioLegend). Samples were then centrifuged for 8 min at 4˚C and 400g, the supernatant was removed and cells were resuspended and incubated with FC-block (BioLegend 101320, 1 µl per 1 × 106 cells). Cells were then washed again and resuspended in PBS ready for counting and antibody staining.
Flow cytometry
Cells isolated from tissues were resuspended (~1 × 107 cells per ml) and incubated for 30 min at room temperature with fluorochrome-conjugated antibodies (Supplementary Table 1) in 100 μl of flow cytometry buffer made of PBS containing 0.1% sodium azide (SigmaAldrich) and 1% FBS. For intracellular marker staining, cells were fixed and permeabilized for 30 min at 4 °C using a fixation–permeabilization kit (eBioscience), washed in 1× permeabilization buffer (eBioscience) and stained with fluorochrome-conjugated antibodies. A final wash with 1× permeabilization buffer was performed, centrifuged and resuspended in 200 μl of flow cytometry buffer. Cell viability was assessed using incubation with viability dyes (Supplementary Table 1). Samples were analyzed on FACSAriaIII (BD Biosciences) running FACSDiVa v.8.0 software (BD Biosciences). CD3 beads (Miltenyi) were routinely used to calibrate the cytometer. Single stain and fluorescence minus one control were acquired for compensation and precise gating (Extended Data Fig. 1, gating strategies). Compensation was automatically calculated, and samples were analyzed using FlowJo software (version 10, FlowJo).
Histology
Hearts were collected into buffered formalin (Sigma Aldrich). All histological processing was carried out by the Bart’s Cancer Institute Histology Core Facility (Masson’s trichrome staining). Stained cardiac sections were scanned using a Nanozoom panoramic scanner (40×). Images (whole heart sections) were analyzed using ImageJ (National Institute of Health).
Electron microscopy
Left ventricle tissue samples were collected from db/db, AZD1656-treated db/db and lean control mice (n = 3 per group) and placed into phosphate-buffered glutaraldehyde (4%) for fixing71. Samples were embedded in Araldite resin (Agar Scientific) and ultrathin sectioned (Reichert-Jung Ultracut E Ultramicrotome, Leica) for TEM analysis in JEM1400F (JEOL) at 120 kV by Transmission Electron Microscope Facility, School of Biological and Behavioural Sciences. Image analysis was completed using Fiji Image J software, and mitochondrial cristae surface density was calculated on the basis of previously described methods75,76.
Plasma analysis
Blood samples were collected at the time of experimental endpoint into heparinized tubes. Plasma biochemical profiling was carried out by the MRC Mouse Biochemistry Laboratory (Addenbrookes NHS Hospital, Cambridge). Circulating plasma cytokines were assessed using the XXL mouse cytokine array kit (Biotechne).
RNA sequencing and bioinformatic analysis
RNA extracted from the snap-frozen hearts was analyzed by massive analysis of cDNA End (MACE-Seq). Rapid MACE-seq was used to prepare 3′ RNA sequencing libraries. Samples of 100 ng of DNA-depleted RNA were used for library preparation, using the Rapid MACE-Seq kit (GenXPro, Germany). Fragmented RNA underwent reverse transcription using barcoded oligo(dT) primers containing TrueQuant unique molecular identifiers, followed by template switching. PCR-amplified libraries were purified by solid-phase reversible immobilization beads, and subsequent sequencing was performed using the Illumina platform NextSeq 500. Unprocessed sequencing reads were adapter-trimmed and quality-trimmed using Cutadapt (version 3.4 (ref. 77)). Deduplication based on unique molecular identifiers (UMIs) was performed using in-house tools. FastQC (0.11.9 (ref. 78)), was used to assess the quality of sequencing reads. MultiQC (version 1.16 (ref. 79)), was used to create a single report visualizing output from multiple tools across many samples, enabling global trends and biases to be quickly identified. MACE-Seq was annotated, reads were quantified and P values for differential expression were generated by GenXPro.
Visual representations of the DEG dataset were performed using the Python programming language (version 3. 9.7) as well as libraries and packages, including Matplotlib (version 3.4.3) for data visualization, Pandas (version 1.3.4) for data management, NumPy (version 1.20.3) for numerical computations and Jupyter Notebook (version 6.4.5) for interactive code development. The functional enrichment analysis was performed using the g:Profiler (version e108_eg55_p17_0254fbf) with g:SCS multiple testing correction method applying a significance threshold of 0.05 (ref. 80). To determine enriched pathways and ontologies in all analysis comparisons, both Ingenuity Pathway Analysis (IPA; Ingenuity) and Metascape comparison analysis (https://metascape.org)81 was performed on all genes. Comparisons were db/db_vs control (2,740 DE genes) and AZ_vs_db/db (1,271 DE genes). Both metascape and IPA utilize hypergeometric tests and Benjamini–Hochberg P value correction to identify all ontology and pathway terms that contain a greater number of genes in common with an input list than expected by chance81.
Data analysis and statistics
Data are presented as mean ± s.e.m. Comparisons between groups were performed by Student’s t-test (Gaussian data distribution), two-way ANOVA with Bonferroni’s correction for multiple comparison and one-way ANOVA using Bonferroni’s correction for multiple comparisons when applicable. The normality of the data distribution was examined using Shapiro–Wilk’s normality test. Statistical analysis was performed using GraphPad Prism (v10.3.1) software. Data analysis and visualization was conducted using R Studio (version 2022.12.0 Build 353). Partial least-squares discriminant analysis (PLS-DA) was conducted using the R package mdatools. PCA was conducted with normalized intensities lipidomics data using the PCA function included in the factoextra package.
Flux estimations (CardioNet) were compared between experimental groups using Wilcoxon rank-sum test, and adjusted P values were computed using the Bonferroni correction. Differences were considered significant when P < 0.05.
To assess the E/A ratio across groups, we employed a quantization approach due to the observed nonlinear (U-shaped) association between low and high E/A values with adverse outcomes, which is inconsistent with a linear relationship. Specifically, both low and high E/A values are known to be indicative of diastolic dysfunction82, which suggests a threshold effect (in which highly elevated E/A in the presence of disease is sometimes termed ‘pseudonormal’)83.
We therefore discretised the observed E/A data into distinct categories, designating values more than three sample s.d. above or below the mean of the lean control group as ‘abnormal’ (A); those within the range of ±3 sample s.d. were classified as ‘normal’ (N). This classification was chosen to reflect the non-linear nature of the response; extreme values on either end of the spectrum were associated with pathological conditions and is arguably more appropriate than a simple shift in means (as could be quantified for example through linear modeling and ANOVA-type approaches), which would not adequately capture the complexities of the relationship. We then used Fisher’s exact test for the resulting contingency table, which is particularly suited for categorical data comparisons in cases with small or unbalanced groups, implemented in the RVAideMemoir R package84,85. Multiplicity correction was performed by the FDR correction method of Benjamini–Hochberg86, with corrected P < 0.05 considered significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
RNA-sequencing data are available (open access) on Array Express accession E-MTAB-13849. All datasets supporting the findings of this study, including ex vivo function, morphology, biochemical assays, 1H NMR metabolomic analysis, echocardiographic, flow cytometric and transcriptomic data, are provided in the Source Data. TTC-stained hearts, fibrosis images and mitochondrial electron micrographs are publicly available on figshare (https://doi.org/10.6084/m9.figshare.30823256.v1) (ref. 87). Detailed results from the Cardionet in silico analyses and all lipidomic mass spectrometry data are included in the Supplementary Table 4 and the Source Data.
References
Henson, S. M. & Aksentijevic, D. Senescence and type 2 diabetic cardiomyopathy: how young can you die of old age? Front. Pharmacol. 12, 716517 (2021).
Aksentijevic, D. et al. Mechano-energetic uncoupling in heart failure. Nat. Rev. Cardiol. 22, 773–797 (2025).
Wilding, J. P., Leonsson-Zachrisson, M., Wessman, C. & Johnsson, E. Dose-ranging study with the glucokinase activator AZD1656 in patients with type 2 diabetes mellitus on metformin. Diabetes Obes. Metab. 15, 750–759 (2013).
Kishore, M. et al. Regulatory T cell migration is dependent on glucokinase-mediated glycolysis. Immunity 47, 875–889.e10 (2017).
Chorlton, J. et al. A randomised, double-blind, placebo-controlled, multicentre clinical trial of AZD1656 in diabetic patients hospitalised with COVID-19: the ARCADIA trial—implications for therapeutic immune modulation. EClinicalMedicine 51, 101604 (2022).
Mauro, C. et al. Obesity-induced metabolic stress leads to biased effector memory CD4+(+) T cell differentiation via PI3K p110delta-Akt-mediated signals. Cell Metab. 25, 593–609 (2017).
Marelli-Berg, F. M. & Aksentijevic, D. Immunometabolic cross-talk in the inflamed heart. Cell Stress 8, 240–266 (2019).
Alex, L., Russo, I., Holoborodko, V. & Frangogiannis, N. G. Characterization of a mouse model of obesity-related fibrotic cardiomyopathy that recapitulates features of human heart failure with preserved ejection fraction. Am. J. Physiol. 315, H934–H949 (2018).
Karlstaedt, A., Khanna, R., Thangam, M. & Taegtmeyer, H. Glucose 6-phosphate accumulates via phosphoglucose isomerase inhibition in heart muscle. Circ. Res. 126, 60–74 (2020).
Karlstaedt, A. et al. Oncometabolite d-2-hydroxyglutarate impairs alpha-ketoglutarate dehydrogenase and contractile function in rodent heart. Proc. Natl Acad. Sci. USA 113, 10436–10441 (2016).
Karlstaedt, A. Stable isotopes for tracing cardiac metabolism in diseases. Front. Cardiovasc. Med. 8, 734364 (2021).
Fanti, S. et al. Circulating c-Met-expressing memory T cells define cardiac autoimmunity. Circulation 146, 1930–1945 (2022).
Turbachova, I. et al. The cellular ratio of immune tolerance (immunoCRIT) is a definite marker for aggressiveness of solid tumors and may explain tumor dissemination patterns. Epigenetics 8, 1226–1235 (2013).
Ouchi, N. et al. Sfrp5 is an anti-inflammatory adipokine that modulates metabolic dysfunction in obesity. Science 329, 454–457 (2010).
Aksentijevic, D., Lewis, H. R. & Shattock, M. J. Is rate-pressure product of any use in the isolated rat heart? Assessing cardiac ‘effort’ and oxygen consumption in the Langendorff-perfused heart. Exp. Physiol. 101, 282–294 (2016).
How, O. J. et al. Increased myocardial oxygen consumption reduces cardiac efficiency in diabetic mice. Diabetes 55, 466–473 (2006).
Tomczyk, M. M. & Dolinsky, V. W. The cardiac lipidome in models of cardiovascular disease. Metabolites 10, 254 (2020).
van der Veen, J. N. et al. The critical role of phosphatidylcholine and phosphatidylethanolamine metabolism in health and disease. Biochim. Biophys. Acta 1859, 1558–1572 (2017).
Kallikourdis, M. et al. T cell costimulation blockade blunts pressure overload-induced heart failure. Nat. Commun. 8, 14680 (2017).
Vuononvirta, J., Marelli-Berg, F. M. & Poobalasingam, T. Metabolic regulation of T lymphocyte motility and migration. Mol. Aspects Med. 77, 100888 (2021).
Xia, Y. et al. Role of Treg cell subsets in cardiovascular disease pathogenesis and potential therapeutic targets. Front. Immunol. 15, 1331609 (2024).
Wang, Y. J. et al. Regulatory T cells attenuate chronic inflammation and cardiac fibrosis in hypertrophic cardiomyopathy. Sci. Transl Med. 17, eadq3516 (2025).
Wang, X. et al. Targeting regulatory T cells for cardiovascular diseases. Front. Immunol. 14, 1126761 (2023).
Saadat, S. et al. Pivotal role of TGF-beta/Smad signaling in cardiac fibrosis: non-coding RNAs as effectual players. Front. Cardiovasc. Med. 7, 588347 (2020).
Feuerer, M. et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat. Med. 15, 930–939 (2009).
Vasanthakumar, A. et al. The transcriptional regulators IRF4, BATF and IL-33 orchestrate development and maintenance of adipose tissue-resident regulatory T cells. Nat. Immunol. 16, 276–285 (2015).
Zhuang, R. & Feinberg, M. W. Regulatory T cells in ischemic cardiovascular injury and repair. J. Mol. Cell. Cardiol. 147, 1–11 (2020).
Chaudhry, A. et al. Interleukin-10 signaling in regulatory T cells is required for suppression of TH17 cell-mediated inflammation. Immunity 34, 566–578 (2011).
Garcia-Rivas, G. et al. The role of B cells in heart failure and implications for future immunomodulatory treatment strategies. ESC Heart Fail. 7, 1387–1399 (2020).
Lee, D. S. W., Rojas, O. L. & Gommerman, J. L. B cell depletion therapies in autoimmune disease: advances and mechanistic insights. Nat. Rev. Drug Discov. 20, 179–199 (2021).
Iellem, A. et al. Unique chemotactic response profile and specific expression of chemokine receptors CCR4 and CCR8 by CD4+CD25+ regulatory T cells. J. Exp. Med. 194, 847–853 (2001).
Yamazaki, T. et al. CCR6 regulates the migration of inflammatory and regulatory T cells. J. Immunol. 181, 8391–8401 (2008).
Mauri, C. & Bosma, A. Immune regulatory function of B cells. Annu. Rev. Immunol. 30, 221–241 (2012).
Callender, L. A. et al. Altered nutrient uptake causes mitochondrial dysfunction in senescent CD8+ EMRA T cells during type 2 diabetes. Front. Aging 2, 681428 (2021).
Emmerson, P. J. et al. The metabolic effects of GDF15 are mediated by the orphan receptor GFRAL. Nat. Med. 23, 1215–1219 (2017).
Bookout, A. L. et al. FGF21 regulates metabolism and circadian behavior by acting on the nervous system. Nat. Med. 19, 1147–1152 (2013).
Zhang, X. et al. Angiogenic factor thymidine phosphorylase associates with angiogenesis and lymphangiogenesis in the intestinal-type gastric cancer. Pathology 46, 316–324 (2014).
Prouse, T., Majumder, S. & Majumder, R. Functions of tam receptors and ligands protein S and Gas6 in atherosclerosis and cardiovascular disease. Int. J. Mol. Sci. 25, 12736 (2024).
Lu, J. et al. IGFBP1 increases beta-cell regeneration by promoting alpha- to beta-cell transdifferentiation. EMBO J. 35, 2026–2044 (2016).
Le Page, L. M. et al. Increasing pyruvate dehydrogenase flux as a treatment for diabetic cardiomyopathy: a combined 13C hyperpolarized magnetic resonance and echocardiography study. Diabetes 64, 2735–2743 (2015).
Schroeder, M. A. et al. Determining the in vivo regulation of cardiac pyruvate dehydrogenase based on label flux from hyperpolarised [1-13C]pyruvate. NMR Biomed. 24, 980–987 (2011).
de Jong, A. J., Kloppenburg, M., Toes, R. E. & Ioan-Facsinay, A. Fatty acids, lipid mediators, and T-cell function. Front. Immunol. 5, 483 (2014).
Waring, M. J. et al. Property-based optimisation of glucokinase activators – discovery of the phase IIb clinical candidate AZD1656. Med. Chem. Commun. 3, 1077–1081 (2012).
Ericsson, H. et al. Tolerability, pharmacokinetics, and pharmacodynamics of the glucokinase activator AZD1656, after single ascending doses in healthy subjects during euglycemic clamp. Int. J. Clin. Pharmacol. Ther. 50, 765–777 (2012).
Zhang, Z., Ji, G. & Li, M. Glucokinase regulatory protein: a balancing act between glucose and lipid metabolism in NAFLD. Front. Endocrinol. 14, 1247611 (2023).
Kroon, T. et al. Chronotherapy with a glucokinase activator profoundly improves metabolism in obese Zucker rats. Sci. Transl Med. 14, eabh1316 (2022).
Shang, R. & Rodrigues, B. Lipoprotein lipase and its delivery of fatty acids to the heart. Biomolecules 11, 1016 (2021).
Nevers, T. et al. TH1 effector T cells selectively orchestrate cardiac fibrosis in nonischemic heart failure. J. Exp. Med. 214, 3311–3329 (2017).
Ngwenyama, N. et al. Antigen presentation by cardiac fibroblasts promotes cardiac dysfunction. Nat. Cardiovasc. Res. 1, 761–774 (2022).
Altara, R., Mallat, Z., Booz, G. W. & Zouein, F. A. The CXCL10/CXCR3 axis and cardiac inflammation: implications for immunotherapy to treat infectious and noninfectious diseases of the heart. J. Immunol. Res. 2016, 4396368 (2016).
Chen, R. et al. Macrophages in cardiovascular diseases: molecular mechanisms and therapeutic targets. Signal Transduct. Target. Ther. 9, 130 (2024).
Karatzia, L. et al. New model of experimental diabetic cardiomyopathy using combination of multiple doses of anomer-equilibrated streptozotocin and high-fat diet: sex matters. Diabetes 74, 760–772 (2025).
Murphy, M. P. & O’Neill, L. A. J. A break in mitochondrial endosymbiosis as a basis for inflammatory diseases. Nature 626, 271–279 (2024).
Kumar, V., Narisawa, M. & Cheng, X. W. Overview of multifunctional Tregs in cardiovascular disease: from insights into cellular functions to clinical implications. FASEB J. 38, e23786 (2024).
da Cruz, L. et al. Phase 1 clinical study of an embryonic stem cell-derived retinal pigment epithelium patch in age-related macular degeneration. Nat. Biotechnol. 36, 328–337 (2018).
Mathur, A. et al. The effect of intracoronary infusion of bone marrow-derived mononuclear cells on all-cause mortality in acute myocardial infarction: the BAMI trial. Eur. Heart J. 41, 3702–3710 (2020).
Mathur, A. et al. Five-year follow-up of intracoronary autologous cell therapy in acute myocardial infarction: the REGENERATE-AMI trial. ESC Heart Fail. 9, 1152–1159 (2022).
AZD1656 (AstraZeneca, 2023); https://www.astrazenecaclinicaltrials.com/study/D1020C00010/
Baker, D. J. et al. Chronic glucokinase activator treatment at clinically translatable exposures gives durable glucose lowering in two animal models of type 2 diabetes. Br. J. Pharmacol. 171, 1642–1654 (2014).
Baker, D. J. et al. Characterization of the heterozygous glucokinase knockout mouse as a translational disease model for glucose control in type 2 diabetes. Br. J. Pharmacol. 171, 1629–1641 (2014).
Schneider, J. E. et al. Fast, high-resolution in vivo cine magnetic resonance imaging in normal and failing mouse hearts on a vertical 11.7 T system. J. Magn. Reson. Imaging 18, 691–701 (2003).
Haase, A., Frahm, J., Matthaei, D., Hanicke, W. & Merboldt, K. D. FLASH imaging: rapid NMR imaging using low flip-angle pulses. J. Magn. Reson. 213, 533–541 (2011).
Iveljic, I. et al. Type 2 diabetes worsens the outcome of ischemia/reperfusion in female STEMI patients and female db/db mice with HFpEF cardiometabolic phenotype. Cardiovasc. Diabetol. 24, 243 (2025).
Dodd, M. S. et al. In vivo mouse cardiac hyperpolarized magnetic resonance spectroscopy. J. Cardiovasc. Magn. Reson. 15, 19 (2013).
Ardenkjaer-Larsen, J. H. et al. Increase of signal-to-noise of more than 10,000 times in liquid state NMR. Discov. Med. 3, 37–39 (2003).
Vanhamme, L., van den Boogaart, A. & Van Huffel, S. Improved method for accurate and efficient quantification of MRS data with use of prior knowledge. J. Magn. Reson. 129, 35–43 (1997).
Aksentijevic, D. et al. Intracellular sodium elevation reprograms cardiac metabolism. Nat. Commun. 11, 4337 (2020).
Prag, H. A. et al. Mechanism of succinate efflux upon reperfusion of the ischaemic heart. Cardiovasc. Res. 117, 1188–1201 (2021).
Faulkes, C. G. et al. Naked mole-rats have distinctive cardiometabolic and genetic adaptations to their underground low-oxygen lifestyles. Nat. Commun. 15, 2204 (2024).
LIPID Metabolites and Pathways Strategy (LIPID MAPS, 2003); https://www.lipidmaps.org
Aksentijevic, D. et al. Cardiac dysfunction and peri-weaning mortality in malonyl-coenzyme A decarboxylase (MCD) knockout mice as a consequence of restricting substrate plasticity. J. Mol. Cell. Cardiol. 75, 76–87 (2014).
Karlstadt, A. et al. CardioNet: a human metabolic network suited for the study of cardiomyocyte metabolism. BMC Syst. Biol. 6, 114 (2012).
Gurobi Optimizer Reference Manual (Gurobi Optimization, 2023); https://www.gurobi.com
Ogata, H. et al. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 27, 29–34 (1999).
Nielsen, J. et al. Plasticity in mitochondrial cristae density allows metabolic capacity modulation in human skeletal muscle. J. Physiol. 595, 2839–2847 (2017).
Weibel, E. R., Kistler, G. S. & Scherle, W. F. Practical stereological methods for morphometric cytology. J. Cell Biol. 30, 23–38 (1966).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12 (2011).
Fast QC. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (Barbraham Bioinformatics, 2010).
Ewels, P., Magnusson, M., Lundin, S. & Kaller, M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048 (2016).
Raudvere, U. et al. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 47, W191–W198 (2019).
Zhou, Y. et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 10, 1523 (2019).
Mitter, S. S., Shah, S. J. & Thomas, J. D. A test in context: E/A and E/e’ to assess diastolic dysfunction and LV filling pressure. J. Am. Coll. Cardiol. 69, 1451–1464 (2017).
Nagarakanti, R. & Ezekowitz, M. Diastolic dysfunction and atrial fibrillation. J. Interv. Card. Electrophysiol. 22, 111–118 (2008).
R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2023).
R VA-Core Team RVAideMemoire: testing and plotting procedures for biostatistics. Version 0.9-83-12 https://cran.r-project.org/package=RVAideMemoire (2023).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B 57, 289–300 (1995).
Aksentijevic, D. Targeting immunometabolic pathways with AZD1656 alleviates inflammation and metabolic dysfunction in type 2 diabetic cardiomyopathy. figshare https://doi.org/10.6084/m9.figshare.30823256.v1 (2025).
Acknowledgements
D.A. acknowledges I. Cubela (Barts Cancer Institute, QMUL), H. Toms, R. Buccafusca, N. Kanwal (School of Physical and Chemical Sciences, QMUL), T. Eykyn (King’s College London), G. Mastroianni (School of Biological and Chemical Sciences, QMUL), A. H. Andaloussi (William Harvey Research Institute, QMUL), K. Bianchi and V. Morales (Barts Cancer Institute Metabolic Flux Analysis Facility, QMUL) for technical assistance. D.A. is a recipient of the Wellcome Trust Career Re-Entry Fellowship (221604/Z/20/Z) and acknowledges the following funding sources: British Heart Foundation Accelerator Award fellowship (AA/18/5/34222), Diabetes UK Grant (19/0005973), Bart’s Charity Grants (MRC 0215, G-002145), AstraZeneca UK (study agreement ref 3853), School of Biological and Behavioural Sciences QMUL (HEFCE lectureship). S.M.H. was funded by Diabetes UK 19/0006057, C.G.-K. by Barts and the London Charity MGU0536. D.J.T. was funded by British Heart Foundation Senior Basic Science Research Fellowships (FS/14/17/30634 and FS/19/18/34252). S.A. was funded by the Pelly-Carys Bannister Scholarship, Somerville College, University of Oxford. A.K. was supported by the National Institutes of Health (NIH) (R00-HL-141702 and R01-HL-177461), the Leukemia Research Foundation (New Investigator Award, grant no. 941997) and the Cedars-Sinai Cancer Center through the 2022 Cancer Biology Program CardioOncology Award. M.R.B. acknowledges funding from the NIHR BRC at Barts and The London School of Medicine and Dentistry. L.J.A. acknowledges British Heart Foundation Programme Grant RG/12/4/29426. C.E.O. was sponsored by Barts and The London School of Medicine and Dentistry, Queen Mary University of London. J.C. was supported by a Career Development Fellowship of Versus Arthritis UK (22855). J.J.J.J.M. acknowledges support of a faculty grant from the Novo Foundation (NNF21OC0068683). This work was supported by the Medical Research Council UK (MC_UU_00028/4) and by a Wellcome Trust Investigator award (220257/Z/20/Z) to M.P.M. This research was supported by the MRC Mouse Biochem Lab, Cambridge (Medical Research Council (MC_UU_00014/5)). Figure 1a created with BioRender.com.
Author information
Authors and Affiliations
Contributions
Conceptualization: D.A. Study design: D.A. Experimental work: D.A., S.A., M.Y., J.C., L.K., F.C., C.E.O., L.J.A., C.G.-K., H.A.P., D.J.T. Molecular analyses (RNA sequencing, lipidomics): A.K., M.R.B., S.D., Z.S. Bioinformatic and in silico analyses: A.K., M.R.B., Z.S., F.C., S.D. Data curation and statistical analysis: D.A., S.A., J.J.J.J.M., S.M.H., A.K., D.J.T., M.Y. Figure preparation and visualization: D.A., Z.S., A.K. Manuscript drafting: D.A. Manuscript review and editing: S.A., A.K., M.Y., F.C., J.C., C.E.O., M.R.B., Z.S., L.J.A., C.G.-K., S.D., H.A.P., J.J.J.J.M., C.T., A.J.M.L., M.P.M., D.M.S., S.M.H., D.J.T., D.A. Supervision: D.A., D.J.T., C.T., A.J.M.L., M.P.M. Funding acquisition: D.A., D.M.S.
Corresponding author
Ethics declarations
Competing interests
D.A. has a research study and exclusive license option agreement with AstraZeneca UK to study impact of AZD1656 on T cell trafficking and cardiac metabolism. D.M.S. is an employee and shareholder of AstraZeneca. The other authors declare no competing interests.
Peer review
Peer review information
Nature Cardiovascular Research thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 FACS experiments gating strategies.
A) FACS experiments representative gating strategies in hearts T cell panel B) hearts non-T cell panel C) FACS experiments representative gating strategies in hearts for detection of Tregs (CD4CD25FoxP3) D) FACS experiment gating strategy for the analysis of spleen E) FACS experiment gating strategy for the analysis of 12 week high-fat (HFD) diet obesity model hearts. Source data provided as a Source Data File.
Extended Data Fig. 2 Cardiac Phenotype.
A) 1H NMR spectroscopy metabolomic profile. Fold change db/db vs AZD treated db/db hearts. Data mean ± SEM. Control. n = 8, db/db n = 8, AZD n = 5. Metabolite fold change vs control (control= 1) with propagated error of mean by t-test, two tailed P < 0.05. B) Acute AZD1656 administration has no impact on ex vivo function (n = 4/group), data mean ± SEM or C) metabolomic profile of hearts metabolite fold change vs control (control= 1) with propagated error of mean by t-test, two tailed P < 0.05 D) Electron microscopy assessment of mitochondrial cristae surface density; black scale bar corresponds to 500nm. n = 3/group. Data displayed as mean ± SEM, analysed by one-way ANOVA. n represents biological replicates (individual animals). Source data provided as a Source Data File.
Extended Data Fig. 3 Systemic metabolic profile.
A) Plasma biochemical profiling *P < 0.05, ** P < 0.01, ***P < 0.001 by one way ANOVA, n = 6/group. Data mean ± SEM. Metabolomic profile of db/db vs AZD treated B) liver C) muscles. Metabolomic data presented as fold change db/db vs AZD-treated db/db. Liver: Control n = 6, db/db n = 7, AZD n = 7. Skeletal Muscle: Control n = 6, db/db n = 6, AZD n = 5. Metabolite fold change with propagated standard error of mean by t-test, two tailed P < 0.05. n represents biological replicates (individual animals). Source data provided as a Source Data File.
Extended Data Fig. 4 Impact of AZD1656 treatment on adipose tissue.
Impact of AZD1656 treatment on adipose tissue A) Whole body composition analysis B) representative 1H NMR spectroscopy lipid spectrum with annotations C) adipose tissue lipid constituents quantified using 1H NMR spectroscopy n = 5/group, one way ANOVA, data mean ± SEM. n represents biological replicates (individual animals). Source data provided as a Source Data File.
Extended Data Fig. 5 Circulating cytokine profile.
A) Circulating cytokine profile data min to max (box and whiskers plot). Comparison by ordinary one-way ANOVA. Control n = 4, db/db n = 5, AZD n = 5. *P < 0.05 B)Flow cytometry assessment of the spleen T cell profile. Control n = 5, db/db n = 6 AZD1656 n = 5. n represents biological replicates (individual animals). Source data provided as a Source Data File.
Extended Data Fig. 6 Physiological profile of 12 week HFD-fed model of pre-diabetes.
Physiological profile of 12 week HFD-fed model of pre-diabetes A) body weight B) plasma insulin, P = 0.054 C) plasma free fatty acids D) fasting plasma glucose E) intraperitoneal glucose tolerance test (IPGTT) F) PCA plot of cardiac metabolomic profile analysed by LC/MS G) Myocardial inflammatory cell profile assessed by flow cytometry. n = 6/group, male, C57/BL6 mice, age at endpoint 20 weeks. Flow experiment n = 3/group *P < 0.05 ***P < 0.001 ****P < 0.0001 by t -test (two sample compafrison) or two-way ANOVA subject to Tukey multiple comparisons post hoc test (IPGTT). n represents biological replicates (individual animals). Source data provided as a Source Data File.
Extended Data Fig. 7 G profiler Analysis.
G profiler analysis of the cellular pathways identified from RNA cardiac sequencing data (DEG gene expression) n = 6/group. Raw RNA seq expression data provided in Supplementary Data Table 1. n represents biological replicates (individual animals).
Extended Data Table. 8 Leukocyte Extravasation Pathway Qiagen IPA.
ZD treatment normalizes aberrant myocardial leukocyte extravasation signalling pathway Qiagen Ingenuity Pathway Analysis (IPA) based on RNA sequencing results (n = 6/group). IPA utilise hypergeometric test and Benjamini-Hochberg p- value correction to identify all ontology and pathway terms that contain a statistically greater number of genes in common with an input list than expected by chance. Red indicates altered expression and pathway impairment. Green signifies gene expression normalisation. n represents biological replicates (individual animals).
Extended Data Fig. 9 AZD treatment normalizes myocardial HIF-1a signalling pathway in dbCM.
Qiagen Ingenuity Pathway Analysis (IPA) based on RNA sequencing results (n = 6/group). IPA utilise hypergeometric test and Benjamini-Hochberg p-value correction to identify all ontology and pathway terms that contain a statistically greater number of genes in common with an input list than expected by chance. n represents biological replicates (individual animals).
Extended Data Fig. 10 AZD treatment normalizes myocardial Nrf2 signalling pathway in dbCM.
Qiagen Ingenuity Pathway Analysis (IPA) based on RNA sequencing results (n = 6/group). IPA utilise hypergeometric test and Benjamini-Hochberg p-value correction to identify all ontology and pathway terms that contain a statistically greater number of genes in common with an input list than expected by chance. n represents biological replicates (individual animals).
Supplementary information
Supplementary Tables 1–4 (download XLSX )
Supplementary Table 1: FACS antibodies information. Supplementary Table 2: Echocardiography of control and 12-week high-fat-diet feeding-induced obesity. Supplementary Table 3: Lipidomic comparison P values presented in Fig. 3 statistical analysis of PC fatty acid length (Fig. 3d) and PC_Bonds (Fig. 3e). Supplementary Table 4: Unprocessed LC/MS MS output for lipidomic analysis.
Source data
Source Data Figs. 1, 2, 4 and 5, Source Data Table 1 and Source Data Extended Data Figs. 2, 3, 4, 5 and 6 (download XLSX )
Source data for all the plots in the main text and the extended data figures. Source Data Fig. 1: Unprocessed cardiac function: data CINE MRI, echocardiography, ex vivo perfusions and infarct size quantification. Source Data Fig. 2: Unprocessed metabolomic data 1H NMR spectroscopy of the hearts, unprocessed 13 C DNP Spectroscopy. Source Data Fig. 4: Unprocessed plasma analysis, body composition analysis, 1H NMRs of liver and muscle tissue and liver weight. Source Data Fig. 5: Unprocessed data for plasma cytokines and flow cytometry analysis of cardiac tissue (CD4, Treg, macrophages, dendritic cells, B Cells, neutrophils). Unprocessed data for fibrosis quantification. Source Data Table 1: Unprocessed morphology, CINE MRI and echocardiography data. Source Data Extended Data Fig. 2: Unprocessed 1H NMR spectroscopy metabolomic data, ex vivo cardiac function data and mitochondrial morphology assessment. Source Data Extended Data Fig. 3: Unprocessed 1H NMR spectroscopy metabolomic data. Source Data Extended Data Fig. 4: Unprocessed 1H NMR spectroscopy metabolomic data. Source Data Extended Data Fig. 5: Unprocessed cytokine plasma data and flow cytometry T cell assessment of spleen. Source Data Extended Data Fig. 6: Unprocessed morphological data, plasma biochemistry, mass pec cardiac metabolic profile analysis and flow cytometry analysis.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Anderson, S., Karlstaedt, A., Young, M. et al. Targeting immunometabolic pathways with AZD1656 alleviates inflammation and metabolic dysfunction in type 2 diabetic cardiomyopathy. Nat Cardiovasc Res 5, 138–154 (2026). https://doi.org/10.1038/s44161-025-00769-0
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s44161-025-00769-0
This article is cited by
-
Targeting immunometabolism for the treatment of diabetic cardiomyopathy
Nature Cardiovascular Research (2026)
