
Abstract
Multidrug resistance in the pathogenic fungus Candida glabrata is a growing global threat. Here, we study mechanisms of multidrug resistance in this pathogen. Exposure of C. glabrata cells to micafungin (an echinocandin) leads to the isolation of a mutant exhibiting resistance to echinocandin and azole antifungals. The drug-resistant phenotype is due to a non-synonymous mutation (R70H) in gene IPI1, which is involved in pre-rRNA processing. Azole resistance in the ipi1R70H mutant depends on the Pdr1 transcription factor, which regulates the expression of multidrug transporters. The C. glabrata Ipi1 protein physically interacts with the ribosome-related chaperones Ssb and Ssz1, both of which bind to Pdr1. The Ipi1-Ssb/Ssz1 complex inhibits Pdr1-mediated gene expression and multidrug resistance in C. glabrata, in contrast to Saccharomyces cerevisiae where Ssz1 acts as a positive regulator of Pdr1. Furthermore, micafungin exposure reduces metabolic activity and cell proliferation in the ipi1R70H mutant, which may contribute to micafungin tolerance.
Similar content being viewed by others

Introduction
Bloodstream fungal infections are largely caused by Candida spp. and are associated with high mortality rates. Approximately 750,000 cases of invasive candidiasis occur annually worldwide, with a mortality rate of 46–75% in the overall infected population and ~40% in patients receiving antifungal therapies1,2. Because therapeutic agents for candidiasis are limited in number and quality and are mainly echinocandins or azole antifungals, with liposomal amphotericin B as an alternative, the increasing emergence of multidrug (azole- and echinocandin-class antifungals) resistant Candida glabrata has become a critical threat in clinical settings3,4,5. The reported rates of multidrug resistance (MDR) are as high as 25%–30% in C. glabrata strains2,6,7,8. Prior drug exposure, especially at insufficient dosages, has been directly linked to the development of antifungal resistance9,10.
Azole antifungals inhibit the biosynthesis of ergosterol, the major sterol in the cell membrane, by targeting lanosterol 14α-demethylase, encoded by ERG1111. The key mechanisms of acquired azole resistance are mutations in ERG11 and activation of the ATP-binding cassette (ABC) transporters, Cdr1 and Cdr2, under the transcriptional regulation of pleiotropic drug resistance 1 (Pdr1). Gain-of-function mutations in PDR1 lead to the activation of Pdr1 signaling and such mutations are often found in azole-resistant C. glabrata clinical isolates. Echinocandin-class antifungals, such as micafungin and caspofungin, inhibit the biosynthesis of β-1,3-D-glucan, a critical cell wall component, and are currently recommended as a first-line therapy for candidemia caused by C. glabrata12,13. However, the increasing use of echinocandins is associated with increased proportions of echinocandin-resistant Candida species14,15. Furthermore, Alexander et al. reported that prior echinocandin therapy was significantly associated with resistance to both echinocandin and fluconazole3. The predominant mechanism of echinocandin resistance in C. glabrata involves hotspot mutations in FKS1 and/or FKS2, which encode the catalytic subunits of β-1,3-D-glucan synthase. However, many C. glabrata clinical isolates exhibiting echinocandin resistance with no FKS mutations have also been reported, and the molecular basis of decreased echinocandin susceptibility is not fully understood3,9,16,17,18,19. For example, among the 77 C. glabrata isolates in the US displaying either resistance or intermediate susceptibility to one or more echinocandins, 27 isolates (35.1%) had wild-type FKS genotypes, and over one-third of the echinocandin resistant isolates were also resistant to fluconazole19. Similar results were also recently reported in Germany9.
In this study, we experimentally developed a multidrug-resistant C. glabrata strain by in vitro exposure to increasing concentrations of micafungin. Here, we report the mechanisms of MDR induced by a single amino acid substitution in C. glabrata.
Results
In vitro exposure to micafungin induced MDR in C. glabrata
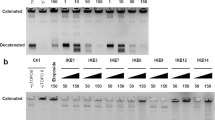
The wild-type C. glabrata strain CBS138 could not grow on agar plates containing 0.03 µg/mL micafungin (Fig. 1a). However, some cells grown on the plate containing 0.015 µg/mL micafungin grew in the presence of 0.03 µg/mL micafungin, suggesting that those cells withstood micafungin pressure and acquired adaptive resistance after exposure to a low concentration of the drug. One of the micafungin-exposed C. glabrata strains was named Cg50. Strain Cg51 was obtained by repeated subculturing of strain Cg50 in the absence of micafungin for 20 days. In spot dilution assays, both Cg50 and Cg51 displayed similarly decreased susceptibility to micafungin; therefore, this phenotype appeared irreversible (Fig. 1b). Antifungal susceptibility was examined using the broth microdilution method (Supplementary Table 1). Increased micafungin MIC was observed for both Cg50 and Cg51 compared to that of the parent strain (0.125 µg/mL vs. <0.03 µg/mL). The MIC of anidulafungin was also slightly increased for Cg50 and Cg51, whereas the susceptibility to caspofungin, flucytosine, and amphotericin B did not change. Surprisingly, both strains were highly resistant to multiple azole antifungals, including fluconazole, itraconazole, and voriconazole, without pre-exposure to any azole antifungals (Fig. 1b, Supplementary Table 1). These results indicated that increased resistance to both micafungin and azole antifungals could be induced by exposure to low concentrations of micafungin in C. glabrata.

a A schema representing sequential exposure of C. glabrata wild-type (CBS138) cells to increasing concentrations of micafungin. C. glabrata cells were spread on YPD plates containing micafungin at the indicated concentrations, incubated at 30 °C for 2 days, and subjected to antifungal susceptibility tests. b, c Spot dilution assay. C. glabrata strain Cg50 was obtained from a plate containing micafungin at the concentration of 0.03 µg/mL as described in (a). Strain Cg51 was obtained after the repeated subculture of Cg50 in YPD broth without micafungin for 20 days. An ipi1R70H mutant was constructed as described in the methods. Serial 10-fold dilutions of C. glabrata cells were spotted onto an SC plate containing an antifungal agent at the indicated concentrations. Plates were incubated at 30 °C or the indicated temperatures for 2 days. All susceptibility tests were performed on at least three separate occasions. d Doxycycline-mediated transcriptional repression of IPI1 resulted in a growth defect in C. glabrata. Logarithmic-phase cells of the C. glabrata wild-type (ACG22) and tet-IPI1 strain, in which the native IPI1 promoter was replaced with the tetracycline regulatable promoter, were adjusted to ~ 1 × 105 cells/mL in SD broth and incubated at 30 °C for 24 h with or without 20 µg/mL of the tetracycline analog doxycycline (Dox). Data are expressed as mean ± standard deviation for biological triplicates (****P < 0.0001; ns, not significant; one-way ANOVA with Dunnett’s multiple comparison test). The experiment was repeated twice with similar results. Source data are provided as a Source Data file. e Northern blotting. Logarithmic-phase C. glabrata cells grown at 30 °C were subsequently incubated at 42 °C, a non-permissive temperature for the ipi1R70H mutant. Total RNA was extracted from each strain at the indicated time points. Top, northern hybridization was performed using an ITS2 probe. Bottom, total RNA was analyzed using 1.0% agarose gel with ethidium bromide, and 25S and 18S rRNA are indicated. The experiment was repeated twice with similar results. Source data including uncropped and unprocessed scans with molecular weight markers are provided as a Source Data file.
Identification of a mutation responsible for MDR in C. glabrata
The irreversible phenotypes of Cg51 seem to reflect mutational resistance rather than a transient stress response. Whole-genome sequencing of Cg51 identified 853 single nucleotide variants compared with the parent strain CBS138 and 103 mutations in the coding regions of 46 genes (Supplementary Data 1). However, none of the mutations identified in Cg51 are known to cause antifungal resistance. Further phenotypic analyses of Cg50 and Cg51 revealed that both the strains could not grow at 41 °C (Fig. 1b). Therefore, we conducted a complementation assay using a C. glabrata cDNA library to identify a gene that could rescue growth at 41 °C. Among the 22 transformants examined, 20 strains had a plasmid containing IPI1 (CAGL0M01210g), and the other two strains had RIM11 (CAGL0L06820g). RIM11 rescued cell growth at 41 °C but did not change the susceptibilities to micafungin and azole, whereas IPI1 recovered these three phenotypes to wild-type levels. Therefore, the following analyses focused on IPI1.
Whole-genome sequencing revealed that the IPI1 gene of Cg51 had a single nucleotide mutation (G209A), which changed arginine to histidine at the 70th amino acid (R70H) of Ipi1. This mutation was confirmed by direct sequencing of IPI1 amplified from Cg50 and Cg51. The three phenotypes (micafungin resistance, azole resistance, and growth defect at high temperature) of Cg51 were restored to the wild-type levels by introducing an intact IPI1 into Cg51. Furthermore, to rule out the possibility that other coexisting mutations in the genome of Cg51 might be involved in these phenotypic changes, we performed site-directed mutagenesis to introduce only the IPI1 G209A mutation into the wild-type strain, and the resulting strain was named the ipi1R70H mutant. All phenotypes observed in Cg51 were reproduced in the ipi1R70H mutant (Fig. 1c). Therefore, we conclude that the R70H substitution in Ipi1 was responsible for the three phenotypes observed in Cg50 and Cg51.
Ipi1 is required for cell growth and ITS2 processing in C. glabrata
In Saccharomyces cerevisiae, Ipi1 (involved in processing ITS2) is a component of the Rix1 complex required to process ITS2 sequences from 35S pre-rRNA and is essential for cell viability20,21,22. We constructed a C. glabrata tet-IPI1 strain in which IPI1 was expressed under the tetracycline regulatable promoter. The tetracycline analog doxycycline did not interfere with the growth of C. glabrata wild-type cells (Fig. 1d). Growth of tet-IPI1 cells was also comparable to that of the wild-type cells in the absence of doxycycline. However, tet-IPI1 cells could not grow in the presence of doxycycline. These results indicate that doxycycline-mediated transcriptional repression of IPI1 caused a growth defect, and therefore, Ipi1 plays an important role in cell growth in C. glabrata. Although 42 °C was the non-permissive temperature for the ipi1R70H mutant, the ipi1R70H cells survived for several hours after the incubation temperature was shifted from 30 °C to 42 °C, and RNA was detectable. ITS2 is an intermediate product of rRNA processing, and digestion of this region is necessary to complete the processing; however, the ipi1R70H mutant failed to digest it, resulting in the increased accumulation of unprocessed ITS2 compared to the wild-type strain (Fig. 1e). These results suggested that the growth defect of the C. glabrata ipi1R70H mutant at elevated temperatures was because of incompetent rRNA processing.
Exploration of azole resistance mechanisms
The strain Cg51 harbored no mutations in the ERG genes involved in ergosterol biosynthesis, such as ERG11 and ERG3 (Supplementary Data 1). The sterol assay revealed that ergosterol levels in the ipi1R70H mutant were higher than those in the wild-type strain regardless of the presence or absence of fluconazole (Fig. 2a). Treatment with 8 µg/mL fluconazole decreased ergosterol content in the wild-type strain (P < 0.01) but not in the ipi1R70H mutant (P ≥ 0.05). Treatment with fluconazole significantly increased lanosterol content in both strains; however, the accumulation level of lanosterol was approximately one-third less in the ipi1R70H mutant than in the wild-type strain. Although transcriptional regulation of ERG genes does not necessarily correlate with ergosterol levels23, the expression patterns of ERG11 appeared to reflect the accumulation of lanosterol in this study (Fig. 2b). These results indicated that fluconazole interfered with lanosterol demethylase (the target enzyme of azoles) in the wild-type strain, but this effect was negligible in the ipi1R70H mutant, implying a lower intracellular concentration of the drug in the mutant. We performed quantitative reverse transcription PCR (qRT-PCR) to examine the expression of azole efflux pump genes, including CDR1 and CDR2, and their transcriptional regulator PDR1, in Cg50 and two different clones of the ipi1R70H mutant (Fig. 2c). CDR1 expression was 12-to 15-fold higher in the Cg50 and ipi1R70H strains than in the wild-type strain; the expression levels of CDR2 and PDR1 were also slightly ( ~ 2.5-fold) higher in the mutants. Previous studies have suggested that C. glabrata Cdr1 is a more efficient transporter than Cdr2 because Cdr1 induces higher levels of drug resistance when the two genes are expressed in S. cerevisiae24,25. We hypothesized that the ipi1R70H mutation would activate CDR1 transcription and induce azole resistance in C. glabrata.

a Sterol analysis. C. glabrata wild-type (WT) and ipi1R70H strains were grown in synthetic defined (SD) broth or SD broth with 8 μg/mL fluconazole ( + FLC). Sterol contents were analyzed by reverse-phase HPLC as described in the methods. The means and standard errors for three independent experiments are shown (ns, not significant; ordinary one-way ANOVA with Sidak’s multiple comparison test). b qRT-PCR. C. glabrata wild-type (WT) and the ipi1R70H mutant were grown as described in (a) and total RNA was extracted. mRNA abundance of ERG11 was measured by qRT-PCR and normalized using ACT1 as an internal control. Data are expressed as the means ± standard deviations (ns, not significant; ordinary one-way ANOVA with Sidak’s multiple comparison test). qRT-PCR was repeated on three independent occasions with similar results. c qRT-PCR. Total RNA was extracted from C. glabrata wild-type, Cg50, and two different clones of ipi1R70H mutant strains. mRNA abundance was measured by qRT-PCR and normalized using ACT1 as an internal control. Expression of wild-type strain was defined as 1 in each assay, and relative mRNA abundances in other strains were calculated. Data are expressed as the means ± standard deviations (ordinary one-way ANOVA with Dunnett’s multiple comparison test). qRT-PCR was repeated on three independent occasions with similar results. Source data for panels (a–c) are provided as a Source Data file. d Rhodamine 6 G (R6G) accumulation assay. Intracellular concentration of the fluorescent dye R6G, a substrate of azole efflux pumps, was measured by flow cytometry in three different wild-type strains, two different clones of the ipi1R70H mutant, and a mutant lacking Cdr1 efflux pump (cdr1Δ). Fluorescence intensity of R6G is shown on the X-axis. Left panel: Cells were incubated with R6G under normal growth conditions to examine the activity of R6G efflux. Right panel: Cells were exposed to R6G under the de-energized condition to examine the levels of R6G infused passively into the cells. These experiments were repeated twice with similar results.
To examine whether the increased mRNA levels of CDR1 translated into increased efflux pump activity, intracellular accumulation of the fluorescent dye Rhodamine 6 G (R6G), a known substrate of the Cdr1 efflux pump26, was measured by flow cytometry. While the cdr1Δ mutant displayed increased R6G accumulation, the ipi1R70H mutants showed decreased intracellular concentrations of R6G compared to the wild-type strain (Fig. 2d). When cells were deenergized, the R6G passive infusion levels were similar in all the tested strains. These results suggest that drug penetration into the cells was not affected, but drug efflux was activated by the increased expression of CDR1 in the ipi1R70H mutants.
To confirm the contribution of Cdr1 and Pdr1 to antifungal resistance in the ipi1R70H mutant, we generated mutant strains lacking either CDR1 or PDR1 in wild-type and ipi1R70H backgrounds and examined their susceptibility to fluconazole and voriconazole. As expected, the loss of CDR1 or PDR1 resulted in increased azole susceptibility in both the wild-type and ipi1R70H mutant backgrounds (Fig. 3a). Although the ipi1R70H mutation in the cdr1∆ and pdr1∆ mutants slightly decreased the susceptibility to low concentrations of fluconazole (e.g. fluconazole 4 and 8 µg/mL), the cdr1∆/ipi1R70H and pdr1∆/ipi1R70H mutants remained much more susceptible to fluconazole than the wild-type strain. The ipi1R70H mutation did not affect voriconazole susceptibility in the cdr1∆ and pdr1∆ mutants. Although these C. glabrata cells grew slightly better at 37 °C than at 30 °C, azole susceptibility phenotypes were similar at both temperatures (Fig. 3a, Supplementary Fig. 1). Collectively, these results indicate that activation of the azole efflux pump, mediated by Pdr1-Cdr1 signaling, is the key mechanism of azole resistance in the ipi1R70H mutant.

a Effects of CDR1 and PDR1 deletion on azole susceptibility were examined using a spot dilution assay in the wild-type and ipi1R70H backgrounds. Logarithmic-phase cells of the C. glabrata strains were serially diluted and spotted on SC plates containing fluconazole or voriconazole at the indicated concentrations. Plates were incubated at 30 °C for 2 days. b Pdr1 P927S, which is known as a gain-of-function mutation in C. glabrata, was introduced in the wild-type and ipi1R70H backgrounds. A spot dilution assay was performed as described above except that plates were incubated at 37 °C. All susceptibility tests were performed on at least three separate occasions.
Activation mechanisms of the Pdr1 signaling in the ipi1 R70H mutant
Although gain-of-function mutations in the transcription factor Pdr1 (e.g., Pdr1 P927S) upregulate the expression of its target genes in C. glabrata26,27,28, no PDR1 mutation was detected in strains Cg50 and Cg51. In this study, we constructed a pdr1P927S mutant in the wild-type and ipi1R70H backgrounds using the CRISPR-Cas9 technique29. The pdr1P927S mutant was slightly more resistant to fluconazole and voriconazole than the ipi1R70H mutant, and the pdr1P927S/ipi1R70H mutant was more resistant to both azoles than each single mutant strain (Fig. 3b).
Loss of the mitochondrial genome also induces constitutive activation of Pdr1-Cdr1 signaling, leading to azole resistance in C. glabrata26. However, the ipi1R70H mutant did not exhibit typical features of cells with mitochondrial DNA deficiency (so-called “petite phenotype”), such as the inability to grow in medium with non-fermentable carbon sources, the absence of mitochondrial staining, and a decrease in the copy number of mitochondrial DNA (mtDNA) (Supplementary Fig. 2). These results indicated that activation of Pdr1 signaling was not associated with mitochondrial dysfunction in the ipi1R70H mutant.
To obtain clues for understanding the mechanism of Pdr1 activation in the ipi1R70H mutant, we performed immunoprecipitation (IP) and mass spectrometry for Ipi1-binding factors and found that Rix1/Ipi2, Ipi3, and Ssb proteins specifically bound to wild-type Ipi1 but not to Ipi1R70H (Supplementary Fig. 3). Rix1/Ipi2 and Ipi3 were expected because Ipi1 is a component of the Rix1 (Ipi1-Rix1/Ipi2-Ipi3) complex in S. cerevisiae21. Ssb is a ribosome-bound Hsp70 chaperone that regulates translation efficiency by interacting with nascent polypeptide chains in S. cerevisiae30,31. Ssb is encoded by two nearly identical isoforms, SSB1 and SSB2, in C. glabrata (Candida genome database), similar to that in S. cerevisiae. To confirm the physical interaction between Ipi1 and Ssb, these proteins were tagged with three tandem repeats of FLAG (3 × FLAG) and HA, respectively, and subjected to sequential immunoprecipitation and immunoblotting (IB). Ssb-HA was detected specifically in the IP fraction of Ipi1-3 × FLAG but not in the mock IP fraction (Fig. 4a). In addition, since Ssz1 is a ribosome-related non-canonical Hsp70 chaperone interacting with Ssb in S. cerevisiae31,32, we also constructed an endogenously expressed Ssz1-HA and found that it specifically bound to Ipi1-3 × FLAG in C. glabrata (Fig. 4b). Furthermore, Ssb and Ssz1 were also bound to each other (Fig. 4c). These results suggested that Ipi1, Ssb, and Ssz1 formed a protein complex in C. glabrata. Next, we generated a strain containing PDR1 tagged with 3×FLAG and confirmed that Ssb-HA specifically bound to Pdr1-3 × FLAG (Fig. 4d). Although Ssz1 was nonspecifically selected in the flag pulldown, much larger amounts of Ssz1 were detected in the presence of Pdr1-3×FLAG (Fig. 4e). Collectively, these results suggest that Ssb and Ssz1 physically interact with Ipi1 and Pdr1 and with each other in C. glabrata.

Each protein C-terminally tagged with three tandem repeats of FLAG (3 × FLAG) or HA was expressed. Logarithmic-phase cells were lysed, and total protein was extracted. FLAG-tagged proteins were immunoprecipitated (IP) using an anti-FLAG antibody. Proteins were separated by SDS-PAGE, and immunoblotted (IB) using anti-FLAG or HA antibodies. a Ipi1-3×FLAG and Ssb-HA, (b) Ipi1-3 × FLAG and Ssz1-HA, (c) Ssb-3 × FLAG and Ssz1-HA, (d) Pdr1-3×FLAG and Ssb-HA, and (e) Pdr1-3 × FLAG and Ssz1-HA were analyzed. These experiments were repeated twice with similar results. Source data for all panels, including uncropped and unprocessed scans with molecular weight markers, are provided as a Source Data file.
Gene deletion strains were constructed to investigate the roles of Ipi1-intaracting proteins (Ssb and Ssz1) in antifungal resistance. Both SSB1 and SSB2 were deleted for the complete deletion of Ssb (named ssb∆). Single gene deletions of either SSB1 or SSB2 did not affect cell growth. SSB1, SSB2, and SSZ1 were not essential for cell viability. However, the ssb∆ and ssz1∆ strains grew slower than the wild-type strain on drug-free SC plates at both 30 °C and 37 °C (Fig. 5a, b, Supplementary Fig. 4a, b). Despite this growth defect, growth of the ssb∆ and ssz1∆ strains was not impaired by micafungin, fluconazole, and voriconazole at the concentrations tested. In contrast, growth of the wild-type and ssb1∆, ssb2∆ single deletants was severely inhibited. The reintroduction of intact SSB1 or SSZ1 into the corresponding deletion strains recovered these phenotypes to the wild-type levels. The additional ipi1R70H mutation did not affect the antifungal susceptibility of the ssb∆ and ssz1∆ strains (Fig. 5c, d, Supplementary Fig. 4c, d). The expression of CDR1, CDR2, and PDR1 increased in the ssb∆ and ssz1∆ strains (Fig. 5e, f), similar to that in the ipi1R70H mutant. Collectively, the ipi1R70H mutation did not have any additive effects on antifungal susceptibility and the expression of CDR1, CDR2, and PDR1 in the ssb∆ and ssz1∆ strains, suggesting that Ipi1, Ssb, and Ssz1 function in the same pathway to induce Pdr1 activation and antifungal resistance.

a–d Logarithmic-phase C. glabrata cells were serially 10-fold diluted and spotted onto SC plates containing micafungin, fluconazole, or voriconazole at the indicated concentrations. Plates were incubated at 30 °C for 96 h. e, f Expression levels of CDR1, CDR2, and PDR1 are elevated in the ssb∆ and ssz1∆ strains. qRT-PCR was performed as described in the methods section and in Fig. 2b. Data are expressed as the means ± standard deviations (*P < 0.05; ***P < 0.001; ****P < 0.0001, ordinary one-way ANOVA with Dunnett’s multiple comparison test). qRT-PCR was repeated on three independent occasions with similar results. Source data for panels (e) and (f) are provided as a Source Data file.
Exploration of micafungin resistance mechanisms
Hotspot mutations in FKS1 and FKS2 are the predominant cause of echinocandin resistance in C. glabrata3,33. However, no mutations were detected in any of the FKS genes (FKS1, FKS2, and FKS3) in strains Cg50 and Cg51. The ipi1R70H strain exhibited decreased susceptibility to all echinocandins tested in this study (micafungin, caspofungin and anidulafungin) (Supplementary Fig. 1). As echinocandins are not a substrate of C. glabrata Cdr134, the echinocandin susceptibilities of the wild-type and ipi1R70H strains were not affected by the deletion of CDR1. Although loss of Pdr1 slightly increased echinocandin susceptibility, the ipi1R70H mutation decreased echinocandin susceptibility even in the pdr1∆ background. Overall, loss of Cdr1 and Pdr1 had no or had marginal effects on echinocandin susceptibility in C. glabrata.
In Candida albicans, cells with thicker cell walls are less susceptible to echinocandins35. We examined the C. glabrata cells using transmission electron microscopy; however, no apparent difference in cell morphology, including cell wall thickness, was observed between the wild-type and ipi1R70H mutant (Supplementary Fig. 5a). The amount of total cell wall and the contents of β-1,3-D-glucan, β-1,6-D-glucan, and chitin were comparable between the wild-type and ipi1R70H strains (Supplementary Fig. 5b). In response to cell wall stress, C. glabrata activates the cell wall integrity pathway, which is mediated by the MAP kinase Slt2. The activation status of this signaling pathway is usually determined based on the phosphorylation levels of Slt2. C. glabrata cells were exposed to various concentrations of echinocandins and total protein was extracted. Western blot analysis showed that the phosphorylation levels of Slt2 in the ipi1R70H mutant were similar to or lower than those in the wild-type strain (Supplementary Fig. 5c).
Echinocandins exert fungicidal effects on actively growing cells but not on cells in the non-proliferative state36,37. In the course of our experiments, we noticed that echinocandin-resistant cells tended to grow slower than echinocandin-susceptible cells. Therefore, the C. glabrata strains used in this study were divided into two groups (Fig. 6a): group S (wild-type level susceptibility to echinocandins) and group R (echinocandin-resistant compared to the wild-type strain) according to the results of antifungal susceptibility assays (Supplementary Fig. 1 and Supplementary Fig. 4). Cell growth of the strains in group R was significantly slower than that of the strains in group S at the exponential growth phase (4–12 h), and the cells in both groups reached a similar level of cell density at 24–48 h (Fig. 6b). To further investigate the cellular response to micafungin, total RNA was extracted from control and micafungin-treated cells and subjected to RNA-seq analysis. In response to micafungin exposure, 372 and 337 genes were upregulated in the wild-type and ipi1R70H strains, respectively, and a total of 233 genes were commonly upregulated in both strains ( > 2-fold change in expression, adjusted P-value of <0.05) (Fig. 7). The expression levels of genes involved in cell wall organization, including SLT2, were commonly increased in both the wild-type and ipi1R70H strains (Supplementary Data 2). Gene ontology (GO) terms enriched in genes that were upregulated only in the wild-type strain were predominantly associated with RNA metabolic process including RNA processing and modification (Fig. 7 and Supplementary Data 2). In contrast, no significant ontology term could be found for genes upregulated only in the ipi1R70H mutant. Intriguingly, a significant set of genes belonging to mitochondrial function, including mitochondrial respiratory chain complex assembly and mitochondrion organization, was downregulated in response to micafungin exposure only in the ipi1R70H mutant. These results including the growth phenotypes and gene expression profiles are consistent with the idea that that antifungal activity of echinocandins depends on the metabolic/proliferative state of fungal cells.

a Growth curves of C. glabrata wild-type and various mutant strains. C. glabrata cells were grown in SC broth at 37 °C with agitation at 250 rpm. Based on the echinocandin susceptibility, C. glabrata strains were classified into two groups: group S (echinocandin-susceptible [wild-type level susceptibility to echinocandins]) and group R (echinocandin-resistant compared to the wild-type strain), shown as blue and red lines, respectively. The growth curves represent the mean of biological duplicates. b Optical densities at 600 nm (OD 600 nm) were compared at the indicated time points between the groups S and R (n = 7, each). Data are expressed as the means ± standard deviations. Individual dots represent the mean of biological duplicates for each strain as indicated in Fig. 6a. Statistical analysis was performed using Mann–Whitney U test. ns, not significant. Source data for all panels are provided as a Source Data file.

C. glabrata cells were exposed to 1 µg/mL of micafungin in SC broth at 37 °C for 1 h. RNA-seq analysis was performed as described in the materials and methods. Venn diagrams show the number of differentially expressed genes in cells treated with micafungin relative to untreated controls (>2-fold change in expression, adjusted P -value of <0.05, DESeq2 analysis applying a two-sided test for each gene with multiple comparison adjustments using the Benjamini-Hochberg method). Representative Gene Ontology (GO) terms are shown (P < 0.05, ~5 terms in order from the term with the lowest P-value, one-sided binomial test with multiple comparison adjustments using the Bonferroni method). Note that no statistically significant term was found for genes upregulated only in the ipi1R70H mutant. Source data are provided in Supplementary Data 2.
Involvement of calcineurin in MDR induced by the ipi1 R70H mutation
Calcineurin is a serine-threonine-specific protein phosphatase required for echinocandin and azole resistance in C. glabrata38. Calcineurin is a heterodimer consisting of a catalytic subunit A (Cna1) and a Ca2+-binding regulatory subunit B (Cnb1), and the association between the two subunits is necessary for phosphatase activity. To examine whether calcineurin is involved in the antifungal resistance induced by the ipi1R70H mutation, we deleted CNA1 in the wild-type and ipi1R70H backgrounds. The cna1∆ mutant was highly susceptible to fluconazole as expected (Fig. 8a). The ipi1R70H mutation induced fluconazole resistance even in the cna1∆ mutant. However, the ipi1R70H mutant was more resistant to fluconazole than the cna1∆/ipi1R70H mutant. These susceptibility phenotypes were reproduced by the deletion of CNB1 in the wild-type and ipi1R70H backgrounds. In addition, similar results were obtained for echinocandin susceptibility. Both the cna1∆ and cnb1∆ mutants were highly susceptible to all the echinocandins tested. The ipi1R70H mutation decreased echinocandin susceptibility even in the absence of calcineurin. However, similar to the findings in the fluconazole susceptibility described above, echinocandin resistance was not fully induced by the ipi1R70H mutation in the strains lacking calcineurin. In a previous study, caspofungin exposure increased the expression of FKS2 encoding the target enzyme of echinocandins, and this effect was partially blocked by the calcineurin inhibitor FK506 in C. glabrata39. Similarly, the present study demonstrated that FKS2 expression was increased following micafungin exposure, and it was partially impaired by the loss of calcineurin in both the wild-type and ipi1R70H strains (Fig. 8b).

a Either calcineurin catalytic subunit A (CNA1) or regulatory subunit B (CNB1) was deleted in the wild-type and ipi1R70H backgrounds, and the antifungal susceptibility was examined using a spot dilution assay. Logarithmic-phase cells of the C. glabrata strains were serially diluted and spotted on SC plates containing an antifungal agent at the indicated concentrations. Plates were incubated at 37 °C for 2 days. b Expression level of FKS2 in the presence and absence of micafungin was determined by qRT-PCR. Logarithmic-phase cells of the C. glabrata strains were exposed to 1 µg/mL of micafungin in SC broth at 37 °C for 1 h. qRT-PCR was performed as described in the materials and methods and in Fig. 2b. Individual dots are shown for biological replicates (n = 6, each). The means ± standard deviations are shown (ns, not significant; Kruskal-Wallis test with Dunnett’s multiple comparison test). c C. glabrata cell survival was assessed before and after exposure to micafungin. Logarithmic-phase cells of the C. glabrata strains were exposed to 0.2 µg/mL of micafungin in SC broth at 37 °C. The CFUs were determined at the indicated time points, and percent CFU was calculated relative to the CFU prior to micafungin exposure. Data are presented as mean values ± standard deviations. The experiments shown in panels (a–c) were repeated on three independent occasions with similar results. Source data for panels (b) and (c) are provided as a Source Data file.
For fungicidal drugs, such as echinocandins against Candida species, antifungal tolerance is defined as the ability of Candida cells to survive drug exposure for a longer time without an increase in MIC40. In the time-kill assay (Fig. 8c), treatment with micafungin decreased the viable cell counts of the wild-type strain over time and exerted a potent fungicidal effect against the cna1∆ mutant. In contrast, the fungicidal effect of micafungin was incomplete for the ipi1R70H mutant. Although a temporary reduction in the number of viable cells was observed, the ipi1R70H mutant cells survived for a longer time than the wild-type cells and the number of viable cells at 24 h was slightly increased compared to that at 8 h. The cna1∆/ipi1R70H double mutant exhibited a phenotype intermediate between the cna1∆ and ipi1R70H single mutant strains. Similar results were observed in the assays using different concentrations of micafungin (Supplementary Fig. 6). Consistent with the findings described above, loss of calcineurin partially diminished echinocandin tolerance of the ipi1R70H mutant.
Effects of the ipi1 R70H mutation on virulence in C. glabrata
As the acquisition of MDR without compromising pathogenicity is a serious threat, we examined effects of the ipi1R70H mutation on key virulence traits of C. glabrata. Adhesion to host tissues or medical devices is the primary step in the process of infection. Elm1 is a serine/threonine kinase involved in the fungal cell wall organization, and loss of Elm1 results in increased adhesion in C. glabrata41. In our adhesion assay, the elm1∆ mutant highly adhered to agar plates as expected, whereas there was no apparent difference in adhesion capacity between the wild-type and the ipi1R70H strains (Supplementary Fig. 7).
Phagocytes are the first line of host defense against Candida infections. We evaluated the resistance of the C. glabrata cells to phagocytosis by counting viable cells after incubation with murine RAW 264 macrophages. As shown in Fig. 9a, there was no significant difference in susceptibility to macrophage killing between the wild-type and the ipi1R70H strains. We also examined the virulence of the ipi1R70H mutant using two different in vivo models. In a mouse model of disseminated candidiasis, the fungal burdens in the kidney, spleen, and liver were comparable between mice infected with the wild-type strain and those infected with the ipi1R70H mutant (Fig. 9b). In the silkworm infection model, the survival rates of larvae infected with the ipi1R70H mutant (no-drug control) were similar to those of larvae infected with the wild-type strain (Table 1). Colony forming units (CFUs) of the ipi1R70H mutant recovered from larvae with no drug treatment were also comparable with those of the wild-type strain (Fig. 9c, d). These results suggested that the ipi1R70H mutation did not attenuate the virulence of C. glabrata.

a Susceptibility of the C. glabrata ipi1R70H mutant to macrophage killing. Logarithmic-phase cells of C. glabrata wild-type strain (CBS138) and the ipi1R70H mutant were cocultured with murine RAW 264 macrophages at 37 °C for 2 h. Percent killing is expressed as the percent reduction of CFU recovered from cocultures compared with the CFU from control cultures (C. glabrata cells without macrophages). All data obtained from the two independent experiments were compared between the wild-type and ipi1R70H strain groups (n = 14, each). The difference was not statistically significant (ns, not significant; two-tailed Mann–Whitney U test). b A mouse model of disseminated candidiasis. Immunocompetent mice were intravenously inoculated with 8 × 107 cells of either C. glabrata wild-type or ipi1R70H strain (n = 9 for the wild-type strain and n = 10 for the ipi1R70H mutant). Bilateral kidneys, spleen, and liver were excised 7 days after injection. Appropriate dilutions of organ homogenates were plated, and the numbers of CFU were counted after 2 days of incubation at 30 °C. Numbers of recovered CFU from each organ are indicated for individual mice in the scatter plots. The geometric mean is shown as a bar. ns, not significant (two-tailed Mann–Whitney U test). The representative data of two independent experiments are shown. c, d A silkworm infection model. Larvae were inoculated with 2.0–2.5 × 107 cells of each C. glabrata strain and injected with an echinocandin-class antifungal agent (micafungin, caspofungin, or anidulafungin) dissolved in saline or saline alone (50 μL). Twenty-four hours later, hemolymph was collected and spread on YPD plates to calculate CFU. The number of larvae is shown in parenthesis. Data were analyzed and presented as described above (ns, not significant; two-tailed Mann–Whitney U test). Source data for all panels are provided as a Source Data file.
In vivo treatment efficacy of echinocandins for the ipi1 R70H mutant
The efficacy of micafungin treatment was evaluated by monitoring survival and CFU in a silkworm infection model (Table 1 and Fig. 9c, d). Larvae infected with the wild-type and ipi1R70H strains were treated with micafungin at two concentrations (1 and 4 µg micafungin/larva). Low-dose micafungin (1 µg/larva) was efficacious for larvae infected with the wild-type strain but not for those infected with the ipi1R70H mutant; survival rates at 48 h after infection were 73.3% (11/15) and 0% (0/14), respectively (Table 1). CFUs of the wild-type strain were also significantly lower than those of the ipi1R70H mutant after treatment with low-dose micafungin (Fig. 9c). A high dose (4 µg/larva) of micafungin was efficacious for both wild-type and ipi1R70H strains (Table 1 and Fig. 9c). Similarly, the treatment efficacy of caspofungin and anidulafungin for larvae infected with the wild-type and ipi1R70H strains was evaluated (Table 1 and Fig. 9d). Collectively, the ipi1R70H mutant exhibited a decreased but dose-dependent susceptibility to all the three echinocandins in the silkworm infection model.
Discussion
In this study, an ipi1R70H mutation appeared to be induced by chance during the growth of C. glabrata wild-type cells in the presence of low concentrations of micafungin. Intriguingly, this single amino acid mutation induced resistance to two distinct antifungal classes, echinocandin and azole antifungals. Activation of drug efflux mediated by Pdr1-Cdr1 signaling was the primary mechanism for azole resistance in the ipi1R70H mutant. Further analyses revealed a physical and functional association between Ipi1 and Pdr1 signaling via ribosome-associated chaperones Ssb/Ssz1 in C. glabrata. The present study demonstrated echinocandin tolerance of the ipi1R70H mutant in vitro and in vivo and suggested a possible link between the metabolic/proliferative state of C. glabrata cells and echinocandin susceptibility. The ipi1R70H mutation induced little fitness cost in C. glabrata and did not affect virulence in vivo.
Multiple interfaces control Pdr1 signaling in C. glabrata42. Vu et al. reported that reduction of ergosterol (e.g., blocks in the ergosterol biosynthesis pathway) elevates Pdr1 activity in C. glabrata even in the absence of azole antifungals43. However, this was not the case with the ipi1R70H strain because the ergosterol level was increased, not decreased, in the ipi1R70H mutant compared to that in the wild-type strain. The susceptibility phenotypes of the pdr1∆/ipi1R70H mutant to low concentrations of fluconazole suggested a minor role for Ipi1 in fluconazole resistance outside of the Pdr1 pathway. The increase in ergosterol content may have been marginally involved in fluconazole resistance by a mechanism other than Pdr1 activity in the ipi1R70H mutant, although its involvement in voriconazole resistance was unlikely (Fig. 3a). Neither the gain-of-function mutations in PDR1 nor mitochondrial dysfunction were relevant to the activation mechanism of Pdr1 signaling in the ipi1R70H mutant. The present study revealed the physical and functional interaction of Ipi1 with Ssb and Ssz1. This study also suggested that the Ipi1-Ssb-Ssz1 complex functions as a negative regulator of Pdr1 in C. glabrata. The ipi1R70H mutation disrupts the interaction of Ipi1 with Ssb and Ssz1, releasing the negative regulation of Pdr1 and leading to the constitutive activation of azole efflux pumps downstream of Pdr1 signaling.
Intriguingly, Ssz1 acts as a positive regulator of Pdr1 in S. cerevisiae44,45; therefore, the Hsp70-based machinery observed in C. glabrata has the opposite effect to that in S. cerevisiae. This scenario is partially supported by the mutant phenotype of the J protein Jjj1, a member of the DnaJ/Hsp40 family involved in rRNA processing by stimulating Hsp70 chaperone activity46. Deletion of JJJ1 results in increased susceptibility to azoles in S. cerevisiae47 but decreased fluconazole susceptibility and increased Pdr1-mediated target gene expression in C. glabrata48. These findings suggest a mode of activation by gain-of-function mutations in Pdr1, involving the release of Pdr1 binding with negative regulators such as Jjj1, Ssb, and Ssz1. Although multiple gain-of-function mutations including P927S have been reported for C. glabrata Pdr1, which mutations affect interactions with which regulators remains unclear. The ipi1R70H and pdr1P927S mutations had additive effects on azole resistance. In addition, Vu et al. reported that calcineurin is required for normal activity of gain-of-function forms of Pdr149. The ipi1R70H mutation conferred fluconazole resistance even in the absence of calcineurin. However, calcineurin was required for the full induction of fluconazole resistance by the ipi1R70H mutation. These results suggest that multiple positive and negative inputs are involved in the regulation of Pdr1 activity. Although complex interactions of ribosome-associated Hsp70 chaperones, including Ssb and Ssz1, have been extensively studied in S. cerevisiae50, the detailed molecular mechanisms of Pdr1 regulation by these molecules should be studied directly in C. glabrata.
Candida glabrata strains with acquired MDR to azoles and echinocandins have been isolated even after short-term antifungal treatment2,6,7,8. In vitro exposure to echinocandins also results in easy development of resistance, mainly through induction of FKS mutations51,52,53. Shields et al. reported that among the three echinocandins, induced mutational frequencies of resistance were the highest for caspofungin and lowest for micafungin54. In the present study, treatment of C. glabrata cells with micafungin resulted in decreased micafungin susceptibility independent of FKS mutation. Similarly, clinical isolates of C. glabrata and Aspergillus fumigatus have increased echinocandin MIC values without an FKS mutation55,56. These observations highlight the clinical importance of FKS mutation-independent mechanisms for echinocandin resistance. C. glabrata cells withstand drug pressure, through drug tolerance or adaptation, by activating stress response pathways, such as the cell wall integrity pathway, and they subsequently acquire stable genetic resistance, which relies on FKS mutations57. However, the phosphorylation levels of Slt2 after exposure to echinocandins suggested that the activation level of the cell wall integrity pathway in the ipi1R70H mutant was not higher than that in the wild-type strain. The C. glabrata ipi1R70H mutant did not have the key mechanisms of echinocandin resistance including mutations in FKS genes, increased cell-wall thickness, and alterations in cell wall composition (e.g. increased chitin content by the activation of the cell-wall integrity pathway).
The growth phenotypes and genome-wide expression profiles of the C. glabrata strains suggested a possible link between the metabolic/proliferative state and echinocandin susceptibility. In response to micafungin exposure, while the wild-type cells tend to facilitate cell growth by up-regulating genes involved in RNA processing, the ipi1R70H mutant cells repressed the metabolic/proliferative state by down-regulating genes involved in ribosome biogenesis and mitochondrial organization in agreement with previous findings that acute mitochondrial inhibition enhances echinocandin tolerance58,59. Garcia-Rubio et al. demonstrated that chemical inhibition of mitochondrial complexes I and IV increased resistance to echinocandin-mediated cell killing, whereas mutants lacking multiple mitochondrial components are hypotolerant to echinocandins, indicating the complexity of mitochondrial involvement in micafungin tolerance59. Cell death upon echinocandin exposure is not attributed to accumulation of reactive oxygen species per se; however, it is affected by mitochondrial status downstream of the echinocandin-Fks interaction.
Calcineurin plays important roles in compensatory responses to various stresses including cell wall damage to promote fitness, cell survival, and proliferation38,60. Phenotypic analysis of the ipi1R70H and calcineurin mutants suggested that the ipi1R70H mutation decreased echinocandin susceptibility through a mechanism different from that mediated by calcineurin. However, calcineurin was required for the full induction of echinocandin resistance by the ipi1R70H mutation. Transcriptional activation of FKS2 by calcineurin is linked to echinocandin resistance in C. glabrata39. Both in the wild-type and ipi1R70H strains, FKS2 expression was similarly increased following micafungin exposure, and this effect was partially impaired by the loss of calcineurin.
Although the acquisition of drug resistance is often accompanied by a reduction in in vivo fitness in pathogenic fungi6,61, the ipi1R70H mutation induced MDR associated with a trivial fitness cost in C. glabrata. The ipi1R70H mutation alone did not affect cell wall components and thickness, adhesion capacity, resistance to macrophage killing, and survival in mouse and silkworm infection models, suggesting that the ipi1R70H mutant maintained the normal pathogenicity of C. glabrata. Future studies using clinical isolates are warranted to evaluate the clinical relevance of the resistance mechanisms identified in the present study.
Methods
Media, reagents, and culture conditions
Candida glabrata cells were routinely propagated at 30 °C in yeast extract-peptone-dextrose (YPD) medium (1% yeast extract, 2% peptone, and 2% dextrose; Difco Laboratories, Detroit, MI), synthetic defined (SD) medium (2% dextrose and 0.67% yeast nitrogen base without amino acids; Difco Laboratories), synthetic complete medium (SC), SC lacking tryptophan (SC-trp), or uracil (SC-ura) medium (59), unless otherwise indicated. SC medium containing non-fermentable carbon sources (3% glycerol and 2% ethanol) instead of dextrose was used to examine petite growth phenotype. Micafungin (Astellas, Tokyo, Japan) and fluconazole (Sigma-Aldrich, St. Louis, MO) were dissolved in distilled water, and voriconazole (Pfizer, New York, NY) was dissolved in dimethyl sulfoxide (DMSO). Antifungal agents were stored at −20 °C until use.
Isolation of micafungin-resistant C. glabrata by sequential exposure to step-up dosage of micafungin
Logarithmic-phase cells of echinocandin-susceptible C. glabrata wild-type strain CBS138 were incubated on YPD plates with increasing concentrations of micafungin. Some colonies appeared on a plate containing 0.015 µg/mL micafungin but not in the presence of 0.03 µg/mL micafungin. Subsequently, a single colony was picked from the plate containing 0.015 µg/mL micafungin, suspended in distilled water, and then spread on a plate containing 0.03 µg/mL micafungin. The plates were incubated at 30 °C for 2 days.
Drug susceptibility assay
Spot dilution tests were performed as previously described38. Briefly, logarithmic-phase cells grown in SC, SC-trp, or SC-ura broths were harvested and adjusted to a 2 × 107 cells/mL concentration. Serial 10-fold dilutions were prepared, and 5 µL of each dilution was spotted onto an agar plate (SC, SC-trp, or SC-ura) containing the test compound at the desired concentration. The plates were incubated at 30 °C for 48–96 h unless otherwise indicated. DMSO alone did not interfere with cell growth at the final concentration used in this study.
MICs of antifungal agents were determined by the EUCAST broth dilution method (https://www.eucast.org/astoffungi/methodsinantifungalsusceptibilitytesting/susceptibility_testing_of_yeasts/) and confirmed using the Sensititre YeastOne colorimetric susceptibility test (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer’s instructions. Candida parapsilosis ATCC 22019 and Candida krusei ATCC 6258 were used as the quality control strains. Clinical breakpoints established by EUCAST were applied for the categorization of antifungal resistance in C. glabrata: MIC > 0.03 mg/L for micafungin, MIC > 0.06 mg/L for anidulafungin, and MIC > 16 mg/L for fluconazole.
A time-kill assay was performed as previously described38. Briefly, logarithmic-phase cells grown in SC broth were adjusted to a 1 × 105 cells/mL and exposed to micafungin at the concentrations of 0.2 and 0.5 μg/mL with agitation at 250 rpm for 24 h. An aliquot of 100 μL of appropriate dilutions was spread on YPD plates in triplicate to determine viable cell counts. The CFUs at each time point were determined, and the percent CFU was calculated relative to that prior to micafungin exposure. All susceptibility tests were performed on at least three separate occasions to ensure reproducibility.
Whole-genome sequencing
Whole-genome sequencing of the C. glabrata strain Cg51 was performed by Hokkaido System Science (https://www.hssnet.co.jp/index_e.htm). Briefly, the whole-genome fragment library was constructed using the TruSeq DNA Sample Prep Kit (Illumina, Tokyo, Japan) and sequenced on the Illumina HiSeq2000 platform using single reads (100 bp). Data from a total of 69,677,003 reads were selected by Illumina chastity filtering (% pass-filter was 96.66% and % of ≥ Q30 high-quality bases was 96.96%). After adapter trimming with Cutadapt (https://doi.org/10.14806/ej.17.1.200), the resulting 69,269,648 read data were used for mapping with the CBS138 reference genome obtained from the Candida genome database (http://www.candidagenome.org).
Complementation assay using the C. glabrata cDNA library
Total RNA was isolated from the wild-type strain CBS138, and poly(A) RNA was purified using the Oligotex-dT30<Super> mRNA Purification Kit (Takara Bio Inc., Shiga, Japan) to construct a C. glabrata cDNA library. The poly(A) RNA quality was verified using an Agilent Bioanalyzer (Agilent, Tokyo, Japan). cDNA was synthesized from poly(A) + RNA using an oligo(dT)18 anchor primer containing 5′-BamHI and 3’-XhoI sites, digested with BamHI and XhoI, and directionally cloned into the BamHI-SalI site of pCgACT-P. This primary library was introduced into E. coli DH10B by electroporation (1.5 kV, 200 Ω, 25 μF) using an E. coli Pulser (Bio-Rad, Hercules, CA). The resulting cDNA library contained ~1.05 × 106 clones in E. coli DH10B at a concentration of 5.0 × 109 CFUs/mL.
The cDNA library was extracted using the PureYield Plasmid Maxiprep System (Promega, Tokyo, Japan) and resuspended in distilled water at a concentration of 1 mg/mL. Subsequently, 3 mL of logarithmic-phase C. glabrata Cg50-trp1 cells (5 × 107 cells/mL) were transformed with 10 μL of cDNA library (1 mg/mL) using the lithium acetate method as described previously62. Transformants were selected based on tryptophan prototrophy and growth capacity at 41 °C.
Strain and plasmid construction
The C. glabrata strains, primers, and plasmids used in this study are listed in Supplementary Tables 2, 3, and 4, respectively. All plasmids constructed using PCR products were verified by sequencing before use. Sequence information of the C. glabrata genes was obtained from the Candida genome database. To introduce an R70H mutation into a C. glabrata wild-type strain, a G209A mutation was generated in CgIPI1-cMyc-TRP1-3’UTR/pBSK using the KOD-Plus-Mutagenesis Kit (Toyobo, Osaka, Japan) and mutagenic primers CgIPI-F208mut and CgIPI-R207mut, yielding CgIPI1(R70H)-cMyc-TRP1-3’UTR/pBSK. Strain 2001T was transformed with the mutagenesis construct amplified from CgIPI1 (R70H)-cMyc-TRP1-3’UTR/pBSK using the primer pair CgIPI-F1 and CgIPI-R401FL, yielding strain ipi1R70H. Successful introduction of the G209A mutation was confirmed by direct sequencing of the IPI1 gene.
The IPI1 native promoter in ACG22 was replaced with a tetracycline-responsive promoter to construct a C. glabrata tet-IPI1 strain, as described previously63. The tetracycline analog doxycycline turns off the tet-promoter function in C. glabrata. Candida glabrata deletion strains were constructed using a one-step PCR-based technique as described previously64. Briefly, a deletion construct was amplified from either pBSK-HIS or pBSK-TRP using primers tagged with 100 bp sequences homologous to the flanking regions of the target open reading frame (ORF). Candida glabrata parent strains were subsequently transformed with the deletion construct, and the resulting transformants were selected using tryptophan or histidine prototrophy, as appropriate. The transformation of C. glabrata was performed using the lithium acetate protocol unless otherwise noted62. Successful homologous recombination was verified by diagnostic PCR, and the absence of mRNA expression of target genes was confirmed by qRT-PCR.
To construct a Pdr1 gain-of-function allele, a C2779T mutation was introduced using the CRISPR-Cas9 technique as described previously29 except donor DNA and CRISPR RNA (crRNA) preparation (Supplementary Fig. 8). Briefly, In-Fusion PCR was performed to generate the donor DNA cassette containing the NAT1 marker between the ORF and 3’ UTR of PDR1. Guide RNA (gRNA) was made by incubating the crRNA and universal trans-activating crRNA (4 μM each) at 95 °C for 5 min and mixed with Cas9 nuclease (4 μM) in a 1.2:1 ratio to create gRNA/Cas9 complex. C. glabrata wild-type and ipi1R70H strains were transformed with the mixture of gRNA/Cas9 and donor DNA using electroporation (Gene Pulser Xcell, Bio-Rad, Hercules, CA, USA). Transformants were incubated in YPD broth overnight and then plated on YPD agar containing nourseothricin at 37 °C. The desired C2779T substitution in PDR1 without an off-target mutation was confirmed by DNA sequencing.
Deletion strains of SSB1 and SSZ1 were transformed with pGRB2.2-SSB1, pCgACTP-SSZ1, and each empty vector, to construct SSB1- and SSZ1-add backed, and control strains, respectively. Transformants were selected based on uracil or tryptophan prototrophy and verified using qRT-PCR. Either FLAG- or HA-tagged Ssb and Ssz1 were expressed by integrating these tag sequences at the C-terminus of the SSB1 and SSZ1 ORFs in the genome. The strains were constructed using the one-step PCR-based technique described above. Transformed PCR products, including SSB1 or SSZ1 ORF, tag sequences, C. glabrata HIS3 or S. cerevisiae URA3 genes, and SSB1 or SSZ1 3′UTR, were amplified from pBSK-HIS-SSB1-HA, pBSK-HIS-SSZ1-HA, or pBSK-URA-SSB1-3×FLAG. C-terminally FLAG-tagged Ipi1 and Pdr1 were expressed using exogenously transformed pCgACTP-IPI1-3×FLAG and pCgACTP-PDR1-3×FLAG, respectively. Growth curves of the C. glabrata strains used in this study are shown in Supplementary Fig. 9. Spot dilution assays confirmed that these C-terminal fusion proteins retained normal activities (Supplementary Fig. 10).
qRT-PCR
qRT-PCR was performed as previously described64. Briefly, total RNA of logarithmic-phase cells grown in SC, SC-trp, or SC-ura broth was extracted using the RNeasy mini kit (Qiagen, Valencia, CA) and reverse-transcribed using a QuantiTect Reverse Transcription kit (Qiagen) to synthesize first-strand cDNA. Further, 3 µL of the resulting cDNA was used as the template for individual PCR with a QuantiTect SYBR Green PCR kit (Qiagen) and gene-specific primers. qRT-PCR was carried out in a 96-well plate using the QuantStudio 12 K Flex System (Thermo Fisher Scientific). The mRNA abundance of target genes was normalized to that of ACT1. RNA extraction and qRT-PCR were performed in triplicate in three independent experiments.
qPCR of mtDNA
The relative mtDNA copy number was determined by quantitative PCR (qPCR) as described previously65. DNA was extracted using the Quick-DNA Fungal/Bacterial Miniprep Kit (Funakoshi, Tokyo, Japan). mtDNA (COX1) and nuclear DNA (ACT1) were amplified with primer pairs qPCR-COX1-F/R and CgACT1-F/ R, respectively. DNA extraction and qPCR were performed in triplicate in three independent experiments.
Flow cytometric analysis of the intracellular concentration of rhodamine 6 G
The intracellular concentration of the fluorescent dye rhodamine 6 G (Sigma-Aldrich), a substrate of azole efflux transporters26, was measured by flow cytometry. A mutant lacking the Cdr1 efflux pump (cdr1Δ) was used as the control. C. glabrata cells (2.0 × 107 cells/mL) were pre-cultured in 5 mL of SC medium at 30 °C for 3.5 h. Then, rhodamine 6 G was added at a final concentration of 0 or 10 µM. After 2 h of incubation at 30 °C with constant shaking, the culture medium was cooled on ice to stop the transport of rhodamine 6 G and then 10-fold diluted in cold sterile phosphate-buffered saline (PBS) at pH 7.4. The suspension was used for flow cytometric analysis.
Intracellular concentration of rhodamine 6 G under de-energized conditions was measured to evaluate the passive infusion of rhodamine 6 G. The yeast overnight culture in SC medium was washed three times with PBS and resuspended in 5 mL of PBS at ~1.0 × 108 cells/mL. To inhibit glycolysis, 2-deoxy-D-glucose was added to the cell suspension at a final concentration of 5 mM. After 2 h of incubation at 30 °C with constant shaking, cells were washed once with PBS and resuspended in 1 mL of PBS with rhodamine 6 G at a final concentration of 10 µM. After 15 min of incubation on ice, the cell suspension was 10-fold diluted in cold sterile PBS and immediately used for flow cytometry analysis. The fluorescence of 20,000 cells was measured with an LSR II cytometer (BD Biosciences, San Jose, CA) using a PE filter. Data were analyzed using FlowJo software 7.6.5 (TreeStar Inc., Ashland, OR).
Northern blotting
Logarithmic-phase C. glabrata cells were cultured at 30 °C in a YPD medium and incubated at 42 °C. Total RNA was extracted at the time points of 0, 2, and 6 h. Northern blotting was performed using the DIG Northern Starter Kit (Roche, Basel, Switzerland), according to the manufacturer’s instructions. C. glabrata internal transcribed spacer 2 (ITS2) sequences (230 nt, GenBank: U70498.1) were used as probes.
Immunoblotting
To explore Ipi1-binding factors, whole protein was extracted from CBS138 and the ipi1R70H mutant, and silver staining was performed with standard procedures using a silver staining kit for mass spectrometry (SP-4020; APRO Science Group, Tokushima, Japan). This experiment was repeated twice on independent occasions. The samples obtained from the second experiment were subjected to the following mass spectrometry. Mass spectrometry was performed by APRO Life Science Institute (Tokushima, Japan). The data were analyzed using the NCBI database and the Scaffold viewer (http://www.proteomesoftware.com/products/scaffold-5).
Anti-FLAG (F1804; Sigma-Aldric), anti-HA (#901513; BioLegend, San Diego, CA), and anti-mouse IgG-hrp (NA931; GE Healthcare, Pittsburgh, PA) antibodies were purchased. HA-tagged Ssb was expressed by integrating the HA sequence at the C-terminus of the SSB1 ORF in the genome. Ipi1 C-terminally tagged with 3×FLAG was expressed from a plasmid under the control of the S. cerevisiae PGK1 promoter because the endogenous expression levels of Ipi1 were too low to analyze by IP/IB. Logarithmic-phase C. glabrata cells were lysed using the Minute Total Protein Extraction Kit for Microbes with Thick Cell Walls (Invent Biotechnologies, Plymouth, MN) according to the manufacturer’s instructions. Lysates were separated by SDS–PAGE and transferred to polyvinylidene difluoride membranes (Bio-Rad). Each protein was detected using the indicated antibodies, an enhanced chemiluminescent substrate (Thermo Fisher Scientific), and ChemiDoc Touch imaging system (Bio-Rad).
For Slt2 western blotting, logarithmic-phase cells were washed twice with ice-cold deionized water, suspended in 200 μL of homogenizing buffer (50 mM Tris [pH 7.5] and 1 mM EDTA) containing 1 × PhosSTOP (Roche, Switzerland) and 1 × Complete (EDTA-free; Roche), and lysed by vigorous vortexing with glass beads. Cell debris and unbroken cells were removed by centrifugation (13,000 × g) at 4 °C for 10 min. The protein content was quantified using an XL-Bradford protein assay kit (Integrale, Tokushima, Japan) according to the manufacturer’s instructions. Next, 40 μg of the total protein was resolved by SDS-PAGE on a 10% gel and transferred onto PVDF membranes (PALL Life Science), which were then blocked using Blocking One-P (Nacalai Tesque) for 1 h at 20–23 °C. Immunoblotting was conducted using an anti-phosphorylated-p44/42 MAP kinase antibody (#4370; Cell Signaling Technology, Danvers, MA) at a dilution of 1:10,000 in TBS and 0.1% Tween 20 for 1 h at room temperature. HRP-linked anti-rabbit IgG (W4011; Promega) was used as the secondary antibody, and the blots were developed using an ECL plus western blotting detection system (GE Healthcare). The assays were independently repeated at least twice.
Transmission electron microscopy
Candida glabrata cells (4 × 107 cells per sample) were pelleted by centrifugation at 1,000 × g, gently washed with 0.1 M MOPS buffer (pH 7.0), and resuspended in the fixative solution (2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer, pH 7.4) to fill the microfuge tube completely. The cell morphology was examined using a transmission electron microscope at Rocky Mountain Labs/NIAID/NIH (Hamilton, MT).
Analysis of cell wall contents
Measurements of the amount of total cell wall, preparation of the alkali-insoluble fraction, and analysis of cell wall composition were performed as described previously66. Candida glabrata cells were cultured in a liquid SC medium at 37 °C until they reached the logarithmic phase. Next, the cells were washed three times with distilled water, collected by centrifugation, and extracted using 1% NaOH at 100 °C for 24 h. The supernatant obtained was neutralized, dialyzed using deionized water, and lyophilized. The pellet obtained was washed twice with distilled water and extracted using 0.5 M acetic acid at 80 °C for 24 h. The pellet was washed twice with distilled water and lyophilized. The former was defined as the alkali-soluble fraction and the latter as the alkali-insoluble fraction. The sugar content of each fraction was determined using the phenol H2SO4 method.
The lyophilized alkali-insoluble fraction (1.0 mg) was suspended in 10 mM Tris-HCl (pH 7.4) containing 1.0 mg/mL Zymolyase-100T (Nacalai Tesque, Kyoto, Japan) and incubated at 37 °C for 24 h to determine the alkali-insoluble β-glucan content. The precipitate obtained was removed by centrifugation at 15,500 × g for 10 min, and half of the supernatant was dialyzed overnight against 10 mM Tris-HCl (pH 7.4). The hexose content was determined using the phenol H2SO4 method.
To determine chitin content, 20 mg of the lyophilized alkali-insoluble fraction was hydrolyzed using 1 M H2SO4 at 100 °C for 4 h and neutralized. The precipitate obtained was removed by centrifugation, and the supernatant was dissolved in 2 mL of deionized water after evaporation to dryness. Next, 500 μL of the solution was added to 1 mL of acetylacetone solution (10% [v/v] acetylacetone in 1.25 M sodium carbonate) and incubated at 90 °C for 1 h, followed by addition of 10 mL of 100% ethanol and 1 mL of Reissig reagent (1% 4-dimethylaminobenzaldehyde, 1.25% [v/v] HCl in glacial acetic acid). Chitin content was determined by measuring the absorbance of glucosamine at 490 nm.
Sterol analysis
Sterol analysis was performed as previously described67. Logarithmic-phase cells of C. glabrata wild-type (CBS138) and ipi1R70H strains were adjusted to 1.0 × 105 cells/mL and grown in SD broth with or without sub-MIC (8 μg/mL) of fluconazole until the cell concentration reached 2.0 × 108 cells/mL. C. glabrata cell pellets (~1.0 × 109 cells) were harvested, washed twice with 0.5% Tween-80, and then rewashed with and resuspended in 1% NaCl. Ten micrograms of sitosterol dissolved in chloroform were added as an internal control. Total lipids were extracted using the Bligh-Dyer method (66) and saponified with 0.4 M methanolic KOH68. The extracted lipids were dried under N2 gas and dissolved in 100 μl of chloroform/methanol (2:1). Ten microliters of the aliquot were injected onto a C-18 column (COSMOSIL 5C18-AR-II 4.6 × 150 mm) and analyzed by reverse-phase high-performance liquid chromatography (HPLC) using an Alliance HPLC system with a Waters 2695 separation module and 2489 UV/visible detector (Waters Corporation, Milford, MA). The mobile phase consisted of 99.5% methanol and 5 mM CH3COONH4. The elution of compounds was automatically monitored by absorption at 210 nm. For sterol quantification, standard curves were prepared using commercially available sterols (ergosterol, lanosterol, sitosterol, and squalene; Sigma-Aldrich). The dynamic range of the sterol standard curve was 10 ng–6 µg per injection. All experiments were performed in triplicate on three independent occasions.
RNA-sequencing (RNA-seq)
Logarithmic-phase cells of C. glabrata wild-type (CBS138) and ipi1R70H strains were exposed to 1 µg/mL of micafungin in SC broth at 37 °C for 1 h. Total RNA was extracted using the RNeasy mini kit (Qiagen). RNA sequence library preparation, sequencing, mapping, gene expression, and GO enrichment analysis were performed by DNAFORM (Yokohama, Kanagawa, Japan). Qualities of total RNA were assessed by Bioanalyzer (Agilent) to ensure that the RNA integrity number (RIN) was over 7.0. After poly (A) + RNA enrichment by the NEBNext Poly(A) mRNA Magnetic Isolation Module (New England BioLabs), double-stranded cDNA libraries (RNA-seq libraries) were prepared using SMARTer stranded RNA-Seq kit (Clontech) and MGIEasy Universal Library Conversion Kit (MGI Tech) according to the manufacturer’s instruction. RNA-seq libraries were sequenced using paired end reads (150 nt of read1 and read2) on a DNBSEQ-G400RS instrument (MGI Tech). The obtained raw reads were trimmed and quality-filtered using the Trim Galore! (version 0.6.7), Trimmomatic (version 0.39), and cutadapt (version 3.7) software. Trimmed reads were then mapped to the C. glabrata CBS138 genome using STAR (version 2.7.10a). Reads of annotated genes were counted using featureCounts (version 2.0.1). FPKM values were calculated from mapped reads by normalizing to total counts and the transcript. Differentially expressed genes were detected using the DESeq2 package (version 1.20.0). The list of differentially expressed genes detected by DESeq2 (base mean > 1 and fold-change <0.5, or base mean > 1 and fold-change > 2) was used for GO enrichment analysis by the Candida Genome Database GO Term Finder (http://www.candidagenome.org/cgi-bin/GO/goTermFinder). The RNA-seq data that support the findings of this study have been deposited in NCBI’s Gene Expression Omnibus with the GEO Series accession number GSE255839.
Adherence assay
Adhesion capacity of C. glabrata cells to an agar plate was evaluated as described previously41. Logarithmic phase C. glabrata cells were grown in YPD medium at 37 °C, washed twice with distilled water, and adjusted to 1.0 × 107 cells/mL. Five microliters of the cell suspension were spotted onto YPD agar plates and incubated at 37 °C for 10 days. Colonies were washed with distilled water and photographed. C. glabrata elm1Δ strain was used as a positive control. The assay was performed twice on independent occasions.
Macrophage killing assay
Murine RAW 264 macrophages were prepared as previously described69. Briefly, macrophages were cultured at 37 °C in 5% CO2 in Dulbecco’s Modified Eagle Medium (DMEM, Sigma-Aldrich) supplemented with 10% fetal bovine serum (Life technologies, Japan) and 1% penicillin and streptomycin (Sigma-Aldrich), Macrophages were scraped with trypsin-EDTA (0.25%) phenol red (Invitrogen, Carlsbad, CA) and rinsed in DMEM. To examine the susceptibility of C. glabrata cells to macrophage killing, equal volumes (0.5 mL) of macrophages (8.0 × 104 cells/mL) and C. glabrata cell suspensions (4.0 × 103 cells/mL) were mixed in 1.5 mL microtubes (20:1 ratio) and co-cultured with rotation at 37 °C for 2 h. The cultures were sonicated, diluted, and spread on YPD plates to count the viable cells. CFU data were obtained from the following four groups: C. glabrata wild-type strain (CBS138) with and without RAW 264 macrophages (n = 7, each) and the ipi1R70H mutant with and without RAW 264 macrophages (n = 7, each). The experiment was repeated twice on independent occasions. The percentage of killing ratios was calculated as [1 - (CFUs from co-culture tubes / CFUs from control tubes)] for all data obtained from the two experiments. Percent killing by RAW 264 macrophages was compared between the wild-type and ipi1R70H strain groups (n = 14, each) using the Mann–Whitney U test. A P-value of <0.05 was considered statistically significant.
Ethics statement
All animal experiments were performed in full compliance with the Guide for the Care and Use of Laboratory Animals (National Research Council, National Academy Press, Washington DC, 2011) and all institutional regulations and guidelines for animal experimentation after pertinent review and approval by the Institutional Animal Care and Use Committee of Nagasaki University (protocol number 1906121536).
In vivo experiments
Animal experiments were performed as previously described38,70. Briefly, to prepare cells for injection, logarithmic-phase C. glabrata cells were harvested, washed, resuspended in sterile saline, and adjusted to 4 × 108 cells/mL after counting the number of cells using a hemocytometer. The actual CFU in the inocula was confirmed by plating serial dilutions of the cell suspension onto YPD plates. Eight-week-old female BALB/c mice (Charles River Laboratories Japan Inc., Japan) were housed under a standard 12 h light/12 h dark cycle condition at a temperature of 23 °C and a humidity of 50%. Food and water were available ad libitum. The mice were injected with 0.2 mL of the C. glabrata cell suspension via the lateral tail vein (n = 9 for the wild-type strain CBS138 and n = 10 for the ipi1R70H mutant). The mice were euthanized through carbon dioxide overexposure 7 days after injection, and the spleen, liver, and bilateral kidneys were excised. None of the mice died before euthanasia. Appropriate dilutions of organ homogenates were plated on YPD plates. Colonies were counted 2 days after incubation at 30 °C, and the CFUs per gram of the organ were calculated. We sequenced the PCR products obtained from 15 colonies (five colonies recovered from each organ) and confirmed the presence of G209A mutation in IPI1 in all the samples examined.
For the silkworm infection model, silkworm eggs were purchased from Ehime-Sanshu Co., Ltd. (Ehime, Japan), disinfected, and hatched at 25–27 °C. Silkworms were fed an antibiotic-containing artificial diet, SilkMate 2S (Nihon-Nosan Co., Ltd., Yokohama, Japan). Hyperglycemia was induced by SilkMate 2S containing 10% glucose on the first day of fifth-instar larvae (day 1). On day 2 of fifth-instar larvae, 50 µL of the C. glabrata cell suspension (2.0–2.5 × 107 cells) was inoculated into the hemolymph through the dorsal surface of the larvae using a 1 mL tuberculin syringe with a 27-gauge needle. An echinocandin dissolved in saline or saline alone (50 μL) was injected into the silkworm hemolymph on days 2 and 3. Infected larvae were maintained at 37 °C without feeding. The survival rate was determined at 24 and 48 h after infection (days 3 and 4). For the CFU assay, another set of larvae was prepared using the protocol described above. On day 3, 2 μL of hemolymph was collected, immediately diluted with 48 µL of saline, and spread on YPD agar plates. The number of colonies was counted to calculate the CFUs/µL of hemolymph. The in vivo experiments were conducted on two separate occasions to ensure reproducibility.
Statistical analysis
Statistical analysis was performed using GraphPad Prism version 10 (GraphPad Software, Boston, MA). Sterol contents were compared using ordinary one-way ANOVA with Sidak’s multiple comparison test. Results of qRT-PCR were compared using ordinary one-way ANOVA with either Sidak’s multiple comparison test or Dunnett’s multiple comparison test. Differential expression analysis of the RNA-seq data was performed using DESeq2, applying a two-sided test for each gene. Multiple comparison adjustments were made using the Benjamini-Hochberg method to control the false discovery rate, with results reported as adjusted P-values. Significant differential expression was determined based on a threshold of log2 Fold change > 1 and adjusted P-value < 0.05. GO enrichment analysis was performed using a one-sided binomial test with multiple comparison adjustments. The Bonferroni method was used to control the false discovery rate. Corrected P-values are reported, with significant terms determined at a corrected P-value threshold of <0.05. Mann–Whitney U test was used to compare differences in the CFUs between two groups. Survival of larvae was compared using the log-rank (Mantel-Cox) test. Statistical significance was set at P < 0.05.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The RNA-seq data discussed in this study have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE255839. All other data supporting the findings of this study are available within the article (including its supplementary information files). Source data are provided with this paper.
References
Brown, G. D. et al. Hidden killers: human fungal infections. Sci. Transl. Med. 4, 165rv113 (2012).
Kullberg, B. J. & Arendrup, M. C. Invasive candidiasis. N. Engl. J. Med 373, 1445–1456 (2015).
Alexander, B. D. et al. Increasing echinocandin resistance in Candida glabrata: clinical failure correlates with presence of FKS mutations and elevated minimum inhibitory concentrations. Clin. Infect. Dis. 56, 1724–1732 (2013).
Pappas, P. G., Lionakis, M. S., Arendrup, M. C., Ostrosky-Zeichner, L. & Kullberg, B. J. Invasive candidiasis. Nat. Rev. Dis. Prim. 4, 18026 (2018).
Perlin, D. S., Rautemaa-Richardson, R. & Alastruey-Izquierdo, A. The global problem of antifungal resistance: prevalence, mechanisms, and management. Lancet Infect. Dis. 17, e383–e392 (2017).
Arendrup, M. C. & Perlin, D. S. Echinocandin resistance: an emerging clinical problem? Curr. Opin. Infect. Dis. 27, 484–492 (2014).
Jensen, R. H. et al. Posttreatment antifungal resistance among colonizing candida isolates in candidemia patients: results from a systematic multicenter study. Antimicrob. Agents Chemother. 60, 1500–1508 (2015).
Prigent, G. et al. Echinocandin resistance in Candida species isolates from liver transplant recipients. Antimicrob. Agents Chemother. 61, e01229 (2017).
Aldejohann, A. M., Herz, M., Martin, R., Walther, G. & Kurzai, O. Emergence of resistant Candida glabrata in Germany. JAC Antimicrob. Resist 3, dlab122 (2021).
Shields, R. K., Nguyen, M. H., Press, E. G. & Clancy, C. J. Abdominal candidiasis is a hidden reservoir of echinocandin resistance. Antimicrob. Agents Chemother. 58, 7601–7605 (2014).
Sanglard, D., Coste, A. & Ferrari, S. Antifungal drug resistance mechanisms in fungal pathogens from the perspective of transcriptional gene regulation. FEMS Yeast Res 9, 1029–1050 (2009).
Cornely, O. A. et al. ESCMID* guideline for the diagnosis and management of Candida diseases 2012: non-neutropenic adult patients. Clin. Microbiol. Infect. 18, 19–37 (2012).
Pappas, P. G. et al. Clinical practice guideline for the management of candidiasis: 2016 update by the infectious diseases society of America. Clin. Infect. Dis. 62, e1–e50 (2016).
Blanchard, E. et al. Prior caspofungin exposure in patients with hematological malignancies is a risk factor for subsequent fungemia due to decreased susceptibility in Candida spp.: a case-control study in Paris, France. Antimicrob. Agents Chemother. 55, 5358–5361 (2011).
Lortholary, O. et al. Recent exposure to caspofungin or fluconazole influences the epidemiology of candidemia: a prospective multicenter study involving 2,441 patients. Antimicrob. Agents Chemother. 55, 532–538 (2011).
Beyda, N. D. et al. FKS mutant Candida glabrata: risk factors and outcomes in patients with candidemia. Clin. Infect. Dis. 59, 819–825 (2014).
Delliere, S. et al. Fluconazole and echinocandin resistance of Candida glabrata correlates better with antifungal drug exposure rather than with MSH2 mutator genotype in a french cohort of patients harboring low rates of resistance. Front. Microbiol. 7, 2038 (2016).
Matsumoto, E. et al. Candidemia surveillance in Iowa: emergence of echinocandin resistance. Diagn. Microbiol. Infect. Dis. 79, 205–208 (2014).
Pham, C. D. et al. Role of FKS mutations in Candida glabrata: MIC values, echinocandin resistance, and multidrug resistance. Antimicrob. Agents Chemother. 58, 4690–4696 (2014).
Galani, K., Nissan, T. A., Petfalski, E., Tollervey, D. & Hurt, E. Rea1, a dynein-related nuclear AAAATPase, is involved in late rRNA processing and nuclear export of 60 S subunits. J. Biol. Chem 279, 55411–55418 (2004).
Krogan, N. J. et al. High-definition macromolecular composition of yeast RNA-processing complexes. Mol. Cell 13, 225–239 (2004).
Peng, W. T. et al. A panoramic view of yeast noncoding RNA processing. Cell 113, 919–933 (2003).
Bhakt, P., Raney, M. & Kaur, R. The SET-domain protein CgSet4 negatively regulates antifungal drug resistance via the ergosterol biosynthesis transcriptional regulator CgUpc2a. J. Biol. Chem. 298, 102485 (2022).
Sanglard, D., Ischer, F., Calabrese, D., Majcherczyk, P. A. & Bille, J. The ATP binding cassette transporter gene CgCDR1 from Candida glabrata is involved in the resistance of clinical isolates to azole antifungal agents. Antimicrob. Agents Chemother. 43, 2753–2765 (1999).
Wada, S. et al. Candida glabrata ATP-binding cassette transporters Cdr1p and Pdh1p expressed in a Saccharomyces cerevisiae strain deficient in membrane transporters show phosphorylation-dependent pumping properties. J. Biol. Chem. 277, 46809–46821 (2002).
Tsai, H. F., Krol, A. A., Sarti, K. E. & Bennett, J. E. Candida glabrata PDR1, a transcriptional regulator of a pleiotropic drug resistance network, mediates azole resistance in clinical isolates and petite mutants. Antimicrob. Agents Chemother. 50, 1384–1392 (2006).
Caudle, K. E. et al. Genomewide expression profile analysis of the Candida glabrata Pdr1 regulon. Eukaryot. Cell 10, 373–383 (2011).
Ni, Q. et al. CgPDR1 gain-of-function mutations lead to azole-resistance and increased adhesion in clinical Candida glabrata strains. Mycoses 61, 430–440 (2018).
Kiyohara, M. et al. Evaluation of a novel FKS1 R1354H mutation associated with caspofungin resistance in Candida auris using the CRISPR-Cas9 system. J. Fungi (Basel) 9, 529 (2023).
Chiabudini, M., Conz, C., Reckmann, F. & Rospert, S. Ribosome-associated complex and Ssb are required for translational repression induced by polylysine segments within nascent chains. Mol. Cell Biol. 32, 4769–4779 (2012).
Conz, C. et al. Functional characterization of the atypical Hsp70 subunit of yeast ribosome-associated complex. J. Biol. Chem 282, 33977–33984 (2007).
Gautschi, M. et al. RAC, a stable ribosome-associated complex in yeast formed by the DnaK-DnaJ homologs Ssz1p and zuotin. Proc. Natl. Acad. Sci. USA 98, 3762–3767 (2001).
Perlin, D. S. Resistance to echinocandin-class antifungal drugs. Drug Resist Updat 10, 121–130 (2007).
Niimi, K. et al. Overexpression of Candida albicans CDR1, CDR2, or MDR1 does not produce significant changes in echinocandin susceptibility. Antimicrob. Agents Chemother. 50, 1148–1155 (2006).
Plaine, A. et al. Functional analysis of Candida albicans GPI-anchored proteins: roles in cell wall integrity and caspofungin sensitivity. Fungal Genet Biol 45, 1404–1414 (2008).
Arastehfar, A. et al. Macrophage internalization creates a multidrug-tolerant fungal persister reservoir and facilitates the emergence of drug resistance. Nat. Commun. 14, 1183 (2023).
Sumiyoshi, M. et al. Novel and potent antimicrobial effects of caspofungin on drug-resistant Candida and bacteria. Sci. Rep. 10, 17745 (2020).
Miyazaki, T. et al. Roles of calcineurin and Crz1 in antifungal susceptibility and virulence of Candida glabrata. Antimicrob. Agents Chemother. 54, 1639–1643 (2010).
Katiyar, S. K. et al. Fks1 and Fks2 are functionally redundant but differentially regulated in Candida glabrata: implications for echinocandin resistance. Antimicrob. Agents Chemother. 56, 6304–6309 (2012).
Yang, F. & Berman, J. Beyond resistance: antifungal heteroresistance and antifungal tolerance in fungal pathogens. Curr. Opin. Microbiol 78, 102439 (2024).
Ito, Y. et al. Roles of Elm1 in antifungal susceptibility and virulence in Candida glabrata. Sci. Rep. 10, 9789 (2020).
Moye-Rowley, W. S. Multiple interfaces control activity of the Candida glabrata Pdr1 transcription factor mediating azole drug resistance. Curr. Genet 65, 103–108 (2019).
Vu, B. G., Thomas, G. H. & Moye-Rowley, W. S. Evidence that ergosterol biosynthesis modulates activity of the Pdr1 transcription factor in Candida glabrata. mBio 10, e00934–19 (2019).
Hallstrom, T. C., Katzmann, D. J., Torres, R. J., Sharp, W. J. & Moye-Rowley, W. S. Regulation of transcription factor Pdr1p function by an Hsp70 protein in Saccharomyces cerevisiae. Mol. Cell Biol. 18, 1147–1155 (1998).
Prunuske, A. J., Waltner, J. K., Kuhn, P., Gu, B. & Craig, E. A. Role for the molecular chaperones Zuo1 and Ssz1 in quorum sensing via activation of the transcription factor Pdr1. Proc. Natl. Acad. Sci. USA 109, 472–477 (2012).
Walsh, P., Bursac, D., Law, Y. C., Cyr, D. & Lithgow, T. The J-protein family: modulating protein assembly, disassembly and translocation. EMBO Rep 5, 567–571 (2004).
Gillies, A. T., Taylor, R. & Gestwicki, J. E. Synthetic lethal interactions in yeast reveal functional roles of J protein co-chaperones. Mol. Biosyst. 8, 2901–2908 (2012).
Whaley, S. G. et al. Jjj1 Is a negative regulator of Pdr1-mediated fluconazole resistance in Candida glabrata. mSphere 3, e00466–17 (2018).
Vu, B. G., Simonicova, L. & Moye-Rowley, W. S. Calcineurin is required for Candida glabrata Pdr1 transcriptional activation. mBio 14, e0241623 (2023).
Lee, K. et al. Pathway of Hsp70 interactions at the ribosome. Nat. Commun. 12, 5666 (2021).
Bordallo-Cardona, M. A. et al. In vitro exposure to increasing micafungin concentrations easily promotes echinocandin resistance in Candida glabrata isolates. Antimicrob. Agents Chemother. 61, e01542–16 (2017).
Ksiezopolska, E. et al. Narrow mutational signatures drive acquisition of multidrug resistance in the fungal pathogen Candida glabrata. Curr. Biol. 31, 5314–5326.e5310 (2021).
Rivero-Menendez, O. et al. Clinical and laboratory development of echinocandin resistance in Candida glabrata: molecular characterization. Front Microbiol 10, 1585 (2019).
Shields, R. K. et al. Spontaneous mutational frequency and FKS mutation rates vary by echinocandin agent against Candida glabrata. Antimicrob. Agents Chemother. 63, 1870 (2019).
Arendrup, M. C. et al. Breakthrough Aspergillus fumigatus and Candida albicans double infection during caspofungin treatment: laboratory characteristics and implication for susceptibility testing. Antimicrob. Agents Chemother. 53, 1185–1193 (2009).
Satish, S. et al. Stress-induced changes in the lipid microenvironment of beta-(1,3)-d-glucan synthase cause clinically important echinocandin resistance in Aspergillus fumigatus. mBio 10, e00779–19 (2019).
Healey, K. R. & Perlin, D. S. Fungal resistance to echinocandins and the MDR phenomenon in Candida glabrata. J. Fungi (Basel) 4, 11128 (2018).
Arastehfar, A. et al. Overlooked Candida glabrata petites are echinocandin tolerant, induce host inflammatory responses, and display poor in vivo fitness. mBio 14, e0118023 (2023).
Garcia-Rubio, R. et al. Multifactorial role of mitochondria in echinocandin tolerance revealed by transcriptome analysis of drug-tolerant cells. mBio 12, e0195921 (2021).
Pavesic, M. W. et al. Calcineurin-dependent contributions to fitness in the opportunistic pathogen Candida glabrata. mSphere 9, e0055423 (2024).
Arastehfar, A. et al. The quiet and underappreciated rise of drug-resistant invasive fungal pathogens. J. Fungi (Basel) 6 https://doi.org/10.3390/jof6030138 (2020).
Cormack, B. P. & Falkow, S. Efficient homologous and illegitimate recombination in the opportunistic yeast pathogen Candida glabrata. Genetics 151, 979–987 (1999).
Nakayama, H. et al. A controllable gene-expression system for the pathogenic fungus Candida glabrata. Microbiol. (Read.) 144, 2407–2415 (1998).
Miyazaki, T., Nakayama, H., Nagayoshi, Y., Kakeya, H. & Kohno, S. Dissection of Ire1 functions reveals stress response mechanisms uniquely evolved in Candida glabrata. PLoS Pathog 9, e1003160 (2013).
Zhou, J., Liu, L. & Chen, J. Mitochondrial DNA heteroplasmy in Candida glabrata after mitochondrial transformation. Eukaryot. Cell 9, 806–814 (2010).
Tanaka, Y. et al. KRE5 suppression induces cell wall stress and alternative ER stress response required for maintaining cell wall integrity in Candida glabrata. PLoS One 11, e0161371 (2016).
Hosogaya, N. et al. The heme-binding protein Dap1 links iron homeostasis to azole resistance via the P450 protein Erg11 in Candida glabrata. FEMS Yeast Res 13, 411–421 (2013).
Lewis, T. A., Rodriguez, R. J. & Parks, L. W. Relationship between intracellular sterol content and sterol esterification and hydrolysis in Saccharomyces cerevisiae. Biochim. Biophys. Acta 921, 205–212 (1987).
Hirayama, T. et al. Clinical and microbiological characteristics of Candida guilliermondii and Candida fermentati. Antimicrob. Agents Chemother. 62, e02528–17 (2018).
Miyazaki, T. et al. Role of the Slt2 mitogen-activated protein kinase pathway in cell wall integrity and virulence in Candida glabrata. FEMS Yeast Res 10, 343–352 (2010).
Acknowledgements
T.M. would like to offer my sincere thanks to my great mentor Dr. John E. Bennett (NIAID/NIH). He has guided us over the years not only in this research project but also in many other studies related to infectious diseases and medical mycology. This work was partially supported by the Research Program on Emerging and Re-emerging Infectious Diseases from the Japan Agency for Medical Research and Development (AMED) to T.M. and S.K. (grant number JP21fk0108094 to T.M. and S.K., and JP24fk0108700 to T.M. and T. H.), the Japan Society for the Promotion of Science (JSPS) KAKENHI Grant Number JP19K07540 to T.M., and a research grant from MSD K.K., a subsidiary of Merck Sharp & Dohme Corp., in accordance with the Investigator-Initiated Studies Program of Merck Sharp & Dohme Corp. to T.M. (#57467). The financial supporter had no role in the study design, data collection and analysis, interpretation of the results, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
T.M. conceptualized the study. T.M. and S.K. obtained the funding. T.M., S.S., Y.N., H.Na., A.M., Y.T., Y.M., T.I., H.Ni., N.N., M.S, and T.H. performed experiments, analyses, and visualization. T.M., S.K., and H.M. supervised the study. T.M., S.S., H.Na., and Y.T. wrote the original manuscript, and all authors reviewed the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
T.M. has received a research grant from MSD for this project. T.M. and H.M. have received research grants and lecture honoraria from MSD, Astellas, and Pfizer outside this work. The sponsors and pharmaceutical companies had no role in the study design; collection, analysis, and interpretation of the data; and writing of the manuscript. The other authors have no competing interests.
Peer review
Peer review information
Nature Communications thanks Scott Moye-Rowley and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Miyazaki, T., Shimamura, S., Nagayoshi, Y. et al. Mechanisms of multidrug resistance caused by an Ipi1 mutation in the fungal pathogen Candida glabrata. Nat Commun 16, 1023 (2025). https://doi.org/10.1038/s41467-025-56269-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-56269-z
