
Abstract
Atopic dermatitis (AD) is a skin disease exhibiting clinical and molecular heterogeneity, thereby jeopardizing the development of personalized treatments. Here we pursue a cross-sectional and longitudinal cohort analysis of 951 whole-skin samples, employing an unsupervised decomposition analysis to link gene expression profiles to disease severity, six distinct skin phenotypes, and blood cytokines representative of given endotypes. Specifically, type 2 and type 17 responses are associated with major skin phenotypes such as erythema and induration, while type 1 response is upregulated in lichen amyloidosis of AD patients. Longitudinal analysis of patients treated with dupilumab finds sustained gene signatures related to type 17 response in lesional skin and upregulated transcription factors in non-lesional skin of patients with poor treatment outcomes. Lastly, several extracellular matrix organization-associated genes are correlated with clinical severity and treatment response to dupilumab. Our findings thus provide potential skin and blood biomarkers for assessing endotypes and therapeutic responses in AD to pave the way for personalized medicine.
Similar content being viewed by others

Introduction
The skin is a complex organ that covers the entire body, made up of various cells such as keratinocytes, fibroblasts, immune cells, nerve cells, and skin appendage cells. Maintaining healthy skin requires a delicate balance among the skin barrier, immune responses, and sensory perception. Disruptions in these functions can lead to various skin disorders. One common and troublesome condition is atopic dermatitis (AD). While not typically life-threatening, AD causes persistent itching and chronic inflammation, significantly impacting patients’ quality of life. AD involves complex interactions between structural and inflammatory cells in the skin, leading to barrier dysfunction and diverse inflammatory responses1,2. The symptoms of AD vary widely among patients, with different areas of the skin affected in different ways3. This variability makes it challenging to understand AD’s pathogenesis and develop precise treatments. Therefore, it is crucial to study the relationship between the diverse symptoms of AD and its underlying molecular features.
Past research on other inflammatory diseases has used large-scale human data to uncover complex pathogeneses4,5. However, similar efforts in AD using blood biomarkers have shown only modest results2,6. Though potentially more informative, skin tissue studies are limited by the invasiveness and scarring associated with whole-skin sampling. Less invasive methods, such as blistering and tape stripping, have been used to study the outer layer of the skin; however, these methods may fail to capture important information derived from deeper skin layers including signals from dermal fibroblasts and macrophages1,7,8,9.
AD also has a “waxing and waning” nature, with unpredictable flare-ups even during effective treatments10,11. In personalized medicine, understanding the skin’s immune state, especially in lesions that do not respond to treatment, is essential. Long-term sequential sampling with detailed clinical data is needed to capture the changes in skin transcriptomic profiles during these flare-ups. Among new treatments, dupilumab, a monoclonal IL-4 receptor antagonist, is the mainstay treatment for severe AD owing to its significant efficacy and safety12,13,14. Understanding why some patients do not respond well to dupilumab is crucial for improving clinical practices.
This study investigates the diverse molecular pathways of AD through a large cross-sectional and longitudinal cohort study using unsupervised decomposition analysis. We analyze mRNA from full-thickness skin tissue, including both the epidermis and dermis, obtained using minimally invasive 1-mm punch biopsies. This method allows us to collect numerous samples from both affected and unaffected skin areas, as well as tracking changes in patients treated with dupilumab. By applying non-negative matrix factorization (NMF), we identify previously recognized and unrecognized pathological pathways in AD-affected skin linked to clinical symptoms and the impact of dupilumab treatment. These findings offer insights into personalized medicine for AD by improving therapeutic response assessments and disease monitoring.
Results
Keratinocytes and fibroblasts as primary elements of 1-mm punch skin biopsies for RNA sequencing
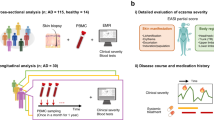
To understand the diverse molecular profiles of AD, we conducted a large-scale cross-sectional and longitudinal cohort study using RNA-seq data from 1-mm punch biopsy skin samples. An overview of the study design is illustrated in Fig. 1. These samples included those from healthy individuals and patients with psoriasis as disease controls. Out of 1061 skin samples, 1036 met the quality criteria for further analysis. This dataset comprised 529 lesional and 422 non-lesional samples from 156 patients with AD, 18 lesional and 18 non-lesional samples from 18 patients with psoriasis, and 49 samples from 26 healthy individuals (Table 1, Supplementary Fig. 1A, B, Supplementary Tables 1–3). Treatments included topical steroids, dupilumab, and other agents. Cross-sectional analyses were limited to samples from patients not treated with dupilumab, while longitudinal analyses were conducted using serial samples from 24 patients treated with dupilumab.

The schematic outlines the process from patient skin sampling to transcriptome analysis and computational identification of disease- and response-related signatures. GC gene cluster, ECM extracellular matrix, IEG immediate early genes.
Before conducting the analyses, we validated the cellular composition of the 1-mm skin samples. The levels of COL1A1 and KRT10 were substantially higher than those of CD3E and CD207 (Fig. 2A), implying that the transcripts from these biopsies predominantly originated from keratinocytes and fibroblasts. In the skin, keratinocytes and fibroblasts function as immune sentinels, producing various immune mediators in response to distinct inflammatory signals1,2,15. Thus, we inferred that our dataset indirectly reflected the immune status of the samples through the gene expression features of keratinocytes and fibroblasts.

A Density plot depicting representative genes associated with cellular components in the skin. The 1-mm punch whole-skin sample used for mRNA-seq included abundant keratinocyte- and fibroblast-derived transcriptomes. 1036 samples were used for analysis. B Heatmap illustrating 2368 highly-weighted genes in metagenes obtained from the decomposition analysis using NMF. Each value represents the gene weights, and the representative genes of each metagene are indicated in the right column. The bubble plot below the heatmap depicts the scaled gene weights (z-scoreg) of the genes that are representative of cellular components. 1036 samples were used for analysis. C The bottom heatmap represents the statistical significance of the comparison between normal samples and lesional AD, non-lesional AD, lesional psoriasis, and non-lesional psoriasis samples, tested by Wilcoxon test. The p values were corrected using the Bonferroni method for multiple comparisons. Blue indicates lower expression, and red indicates significantly higher expression than that in normal samples. Statistically insignificant results are highlighted in gray. All tests were two-sided. n (samples) = 49 (normal), 246 (lesional AD), 170 (non-lesional AD), 18 (lesional psoriasis), and 18 (non-lesional psoriasis). Source data are provided as a Source Data file. AD atopic dermatitis, Pso psoriasis.
Decomposition using NMF depicts disease-related metagenes and sample batch effects
A differentially expressed gene analysis comparing AD and psoriasis skin with healthy skin revealed genes associated with epidermal barrier and immunological dysfunction, consistent with previous studies (Supplementary Fig. 2A, Supplementary Data 1)2,8,16,17,18,19. Unsupervised hierarchical clustering identified sample biases in gene expression due to the inclusion of skin appendages, such as sebaceous and sweat glands (Supplementary Fig. 2B).
Therefore, to exclude sample biases and unravel the molecular heterogeneity in AD skin, we used NMF, an unsupervised decomposition method. NMF decomposes high-dimensional gene expression data into a smaller number of gene sets in an unbiased manner20. This method uncovers hidden co-expression profiles and reveals novel genes that correlate with recognized genes, providing insights into previously unrecognized gene interactions underlying the biology of complex diseases. Applying NMF to our data, we identified 29 gene sets, which we henceforth refer to as “SKIn-Tissue derived Metagenes” (SKITm), hereafter referred to as SKITm1 through SKITm29 (Fig. 2B and Supplementary Fig. 2C).
Genes with high weights in each metagene, defined by a z-score >3 (referred to as z-scoreg), were considered key representative gene signatures for their respective metagenes (Fig. 2B, Supplementary Fig. 3, Supplementary Data 2 and 3). Genes highly weighted specifically in only one metagene were extracted as “marker” genes, serving as candidate biomarkers of each metagene (see “Methods” for details). To understand the biological implications of the metagenes, we verified the highly weighted genes using knowledge-based analytical approaches in reference to previous studies (descriptions of the top 10 highly-weighted genes and marker genes for all metagenes are presented in Supplementary Data 4. An example of enrichment analysis for SKITm10 is shown in Supplementary Fig. 4, Supplementary Tables 4 and 5)17,19,21,22,23,24,25,26,27,28. Additionally, the expression levels of key cytokines representing type 2 and type 17 inflammation (IL13, CCL13, CCL18, CCL26, IL17A, IL17C, IL17F, IL23A, CCL20) were quantified by quantitative real-time PCR in 33 AD lesional samples and their correlation with the expression levels of key metagenes was analyzed. As a result, IL13, CCL13, and CCL26 expression levels showed significant correlations with SKITm17, IL17A, IL23A, and CCL20 were significantly correlated with SKITm10, and IL17C was significantly correlated with SKITm15, supporting the biological relevance of the metagenes (Fig. 3).

The expression levels of cytokines, quantified by qRT-PCR, were correlated with metagene expression levels derived from RNA-seq data, both obtained from the same set of skin samples (n = 33). The y-axis represents the cytokine levels quantified by qRT-PCR, and the x-axis represents the metagene expression levels derived from the decomposition analysis of RNA-seq data. The r and p indicate the correlation coefficient and the significance obtained through Spearman’s correlation analysis, respectively. The linear regression line is shown with a blue shaded area indicating the 95% confidence interval of the fitted line. Source data are provided as a Source Data file. *p < 0.01.
We then investigated metagenes associated with sample biases. In addition to the seven metagenes derived from skin appendages and reticulocytes (SKITm22–27, SKITm19), we identified four metagenes associated with sample bias, including outlier sample (SKITm14, SKITm20), sample quality such as RIN score and sequence batch effects (SKITm28, SKITm29) (Fig. 2B, Supplementary Figs. 5 and 6A–D). Uniform manifold approximation and projection (UMAP) revealed a distribution that distinctly reflected the disease state when we excluded the expression of sample bias-related metagenes (Supplementary Fig. 7).
Among the 18 metagenes unrelated to sample bias, 12 were differentially expressed in lesional AD or psoriasis samples in the cross-sectional study (Fig. 2C, Supplementary Fig. 8. Patient demographics and sample information are presented in Supplementary Tables 6–8.). Descriptions of highly-weighted genes, marker genes and biological interpretation for the 12 metagenes are presented in Table 2. SKITm1, SKITm17, SKITm4 and SKITm8, with high contributions of genes associated with leukocyte adhesion, type 2 response, and keratinocyte differentiation respectively, were upregulated in AD samples. SKITm10, characterized by inflammatory keratins and keratinocyte-derived genes inducible by type 17 inflammation, and SKITm15, also enriched with genes associated with type 17 response in keratinocytes with marked weight of SPRR2G, were upregulated in AD and psoriasis lesional samples. SKITm9, also characterized by type 17 response, was upregulated in psoriasis only. SKITm11, associated with type 1 response, was also highly expressed in psoriasis. SKITm2, representing an epidermal barrier gene signature, and SKITm16, enriched for genes involved in dermal extracellular matrix (ECM) organization, were both downregulated in AD and psoriasis. SKITm5 and SKITm6, composed of transcription factor genes such as immediate early genes (IEG), were upregulated in AD. Notably, AD and psoriasis exhibited apparent differences in the non-lesional sites. Non-lesional psoriasis samples presented almost the same profiles as normal controls, whereas non-lesional AD samples showed similar but weaker features than lesional AD, with enhanced immunological and barrier disruption. Most of these 12 metagenes correlated with the gene modules from our previous study which included 219 common AD samples (Supplementary Fig. 9)29.
The total expression score of 12 metagenes, representing the overall molecular dysregulation, was significantly higher in lesional and non-lesional AD than in normal samples (Fig. 4A). The metagene expression score correlated with the local and total disease severity (Fig. 4B, C). The expression profiles of representative normal and AD participants with mild, moderate, and severe disease are shown in Supplementary Fig. 10A–H.

A The y-axis represents the metagene expression score, which was calculated by the sum of expression values of 12 disease-associated metagenes, namely, SKITm17, SKITm10, SKITm9, SKITm15, SKITm11, SKITm8, SKITm6, SKITm5, SKITm4, SKITm1, SKITm2 and SKITm16. All the expression values of each sample were divided by the maximum expression value in each metagene for normalization (relative expression values). The expression values for SKITm2 and SKITm16 were then subtracted from 1 before the calculation of the metagene expression score. The relative expression value was used for analysis throughout the cross-sectional and longitudinal analysis. The metagene expression score between normal samples and lesional AD and non-lesional AD were tested by Wilcoxon test. The p values were corrected using the Bonferroni method for multiple comparisons. In box plots, the center line represents the median; the lower and upper bounds of the box represent the 25th and 75th percentiles, respectively; whiskers extend to the smallest and largest values within 1.5× the interquartile range from the lower and upper quartiles. N (samples) = 49 (normal), 246 (lesional AD), and 170 (non-lesional AD). *p < 0.01. B The y-axis represents the local severity score, calculated by the sum of clinical severity scores of erythema, papulation/induration, excoriation, and lichenification of the sampled sites. n (samples) = 49 (normal), 235 (lesional AD), and 159 (non-lesional AD). *p < 0.01. C The y-axis represents the total disease severity (EASI). n (samples) = 49 (normal), 234 (lesional AD), and 156 (non-lesional AD). *p < 0.01. For (B, C), the severity scores of normal samples were set to 0. The r and p indicate the correlation coefficient and the significance obtained through Spearman’s correlation analysis, respectively. The linear regression line is shown with a gray shaded area indicating the 95% confidence interval of the fitted line. D Heatmap depicting p values obtained from a regression analysis of the severity of skin phenotypes (erythema, papulation/induration, excoriation, lichenification), the overall severity (EASI), and pruritus score. Results from PN and LA lesions were compared with those of non-lesional samples from the same patients using a paired t-test. Red and blue indicate a significant positive and negative association with metagene expression and the severity of symptoms, respectively. Statistical significance was assessed using a two-sided test. Sample sizes: erythema/papulation/excoriation/lichenification (n = 212), LA (n = 12), PN (n = 16), pruritus (lesional, n = 230; non-lesional, n = 151), EASI (lesional, n = 235; non-lesional, n = 159). E Heatmap displaying p values obtained from regression analysis of plasma/serum cytokine levels. Statistical significance of regression coefficients was assessed using a two-sided test. EDN eosinophil-derived neurotoxin, TSLP thymic stromal lymphopoietin. Sample sizes: lesional, n = 224 samples (206 for CCL17); non-lesional, n = 148 (140 for CCL17). For (D, E), no adjustments for multiple comparisons were made, as the analyses were exploratory in nature. Source data are provided as a Source Data file.
Collectively, our unsupervised decomposition analysis revealed a collection of gene clusters, including both established and unrecognized gene clusters within the context of previous inflammatory skin studies.
Different gene co-expression patterns contribute to various skin phenotypes and disease severity
In the cross-sectional study, we explored the correlation between metagenes and local symptoms (erythema, papulation/induration, excoriation, lichenification), as well as disease severity indicators, including Eczema Area and Severity Index (EASI; a measure of disease severity), and pruritus (patient-oriented pruritus severity score), using a mixed-effects regression model (Eq. (1)). We also evaluated metagene expression in prurigo nodules (PN) and lichen amyloidosis (LA), rare chronic subtypes of AD skin symptoms30, and compared them with non-lesional samples from the same patients (PN: eight patients; LA: six patients) (Supplementary Fig. 1B). Each metagene was associated with distinct skin phenotypes (Fig. 4D and Supplementary Fig. 11A). Specifically:
-
Erythema: Negatively associated with SKITm2 and SKITm16 and positively associated with SKITm17, SKITm10, and SKITm5.
-
Papulation/Induration: Negatively associated with SKITm2 and SKITm16, and positively associated with SKITm17, SKITm10, and SKITm9.
-
Excoriation: Displayed marked SKITm10, SKITm8, and SKITm1 expression. Particularly, high SKITm1 expression suggested leukocyte induction through scratching.
-
Lichenification: Uniquely associated with SKITm8.
-
PN: Associated with SKITm2, SKITm17, SKITm10, and SKITm8.
-
LA: Exhibited significant expression of SKITm11 and SKITm8 metagenes. Notably, SKITm11 expression was distinct in LA.
Disease severity (EASI) and pruritus score were negatively associated with SKITm2 and SKITm16, and positively associated with SKITm17 and SKITm10. Even non-lesional sites displayed a weak but significant association with disease severity and pruritus; EASI was associated with SKITm10, SKITm8, SKITm5 and SKITm4, whereas pruritus was associated with SKITm17 and SKITm15 (Fig. 4D and Supplementary Fig. 11B).
These results suggested that the 1-mm skin sample serves as a reliable indicator of both local skin phenotypes and overall severity in AD patients. Our findings revealed distinct metagenes associated with various local skin symptoms, highlighting their differential significance in lesions and non-lesions.
Blood cytokine levels are associated with multiple skin metagenes
We observed that most blood cytokines were associated with the expression of multiple metagenes in the skin (Fig. 4E) using a mixed-effects regression model (Eq. (2)). Notably, CCL27, IL-22, and TNF-α levels in the blood were positively associated with SKITm17, SKITm10, SKITm2, and SKITm16 in lesional sites, with CCL27 exhibiting the highest level of significance. CCL17 (thymus and activation-regulated chemokine, TARC) showed significant yet weaker associations with SKITm17, SKITm10, and SKITm2. At lesional sites, eosinophil-derived neurotoxin (EDN) was associated with SKITm17, a type 2 response metagene, but not with other inflammation-related metagenes. Conversely, blood levels of IL-17 and CCL20, cytokines associated with type 17 inflammation31, positively correlated with SKITm10, validating the metagene as a “type 17 response-related metagene.” Notably, CCL20 exhibited a stronger and more significant association with SKITm10 than with the IL-17 level. Furthermore, the expression levels of non-lesional SKITm15, SKITm5, and SKITm4 were associated with more blood cytokines than those of lesional samples. Among blood clinical biomarkers, LDH demonstrated the highest association with the expression of most skin metagenes and disease severity, whereas eosinophil counts were specifically related to SKITm17, a type 2 inflammation response metagene (Supplementary Fig. 12) (Eq. (3)).
These results suggest that while most blood cytokines are related to multiple metagenes and overall severity, EDN, IL-17, and CCL20 could potentially be utilized to assess immunophenotypes in AD lesions.
Correlation between dupilumab treatment outcomes and baseline metagene expression
In the longitudinal analysis, we examined skin samples from 24 patients sampled monthly during dupilumab treatment, with patient demographics provided in Supplementary Table 9. Because dupilumab can induce facial erythema with a distinct etiology from pre-existing AD, we evaluated the treatment efficacy by modified EASI (mEASI), which is calculated by excluding the scores of the head and neck from the total EASI score32,33,34,35,36 (clinical photo of a patient who developed dupilumab-associated facial erythema is presented in Supplementary Fig. 13). Patients were classified into three groups based on mEASI during treatment: “early” responders, who achieved a mild disease level (defined by mEASI ≤ 6.3) within the first month and maintained that level for 6 months; “poor” responders, who did not achieve the mild level during the entire 6-month study period; and “intermediate” responders, who did not meet the criteria for either “early” or “poor” responders (Fig. 5A). These response trends persisted for over a year for most patients (Supplementary Fig. 14).

A Time-course of disease severity (modified Eczema Area and Severity Index: mEASI) in patients treated with dupilumab (n = 24 patients). The blue line represents 6.3, which is the cutoff for mild disease severity in the mEASI. B Correlation between the disease severity score (mEASI) and treatment response groups before treatment. The x-axis represents the responder group: +++ “early responders,” ++ “intermediate responders,” and + “poor responders.” n = 5 (early responders), 11 (intermediate responders), 8 (poor responders). Correlation between treatment outcomes and the expression of skin metagenes (C), skin marker genes (D), and blood cytokines (E) before treatment. n (samples) = 19 (lesional AD skin), 15 (non-lesional AD skin), 19 (blood). In (C, D), the red and blue dots represent lesional and non-lesional samples, respectively. In (B–D), in box plots, the center line represents the median; the lower and upper bounds of the box represent the 25th and 75th percentiles, respectively; whiskers extend to the smallest and largest values within 1.5× the interquartile range from the lower and upper quartiles. The p values indicate the significance obtained using Spearman’s correlation analysis. *p < 0.01, †p < 0.05. Source data are provided as a Source Data file.
We then investigated the clinical and molecular features of the response groups before treatment. Clinically, the pretreatment mEASI significantly differed among the three groups, with lower disease severity being associated with a better response to dupilumab (Fig. 5B). Skin transcriptomic analysis revealed a significant correlation between lesional SKITm16 expression and treatment responses (p = 6.3e-03). Additionally, marginal associations were observed between lesional SKITm10 expression and non-lesional SKITm5 and SKITm4 expression (p = 2.5e-02, 3.6e-02, 1.8e-02, respectively, Fig. 5C).
To identify potentially useful standalone biomarkers, we further assessed the pretreatment expression levels of SKITm16, SKITm10, and SKITm5 marker genes. SKITm4, lacking marker genes, was excluded from this analysis. We found significant associations with treatment outcomes in lesional samples for three SKITm16 genes (OGN, RECK, PDGFRL) and one SKITm10 gene (FBLIM1). In non-lesional samples, significant associations were found for SKITm5 genes (TOB2, EGR1, MCL1, CBX4, IER2, PLK2, CSRNP1, EGR3, PMAIP1) (p < 0.01, Fig. 5D). Supplementary Fig. 15 displays marker genes with marginal correlations (p < 0.05). For these analyses, five patients who were receiving cyclosporine at baseline were excluded, and four patients with no non-lesional sites on their trunks were excluded from the non-lesional skin analysis.
Pretreatment levels of blood cytokines were also analyzed, which showed significant associations with treatment outcomes for IL-22 (p = 2.4e-03) and IL-18 (p = 1.1e-03), while CCL20 exhibited marginal significance (p = 5.0e-02) (Fig. 5E and Supplementary Fig. 16). Although IL-17 did not exhibit a significant correlation, IL-17 blood levels in most early responders were nearly zero. EDN, associated with skin SKITm17 expression (type 2 immune response) in the cross-sectional analysis, was not significantly associated with treatment responses. To further evaluate biomarkers for treatment effect, we validated the correlation between pretreatment levels of skin and blood biomarkers and disease severity after treatment. We found that pretreatment levels of lesional SKITm10, non-lesional SKITm15, plasma IL-22, and IL-18 correlated with the average severity score after 5 and 6 months of treatment (Fig. 6A–C).

Analysis of pretreatment levels of (A) skin metagene expression, (B) marker gene expression of SKITm10, and (C) blood cytokines. The x-axis represents the averaged values of AD disease severity score (mEASI) after 5 and 6 months of dupilumab treatment, and the r and p indicate the correlation coefficient and the significance obtained through Spearman’s correlation analysis, respectively. n (samples) = 19 (lesional AD skin), 15 (non-lesional AD skin), 19 (blood). *p < 0.01, †p < 0.05. Source data are provided as a Source Data file.
Dupilumab suppresses type 2 inflammation and restores the epidermal barrier and ECM-related genes
We evaluated the chronological effect of dupilumab on skin mRNA profiles (Fig. 7A) using a mixed-effects regression model (Eq. (4)). Average metagene expression levels in the 24 patients during treatment were compared with respective pretreatment levels. The most notable changes were observed in SKITm17 (associated with a type 2 immune response) and SKITm16 (ECM-related gene signature) in both lesional and non-lesional samples, with expression levels almost shifting to the normal range after 3 months. Lesional SKITm2 expression also recovered slowly but did not reach the normal range. SKITm10, SKITm6, and SKITm5 in the lesional samples and SKITm4 in the non-lesional samples showed a partial response. SKITm11 expression was low among the sampled patients, showing no change in the first 6 months of dupilumab treatment. Blood CCL27, CCL17, IL-22, IL-16, and IL-18 levels consistently decreased (Supplementary Fig. 17) (Eq. (5)). These results confirmed the strong effect of dupilumab in suppressing type 2 inflammation and restoring the epidermal barrier. Additionally, the findings suggested that dupilumab also helps restore the pathological gene expression in fibroblasts, while the effect on type 17 response in the skin is limited.

A Average expression of skin metagenes during dupilumab treatment in 24 patients. The p values indicate the significance of the differences in metagene expression compared with the pretreatment level (month 0) as determined by a linear mixed-effects regression analysis. n = 24 (AD), 26 (normal). *p < 0.01. P values were corrected using the Bonferroni method for multiple comparisons. Source data are provided as a Source Data file. B Averaged transition in skin metagene expression among different responder groups. In (A, B), the mean and SEM (standard error of the mean) are presented as red and blue points and bars, respectively, for lesional and non-lesional AD skin samples. Green highlighted shadows represent the 95% confidence interval (CI) for the mean expression in healthy individuals. n = 5 (early responders), 11 (intermediate responders), 8 (poor responders).
Long-course sampling demonstrates skewed inflammation in patients with poor responses
Our subsequent interest was to investigate skin immunophenotypes under dupilumab treatment. Among the three responder groups, the average transition in skin metagene expression exhibited notable patterns (Fig. 7B). Specifically, SKITm17, associated with a type 2 immune response, was strongly suppressed across all three responder groups. Conversely, SKITm10, associated with a type 17 response, was consistently elevated in the poor-response group. Furthermore, SKITm15 and SKITm5 expression were higher in non-lesional sites of poor responders during treatment (Fig. 7B and Supplementary Fig. 18). The average transition in blood cytokine/chemokine levels was not notably different among the responder groups (Supplementary Fig. 19).
The residual expression of lesional SKITm10 in poor responders persisted over the long term. In the longitudinal analysis, eight patients were sampled sequentially for over 1 year; two patients were classified into the early, two into the intermediate, and four into the poor-response groups in the previous analysis. The transition in metagene expression and its relative expression compared with those at baseline in poor responders are presented in Fig. 8A, B, respectively (results of all 12 metagenes and blood cytokines are presented in Supplementary Figs. 20 and 21). SKITm17 expression was suppressed in almost all patients throughout dupilumab therapy, with the exception of Pt12, where SKITm17 increased when the dosing interval was extended from every other week to once per month. Conversely, poor responders exhibited skewed expressions toward SKITm10. Skewing toward type 17 response-associated cytokines was less evident in the blood (Fig. 8B).

A Dynamics of skin metagene expression and blood cytokines in individual patients during the long-term course of dupilumab treatment. Green highlighted shadows represent the 95% CI for the mean expression in healthy individuals. B Transition in relative values compared with the baseline skin metagene expression and blood cytokines in four poorly responding patients. C Clinical images of the four poorly responding patients before and after treatment. No apparent changes in clinical phenotypes were observed during the course of treatment.
Further observation of individual patients revealed notable results. Two poorly responsive patients (Pt21 and Pt23) showed persistently high expression of SKITm5 (IEGs) at non-lesional sites (Fig. 8A); both patients had common clinical phenotypes of generalized erythema (Fig. 8B). Pt17 had extensive areas of LA, consistently observed in the longitudinal study. These results suggested that LA eruptions were persistently associated with high SKITm11 expression (indicating type 1 inflammation). Notably, SKITm10 expression, initially low before dupilumab treatment, increased in LA lesions following dupilumab initiation. No patients developed any evident new skin phenotypes throughout treatment (Supplementary Fig. 22). Finally, the marker genes showed trends similar to those of metagene expression in eight patients (Supplementary Fig. 23), highlighting the potential utility of marker genes for disease monitoring and as additional therapeutic targets in poor responders to dupilumab.
Discussion
Human clinical research often struggles to balance invasiveness and information richness. To address this, we profiled gene expression in large-scale whole-skin samples using an unbiased approach to explore the molecular heterogeneity of AD. Consequently, our 1-mm punch method yielded ample samples for comprehensive analyses with minimal invasion. Additionally, unsupervised decomposition successfully captured various keratinocyte and fibroblast gene expression profiles, revealing both recognized and unrecognized gene signatures underlying AD pathogenesis. This is the first comprehensive study to examine the gene expression profiles associated with local AD phenotypes, overall severity, and treatment outcomes.
Conventional analytical approaches often fall short due to disease population diversity and potential sampling bias. Techniques such as gene set variation analysis and CYBERSORT have been used to assess inflammatory pathways in AD2,8,37; however, detailed reference data for various cell populations in the skin are limited. Single-cell studies can identify novel cell populations1, but comprehensive understanding requires many patient samples, and single-cell analysis is typically performed on a limited cohort.
We overcame these challenges using unsupervised learning with NMF to profile numerous whole-skin samples in an unbiased manner, revealing the molecular heterogeneity of AD and responses to different inflammatory signals. NMF, a potent decomposition method widely used in bioinformatics38,39, extracts gene sets that are co-expressed in the original data, providing a low-rank approximation of high-dimensional expression data. The gene sets, or metagenes, obtained by NMF, have the following features: (1) genes with high weights across multiple metagenes uncover the contribution of single genes to multiple pathways, and (2) genes that are highly weighted in a single metagene serve as specific biomarkers for each pathway. For example, KRT17 is upregulated in both normal sebaceous glands and interfollicular keratinocytes in psoriasis40, highlighting the need for caution when evaluating the disease state solely based on the gene expression levels per se. In addition, genes such as FLG, FLG2, and LOR, widely recognized as skin-barrier-related genes, exhibited high weights in multiple metagenes, suggesting their involvement in various skin processes. This may explain why the epidermal barrier metagene (SKITm2) did not significantly correlate with the skin modules in our previous study using weighted gene co-expression network analysis (WGCNA), where each gene is clustered to a single module (Supplementary Fig. 9)29. The identified marker genes specific for each metagene serve as potential clinical biomarkers for assessing diverse skin barrier and inflammatory states, as well as for future precision medicine in AD.
Thus, NMF uncovered a significant collection of gene signatures in AD skin tissue reflecting responses to different inflammatory signals, with minimal influence from sample bias related to skin appendages and technical factors.
In previous studies, gene expression signatures in AD have been compared among lesional, non-lesional, and normal skin, but their relevance to different AD eruptions has been less explored. Our study clarifies the contribution of “AD-associated” molecular profiles to various AD skin phenotypes. The following discusses the metagenes identified by NMF, comprising genes involved in linked to type 2, 17, and 1 inflammatory responses, as well as barrier-related genes.
The SKITm17 metagene, associated with type 2 response, correlated strongly with clinical signs of AD, including local symptoms, severity, and pruritus. Signature genes included key chemokines such as CCL18 and CCL13 produced by macrophages and MMP12 expressed in DCs, and genes such as COL6A5, POSTN, CCL2, and CCL19 expressed in a recently reported unique fibroblast population in AD skin1,41. Th2-responding fibroblasts expressing COL6A5, POSTN, and IL13RA1 have also been identified in eosinophilic fasciitis in humans42. Our findings emphasize the crucial role of fibroblasts and macrophages in the skin with type 2 inflammation.
Multiple metagenes, including SKITm9, SKITm10, and SKITm15, shared common high-weight genes known to be inducible by type 17 inflammation in keratinocytes17,19,21,22,23,24. Our findings revealed distinct correlations among these metagenes, marker genes, and clinical parameters. SKITm10 was linked to lesional sites, while SKITm15 was associated with non-lesional sites. The involvement of these metagenes in our samples may be influenced by race, as all participants were Japanese, a population shown to have type 17 polarization in AD18.
The SKITm11 metagene, linked to LA, a chronic AD type, includes genes implicated in type 1 inflammation. In AD, genes such as MX1, STAT1, CXCL9, and CXCL10 have been implicated in type 1 inflammation with “chronic” lesions and African-American AD; however, other studies have shown conflicting results37,43,44. The inconclusive results in these studies may stem from inadequate consideration of local skin phenotypes and inconsistent definitions of “chronic disease.” Discrepancies arise, as some define “chronic AD” based on the disease duration (e.g., childhood-onset adult AD)44, whereas others focus on the local skin lesion duration (e.g., >72 h)43. By examining eruption types, we confirmed the clinical significance of type 1 inflammation in AD skin rather than relying on divergent definitions across studies.
Within the barrier-related metagene SKITm2, we identified unrecognized genes as potential biomarkers for barrier disruption. The high-weighted genes in SKITm2, such as FLG, FLG2, and LOR, are well-known as genetic risk factors in AD7,45. Nevertheless, their utility as universal biomarkers for barrier function has not been shown46,47. Here, within the barrier-related metagene SKITm2, we identified unrecognized genes, namely PHYHIP, UGT3A2, CACNA1H, and BTC, as potential biomarkers for barrier disruption. Their roles in skin barrier functions, however, remain to be explored48. Our emphasis on potential markers resulting from unbiased analysis warrants a comprehensive evaluation of skin barriers in AD, transcending conventional gene markers rooted in one-sided hypotheses.
SKITm8, linked to epidermal terminal differentiation in the granular layer49,50, was upregulated in chronic AD phenotypes, such as lichenification, PN, and LA resulting from repetitive scratching51. These eruptions also share common histological features, such as hyperkeratosis and hypergranulosis52. HRNR, a paralog of FLG2 and a marker gene of SKITm8, has shown conflicting expression patterns in AD lesions in previous studies53,54, highlighting the importance of considering eruption types. Thus, genes in SKITm8 may reflect “abnormal” epidermal differentiation following scratching in AD skin.
Our investigation on non-lesional sites indicated distinctive pathology in AD compared with that of psoriasis. In AD, even clinically asymptomatic skin exhibits barrier dysfunctions and subclinical inflammation55,56, whereas, in psoriasis, non-lesional skin resembles healthy control skin57. This distinction emphasizes the importance of evaluating non-lesional sites in AD.
This study revealed unique associations between detailed clinical symptoms and conventional AD-associated genes. Unlike previous transcriptomic studies that often lacked comprehensive clinical data, our study included this crucial information, allowing us to uncover the intricate relationships between molecular profiles and clinical manifestations. This integrated approach underscores the importance of detailed clinical data in understanding the heterogeneous nature of diseases, such as AD, and provides consistent, accurate insights into complex disease pathogenesis.
Beyond the reidentification of known AD-associated genes, our unbiased approach revealed two significant gene clusters not well-recognized in previous AD studies: IEG-related genes (SKITm5) and fibroblast-derived genes associated with ECM organization (SKITm16)58.
The role of IEGs in AD pathogenesis is not well understood; however, in vitro studies suggest their association with “tissue-based memory.” IEGs are transcription factors rapidly and transiently transcribed in response to acute stress or proliferation signals, with expression levels that are sensitive to timing59,60. Repetitive IL-13 stimulation in alveolar epithelial cells leads to sustained IEG activation and hyper-reactivity, resulting in increased cytokine and chemokine production61, implying a role in chronic type 2 inflammation in asthma, known as “tissue-based memory”62. Our results suggest that IEG-related genes (SKITm5) in non-lesional sites could serve as biomarkers for disease severity and dupilumab treatment outcomes (Figs. 4D and 5C). This supports the idea that IEG expression may reflect “latent inflammation” in non-lesional AD skin. Additionally, the inclusion of epidermal growth factor receptor ligands in the same metagene suggests underlying activation of keratinocytes.
SKITm16, comprising fibroblast-derived genes, showed a strong negative association with disease severity and dupilumab treatment outcomes. Skin fibroblasts are crucial in ECM organization and immune regulation63, but the molecular features of hypofunctional fibroblasts and their clinical impact on skin diseases are largely unknown. Single-cell studies have highlighted fibroblast heterogeneity in normal skin58, yet their utility as biomarkers in diseased skin remains uncertain. Genes in SKITm16, specifically fibroblast-derived genes, have been linked to other skin diseases. For example, CCDC80 was identified in a genome-wide association study of AD64, and LOX, overexpressed in the lungs and skin of patients with systemic sclerosis65, was associated with dupilumab treatment outcomes in our study. Downregulation of LOX expression impairs ECM cross-linking, affecting its structural integrity and stability63. RECK and PI16, expressed in endothelial cells, inhibit MMP2 involved in ECM breakdown and are suppressed by inflammatory cytokines such as IL-6 in human endothelial cells66, suggesting their responsiveness under inflammatory conditions. The downregulation of RECK is also linked to poor prognosis in multiple cancers66,67. These findings highlight the significant role of impaired ECM organization in the inflammatory pathogenesis of AD and suggest that these genes could be important biomarkers for assessing disease severity and treatment responses.
To achieve personalized AD therapy, understanding both the immunophenotypes and personal molecular signatures under targeted treatments is essential. Identifying pretreatment molecular markers can help predict dupilumab responses, while understanding treatment dynamics can inform alternative strategies.
In the longitudinal study, we found that four metagenes (SKITm10, SKITm16, SKITm5, and SKITm4) and clinical severity were associated with treatment response to dupilumab. Notably, the expression of SKITm10 and its marker genes, linked to type 17 response, were associated with limited treatment outcomes, unlike those linked to barrier impairment and type 2 inflammation (Fig. 5C). Of note, the pretreatment severity of patients was strongly correlated with an increased type 17/type 2 ratio, characterized by skewed expression of SKITm10 relative to SKITm17, and severe patients who exhibited a pure type 2 profile were rare (Supplementary Fig. 24). Given that many biomarkers related to dupilumab response were confounded by disease severity, further investigation is necessary to assess their independence from disease severity.
Temporal analysis of the cohort revealed that dupilumab had the most robust effect on SKITm17, a type 2 response-related metagene, consistent with previous reports2,41. This effect was continuous for over a year, with recovery observed in barrier-related and ECM organization-related metagenes, SKITm2 and SKITm16.
In contrast, poor responders exhibited skewed expression of SKITm10, characterized by high weights of keratinocyte-derived genes responsive to type 17 inflammation (Figs. 7B, 8A, B). The potential skewing toward a Th17 response under Th2 pathway blockade has been reported68,69, while other reports have been inconsistent2,70,71,72. Our study’s unique features, including shorter sampling intervals and stratified analysis, provided new insights. This suggests that patients with inadequate dupilumab response might benefit from JAK inhibitors, known to downregulate type 1, 2, and 17 responses73.
Furthermore, sustained upregulation of IEGs (SKITm5 metagene) in some poor responders suggests an additional therapeutic target. Recently developed topical drugs that inhibit the nuclear transport of transcription factors, such as AP-1, a major component of IEGs74, might aid in further disease control for those patients with sustained IEG expression during systemic treatment. Our findings help identify potential genetic biomarkers to stratify populations that would benefit from IEG-targeted therapy in AD.
Our study had some limitations. For example, all participants were Japanese, warranting further research on other races. Additionally, we could not directly evaluate immune cells, presumably due to their small numbers in the samples. Nevertheless, our unbiased approach, fortified by minimally invasive skin sampling and integrative analysis with comprehensive unsupervised decomposition method and detailed clinical information, helped elucidate the complex links between heterogeneous molecular signatures and clinical AD features in skin tissue. The cross-sectional analysis highlighted the distinct clinical significance of both recognized and previously unrecognized gene sets in AD skin. The longitudinal analysis identified potential biomarkers and additional targets for dupilumab treatment. The marker genes identified offer a robust framework for assessing each pathway, with potential applications across various study settings, including other datasets and small-sample-size research. Altogether, our findings provide comprehensive insights into the complex molecular heterogeneity of AD, paving the way for future rational use of molecular-targeted treatments in personalized medicine.
Methods
Patients and sample collection
The study protocol was approved by the Keio University School of Medicine Ethics Committee (Approval Numbers: 20160377, 20150325, and 20130384), Nippon Medical School Ethics Committee (29-03-916), Tokyo Teishin Hospital (1054), and RIKEN Yokohama Ethics Review Committee (Approval Number H28-24), aligning with the Declaration of Helsinki. Patients with AD, plaque-type psoriasis visiting Keio University Hospital, Nippon Medical School Hospital, and Tokyo Teishin Hospital between 2018 and 2022 were invited to participate. AD was diagnosed according to the criteria of Hanifin and Rajka75, and psoriasis diagnosis was based on clinical symptoms or previously obtained skin histopathology, reviewed by at least two board-certified dermatologists. Normal control skin was obtained from healthy volunteers who were recruited at Keio University Hospital, defined as individuals with no inflammatory skin diseases. Exclusion criteria included age <15 years, pregnancy, and a history of lidocaine allergy, bleeding disorders, and complications of other inflammatory skin diseases. Written informed consent was obtained from all participants: 160 patients with AD, 18 patients with psoriasis, and 26 healthy volunteers (Supplementary Fig. 1). Participants who underwent skin sampling were compensated with 10,000 JPY. The sex (biological attribute) of the participants was determined based on self-reporting.
A 1-mm punch biopsy (Kai Medical, Japan, #BPP-10F) was used for skin sampling, using local anesthesia with topical lidocaine/prilocaine cream (EMLA cream; Sato Seiyaku, Tokyo, Japan, #1219800N1023) or 1% lidocaine injection (Xylocaine Injection Polyamp 1%; Sandoz Pharma K.K., Tokyo, Japan, #1214400A7055). Lesional and non-lesional samples were obtained based on consent, and “lesional” samples were defined by the presence of one or more skin symptoms: erythema, papulation/induration, excoriation, and lichenification. Sampling sites were primarily selected based on the most severe or characteristic lesions of the posterior trunk in patients with AD or psoriasis. Sampling was performed at multiple time points after obtaining patient consent.
During sampling, we avoided hair follicles by selecting sampling sites through dermoscopy, a microscopy tool commonly used in dermatological clinical practice. This precaution was adopted to mitigate potential confounding effects on the transcriptomic data. However, in some patients, hair follicles were undetectable, likely owing to edema or thickening of the epidermis. To acquire control data, we collected two skin samples from healthy volunteers: one from a non-hair follicle site and the other from a hair follicle site.
Each sampling site was rated from 0 to 3 based on the local severity of erythema (redness), papulation/induration (swelling of the skin), excoriation (erosion due to scratching), and lichenification (leathery thickening of the skin with exaggerated skin markings), following the EASI scoring system for AD patients76. Excoriated samples were obtained from epithelialized skin adjacent to excoriated erosions to avoid bias arising from a lack of epidermis. Prurigo nodules (PN, hard bumps) and lichen amyloidosis (LA, discrete, hard, small reddish-brown papules), which are rare chronic subtypes of AD skin symptoms30, were also evaluated. Examples of skin symptom annotations are presented in Supplementary Fig. 1B.
For AD patients, the severity of the whole body was assessed using EASI, and the patient-oriented pruritus score was recorded at the time of sampling. The Modified EASI (mEASI) was calculated by excluding the scores of the head and neck from the total EASI score32. Upon EASI scoring, PN was assessed as lichenification scores in accordance with the Harmonizing Outcome Measures for Eczema (HOME) guidance76. LA, another subtype of chronic AD skin symptoms, was also scored as lichenification. The suggested EASI range for a “mild” condition is 0–7, 7.1–21.0 for “moderate,” and 21.1–50 for “severe,” based on previous literature33. Therefore, the “mild” mEASI was set at 6.3, calculated as 7×64.8/72 (based on the maximum score of EASI being 72 and that of mEASI being 64.8). The diagnosis of skin diseases, evaluation of severity scores, and selection of sampling sites involved at least two board-certified dermatologists.
Medications for AD, psoriasis vulgaris, and other complications within 1 week of skin sampling were recorded. AD patient treatments included topical moisturizers, steroids, tacrolimus, JAK inhibitors, oral cyclosporine, nemolizumab, and dupilumab. Psoriasis patient treatments included topical steroids, topical vitamin D analogs, TNF-alpha inhibitors, and IL-17 inhibitors. Topical treatments were marked as “used” if the ointment had been applied to the sampled area within 1 week of sampling. Dupilumab was administered to adults with severe AD meeting the indication criteria under the Japanese national insurance; these included a body surface area percentage >10%, a Validated Investigator Global Assessment score higher than 3, and EASI > 16, despite a history of treatment with topical steroids (strong class or higher) or topical calcineurin inhibitors for at least the last 6 months, or patients with hypersensitivity to topical medications, rendering it difficult to continue topical treatments.
RNA extraction and sequencing
Samples for RNA transcriptome analysis were immediately submerged and stored in RNAlater solution (Thermo Fisher Scientific, Waltham, MA, USA, #AM7020) at 4 °C and subsequently frozen at −80 °C for storage. RNA was extracted from the samples using TRIzol (Thermo Fisher Scientific, Waltham, MA, USA, #15596026) and the Direct-Zol RNA MiniPrep Kit (ZYMO RESEARCH, Irvine, CA, USA, #R2052) following the manufacturer’s protocol. For quality control, the RNA integrity number (RIN) was calculated using an RNA Pico 6000 kit (Agilent Technologies, Santa Clara, CA, USA, #5067-1513) and Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA, #G2939A) with 2100 Expert software (version B.02.07; Agilent Technologies, Santa Clara, CA, USA), following the manufacturer’s instructions. The RNA concentration was measured using a Qubit fluorometer (Thermo Fisher Scientific, Waltham, MA, USA). Samples with RNA concentrations below the measurement range of the Qubit fluorometer (below 1 ng/μl) were measured using a Quantus Fluorometer (Promega Benelux, Leiden, the Netherlands) or estimated from the electrophoresis results using the bioanalyzer. The libraries were prepared using a NEBNext Ultra RNA Library Prep Kit for Illumina (New England Biolabs, Ipswich, MA, USA, #E7530). A total of 10 ng of RNA (for smaller samples, 1 ng) was used for library preparation (18 or 21 PCR cycles, depending on the amount of extracted RNA). Sequencing was performed using an Illumina HiSeq 1500 or 2500 to obtain 15–20 million reads using a 50 bp single-end read configuration. Reads were aligned to the Ensembl GRCh38 human genome assembly using STAR77, and gene counts and Transcripts Per Kilobase Million (TPM) were calculated using the R package Rsubread78. The quality data for RNA-sequence has been shown in Supplementary Data 5.
Sample filtering and patient demographics
We excluded low-quality samples from the analysis, specifically samples with low expression of representative structural genes from the stratum corneum, epidermis, and dermis were excluded. Specifically, we excluded samples with counts fewer than 101.5 TPM for CDSN (used as a marker of the stratum corneum of the epidermis) or 103 TPM for KRT10 (used as a marker of the spinous layer of the epidermis). No outliers were observed in the distribution of COL1A1 (used as a dermal fibroblast marker). The criteria were set based on gene distribution (Supplementary Fig. 25). In addition, we removed three samples with a read count lower than 106.5 and seven samples with a RIN (an index of RNA degradation) lower than 2 or unmeasurable. To evaluate the sample quality of samples with lower RIN scores (<6), we conducted a correlational analysis of disease severity (EASI) and metagene expression in AD samples, stratified by RIN scores of the samples (<6 and ≥6). The results demonstrated a consistent trend across both RIN groups, with similar correlational relationships to disease severity (Supplementary Fig. 26). Notably, the correlations involving the four key metagenes (SKITm2, SKITm16, SKITm17, SKITm10) in the samples with low RIN score (<6) exhibited the same trend as observed in the regression analysis which used all samples with RIN scores between 2 and 10 (Fig. 4D).
Based on these criteria, 25 of the 1061 samples were excluded. Thus, for further investigation, we used 1036 samples: 951 AD samples (529 lesional and 422 non-lesional samples of 156 patients), 36 psoriasis samples (18 lesional and 18 non-lesional samples of 18 patients), and 49 normal control samples (26 healthy individuals). The patient and sample demographics are presented in Table 1. In addition, 43 patients with AD and two healthy volunteers were sampled multiple times on different dates (Supplementary Table 2). Table 1 presents the age and severity at the time of initial sampling for patients from whom samples were collected at multiple time points. Detailed information on the sampled regions and local symptoms is summarized in Supplementary Tables 1 and 3.
mRNA expression analysis
All bioinformatic and statistical analyses were conducted using R (version 3.6.3)79. Figures were created using the ggplot2 package (version 3.3.2)80, pheatmap package (version 1.0.12)81, ComplexHeatmap (version 2.2.0)82, VennDiagram (version 1.7.3)83, or the pals package (version 1.6)84. Gene counts were normalized based on a variance-stabilizing transformation (vst) using the DESeq2 R package (version 1.26.0)85. The normalization of gene counts and differentially expressed gene analysis was also performed using DESeq2 R package. Each sample was assigned to one of the five categorical variables according to the disease and presence of symptoms (“lesional AD,” “non-lesional AD,” “lesional psoriasis,” “non-lesional psoriasis,” and “normal”). This variable was included in the design option of the DEseq function, followed by the vst function. The differentially expressed genes were defined as those with an adjusted p < 0.01 and a log fold change greater than 1 or less than -1. For hierarchical clustering using all samples, we selected the 100 genes with the highest variance in the dataset (Supplementary Fig. 2B). Clustering was performed using the ward. D2 method, applying the Euclidean distance.
Quantitative real-time PCR (qRT-PCR)
qRT-PCR was performed to quantify the expression levels of IL17A, IL17C, IL17F, IL23A, CCL20, IL13, CCL13, CCL18, CCL26. Of the samples used in the cross-sectional analysis, 33 samples of lesional AD skin were used for analysis due to considerations of sample quality and the availability of remaining material for PCR analysis. Total RNA (50–100 ng) was reverse-transcribed into complementary DNA (cDNA) using the SuperScript VILO cDNA Synthesis Kit (Thermo Fisher Scientific, #11754050) in a reaction volume of 20 µL, according to the manufacturer’s instructions. TaqMan assays were performed using the TaqMan Fast Advanced Master Mix for qPCR (Thermo Fisher Scientific, #4444556) in a reaction volume of 20 µL, containing 0.2 µL of the reverse transcription product. The details of the TaqMan Gene Expression Assays used in this study are provided in Supplementary Table 10. Reactions were run on a QuantStudio 12 K Flex Real-Time PCR System (Thermo Fisher Scientific) under the recommended cycling conditions. Gene expression data were analyzed using ExpressionSuite Software v1.3 (Thermo Fisher Scientific) following the ΔΔCt method. Ct values of the target genes were normalized to that of the reference gene (HPRT1) and expressed relative to the reference sample. Correlational analysis with metagene expression derived from RNA-seq analysis was performed using Spearman’s correlation method (Fig. 3).
Blood cytokine levels
CCL17 levels were measured in serum samples, and other cytokines were measured in plasma samples. IL-17 levels were measured using a fully automatic, highly sensitive immune analyzer (HI-1000; Sysmex Corp., Hyogo, Japan). Levels of other plasma cytokines (CCL27, IL-4, IL-5, EDN, CCL20, IL-17, IL-22, CXCL9, IL-16, IL-18, CCL5, MMP9, IL-6, TNF-α, TSLP, IL-10) were measured using a fully automatic immune analyzer (HISCL-5000 or HISCL-800; Sysmex Corp., Hyogo, Japan)86,87. The normal serum CCL17 level range was obtained from a previous study88. Since the distribution of cytokine levels was right-skewed, we used log- logarithmically transformed values for analysis to correct skewness and enhance data visualization and interpretability (Supplementary Fig. 27). A constant of 0.1 was added before the log transformation to prevent undefined values at 0 pg/mL.
NMF and rank estimate
NMF is mathematically formulated as the problem of decomposing a two-dimensional matrix A, as the product of two matrices, W and H. Unlike PCA methods, NMF does not require an orthogonality assumption based on the matrix W. In this study, A was the original RNA expression N×M matrix (N genes and M samples). Matrix W had a size of N×k, where each of the k columns represents a metagene, and element wij represents the weight of gene i for metagene j. Matrix H had a size of k×M, where each of the M columns represents the metagene expression pattern of a sample, and element hij represents the expression values of metagene i in sample j.
We performed NMF with the NNLM R package89 using the vst-normalized data. Before the analysis, we filtered genes with a variance less than 0.1 or a mean less than 5.5 to exclude insignificant genes and genes potentially related to measurement noise. Additionally, Y-chromosome genes were excluded from the dataset to avoid sex-biased results. Consequently, we used the expression data of 11,270 genes from 1036 samples for NMF analysis. The minimum expression value was set to zero by subtracting the minimum value of each gene from the expression value of each gene. We estimated the optimal rank k according to the masking approach90,91. Specifically, 1% of elements were randomly deleted from the original matrix and then imputed with NMF through ranks 1 to 50. We then compared the imputed values with the original values by calculating the mean squared error (MSE) between the values to evaluate the reconstruction errors. The evaluation procedure was performed as follows:
-
1.
Randomly select 1% of elements to be deleted.
-
2.
Substitute NA for the selected element of the original matrix A to obtain Adel.
-
3.
Decompose Adel into Wdel and Hdel using the nnmf function with default parameters in the NNLM package.
-
4.
Compute the product of Wdel and Hdel to obtain the reconstructed matrix Arecon.
-
5.
Compute the MSE between the original matrix A and the reconstructed matrix Arecon for the deleted elements only.
The aforementioned steps were repeated 20 times for each rank, and the median reconstruction error for ranks 1–50 was used to determine the optimal rank k (Supplementary Fig. 2C). Next, among the optimal ranks of 25–34 that presented the smallest median errors, k = 29 was chosen as the factorization rank for our dataset. Finally, we decomposed the dataset based on the optimal rank k using the nnmf function with max.iter = 10,000 to calculate the metagenes and the loading matrix.
The “metagene” matrix resulted in 11,270 rows and 29 columns, with each value wij, which was the weight of gene i in metagene j. The “loading” matrix had 29 rows and 1036 columns; value hij represented the expression level of metagene i in sample j. The distribution of gene weights and expression levels in each metagene are shown in Supplementary Figs. 3 and 5.
We declared results with a p < 0.01 as significant throughout the analysis after NMF. As R does not store values smaller than 2.225074e-308 (obtained based on.Machine$double.xmin), an output p value of 0.e + 00 by R was denoted as a p < 2.2e-308 in the figures and text.
Definition of “highly-weighted” and “marker” genes
To interpret the cellular components and pathways associated with the metagenes, we defined the “highly-weighted” genes in the metagene matrix, which were evaluated from the right-skewed distribution (Supplementary Fig. 3), as genes with z-scores > 3. These were obtained by z-normalizing the weights of a metagene (z-scoreg). Of the 11,270 genes, 2368 were selected as “highly weighted” genes. The raw weights of the highly weighted genes within the 29 metagenes are as a heatmap in Fig. 2B. The z-scoreg of the highly-weighted genes within the 29 metagenes are presented in Supplementary Data 2 and 3. The maximum number of “highly-weighted” genes in a metagene was 263, with a minimum of 57, averaging 137.
Furthermore, to search for candidate biomarkers of each metagene, we searched for “marker” genes specifically included in each metagene. The z-score (z-scorem), a measure of the specificity of a particular metagene, was calculated by z-normalizing the z-scoreg again across the metagene. We extracted genes with a z-scorem > 3 that were highly correlated (Pearson’s correlation coefficient > 0.7, p < 0.01) with metagene expression in all skin samples (Supplementary Fig. 28). These genes were defined as “marker” genes. SKITm3, SKITm4, SKITm6, SKITm13, SKITm14, and SKITm20 had no marker genes that met these criteria. The top 10 “highly-weighted” and “marker” genes are presented in Supplementary Data 4.
As for the biological roles of the metagenes, we verified them using knowledge-based approaches in reference to previous in vivo and in vitro studies based on their documented roles, involvement, or activation in skin disorders. For example, SKITm9, SKITm10, and SKITm15, had high weights of genes activated in the skin through IL-17 or IL-22 and related cytokine stimulation. The representative enrichment analysis of the highly weighted genes in SKITm10 with KEGG pathway analysis performed with enrichR92 and Fisher’s exact test are presented in Supplementary Fig. 4 and Supplementary Table 4. The KEGG pathway enrichment analysis included “IL-17 signaling pathway” in the top 10 enriched KEGG pathways (Supplementary Fig. 4). Additionally, we used Fisher’s exact test to evaluate the significance of the overlap between highly weighted genes (Supplementary Table 4) and marker genes (Supplementary Table 5) with genes reportedly induced by IL-17 and its related cytokines in keratinocytes21,25,26,27,28. We constructed a 2 × 2 contingency table to calculate p values and odds ratios.
Statistical analysis of the sample quality and sequence batch
We further studied the association between metagene expression and the symptoms or treatment effects using relative expression values, which were calculated by dividing the expression value of each sample by the maximum expression value in each metagene. The relative expression value was used for analysis throughout the cross-sectional and longitudinal analysis.
Pearson’s correlation coefficients were used to evaluate the correlation between metagene expression and the RIN, total read counts, and the amount of extracted RNA (Supplementary Fig. 6A–C). The amount of RNA was too low to be measured in seven samples, and these samples were thus excluded from this analysis. The RNA amount and total read counts were log10-transformed. Metagenes with correlation coefficients >0.3 or <−0.3 and a p < 0.01 were considered relevant.
The sequence batch effect was also assessed because RNA sequencing was carried out in nine separate sessions owing to the large number of samples. For this analysis, we compared two linear regression models fitted to the data, specifically model 1 with sequence sessions (SQ1–SQ9) as an explanatory variable and model 2 without these sessions. The models were built using a linear model (lm function) and compared by performing an ANOVA test (anova function). To evaluate the effect of sequence sessions on metagene expression depending on the patient background, both models were controlled for sex, the use of cyclosporine, months used after the initiation of dupilumab (0 if not on dupilumab, 1–5 for the first 5 months, and ≥6 months as month 6), disease activity, and the calendar month of the sampled date. The p values in these analyses were corrected using the Bonferroni method for multiple comparisons (Supplementary Fig. 6D).
In summary, SKITm28 was weakly correlated with the RIN (Supplementary Fig. 6A, r = −0.32, p < 2.2e-308). SKITm29 was negatively correlated with the read counts (Supplementary Fig. 6B, r = −0.49, p < 2.2e-308). In addition, SKITm28 and SKITm29 were most significantly associated with sequence sessions (Supplementary Fig. 6D). Thus, we considered SKITm28 and SKITm29 to be related to the sample quality and sequence batch. SKITm14 and SKITm20 were highly expressed in only one sample each (Supplementary Figs. 5 and 6); therefore, we assumed that these two samples were outliers owing to unknown sample bias. The expression of three metagenes (SKITm2, SKITm7, and SKITm10) was correlated substantially with the amount of RNA extracted (Supplementary Fig. 6C). However, the expression of these metagenes did not correlate with the RIN (Supplementary Fig. 6A), and thus, they were considered independent of sample quality.
To evaluate the effect of sample biases on the expression data, we conducted a UMAP analysis using the expression of all metagenes (Supplementary Fig. 7A), as well as a subset of metagenes that was considered unrelated to sample bias (Supplementary Fig. 7B). Specifically, for Supplementary Fig. 7B, we excluded data corresponding to metagenes associated with skin appendages and reticulocyte inclusion (SKITm22–27, SKITm19), outlier samples (SKITm14, SKITm20), and samples affected by quality issues and sequence batch effects (SKITm28, SKITm29). The UMAP analysis was performed using the umap function from the uwot package (version 0.1.8)93.
The association between skin metagenes identified in this study and transcriptomic modules in our prior study was verified as follows29. The previous study was conducted on 315 skin samples, including lesional, non-lesional AD, and normal samples. Of these, 219 samples were common to the present study. First, we assessed the similarity between the highly weighted genes in the 29 metagenes and the highly contributing genes in the 21 skin gene modules using the Jaccard index, which was calculated by dividing the intersection by the union of the genes (Supplementary Fig. 9A). Next, we calculated Spearman’s correlation between the weights of common genes included in the skin module with the highest Jaccard index and the corresponding skin metagene (Supplementary Fig. 9B). For better interpretability, negative weights of skin modules were converted to positive values, and the minimum value was shifted to zero before calculation.
Analysis of association among metagene expression in disease, clinical symptoms, and disease severity in the cross-sectional study
In the cross-sectional study, we analyzed the association between metagene expression levels and the disease severity and local severity of various types of eruptions. Samples from patients not receiving dupilumab treatment were used in this analysis (lesional AD, n = 246; non-lesional AD, n = 170; lesional psoriasis, n = 18; non-lesional psoriasis, n = 18; normal, n = 49; patient demographics are presented in Supplementary Tables 6–8).
Metagene expression between diseases (normal vs. lesional AD, normal vs. non-lesional AD, normal vs. lesional psoriasis, and normal vs. non-lesional psoriasis) was tested using the Wilcoxon test. P values were corrected using the Bonferroni method for multiple comparisons (Fig. 2C and Supplementary Fig. 8). We determined a negative or positive association based on the subtraction of the expected value from the statistical value, with a negative or positive sign indicating the direction of the association. To assess the effect of skin symptom severity (EASI, pruritus score, local severity of erythema, papulation/induration, excoriation, and lichenification) on metagene expression in the sampled skin, we used a linear mixed-effects regression model with random intercepts for each patient. The following model was used for this purpose:
where α is the overall intercept, β1 is the fixed effect coefficient for the skin symptom levels, β2 to βk are the fixed coefficients for the covariates (age, sex, and treatments; oral cyclosporine, topical steroids, tacrolimus, and moisturizers within 1 week of sampling, as categorical values of 0 or 1), μ represents the random intercept for the patients, and ε is the residual term. We used patient ID as a random effect variable because 23 patients were sampled at least twice, and samples from 59 and three patients were obtained simultaneously from more than two regions at the lesional and non-lesional sites, respectively. The use of topical JAK inhibitors and nemolizumab was not included in the regression model because of the small sample size (four samples from two patients on a topical JAK inhibitor and two samples from one patient on nemolizumab). In the analysis of PN and LA, which are rare skin symptoms of AD, we compared metagene expression in lesional and non-lesional sites obtained from the same patients (PN, eight patients; LA, six patients) using a paired t-test. The results are presented in Fig. 4D and Supplementary Fig. 11. The effects of blood cytokine levels and clinical biomarkers on metagene expression were analyzed using the following model:
where β1 represents the fixed effect coefficient for the blood cytokine levels (adding 0.1 and log10-transformed). The same fixed effect variables in β2 to βk were used in the analysis of the skin symptoms. The results are summarized in Fig. 4E and Supplementary Fig. 12. The 95% confidence interval (CI) of predicted values in Supplementary Fig. 11 was visualized using the ggpredict function in the ggeffects package (version 1.1.2)94.
In the analysis of expression profiles of samples with the same local severity and phenotype obtained from the same patients (n = 7) at different time points (Supplementary Fig. 29), the local severity was assessed by the total score of erythema, lichenification, and induration/papulation. The clinical phenotypes that exhibited distinctive expression profiles (excoriation, PN, and LA) were not included to exclude the effect of the clinical phenotypes on the expression profiles. In addition, patients on systemic treatment were not included to exclude treatment effects. A paired Wilcoxon test was used to assess the difference in the metagene expression values.
Transition in skin metagenes and blood cytokines in the longitudinal study of patients treated with dupilumab
In the study involving 24 patients treated with dupilumab, we employed a linear mixed model with random intercepts for each patient. Twenty-four patients from whom a sample could be obtained on the day of dupilumab initiation (month 0) and more than four times in the first sequential 6 months based on calendar months (months 1–6) were included in the analysis. A window of ±7 days was allowed for all time points. The patient demographics are presented in Supplementary Table 9. We tested the changes in metagene expression and blood cytokine levels during treatment using the following model:
where β1 is the fixed effect coefficient for the number of months since the start of dupilumab, which was transformed into seven categorical variables (months 0–6), β2 to βk are the fixed coefficients for the treatment covariates (oral cyclosporine, topical steroids, tacrolimus, and moisturizers within 1 week of sampling, as categorical values of 0 or 1), μ represents the random intercept for the patients, and ε is the residual term. All skin samples used in this analysis were obtained from the posterior trunk and, whenever feasible, were sequentially sampled from the same skin phenotype within each patient. In addition, the analysis of skin did not include samples with excoriation scores ≥1, since skin excoriation was observed only sporadically in the patients. P-values were corrected using the Bonferroni method for multiple comparisons. The results of the regression analysis on skin metagene expression and blood cytokine levels are presented in Fig. 7A and Supplementary Fig. 17. The upper and lower limits of the 95% CI for the mean of normal controls were calculated by subtracting or adding 1.96× the standard error to the mean. The linear mixed models employed in all analyses were fitted using the lmer function in the lmerTest package (version 1.1-23)95.
Response to dupilumab treatment
The mEASI reduction rates in dupilumab-treated patients were assessed in 22 patients from whom the EASI was available at baseline and month 6 (data for two patients were unavailable) (Supplementary Table 9). After 6 months of treatment, 14 of 22 patients achieved a mEASI75 (75% improvement in the EASI from baseline), and 20 of 22 patients achieved a mEASI50 (50% improvement in the EASI); no data were available for two patients.
However, the absolute value of severity scores, rather than the reduction rate, has recently been discussed as a better measure for assessing treatment outcomes and long-term disease control in real-world clinical settings96,97,98. Accordingly, we classified the patients into three groups based on their absolute severity scores during treatment: the “early” responders achieved and maintained a mild disease level (defined by mEASI ≤ 6.3) within the first month and throughout 6 months of dupilumab treatment, “poor” responders never reached the mild level during the entire 6-month study, while “intermediate” responders did not meet the criteria for “early” or “poor” (Fig. 5A).
We then assessed differences in pretreatment metagene expression and blood biomarkers among the three patient clusters using Spearman’s correlation analysis. For this analysis, response categories were converted to numeric variables (“early” = 1, “intermediate” = 2, “poor” = 3), and correlations were calculated with respect to the expression values of metagenes, marker genes, and blood cytokine levels (Fig. 5C–E, Supplementary Figs. 15 and 16). We also evaluated the correlation between disease severity scores post-dupilumab treatment and pretreatment levels of skin and blood biomarkers using Spearman’s correlation (Fig. 6). Marker genes from SKITm15, which did not exhibit significant results, are not included.
Five patients receiving cyclosporine at the start of dupilumab treatment were excluded from the correlation analysis. Additionally, four patients with no non-lesional regions in the posterior trunk area were excluded from the correlation analysis of non-lesional samples.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
New unique reagents were not generated in this study. The RNA-seq data generated in this study have been deposited in the National Bioscience Database Center (NBDC) Human Database under accession code JGAS000780. The RNA-seq data are available under restricted access for issue on privacy in informed consent by participants, access can be obtained by application for hum0413 at the NBDC. All data are included in the Supplementary Information or available from the authors. The raw numbers for charts and graphs are available in the Supplementary Information or the Source Data file whenever possible. Source data are provided with this paper.
Code availability
This paper does not report the original code.
References
He, H. et al. Single-cell transcriptome analysis of human skin identifies novel fibroblast subpopulation and enrichment of immune subsets in atopic dermatitis. J. Allergy Clin. Immunol. 145, 1615–1628 (2020).
Clayton, K. et al. Machine learning applied to atopic dermatitis transcriptome reveals distinct therapy-dependent modification of the keratinocyte immunophenotype. Br. J. Dermatol. 184, 913–922 (2021).
Silvestre Salvador, J. F., Romero-Pérez, D. & Encabo-Durán, B. Atopic dermatitis in adults: a diagnostic challenge. J. Investig. Allergol. Clin. Immunol. 27, 78–88 (2017).
Soret, P. et al. A new molecular classification to drive precision treatment strategies in primary Sjögren’s syndrome. Nat. Commun. 12, 1–18 (2021).
Banchereau, R. et al. Personalized immunomonitoring uncovers molecular networks that stratify lupus patients. Cell 165, 551–565 (2016).
Thijs, J. L. et al. Moving toward endotypes in atopic dermatitis: identification of patient clusters based on serum biomarker analysis. J. Allergy Clin. Immunol. 140, 730–737 (2017).
Guttman-Yassky, E. et al. Use of tape strips to detect immune and barrier abnormalities in the skin of children with early-onset atopic dermatitis. JAMA Dermatol. 155, 1358–1370 (2019).
He, H. et al. Tape strips detect distinct immune and barrier profiles in atopic dermatitis and psoriasis. J. Allergy Clin. Immunol. 147, 199–212 (2021).
Rojahn, T. B. et al. Single-cell transcriptomics combined with interstitial fluid proteomics defines cell type–specific immune regulation in atopic dermatitis. J. Allergy Clin. Immunol. 146, 1056–1069 (2020).
Margolis, J. S., Abuabara, K., Bilker, W., Hoffstad, O. & Margolis, D. J. Persistence of mild to moderate atopic dermatitis. JAMA Dermatol. 150, 593–600 (2014).
Hendricks, A. J., Lio, P. A. & Shi, V. Y. Management recommendations for dupilumab partial and non-durable responders in atopic dermatitis. Am. J. Clin. Dermatol. 20, 565–569 (2019).
AbbVie. Rinvoq: Prescribing Information. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/211675s000lbl.pdf (2019).
Pfizer. Cibinqo: Prescribing Information. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/213871s000lbl.pdf (2022).
Drucker, A. M. et al. Systemic immunomodulatory treatments for atopic dermatitis: update of a living systematic review and network meta-analysis. JAMA Dermatol. 158, 523–532 (2022).
Nestle, F. O., Di Meglio, P., Qin, J.-Z. & Nickoloff, B. J. Skin immune sentinels in health and disease. Nat. Rev. Immunol. 9, 679–691 (2009).
Möbus, L. et al. Atopic dermatitis displays stable and dynamic skin transcriptome signatures. J. Allergy Clin. Immunol. 147, 213–223 (2021).
Cheng, J. B. et al. Transcriptional programming of normal and inflamed human epidermis at single-cell resolution. Cell Rep. 25, 871–883 (2018).
Noda, S. et al. The Asian atopic dermatitis phenotype combines features of atopic dermatitis and psoriasis with increased TH17 polarization. J. Allergy Clin. Immunol. 136, 1254–1264 (2015).
Tian, S. et al. Meta-analysis derived (MAD) transcriptome of psoriasis defines the ‘core’ pathogenesis of disease. PLoS ONE 7, e44274 (2012).
Zhao, L., Zhao, H. & Yan, H. Gene expression profiling of 1200 pancreatic ductal adenocarcinoma reveals novel subtypes. BMC Cancer 18, 603 (2018).
Tollenaere, M. A. X. et al. Signalling of multiple interleukin (IL)-17 family cytokines via IL-17 receptor A drives psoriasis-related inflammatory pathways. Br. J. Dermatol. 185, 585–594 (2021).
Sestito, R. et al. STAT3-dependent effects of IL-22 in human keratinocytes are counterregulated by sirtuin 1 through a direct inhibition of STAT3 acetylation. FASEB J. 25, 916–927 (2011).
Lambert, S., Swindell, W. R., Tsoi, L. C., Stoll, S. W. & Elder, J. T. Dual role of Act1 in keratinocyte differentiation and host defense: TRAF3IP2 silencing alters keratinocyte differentiation and inhibits IL-17 responses. J. Invest. Dermatol. 137, 1501–1511 (2017).
Wolk, K. et al. IL-22 regulates the expression of genes responsible for antimicrobial defense, cellular differentiation, and mobility in keratinocytes: a potential role in psoriasis. Eur. J. Immunol. 36, 1309–1323 (2006).
Nograles, K. E. et al. Th17 cytokines interleukin (IL)-17 and IL-22 modulate distinct inflammatory and keratinocyte-response pathways. Br. J. Dermatol. 159, 1092–1102 (2008).
Swindell, W. R. et al. Heterogeneity of inflammatory and cytokine networks in chronic plaque psoriasis. PLoS One 7, e34594 (2012).
Gupta, R. K. et al. TWEAK functions with TNF and IL-17 on keratinocytes and is a potential target for psoriasis therapy. Sci. Immunol. 6, eabi8823 (2021).
Jiang, Y. et al. Suppression of TCF4 promotes a ZC3H12A-mediated self-sustaining inflammatory feedback cycle involving IL-17RA/IL-17RE epidermal signaling. JCI Insight 9, e172764 (2024).
Sekita, A. et al. Multifaceted analysis of cross-tissue transcriptomes reveals phenotype-endotype associations in atopic dermatitis. Nat. Commun. 14, 6133 (2023).
Patruno, C., Potestio, L. & Napolitano, M. Clinical phenotypes of adult atopic dermatitis and related therapies. Curr. Opin. Allergy Clin. Immunol. 22, 242–249 (2022).
Furue, K., Ito, T., Tsuji, G., Nakahara, T. & Furue, M. The CCL20 and CCR6 axis in psoriasis. Scand. J. Immunol. 91, e12846 (2020).
Nakagawa, H. et al. Phase 2 clinical study of delgocitinib ointment in pediatric patients with atopic dermatitis. J. Allergy Clin. Immunol. 144, 1575–1583 (2019).
Leshem, Y. A., Hajar, T., Hanifin, J. M. & Simpson, E. L. What the Eczema Area and Severity Index score tells us about the severity of atopic dermatitis: an interpretability study. Br. J. Dermatol. 172, 1353–1357 (2015).
Jang, D. H. et al. A 52 weeks dupilumab treatment for moderate to severe atopic dermatitis in Korea: long-term efficacy and safety in real world. Sci. Rep. 11, 23539 (2021).
Okiyama, N. et al. Successful topical treatment with ketoconazole for facial rashes refractory to dupilumab in patients with atopic dermatitis: case reports. J. Eur. Acad. Dermatol. Venereol. 34, e474–e476 (2020).
Jo, C. E. et al. Facial and neck erythema associated with dupilumab treatment: a systematic review. J. Am. Acad. Dermatol. 84, 1339–1347 (2021).
Wongvibulsin, S. et al. Transcriptomic analysis of atopic dermatitis in African Americans is characterized by Th2/Th17-centered cutaneous immune activation. Sci. Rep. 11, 11175 (2021).
Brunet, J.-P., Tamayo, P., Golub, T. R. & Mesirov, J. P. Metagenes and molecular pattern discovery using matrix factorization. Proc. Natl. Acad. Sci. USA 101, 4164–4169 (2004).
Johnson, J. A. I. et al. Inferring cellular and molecular processes in single-cell data with non-negative matrix factorization using Python, R and GenePattern Notebook implementations of CoGAPS. Nat. Protoc. 18, 3690–3731 (2023).
Yang, L., Zhang, S. & Wang, G. Keratin 17 in disease pathogenesis: from cancer to dermatoses. J. Pathol. 247, 158–165 (2019).
Bangert, C. et al. Persistence of mature dendritic cells, TH2A, and Tc2 cells characterize clinically resolved atopic dermatitis under IL-4Rα blockade. Sci. Immunol. 6, eabe2749 (2021).
Boothby, I. C. et al. Early-life inflammation primes a T helper 2 cell-fibroblast niche in skin. Nature 599, 667–672 (2021).
Gittler, J. K. et al. Progressive activation of TH2/TH22 cytokines and selective epidermal proteins characterizes acute and chronic atopic dermatitis. J. Allergy Clin. Immunol. 130, 1344–1354 (2012).
Sanyal, R. D. et al. Atopic dermatitis in African American patients is TH2/TH22-skewed with TH1/TH17 attenuation. Ann. Allergy Asthma Immunol. 122, 99–110.e6 (2019).
Martin, M. J. et al. Genetics and epigenetics of atopic dermatitis: an updated systematic review. Genes 11, 442 (2020).
Rios, C. et al. A multi-omics evaluation of the non-lesional skin surface identifies atopic dermatitis with food allergy (AD FA+) as a unique endotype. J. Allergy Clin. Immunol. 143, AB125 (2019).
Nemoto-Hasebe, I. et al. Clinical severity correlates with impaired barrier in filaggrin-related eczema. J. Invest. Dermatol. 129, 682–689 (2009).
Peng, G. et al. Experimental and clinical evidence suggests that treatment with betacellulin can alleviate Th2-type cytokine-mediated impairment of skin barrier function. Int. J. Mol. Sci. 23, 11520 (2022).
Köten, B. et al. RNase 7 contributes to the cutaneous defense against Enterococcus faecium. PLoS One 4, e6424 (2009).
Wang, S. et al. Single cell transcriptomics of human epidermis identifies basal stem cell transition states. Nat. Commun. 11, 1–14 (2020).
Pugliarello, S., Cozzi, A., Gisondi, P. & Girolomoni, G. Phenotypes of atopic dermatitis. J. Dtsch. Dermatol. Ges. 9, 12–20 (2011).
Belzberg, M. et al. Prurigo nodularis is characterized by systemic and cutaneous T helper 22 immune polarization. J. Investig. Dermatol. 141, 2208–2218.e14 (2021).
Henry, J. et al. Hornerin is a component of the epidermal cornified cell envelopes. FASEB J. 25, 1567–1576 (2011).
Trzeciak, M. et al. Expression profiles of genes encoding cornified envelope proteins in atopic dermatitis and cutaneous T-cell lymphomas. Nutrients 12, 862 (2020).
McAleer, M. A. et al. Systemic and stratum corneum biomarkers of severity in infant atopic dermatitis include markers of innate and T helper cell-related immunity and angiogenesis. Br. J. Dermatol. 180, 586–596 (2019).
Suárez-Fariñas, M. et al. Nonlesional atopic dermatitis skin is characterized by broad terminal differentiation defects and variable immune abnormalities. J. Allergy Clin. Immunol. 127, 954–64.e1–4 (2011).
Guttman-Yassky, E., Krueger, J. G. & Lebwohl, M. G. Systemic immune mechanisms in atopic dermatitis and psoriasis with implications for treatment. Exp. Dermatol. 27, 409–417 (2018).
Ascensión, A. M., Fuertes-Álvarez, S., Ibañez-Solé, O., Izeta, A. & Araúzo-Bravo, M. J. Human dermal fibroblast subpopulations are conserved across single-cell RNA sequencing studies. J. Investig. Dermatol. 141, 1735–1744.e35 (2021).
Golebski, K. et al. High degree of overlap between responses to a virus and to the house dust mite allergen in airway epithelial cells. PLoS ONE 9, e87768 (2014).
Tatebe, K. et al. Response network analysis of differential gene expression in human epithelial lung cells during avian influenza infections. BMC Bioinform. 11, 1–15 (2010).
Liu, W. et al. Establishment of extracellular signal-regulated kinase 1/2 bistability and sustained activation through Sprouty 2 and its relevance for epithelial function. Mol. Cell. Biol. 30, 1783–1799 (2010).
Christianson, C. A. & Alam, R. Mechanisms of sustained signalling in asthma. Curr. Opin. Allergy Clin. Immunol. 13, 100 (2013).
Pfisterer, K., Shaw, L. E., Symmank, D. & Weninger, W. The extracellular matrix in skin inflammation and infection. Front Cell Dev. Biol. 9, 682414 (2021).
Hirota, T. et al. Genome-wide association study identifies eight new susceptibility loci for atopic dermatitis in the Japanese population. Nat. Genet. 44, 1222–1226 (2012).
Nguyen, X.-X. et al. Lysyl oxidase directly contributes to extracellular matrix production and fibrosis in systemic sclerosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 320, L29–L40 (2021).
Russell, J. J. et al. Reversion inducing cysteine rich protein with Kazal motifs and cardiovascular diseases: the RECKlessness of adverse remodeling. Cell. Signal. 83, 109993 (2021).
Dong, Z.-R. et al. RECK expression is associated with angiogenesis and immunogenic tumor microenvironment in hepatocellular carcinoma, and is a prognostic factor for better survival. J. Cancer 12, 3827–3840 (2021).
Dimitrov, D., Al Adawi, M., Abdelhadi, Z. & Jafferany, M. Exacerbation of psoriasis after initiation of dupilumab in atopic dermatitis patient. Dermatol. Ther. 33, e13572 (2020).
Schrom, K. P., Kobs, A. & Nedorost, S. Clinical psoriasiform dermatitis following dupilumab use for autoeczematization secondary to chronic stasis dermatitis. Cureus 12, e7831 (2020).
Guttman-Yassky, E. et al. Dupilumab progressively improves systemic and cutaneous abnormalities in patients with atopic dermatitis. J. Allergy Clin. Immunol. 143, 155–172 (2019).
Bakker, D. S. et al. Early and long-term effects of dupilumab treatment on circulating T-cell functions in patients with moderate-to-severe atopic dermatitis. J. Investig. Dermatol. 141, 1943–1953.e13 (2021).
Imai, Y., Kusakabe, M., Nagai, M., Yasuda, K. & Yamanishi, K. Dupilumab effects on innate lymphoid cell and helper T Cell populations in patients with atopic dermatitis. JID Innov. 1, 100003 (2021).
Bissonnette, R. et al. The oral Janus kinase/spleen tyrosine kinase inhibitor ASN002 demonstrates efficacy and improves associated systemic inflammation in patients with moderate-to-severe atopic dermatitis: results from a randomized double-blind placebo-controlled study. Br. J. Dermatol. 181, 733–742 (2019).
Liu, Y. et al. Genomic control of inflammation in experimental atopic dermatitis. Sci. Rep. 12, 18891 (2022).
Hanifin, J. & Rajka, G. Diagnostic features of atopic dermatitis. Acta Derm. Venereol. 92, 44–47 (1980).
Hanifin, J. M. et al. The eczema area and severity index—a practical guide. Dermatitis 33, 187–192 (2022).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Liao, Y., Smyth, G. K. & Shi, W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 47, e47 (2019).
R Core Team. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria (2019). Available at: https://www.R-project.org/.
Wickham, H. ggplot2: Elegant Graphics for Data Analysis. (Springer, 2016).
Kolde, R. Pretty Heatmaps. R package version 1.0.12. Available at: https://CRAN.R-project.org/package=pheatmap (2019).
Gu, Z., Eils, R. & Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849 (2016).
Chen, H. VennDiagram: Generate High-Resolution Venn and Euler Plots. R package version 1.6.20. Available at: https://CRAN.R-project.org/package=VennDiagram (2018).
Wright, K. pals: Color Palettes, Colormaps, and Tools to Evaluate Them. R package version 1.6. Available at: https://CRAN.R-project.org/package=pals (2019).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Hasegawa, T. et al. Development of a new HISCL automated CXCL9 immunoassay. Sci. Rep. 13, 5342 (2023).
Hasegawa, T. et al. Type 1 inflammatory endotype relates to low compliance, lung fibrosis, and severe complications in COVID-19. Cytokine 148, 155618 (2021).
Tamaki, K. et al. Serum TARC/CCL 17 levels as a disease marker of atopic dermatitis. Jpn. J. Dermatol. B 116, 27–39 (2006).
Lin, X. & Paul C. Boutros. NNLM: Fast and Versatile Non-Negative Matrix Factorization. R package version 0.4.4. Available at: https://github.com/linxihui/NNLM (2020).
Ikeda, K. et al. Detecting time-evolving phenotypic components of adverse reactions against BNT162b2 SARS-CoV-2 vaccine via non-negative tensor factorization. iScience 25, 105237 (2022).
Lin, X. & Boutros, P. C. Optimization and expansion of non-negative matrix factorization. BMC Bioinforma. 21, 7 (2020).
Jawaid, W. enrichR: Provides an R Interface to ‘Enrichr’. R package version 3.2. Available at: https://CRAN.R-project.org/package=enrichR (2023).
Melville, J. uwot: The Uniform Manifold Approximation and Projection (UMAP) Method for Dimensionality Reduction. R package version 0.1.8. Available at: https://CRAN.R-project.org/package=uwot (2020).
Lüdecke, D. ggeffects: tidy data frames of marginal effects from regression models. J. Open Source Softw. 3, 772 (2018).
Kuznetsova, A., Brockhoff, P. B. & Christensen, R. H. B. lmerTest package: tests in linear mixed effects models. J. Stat. Softw. 82, 1–26 (2017).
Seidl, U. et al. Absolute Psoriasis Area and Severity Index as a valuable marker to determine initial treatment response in psoriasis patients treated with guselkumab in routine clinical care. Dermatol. Ther. 35, e15193 (2022).
Thyssen, J. P. et al. Baricitinib provides rapid and sustained improvements in absolute EASI and SCORAD outcomes in adults with moderate-to-severe atopic dermatitis. J. Dermatol. Treat. 34, 2216322 (2023).
Ferrucci, S. et al. Predictive factors of early response to dupilumab in patients with moderate-to-severe atopic dermatitis. J. Clin. Med. 12, 6575 (2023).
Acknowledgements
We are grateful to all the participants for their cooperation in this research. We thank Y. Yano, H. Maeo, S. Shibata, M. Tanaka, Y. Toriumi, and S. Saeki for supporting patient sampling; Y. Hasegawa for mRNA extraction and quality assessment and qRT-PCR analysis; and R. Ohashi, A. Hananoe, M. Otsuka, E. Okutsu, A. Kobayashi, A. Sugimoto, and T. Takemori for supporting the maintenance of patient samples and data storage. We thank D. Karalis and F. Zhao of Sysmex R&D Centre Europe GmbH; K. Yoshida, T. Oyama, T. Kaji, and S. Hazama of Sysmex Corporation for plasma sample analysis; and H Lunding, K. Oda, and A. Nehrmann of Sysmex R&D Centre Europe GmbH for the establishment of reagents for immunoassays. This study was supported by the Japan Agency for Medical Research and Development (AMED) (JP16ek0410028, JP19ek0410058 and JP22ek0410098 awarded to M.A., JP17ek0410046 and JP20ek0410079 awarded to H. Koseki, JP23ek0410118 awarded to H. Kawasaki, and 243fa627003h0003 awarded to E.K.), the Japan Science and Technology Agency (JST) support program for starting up innovation hubs (JPMJIH1504 awarded to K.S.) and Moonshot R&D Grants (JPMJMS2025 awarded to E.K.), and the Japan Society for the Promotion of Science (JSPS) KAKENHI (22H04994 awarded to M.A., 21K08338 awarded to H. Kawasaki and 22K08391 awarded to A.F.N.) and Core-to-Core Program (JPJSCCA20200006 awarded to E.K.), and the Maruho Takagi Dermatology Foundation awarded to H. Kawasaki. We would like to thank Editage for editing and reviewing this manuscript for the English language.
Author information
Authors and Affiliations
Contributions
Conceptualization: H. Kawasaki, H. Koseki, E.K., M.A. Formal analysis: A.F.N., E.K. Resources: A.F.N., H. Kawasaki, K.Y., S.O., H.S., T.E. Writing—Original Draft: A.F.N. Writing—Review & Editing: A.F.N., H. Kawasaki, K.Y., S.O., K.T., T. Etoh, T. Ebihara, H.S., T.H., J.Y., J.S., O.O., A.S., T.M., K.A., H. Koseki, K.S., E.K., M.A. Visualization: A.F.N., E.K. Funding Acquisition: A.F.N., H. Kawasaki, K.S., H. Koseki, M.A. Project Administration: M.A.
Corresponding authors
Ethics declarations
Competing interests
M.A. has received research support and funds (grants paid to his institution) from Maruho, Ono, Torii, Sato, and Taiho. H. Koseki has received research funds (grants paid to his institution) from Maruho and Kao. H. Kawasaki received research funds (grants paid to his institution) from Torii. H.S. has received research support and funds (grants paid to his institution) from Taiho, Torii, and Tokiwa and received lecture fees from Maruho, Sanofi, AbbVie, and Torii. T. Etoh received lecture fees from Maruho. T.H. is employed by Sysmex R&D Center Europe GmbH. All other authors declare no conflicts of interest.
Peer review
Peer review information
Nature Communications thanks Kilian Eyerich and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Fukushima-Nomura, A., Kawasaki, H., Yashiro, K. et al. An unbiased tissue transcriptome analysis identifies potential markers for skin phenotypes and therapeutic responses in atopic dermatitis. Nat Commun 16, 4981 (2025). https://doi.org/10.1038/s41467-025-59340-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-59340-x
