
Abstract
Biomedical interventions that block the transmission of Plasmodium falciparum (Pf) from humans to mosquitoes may be critical for malaria elimination. Pfs48/45, a gamete-surface protein essential for Pf development in the mosquito midgut, is a target of clinical-stage transmission-blocking vaccines and monoclonal antibodies (mAbs) that disrupt Pf transmission to mosquitoes. Antibodies directed to domain 3 of Pfs48/45 have been structurally and functionally described; however, in-depth information about other inhibitory epitopes on Pfs48/45 is currently limited. Here, we present a cryo-electron microscopy structure of full-length Pfs48/45 in complex with potent human mAbs targeting all three domains. Our data indicate that although Pfs48/45 domains 1 and 2 are rigidly coupled, there is substantial conformational flexibility between domains 2 and 3. Characterization of mAbs against domain 1 revealed the presence of a conformational epitope class that is largely conserved across Pf field isolates and is associated with recognition by potent antibodies. Our study provides insights into epitopes across full-length Pfs48/45 and has implications for the design of next-generation malaria interventions.
Similar content being viewed by others


Main
The severe burden of malaria is evident from the 597,000 deaths worldwide in 2023, predominantly in children under the age of 5 in sub-Saharan Africa1. The deadliest form is caused by Pf parasites that are transmitted to humans by Anopheles mosquitoes2. The development of effective vaccines and mAbs to combat malaria is challenged in part by the transient presentation of immunogenic epitopes throughout the parasite’s complex lifecycle3,4. Modestly effective, pre-erythrocytic-stage vaccines have been approved by the World Health Organization (WHO)5,6, and pre-erythrocytic-stage mAbs are currently advancing through clinical trials for malaria prophylaxis7,8. Targeting antigens expressed by parasites during their asexual blood stage9 and sexual mosquito stage3 provides additional opportunities for biomedical interventions. For example, vaccines and mAbs preventing the transmission of sexual-stage gametocytes from humans back to mosquitoes exploit a bottleneck that presents a critical opportunity to end the chain of transmission. Transmission-blocking vaccines (TBVs) elicit a humoral response against sexual-stage antigens that are required for parasite development in the mosquito midgut and represent a complementary approach to pre-erythrocytic stage vaccines towards malaria elimination10,11.
The Pfs48/45 protein is a leading candidate for the development of TBVs and mAbs. Pfs48/45 is expressed during gametocytogenesis, and its ortholog in Plasmodium berghei, Pbs48/45, is essential for male gamete fertility12. Pfs48/45 is attached to the surface of the parasite membrane through a glycophosphatidylinositol (GPI)-linked anchor13. Clinical development of Pfs48/45-based TBVs has been hindered by the inherently unstable nature of this protein14,15. To address this, the leading Pfs48/45-based TBV candidates are composed of domain 3 (D3) genetically conjugated to the stabilizing carrier protein R0 (R0.6C) or the pro-domain of Pfs230 (ProC6C). These promising candidates have advanced to phase I clinical trials for evaluation of humoral immunogenicity (NCT04862416, PACTR202201848463189, ISRCTN13649456, NCT05400746)16. Recent preclinical studies have also demonstrated additional opportunities for improved immune responses using other designs. For example, two studies have reported the rational design of stabilizing substitutions in D3 that conferred enhanced protein expression and stability, leading to increased potency of the humoral response in animal studies compared with the wild-type protein17,18.
TB31F is a highly potent clinical-stage mAb directed against Pfs48/45, which was derived from humanization of the rat mAb 85RF45.1 (ref. 19). The structural and functional elucidation of TB31F bound to Pfs48/45 D3 has provided the molecular rationale for clinical evaluation of TB31F and further improved biochemical characterization of Pfs48/45 (ref. 20). Moreover, the rational design of D3 was the result of insights into the structure–function relationship of TB31F (ref. 20) against Pfs48/45 D3, thus highlighting the value of characterizing mAb epitopes at the molecular level to enable protein engineering of next-generation vaccine candidates.
Antibodies targeting all the domains of Pfs48/45 have been identified with varying degrees of transmission-reducing activity (TRA), yet the relationship between epitope and antibody potency remains poorly understood, particularly for D1 and D2 (refs. 21,22). For example, from a panel of 16 mAbs derived from mice immunized with recombinant full-length Pfs48/45 expressed from Drosophila melanogaster S2 cells, only one mAb was detected that possessed significant TRA when assessed at 350 µg ml–1 (ref. 16). In a follow-up study, the D2-targeting mAb 10D8, which possesses weak TRA, was observed to bind competitively with low-potency mAb 6A10 and more potent mAb 1F10 (ref. 23). Crystal structures of full-length Pfs48/45 bound by 10D8 and two D3-directed Fabs unveiled the 10D8 epitope at an interface opposite from those of the two more-potent mAbs targeting D3. However, it remains unclear how D2-directed mAbs interact with Pfs48/45 more broadly16,23. Moreover, although we and others have reported that mAbs targeting D1 or D2 can also possess TRA, the structural elucidation of any D1-targeting mAbs has not been previously reported20,22,24.
In this study, we present functional and structural insights into human mAbs that target D1, D2, and D3 of Pfs48/45. Using protein engineering and cryo-electron microscopy (cryo-EM), we determined the structure of full-length Pfs48/45 in complex with the fragment antigen-binding regions (Fabs) RUPA-58 (D1) and RUPA-154 (D2), and the previously described D3-targeting Fabs TB31F and RUPA-44 (ref. 22). We report an extended conformation for full-length Pfs48/45, with a rigid D1–D2 interface but discernible flexibility between D2 and D3. Focused refinement of the D1–D2 segment of Pfs48/45 resulted in a 3.3-Å-resolution map that revealed previously undescribed highly potent and moderately potent epitopes on D1 and D2, respectively. Our study, using antibodies against Pfs48/45 across all its domains, unveils the structure–function relationships underlying TRA.
Results
Selection of D1-, D2- and D3-targeting human antibodies
We previously characterized a panel of human mAbs22 directed against full-length Pfs48/45 and a potent humanized mAb against D3 (ref. 20). From these, we selected for further characterization a set of mAbs with significant TRA that bind with high affinity to conformational epitopes on different domains of Pfs48/45: D1 (RUPA-58), D2 (RUPA-154), and D3 (RUPA-44 and TB31F) (Extended Data Fig. 1a).
mAbs to D1 and D3 showed potent TRA in the standard membrane-feeding assay (SMFA), with 80% inhibitory concentration (IC80) values of 7.5, 11.0, and 0.86 µg ml–1 for RUPA-58, RUPA-44, and TB31F, respectively (Fig. 1a). D2-targeting RUPA-154 showed moderate TRA, with an IC80 of 103.1 µg ml–1 (Fig. 1a). All mAbs showed high apparent affinity for female gametes in surface immunofluorescence assays (SIFAs), with half-maximal effective concentration (EC50) values ranging from 0.009 to 0.31 µg ml–1 (Fig. 1b). The strong binding of RUPA-154 to female gametes (EC50 = 0.14 µg ml–1) contrasts with its moderate TRA and suggests that TRA and binding to female gametes are not directly correlated. Importantly, our previous study indicated that all four functionally-active antibodies can bind non-overlapping epitope bins22 and could therefore be used to assemble a complex with full-length Pfs48/45.

a, TRA of mAbs in SMFA with Pf NF54 and Anopheles stephensi mosquitoes. Multiple concentrations of each mAb were tested in four to six separate SMFA experiments that are represented by different symbols. IC80 values were calculated using linear regression analysis and are shown above each graph, including 95% confidence intervals. TRA values above 99.5% were not included in the analyses. b, Binding of mAbs to live female Pf NF54 gametes by flow cytometry. Data points are the means of two experiments with three technical replicates each. Data from each experiment were normalized for the mean fluorescence intensity (MFI) of each mAb at the highest concentration tested. Bars represent the s.e.m. Non-linear regression curves were fitted to calculate EC50 binding values; EC50 values are shown above each graph with 95% confidence intervals. c, Construct schematics and size-exclusion chromatography profiles of full-length Pfs48/45, stabilized Pfs48/45 mAgE2 (with p.G397L, p.H308Y, p.I402V, p.S361R and p.D373S), and stabilized Pfs48/45 mAgE2 linked to a scTB31F Fab (Pfs48/45 mAgE2-scTB31F, top; abbreviated as Pfs48/45-scTB31F hereafter). Y-axis indicates milli-absorbance units (mA.U.) d, BLI binding curves of RUPA-58, RUPA-154, and RUPA-44 Fabs to Pfs48/45-scTB31F.
Engineering of a Pfs48/45 construct for structure determination
Achieving recombinant expression of full-length Pfs48/45, with its native GPI anchor removed (amino acids (aa) 28–428), in HEK293 cells was challenging owing to poor expression and a tendency to aggregate (Fig. 1c). We have recently demonstrated that stabilizing substitutions in D3 can improve expression of properly folded D3 compared with the wild-type protein17. To obtain a sufficient quantity of well-behaved full-length Pfs48/45, we introduced five D3-stabilizing substitutions (mAgE2-G397L H308Y I402V S361R D373S)17 in the full-length construct. This approach improved protein yield considerably, but still resulted in heterogeneity. To further ameliorate the construct, we fused the carboxy terminus of the stabilized full-length Pfs48/45 with a 21-aa flexible linker to the heavy chain (HC) of the single chain (sc) Fab of TB31F. The yield of the resulting construct, Pfs48/45-scTB31F, was ~30% higher than that of stabilized Pfs48/45 without TB31F, and the proportion of misfolded oligomers was reduced (Fig. 1c and Extended Data Fig. 1b,c). Moreover, Pfs48/45-scTB31F was recognized with nanomolar or sub-nanomolar binding affinity by RUPA-44, RUPA-154 and RUPA-58 Fabs, as measured by bio-layer interferometry (BLI), confirming proper folding of all three domains of Pfs48/45 in this engineered construct (Fig. 1d).
Cryo-EM structure determination of full-length Pfs48/45
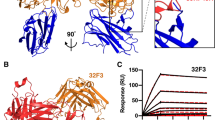
We used Pfs48/45-scTB31F in complex with D1-binding RUPA-58 Fab and D3-binding RUPA-44 Fab for cryo-EM studies. Initial analysis of the collected dataset revealed a highly preferred orientation (Extended Data Figs. 2a and 3a–e). The addition of D2-binding RUPA-154 Fab to the quaternary complex to form a quinary complex did not initially improve the distribution of views (Extended Data Fig. 3f–j and Supplementary Fig. 1a). Further enriching the diversity of particle orientations and improving particle alignment was challenging owing to the extended, narrow shape and flexibility of the complex, so we subsequently collected a dataset on the Pfs48/45-scTB31F:RUPA-44:RUPA-58:RUPA-154 complex with the specimen stage tilted to 40° and 55°. This approach resulted in an improved distribution of views and alignment of particles (Extended Data Figs. 2b and 3k–o and Supplementary Fig. 1b). Combining particles from the datasets collected at the 0°, 40° and 55° tilts and subsequent flexible refinement in cryoSPARC v4 (ref. 25) resulted in a 3.9-Å-resolution map of the complex (Fig. 2a, Extended Data Figs. 2b and 3o, and Supplementary Fig. 1c,e). This map enabled fitting of the X-ray crystal structure of D3 bound to the RUPA-44 Fab (PDB ID: 7UXL (ref. 22)), as well as the TB31F Fab (PDB ID: 6E63 (ref. 20); Fig. 2a and Extended Data Fig. 4a). To further enhance the resolution of the less-characterized D1–D2 segment of Pfs48/45, the Pfs48/45 D1–D2 and RUPA-58 and RUPA-154 variable regions were locally refined, resulting in a 3.3-Å-resolution map of this region (Fig. 2b,c, Extended Data Figs. 2b and 3p–r, Supplementary Fig. 1d,f, and Table 1). The quality of the locally refined map of the Pfs48/45-scTB31F:RUPA-44:RUPA-58:RUPA-154 complex was sufficient to unambiguously build a molecular model for the majority of the RUPA-58 and RUPA-154 variable regions and ~80% of the Pfs48/45 D1 and D2 residues (Fig. 2c and Extended Data Fig. 4b,c). Together, the cryo-EM data led to detailed molecular insights into the Pfs48/45 structure and the epitopes it contains across its three domains.

a, A cryo-EM map of Pfs48/45-scTB31F:RUPA-44:RUPA-58:RUPA-154 complex at 3.9-Å resolution. b, A locally refined cryo-EM map of D1, D2, and the RUPA-58 and RUPA-154 variable regions at 3.3-Å resolution. c, Details of the atomic model fit into the locally refined cryo-EM map. The cryo-EM density is shown as black mesh. D1, D2, and D3 are colored light blue, blue, and navy, respectively, and RUPA-58, RUPA-154, RUPA-44, and TB31F are colored white, yellow, orange, and red, respectively.
Flexibility within full-length Pfs48/45
Although D1 and D2 are closely situated and form a well-defined interface, D3 is positioned farther away and is connected to D2 through a 5-aa flexible linker (aa 290–294; Fig. 3a). Three-dimensional (3D) variability analysis of the Pfs48/45-scTB31F:RUPA-44:RUPA-58:RUPA-154 map indicates that D3 rotates approximately 7° relative to D1 and D2 (Fig. 3b and Supplementary Video 1). Notably, comparison of the quaternary complex Pfs48/45-scTB31F:RUPA-44:RUPA-58 revealed a larger angle between D2 and D3 than in the quinary complex Pfs48/45-scTB31F:RUPA-44:RUPA-58:RUPA-154 (~130° and ~90°, respectively), although the angle between D1 and D2 remained unchanged (Fig. 3c). The previously reported crystal structure of Pfs48/45 in complex with Fabs 10D8 and 85RF45.1 (ref. 23) revealed that the three domains of Pfs48/45 formed a compact, disc-like conformation, with an angle between D2 and D3 of ~60° (Fig. 3c). Alignment of the Pfs48/45-scTB31F:RUPA-44:RUPA-58:RUPA-154 and Pfs48/45:10D8:85RF45.1 models reveals that individual domains have almost identical folds, with the major difference coming from the position of D3, which is displaced by ~40 Å between models (Fig. 3c,d). The D1–D2 interface remains consistent between the Pfs48/45-scTB31F:RUPA-44:RUPA-58:RUPA-154 and Pfs48/45–10D8:85RF45.1 models, with buried surface areas (BSAs) of 837 and 825 Å2, respectively. However, owing to the compact fold of Pfs48/45 in the Pfs48/45:10D8:85RF45.1 cocrystal structure, there are additional extensive interactions between D2 and D3 (BSA of 969 Å2) and D1 and D3 (BSA of 216 Å2); these are absent in the Pfs48/45-scTB31F:RUPA-44:RUPA-58:RUPA-154 structure. Taken together, these structural data reveal a large possible range of motion at the D2–D3 hinge region inherent to the full-length Pfs48/45 structure, exceeding previous descriptions23.

a, A composite model of Pfs48/45 in the Pfs48/45-scTB31F:RUPA-44:RUPA-58:RUPA-154 complex reveals an extensive interface between D1 and D2. The 5-aa linker (residues 290–294) between D2 and D3 is highlighted in red. b, The flexible linker between D2 and D3 results in continuous rotation of the D1–D2 region versus D3 by ~7°, as revealed by 3D variability analysis. c, Top, a ccomparison of Pfs48/45 models from the Pfs48/45-scTB31F:RUPA-44:RUPA-58:RUPA-154 complex (left), the Pfs48/45-scTB31F:RUPA-44:RUPA-58 complex (middle), and the Pfs48/45:10D8 cocrystal structure (right, PDB ID: 7ZXG) reveals considerable changes in angles between D2 and D3. Bottom, D3 displacement between the Pfs48/45-scTB31F:RUPA-44:RUPA-58:RUPA-154 complex (purple), the Pfs48/45-scTB31F:RUPA-44:RUPA-58 (magenta) complex, and the Pfs48/45:10D8 cocrystal structure (pink) is depicted by red dashed lines. The angle and displacement between domains was calculated with angle_between_domains command in Pymol36. The red dots and lines denote the centers of mass of individual domains and distances between them, respectively. d, Backbone r.m.s.d. between Pfs48/45 in the Pfs48/45-scTB31F:RUPA-44:RUPA-58:RUPA-154 complex and the Pfs48/45:10D8 cocrystal structure. Models were aligned either to D2 (left) or D3 (right) using PyMOL36, and r.m.s.d. values were calculated using UCSF Chimera37.
Delineation of the D2 epitope recognized by the RUPA-154 monoclonal antibody
RUPA-154 interacts predominantly with three D2 β-strands (212–217 aa, 271–275 aa, and 282–288 aa) and adjacent residues, as well as the loop spanning residues 226–247. The interactions of RUPA-154 contacts with Pfs48/45 are mediated mainly through lambda chain complementarity-determining regions (LCDRs) 1 and 2, as well as heavy chain complementarity-determining region 3 (HCDR3), with BSAs of 579 Å2 and 315 Å2 by the light chain (LC) and heavy chain (HC), respectively (Fig. 4a). Three HCDR3 tyrosine residues (H.Y100B, H.Y100C and H.Y100D) contribute 25% of the overall BSA and are optimally positioned across the D2 surface to form multiple contacts with Pfs48/45 residues Val 209, Ser210, Val212, Glu214, Lys239, Glu240, Asn270 and Val285 (Fig. 4b). Although there is a high number of somatic hypermutations (SHMs) in RUPA-154 (16 in the HC and 6 in the LC; Supplementary Fig. 2a), only two of these residues seem to be involved in antigen recognition (LC N51 and I52). The LC I52 SHM results in the optimal positioning of the Ile side chain to form contacts with the side chains of Pfs48/45 Asn211, Val212, Asn281 and Lys283 (Fig. 4b).

a, The interactions between RUPA-154 and the surface of D2, as delineated by locally refined cryo-EM map of Pfs48/45-scTB31F:RUPA-44:RUPA-58:RUPA-154 complex. The surface representation of D2 was generated from the atomic model. b, Details of interactions between RUPA-154 and D2. D2, RUPA-154 HC, and RUPA-154 LC residues are shown in blue, gray, and white, respectively. HCDR1, HCDR2, and HCDR3 residues are shown in pale yellow, yellow, and orange, and LCDR1, LCDR2, and LCDR3 residues are shown in pink, red, and brown, respectively. Fab residues are annotated with H or L letters to indicate the heavy and lambda light chain, respectively. The surface representation of D2 was generated from the atomic model. c, A heatmap of results from binding competition experiments. High signal responses for the second binding event (white) represent low competition, whereas low signal responses (black) correspond to high competition. For c and d, blue and yellow correspond to mAbs with moderate to strong (IC80 < 110 μg ml–1) and no to weak (IC80 > 110 μg ml–1) activity, respectively22,23. d, The binding kinetics of D2-directed Fabs determined using BLI with immobilized Pf D1D2 antigen. Binding affinity (Kd) is indicated along the diagonal lines. kon, association constant; koff, dissociation constant. e–i, The interfaces between Pfs48/45 (blue) and RUPA-154 (yellow/red), shown as ribbons. Key contacts are depicted as sticks and Pfs48/45 residues, and polymorphisms are labelled (e). BLI binding curves of RUPA-154 bound to WT D1D2 (f), D1D2-V212I (g), and D1D2-K239T (h). RUPA-58 binding to D1D2-K239T was used as a control (i).
Superimposing RUPA-154 onto the previously determined structure of 10D8 bound to Pfs48/45 (ref. 23) reveals that they bind adjacent epitopes (Extended Data Fig. 5a–c). The Pfs48/45 residues Val209, Ser210, Glu214, Glu216, Val285, and Lys287 contact both antibodies, and there are clashes between 10D8 and the framework region of the RUPA-154 HC (Extended Data Fig. 5a–c). This finding aligns with the results of epitope binning assays, in which RUPA-154 and 10D8 were shown to compete (Fig. 4c,d). An overlay of 10D8 on the cryo-EM map of Pfs48/45-scTB31F:RUPA-44:RUPA-58:RUPA-154 revealed that 10D8 binding would be occluded owing to steric clashes with D3 when Pfs48/45 adopts this conformation (Extended Data Fig. 5a–c). Variations in accessibility across conformations could explain why 10D8 is less potent than RUPA-154, despite both binding to a similar region of D2.
We assessed the prevalence of non-synonymous single-nucleotide polymorphisms (SNPs) occurring in Pfs48/45, using genomic sequence data from 7,113 Pf field isolates collected from 73 locations in Africa, Asia, America, and Oceania (MalariaGen26). We identified 14 non-synonymous SNPs in D2 across Pf field isolates (Supplementary Table 1). Of these polymorphisms, only two are present in the RUPA-154 epitope, p.V212I (allele frequency = 0.02 %) and p.K239T (allele frequency = 0.03%) (Fig. 4e). Binding kinetics experiments with RUPA-154 Fab show that although it retains nanomolar binding affinity to the V212I mutant, the other low-frequency SNP, p.K239T, does prevent RUPA-154 binding (Fig. 4f–i). The drastic impact of this mutation is likely due to K239 forming a salt bridge at the antibody–antigen interface and its high BSA (108 A2). Together, these results indicate that the RUPA-154 epitope is highly conserved, with rare polymorphisms that can affect binding by an inhibitory mAb.
To further elucidate the human humoral response to D2, we selected several D2-targeting antibodies with varying potencies for biophysical studies. In addition to RUPA-154 (IC80 = 103.1 μg ml–1) and 10D8 (TRA of ~56% at 375 µg ml–1)11, we investigated the highest-potency D2 antibody, RUPA-160, and two other low-potency antibodies, RUPA-111 and RUPA-43. RUPA-160 has a TRA of ~80% at 10 μg ml–1, whereas RUPA-111 and RUPA-43 did not demonstrate TRA at 100 μg ml–1 (ref. 22). Binding kinetics experiments with a recombinant D1–D2 construct revealed that these antibodies bind with nanomolar affinities ranging from 0.4 nM to 8.1 nM (Fig. 4d). These results indicate that binding affinity to the recombinant antigen is not directly correlated with TRA. Epitope binning assays revealed that all selected antibodies compete, except for 10D8 and RUPA-111 (Fig. 4c). These two mAbs compete with RUPA-154, RUPA-160, and RUPA-43, but not with each other (Fig. 4c). Given that antibodies with low to moderate TRA bind to overlapping positions on D2, we propose that variations in potency for this epitope class could be due to slight differences in angles of approach or contacts with specific Pfs48/45 residues.
Delineation of the D1 epitope recognized by potent RUPA-58 monoclonal antibody
An initial model of the RUPA-58 variable domain was created by manually docking the 3.09-Å-resolution structure of ligand-free RUPA-58, obtained from X-ray crystallography (Table 2), into the locally refined cryo-EM map of Pfs48/45-scTB31F:RUPA-44:RUPA-58:RUPA-154. The potent RUPA-58 epitope includes parts of Pfs48/45 D1 loops, specifically residues 86–115 and 130–140. Kappa chain complementarity-determining region 1 (KCDR1), KCDR3, and HCDR1–HCDR3 form extensive interactions with D1, with a total BSA of 975 Å2 (573 and 402 Å2 from the HC and KC, respectively) (Fig. 5a). Notably, the HCDR3 of this antibody contains a disulfide bond between cysteines H.C97 and H.C100B, which helps optimally position the HCDR3 to interact with the D1 loops spanning residues 86–100 and 131–139. H.Y100C is central to the HCDR3–D1 interaction, contributing 195 Å2 of the BSA and forming numerous contacts with residues at the base of the D1 loop spanning residues 86–100, including the side chains of Phe95, Lys96 and Ile97 (Fig. 5b). Comparing RUPA-58 with its inferred germline precursor, IGHV1-2/IGKV1-39, indicates the presence of 12 SHMs in the HC and 10 in the KC (Supplementary Fig. 2b). Notably, only one SHM (H.N58K) in the HC forms a direct contact with the antigen. From five somatically hypermutated residues in the KC interacting with the antigen, the side chains of K.T31 and K.S50 are optimally positioned to interact with Glu100 and contribute 36 Å2 and 28 Å2 of BSA to the interaction, respectively (Fig. 5b).

a, Interactions between RUPA-58 and the surface of D1, as delineated by the locally refined cryo-EM map of the Pfs48/45-scTB31F:RUPA-44:RUPA-58:RUPA-154 complex. The surface representation of D1 was generated from the atomic model. b, Details of interactions between RUPA-58 and D1. D1, RUPA-58 HC, and RUPA-58 KC residues are shown in light blue, gray, and white, respectively. HCDR1, HCDR2, and HCDR3 residues are shown in pink, magenta, and purple, and KCDR1, KCDR2, and KCDR3 residues are shown in light green, green, and olive, respectively. Fab residues are annotated with H or K letters to indicate heavy and kappa light chain, respectively. The surface representation of D1 was generated from the atomic model. c, Binding kinetics of D1-directed Fabs determined using BLI with immobilized Pf D1D2 antigen. Kd is indicated along the diagonal lines. d, A heatmap of results of binding competition experiments. High signal responses for the second binding event (white) represent low competition, whereas low signal responses (black) correspond to high competition. For c and d, blue and yellow correspond to potent (IC80 < 110 μg ml–1) and non-functional mAbs (IC80 > 110 μg ml–1), respectively22. e, Epitope mapping of RUPA-71 on Pfs48/45 D1D2 (gray, ribbons), as determined by nsEM, HDX-MS, and BLI using a D1D2 construct containing p.N131Q. Left, D1D2 with the RUPA-71 epitope derived from nsEM in coral. Middle, D1D2 with the mutated residue, N131, colored in dark violet and shown as sticks. Right, D1D2 with the peptides displaying significant changes in deuterium uptake between the RUPA-71-bound D1D2 and ligand-free D1D2 as measured by HDX-MS. Small (light red) to large (dark red) decreases in deuterium uptake are shown in red; peptides that showed an increase in deuterium uptake are colored in blue. f, BLI biosensor data depicting the binding of RUPA-71 Fab (Kd = affinity) and RUPA-71 IgG (Kd = apparent affinity) to Pvs48/45–D1D2. g, RUPA-71 (gray) bound to Pvs48/45–D1, created by aligning predicted models of RUPA-71 and Pvs48/45–D1D2 generated by ABodyBuilder and RoseTTAFold38,39 to PfsD1D2 from the nsEM model. Conservation of residues in PvsD1 are shown as a gradient with identical residues in dark magenta, highly conserved residues in orchid, semi-conserved residues in pink, and non-conserved residues in light gray.
Thirteen non-synonymous SNPs were identified in D1, all displaying very rare allele frequencies ranging from 0.007% to 0.036%. Among these, only p.S90R, p.K101T, and p.N138I are located at positions that are involved in RUPA-58 binding (Extended Data Fig. 5d). To assess the impact of these substitutions on RUPA-58 binding, we conducted binding kinetics experiments with recombinant D1–D2 constructs containing these SNPs. These results showed that RUPA-58 retains nanomolar affinity to all three constructs, with dissociation constant (Kd) values of 1.1 nM, 1.5 nM, and 3.7 nM for D1D2-K101T, D1D2-S90R, and D1D2-N138I, respectively (Extended Data Fig. 5e–h). Notably, the N138I mutant exhibited a higher off-rate for RUPA-58 binding, likely owing to a loss of hydrogen bonding with the HCDR3 residue H.S100A. This analysis demonstrates that the RUPA-58 epitope is well-conserved across different strains of Pf, and any SNPs documented to date would minimally impact potent recognition mediated by this mAb.
Characterization of epitopes on Pfs48/45 D1 targeted by human monoclonal antibodies
Several other D1-targeting antibodies have demonstrated TRAs of over 80% at 10 μg ml–1 (ref. 22) but their epitopes have yet to be mapped at the molecular level. To continue our delineation of antibody-mediated inhibition mediated by recognition of Pfs48/45 D1, we selected additional potent D1 antibodies to analyze: RUPA-119, RUPA-46, RUPA-116, RUPA-75, RUPA-71, RUPA-94, and RUPA-155. We also picked a range of low-potency antibodies that did not demonstrate TRA at 100 μg ml–1, including RUPA-122, RUPA-114, RUPA-77, and RUPA-78 (ref. 22). To determine whether there was any variation in binding affinities between these different mAb classes, we measured the Kd values of each Fab using BLI. These results revealed a wide spread of binding affinities, ranging from <1 pM to >100 nM (Fig. 5c). Kd values were not directly associated with potency; for example, the non-inhibitory mAb RUPA-122 had higher affinity to the recombinant antigen than did the potently inhibitory mAb RUPA-46.
Through a series of competition assays, we found that RUPA-58, for which we obtained the cryo-EM structure, competes directly with RUPA-94, outlining the first epitope bin of Pfs48/45 D1 (Fig. 5d). RUPA-119, RUPA-46, RUPA-116, RUPA-75, RUPA-71, and RUPA-155 all compete with each other, revealing a second well-defined bin associated with high-potency mAbs (Fig. 5d). Given that all of these antibodies in the second bin, except RUPA-46, bind to linear epitopes22, these mAbs likely bind to a similar stretch of Pfs48/45 residues. Additionally, we observed that RUPA-46 and RUPA-71 could partially compete with antibodies in this bin as well as RUPA-58, signifying that the second potent D1 epitope bin is likely adjacent to the RUPA-58 epitope (Fig. 5d). The lower-potency antibodies, RUPA-114 and RUPA-122, did not extensively compete with any other antibody, suggesting there is a potential third epitope on Pfs48/45 D1 (Fig. 5d).
To identify the highly potent linear bin 2 epitope associated with the high-potency D1 antibodies, we conducted negative-stain electron-microscopy (nsEM) experiments using RUPA-71 (Fig. 5e and Extended Data Fig. 6a). A 3D reconstruction of the RUPA-71:RUPA-154:D1D2 complex revealed that the RUPA-71 binding site maps to the region of D1 containing residues 85–92 and 131–139 (Fig. 5e). The epitope of RUPA-71 was further delineated using hydrogen/deuterium exchange mass spectrometry (HDX-MS). A comparison of deuterium uptake in the D1D2:RUPA-71 complex and ligand-free D1D2 revealed significant reductions in deuterium uptake in the 46–51, 85–102, 124–141, 188–195, and 196–208 peptides upon RUPA-71 binding, with 124–141 showing the largest change (Fig. 5e and Extended Data Fig. 7). A comparison of RUPA-71 with the structure of Pfs48/45-bound RUPA-58 indicated that these antibodies bind to nearby, partially overlapping epitopes, consistent with our BLI competition data (Extended Data Fig. 6b and Fig. 5d). BLI experiments with the point mutant D1D2-N131Q construct further validated the nsEM and HDX-MS epitope mapping, wherein p.N131Q resulted in a faster off-rate for RUPA-71 (Fig. 5e and Extended Data Fig. 8). This effect was even more pronounced for RUPA-116, RUPA-119, RUPA-75, RUPA-46, and RUPA-155, indicating that the binding sites of these antibodies, which compete with one another, are likely in proximity of N131 (Extended Data Fig. 8).
Interestingly, affinity measurements to the Plasmodium vivax (Pv) ortholog of D1D2 revealed that one of the potent D1 antibodies, RUPA-71, is capable of cross-reactive binding across the two Plasmodium species, binding to Pv D1D2 with an affinity of 2.2 μM as a Fab and with an avidity-boosted apparent affinity of 12.5 nM as an IgG (Fig. 5f and Extended Data Fig. 9a–g). Pfs48/45 and Pvs48/45 share 55% and 75% amino acid sequence identity and similarity27, respectively, and a comparison of residues revealed good conservation between the Pv D1D2 and Pf D1D2 proteins in the putative RUPA-71 binding site (Fig. 5g and Extended Data Fig. 9h). Together, these results highlight a degree of conservation for the RUPA-71 epitope that allows cross-reactivity not only across Pf field isolates, but also with the Pv ortholog.
Discussion
Pfs48/45 is a promising TBV candidate and a target for mAbs with potent TRA, including the TB31F mAb, which is to date the transmission-blocking mAb with the highest potency and is well-tolerated in humans19. To enable the molecular characterization of inhibitory epitopes on all domains of Pfs48/45, here we designed a full-length construct that incorporates previously described stabilizing mutations within D3 (ref. 17) and combines these with the genetic fusion to a scFab fragment of the D3-targeting mAb TB31F. As a result of this construct engineering, we derived a high-resolution cryo-EM structure of an antibody–antigen complex formed by full-length Pfs48/45 bound simultaneously by four inhibitory antibodies to all three Pfs48/45 domains.
Previously reported crystal structures of full-length Pfs48/45 bound by antibodies to either D2 or D3 demonstrated that the three domains of Pfs48/45 could adopt a disc-like conformation that is highly similar to a model predicted by AlphaFold23 (Figs. 3c and 6a). By contrast, our cryo-EM structures reveal a notable difference in the positioning of D3 relative to D2 compared with the crystal structures of full-length Pfs48/45 (PDB IDs: 7ZXG and 7ZXF), suggesting a ~40° range of motion in the hinge region between D2 and D3 (Fig. 6b,c). In addition to intrinsic flexibility, differences in the interdomain dispositions could be related to the experimental conditions from which the structures were determined (that is, crystal or vitreous ice) or might also be influenced by the binding of antibodies at specific interfaces. Furthermore, although D3 possesses an overall electronegative surface potential, both D1 and D2 possess a partially electropositive surface (Extended Data Fig. 10). The electronegative plasma membrane could be an additional factor that influences Pfs48/45 domain orientation on the parasite surface, which is absent in the present molecular description. An analysis of available structures of full-length Pfs48/45 provides valuable insights for immunogen design. Because the D1–D2 interface is occluded in all described conformations of Pfs48/45, immunization with those individual domains might be suboptimal, potentially leading to antibodies elicited against interface regions occluded in the full-length protein. It is likely that D1–D2-type constructs might make for superior immunogens. However, D3 seems to be accessible to antibody binding from all sides, except the membrane-proximal side, making it a potentially viable candidate for a viable individual domain-type immunogen. Future studies examining the conformational dynamics of Pfs48/45 on the parasite surface and the interplay with native binding partners, such as Pfs230, could clarify the functional determinants and biological implications of this flexibility.

a–c, Models of Pfs48/45 derived from the Pfs48/45:10D8 cocrystal structure (PDB ID: 7ZXF) (a), the Pfs48/45-scTB31F:RUPA-44:RUPA-58:RUPA-154 complex (b), and the Pfs48/45-scTB31F:RUPA-44:RUPA-58 complex (c) are depicted, with a carboxy terminal linker connecting D3 to GPI anchor (black). The plasma membrane is shown in gray. D1, D2, and D3 are colored light blue, blue, and navy, respectively. Epitopes of TB31F (red), RUPA-58 (white), RUPA-44 (orange), and RUPA-71 (green) are accessible in all above conformations; the epitope of RUPA-154 (yellow) is not accessible in the conformation of the Pfs48/45-scTB31F:RUPA-44:RUPA-58 complex. The overlay between epitopes of RUPA-58 and RUPA-71 is depicted in light green.
Local refinement of the D1D2:RUPA-58:RUPA-154 region in the cryo-EM map produced a 3.3-Å-resolution reconstruction that revealed the epitopes of RUPA-58 and RUPA-154 at the atomic level. These data provide molecular insights into human antibody-mediated inhibition of gametes by binding outside the well-established potent epitopes on Pfs48/45 D3. All D2-directed antibodies evaluated in this study were found to compete with RUPA-154, suggesting that they bind to overlapping epitopes. Before the present study, the only structurally characterized D2-directed mAb was the murine antibody 10D8, which like RUPA-154 also possesses low TRA. Although RUPA-154 and the murine antibody 10D8 compete for binding to Pfs48/45, structural comparisons show slight differences in their epitopes and angles of approach, which might or might not accommodate binding to Pfs48/45 in different domain conformations. From this observation emerges the hypothesis that certain Pfs48/45-D2 epitopes could be transiently occluded by D3, potentially leading to reduced inhibitory potency. However, given that RUPA-154 binds strongly to female gametes in SIFAs, the modest potency of this antibody could arise from factors other than the low accessibility of its epitope. An alternative hypothesis suggests that its cognate binding partners, such as Pfs230 (ref. 28) and PfCCp29,30, might compete for access to Pfs48/45 interfaces; however, the molecular details of these interactions are not yet understood. Indeed, this phenomenon has also been proposed to explain why antibodies to the blood-stage vaccine antigen, PfRH5, which target the CyRPA interface, do not exhibit growth inhibitory activity31.
Our cryo-EM work also provides molecular insights into antibodies to Pfs48/45 D1 that can possess higher TRA than can those to D3. RUPA-58 competed with RUPA-94, RUPA-78, and RUPA-77, demonstrating a range of TRAs in the first epitope bin. An nsEM 3D reconstruction of the RUPA-71:RUPA-154:D1D2 complex, in combination with results of binding competition assays, revealed the putative location of a second inhibitory binding site on D1, and the location of this potent epitope was further delineated by HDX-MS mapping experiments. In addition, mutagenesis experiments supported this binding site, showing that p.N131Q negatively impacts the binding of all the antibodies in this epitope bin. Notably, p.N131Q eliminated a putative N-linked glycosylation sequon and consequently removed an N-linked glycan at this position when the construct is expressed in HEK293 cells, as was the case in our studies. The presence and extent of N-glycosylation in Plasmodium is still heavily debated32. On the basis of genomic analyses and metabolic-labeling experiments, it seems that parasites could be capable of producing short N-glycans of no more than two N-acetylglucosamine moieties33. Given that the human antibodies studied here are the result of natural exposure, are highly potent, and can bind to the gamete surface, these data suggest that the glycan at N131 could be naturally occurring in Pf, although an alternative hypothesis is that substitution of this glycan distorts the local conformation of this epitope bin. Further studies characterizing possible post-translational modifications on Pfs48/45 derived directly from the pathogen will be necessary to fully explore the nature of this highly potent epitope. Recent advances in potent chromatographic and mass-spectrometric techniques have made it feasible to investigate carbohydrate repertoires, evident by the progress made in characterization of heavily glycosylated HIV-1 envelope spike protein (Env)34.
Intriguingly, BLI experiments demonstrated that RUPA-71 can bind both Pf D1D2 and Pv D1D2, supporting the potential for interventions seeking cross-species Plasmodium transmission-blocking activity against the two major species circulating worldwide27. Overall, our data support incorporation of highly conserved Pfs48/45 D1 epitopes, now resolved at molecular level, into next-generation transmission-blocking immunogen designs. Human-derived mAbs to D1 could also become valuable additions to TB31F-mAb-based interventions and put additional pressure on the parasite at the bottleneck of transmission by conveying further resilience against any mutational escape potential. Future studies on larger cohorts of mAbs will continue to shed light on relationships between Pfs48/45 epitopes and antibody potency. Epitope recognition by polyclonal antibodies can be further elucidated by techniques like cryoelectron microscopy-based polyclonal epitope mapping (cryoEMPEM)35—a powerful method for rapid and high-resolution structural characterization of antibody–antigen complexes, without the need for isolation of mAbs. CryoEMPEM holds promise for future structural studies to provide molecular insights into epitope recognition by polyclonal antibody responses elicited by infection or vaccination trials, and should be considered as part of the evaluation of immunological outcomes.
Methods
Domain specificity by ELISA
The domain specificity of mAbs was tested using ELISA, as previously described22. Nunc MaxiSorpTM 96-wells plates (Thermo Fisher Scientific) were coated overnight at 4 ˚C with 0.5 µg ml–1 recombinant full-length Pfs48/45 and fragments containing D1–D2, D2–D3, and D3. Plates were blocked with 5% skimmed milk in PBS for 1 h at room temperature, washed, and then incubated with 10 µg ml–1 mAb in 1% milk in PBST for 3 h at room temperature. Plates were washed and then incubated with 1:60,000 HRP-conjugated goat anti-human-IgG antibody (Pierce, cat. no. 31412) in 1% milk in PBST for 1 h. Plates were washed again, 100 µl 3,3′,5,5′-tetramethylbenzidine was added, and after approximately 20 min, the reaction was stopped with 0.2 M H2SO4. Absorbance was measured at 450 nm. Monoclonal antibodies were considered positive if the absorbance was higher than the mean absorbance plus three s.d. of six negative mAbs.
Surface immunofluorescence assay
N-acetyl-glucosamine-treated 16-day-old Pf NF54 gametocytes were collected by centrifugation for 10 min at 2,000g. Pelleted gametocytes were resuspended in fetal bovine serum (FBS) and activated on a roller bank for 45 min at room temperature. The cells were then pelleted by centrifugation for 10 min at 2,000g at 4˚C, resuspended in PBS, and loaded on an 11% Accudenz (Accurate Chemical) cushion; this was followed by centrifugation for 30 min at 7,000g at 4 ˚C. The top layer, containing activated female gametes, was collected and gametes were then washed with PBS before incubation with mAbs. mAbs diluted in PBS + 2% FBS + 0.02%, and sodium azide were mixed with 50,000 gametes and incubated for 1 h at room temperature. Thereafter, gametes were washed three times with PBS, followed by 30 min incubation at room temperature with 1:200 Alexa-Fluor-488-conjugated anti-human-IgG (Invitrogen, cat. no. A-11013) and 1:1,000 eBioscience Fixable Viability Dye eFluor 780 (Invitrogen, cat. no. 65-0865-14). Cells were finally washed three times with PBS and resuspended in PBS. mAb binding to gametes was assessed by measuring fluorescence of at least 2,000 single live gametes on a Gallios 10-color system (Beckman Coulter) followed by analyses in FlowJo (BD Biosciences).
Standard membrane feeding assay
SMFA experiments used cultured Pf NF54 gametocytes and oocyst counts as readout, as previously described40. mAbs were diluted in FBS and mixed with cultured gametocytes and normal human serum as a source of active complement. The mAb concentrations presented refer to the final concentrations in the total volume of blood meals. These blood meals were fed to 4- to 6-day-old female A. stephensi mosquitoes (Nijmegen colony). For each condition, 20 fully-fed mosquitoes were analyzed. TRA was calculated as the reduction in oocysts compared to a negative control. IC80 values were calculated by linear regression analysis on the log10-transformed ratio of mean oocyst count in control and test sample, and the square root of the antibody concentration41. The 95% confidence intervals of the IC80 values were calculated using the delta method19. SMFA data analyses were done in R (version 4.1.2).
Expression and purification of Fabs
The variable light and heavy chains of the Fabs used in these studies were gene-synthesized and cloned (GeneArt) into custom pcDNA3.4 expression vectors, located immediately upstream of constant domains. Fab HC and Fab LC plasmids were cotransfected at a 2:1 ratio into FreeStyle HEK 293-F or 293-S cells (Thermo Fisher Scientific) at a cell density of 0.8 × 106 cells ml–1 for transient expression using FectoPRO DNA transfection reagent (Polyplus). Cells were cultured in Gibco FreeStyle HEK 293 Expression Medium for 7 days, and supernatants were isolated by centrifugation and filtered through a 0.22-μm membrane. Secreted Fabs were purified using affinity chromatography with either a HiTrap KappaSelect or LambdaSelect column (Cytiva) with 1× PBS and 100 mM glycine at pH 2.2 as wash and elution buffers, respectively. Fabs were further purified by cation-exchange chromatography using a MonoS column (Cytiva) in a buffer of 20 mM sodium acetate at pH 5.6 across a 0–1 M KCl gradient for elution. Fabs expressed in HEK 293-S cells underwent subsequent treatment with EndoH.
Expression and purification of Pfs48/45 proteins
A full-length Pfs48/45 construct (aa 28–428, with N-linked glycan sequons intact) integrating stabilizing mutations to D3 (p.G397L, p.H308Y, p.I402V, p.S361R and p.D373S17) was fused at the carboxy terminus using a 7×GGS linker to the HC portion of a single chain Fab of TB31F with a 21-aa-long flexible linker. The scFab of TB31F contained a 14×GGGGS linker between the HC and LC. A Pfs48/45 construct containing only domains 1 and 2 (aa 27–294) was also generated containing the native sequences or any SNPs located with the RUPA-58 or RUPA-154 epitopes to interrogate binding kinetics. To all constructs, a signal peptide sequence was added to the N terminus and a 6×His tag was added to the carboxy terminus, followed by gene synthesis and cloning into a pcDNA3.4 expression vector (GeneArt). Stabilized Pfs48/45 linked scTB31F was recombinantly expressed in HEK 293-F or 293-S cells and purified with a HisTrap FF column (GE Healthcare), using TBS at pH 7.0 with a linear elution gradient of imidazole up to 500 mM, and further purified using size-exclusion chromatography with a Superdex 200 Increase column (GE Healthcare) in TBS at pH 7.0. The Pfs48/45-scTB31F:RUPA-44:RUPA-58 and Pfs48/45-scTB31F:RUPA-44:RUPA-58:RUPA-154 complexes were formed by incubating proteins together for 30 min to overnight at 4 oC and subsequently purifying the complex using size-exclusion chromatography with a Superose 6 Increase column (GE Healthcare) in TBS pH 7.0.
Biolayer interferometry binding studies
Direct binding kinetics measurements were conducted at 25 °C using an Octet RED96 instrument (Sartorius ForteBio). Fabs and purified recombinant Pfs48/45 were diluted in kinetics buffer (PBS, pH 7.4, 0.01% (wt/vol) BSA, and 0.002% (vol/vol) Tween-20) as previously described22. Binding kinetics parameters were determined using Ni-NTA or HIS1K biosensors loaded with 15 μg ml–1 of purified His-tagged Pfs48/45 protein followed by a 30-s baseline and an association phase in serially diluted Fab or IgG protein. Biosensors were then dipped into wells containing kinetics buffer for a dissociation step. Data analysis was performed using the Octet software (Sartorius ForteBio, version 8.2.0.7) and the sensograms were fit using a 1:1 binding model. Binding competition assays were performed by loading 15 μg ml–1 of 6×His-tagged antigen to reach a BLI signal response of 1.0 nm, followed by a 30-s baseline step. The antigen-loaded biosensors were then dipped into wells containing the first antibody (Fab 1) at 50 μg ml–1 for 300 s; then, sensors carrying Fab 1–antigen complexes were dipped into wells containing the second antibody (Fab 2) at 50 μg ml–1 for an additional 300 s. Data analysis was performed using the Octet software (Sartorius ForteBio, version 8.2.0.7) and the binding competition was quantified as the percentage ratio of each antibody’s BLI signal at each stage of the two-step binding sequence (that is, Fab2/Fab1 measured in nm).
Single-nucleotide polymorphism detection
SNPs were obtained from the MalariaGEN Catalogue of Genetic Variation in Pf version 6.0 (ref. 26). Genotype calls for chromosome 13 (Pf3D7_13_v3) were downloaded (ftp://ngs.sanger.ac.uk/production/malaria/pfcommunityproject/Pf6/Pf_6_vcf/Pf_60_public_Pf3D7_13_v3.final.vcf.gz) and Bcftools42 (version 1.15.1) was used to subset calls between nucleotide positions 1,875,452 and 1,878,087 that coincide with Pfs48/45 and calculate the allele frequency of all polymorphism occurrences. SNPs calls below MalariaGEN’s quality filter (Low_VQSLOD) and all silent mutations were removed.
Cryo-EM data collection and image processing
The Pfs48/45-scTB31F:RUPA-44:RUPA-58 and Pfs48/45-scTB31F:RUPA-44:RUPA-58:RUPA-154 complexes were concentrated to 0.7 mg ml–1 and 3.0 µl of the sample was deposited on homemade holey gold grids43, which were glow-discharged in air for 15 s before use. All samples were blotted for 3.0 s and subsequently plunge-frozen in liquid ethane using a Leica EM GP2 Automatic Plunge Freezer (maintained at 4 °C and 100% humidity). Data collection was performed on a Thermo Fisher Scientific Titan Krios G3 operated at 300 kV with a Falcon 4i camera automated with the EPU software. A nominal magnification of ×75,000 (with a calibrated pixel size of 1.03 Å) and defocus range between 0.8 and 1.5 μm were used for data collection.
Exposures were collected for 8.8 s (Pfs48/45-scTB31F:RUPA-44:RUPA-58:RUPA-154 complex) or 8.3 s (Pfs48/45-scTB31F:RUPA-44:RUPA-58 complex) as videos with 30 frames with a camera exposure rate of ∼6 e− per pixel per second, and total exposure of ~49 electrons per Å2. A total of 6,850 raw videos were obtained for the Pfs48/45-scTB31F:RUPA-44:RUPA-58 complex and 7,749 for the Pfs48/45-scTB31F:RUPA-44:RUPA-58:RUPA-154 complex (1,529, 3,435, and 2,785 raw videos were obtained with no stage tilt, 40° and 55° stage tilts, respectively).
Image processing was carried out in cryoSPARC v3 and v4 (ref. 25). Initial specimen movement correction, exposure weighting, and CTF parameter estimation were done using patch-based algorithms. Micrographs were sorted on the basis of CTF fit resolution, and only micrographs with a score lower than 7.0 Å (videos obtained with 55° stage tilt) or 5.0 Å (all other videos) were accepted for further processing. Manual particle selection was performed on 30 micrographs to create templates for template-based picking. For the Pfs48/45-scTB31F–RUPA-44–RUPA-58 complex, 3,904,349 particle images were selected by template picking and individual particle images were corrected for beam-induced motion with the local motion algorithm44. Particle images of intact Pfs48/45-scTB31F:RUPA-44:RUPA-58 complex were separated from dissociated components through two-dimensional (2D) classification and several rounds of heterogeneous refinement. Then, 133,106 particle images were selected for non-uniform refinement45 with no symmetry applied, which resulted in a 4.3-Å resolution map of the Pfs48/45-scTB31F:RUPA-44:RUPA-58 complex, estimated from the gold-standard refinement with correction of the Fourier shell correlation (FSC) for masking effects.
For the Pfs48/45-scTB31F:RUPA-44:RUPA-58:RUPA-154 complex, videos obtained at no tilt and 40° and 55° stage tilts were initially processed separately. Particle images were selected by template picking and individual particle images were corrected for beam-induced motion with the local motion algorithm44. Initial model of the Pfs48/45-scTB31F:RUPA-44:RUPA-58:RUPA-154 complex was obtained through the ab initio algorithm in cryoSPARC v4. Particle images of intact Pfs48/45-scTB31F:RUPA-44:RUPA-58:RUPA-154 complex were separated from dissociated components through several rounds of heterogeneous refinement, resulting in 204,735, 309,360, and 127,262 particle images from videos obtained with no tilt, 40° and 55° stage tilts, respectively. To alleviate the effects of preferred orientation, 2D classification was performed on a combined particles set, followed by rebalancing of 2D classes. The resulting 306,744 particle images were selected for non-uniform refinement45 with no symmetry applied, which resulted in a 3.8-Å-resolution map of the Pfs48/45-scTB31F:RUPA-44:RUPA-58:RUPA-154 complex. Densities for peripheral regions of the map were further improved with the flexible refinement algorithm in cryoSPARC v4, resulting in a 3.9-Å-resolution map. To improve interpretability, the final map was processed in EMReady2 (ref. 46) with a custom soft mask.
Local refinement was performed on all particle images with a mask including the D1 and D2 domains of Pfs48/45 and variable regions of RUPA-58 and RUPA-154 Fabs, which resulted in a 3.3-Å-resolution map of this region. EMReady2 (ref. 46) was used to postprocess the locally refined map to improve map interpretability and facilitate model building. To visualize flexibility in the Pfs48/45-scTB31F:RUPA-44:RUPA-58:RUPA-154 cryo-EM map, 3D variability analysis was performed47.
An initial model for Pfs48/45:RUPA-58:RUPA-154 was created by manually docking crystal structures of Pfs48/45 D1 and 2 (PDB ID: 7ZXF ref. 23), the variable domain of RUPA-58 Fab derived from the crystal structure (Table 2)22 and the predicted variable domain of RUPA-154 Fab48 into the cryo-EM map with UCSF Chimera37, followed by manual building with Coot49. All models were refined using phenix.real_space_refine50 with secondary structure and geometry restraints. The final models were evaluated by MolProbity51. The figures were prepared with UCSF Chimera37 and UCSF ChimeraX52. Interactions in the Pfs48/45:RUPA-58:RUPA-154 model were identified by PDBePISA53. Electrostatic surface potentials were calculated using the Adaptive Poisson-Boltzmann Solver (APBS) plug-in in PyMOL36 and images were generated using PyMOL (The PyMOL Molecular Graphics System, v2.3.4, Schrödinger)36. Antibody numbering is according to the Kabat scheme.
X-ray crystallography
A sample of recombinant RUPA-58 Fab at 10 mg ml–1 in TBS at pH 7.0 was added to sitting-drop crystallization trays and mixed with reservoir solution at a 1:1 ratio. RUPA-58 crystals grew in 0.2 M sodium acetate, 30% PEG 8000, 0.1 M sodium cacodylate at pH 6.5, and 20% ethylene glycol. Data were collected at the 23-ID-D beamline at the Advanced Photon Source (APS). Data processing was done using XDS54 and Xprep55, and Phaser56 was used for molecular replacement using a Fab from PDB: 4YK4 as a search model57. The crystal structure was then built and refined using phenix.refine58 and Coot49 accessed through SBGrid59.
Negative-stain electron microscopy and processing
Purified RUPA-71 Fab, RUPA-154 Fab, and Pfs48/45-D1D2 were put in a complex at a ratio of 1.5:1.5:1, respectively. Intact complex was purified from excess Fabs using a Superdex 200 column in TBS at pH 7.0. Then 5 μg ml–1 of complex was applied onto homemade carbon film-coated grids (previously glow-discharged in air for 15 s) and stained with 2% uranyl formate. Micrographs were collected using a Hitachi HT7800 transmission electron microscope operating at 120 kV, ×80,000 magnification with a pixel size of 1.83 Å per pixel. Image processing, particle extraction, 2D classes, and 3D reconstruction were done using Relion 3.1 (ref. 60) and cryoSPARC v3 (ref. 25).
HDX-MS
Protein samples were EndoH treated, further purified as described above, then concentrated to 100 μM. Hydrogen/deuterium exchange was initiated by mixing 2.5 μl of protein with 37.5 μl of D2O buffer (10 mM phosphate in D2O, pH 7.0), and incubated for various periods of time (10 s, 100 s, 1,000 s, and 10,000 s) at 4 °C. The exchange was quenched by adding 40 μl of chilled (4 °C) quench buffer (100 mM phosphate, pH 2.5, 2 M urea, 1 M TCEP) and snap frozen on dry ice at the indicated time points. For non-deuterated (ND) samples, 2.5 μl of purified protein was mixed with 37.5 μl of H2O buffer (10 mM phosphate in H2O, pH 7.0), and quenched and frozen in the same manner. Quenched samples were thawed and mixed with pepsin in a 1:1 (wt/wt) ratio for 5 min for offline digestion and passed through an immobilized pepsin column (2.1 × 30 mm; Waters) at a flow rate of 100 μl min–1 with 0.1% formic acid and 2% acetonitrile in H2O at 20 °C for online peptic digestion. Peptide fragments were subsequently collected on a C18 VanGuard trap column (2.1 mm × 5 mm; Waters) for desalting with 0.1% formic acid and 2% acetonitrile in H2O and then separated by ultra-pressure liquid chromatography using an Acquity UPLC C18 column (1.7 μm, 1.0 × 100 mm; Waters) at a flow rate of 40 μl min–1 with an acetonitrile gradient from 5% B to 90% B over 10 min. The mobile phase A consisted of 0.1% formic acid in H2O, and solvent B consisted of 0.1% formic acid in acetonitrile. To minimize the back-exchange of deuterium to hydrogen, the entire system was maintained at pH 2.5 and 0 °C (except pepsin digestion was performed at 20 °C) during the analysis. Mass spectra were analyzed by SYNAPT G2-Si quadrupole-time of flight (Q-TOF) equipped with a standard electrospray ionization (ESI) source in MSE mode (Waters) with positive ion mode. The capillary, cone, and extraction cone voltages were set to 3 kV, 30 V, and 4 V, respectively. Source and desolvation temperatures were set to 80 °C and 150 °C, respectively. Trap and transfer collision energies were set to 6 V, and the trap gas flow was set to 2.0 ml min–1. Sodium iodide solution (2 µg µl–1) was utilized to calibrate the mass spectrometer. [Glu1]-Fibrinopeptide B solution (200 fg µl–1) in methanol:water (50:50 (vol/vol) + 1% acetic acid) was utilized for the lock-mass correction of the spectra and the ions at a m/z of 785.8427 with a mass window of ±0.5 Da were monitored at scan time 0.1 s. Ar gas was used for collision induced dissociation (CID). For HDMSE and UDMSE measurements, the instrument was operated in ion mobility mode. The T-wave was operated with a wave height of 40 V and a wave velocity ramp from 850 to 350 m s–1.
Peptide identification and HDX-MS data processing
Mass spectra were acquired in the range of 50–2,000 m/z for 10 min. Methionine oxidation and delta mass of N-acetylglucosamine were assigned as variable modifications to identify peptic peptides from the non-deuterated samples using ProteinLynx Global Server 3.0.3 (Waters). Protein identification criteria were set as the detection of at least one fragment per protein, three fragments per peptide, and at least one peptide per protein. PLGS search results and deuterium exchange measurements were imported into DynamX v. 3.0 (Waters). A minimum of 0.03 products per amino acid, and a precursor mass error of less than 25 ppm, were established as the settings in DynamX. The centroid of isotopic distributions in deuterium uptake were analyzed and curated as required. Because proteins aggregated in the full deuteration condition, the analyses are represented as the comparison between different states of protein without back exchange correction. All the data were derived from at least three independent experiments. The significance threshold was set as the 95% confidence interval, calculated from the pooled s.d. for all peptides across all time points for both states. The detailed HDX-MS results are summarized in the Supplementary Table 2, following Masson et al.’s guidelines61.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
X-ray crystallography and cryoEM structures are accessible from the Protein Data Bank and Electron Microscopy Data Bank under IDs: EMD-41821 (Pfs48/45-scTB31F–RUPA-44–RUPA-58–RUPA-154 complex), EMD-41822 (locally refined map of Pfs48/45-scTB31F–RUPA-44–RUPA-58–RUPA-154 complex), 8U1P (Pfs48/45–RUPA-58–RUPA-154 model), and 8U70 (RUPA-58 Fab crystal structure). Data and materials can be obtained from the corresponding authors upon reasonable request. Source data are provided with this paper.
References
WHO. World Malaria Report 2024 (WHO, 2024).
Hall, B. F. & Fauci, A. S. Malaria control, elimination, and eradication: the role of the evolving biomedical research agenda. J. Infect. Dis. 200, 1639–1643 (2009).
Duffy, P. E. Transmission-blocking vaccines: harnessing herd immunity for malaria elimination. Expert Rev. Vaccines 20, 185–198 (2021).
Duffy, P. E. Current approaches to malaria vaccines. Curr. Opin. Microbiol. 70, 102227 (2022).
Mahase, E. WHO recommends second vaccine for malaria prevention in children. Brit. Med. J. 383, 2291 (2023).
Laurens, M. B. RTS,S/AS01 vaccine (MosquirixTM): an overview. Hum. Vaccines Immunother. 16, 480–489 (2020).
Wu, R. L. et al. Low-dose subcutaneous or intravenous monoclonal antibody to prevent malaria. N. Engl. J. Med. 387, 397–407 (2022).
Kayentao, K. et al. Safety and efficacy of a monoclonal antibody against malaria in Mali. N. Engl. J. Med. 387, 1833–1842 (2022).
Silk, S. E. et al. Superior antibody immunogenicity of a viral-vectored RH5 blood-stage malaria vaccine in Tanzanian infants as compared to adults. Med 4, 668–686 (2023).
Wu, Y. et al. Phase 1 trial of malaria transmission blocking vaccine candidates Pfs25 and Pvs25 formulated with montanide ISA 51. PLoS ONE 3, e2636 (2008).
Healy, S. A. et al. Pfs230 yields higher malaria transmission-blocking vaccine activity than Pfs25 in humans but not mice. J. Clin. Invest. 131, e146221 (2021).
van Dijk, M. R. et al. A central role for P48/45 in malaria parasite male gamete fertility. Cell 104, 153–164 (2001).
Kocken, C. H. M. et al. Cloning and expression of the gene coding for the transmission blocking target antigen Pfs48/45 of Plasmodium falciparum. Mol. Biochem. Parasitol. 61, 59–68 (1993).
Outchkourov, N. S. et al. Correctly folded Pfs48/45 protein of Plasmodium falciparum elicits malaria transmission-blocking immunity in mice. Proc. Natl Acad. Sci. USA 105, 4301–4305 (2008).
Theisen, M., Jore, M. M. & Sauerwein, R. Towards clinical development of a Pfs48/45-based transmission blocking malaria vaccine. Expert Rev. Vaccines 16, 329–336 (2017).
Lennartz, F. et al. Structural basis for recognition of the malaria vaccine candidate Pfs48/45 by a transmission blocking antibody. Nat. Commun. 9, 3822 (2018).
McLeod, B. et al. Vaccination with a structure-based stabilized version of malarial antigen Pfs48/45 elicits ultra-potent transmission-blocking antibody responses. Immunity 55, 1680–1692.e8 (2022).
Dickey, T. H. et al. Design of a stabilized non-glycosylated Pfs48/45 antigen enables a potent malaria transmission-blocking nanoparticle vaccine. NPJ Vaccines 8, 20 (2023).
van der Boor, S. C. et al. Safety, tolerability, and Plasmodium falciparum transmission-reducing activity of monoclonal antibody TB31F: a single-centre, open-label, first-in-human, dose-escalation, phase 1 trial in healthy malaria-naive adults. Lancet Infect. Dis. 22, 1596–1605 (2022).
Kundu, P. et al. Structural delineation of potent transmission-blocking epitope I on malaria antigen Pfs48/45. Nat. Commun. 9, 4458 (2018).
Roeffen, W. et al. Plasmodium falciparum: production and characterization of rat monoclonal antibodies specific for the sexual-stage Pfs48/45 antigen. Exp. Parasitol. 97, 45–49 (2001).
Fabra-García, A. et al. Highly potent, naturally acquired human monoclonal antibodies against Pfs48/45 block Plasmodium falciparum transmission to mosquitoes. Immunity 56, 406–419.e7 (2023).
Ko, K.-T. et al. Structure of the malaria vaccine candidate Pfs48/45 and its recognition by transmission blocking antibodies. Nat. Commun. 13, 5603 (2022).
Stone, W. J. R. et al. Unravelling the immune signature of Plasmodium falciparum transmission-reducing immunity. Nat. Commun. 9, 558 (2018).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
MalariaGEN et al. An open dataset of Plasmodium falciparum genome variation in 7,000 worldwide samples. Wellcome Open Res. 6, 42 (2021).
Cao, Y., Bansal, G. P., Merino, K. & Kumar, N. Immunological cross-reactivity between malaria vaccine target antigen P48/45 in Plasmodium vivax and P. falciparum and cross–boosting of immune responses. PLoS ONE 11, e0158212 (2016).
Kumar, N. Target antigens of malaria transmission blocking immunity exist as a stable membrane bound complex. Parasite Immunol. 9, 321–335 (1987).
Pradel, G. et al. A multidomain adhesion protein family expressed in Plasmodium falciparum is essential for transmission to the mosquito. J. Exp. Med. 199, 1533–1544 (2004).
Simon, N., Kuehn, A., Williamson, K. C. & Pradel, G. Adhesion protein complexes of malaria gametocytes assemble following parasite transmission to the mosquito. Parasitol. Int. 65, 27–30 (2016).
Ragotte, R. J. et al. Heterotypic interactions drive antibody synergy against a malaria vaccine candidate. Nat. Commun. 13, 933 (2022).
Goerdeler, F., Seeberger, P. H. & Moscovitz, O. Unveiling the sugary secrets of plasmodium parasites. Front. Microbiol. 12, 712538 (2021).
Bushkin, G. G. et al. Suggestive evidence for darwinian selection against asparagine-linked glycans of Plasmodium falciparum and Toxoplasma gondii. Eukaryot. Cell 9, 228–241 (2010).
Crispin, M., Ward, A. B. & Wilson, I. A. Structure and immune recognition of the HIV glycan shield. Annu. Rev. Biophys. 47, 499–523 (2018).
Antanasijevic, A. et al. Polyclonal antibody responses to HIV Env immunogens resolved using cryoEM. Nat. Commun. 12, 4817 (2021).
The PyMOL Molecular Graphics System (Schrodinger, 2015).
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Abanades, B., Georges, G., Bujotzek, A. & Deane, C. M. ABlooper: fast accurate antibody CDR loop structure prediction with accuracy estimation. Bioinforma. Oxf. Engl. 38, 1877–1880 (2022).
Baek, M. et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 373, 871–876 (2021).
Ponnudurai, T. et al. Infectivity of cultured Plasmodium falciparum gametocytes to mosquitoes. Parasitology 98, 165–173 (1989).
Miura, K. et al. Qualification of standard membrane-feeding assay with Plasmodium falciparum malaria and potential improvements for future assays. PLoS ONE 8, e57909 (2013).
Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinforma. Oxf. Engl. 27, 2987–2993 (2011).
Marr, C. R., Benlekbir, S. & Rubinstein, J. L. Fabrication of carbon films with 500nm holes for cryo-EM with a direct detector device. J. Struct. Biol. 185, 42–47 (2014).
Rubinstein, J. L. & Brubaker, M. A. Alignment of cryo-EM movies of individual particles by optimization of image translations. J. Struct. Biol. 192, 188–195 (2015).
Punjani, A., Zhang, H. & Fleet, D. J. Non-uniform refinement: adaptive regularization improves single-particle cryo-EM reconstruction. Nat. Methods 17, 1214–1221 (2020).
He, J., Li, T. & Huang, S.-Y. Improvement of cryo-EM maps by simultaneous local and non-local deep learning. Nat. Commun. 14, 3217 (2023).
Punjani, A. & Fleet, D. J. 3D variability analysis: resolving continuous flexibility and discrete heterogeneity from single particle cryo-EM. J. Struct. Biol. 213, 107702 (2021).
Dunbar, J. et al. SAbPred: a structure-based antibody prediction server. Nucleic Acids Res. 44, W474–W478 (2016).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010).
Afonine, P. V. et al. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. D Struct. Biol. 74, 531–544 (2018).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 (2010).
Goddard, T. D. et al. UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci. Publ. Protein Soc. 27, 14–25 (2018).
Krissinel, E. & Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 (2007).
Kabsch, W. XDS. Acta Crystallogr. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 (2010).
XPREP. X-ray data Preparation and Reciprocal space Exploration Program. (Bruker AXS, 2014).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007).
Schmidt, A. G. et al. Viral receptor-binding site antibodies with diverse germline origins. Cell 161, 1026–1034 (2015).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010).
Morin, A. B. et al. Cutting edge: collaboration gets the most out of software. eLife 2, e0145 (2013).
Scheres, S. H. W. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012).
Masson, G. R. et al. Recommendations for performing, interpreting and reporting hydrogen deuterium exchange mass spectrometry (HDX-MS) experiments. Nat. Methods 16, 595–602 (2019).
Acknowledgements
We are grateful to J. Di Trani and S. Benlekbir for help and advice regarding cryo-EM specimen preparation. We thank L. Pelser, A. Pouwelsen, J. Kuhnen, J. Klaassen, and S. Mulder for assistance with mosquito dissections. This work was undertaken, in part, thanks to funding from the Canadian Institutes for Health Research (428410) and was supported by the CIFAR Azrieli Global Scholar program (J.-P.J.), the Ontario Early Researcher Award program (J.-P.J.), and the Canada Research Chair program (J.L.R., J.-P.J.). I.K. and D.I. were supported by SickKids Restracomp Fellowships. S.H. was supported by the Canada Graduate Scholarships Doctoral Award. M.R.I. and M.M.J. are supported by the Netherlands Organisation for Scientific Research (Vidi fellowship number 192.061). X-ray diffraction experiments were performed at GM/CA@APS, which has been funded in whole or in part with federal funds from the National Cancer Institute (ACB-12002) and the National Institute of General Medical Sciences (AGM-12006). The Eiger 16M detector was funded by an NIH–Office of Research Infrastructure Programs High-End Instrumentation grant (1S10OD012289-01A1). This research used resources of the Advanced Photon Source, a US Department of Energy (DOE) Office of Science user facility operated for the DOE Office of Science by Argonne National Laboratory under contract DE-AC02-06CH11357. Cryo-EM data were collected at the Toronto High-Resolution High-Throughput cryo-EM facility, and biophysical data were acquired at the Structural & Biophysical Core facility at SickKids, supported by the Canada Foundation for Innovation and Ontario Research Fund. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the paper.
Author information
Authors and Affiliations
Contributions
I.K., D.I., S.H., M.M.J., and J.-P.J. designed the experiments and prepared the original draft. D.I., S.H., A.S., and B.M. cloned the constructs, expressed and purified the proteins, and performed BLI measurements. I.K. collected and processed the cryo-EM data. S.H. and D.I. collected and processed X-ray crystallography data. M.R.I., K.T., R.S., M.v.d.V.-B., and G.-J.v.G. performed ELISA, SIFA, and SMFA experiments. A.K. and W.-K.L. performed MS experiments, and H.R.K. analyzed and interpreted the data. J.L.R., M.M.J., and J.-P.J. obtained funding to support the work and provided supervision. All authors contributed to writing, review, and editing of the final paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Structural & Molecular Biology thanks Nirbhay Kumar, Wai-Hong Tham and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Katarzyna Ciazynska, in collaboration with the Nature Structural & Molecular Biology team. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 mAb domain specificity and construct engineering of a biochemically tractable full-length Pfs48/45.
(a) Domain specificity of mAbs as determined by ELISA with full-length Pfs48/45 and fragments thereof. Bar heights are means of three technical replicates with individual values represented as dots and error bars indicate S.E.M. D3-specific mAbs TB31F and RUPA-44 only bind to D3-containing fragments, D2-specific mAb RUPA-154 only to D2-containing fragments and RUPA-58 only to D1-containing fragments. The threshold for positivity is set as the mean of six negative controls + 3 * SD and is indicated by a dashed line. (b) Experimental replicate size-exclusion chromatography profiles of full-length Pfs48/45, stabilized Pfs48/45 mAgE2, and Pfs48/45 mAgE2 linked to a single chain TB31F Fab (Pfs48/45 mAgE2-scTB31F, top panel; abbreviated as Pfs48/45-scTB31F) with molecular weight standards (bottom panel.) (c) SDS-PAGE analysis of peaks indicated in (b) under non-reducing conditions (-) and reduced conditions (+) using β-mercaptoethanol (BME). The gel is stained with Coomassie brilliant blue and the expected molecular weights for each construct are provided. This gel was produced twice with similar results. Source data for panels a and c are available online.
Extended Data Fig. 2 CryoEM processing workflow in cryoSPARC v3/v4 and EMReady2.
(a) Pfs48/45-scTB31F:RUPA-44:RUPA-58 complex dataset. (b) Pfs48/45-scTB31F:RUPA-44:RUPA-58:RUPA-154 complex dataset.
Extended Data Fig. 3 CryoEM analysis of the Pfs48/45-scTB31F:RUPA-44:RUPA-58 and Pfs48/45-scTB31F:RUPA-44:RUPA-58:RUPA-154 complexes.
(a) Representative cryoEM micrograph (from 1529 collected) of the Pfs48/45-scTB31F:RUPA-44:RUPA-58 complex. (b) Selected 2D class averages of the Pfs48/45-scTB31F:RUPA-44:RUPA-58 complex. (c) Gold standard Fourier shell correlation (GSFSC) curves for the final 3D non-uniform refinement of the Pfs48/45-scTB31F:RUPA-44:RUPA-58 complex in cryoSPARC v3. (d) Viewing direction distribution of the Pfs48/45-scTB31F:RUPA-44:RUPA-58 complex dataset. (e) Local resolution (Å) plotted on the surface (left panels) and center (right) of the cryoEM map of the Pfs48/45-scTB31F:RUPA-44:RUPA-58 complex. (f) Representative cryoEM micrograph (from 3435 collected) of the Pfs48/45-scTB31F:RUPA-44:RUPA-58:RUPA-154 complex acquired with no tilt applied. (g) Selected 2D class averages of the Pfs48/45-scTB31F:RUPA-44:RUPA-58:RUPA-154 complex. (h) GSFSC curves for the final 3D non-uniform refinement of the Pfs48/45-scTB31F:RUPA-44:RUPA-58:RUPA-154 in cryoSPARC v4. (i) Viewing direction distribution of the Pfs48/45-scTB31F:RUPA-44:RUPA-58:RUPA-154 complex dataset. (j) Local resolution (Å) plotted on the surface (top panels) and center (bottom) of the Pfs48/45-scTB31F:RUPA-44:RUPA-58:RUPA-154 complex cryoEM map. (k) Representative cryoEM micrograph (from 2785 collected) of the Pfs48/45-scTB31F:RUPA-44:RUPA-58:RUPA-154 complex collected under 40o tilt. (l) Selected 2D class averages of the Pfs48/45-scTB31F:RUPA-44:RUPA-58:RUPA-154 complex. (m) GSFSC curves for the final 3D non-uniform refinement (top) and the final 3D flexible refinement (bottom) of the Pfs48/45-scTB31F:RUPA-44:RUPA-58:RUPA-154 complex in cryoSPARC v4. (n) Viewing direction distribution of the Pfs48/45-scTB31F:RUPA-44:RUPA-58 complex dataset. (o) Local resolution (Å) plotted on the surface of the flexibly refined Pfs48/45-scTB31F:RUPA-44:RUPA-58:RUPA-154 complex cryoEM map (top panels) and on the surface (bottom left panels) and center (bottom right panel) of flexibly refined Pfs48/45-scTB31F:RUPA-44:RUPA-58:RUPA-154 complex cryoEM map after processing with EMReady2. (p) GSFSC curves for the final locally refined map of the D1D2:RUPA-58:RUPA-154 region in cryoSPARC v4. (q) Local resolution (Å) plotted on the surface of the locally refined cryoEM map of D1D2:RUPA-58:RUPA-154 region. (r) Local resolution (Å) plotted on the surface (left panels) and center (right) of the locally refined cryoEM map of D1D2:RUPA-58:RUPA-154 region further enhanced with EMReady2. All scale bars are 50 nm.
Extended Data Fig. 4 Comparison of the D3-scTB31F:RUPA-44 region of the Pfs48/45-scTB31F:RUPA-44:RUPA-58:RUPA-154 cryoEM map with X-ray structures of D3-TB31F (PDB ID: 6E63) and D3-RUPA-44 (PDB ID: 7UXL) (a) and details of D1D2:RUPA-58:RUPA-154 model fit into the locally refined cryoEM map (b-c).
(a) Models of D3-TB31F and D3-RUPA-44 were fitted individually into cryoEM map of the Pfs48/45-scTB31F:RUPA-44:RUPA-58:RUPA-154 complex using the ‘fit in map’ command in USCF Chimera. TB31F, RUPA-44 and D3 models are depicted in red, orange and navy, respectively, while the D1-D2-RUPA-58:RUPA-154 region of cryoEM map is shown as a white outline. (b) Low-pass filtered (7 Å) locally refined cryoEM map of D1-D2-RUPA-58:RUPA-154 region (grey) with models of D1, D2, RUPA-58 and RUPA-154 colored in light blue, blue, white and yellow, respectively. (c) Details of atomic model fit into the locally refined cryoEM map. CryoEM density is shown as black mesh, D1 and D2 are colored light blue and blue, respectively.
Extended Data Fig. 5 Comparison of the RUPA-154 and 10D8 binding sites on full-length Pfs48/45 from the cryoEM structure (a-c) and conservation of the RUPA-58 and RUPA-154 binding sites across Pf isolates (d-h).
(a) The variable domains of 10D8 and RUPA-154 bound to D1D2 with RUPA-154 (tan), 10D8 (dark gray), and shared (coral) D2 contact residues indicated with RUPA-154 (tan), 10D8 (dark gray), and shared (coral) D2 contact residues indicated. (b-c) Fabs RUPA-154 (b) and 10D8 (c) bound to full-length Pfs48/45. RUPA-154 (yellow) and 10D8 (grey) are depicted as ribbons and Pfs48/45 (blue) is represented as a surface. (d) Interfaces between Pfs48/45 (white) and RUPA-58 (pink) shown as ribbons, with D1 residues with polymorphisms labelled. (e-h) BLI binding curves of RUPA-58 bound to D1D2 WT (e) and mutants S90R (f), K101T (g), and N138I (h). Source data for panels e-h are available online.
Extended Data Fig. 6 NsEM epitope mapping of the RUPA-71 binding site on Pfs48/45 D1D2 (a) and comparison of the RUPA-58 and putative RUPA-71 binding sites on D1D2 (b).
(a) 2D classes and a 3D reconstruction of the RUPA-154:RUPA-71:Pfs48/45-D1D2 complex generated using nsEM. A predicted structure of RUPA-71 generated from Abodybuilder2 and the RUPA-154-D1D2 portion of the full-length Pfs48/45-Fab cryoEM structure were used to model this complex. RUPA-71 (dark violet), RUPA-154 (yellow), and D1D2 (blue) are depicted as ribbons within the nsEM map. The putative epitope of RUPA-71 is shown in coral. (b) RUPA-58 (light gray) and RUPA-71 (dark violet) are depicted with ribbons and surface bound to Pfs48/45-D1D2 (blue) which is represented as ribbons.
Extended Data Fig. 7 HDX analysis and epitope mapping on Pfs48/45 D1D2.
(a) Differential uptake plot representing the differences in deuterium uptake between free and RUPA71 bound states of Pfs48/45 D1D2. Each bar represents a peptic peptide of Pfs48/45 D1D2 (ordered from N- to C-terminus based on the first amino acid of each peptic peptides). The deuterium uptake differences for each peptide at different time points are shown in orange (10 s), red (100 s), blue (1000 s) and black (10,000 s). The height of the vertical gray bar represents the sum of the mass differences for each peptide between the two states at each time point. The gray shades represent the differential uptake errors for each peptide, and the dashed horizontal lines represent the threshold which the uptake differences between the two states are considered significant if exceeded. This threshold corresponds to the 95% confidence interval, calculated from the pooled standard deviations for all peptides across all time points which is ±0.3 Da. The red and blue boxes highlight the regions with decreased and increased deuterium uptake in the bound state, respectively. (b) Deuterium uptake plots of the selected peptides of the regions that show significant HDX changes. Error bars represent mean ± S.E.M of three independent experiments.
Extended Data Fig. 8 Mutation N131Q reduces binding of potent Pfs48/45 domain 1 antibodies.
BLI data of RUPA-71 (a), RUPA-116 (b), RUPA-119 (c), and RUPA-75 (d), RUPA-155 (e), and RUPA-46 (f) binding to wild-type Pfs48/45-D1D2 and a D1D2 construct containing a N131Q point mutation. This mutation removes a putative N-linked glycosylation sequon. RUPA-58 was used as a control for proper folding of the D1D2 N131Q mutant (g). (h) A model of RUPA-71 (dark violet) binding to D1D2 (blue) generated using nsEM with a glycan at position N131 modeled in coral, which would result from expression of the D1D2 construct in mammalian cells. Source data for panels a-g are available online.
Extended Data Fig. 9 Binding of potent Pfs48/45 D1 IgGs to Pv D1D2 and conservation of P48/45 across Pf and Pv.
BLI data of RUPA-58 IgG (a), RUPA-116 IgG (b), RUPA-119 IgG (c), and RUPA-75 IgG (d), RUPA-155 IgG (e), RUPA-46 IgG (f), and RUPA-94 IgG (g) binding to Pfs48/45-D1D2 and Pvs48/45-D1D2 (KD = apparent affinity). (h) Sequence alignment with identical, highly conserved, semi-conserved, and non-conserved residues indicated in dark magenta, orchid, pink, and light gray, respectively. The putative epitope of RUPA-71 is indicated with a coral bar above the sequence. Source data for panels a-g are available online.
Extended Data Fig. 10 Electrostatic surface potential representations of the three observed Pfs48/45 interdomain conformers, as depicted in Fig. 6.
Models of Pfs48/45 derived from the Pfs48/45-:10D8 cocrystal structure (PDB ID: 7ZXF) (a), the Pfs48/45-scTB31F:RUPA-44:RUPA-58:RUPA-154 complex (b), and the Pfs48/45-scTB31F:RUPA-44:RUPA-58 complex (c) are depicted with surface color indicating electrostatic potential ranging from −5kT/e (red) to +5kT/e (blue). In general, D3 possesses an electronegative surface potential, while D1 and D2 possess a mixed electronegative and electropositive surface.
Supplementary information
Supplementary Information
Supplementary Figs. 1 and 2.
Supplementary Tables 1 and 2
Supplementary Table 1. Non-synonymous single nucleotide polymorphisms in Pfs48/45. Supplementary Table 2. Summary of continuous labeling HDX-MS data of Pfs48/45 D1D2 upon RUPA71 binding.
Supplementary Video 1
3D variability analysis of the Pfs48/45-scTB31F–RUPA-44–RUPA-58–RUPA-154 complex.
Source data
41594_2025_1532_MOESM6_ESM.xlsx
Source Data Figs. 1, 4, and 5, and Extended Data Figs. 5, 8, and 9. Raw SMFA data. SIFA source data. BLI data. Source Data Extended Data Fig. 8. BLI data. Source Data Extended Data Fig. 9. BLI data
Source Data Extended Data Fig. 1
Uncropped SDS–PAGE.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kucharska, I., Ivanochko, D., Hailemariam, S. et al. Structural elucidation of full-length Pfs48/45 in complex with potent monoclonal antibodies isolated from a naturally exposed individual. Nat Struct Mol Biol 32, 1396–1407 (2025). https://doi.org/10.1038/s41594-025-01532-6
Received:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/s41594-025-01532-6
