
Abstract
Solute carrier family 37 member 4 (SLC37A4) is known to regulate glucose-6-phosphate transport from cytoplasm to the lumen of the endoplasmic reticulum, which serves to maintain glucose homeostasis. Glycogen storage disease type 1b (GSD1b) is caused by a mutation of SLC37A4, leading to a glycogenolysis defect. Although GSD1b cases are known to be complicated by periodontitis, the etiological molecular basis remains unclear. The present study investigated the effects of SLC37A4 on gingival barrier function. Examinations of immortalized human gingival epithelial (IHGE) cells showed SLC37A4 localized in the endoplasmic reticulum. SLC37A4 knockout decreased expression of JAM1, a tight junction-related protein, in IHGE cells. Using in silico analysis to investigate potential transcription factor binding sites, H6 family homeobox 3 (HMX3) was shown to be related to JAM1 expression. In HMX3-knockdown IHGE cells, JAM1 expression was markedly suppressed. Furthermore, HMX3 was scarcely detected in SLC37A4-knockout cells, while HMX3 overexpression restored JAM1 expression in those cells. Finally, using a three-dimensional multilayered gingival epithelial tissue model, knockout of SLC37A4 was also found to increase permeability to lipopolysaccharide and peptidoglycan, which was dependent on JAM1 expression. Specific downregulation of HMX3 by SLC37A4 and the consequent decrease in JAM1 expression provides findings indicating a molecular basis for the reduction in barrier function of gingival epithelial tissues in GSD1b cases.
Similar content being viewed by others

Introduction
Glycogen storage disease type 1b (GSD1b) is an autosomal recessive disease characterized by hypoglycemia, excessive glycogen accumulation in the liver and kidneys, and abnormal metabolic serum profiles1. Overall, GSD1b incidence is estimated to occur in 1 per 500,000 live births2. The prognosis of untreated GSD1b patients was previously very poor and many died at a young age, though recently better methods for early diagnosis and appropriate treatment have improved the situation, and most now live to adulthood3.
Glucose is the most important source of energy metabolism in all organisms. Upon cellular uptake, the majority of glucose is phosphorylated at the hydroxy group on carbon 6, resulting in glucose 6-phosphate (G6P). Nearly 50 years ago, a GSD1b patient was shown to have an impaired G6P transport system4. When cells require energy or carbon skeletons for synthesis, G6P is targeted for glycolysis. Solute carrier family 37 member 4 (SLC37A4) located in 11q23.3 has been identified as the gene responsible for GSD1b5,6,7. SLC37A4 is a transmembrane protein located in the endoplasmic reticulum membrane8, whose biological function is translocation of G6P from cytoplasm into the ER lumen, where it is hydrolyzed to glucose and phosphate. Glucose supplement therapy is the only method available to maintain normal blood glucose and prevent hypoglycemia in patients with glycogenic diseases treated with nasogastric tubes or a gastrostomy1.
Several different systemic genetic disorders can cause loss of periodontal attachment and alveolar bone9. GSD1b patients also often suffer from severe periodontitis9,10, with symptoms such as deep periodontal pocketing with bleeding on probing and generalized severe horizontal alveolar bone loss11. However, it remains unknown whether SLC37A4 dysfunction is related to initiation of diseases that affect periodontal tissues.
Periodontitis is a chronic inflammatory disease with effects on the periodontium and its etiology is multifactorial, including chronic infection by commensal periodontal pathogens in contact with periodontal tissue12. Among cell adhesion molecules, junctional adhesion molecule 1 (JAM1), a member of the immunoglobulin superfamily13, is expressed in gingival epithelial tissues and serves to protect epithelial barrier function14. Recent studies have reported that JAM1 is degraded by Porphyromonas gingivalis, a periodontal pathogen, which leads to breakdown of the epithelial barrier against bacterial virulence factors such as lipopolysaccharide (LPS) and peptidoglycan (PGN)15. In GSD1b patients, glucose supplement therapy does not resolve periodontal disease, thus there are likely other factors related to SLC37A4 deficiency, including breaching of the barrier function of the gingival epithelium that serves to protect against bacterial components.
The present study was performed to examine the effects of SLC37A4 deficiency on gingival epithelial barrier function. In previous studies, neutrophils have been the main host cells analyzed for GSD1b, while use of immortalized human epithelial cells in the present experiments enabled establishment of genetically engineered cell lines and production of syndrome-specific tissue. Results obtained using genome editing with clustered regularly interspaced short palindromic repeats (CRISPR)-associated protein 9 (CRISPR-Cas9) and short hairpin RNA (shRNA) systems in combination with a three-dimensional tissue model are presented. They provide insight into the barrier function-subversive processes of SLC37A4 deficiency, which may explain, at least in part, the increased propensity for bacterial infections not only in association with GSD1b but also other systemic diseases associated with periodontitis.
Results
SLC37A4 localized in endoplasmic reticulum of gingival epithelial cells
Investigations of SLC37A4 localization in clinical human tissue specimens have been reported16,17,18, though, to the best of our knowledge, that has not been studied with use of gingival epithelial cells. Results of the present reverse transcription (RT)-polymerase chain reaction (PCR) tests revealed mRNA expression of SLC37A4 in immortalized human gingival epithelial (IHGE) cells (Fig. 1a), while SLC37A4 endogenous protein expression was also confirmed with use of three different antibodies (Supplementary Fig. 1). To reveal more details regarding localization, IHGE cells expressing Myc-tagged SLC37A4 protein and organelle markers, including enhanced green fluorescent protein (EGFP)-SEC61β (marker for endoplasmic reticulum membrane protein), TOMM20 (marker for mitochondria outer membrane), TGN46 (marker for trans-Golgi), and LAMP1 (marker for lysosome), were analyzed using confocal microscopy. As shown in Fig. 1b-e and Supplementary Fig. 2a-h, Myc signaling was co-localized with EGFP signaling, but scarcely observed with TOMM20, TGN46, or LAMP1, suggesting that SLC37A4 localizes in endoplasmic reticulum of gingival epithelial cells.

SLC37A4 localization in human gingival epithelial cells. (a) RT-PCR analysis of SLC37A4 gene in IHGE cells. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as the control. (b) IHGE cells expressing Myc-SLC37A4 and EGFP-SEC61β (green) were fixed, stained with anti-Myc (magenta: Alexa Fluor 555), and analyzed by confocal microscopy. The area enclosed by the white square area showing the periodontal pocket in the upper panel is magnified in the lower panel. (c-e) IHGE cells expressing Myc-SLC37A4 were fixed, stained with anti-Myc (green: FITC) and either anti-TOMM20 (magenta: Alexa Fluor 555 in c), anti-TGN46 (magenta: Alexa Fluor 555 in d), or anti-LAMP1 (magenta: Alexa Fluor 555 in e), then analyzed by confocal microscopy. The white square areas showing the periodontal pocket in the left panels are magnified in the right panels. Scale bars, 10 μm. See also Supplementary Fig. 1.
To assess the contribution of microbial products, IHGE cells were treated with LPS and PGN, then the mRNA level of SLC37A4 was analyzed using quantitative real-time PCR (qRT-PCR). At three hours after administration, SLC37A4 expression was decreased by LPS, while that was increased by PGN (Supplementary Fig. 3), suggesting possible modulation of LPS and PGN by SLC37A4 expression in gingival epithelial cells.
SLC37A4 expression in gingival epithelial cells related to JAM1 expression
To assess the contribution of SLC37A4 to gingival epithelial cell phenotype, an SLC37A4 knockout IHGE cell line was established using the CRISPR/Cas9 system. Additionally, to avoid off-target effects, an SLC37A4 knockdown cell line was also established using an expression vector coding small hairpin RNA (shRNA) against SLC37A4 or firefly luciferase (shLuc) as a control. Confirmation of SLC37A4 knockout and knockdown in IHGE cells was based on immunofluorescence (Fig. 2a) and qRT-PCR findings (Supplementary Fig. 4), respectively. To assess the effects of SLC37A4 knockout or knockdown on organelle phenotype in gingival epithelial cells, morphologic changes in endoplasmic reticulum, mitochondria, and cis-Golgi were analyzed following staining with concanavalin A, anti-TOMM20, and anti-GM130, respectively. The findings obtained indicated a partial aggregation pattern in endoplasmic reticulum (Fig. 2b, Supplementary Fig. 5a) and mitochondrial fragmentation (Fig. 2c, Supplementary Fig. 5b). On the other hand, cis-Golgi localization was negligibly altered by SLC37A4 knockdown (Supplementary Fig. 5c), suggesting that the effects of SLC37A4 loss on endoplasmic reticulum and mitochondria are selective.

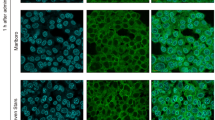
Effects of SLC37A4 knockout on phenotype of IHGE cells. (a) IHGE WT or ΔSLC37A4 cells were fixed, stained with DAPI (cyan) and rabbit polyclonal anti-SLC37A4 (gray: Alexa Fluor 555, Proteintech 20,612–1-AP), and analyzed by confocal microscopy. (b-d) IHGE WT or ΔSLC37A4 cells were fixed, stained with DAPI (cyan) and either concanavalin A (gray: Alexa Fluor 594), rabbit polyclonal anti-TOMM20 (gray: Alexa Fluor 555 in c), or mouse monoclonal anti-JAM1 (gray: Alexa Fluor in d), then analyzed by confocal microscopy. The white square areas in the upper panels of b and c are magnified in the lower panels. Arrows in knockout cells indicate a pattern different from WT cells. Scale bars, 5 μm.
Under this condition, SLC37A4 knockdown was shown to decrease JAM1 mRNA (Supplementary Fig. 6) and protein (Supplementary Fig. 7) levels in IHGE cells. Additionally, confocal microscopic imaging revealed loss of JAM1 on the surface of SLC37A4-knockout as well as knockdown cells (Fig. 2d, Supplementary Fig. 8). Notably, negligible effects of SLC37A4 knockout were observed in regard to mRNA level (Supplementary Fig. 9a) and also localization (Supplementary Fig. 9b) of the JAM family protein coxsackie and adenovirus receptor, which is also involved in gingival epithelial barrier function19. Additionally, to examine the effects of SLC37A4 loss on potential biomarkers for periodontitis, the mRNA level of dickkopf-related protein 1 (DKK1)20,21 in IHGE WT cells and ΔSLC37A4 was analyzed, which showed that DKK1 expression was negligibly altered by SLC37A4 knockout (Supplementary Fig. 10). Together, these findings indicate that SLC37A4 expression is selectively involved in JAM1 gene expression in gingival epithelial cells.
HMX3 involved in SLC37A4-mediated JAM1 expression in gingival epithelial cells
To determine the transcription factor involved in SLC37A4-mediated JAM1 expression, a human gene database (GeneCards, Weitzmann Institute of Science) was utilized, which revealed the following four candidate transcription factors; nuclear factor IA (NFIA), HMX3, GATA binding protein 1 (GATA1), and paired box 2 (PAX2). qRT-PCR was then performed to reveal expressions of these four genes in gingival epithelial cells, with the results showing that NFIA and HMX3 were mainly expressed (Fig. 3a). We then confirmed that knockout of SLC37A4 decreased mRNA levels of HMX3 (Fig. 3b), though the effects were quite limited on NFIA (Fig. 3c) and, accordingly, JAM1 (Fig. 3d) in IHGE cells.

HMX3 involved in SLC37A4 knockout-reduced JAM1 expression in IHGE cells. (a) NFIA, HMX3, GATA1, and PAX2 mRNA expression in IHGE cells, with results expressed as fold change relative to NFIA and shown as the mean of five technical replicates (bars). ND, not detected after 45 cycles of qRT-PCR. (b, d) HMX3 (b) and JAM1 (d) mRNA expression in IHGE WT and SLC37A4 knockout cells with HA-HMX transient transfection, with results expressed as fold change relative to WT cells and shown as the mean of five technical replicates (bars). Significant differences were determined using a two-tailed t test (closed-testing procedure). (c) NFIA mRNA expression in IHGE WT and SLC37A4 knockout cells, with results expressed as fold change relative to WT cells and shown as the mean of five technical replicates (bars). Significant differences were determined using a two-tailed t test. (e, f) HMX3 (e) and SLC37A4 (f) mRNA expression in IHGE cells with HA-HMX transient transfection, with results expressed as fold change relative to no transfection (Control) and shown as the mean of five technical replicates (bars). Significant differences were determined using a two-tailed t test. *p < 0.05. β-ACTIN was used as the internal control. The results shown are representative of two biological replicates. Dot plots display data values for each experimental condition.
To assess the effects of HMX3 on JAM1 reduction induced by SLC37A4 knockout, IHGE cells lacking SLC37A4 were transfected with plasmid coding hemagglutinin (HA)-HMX3 and JAM1 expression was evaluated. Confocal microscopic images revealed that HA-tagged HMX3 was appropriately localized in both wild-type (WT) (Supplementary Fig. 11a) and SLC37A4-knockout cells (Supplementary Fig. 11b), while loss of JAM1 was also restored by HMX3 overexpression (Fig. 3b, 3d). Notably, HMX overexpression in WT cells did not increase JAM1 (Fig. 3b, d) nor SLC37A4 (Fig. 3e, f) expression, suggesting that the SLC37A4-HMX3-JAM1 cascade is a one-way process. These results collectively indicate that HMX3 is involved in SLC37A4-mediated JAM1 expression in gingival epithelial cells.
HMX3 involved in JAM1 expression in gingival epithelial cells
Next, the effects of HMX3 depletion on JAM1 expression were examined. RT-PCR results revealed expression of HMX3 mRNA at a certain level in IHGE cells (Fig. 4a). An expression vector coding shRNA against HMX3 (shHMX3 #644 and shHMX3 #737) was constructed, which showed stable expression in IHGE cells. Knockdown of HMX3 in each cell line was confirmed by qRT-PCR (Fig. 4b), and under that condition it was confirmed that knockdown of HMX3 decreased mRNA (Fig. 4c) and protein (Fig. 4d) levels of JAM1 in IHGE cells. Furthermore, JAM1 was no longer found on the surface of HMX3-knockdown cells (Fig. 4e). On the other hand, SLC37A4 expression was negligibly altered by HMX3 knockdown (Supplementary Fig. 12), consistent with the previous finding suggesting a one-way SLC37A4-HMX3 cascade. These results indicate that HMX3 expression is involved in JAM1 gene expression in gingival epithelial cells.

HMX3 knockdown results in decreased JAM1 expression in IHGE cells. (a) RT-PCR analysis of HMX3 gene in IHGE cells. β-ACTIN was used as the control. (b, c) HMX3 (b) and JAM1 (c) mRNA expression in IHGE cells stably expressing shLuc or shHMX3 (#644 or #737) is shown as fold change relative to shLuc-expressing cells, and presented as the mean of five technical replicates (bars). Significant differences were determined using Dunnett’s test. *p < 0.05. The results shown are representative of two biological replicates. (d) Lysates of IHGE cells expressing indicated shRNA were analyzed by immunoblotting with the indicated antibodies. (e) IHGE cells stably expressing shLuc or shHMX3 (#644 or #737) were fixed, then stained with DAPI (cyan) and mouse monoclonal anti-JAM1 (gray: Alexa Fluor 555), then analyzed by confocal microscopy. Scale bars, 10 μm.
Epithelial barrier function regulated by SLC37A4 in JAM1-dependent manner
A two-layered culture system was employed to assess the role of SLC37A4 (Fig. 5a). It was confirmed that SLC37A4 knockout increased the permeation of fluorescein isothiocyanate (FITC)-labeled 4-, 40-, or 70-kDa dextran into the IHGE cell layers (Supplementary Fig. 13a-c), suggesting that SLC37A4 is involved in blocking gingival epithelial cell permeability. To eliminate off-target effects of the knockout system, SLC37A4-knockout cells additionally expressing JAM1 were generated, and sufficient compensation of cell surface JAM1 was confirmed (Fig. 5b). Permeability assay findings obtained under this condition indicated that SLC37A4 depletion increased permeation of FITC-labeled P. gingivalis LPS (Fig. 5c) and PGN (Fig. 5d) in SLC37A4-knockout cells, which was abrogated by JAM1 overexpression.

JAM1 involved in SLC37A4 knockout-disturbed barrier against LPS and PGN in IHGE cells. (a) Schematic image of culture-insert system. A monolayer of IHGE WT or SLC37A4 knockout cells along with stable expression of HA-inserted JAM1 was cultured in the upper compartment. Fluorescent tracers were added and culturing was performed for 30 min, after which culture medium from the lower compartment was analyzed using spectrometry. (b) Representative confocal microscopic images of IHGE WT or SLC37A4 knockout cells along with stable expression of HA-inserted JAM1. DAPI (cyan), anti-JAM1 (magenta: Alexa Fluor 555), anti-HA (gray: Alexa Fluor 647). (c, d) Permeability to FITC-P. gingivalis LPS (c) or FITC-S. aureus PGN (d) in indicated IHGE cells. Results are expressed as fold change relative to WT cells and presented as the mean ± SD of eight technical replicates. *p < 0.05, two-tailed t test (closed-testing procedure). The results shown are representative of two biological replicates.
Finally, 3D-tissue model WT and ΔSLC37A4 cells with or without overexpression of JAM1 were generated using a previously reported cell accumulation technique15 (Fig. 6a). Construction of gingival epithelial tissues was confirmed using confocal microscopy (Fig. 6b). The tissues were treated with FITC-labeled P. gingivalis LPS or S. aureus PGN, then subjected to permeability assays. Three hours after administration, permeability to both FITC-labeled P. gingivalis LPS (Fig. 6c) and S. aureus PGN (Fig. 6d) was significantly increased by knockout of SLC37A4 in gingival epithelial tissues, while that increase was abrogated by JAM1 overexpression. These findings indicate that JAM1 is involved in SLC37A4 knockout-mediated permeability of gingival epithelium to LPS and PGN.

SLC37A4 knockout dampens epithelial barrier function of gingival epithelial tissues. (a) Schematic image of culture-insert system. WT or ΔSLC37A4 gingival epithelial tissues with or without overexpression of JAM1 were cultured in the upper compartments. FITC-labeled tracers were then added to culture medium in each upper compartment. Following 3 h of incubation, transmission of a tracer from the upper to lower compartment was analyzed by spectrometry. (b) Confocal microscopic cross-sectional images of 3D-tissue model of IHGE cells. WT and ΔSLC37A4 gingival epithelial tissues with or without overexpression of JAM1 on coverslips were fixed, stained with DAPI (cyan) and Alexa Fluor 633-conjugated phalloidin (gray), and analyzed using confocal microscopy. Scale bars, 30 μm. (c, d) Permeability of gingival epithelial tissues to FITC-P. gingivalis LPS (c) or S. aureus PGN (d). Results expressed as fold change relative to the control (WT tissues) were obtained and are presented as the mean ± SD of eight technical replicates. *p < 0.05, two-tailed t test (closed-testing procedure). The results shown are representative of two biological replicates.
Discussion
This study is the first to present findings of a tight junction-related protein downregulated in genetic disorders related to periodontitis, leading to loss of barrier function of human gingival epithelial tissues. It was shown that loss of SLC37A4 decreased JAM1 gene expression, which is different from the cause of JAM1 dysfunction, i.e., degradation by periodontal pathogens15. In another study, gingival epithelial function was shown to be damaged by abnormal localization of JAM1 due to cigarette smoke extract14. Additionally, the present findings are related to a molecular basis that is different than found in those prior studies.
LPS translocation within inflammatory lesions in gingival tissue of patients with leukocyte adhesion deficiency, another genetic disorder associated with periodontitis, has also been reported22, supporting the present speculation that a dysregulated barrier function in gingival epithelial tissue may be a key factor for periodontitis etiology. Lysozyme in saliva is generally known to degrade the cell wall components of bacteria, including PGN, and to inactivate LPS23. Should an increase in epithelial permeability related to LPS be shown to be a common periodontal risk factor, then identification of host factors that neutralize bacterial toxins as well as LPS in turn may be an important area of research related to genetic disorders associated with periodontal disease. On the other hand, the relationship of GSD1b-associated microbial communities in dental plaque with local inflammatory response remains unknown. Use of in vitro tissue reconstruction, including gingival subepithelial tissue, seems to be a promising method for determining whether specific flora or trans-threshold transmission of bacterial toxins are causative factors for GSD1b-associated periodontal disease.
The present results revealed a novel transcription factor regulating JAM1 gene expression in gingival epithelial cells. A de novo microdeletion of chromosome 10q26.11q26.13, where HMX3 is located, was reported to cause inner ear malformations24, leading to congenital hearing loss, while GSD1b has also been shown to be associated with sensorineural hearing loss25,26. Hence, the common occurrence of hearing loss in both GSD1b and HMX3 deficiency cases strongly supports the SLC37A4-HMX3 axis identified in the present study. In addition, a deficiency of SLC37A4 was found to disturb energy homeostasis in the endoplasmic reticulum (ER) caused by loss of endogenous glucose production, leading to ER stress27. Other studies have found that the deafness condition associated with impaired ER stress is caused by gene mutations in Wolframin ER transmembrane glycoprotein (Wolframin syndrome)28 and gap junction protein beta 2 (Bart-Pumphrey syndrome)29. Hence, the relationship between ER stress and hearing loss related to these syndromes may involve HMX3 gene function.
Regarding upstream of the HMX3 pathway, analysis of the GeneCards database (https://www.genecards.org/cgi-bin/carddisp.pl?gene=HMX3) showed predicted transcription factors, including nuclear factor kappa b and tumor protein p53, which are also predicted by the TFLink gateway (https://tflink.net/protein/a6nht5/), suggesting that rescue experiments (gene expression, nuclear translocation, post-translational modifications, intracellular degradation, etc.) performed using these two transcription factors or related proteins in SLC37A4 knockout cells may help to elucidate how SLC37A4 regulates HMX3 expression. Currently, there is no fundamental treatment for GSD1b and dietary therapy is the primary choice, though it is not capable of resolving all related complications. While clinical trials of adeno-associated virus and mRNA therapy for GSD1b have been performed, problems have been noted, such as off-target effects on host genes, dosing limitations, and difficulty with blood glucose control. It is considered that even when SLC37A4 mutations cannot be corrected, increasing HMX3 expression may be an alternative means to alleviate acquired complications in GSD1b cases.
GSD1b has also been found to be associated with recurrent mucositis and ulcers in the oral cavity30, chronic inflammatory bowel disease (IBD)-like colitis31, and Crohn’s disease32, as well as nose bleeding due to platelet dysfunction33. Furthermore, JAM1, which was first identified in platelets34, has been reported to be involved in platelet adhesion to endothelial cells35 and growth of platelet aggregation36. Since dysfunctions of SLC37A4 and JAM1 are known to be related to bowel and platelet diseases, the association between these two genes is considered to be robust. It has also been reported that granulocyte colony stimulating factor (G-CSF) therapy in GSD1b patients has possible side-effects, such as acute myeloid leukemia with shortened telomeres2 and giant cell tumor development37. To better understand symptoms associated with G-CSF-based therapy in GSD1b patients, analysis of JAM1 as a key factor is considered to be important.
Although GSD1b is generally associated with neutropenia, SLC37A4 mutations in patients without neutropenia have also been reported38,39,40. To promote differentiation and proliferation of neutrophils, patients with GSD1b usually receive treatment with G-CSF, which limits but does not eliminate infections41. These reports suggest that the gingival epithelium has a crucial role in the etiology of periodontal diseases, including those accompanied with GSD1b. As for systemic effects, GSD1b patients are known to be complicated by hyperuricemia, hyperlipidemia, and lactic acidemia1, thus typically receive long-term antibiotic and/or recombinant protein therapy to prevent infection. To exclude such systemic effects on gingival epithelial tissues, the cell accumulation technique used in the present study allowed for analyses of the effects of the SLC37A4 mutation alone on barrier function. In future studies, disease-specific tissue models may be useful for providing a better understanding of the pathogenesis of not only periodontitis, but also diseases that accompany congenital genetic disorders.
Experimental procedures
Cell culture
This study was performed according to the principles of the Declaration of Helsinki. All human subjects who participated provided informed consent to the study protocol, which was reviewed and approved by the ethics committee of Osaka University Graduate School of Dentistry (R2-E8-1).
IHGE cells (epi 4, kindly provided by Shinya Murakami, Osaka University) were maintained in Humedia KG-2 (Kurabo), as previously described42. Three-dimensional cultures of IHGE cells were performed as previously described15,43, with some modification. Briefly, IHGE cells were collected by centrifugation, then after trypsinization were incubated for 3 min with 0.2 mg mL-1 fibronectin (Sigma-Aldrich) in 0.1 mg mL-1 of gelatin solution (Nacalai Tesque) for 3 min. After three immersion steps, fibronectin/collagen nanofilms were coated onto single-cell surfaces. For tissue morphological analysis, a total of 2 × 106 cells coated with fibronectin/collagen were seeded onto coverslips coated with vitronectin solution (A14700, Invitrogen) diluted 1/100 (v/v) in phosphate-buffered solution (PBS) in 24-well plates (Iwaki). After 36 h of incubation, tissues were subjected to experiments, then fixed and analyzed using a confocal microscope (TCS SP8; Leica Microsystems). For permeability experiments, a total 1 × 106 cells with fibronectin/collagen were seeded into 24-well cell culture inserts (353,096, Corning).
Antibodies, plasmids, and reagents
Antibodies, plasmids, and reagents used in this study are presented in Supplementary Table 1.
RT-PCR for SLC37A4 and HMX3
Reverse transcription reactions were performed using ReverTra Ace qPCR RT Master Mix (Toyobo). PCR was done with Go-Taq (Promega). Primer sequences are listed in Supplementary Table 2.
Transient transfection
Plasmid-encoding Myc–tagged SLC37A4 was produced using PCR from a constructed vector (MHS6278-202,832,047; Horizon Discovery), then inserted into a pCMV-Myc vector (Clontech) using exogenously added EcoRI and KpnI sites. A plasmid encoding HA-tagged HMX3 was produced using PCR from the constructed vector (RC225531L4; OriGene) using exogenously added KpnI and NotI sites. A plasmid encoding EGFP-SEC61β or HA-inserted JAM1 was constructed as previously described15. All PCR products and mutations were confirmed by sequencing (FASMAC). Transfection was performed using FuGENE 6 Transfection Reagent (Promega).
Immunocytochemistry and immunoblotting
Immunocytochemistry and immunoblotting were performed as previously described15,44. Antibodies were obtained, then diluted 1/400 (v/v) in PBS for immunostaining and 1/20,000 (v/v) in PBS with 0.1% (v/v) Tween 20 (16,021,211, Fujifilm) for immunoblotting. Endoplasmic-reticulum staining was performed using concanavalin A (1 mg mL-1, Invitrogen) after dilution 1/400 (v/v) in PBS. Immunoreactive bands were detected using Pierce ELC Western Blotting Substrate (Thermo Scientific) and ChemiDoc XRS (Bio Rad), then images were acquired using the Quantify One software package (Bio-Rad). Confocal microscopic images were acquired with a confocal laser microscope (TCS SP8; Leica Microsystems) using a 64 × oil-immersion objective lens with a numerical aperture of 1.4, then analyzed using the Application Suite X software package (Leica Microsystems, https://www.leica-microsystems.com/products/microscope-software/p/leica-las-x-ls/downloads/?country=GB).
RNA interference
Plasmid encoding shRNA was constructed by ligation of linear DNA (Sigma-Aldrich) into pSIREN-RetroQ (Clontech). Plasmids pSIREN-RetroQ-shSLC37A4 #903, #994, and #1152, and pSIREN-RetroQ-shHMX3 #644 and #737 were used for generation of the siRNA duplex (target sequences: 5’-GAGCAGAATGGTGAGAAGTTTGT-3’, 5’-GTGAATTACTTTGCCTTATTTGA-3’, 5’-GTGAAGAAAGGTTACATCAAAGC-3’, 5’-GCGCTGAAAGTCCAGAGAAGAAG-3’, and 5’-TCGACATGAAGCGCTATCTGAGC-3’, respectively) in the cells. Plasmid pSIREN-RetroQ-shLuc was produced as previously described15,45. IHGE cells were transfected with shRNA-encoding plasmid using FuGENE 6 (Promega). Seventy-two hours after transfection, cells stably expressing shRNA were selected with puromycin (2 μg mL-1).
Quantitative real-time PCR
Quantitative real-time PCR (qRT-PCR) was performed as previously described15. Briefly, total RNA was extracted from IHGE cells using an RNeasy Micro Kit (Qiagen) and complementary DNA was synthesized using ReverTra Ace qPCR RT Master Mix (Toyobo). Real-time PCR was performed using a Rotor Gene Q cycler (Qiagen) with THUNDERBIRD SYBR qPCR Mix (Toyobo). Primer sequences are shown in Supplementary Table 2. Amplicon level in each sample was normalized against the corresponding level of β-ACTIN mRNA content using the 2−ΔΔCt method. The specificity of each pair of primers was confirmed by qRT-PCR using a template with or without reverse transcription (Supplementary Fig. 14).
Establishment of SLC37A4 KO IHGE cells
Using a CRISPR/Cas9 Genome Knockout Kit (Origene) designed to target the human SLC37A4 gene (KN404879), the target sequence was human SLC37A4, 5’-GGGATCTCTTCCACCAATGA-3’ (upstream of exon 1). This guide RNA sequence was designed to insert a puromycin-resistant gene along with a termination codon in the exon 1 region of the gene. IHGE cells were transfected using FuGENE6 (Promega) with the guide vector and linear donor. Seventy-two hours after transfection, knockout cells were selected with puromycin (2 μg mL-1; InvivoGen). Clones with mutations in both alleles were identified by genomic DNA sequencing and immunoblotting.
SLC37A4 KO IHGE cells stably expressing HA-inserted JAM1 were generated using the following procedures. A plasmid-encoding HA-inserted JAM1 was constructed with cloning PCR products amplified from the pCMV plasmid15 into pBApo-EF1α NEO (Takara). The pBApo-EF1α NEO HA-inserted JAM1 plasmid was used for overexpression of cDNA in IHGE cells. SLC37A4 KO IHGE cells stably expressing JAM1 were selected with G418 (200 μg mL-1) (InvivoGen).
Prediction of transcription factor binding sites in JAM1 gene promoter
The top transcription factor binding sites in the JAM1 gene promoter were analyzed using the GeneCards database (https://www.genecards.org/cgi-bin/carddisp.pl?gene=F11R). In addition, the TFLink gateway (https://tflink.net/protein/q9y624/) was referred to for confirmation of the predicted associations of the HMX and NF transcription factors with the JAM1 promoter.
Epithelial barrier functional assay
FITC-tracers were prepared using a previously described method15. To assess barrier function, in vitro epithelial permeability assays were also performed with 12-well cell culture inserts (353,180; Corning), as described in that study. Culture media containing transferred tracers in the lower compartment were analyzed without dilution using a black plate (OptiPlate, Revvity Health Sciences) and a Wallac 1420 ARVO X Multilabel Counter (PerkinElmer) for determining the fluorescence intensity of FITC. Data obtained were analyzed using the WorkOut Plus software package (PerkinElmer).
Statistical analysis
P values were determined using a two-tailed t test and Dunnett’s test with the Excel software package (Microsoft), with p < 0.05 considered to indicate significance.
Data availability
Data supporting the findings presented in this study are available from the corresponding author upon reasonable request.
References
Kishnani, P. S. et al. Diagnosis and management of glycogen storage disease type I: a practice guideline of the American college of medical genetics and genomics. Genet. Med. 16, e1. https://doi.org/10.1038/gim.2014.128 (2014).
Li, A. M. et al. Prolonged granulocyte colony stimulating factor use in glycogen storage disease type 1b associated with acute myeloid leukemia and with shortened telomere length. Pediatr. Hematol. Oncol. 35, 45–51 (2018).
Bali DS, El-Gharbawy A, Austin S, Pendyal S, Kishnani PS. (2006) Glycogen Storage Disease Type I. Apr 19 [Updated 2021 Oct 14]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1312/
Narisawa, K., Igarashi, Y., Otomo, H. & Tada, K. A new variant of glycogen storage disease type I probably due to a defect in the glucose-6-phosphate transport system. Biochem. Biophys. Res. Commun. 83, 1360–1364 (1978).
Gerin, I., Veiga-da-Cunha, M., Achouri, Y., Collet, J. F. & Van Schaftingen, E. Sequence of a putative glucose 6-phosphate translocase, mutated in glycogen storage disease type Ib. FEBS Lett. 419, 235–238 (1997).
Veiga-da-Cunha, M. et al. A gene on chromosome 11q23 coding for a putative glucose- 6-phosphate translocase is mutated in glycogen-storage disease types Ib and Ic. Am. J. Hum. Genet. 63, 976–983 (1998).
Annabi, B. et al. The gene for glycogen-storage disease type 1b maps to chromosome 11q23. Am. J. Hum. Genet. 62, 400–405 (1998).
Pan, C. J. et al. SLC37A1 and SLC37A2 are phosphate-linked, glucose-6-phosphate antiporters. PLoS One 6, 23157. https://doi.org/10.1371/journal.pone.0023157 (2011).
Albandar, J. M., Susin, C. & Hughes, F. J. Manifestations of systemic diseases and conditions that affect the periodontal attachment apparatus: Case definitions and diagnostic considerations. J. Clin. Periodontol. 45(Suppl 20), S171–S189 (2018).
Barrett, A. P., Buckley, D. J. & Katelaris, C. H. Oral complications in type 1B glycogen storage disease. Oral Surg. Oral Med. Oral. Pathol. 69, 174–176 (1990).
Ma, R. et al. Glycogen storage disease Ib and severe periodontal destruction: A case report. Dent. J. (Basel). 6, 53. https://doi.org/10.3390/dj6040053 (2018).
Lamont, R. J., Miller, D. P. & Bagaitkar, J. Illuminating the oral microbiome: cellular microbiology. FEMS Microbiol. Rev. 47, 045. https://doi.org/10.1093/femsre/fuad045 (2023).
Mandell, K. J., McCall, I. C. & Parkos, C. A. Involvement of the junctional adhesion molecule-1 (JAM1) homodimer interface in regulation of epithelial barrier function. J. Biol. Chem. 279, 16254–16262 (2004).
Yamaga, S. et al. Cigarette smoke extract impairs gingival epithelial barrier function. Sci. Rep. 13, 9228. https://doi.org/10.1038/s41598-023-36366-z (2023).
Takeuchi, H. et al. Porphyromonas gingivalis induces penetration of lipopolysaccharide and peptidoglycan through the gingival epithelium via degradation of junctional adhesion molecule 1. PLoS Pathog. 15, e1008124. https://doi.org/10.1371/journal.ppat.1008124 (2019).
Gerin, I., Veiga-da-Cunha, M., Achouri, Y., Collet, J. F. & Van Schaftingen, E. Sequence of a putative glucose 6-phosphate translocase, mutated in glycogen storage disease type Ib. FEBS Lett. 419(2–3), 235–238 (1997).
Torabidastgerdooei, S., Roy, M. E. & Annabi, B. A molecular signature for the G6PC3/SLC37A2/SLC37A4 interactors in glioblastoma disease progression and in the acquisition of a brain cancer stem cell phenotype. Front. Endocrinol. (Lausanne). 16(14), 1265698. https://doi.org/10.3389/fendo.2023.1265698 (2023).
Ihara, K., Nomura, A., Hikino, S., Takada, H. & Hara, T. Quantitative analysis of glucose-6-phosphate translocase gene expression in various human tissues and haematopoietic progenitor cells. J. Inherit. Metab. Dis. 23(6), 583–592 (2000).
Takeuchi, H. et al. Porphyromonas gingivalis induces penetration of lipopolysaccharide and peptidoglycan through the gingival epithelium via degradation of coxsackievirus and adenovirus receptor. Cell Microbiol. 23, e13388. https://doi.org/10.1111/cmi.13388 (2021).
Napimoga, M. H. et al. Involvement of the Wnt-β-catenin signalling antagonists, sclerostin and dickkopf-related protein 1, in chronic periodontitis. J. Clin. Periodontol. 41, 550–557. https://doi.org/10.1111/jcpe.12245 (2014).
Azab, E., Attia, A., Yaghmoor, W., Aldahlawi, S. & Youssef, A. R. The Impact of nonsurgical periodontal therapy on serum levels of dickkopf-related protein-1 in smokers and nonsmokers with periodontitis: A prospective comparative study. Clin. Cosmet. Investig. Dent. 14, 191–198. https://doi.org/10.2147/CCIDE.S362801 (2022).
Moutsopoulos, N. M. et al. Subgingival microbial communities in leukocyte adhesion deficiency and their relationship with local immunopathology. PLoS Pathog. 11, e1004698. https://doi.org/10.1371/journal.ppat.1004698.Erratum.In:PLoSPathog.11:e1004860.doi:10.1371/journal.ppat.1004860 (2015).
Ito, H. O., Hirata, M. & Koga, T. Hen egg-white lysozyme inhibits biological activities of lipopolysaccharides from periodontopathic bacteria. J. Periodontal. Res. 32, 295–299. https://doi.org/10.1111/j.1600-0765.1997.tb00537.x (1997).
Sangu, N. et al. A de novo microdeletion in a patient with inner ear abnormalities suggests that the 10q26.13 region contains the responsible gene. Hum. Genome Var. 3, 16008. https://doi.org/10.1038/hgv.2016.8 (2016).
Iwanicka-Pronicka, K. et al. Sensorineural hearing loss in GSD type I patients. A newly recognized symptomatic association of potential clinical significance and unclear pathomechanism. Int. J. Pediatr. Otorhinolaryngol. 151, 110970. https://doi.org/10.1016/j.ijporl.2021.110970 (2021).
Şanlı, M. E. et al. Assessment of auditory functions in patients with hepatic glycogen storage diseases. Turk J. Pediatr. 64, 658–670 (2022).
Skakic, A. et al. CRISPR/Cas9 genome editing of SLC37A4 gene elucidates the role of molecular markers of endoplasmic reticulum stress and apoptosis in renal involvement in glycogen storage disease type Ib. Gene 703, 17–25 (2019).
Gong, Y., Xiong, L., Li, X., Su, L. & Xiao, H. A novel mutation of WFS1 gene leading to increase ER stress and cell apoptosis is associated an autosomal dominant form of Wolfram syndrome type 1. BMC Endocr. Disord. 21(1), 76. https://doi.org/10.1186/s12902-021-00748-z (2021).
Iossa, S., Marciano, E. & Franzé, A. GJB2 gene mutations in syndromic skin diseases with sensorineural hearing loss. Curr. Genom. 12(7), 475–785 (2011).
Salapata, Y., Laskaris, G., Drogari, E., Harokopos, E. & Messaritakis, J. Oral manifestations in glycogen storage disease type 1b. J. Oral Pathol. Med. 24, 136–139 (1995).
Yamaguchi, T. et al. Inflammatory bowel disease-like colitis in glycogen storage disease type 1b. Inflamm. Bowel Dis. 7, 128–132 (2001).
Dieckgraefe, B. K., Korzenik, J. R., Husain, A. & Dieruf, L. Association of glycogen storage disease 1b and Crohn disease: results of a North American survey. Eur. J. Pediatr. 161(Suppl 1), S88-92 (2002).
Ozen, H. Glycogen storage diseases: new perspectives. World J. Gastroenterol. 13, 2541–2553 (2007).
Sobocka, M. B. et al. Cloning of the human platelet F11 receptor: a cell adhesion molecule member of the immunoglobulin superfamily involved in platelet aggregation. Blood 95, 2600–2609 (2000).
Babinska, A. et al. Two regions of the human platelet F11-receptor (F11R) are critical for platelet aggregation, potentiation and adhesion. Thromb. Haemost. 87, 712–721 (2002).
Sobocka, M. B. et al. Signaling pathways of the F11 receptor (F11R; a.k.a. JAM-1, JAM-A) in human platelets: F11R dimerization, phosphorylation and complex formation with the integrin GPIIIa. J. Recept. Signal Transduct. Res. 24, 85–105 (2004).
Prasad, R., Estrella, J., Christodoulou, J., McKellar, G. & Tchan, M. C. A Third Case of Glycogen Storage Disease IB and Giant Cell Tumour of the Mandible: A Disease Association or Iatrogenic Complication of Therapy. JIMD Rep. 42, 5–8 (2018).
Kure, S. et al. Glycogen storage disease type Ib without neutropenia. J. Pediatr. 137, 253–256 (2000).
Miltenberger-Miltenyi, G., Szonyi, L., Balogh, L., Utermann, G. & Janecke, A. R. Mutation spectrum of type I glycogen storage disease in Hungary. J. Inherit. Metab. Dis. 28, 939–944 (2005).
Jun, H. S., Weinstein, D. A., Lee, Y. M., Mansfield, B. C. & Chou, J. Y. Molecular mechanisms of neutrophil dysfunction in glycogen storage disease type Ib. Blood 123, 2843–2853 (2014).
Kaczor, M. et al. Clinical characteristics and long-term outcomes of patients with glycogen storage disease type 1b: A retrospective multi-center experience in Poland. Pediatr. Endocrinol. Diabetes. Metab. 28, 207–212 (2022).
Murakami, S. et al. Activation of adenosine-receptor-enhanced iNOS mRNA expression by gingival epithelial cells. J. Dent. Res. 81, 236–240 (2002).
Nishiguchi, A., Yoshida, H., Matsusaki, M. & Akashi, M. Rapid construction of three-dimensional multilayered tissues with endothelial tube network by the cell-accumulation technique. Adv. Mater. 23, 3506–3510 (2011).
Takeuchi, H. & Amano, A. Invasion of gingival epithelial cells by Porphyromonas gingivalis. Methods Mol. Biol. 2210, 215–224 (2021).
Matsunaga, K. et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Biol. 11, 385–396 (2009).
Acknowledgements
We sincerely appreciate the Center for Oral Science, Graduate School of Dentistry, Osaka University, for providing confocal laser microscopy technical support.
Funding
This research was supported by a Scientific Research grant (C), number 19K10085 (to H.T.), and Scientific Research grant (A), number 22H00487 (to A.A.), from the Japan Society for the Promotion of Science. The funders had no role in study design, data collection, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
K.T.: Investigation, Writing review & editing, Visualization. M.R.: Investigation, Writing review & editing. S.N.: Methodology. Y.K.: Investigation. T.T.: Investigation. SY: Resources. E.N.: Resources. A.S.: Resources. M.K.: Resources. M.M.: Methodology. A.A.: Conceptualization, Validation, Resources, Supervision, Funding acquisition, Writing review & editing. H.T.: Conceptualization, Methodology, Validation, Formal analysis, Investigation, Writing original draft, Writing review & editing, Visualization, Supervision, Funding acquisition.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no conflicts of interest related to the contents of this article.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tanigaki, K., Matsumura, R., Sasaki, N. et al. SLC37A4, gene responsible for glycogen storage disease type 1b, regulates gingival epithelial barrier function via JAM1 expression. Sci Rep 14, 24797 (2024). https://doi.org/10.1038/s41598-024-75524-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-75524-9
