
Abstract
Diabetic peripheral neuropathy is characterized by mitochondrial dysfunction, including suboptimal respiration, impaired calcium homeostasis, and accumulation of reactive oxygen species (ROS). Hyperglycemia drives excessive electron donation to the mitochondria, resulting in damaging ROS levels. In peripheral neurons, hyperglycemia also leads to the formation of advanced glycation end-products (AGEs), which, through their membrane receptor (RAGE), trigger autonomic malfunction in diabetes. However, it remains unclear whether RAGE is required to induce mitochondrial abnormalities under hyperglycemia. Thus, we first investigated mitochondrial morphology in autonomic ganglia (superior cervical ganglion, SCG) from streptozotocin (STZ)-induced diabetic mice and found an enhanced proportion of swollen mitochondria with disrupted cristae in wild-type (WT) diabetic mice, but not in RAGE knock-out (KO) diabetic mice. Next, we exposed cultured SCG neurons to high glucose and found fragmentation and an imbalanced traffic represented by an increased proportion of only anterograde moving mitochondria in neurons from WT, but such imbalance was not observed in neurons from RAGE KO mice. Treating WT neurons with the non-membrane permeable RAGE selective inhibitor FPS-ZM1, did not prevent fragmentation, although a non-significant restoring trend was observed. Furthermore, ATP production was unaffected by exposure to high glucose in neurons for WT, and remained unchanged by incubation in FPS-ZM1. Interestingly, neurons from RAGE KO mice had significantly less ATP produced in all conditions than those from WT mice. Lastly, we found RAGE protein in enriched mitochondrial fractions from nerve growth factor (NGF)-transformed PC12 (PC12NGF) cells, as well as the colocalization of RAGE with a mitochondrial marker in cultured SCG neurons. Therefore, our data support that RAGE mediates mitochondrial damage in autonomic neurons under hyperglycemic conditions.
Similar content being viewed by others


Introduction
Diabetic peripheral neuropathy (DPN) is a common disorder contributing to the damage of peripheral nerves during hyperglycemia. The impairment in the peripheral nervous system (PNS) function can manifest as a severe loss of sensory, motor, or autonomic function, depending on the type of neuropathy. Thus, symptoms of DPN can be wide-ranging, and when autonomic nerves are compromised, it leads to multiple organ system abnormalities with widespread clinical manifestations, including orthostatic hypotension, exercise intolerance, gastroparesis, diarrhea, constipation and urinary incontinence1,2,3,4,5,6. However, the most serious forms of autonomic neuropathy are those that can lead to malignant arrhythmogenesis and sudden cardiac death2. Mitochondrial health and bioenergetics play crucial roles in the development and progression of DPN7. Abnormal mitochondrial morphology, including swellings and fragmentation, is frequently observed in the tissues of patients and animal models of diabetes8,9. A correlation has been observed between mitochondrial superoxide production and fragmentation in cell lines, endothelial cells, and peripheral neurons exposed to high glucose10,11,12,13. However, the generation of reactive oxygen species (ROS) is not exclusive to pathological conditions; healthy mitochondria also produce ROS during ATP biosynthesis, primarily at complexes I and III13. The persistent infliction of oxidative damage on mitochondrial DNA, proteins, and lipids leads to further generation of ROS, which in healthy cells are normally buffered by the antioxidant machinery14,15. However, excessive ROS production under hyperglycemic conditions can exceed the capacity of the antioxidant protection systems. Reports from cardiomyocytes and central neurons have linked excessive ROS to mitochondrial permeability transition pore (mPTP) openings, mitochondrial membrane depolarization, and decreased ATP production, which precede cellular dysfunction15,17. In a rat model of type 1 diabetes, the mitochondria in dorsal root ganglion (DRG) neurons exhibited decreased activities of the mitochondrial electron transport chain (ETC) complexes I and IV, along with downregulation of the Krebs cycle enzyme citrate synthase, resulting in suboptimal rates of respiratory activity16,17,18, Additionally, mitochondrial membrane depolarization was observed, which was restored by insulin treatment, supporting a direct link between high glucose and mitochondrial dysfunction19.
Mitochondrial abnormalities have also been reported in autonomic neurons of genetic mouse models of type 1 diabetes, particularly within dystrophic neurites in the superior mesenteric and celiac ganglia. These neurites contained distinctive aggregates of subcellular organelles, predominantly characterized by large numbers of fragmented mitochondria20. We have demonstrated that high glucose-induced ROS accumulation in autonomic neurons from the superior cervical ganglion (SCG) of mice leads to the inactivation of neuronal nicotinic receptors (nAChRs) and depression of autonomic synaptic transmission21,22. Interestingly, these pathological changes in SCG function were dependent on the expression of the receptor for advanced glycation end products (RAGE)23.
Hyperglycemia increases the formation of advanced glycation end products (AGEs), which accumulate and bind to the RAGE receptor in the cell surface. This interaction initiates a cascade of intracellular signaling events through the full-length membrane-bound RAGE; however, many other isoforms of RAGE are expressed such as secretory and soluble variants lacking the membrane domain allowing it to either remain in the cytosol or to be secreted extracellularly24. Substantial evidence supports the notion that ROS production and accumulation under hyperglycemic conditions are linked to signalling through RAGE23,25,26,27,28, potentially damaging mitochondria further and exacerbating ROS production. A direct connection between RAGE and mitochondrial function was established in cancer cells where a cytosolic RAGE isoform was found to translocate to the intermembrane space upon phosphorylation, interacting with the ETC complex I to promote ATP production and cell proliferation29. This raises the question of whether pathological RAGE expression in hyperglycemic conditions could affect mitochondrial function in neurons.
Here, we investigated whether changes in mitochondrial structure, dynamics, and function induced by high glucose in vitro or hyperglycemia in vivo were linked to RAGE expression. We demonstrated that the absence of RAGE expression prevented abnormal mitochondrial morphology and dynamics induced by high glucose. SCG neurons from hyperglycemic RAGE KO mice conserved normal mitochondrial morphology and ultrastructure comparable to those observed in neurons from normoglycemic WT mice. Contrarily, high glucose exposure in RAGE-expressing neurons resulted in a mitochondrial fragmentation phenotype and an imbalance in the retrograde versus anterograde mitochondrial transport. Moreover, we detected RAGE protein in mitochondrial-enriched fractions from nerve growth factor (NGF)-transformed PC12 (PC12NGF) cells. Additionally, the colocalization of RAGE with mitochondrial markers in SCG neurons suggests that RAGE may play a direct role in mitochondrial function. Our findings reveal, for the first time, a direct link between RAGE signalling and mitochondrial dysfunction in the pathogenesis of autonomic neuropathy in diabetes.
Methods
Mice. A colony of RAGE knock-out (RAGE KO) mice on a C57BL/6 background was maintained by breeding heterozygous mice as previously described23,30,31,32,33. Heterozygous mice were generated by back-crossing RAGE KO (homozygous) mice34 with C57BL/6 wild-type (WT) mice. All experiments were based on superior cervical ganglia (SCG) from homozygous (RAGE KO) mice and C57BL/6 (wild type). Heterozygous mice generated from our breeding strategy were solely used as breeding animals and were not used for experimental purposes in this study. Mice were genotyped using genomic DNA and polymerase chain reaction as previously described23. All in vitro experiments were done with neonatal pups (P0–P5) and young adult (4–6 weeks-old) mice were used for streptozotocin (STZ)-induction of hyperglycemia. For STZ treatment, we used a modified version of the JAX Mice (Bar Harbor, Maine, USA) STZ protocol, in which mice receive daily i.p. injections of 50 mg STZ/kg body weight for three consecutive days; age-matched controls receive citrate buffer-only injections. Isoflurane-anesthetized mice were euthanized 12 weeks post injections by carbon dioxide (CO2), and blood glucose measurements were obtained with the help of a CONTOUR ® Glucose Meter (Bayer Inc., Toronto, ON, Canada); animals with blood glucose levels > 15 mM glucose were considered hyperglycemic21,30,32.
This work was approved by the University of Saskatchewan Animal Research Ethics Board (AUP 20090082) and adhered to both the Canadian Council on Animal Care guidelines for humane animal use and the ARRIVE guidelines35.
Electron microscopy analysis
Mice were transcardially perfused with phosphate-buffered saline (PBS) followed by 4% paraformaldehyd (PFA) and 0.25% glutaraldehyde in 0.1 M phosphate buffer (PB; pH 7.4) as previously described36. Next, dissected SCGs were postfixed overnight at 4 °C in the same fixative. Then, the tissue was incubated in blocked and fixed with 1% osmium tetroxide in 0.1 M Phosphate buffer (PB) for contrast. Next, the tissue was transferred to 1% uranyl acetate in 0.1 M PB (pH 6.0) and then embedded in epoxy Durcupan resin. Epoxy blocks were sectioned at 70 nm and stained with 2% uranyl acetate and lead citrate. Images were acquired with a Zeiss EM 109 T transmission electron microscope. Mitochondria within the neuronal cell bodies, which were identified by nuclear organization, were imaged and counted in 3000× magnification and 12,000× magnification.
Primary SCG cultures
Superior cervical ganglion (SCG) neurons were cultured from neonatal (P1-P5) mice as previously described21,22. Briefly, ganglia were removed under sterile conditions and enzymatically dissociated at 37 °C in Hanks balanced salt solution (HBSS) containing trypsin (180–200 U/ml; Worthington, Freehold, NJ, USA) and buffered with HEPES (pH 7.4). The resulting cell suspension was washed twice in serum-containing Leibovitz’s L-15 medium to inactivate the trypsin and plated on laminin-coated glass bottom petri dishes (35 mm) made in-house. The neurons were grown in media consisting of L-15 supplemented with vitamins, cofactors, penicillin–streptomycin, 5 mM glucose, 5% rat erum and NGF (10 ng/ml). Cultures were maintained at 37 °C in a humidified atmosphere of 95% air- % CO2 nd fed every 4 days with growth media. To eliminate non-neuronal cells, cultures were treated with cytosine arabinoside (10 µM; Sigma, St. Louis, MO, USA) from days 2 to 4. Cultured neurons were exposed to culture media containing control glucose (5 mM) or high glucose (25 mM) for 7 to 10 days before data collection21,23,30,31.
Mitochondrial labelling and live imaging
To visualize mitochondria within neurites, we used two methods: (1) Transfection of cultured neurons with a pCDNA3 vector containing the enhanced green fluorescent protein (EGFP) fused to a mitochondrial signal peptide from the human cytochrome C (MITO-EGFP)37 using Lipofectamin 2000 (Invitrogen). To monitor and quantify mitochondrial movement along neurites, we generated time-lapse imaging at a 100 μm distance from the cell body and performed kymograph analysis of MITO-EGFP movement as previously described38,39. Briefly, transfected neurons expressing MITO-EGFP were visualized under an Axio Observer inverted epifluorescent microscope equipped with an AxioCam HR camera (Zeiss) and driven by AxioVision 4.8.2 software (Zeiss). The excitation wavelength used was 450–480 nm generated with a Colibri 2.0 LED illumination system (Zeiss), and the emission wavelength collected was 510–550 nm. Cultures were continuously viewed under a 63x objective (1.4 numerical aperture) Plan Neofluor oil-immersion objectives (Zeiss) at 37oC using a combined heating stage and 5% CO2 chamber (Harvard Apparatus). Cells were imaged 24–48 h after transfection for 15 min. Particles moving from the cell body towards the end of the neurite were considered anterograde, and from the ends towards the cell bodies were considered retrograde. We generated kymographs and extracted averaged speed, distance, and directionality for analysis. Mitochondrial morphology was assessed by measuring their length and coverage (density) in a 50 μm section of a given neurite. (2) In some experiments, we labelled mitochondria in live cultured SCG neurons using MitoTracker™ Green FM (Thermo Fisher Scientific). Briefly and as recommended by the supplier, MitoTracker was diluted in anhydrous DMSO (Sigma), and cells were incubated at 37oC for 45 min at a final concentration of 200 nM. After washing with fresh 1x PBS, images were captured and processed using the same imaging system as in1.
ATP determination in cultured neurons
To determine ATP production in cultured SCG neurons, we used an ATP Determination Kit (Molecular Probes, Thermo Fisher Scientific). Measurements were carried out as recommended by the supplier. Briefly, we used a Pierce ™ White Opaque 96-well plate for both the standard curve determination and sample analysis. The standard curve was generated for ATP concentrations of 0 to 600 nM. To determine ATP production in neuronal samples, neurons were first lysed by incubating for 20 min at room temperature (RT) in Cell lysis buffer (in mM: 10 Tris pH 7.5, 100 NaCl, 1 EDTA, and 0.2% Triton X-100). Next, a 10 µL aliquot of the lysate was combined with 90 µL of the standard reaction solution. The luminescent reaction was measured using a GloMax Microplate Luminometer (Promega Corporation, Wisconsin, USA). ATP production was quantified in cultured SCG neurons maintained under control or high glucose conditions. Cultures were generated from RAGE KO and WT mice, and in some cases, neurons from WT mice were incubated with the RAGE inhibitor FPS-ZM1 (10 µM) for 72 h ours30,40 before ATP measurements. Each plate containing cultured neurons was read three times (replicas), and those replicas were averaged per plate. In each run, a blank was also included and subtracted from each experimental value. To determine the mitochondrial ATP production, we subtracted the average ATP generated in WT and RAGE KO cultures incubated with sodium cyanide (1.5 mM, NaCN) for 15 min at 37 °C41,42. Lastly, the mitochondrial ATP production was normalized to the number of cells per area.
Transformation of PC12 cells (PC12NGF by NGF treatment
Adherent PC12 cells (ATCC® CRL-1721.1™) were cultured in F-12 K growth media (2.5% fetal bovine serum, 15% horse serum; PC12 GM). After establishing the cell line in T75 flasks (15mL PC12 GM per flask, which was changed every 5–6 days and passed at 80% confluency), we used a modified version of the previously described protocol43 for the NGF-mediated transformation of PC12. Briefly, we used 50ng/mL NGF (Alomone Labs) for 9 days. Transformation of PC12 cells was confirmed by Western blotting. PC12NGF cells were maintained in NGF-containing media containing either the control condition (5 mM glucose) or high glucose (25 mM) for 10 days. Next, PC12NGF cells were collected for mitochondrial isolation and Western blotting.
Protein sample Preparation and Immunoblotting
PC12NGF cell cultures were maintained for 1–2 weeks in control and high-glucose conditions. For whole cell extracts, the cultures were lysed using a 1% Nonidet P-40 (NP-40) lysis buffer with 1x Halt™ Protease and Phosphatase inhibitor cocktail (ThemoFisher Scientific). For subcellular fractionation, cell cultures were processed using a Mitochondrial Isolation Kit for Cultured Cells (ThemoFisher Scientific). The total protein concentration in each of the samples/fractions was quantified using a BCA protein assay kit (ThemoFisher Scientific). Equal amounts of protein were loaded per group, separated on a 4–15% Tris-glycine polyacrylamide precast gel (TGX, Bio-Rad Laboratories, Hercules, CA, USA) and electroblotted onto a 0.2 μm Polyvinylidene Fluoride (PVDF) membrane (Bio-Rad Laboratories, Hercules, CA, USA). Membranes were blocked with 5% Bovine Serum Albumin (BSA) for 1 h at room temperature and incubated overnight at 4 °C with the following primary antibodies diluted in 1% BSA: rabbit-anti Neurofilament-Light Chain (1:1000; NF-L; Abcam), rabbit-anti Microtubule-Associated Protein-2 (1:1000; MAP2; Abcam), rabbit anti-RAGE (1:1000; Abcam), rabbit anti-VDAC (1:1000, Allomone), mouse anti-Tubulin (1:5000, Abcam); followed by horseradish peroxidase (HRP)-conjugated goat anti-rabbit or goat anti-mouse secondary antibodies (1:5000; Bio-Rad Laboratories, Hercules, CA, USA) diluted in 1% BSA. Protein signals were visualized using enhanced chemiluminescence reagents (Bio-Rad Laboratories, Hercules, CA, USA) and quantified by densitometry using ImageJ software (NIH, Bethesda, MD, USA).
Immunocytochemistry and confocal imaging
Primary SCG cultures were briefly washed with warmed 1x PBS and fixed for 15 min with 4% paraformaldehyde (Thermo Fisher Scientific) at room temperature (RT), followed by 3 x washed with PBS and blocked with 5% horse serum in 1% BSA for 1 h. Next, cultures were processed for sequential immunostaining. First, cultures were incubated overnight at 4 °C with rabbit anti-RAGE antibody (1:100, Abcam), followed by incubation with mouse Alexa fluor 647 rabbit anti-ATP synthase subunit beta (ATPB; 1:100 Abcam) and an Alexa fluor 488 donkey anti-rabbit antibody (1:100; Thermo Fisher Scientific) for one hour at RT, washed 3 x with PBS and covered with ProLong Gold Antifade medium (Thermo Fisher Scientific). All antibodies were diluted in 1% BSA. For control experiments, we used the same procedures omitting the primary antibodies (anti-RAGE and anti-ATPB) and confirmed the lack of detectable fluorescence signal after staining with Alexa fluor 488 antibody.
The cultures were imaged with a Nikon Ti2E spinning disk confocal microscope with a 60X oil immersion objective (Nikon Canada Inc., Canada). The co-localization of RAGE and ATPB fluorescence was determined by Pearson’s and Mander’s coefficients. To calculate the Person’s coefficient and the Mander’s coefficients, we used the Just Another Colocalization plugin (JACOP) through the Image J software (NIH; Bethesda, MD, USA). To differentiate between “background” and “foreground” we calculated the threshold in each of the channels using Otsu’s method.
Statistics
To determine the statistical significance between normal and abnormal mitochondria, we used Chi-square test (χ2) of independence. For statistical significance of changes in mitochondrial size, density and velocity, we used One-Way ANOVA, followed by post hoc comparisons using the Tukey’s or Sidak’s tests. Mitochondrial proportions were compared by the Kruskal-Wallis Test, followed by post hoc comparisons using the Dunn’s Multiple Comparisons test.
All values are reported as mean ± SEM and P < 0.05 as a criterion for significance. All statistical analyses were carried out with the help of Prism 9.0 (GraphPad Software Inc., CA, USA). To determine statistical significance in the immunoblots and immunofluorescence experiments, we compared the two means using parametric Student’s t-test or non-parametric Mann-Whitney U tests. Finally, the average Pearson’s and Mander’s coefficients in each group were compared using Student’s t-test or non-parametric Mann-Whitney U tests.
All data generated by our study is provided within the manuscript and supplementary information files.
Results
The development of abnormal mitochondrial ultrastructure in STZ-induced diabetic mice required RAGE expression
We investigated the effects of hyperglycemia on mitochondrial ultrastructure using transmission electron microscopy (TEM) of ultrathin sections of whole SCGs from control (n = 5) or STZ-induced diabetic (n = 5) WT mice (9.9 ± 0.93 and 30.65 ± 1.10 mM blood glucose, respectively. p < 0.0001) (Fig. 1A-B), and control (n = 5) or hyperglycemic (n = 4) RAGE KO mice (10.33 ± 1.7 and 27.9 ± 1.2 mM blood glucose, respectively. p < 0.0001) (Fig. 1C-D).

Hyperglycemia induces changes in mitochondrial ultrastructure in WT but not in RAGE KO mice. A-B, Transmission electron microscopy (TEM) images were obtained from the SCG of WT (A-B) and RAGE KO (C-D) mice. Mice were either control (CTL, A&C) and STZ-induced diabetic (STZ; B&D). Insets show the magnified area of interest outlined in red. N: nucleus; Cyt: cytoplasm; and M: plasma membrane. Scale bars: 1 μm (A-B), 2 μm (C-D), and for the inset panels 0.2 and 0.4 μm WT and RAGE KO, respectively. (N = 5 for WT CTL, WT STZ and RAGE KO CTL; and N = 4 for RAGE KO STZ).
The SGC neurons from control WT mice exhibited approximately 80% of mitochondria with “normal” ultrastructure, including visible inner and outer membranes and a regular crista arrangement (Fig. 2A-B). In contrast, approximately 50% of mitochondria from STZ-induced diabetic WT mice displayed significant changes in ultrastructure, characterized by swollen and vacuolated mitochondria and abnormal membrane cristae with disorganized and ruptured inner membranes (Fig. 2A-B). Interestingly, SCG neurons from RAGE KO mice in control conditions exhibited fewer abnormal mitochondria than control WT mice (Fig. 2A-B). Moreover, the ultrathin SCG sections from STZ-induced diabetic RAGE KO mice exhibited a significantly higher proportion of normal-looking mitochondria, characterized by the presence of defined inner and outer membranes and organized cristae (Fig. 1B), when compared with hyperglycemic WT neurons.

The SCG of STZ-induced diabetic WT mice had an increased proportion of abnormal mitochondria with respect to RAGE KO mice. A, Quantification of the average proportion of normal to abnormal mitochondria from TEM images of SCG slices from WT and RAGE KO mice, control (CTL) and STZ-induced diabetic (STZ), Chi-square test (χ2) of independence. B, Representative images depicting examples of normal vs. abnormal mitochondria. Scale bar = 500 nm. N = 5 for WT CTL, WT STZ and RAGE KO CTL; and N = 4 for RAGE KO STZ.
RAGE is required for high glucose-dependent mitochondrial fragmentation
Exposure to high glucose in vitro has been shown to induce mitochondrial fragmentation in cultured coronary endothelial cells, sensory neurons, and sympathetic neurites10,11,20,44. We next investigated whether RAGE expression played a role in mitochondrial dynamics and size during high glucose conditions. STZ-induced diabetic mice commonly experience levels of hyperglycemia above 25 mM, as we showed previously30,32 and in the current study. Therefore, to replicate glucose levels in culture consistent with what mice experience in vivo, SGC were maintained in 5 mM glucose as control or 25 mM glucose as high glucose condition. We then quantified the length and dynamics of mitochondria in Mito-eGFP transfected SCG neurons from WT and RAGE KO mice primary cultures, maintained in either control (5 mM glucose) or high glucose (25 mM glucose) media for one week21,23. Mitochondria were considered “mobile” when moving away or towards the cell body, and “stationary” when they showed no directional movement during image acquisition (Fig. 3 A). Interestingly, neurites from WT cultures maintained in high glucose exhibited a significant reduction in the size of stationary mitochondria, while the size of mobile mitochondria remained unchanged (Fig. 3B). Contrarily, the size of both mobile and stationary mitochondria in SCG neurons from RAGE KO mice in high glucose remained unchanged when compared with control (Fig. 3 C).
To investigate whether membrane-bound RAGE was solely responsible for this effect, instead of the less known cytoplasmic RAGE isoforms24, we repeated our experiment in neurons from WT mice under control and high glucose conditions in the presence of the selective and non-permeable RAGE inhibitor (RI) FPS-ZM140. To observe live mitochondria, we used MitoTracker Green and quantified mitochondrial size by measuring the average length of the organelles in a 50 μm neurite section. Consistent with our findings in Fig. 3, maintaining cultured SCG neurons in high-glucose-containing media resulted in mitochondrial fragmentation in wild-type (WT) mice but not in those from RAGE knockout (KO) mice (Fig. 4A-B). Interestingly, and unlike our findings from RAGE KO neurons, incubating neurons for with the RAGE inhibitor did not prevent mitochondrial fragmentation (Fig. 4A-B).

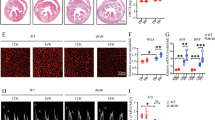
High glucose treatment in vitro caused a reduction in the size of stationary mitochondria in SCG neurons from WT but not from RAGE KO mice. A, Representative examples of epifluorescence images captured from neurites of SCG neurons expressing Mito-eGFP from either WT or RAGE KO mice exposed to control (CTL) or high glucose (HG) conditions. Images are oriented with the cell body to the left and the neurite tip to the right. Scale bar = 20 μm. B-C, Quantification of the mitochondrial size from mobile and stationary organelles in SCG cultures from WT (B) and RAGE KO (C) mice. Means were statistically compared by One-Way ANOVA; *** p < 0.001. (N = 134 WT CTL, 156 WT HG, 70 RAGE KO CTL, and 161 RAGE KO HG).

Mitochondrial fragmentation in high glucose was not prevented by the selective RAGE inhibitor FPS-ZM1. A, Representative examples of epifluorescence images captured from neurites of SCG neurons labeled with MitoTracker Green in either WT or RAGE KO mice, or in WT mice incubated with the selective RAGE inhibitor (RI) FPS-ZM1 for 72 h before measurement. All groups were maintained in control (CTL) or high glucose (HG) conditions. Scale bar = 10 μm. B, Quantification of the mitochondrial size in SCG cultures from WT and RAGE KO mice. Means were statistically compared using one-way ANOVA, F (5, 1908) = 13.87; *** p < 0.0001; **** p < 0.0001. (N = 353 WT CTL, 332 WT CTL + RI, 359 WT HG, 282 WT HG + RI, 297 RAGE KO CTL, 413 RAGE KO HG).
High glucose-induced changes in mitochondrial trafficking that required RAGE signalling
To investigate the effect of high glucose on mitochondrial trafficking, we generated kymographs from live-cell imaging of cultured WT and RAGE KO neurons expressing fluorescent mitochondria (Mito-eGFP). Notably, SCG neurons from WT mice (Fig. 5 A) maintained in high glucose displayed an increase in the proportion of anterograde moving mitochondria, while retrograde and stationary mitochondrial proportion remained unchanged when compared to WT controls (Fig. 5B). Moreover, we observed a significant increase in the density of anterograde moving mitochondria in high glucose but not of retrograde, suggesting an imbalance in mitochondrial transport in high glucose conditions (Fig. 5 C). Mitochondrial velocities in WT neurons under control or high glucose conditions remained unchanged (Fig. 5D). On the other hand, SCG neurons from RAGE KO mice (Fig. 5E), showed no change in the proportion of anterograde, retrograde, and stationary mitochondria (Fig. 5 F). However, we observed a significant increase in the density of both anterograde and retrograde mitochondrial in RAGE KO neurons under high glucose conditions when compared to control (Fig. 5G), suggesting high glucose increases transport in a balanced fashion in neurons lacking RAGE expression. Again, mitochondrial velocities were unchanged by exposing RAGE KO neurons to the high glucose treatment. (Fig. 5H).

RAGE interferes with retrograde mitochondrial transport in high glucose conditions. A, Kymograph analysis from movies neurites of SCG neurons expressing Mito-eGFP (CTL and HG) from WT mice. B, Quantification of the proportion of anterograde (Anter), retrograde (Retro), and stationary (Stat) mitochondria within neurites. C, Mitochondrial density per micron. D-E, Kymographs analysis and quantification of parameters as in A-C in cultured SCG neurons from RAGE KO mice. Scale bar = 20 μm. Means were statistically compared by One-Way ANOVA; * p < 0.05. (N = 64 WT CTL, 37 WT HG, 23 RAGE KO CTL, and 22 RAGE KO HG).
RAGE expression is required for mitochondrial function in SCG neurons
A mitochondrial function of RAGE has been identified in pancreatic cancer cells, in which RAGE enhances complex I activity, ATP production, and, thus, tumour cell proliferation and migration29. However, a mitochondrial role for RAGE outside cancer cells has yet to be reported. To address this, we quantified mitochondrial ATP production, followed by the detection of RAGE protein in neuronal mitochondria. Initially, we measured the intracellular levels of mitochondrial produced ATP (mATP), which we calculated as the total ATP production minus that under the oxidative metabolism blocker (NaCN) after normalizing it to the number of cells per area. The levels of non-mitochondrial ATP produced were not significantly different between the groups and were 13% for WT CTL (1.92 ± 1.41 nM/mm²), 14% for WT HG (2.53 ± 0.34 nM/mm²), 10% for RAGE CTL (0.35 ± 0.163 nM/mm²), and 9% for RAGE HG (0.32 ± 0.128 nM/mm²). Thus, we found that mATP levels in neurons from WT mice were not significantly different in high glucose with respect to control conditions, although there was a non-significant increase trend in neurons exposed to high glucose (WT HG, Fig. 6). This was also the case for cells treated with the RAGE selective inhibitor (RI) FPS-ZM1 (Fig. 6). Interestingly, in neurons from RAGE KO mice we found that although high glucose had no significant effect on mATP produced, these levels were significantly lower from those produced by wild type neurons (Fig. 6). These intriguing results suggested that RAGE expression was linked to mitochondrial ATP production.

RAGE contributes to mitochondrial ATP production in SCG neurons exposed to high glucose. Quantification of mitochondrial ATP (mATP) production in media containing control (CTL) or high glucose (HG) in cultured neurons from WT and RAGE KO mice. Neurons from WT mice were also incubated with the RAGE inhibitor (RI) FPS-ZM1 in both experimental conditions. Means were statistically compared by One-Way ANOVA– (F5,51 = 6.014); ** p < 0.001. (N = 12 WT CTL, 11 WT CTL + RI, 11 WT HG, 12 WT HG + RI, 8 RAGE CTL, 8 RAGE HG).
To further investigate the possible relationship between mitochondrial function and RAGE expression, we next investigated whether RAGE protein was associated with mitochondria in autonomic neurons. Due to limitations in obtaining sufficient mitochondrial fractions from cultured SCG neurons or isolated SCG ganglia, we used PC12NGF as a model of autonomic neurons43. First, we confirmed the transformation of PC12 by NGF, as demonstrated by the upregulation of the neuronal markers NF-L and MAP-2 (Fig. 7A-B). Next, we performed subcellular fractionations from PC12NGF for the detection of RAGE in the cytosol- and the mitochondria-enriched fractions. To identify whether RAGE protein was present and/or enriched in the mitochondrial subcellular fractions after high-glucose treatment, the RAGE cytosolic-enriched fractions were normalized to the soluble protein tubulin (RAGE/tubulin), while the RAGE mitochondrial-enriched fractions were normalized to both the mitochondrial membrane chloride channel VDAC and tubulin (RAGE/VDAC and tubulin) (Fig. 7C-E). Interestingly, we detected the presence of a lower, yet consistent, amount of RAGE protein in the mitochondrial-enriched fractions compared to the cytosolic fraction in PC12NGF cells. However, the mitochondrial RAGE fraction was not affected by glucose levels, as we did not observe any significant changes in RAGE levels when the cells were subjected to high glucose conditions with respect to control (Fig. 7C-E).

Detection of RAGE protein in mitochondria-enriched samples from PC12NGF cells. A-B, Immunoblots and quantification of the level of expression of the neuronal markers MAP-2, neurofilament light chain (NF-L) and tubulin in whole PC12 samples maintained in media without (-NGF) or with NGF added (+ NGF). C, The immunoblots show the levels of RAGE, VDAC and tubulin detected in the cytosol-enriched and mitochondria-enriched samples from PC12 cells transformed by NGF (PC12NGF) maintained in either control (CTL) or high glucose (HG) conditions. C-D, The bar graphs show mean ± SEM levels of each protein after normalization to tubulin or VDAC in cytosol-enriched and mitochondria-enriched samples, respectively. Means were statistically compared by the Mann-Whitney U test; (MAP2: t4 = 2.695; NF-L: t4 = 3.297); * p < 0.05. (N = 3 per group in A and C). For clarity, the blots shown in A and C have been cropped, and the background brightness has been adjusted (complete blots available in Supplementary Data file).
Finally, to evaluate whether the detection of RAGE protein in mitochondrial fractions from PC12NGF was relevant for mitochondria of SCG neurons, we colocalized RAGE protein and the mitochondrial marker ATPB by confocal microscopy in cultured SCG neurons from WT mice (Fig. 8A-B). The colocalization analysis revealed an association of RAGE with ATPB with a Pearson’s correlation coefficient 0.19 ± 0.018 and 0.16 ± 0.019 for control and high glucose conditions, respectively (Fig. 8 C); and for the Mander’s correlation coefficient 0.16 ± 0.015 and 0.28 ± 0.017 for control and high glucose conditions, respectively (Fig. 8D). Again, similar levels of RAGE association with the mitochondrial marker were observed in high glucose treatment compared with control (Fig. 8A-D).

Colocalization of RAGE and ATPB in the neurites of cultured SCG neurons. A-B, Representative images of immunocytochemical colocalization of RAGE and the mitochondrial marker ATPB in neurites of SCG neurons maintained in either control (CTL, A) or high glucose (HG, B). The inset image show a zoomed-in area depicting a mitochondrion and puncta RAGE staining. Scale bar = 2 μm. C-D, Bar graphs show the Pearson’s and Mandel’s coefficients obtained for the colocalization of RAGE and ATPB in the neurites of SCG neurons maintained in CTL or HG conditions. Means were statistically compared by Student’s t-test; p > 0.05. (N = 3 per group).
Discussion
Role of RAGE in mitochondrial morphology in diabetes
Mitochondrial health and function are reflected by their morphology, which can rapidly undergo changes in response to the microenvironment45,47,48,49,50. Mitochondria are highly dynamic organelles that frequently change shape and distribution; in neurons, they have a tubular basic morphology determined by a balanced frequency of fission and fusion events51. Mitochondrial fission involves the dynamin-related protein 1 (Drp1), while fusion events involve the mitofusin proteins (Mnf1 and Mnf2)52. A shift in the mitochondrial fission/fusion balance towards fragmentation has been reported in tissues of diabetic animals. In sensory neurons from the dorsal root ganglia (DRG) of diabetic rodents, there was an increase in the number and shortening of mitochondria44,53. Mitochondrial fission could worsen ischemic brain damage in hyperglycemic rats54, while inhibition of the fission protein Drp1 was beneficial in a rodent model of brain injury during diabetes, which reduced inflammation and neuronal loss, as well as microglia and astrocyte proliferation55. Consistently, cardiomyocytes maintained in high glucose media containing the mitochondrial fusion promoter M1, showed improved mitochondrial function and alleviated parameters of diabetic cardiomyopathy56. The mitochondrial fusion/fission balance is also affected by yet another component of the pathological environment of diabetes: intracellular ROS accumulation. The deleterious effects of diabetes on ROS accumulation and mitochondrial damage have been described in both the peripheral and central nervous systems22,30,57,59,60,61. Furthermore, impaired insulin secretion in pancreatic b cells of a mouse model of type 2 diabetes was linked to both ROS accumulation and mitochondrial fission due to increased Drp1:Mnf2 ratio62.
Our findings are partially in line with previous reports from other mouse models of diabetes, including STZ-induced models. It has been reported that diabetes caused a progressive (followed for up to 6 months) neurite dystrophic phenotype characterized by multivesicular autophagic bodies and small mitochondria in sympathetic neurons63,64. This is consistent with our current report of the deleterious effects of high glucose in cultured SCG neurons. However, it was also reported that those dystrophic changes were observed in the cell bodies of sympathetic ganglia such as superior mesenteric and celiac ganglia but not in those of superior cervical neurons64, which is inconsistent with our study. Unfortunately, we are not able to explain the differences between the studies, other than in the current report, we have looked at the 3-month time point in STZ-induced mice and that even though our mice were from one of the genetic backgrounds reported before (C57BL6J), they were bred in-house for many generations, which may explain genetic variability. Nevertheless, in our study, we observed differences in the SCG perikaryon that were prevented by the absence of RAGE expression in diabetic mice.
A deleterious effect of diabetes on mitochondria along sensory neurites has also been previously reported. In particular, the sciatic nerves of STZ-induced diabetic rats showed an energy deficit characterized by a decrease in the mitochondrial NAD+/NADH ratio and ROS accumulation65. Although our study focuses on the effect of diabetes on the cell bodies of SCG neurons, we emphasize that changes in mitochondrial morphology in hyperglycemia depend on RAGE expression, highlighting a novel role for RAGE signaling in mediating mitochondrial fragmentation. Whether this process occurs directly through the regulation of fusion/fission proteins or as an indirect steric effect on the mitochondrial membrane remains to be investigated.
Signalling through the RAGE axis is another piece of the puzzle contributing to oxidative stress and inflammatory signals in diabetes23,28,30,33,66,68,69. Our own work in autonomic neurons showed that SCG neurons exposed to high glucose conditions displayed RAGE upregulation and inactivation of nicotinic receptors (nAChRs). Interestingly, when SCG neurons were isolated from RAGE KO mice, these deleterious effects of high glucose were absent. We also reported consistent findings in the hippocampus of STZ-induced diabetic mice, in which RAGE expression and activation of the MAP kinase pathways correlated with the downregulation of the GluA1 subunit of AMPA receptors, impaired hippocampal long-term potentiation, and induced cognitive decline evaluated in spatial learning tasks30,32. Again, these effects were not found in RAGE KO mice or WT mice treated with the RAGE inhibitor FPS-ZM130,32. Signalling through the RAGE axis was also connected to mitochondrial dysfunction in diabetes. AGE accumulation was reported to increase mitochondrial ROS production in diabetic nephropathy28,70, and the lack of RAGE expression in high-fat diet-treated mice improved myocardial dysfunction, which was supported by increased antioxidant capacity and partial prevention of mitochondrial respiration defects, fragmentation, and mitochondrial degradation (mitophagy) in cardiomyocytes71.
Therefore, in addition to previously reported effects of RAGE in hyperglycemia affecting neuronal and mitochondrial function, our findings highlight the role of RAGE expression in the induction of swollen mitochondria with disruption of the inner membrane cristae and increased fragmentation during hyperglycemia. Using in vivo and in vitro experimental designs, we show that RAGE expression was required for the ultrastructural changes (Figs. 1 and 2) observed in the SCG of long-term diabetic mice and for the reduction in mitochondrial size (Fig. 3) observed in cultured SCG neurons exposed to short-term high glucose in vitro. Interestingly, incubating cultured SCG neurons with the impermeable RAGE inhibitor FPS-ZM1, did not prevent mitochondrial fragmentation. However, we observed a non-significant recovery trend (Fig. 4B), suggesting that membrane-bound RAGE may contribute to but was not solely responsible for mitochondrial shortening in SCG neurons under high glucose conditions. A role for membrane-bound RAGE in mitochondrial fission and mitophagy has recently been described in cardiomyocytes72 and microglia in the rostral ventrolateral medulla (RVLM)73, respectively. Thus, these findings support an active role of RAGE24 contributing to the mitochondrial morphological changes during diabetes.
Role of RAGE in mitochondrial transport along neurites in diabetes
Mitochondria anterograde and retrograde transport along axons and dendrites relies on the coordinated action of kinesins and dynein molecular motor complexes74,76,77. Mitochondrial anterograde transport is critical for axon growth and delivering healthy mitochondria to axon terminals and dendrites78,80,81, while retrograde transport is involved in returning aged or damaged mitochondria to the soma for degradation by mitophagy78,82,83. Disruption in mitochondrial transport has been linked to neurodegeneration, particularly to amyotrophic lateral sclerosis (ALS), Huntington’s disease, Alzheimer’s (AD), and Parkinson’s disease (PD)84,86,87,88. While defects in either transport direction could be deleterious to the health of neurons, an increasing number of models link defects in retrograde transport with neurodegenerative diseases89. For example, disruption of the dynein-dynactin motor complex, which impairs mitochondrial retrograde transport, leads to motor neuron death and muscle denervation in mice90. In fact, in mice, mutations in the dynein heavy chain display degeneration of motor and/or sensory neurons91. Likewise, a mouse model of AD lacking the SNARE-associated protein snapin showed a loss of synapses due to defective dynein-snapin-mediated retrograde transport that was linked to mitophagosome accumulation92. Our study shows that exposure of SCG neurons from WT mice to high glucose causes a shift in the transport of mitochondria, favouring the anterograde traffic and resulting in unbalanced transport. However, under similar experimental conditions, neurons from RAGE KO mice exhibited increased transport in both directions, maintaining a balanced mitochondrial transport. Therefore, our findings suggest that RAGE expression in high glucose conditions may impair the retrograde transport of mitochondria, possibly leading to the accumulation of damaged organelles at the terminals. An underlying potential mechanism may involve the disruption of axonal transport by ROS93, which we have previously shown accumulates in SCG neurons under high glucose conditions, an event that requires RAGE expression21,22,23.
RAGE detection within the mitochondria of peripheral neurons
A direct link between RAGE and mitochondrial function has been documented in pancreatic cancer cells, in which RAGE activation by the high mobility group box 1 protein (HMGB1) leads to the phosphorylation of a cytosolic RAGE isoform by p-ERK1/2. This phosphorylation is a critical step for RAGE translocation to the mitochondrial intermembrane space, regulation of ATP production and support of tumour growth29. However, a direct role of RAGE in the mitochondria of neurons has not been described. Much less is known about the physiological and pathological role of intracellular RAGE isoforms in the nervous system. In sensory neurons from the DRG, we showed that exposure of these neurons in vitro to lipopolysaccharides (LPS) upregulated full-length membrane-bound RAGE and downregulated a soluble (sRAGE) splice variant (encoded by MmusRAGEv4). These changes in RAGE isoform expression were linked to the sensitization of DRG neurons by inflammatory signals33.
Our findings demonstrate a link between RAGE expression and ATP production in SCG neurons. First, high glucose did not significantly affect mATP production in neurons from WT mice, although a consistent increase trend was observed. This effect was also consistently observed in neurons from WT mice treated with FPS-ZM1; suggesting that the inhibition of membrane-bound RAGE may not be the only contributing factor. These results contrast with ATP production in sensory neurons isolated from adult STZ-induced diabetic rats (3 months), which exhibited an energy deficit due to impaired glycolysis94. The difference may reside in the age of the animals as well as the pre-treatment conditions, since SCG neurons were obtained from healthy P0-P5 pups. Second, in neurons from RAGE KO mice, high glucose had no effect on mATP production but those ATP levels were significantly lower than the ones recorded in neurons from WT mice (Fig. 6). The latter is consistent with our previous report of reduced basal level of ROS in SCG neurons from RAGE KO mice23. These findings suggest a physiological role of RAGE in neuronal metabolism. Third, RAGE was directly associated with mitochondria, since it was detected in the mitochondria-enriched fraction from PC12NGF cells (Fig. 7) and colocalized with a mitochondrial marker in SCG neurons (Fig. 8). Interestingly, our study using both protein analysis and immunocytochemistry, revealed that the colocalization of RAGE with the mitochondrial markers doesn’t seem to be driven by high glucose. However, we can not dismiss the fact that RAGE posttranslational modifications (such as RAGE phosphorylation), rather than its total level, could be modulated/affected by high glucose conditions. We previously reported that high glucose induced the upregulation of membrane RAGE in cultured SCG neurons23, which was not detected in the current study because the mitochondria extraction kit used to generate mitochondria-enriched fractions from PC12NGF cells discarded the subcellular fraction containing the plasma membrane, and thus, membrane-bound RAGE was excluded from the current protein analysis. Thus, our current results do not include membrane-bound RAGE.
In summary, our data suggest that RAGE plays a more complex role in peripheral neurons than previously thought. In the context of long-term diabetes, RAGE expression was associated with mitochondrial damage, whereas the absence of RAGE was protective for mitochondrial ultrastructure. In short-term in vitro experiments, RAGE expression was linked to mitochondrial fragmentation and unbalanced transport along neurites. The lack of significant changes in mATP production under the same experimental conditions suggests that changes in dynamics may serve as compensatory adjustments to cope with energy production in the short term. Our findings also reveal that the metabolic effects of RAGE observed in peripheral neurons may be mediated not only by plasma membrane-bound RAGE but also by intracellular isoforms. We found RAGE to be associated with mitochondria, and the inhibition of RAGE by the non-permeable FPS-ZM1 compound did not prevent/revert the effects of high glucose, supporting the need for mitochondria-associated RAGE in translating many of the neuronal mitochondrial changes induced by hyperglycemia that contribute to the development of peripheral diabetic neuropathy.
Data availability
All data generated by our study is provided within the manuscript and supplementary information files.
References
Maser, R., Mitchell, B., Vinik, A. & Freeman, R. The association between cardiovascular autonomic neuropathy and mortality in individuals with diabetes: a meta-analysis. Diabetes Care. 26 (6), 1895–1901 (2003).
Vinik, A. I., Freeman, R. & Erbas, T. Diabetic autonomic neuropathy. Semin Neurol. 23 (4), 365–372 (2003).
Tang, M., Donaghue, K. C., Cho, Y. H. & Craig, M. E. Autonomic neuropathy in young people with type 1 diabetes: A systematic review. Pediatr. Diabetes. 14 (4), 239–248 (2013).
Pop-Busui, R. Cardiac autonomic neuropathy in diabetes: A clinical perspective. Diabetes Care. 33 (2), 434–441 (2010).
Kuehl, M. & Stevens, M. J. Cardiovascular autonomic neuropathies as complications of diabetes mellitus. Nat Rev Endocrinol. 8(7):405–16. http://www.ncbi.nlm.nih.gov/pubmed/22371159 (2012).
Marcovecchio, M. L., Tossavainen, P. H. & Dunger, D. B. Prevention and treatment of microvascular disease in childhood type 1 diabetes. Br Med Bull. 94 (1):145–64. https://pubmed.ncbi.nlm.nih.gov/20053672/ (2010).
Eid, S. A. et al. New perspectives in diabetic neuropathy. 111 (17):2623–41. http://www.cell.com/article/S0896627323003458/fulltext (2023).
Vanhorebeek, I. et al. Protection of hepatocyte mitochondrial ultrastructure and function by strict blood glucose control with insulin in critically ill patients. Lancet 365 (9453), 53–59 (2005).
Kelley, D. E., He, J., Menshikova, E. V. & Ritov, V. B. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 51 (10), 2944–2950 (2002).
Makino, A., Scott, B. T. & Dillmann, W. H. Mitochondrial fragmentation and superoxide anion production in coronary endothelial cells from a mouse model of type 1 diabetes. Diabetologia. 53 (8):1783–94. http://www.ncbi.nlm.nih.gov/pubmed/20461356 (2010).
Yu, T., Robotham, J. L., Yoon, Y. & S A [Internet]. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc Natl Acad Sci U. 103 (8):2653–8. http://www.ncbi.nlm.nih.gov/pubmed/16477035 (2006).
Vincent, A. M., Brownlee, M. & Russell, J. W. Oxidative stress and programmed cell death in diabetic neuropathy. Ann. N Y Acad. Sci. 959, 368–383 (2002).
Edwards, J. L. et al. Diabetes regulates mitochondrial biogenesis and fission in mouse neurons. Diabetologia 53 (1), 160–169 (2010).
Nakamura, T., Oh, C., Zhang & Lipton, X. SA. Protein S-nitrosylation and oxidation contribute to protein misfolding in neurodegeneration. Free Radic Biol Med. 172:562–77. https://pubmed.ncbi.nlm.nih.gov/34224817/ (2021)
Sarkar, A., Rasheed, M. S. U. & Singh, M. P. Redox Modulation of Mitochondrial Proteins in the Neurotoxicant Models of Parkinson’s Disease. Antioxid Redox Signal. 38 (10–12):824–52. https://www.liebertpub.com/doi/10.1089/ars.2022.0106. (2023).
Eftekharpour, E. & Fernyhough, P. Oxidative stress and mitochondrial dysfunction associated with peripheral neuropathy in type 1 diabetes. Antioxid. Redox Signal. 37 (7–9), 578–596 (2022).
Fernyhough, P., Roy Chowdhury, S. K. & Schmidt, R. E. Mitochondrial stress and the pathogenesis of diabetic neuropathy. Expert Rev Endocrinol Metab. 5 (1):39–49. http://www.ncbi.nlm.nih.gov/pubmed/20729997 (2010).
Chowdhury, S. et al. Mitochondrial respiratory chain dysfunction in dorsal root ganglia of streptozotocin-induced diabetic rats and its correction by insulin treatment. Diabetes 1082–91. http://diabetes.diabetesjournals.org/content/59/4/1082.short (2010).
Fernyhough, P., Huang, T. J. & Verkhratsky, A. Mechanism of mitochondrial dysfunction in diabetic sensory neuropathy. J. Peripher Nerv. Syst. 8 (4), 227–235 (2003).
Schmidt, R. E. Autonomic Neuropathy in Experimental Models of Diabetes Mellitus 1st edn, 126 579–602 Handbook of Clinical Neurology. (2014).
Campanucci, V., Krishnaswamy, A. & Cooper, E. Diabetes depresses synaptic transmission in sympathetic ganglia by inactivating nAChRs through a conserved intracellular cysteine residue. Neuron 66 (6), 827–834. https://doi.org/10.1016/j.neuron.2010.06.010 (2010).
Campanucci, V. A., Krishnaswamy, A. & Cooper, E. Mitochondrial reactive oxygen species inactivate neuronal nicotinic acetylcholine receptors and induce long-term depression of fast nicotinic synaptic transmission. J. Neurosci. 28 (7), 1733–1744. https://doi.org/10.1523/JNEUROSCI.5130-07.2008 (2008).
Chandna, A. R. et al. RAGE mediates the inactivation of nAChRs in sympathetic neurons under high glucose conditions. Eur. J. Neurosci. 41 (3), 341–351 (2015).
López-Díez, R. et al. Complex Tissue-Specific Patterns and Distribution of Multiple RAGE Splice Variants in Different Mammals. Genome Biol Evol. 5 (12):2420–35.http://www.ncbi.nlm.nih.gov/pubmed/24273313 (2013).
Vincent, A. M., Russell, J. W., Low, P. & Feldman, E. L. Oxidative stress in the pathogenesis of diabetic neuropathy. Endocr. Rev. 25 (4), 612–628 (2004).
Li, J. & Schmidt, A. M. Characterization and Functional Analysis of the Promoter of RAGE, the Receptor for Advanced Glycation End Products. Journal of Biological Chemistry. 272 (26):16498–506. http://www.jbc.org/cgi/doi/10.1074/jbc.272.26.16498. (1997).
Tomlinson, D. R. & Gardiner, N. J. Glucose neurotoxicity. Nat Rev Neurosci 9 (1):36–45.http://www.nature.com/doifinder/10.1038/nrn2294
Coughlan, M. T. et al. RAGE-Induced Cytosolic ROS Promote Mitochondrial Superoxide Generation in Diabetes. J Am Soc Nephrol. 20 (4):742. /pmc/articles/PMC2663823/. (2009).
Kang, R. et al. The HMGB1/RAGE inflammatory pathway promotes pancreatic tumor growth by regulating mitochondrial bioenergetics. Oncogene 30 (5):567–77. https://pubmed.ncbi.nlm.nih.gov/23318458/ (2014).
Momeni, Z. et al. RAGE signaling is required for AMPA receptor dysfunction in the hippocampus of hyperglycemic mice. Physiol. Behav. 229, 113255. https://doi.org/10.1016/j.physbeh.2020.113255 (2021).
Lam, D. et al. RAGE-dependent potentiation of TRPV1 currents in sensory neurons exposed to high glucose. Yagihashi S, editor. PLoS One. 13 (2):e0193312. http://www.ncbi.nlm.nih.gov/pubmed/29474476 (2018).
Momeni, Z. et al. Hyperglycemia induces RAGE-dependent hippocampal Spatial memory impairments. Physiol. Behav. 229, 113255. https://doi.org/10.1016/j.physbeh.2020.113255 (2021).
Nair, M. et al. Lipopolysaccharides induce a RAGE-mediated sensitization of sensory neurons and fluid hypersecretion in the upper airways. Sci. Rep. 11 (1), 8336. https://doi.org/10.1038/s41598-021-86069-6 (2021).
Myint, K. M. et al. RAGE control of diabetic nephropathy in a mouse model: effects of RAGE gene disruption and administration of low-molecular weight heparin. Diabetes 55 (9), 2510–2522 (2006).
Percie Du Sertid, N. et al. Reporting animal research: Explanation and elaboration for the ARRIVE guidelines 2.0. https://doi.org/10.1371/journal.pbio.3000411 (2020).
Saez, T. M. M. et al. Kinesin-1-mediated axonal transport of CB1 receptors is required for cannabinoid-dependent axonal growth and guidance. Development. 147 (8). https://pubmed.ncbi.nlm.nih.gov/32265198/ (2020).
Falzone, T. L. et al. Axonal Stress Kinase Activation and Tau Misbehavior Induced by Kinesin-1 Transport Defects. Journal of Neuroscience. 29 (18):5758–67. http://www.jneurosci.org/cgi/doi/10.1523/JNEUROSCI.0780-09.2009. (2009).
Otero, M. G. et al. Proteasome stress leads to APP axonal transport defects by promoting its amyloidogenic processing in lysosomes. J Cell Sci. 131 (11).https://pubmed.ncbi.nlm.nih.gov/29724915/ (2018).
Otero, M. G. et al. Fast axonal transport of the proteasome complex depends on membrane interaction and molecular motor function. J Cell Sci. 127 (7):1537–49. http://www.ncbi.nlm.nih.gov/pubmed/24522182 (2014).
Deane, R. et al. A multimodal RAGE-specific inhibitor reduces amyloid β-mediated brain disorder in a mouse model of alzheimer disease. J. Clin. Invest. 122 (4), 1377–1392 (2012).
Dalal, P. & Romani, A. Adenosine triphosphate depletion by cyanide results in a Na(+)-dependent Mg(2+) extrusion from liver cells. Metabolism. 59 (11):1663–71. https://pubmed.ncbi.nlm.nih.gov/20494376/ (2010).
Kilbride, S. M., Telford, J. E., Davey, G. P. & Complex I Controls Mitochondrial and Plasma Membrane Potentials in Nerve Terminals. Neurochem Res. 46 (1):100–7. https://pubmed.ncbi.nlm.nih.gov/32130629/ (2021).
Das, K. P., Freudenrich, T. M., Mundy, W. R. & Assessment of PC12 cell differentiation and neurite growth: A comparison of morphological and neurochemical measures. Neurotoxicol Teratol. 26 (3):397–406. https://pubmed.ncbi.nlm.nih.gov/15113601/ (2004).
Vincent, A. M. et al. Mitochondrial biogenesis and fission in axons in cell culture and animal models of diabetic neuropathy. Acta Neuropathol. 120 (4), 477–489 (2010).
Sajic, M. Mitochondrial dynamics in peripheral neuropathies. Antioxid. Redox Signal. 21 (4), 601–620 (2014).
Yoon, Y. Sharpening the scissors: mitochondrial fission with aid. Cell. Biochem. Biophys. 41 (2), 193–206 (2004).
Westermann, B. Merging mitochondria matters: cellular role and molecular machinery of mitochondrial fusion. EMBO Rep. 3 (6), 527–531 (2002).
Skulachev, V. P. Mitochondrial filaments and clusters as intracellular power-transmitting cables. Trends Biochem. Sci. 26 (1), 23–29 (2001).
Riva, A., Tandler, B., Loffredo, F., Vazquez, E. & Hoppel, C. Structural differences in two biochemically defined populations of cardiac mitochondria. Am. J. Physiol. Heart Circ. Physiol. 289 (2), H868–H872 (2005).
Hackenbrock, C. R. Ultrastructural bases for metabolically linked mechanical activity in mitochondria. I. Reversible ultrastructural changes with change in metabolic steady state in isolated liver mitochondria. J. Cell. Biol. 30 (2), 269–297 (1966).
Sesaki, H. & Jensen, R. E. Division versus fusion: Dnm1p and Fzo1p antagonistically regulate mitochondrial shape. J. Cell. Biol. 147 (4), 699–706 (1999).
Shan, Z., Fa, W. H., Tian, C. R., Yuan, C. S. & Jie, N. Mitophagy and mitochondrial dynamics in type 2 diabetes mellitus treatment. Aging 14 (6), 2902–2919 (2022).
Russell, J. W., Sullivan, K. A., Windebank, A. J., Herrmann, D. N. & Feldman, E. L. Neurons undergo apoptosis in animal and cell culture models of diabetes. Neurobiol. Dis. 6 (5), 347–363 (1999).
Liu, P., Yang, X., Niu, J. & Hei, C. Hyperglycemia aggravates ischemic brain damage via ERK1/2 activated cell autophagy and mitochondrial fission. Front Endocrinol (Lausanne). 13 . https://pubmed.ncbi.nlm.nih.gov/35992111/ (2022).
Chung, C. L. et al. Therapeutic Effect of Mitochondrial Division Inhibitor-1 (Mdivi-1) on Hyperglycemia-Exacerbated Early and Delayed Brain Injuries after Experimental Subarachnoid Hemorrhage. Int J Mol Sci. 23 (13). https://pubmed.ncbi.nlm.nih.gov/35805932/ (2022).
Ding, M. et al. Mitochondrial fusion promoter restores mitochondrial dynamics balance and ameliorates diabetic cardiomyopathy in an optic atrophy 1-dependent way. Acta Physiol (Oxf). 229 (1). Available from: https://pubmed.ncbi.nlm.nih.gov/31840416/ (2020).
Russell, J. W. et al. High glucose-induced oxidative stress and mitochondrial dysfunction in neurons. FASEB J. 16 (13), 1738–1748 (2002).
Vincent, A. M., Kato, K., McLean, L. L., Soules, M. E. & Feldman, E. L. Sensory neurons and Schwann cells respond to oxidative stress by increasing antioxidant defense mechanisms. Antioxid. Redox Signal. 11 (3), 425–438 (2009).
Vincent, A. M., McLean, L. L., Backus, C. & Feldman, E. L. Short-term hyperglycemia produces oxidative damage and apoptosis in neurons. FASEB J. 19 (6), 638–640 (2005).
Srinivasan, S., Stevens, M. & Wiley, J. W. Diabetic peripheral neuropathy: evidence for apoptosis and associated mitochondrial dysfunction. Diabetes 49 (11), 1932–1938 (2000).
Roy Chowdhury, S. et al. Depressed mitochondrial function and electron transport Complex II-mediated H2O2 production in the cortex of type 1 diabetic rodents. Mol Cell Neurosci. 90 :49–59. https://pubmed.ncbi.nlm.nih.gov/29802939/ (2018).
Cooper, I. D., Brookler, K. H., Kyriakidou, Y., Elliott, B. T. & Crofts, C. A. P. Metabolic phenotypes and step by step evolution of type 2 diabetes: A new paradigm. Biomedicines 9 (7), 800. https://doi.org/10.3390/biomedicines9070800 (2021).
Schmidt, R. E., Parvin, C. A. & Green, K. G. Synaptic ultrastructural alterations anticipate the development of neuroaxonal dystrophy in sympathetic ganglia of aged and diabetic mice. J Neuropathol Exp Neurol. 67 (12):1166–86. https://pubmed.ncbi.nlm.nih.gov/19018240/ (2008).
Schmidt, R. E., Green, K. G., Snipes, L. L. & Feng, D. Neuritic dystrophy and neuronopathy in Akita (Ins2(Akita)) diabetic mouse sympathetic ganglia. Exp Neurol. 216 (1):207–18. https://pubmed.ncbi.nlm.nih.gov/19111542/ (2009).
Obrosova, I. G. et al. An aldose reductase inhibitor reverses early diabetes-induced changes in peripheral nerve function, metabolism, and antioxidative defense. FASEB J. 16 (1):123–5. Available from: https://pubmed.ncbi.nlm.nih.gov/11709499/ (2002).
Bianchi, R., Kastrisianaki, E., Giambanco, I. & Donato, R. S100B Protein Stimulates Microglia Migration via RAGE-dependent Up-regulation of Chemokine Expression and Release. Journal of Biological Chemistry. 286 (9):7214–26. http://www.ncbi.nlm.nih.gov/pubmed/21209080 (2011).
Yan, S. F., Ramasamy, R., Naka, Y. & Schmidt, A. M. Glycation, inflammation, and RAGE: A scaffold for the macrovascular complications of diabetes and beyond. Circ. Res. 93 (12), 1159–1169 (2003).
Ramasamy, R. et al. Advanced glycation end products and RAGE: a common thread in aging, diabetes, neurodegeneration, and inflammation. Glycobiology. 15 (7):16R-28R. http://academic.oup.com/glycob/article/15/7/16R/689769/Advanced-glycation-end-products-and-RAGE-a-common (2005).
Yamamoto, H. et al. RAGE in diabetic nephropathy. Curr. Mol. Med. 7 (8), 752–757 (2007).
Higgins, G. C. & Coughlan, M. T. Mitochondrial dysfunction and mitophagy: the beginning and end to diabetic nephropathy? Br. J. Pharmacol. 171 (8), 1917–1942 (2014).
Yu, Y. et al. Advanced glycation end products receptor RAGE controls myocardial dysfunction and oxidative stress in high-fat fed mice by sustaining mitochondrial dynamics and autophagy-lysosome pathway. Free Radic Biol Med. 112 :397–410. https://pubmed.ncbi.nlm.nih.gov/28826719/ (2017).
Yu, Y. et al. Advanced glycation end products receptor RAGE controls myocardial dysfunction and oxidative stress in high-fat fed mice by sustaining mitochondrial dynamics and autophagy-lysosome pathway. Free Radic Biol. Med. 112, 397–410 (2017).
Zhang, S. et al. HMGB1/RAGE axis mediates stress-induced RVLM neuroinflammation in mice via impairing mitophagy flux in microglia. J Neuroinflammation. 17 (1):1–20. https://jneuroinflammation.biomedcentral.com/articles/10.1186/s12974-019-1673-3 (2020).
Schnapp, B. J. & Reese, T. S. Dynein is the motor for retrograde axonal transport of organelles. Proc Natl Acad Sci U S A. 86 (5):1548–52. https://pubmed.ncbi.nlm.nih.gov/2466291/ (1989).
Hollenbeck, P. J. & Saxton, W. M. The axonal transport of mitochondria. J Cell Sci. 118 (23):5411–9. https://pubmed.ncbi.nlm.nih.gov/16306220/ (2005).
Tanaka, Y. et al. Targeted disruption of mouse conventional kinesin heavy chain, kif5B, results in abnormal perinuclear clustering of mitochondria. 93 (7):1147–58. Available from: https://pubmed.ncbi.nlm.nih.gov/9657148/ (1998).
Pilling, A. D., Horiuchi, D., Lively, C. M. & Saxton, W. M. Kinesin-1 and dynein are the primary motors for fast transport of mitochondria in Drosophila motor axons. Mol. Biol. Cell. 17 (4), 2057–2068 (2006).
Pozo Devoto, V. M. & Falzone, T. L. Mitochondrial dynamics in Parkinson’s disease: a role for α-synuclein? Dis Model Mech. 10 (9):1075–87. http://www.ncbi.nlm.nih.gov/pubmed/28883016 (2017).
Morris, R. L. & Hollenbeck, P. J. The regulation of bidirectional mitochondrial transport is coordinated with axonal outgrowth. J Cell Sci. 104 (3):917–27. https://pubmed.ncbi.nlm.nih.gov/8314882/ (1993).
Ruthel, G. & Hollenbeck, P. J. Response of mitochondrial traffic to axon determination and differential branch growth. J Neurosci. 23 (24):8618–24. https://pubmed.ncbi.nlm.nih.gov/13679431/ (2003).
Spillane, M., Ketschek, A., Merianda, T. T., Twiss, J. L. & Gallo, G. Mitochondria coordinate sites of axon branching through localized intra-axonal protein synthesis. 5 (6):1564–75. https://pubmed.ncbi.nlm.nih.gov/24332852/ (2013).
Cai, Q., Zakaria, H. M. & Sheng, Z. H. Long time-lapse imaging reveals unique features of PARK2/Parkin-mediated mitophagy in mature cortical neurons. Autophagy 8 (6), 976–978 (2012).
Cai, Q., Zakaria, H. M., Simone, A. & Sheng, Z. H. Spatial parkin translocation and degradation of damaged mitochondria via mitophagy in live cortical neurons. Curr. Biol. 22 (6), 545–552 (2012).
Perlson, E., Maday, S., Fu, M., meng, Moughamian, A. J. & Holzbaur, E. L. F. Retrograde Axonal Transport: Pathways To Cell Death? Trends Neurosci. 33 (7):335. /pmc/articles/PMC2902719/. (2010).
Calkins, M. J. & Reddy, P. H. Amyloid beta impairs mitochondrial anterograde transport and degenerates synapses in Alzheimer’s disease neurons. Biochim Biophys Acta. 1812 (4):507–13. https://pubmed.ncbi.nlm.nih.gov/21241801/ (2011).
Krzystek, T. J. et al. Differential mitochondrial roles for α-synuclein in DRP1-dependent fission and PINK1/Parkin-mediated oxidation. Cell. Death Dis. 12 (9), 796. https://doi.org/10.1038/s41419-021-04046-3 (2021).
Pozo Devoto, V. M. & Falzone, T. L. Mitochondrial dynamics in parkinson’s disease: A role for α-synuclein? DMM Disease Models Mech. 10 (9), 1075–1087 (2017).
Holubiec, M. I. et al. Mitochondrial vulnerability to oxidation in human brain organoids modelling alzheimer’s disease. Free Radic Biol. Med. 208, 394–401 (2023).
Bhatti, J. S. et al. Targeting dynamin-related protein-1 as a potential therapeutic approach for mitochondrial dysfunction in Alzheimer’s disease. Biochim Biophys Acta Mol Basis Dis. 1869 (7):166798. https://pubmed.ncbi.nlm.nih.gov/37392948/ (2023).
LaMonte, B. H. et al. Disruption of dynein/dynactin inhibits axonal transport in motor neurons causing late-onset progressive degeneration. 34 (5):715–27. https://pubmed.ncbi.nlm.nih.gov/12062019/ (2002).
Chen, X. J. et al. Proprioceptive sensory neuropathy in mice with a mutation in the cytoplasmic Dynein heavy chain 1 gene. J Neurosci. 27 (52):14515–24. https://pubmed.ncbi.nlm.nih.gov/18160659/ (2007).
Han, S., Jeong, Y. Y., Sheshadri, P., Su, X. & Cai, Q. Mitophagy regulates integrity of mitochondria at synapses and is critical for synaptic maintenance. EMBO Rep. 21 (9). https://pubmed.ncbi.nlm.nih.gov/32627320/ (2020).
Ireland, S. C., Huang, H., Zhang, J., Li, J. & Wang, Y. Hydrogen peroxide induces Arl1 degradation and impairs Golgi-mediated trafficking. Mol Biol Cell. 31 (17):1931–42. https://pubmed.ncbi.nlm.nih.gov/32583744/ (2020).
Aghanoori, M. R. et al. Sensory neurons derived from diabetic rats exhibit deficits in functional Glycolysis and ATP that are ameliorated by IGF-1. Mol. Metab. 49, 101191 (2021).
Funding
This work was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC; RGPIN-2015-03958; RGPIN-2021-04252) to V.A.C. and the University of Saskatchewan’s International Research Partnership Fund to V.A.C. and T.L.F.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by M.G. Otero, N. Henao-Romero, T.M. Krysak, M.T. Vu-Lu, O.O. Morales Morales and Z. Momeni. Y. Yamamoto, provided access to transgenic mice and contributed to the manuscript discussion. The first draft of the manuscript was written by V.A. Campanucci. T.L. Falzone and V.A. Campanucci are the principal investigators directing the two research laboratories participating in the current study and are listed as co-senior authors. All authors read and commented on the manuscript and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
This study was performed in line with the principles of the Canadian Council on Animal Care (CCAC). The Animal Use Protocol (AUP 20090082) involved in the current study is reviewed annually, and approval is granted by the University of Saskatchewan’s Animal Research Ethics Board (AREB).
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Otero, M.G., Henao-Romero, N., Krysak, T.M. et al. Hyperglycemia-induced mitochondrial abnormalities in autonomic neurons via the RAGE axis. Sci Rep 15, 25231 (2025). https://doi.org/10.1038/s41598-025-10933-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-10933-y
