
Abstract
Understanding the biological factors that influence carcass and meat quality traits in Nellore cattle requires a clear interpretation of molecular complexity and its variations at different levels of expression. Hence, this study aimed to elucidate the biological control of carcass and meat quality traits in Nellore cattle by integrating genome-wide association study (GWAS), transcriptomic, and proteomic data, focusing on identifying key genes and pathways. GWAS analysis was performed using weighted single-step GBLUP with two iterations. RNA-Seq and proteomic analyses were performed on 24 muscle samples from animals with divergent adjusted phenotypic values (12 for high and 12 for low), for meat tenderness, ribeye area (REA), marbling, and backfat thickness (BFT). The phenotypic values were adjusted for the systematic effects of contemporary groups and age. Differential expression analyses indicated that genes associated with the promotion of growth processes, such as FRZB, IGFBP5 and SEMA6C, exhibited overexpression within the group characterized by higher meat tenderness that inhibits cellular cycles and growth (RTN4 and RB1) were downregulated. Proteins related to heat shock, structural functions, and metabolic regulation also affected the higher meat tenderness group. For marbling, actin-binding proteins, microtubule-forming proteins, and structural proteins were downregulated, while genes involved in fatty acid composition and synthesis were upregulated, with the key genes and transcripts CAND1, ACTN4, FGFR2, and NCOR2 identified. For BFT, neuronal genes, transcripts, and proteins associated with actin cytoskeleton organization and microtubule formation were found. Key genes related to ubiquitination, regulation of energy metabolism, and tissue remodeling were also identified. These findings provide a better understanding of genes, transcripts, proteins, and metabolic pathways involved in carcass and meat quality traits in Nellore cattle.
Similar content being viewed by others

Introduction
Nellore cattle (Bos taurus indicus) are the main beef cattle breed raised in Brazil due to their heat resistance, ability to thrive on poor-quality forages, and easy calving1. Breeding programs for Nellore cattle have focused on enhancing both productive and reproductive traits, resulting in significant productivity improvements under a tropical production system1,2. Recently, breeding programs have expanded the selection criteria to include carcass merit traits such as hot carcass weight (HCW), ribeye area (REA), backfat thickness (BFT), and marbling score into selection index1,3. These traits directly affect the production system profitability, as they significantly influence both meat yield and quality grade, aligning with industry demands for higher-quality beef1,3. However, efforts to enhance carcass and meat quality through animal selection face notable challenges due to the high costs associated with phenotyping and the dependence on post-slaughter evaluations for these traits4.
Given these challenges, it is essential to understand the genetic mechanisms that control the carcass and meat quality variability in Nellore cattle to meet production standards that align with consumer demands. Meat quality and carcass traits are complex and affected by different biological factors that are not entirely understood5,6,7. Variations in meat and carcass traits have been exploited using genomic information through GWAS5,8 and selection signatures9 to elucidate their genetic architecture. These analyses identified major biological factors influencing meat quality and carcass traits, such as muscle growth differentiation and development and lipid metabolism, as well as pathways like INS/IGF-1, mTOR, and PPARγ that regulate muscle fiber composition, fat deposition and tenderness. In this context, multi-omics integration (i.e., GWAS, Transcriptomics, Proteomics) datasets from the same individuals have been proposed as an approach able to uncover the molecular information flow, offering a comprehensive view of complex traits10,11.
Multi-omics integration has emerged as a promising approach to a better understanding of the molecular architecture of complex traits, offering a comprehensive perspective on the biological pathways that affect phenotypic expression and enabling a better translation from genotypes to phenotypic variability12,13. Over the past decade, genome, transcriptome and proteome data have been separately explored to investigate the molecular basis of carcass and meat traits. Nevertheless, combining these data sources through functional enrichment analysis allows researchers to connect genotypic variations with phenotypic variability more effectively, providing insights into the biological control of these traits5,14,15.
The biological processes regulating meat quality and carcass traits involve complex mechanisms controlled by different genes and genetic variants that influence and explain their variability16. Functional enrichment plays a key role in multi-omics studies by identifying differentially expressed genes, proteins, or molecules and grouping them into biological groups using bioinformatics tools17,18. This approach facilitates the identification of the biological pathways driving trait expression and enhances our understanding of their molecular and cellular basis of phenotypic variability.
Advancements in genetic sequencing techniques have enabled the implementation of large-scale genomic studies, such as genome-wide association studies (GWAS), RNA sequencing (RNA-Seq), and proteomics (LC–MS/MS). These methods provide opportunities to investigate the genetic architecture of complex traits at the molecular level, allowing for a more accurate determination of the factors that influence various aspects of meat quality5,19,20. Thus, as more omics information, such as genetic markers, transcript, and proteome, becomes available, its integration is able to explain the genetic influence on the trait better. Therefore, we hypothesize that combining information from GWAS, RNA-Seq, and proteomic information could help identify key genes and functional markers affecting carcass composition traits. Thus, we aimed to use an integrative biological approach, combining these data sources through functional analyses and identifying the main loci and biological pathways associated with meat quality and carcass traits in Nellore cattle.
Materials and methods
Carcass and meat quality traits
The carcass and meat quality traits were measured in 6,910 young Nellore bulls belonging to four Brazilian commercial breeding programs (DeltaGen, Cia do Melhoramento, Paint—CRV Lagoa, and Nelore Qualitas) that integrate the Nellore Alliance dataset (www.gensys.com.br). The animal procedures used in this research were in accordance with the Animal Care regulations of the São Paulo State University (UNESP), School of Agricultural and Veterinary Science (protocol number 18.340/16).
The database encompasses information for post-mortem carcass traits: ribeye area (REA) and backfat thickness (BFT), and meat quality traits: Warner–Bratzler shear force (meat tenderness) and marbling score. During the growth period, the animals were raised under a grazing production system with mineral supplementation during the dry season and finished in a feedlot system for approximately 90 days. During the feedlot period, the animal received a total mixed ration (corn silage, dry corn grain, sugarcane bagasse, soybean, urea, mineral salt, and potassium chloride). The young bulls measured for carcass and meat quality traits were raised in contemporary groups under the same nutritional management and slaughtered in commercial slaughterhouses with an average age of 706.5 ± 79.6 days.
After the animals’ slaughter, their carcasses were divided in half and weighed. The half-carcasses were then stored in a cooling chamber for 24 to 48 h. Subsequently, the meat was boned, and samples of the Longissimus thoracis muscle (12th and 13th ribs) were collected and frozen for later measurement of the REA, BF, and meat tenderness. Meat tenderness was determined using a Brookfield CT-3 Texture Analyzer (AMETEK Brookfield, Middleborough, MA, United States) following the procedure proposed by Wheeler et al.21. The REA was determined using a quadrant points method (in square centimeters)22 and BF (in millimeters) was measured using a digital caliper. The meat’s marbling was graded using a visual scale based on the USDA Quality and Yield Grade23. It is important to note that the carcasses had not undergone a dry age process, as this is not a standard procedure in Brazil. For more details regarding the animal procedures and traits assessments, see Arikawa et al.5.
Sampling information
The quality control (QC) for REA, BFT, marbling score, and meat tenderness was performed to remove phenotypic information with 3.5 standard deviations above or below the contemporary group (CG) means and maintain CG with at least five animals. After the QC, the phenotypic information was adjusted for the fixed effects (b) of the CG and animal age at slaughter as a linear covariate since the animals were slaughtered at different ages. The adjusted phenotypic information was analyzed using the animal model as follows:
where y is a vector of observations for each trait; b is the vector of fixed effects; a is the vector of additive genetic effect, assumed as \(\mathbf{a}\sim \text{N }(0,\mathbf{A}{\upsigma }_{\text{a}}^{2})\) and e is the residual effect assumed as \(\mathbf{e}\sim \text{N }(0,\mathbf{I}{\upsigma }_{\text{e}}^{2})\). The \(\mathbf{X}\) and Z correspond to the incidence matrices relating y to the fixed effects (b) and the additive genetic effects (a), respectively. The A is the relationship matrix based on the pedigree, \({\upsigma }_{\text{a}}^{2}\) is the additive genetic variance; I is an identity matrix, and \({\upsigma }_{\text{e}}^{2}\) is the residual variance. The CG was defined considering the year and season of birth, herd (at birth, weaning, and yearling), and slaughter date. The birth season was split into two classes: animals born from August to January and from February to July.
The analysis was implemented using blupf90 + software from the blupf90 suite24, and the phenotypic data was adjusted for fixed effects using the predictf90 software24. Thus, a total of 24 animals with extreme values of adjusted phenotype (12 lower and 12 higher) for each trait (meat tenderness, marbling, BFT, and REA) were selected, the number of common and specific animals by traits are shown in Supplementary Figure S1. The muscle samples of selected animals were used for transcriptomic and proteomic analyses. The descriptive statistics regarding the selected animals with extreme values are shown in Table 1.
Transcriptomic analysis
RNA extraction and sequencing
Muscle samples (Longissimus thoracis) were collected immediately after animal slaughter and stored in Falcon tubes (15-mL) with a 5 mL RNA holder (BioAgency, São Paulo, SP, Brazil) at -80 °C until the RNA extraction. Total RNA extraction was done considering 50 mg of muscle using the RNeasy Lipid Tissue Mini Kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s protocol. RNA purity was evaluated by determining the absorbance value in a NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific, Santa Clara, CA, USA, 2007) at wavelengths 260, 280, and 230 nm. The integrity of total RNA was assessed in an Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA, 2009). The concentration of extracted RNA and contamination with genomic DNA was measured in a Qubit® 2.0 Fluorometer (Invitrogen, Carlsbad, CA, USA, 2010).
The RNA-Seq was done on an Illumina HiSeq 2500 System, and the messenger RNA was obtained from the total RNA extracted. The libraries contained, on average, 25 million reads per sample, and each sample was sequenced at both ends (pair-end sequencing), obtaining 100-bp fragments (2 × 100).
Quality control
The quality of RNA-Seq reads was checked using the FASTQC software25, which assessed quality scores, GC and N content, length distributions, duplication, overrepresented sequences, and K-mer content. The control of trimmed reads was performed by removing low-quality bases and adaptor sequences. Contaminating sequencing was removed in two steps. In the first step of data processing, the Atropos software26 was used to remove adapter contamination through two approaches. The first approach, called adapter-match, identifies the best alignment between each adapter and the corresponding read end. The second approach, insert-match, aligns read 1 with the reverse complement (rc) of read 2. If a valid alignment is found, the adapters are matched to the remaining extent26.
Adapter contamination and sequencing errors can increase the occurrence of unaligned reads or incorrect alignments, leading to potential errors in data analysis. To address this, a second software was used to mitigate any remaining sequencing errors and adapter contamination. The PrinSeq software27 was utilized in the second step of data processing. This tool allows for filtering, reformatting, and sequence removal while also providing a statistical summary of the data, similar to FASTQC. In this study, PrinSeq was specifically used to filter and remove sequences generated in the previous step, ensuring that only high-quality reads were used for data analysis, free from low-quality sequences or artifacts that could result in erroneous conclusions.
Sequence alignment
The STAR software28 was used for mapping and aligning the fragments to the bovine reference genome (ARS-UCD1.3), available in the NCBI database (http://www.ncbi.nlm.nih.gov/genome/?term=bos+taurus). For each library, a file with a bam extension that contained the fragments aligned to the reference genome was created.
The transcripts, derived from the reads of the RNA-Seq data that aligned to the reference genome, were assembled using StringTie29. Differentially expressed genes were identified using the DESeq2 program30, which employs raw transcript count data generated by the STAR aligner. Before statistical analysis, the raw RNA-seq counts were normalized using the DESeq2 program, which adjusts for variations in sequencing depth and RNA composition by calculating size factors based on the ratio of individual counts to the geometric mean of each gene. Thus, the normalized RNA-seq information was used to identify quantitative differences in expression levels between experimental groups (high vs low). The p-values were obtained, and the Benjamini–Hochberg test31 was used to calculate the false discovery rate (FDR) at 5% (q-value). The log2 fold change (Log2 FC) with an FDR lower than 0.05 was used to indicate differentially expressed (DE) genes for group comparison groups (high vs low).
Proteomic analysis
The muscle samples were first washed with phosphate-buffered saline solution (PBS) and then weighed and powdered in liquid nitrogen using a mortar and pestle. Protein extraction involved the addition of a buffer solution containing 8 M urea, 0.15 M Tris–HCl buffer (pH 8.8), 0.5% O.G detergent (1-S-octyl-b-D-thioglucopyranoside), and 10 µL of protease inhibitor solution (Sigma–Aldrich, USA). Subsequently, the samples were submitted to three cycles of sonication for 1 min each, followed by centrifugation at 12,000 g for 5 min at 4 °C. Protein quantification was carried out using the Bradford Protein Assay method (Bio-Rad, Hercules, CA, USA)32, using different dilutions of Bovine serum albumin (BSA) as a standard curve. The samples were duplicated in microplates, and the absorbance at 595 nm was measured using a spectrophotometer (Molecular Devices, SpectraMax Plus384).
Sample preparation (50 µg) for mass spectrometry analysis involved three steps: 1—reducing and alkylating the proteins, 2—digesting proteins with trypsin, and 3—cleaning up and desalting the samples. Initially, disulfide bonds were reduced by adding dithiothreitol (DTT) at a ratio of 1 mg of DTT per mg of protein, followed by a 2-h incubation at room temperature. Alkylation was then performed by adding iodoacetamide (IA) at a ratio of 3 mg IA per mg of protein, and the sample was incubated in the dark at room temperature for 1 h. After that, the sample volume was diluted fivefold in a 0.1 M ammonium bicarbonate solution (pH ≥ 8.0) to reach a final volume of 500 µL. Following this, the samples were incubated overnight at 37°C with 1 µg of trypsin (Promega, Madison, WI, USA). Prior to mass spectrometry analysis, clean-up and desalting were conducted using an OASIS HLB 1 cc column as per the manufacturer’s instructions. Subsequently, the samples were dried using a speed vacuum and then subjected to mass spectrometry.
Peptide masses of 1 lg of each sample were obtained using an OrbiTrap fusion mass spectrometer, and the information was automatically processed using the Computational Proteomics Analysis System (CPAS)33. The ProteinProphet tools (https://bioinformaticshome.com/tools/proteomics/descriptions/ProteinProphet.html#gsc.tab=0) were used to screen the proteins from the identified peptides using the Bovine database deposited in UniProt. The protein intensity values were normalized using, first, the total ion current (TIC) normalization to correct for possible variability in instrument performance, followed by a log₂ transformation to stabilize variance and approximate a normal distribution. These normalized spectral counts were then used for the statistical analysis of differentially expressed proteins using the msmstests R package34 considering the negative binomial test considering a multiple test correction for false discovery rate (FDR). This package employs statistical tests for label-free LC–MS/MS data-based spectral counts to identify variations in protein expression between the two biological conditions (low vs high). The proteins were deemed DE when the FDR value was lower than 0.05 for the group comparison (low vs. high).
Functional enrichment
The GWAS analyses were previously performed by Arikawa et al.5 using a total of 23,859 genotyped animals with 435,447 SNP markers after QC. The SNP effects were estimated using the weighted single-step GBLUP (WssGBLUP) method35. The top 10 windows of 1 Mb explained 8.13%, 11.81%, 17.56%, and 14.95% of the additive genetic variance for REA (BTA 3, 4, 5, 9, 10, 12, 13, 14, 20, and 22), BFT (BTA 1, 3, 6, 11, 13, 17, 18, 20 and 29), marbling (BTA 4, 7, 10, 11, 12, 13, 15 and 20), and meat tenderness (BTA 1, 2, 5, 7, 9, 10, 19 and 25). The top 10 windows regions harbored a total of 119, 143, 155, and 92 candidate genes according to the Bos taurus ARS-UCD1.2 assembly as a reference (https://www.ncbi.nlm.nih.gov/gene/?term=bos+taurus; Supplementary Material Tables S1, S2, S3, and S4). Providing insights into the genetic architecture of meat and carcass traits.
The joint functional enrichment analysis was conducted, taking into account the significant gene list from the GWAS, transcriptomics, and proteomics analyses, utilizing the ClueGO plugin of the Cytoscape software program36. The candidate gene list was also submitted to functional enrichment analysis (biological process terms) for each trait, using DAVID 6.8 Functional Annotation Tools37, considering the bovine database as a reference.
Results
Alignment statistics of RNA-Seq data
A total of 375 million reads (2 × 100 bp) and a 25 × sequencing coverage were obtained. On average, 25 million reads were generated per sample, and the overall alignment rate was 96.86% (Supplementary Tables S5, S6, S7, and S8). Principal component analysis showed distinct group formation and differences in gene expression between groups with divergent phenotypes (Supplementary Figure S2).
Analysis of differentially expressed genes
The sign of the log2 fold change, which refers to relative expression and positive values, indicates upregulated genes, while negative values indicate downregulated genes in relation to the high meat tenderness, high REA, high marbling, and high BFT groups. Differential expression analysis between the tenderness and tough meat groups revealed 139 differentially expressed genes, including 78 downregulated and 59 upregulated genes in the meat tenderness group (q-value < 0.05) (Table 2 and Table S9). Ten differentially expressed genes were identified for REA, including 3 downregulated and 7 upregulated genes for the high REA group (Table 3). For marbling, 21 differentially expressed genes were identified, including 7 downregulated and 14 upregulated genes in the high marbling group (Table 4). For BFT, 40 differentially expressed genes were identified, including 15 downregulated and 25 upregulated genes in relation to the high BFT group (Table 5).
Proteomic analysis
Log fold change values were used to describe proteins that were up- and down-regulated in relation to the groups with high vs. low meat tenderness, REA, marbling, and BFT. Positive values indicate an increase in expression, while negative values indicate a decrease in expression relative to the mentioned groups. Proteomic analysis of meat tenderness revealed 87 differentially expressed proteins, including 44 upregulated and 43 downregulated proteins for the meat tenderness group (q-value < 0.05; Supplementary Material Table S10). Considering REA, 75 differentially expressed proteins were identified, including 30 proteins that were upregulated and 45 that were downregulated in relation to the high REA group (q-value < 0.05; Supplementary Material Table S11). For marbling, 48 upregulated proteins in relation to the high marbling group and 35 downregulated proteins were identified for the high marbling score group (q-value < 0.05; Supplementary Material Table S12). For BFT, 86 differentially expressed proteins were identified, including 38 upregulated proteins and 48 downregulated proteins in relation to the high BFT group (q-value < 0.05; Supplementary Material Table S13).
Functional enrichment
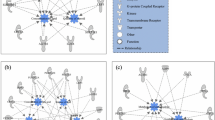
For meat tenderness, we conducted joint functional enrichment analyses using two techniques (ClueGO plug-in of Cytoscape and DAVID 6.8). These analyses identified 14 and 18 significantly enriched Gene Ontology (GO) terms in the biological process category. In general, the transcribed genes and proteins are associated with the following GO terms: protein localization to membrane (GO:0072657); purine-containing compound biosynthetic process (GO:007252); response to heat (GO:0009408); cellular component organization or biogenesis (GO:0071840); negative regulation of growth (GO:0045926), and cytoskeleton organization (GO:0007010; Fig. 1, Supplementary Material Tables S14 and S15). Both transcriptomic and proteomic analyses indicated that the NEXN gene was downregulated in the higher meat tenderness group, while the ZNHIT6 gene was identified as upregulated. Additionally, the joint functional enrichment analysis pointed out key genes and proteins related to the trait variability, such as AUTS2, CAST, RDX, and PSMD, with effects on muscle type fiber and meat quality.

Functional network based on GO terms enrichment analysis for meat tenderness. Genes identified by GWAS are represented by circles, transcripts identified by RNA-Seq are represented by squares, and triangles represent proteins. The pie chart represents the percentage of each GO biological process term identified in the enrichment analysis.
Joint functional enrichment analysis (ClueGO and DAVID) for REA identified 13 GO terms significantly enriched in the biological process category. In general, the transcribed genes and proteins are related to the following GO terms: cytoskeleton organization (GO: 0007010); apoptotic process (GO:0006915); regulation of catalytic activity (GO:0050790); enzyme inhibitor activity (GO:0004857); regulation of proteolysis (GO: 0030162); negative regulation of cellular protein metabolic process (GO:0032269), and protein stabilization (GO:0050821; Fig. 2; Supplementary Material Tables S16 and S17). Both GWAS and proteomic analysis identified the YWHAB gene, which was downregulated in the high REA group. Joint functional enrichment analysis identified two key genes related to REA variability: FBXW7 and CDK7.

Functional network based on GO terms enrichment analysis for ribeye area. Genes identified by GWAS are represented by circles, transcripts identified by RNA-Seq are represented by squares, and triangles represent proteins. The pie chart represents the percentage of each GO biological process term identified in the enrichment analysis.
For marbling, functional enrichment analysis identified 4 and 8 GO terms significantly enriched in the biological process category. Enriched GO terms were related to cytoskeleton organization (GO:0007010), microtubule cytoskeleton organization (GO:0000226), negative regulation of catalytic activity (GO:0043086), and supramolecular fiber organization (GO:0097435; Fig. 3; Supplementary Material Tables S18 and S19). The analysis identified key genes and proteins related to marbling deposition, such as CAND1, ACTN4, YWHAZ, and FGFR2.

Functional network based on GO terms enrichment analysis for marbling. Genes identified by GWAS are represented by circles, transcripts identified by RNA-Seq are represented by squares, and triangles represent proteins. The pie chart represents the percentage of each GO biological process term identified in the enrichment analysis.
Joint enrichment analysis using the ClueGO plug-in and DAVID identified 7 and 12 enriched GO terms, respectively. These GO terms were related to cytoskeleton organization (GO:0007010), cellular component morphogenesis (GO:0032989), microtubule cytoskeleton organization (GO:0000226), and multicellular organismal process (GO:0032501; Fig. 4; Supplementary Material Tables S20 and S21). Three shared genes were identified by transcriptomic and proteomic analyses: HNRNPM, CAMK2B, and INTS3. The HNRNPM was downregulated in proteomic analysis and upregulated in transcriptomic analysis. The two techniques upregulated the CAMK2B gene, while INTS3 was downregulated in proteomic analysis and upregulated in transcriptomic analysis. Joint functional enrichment analysis identified key genes and proteins related to the traits, such as CTNNA2, CAND1, CAMK2D, and PAPPA. In addition, genes, transcripts, and proteins with pleiotropic effects affecting the carcass and meat quality traits were identified considering each specific omics (Fig. 4a–c) and all omics (Fig. 4 d; Table 6).

Venn diagram for common and shared genes, transcripts, and proteins information identified for each GWAS (a), RNA-seq (b) and proteomics (c), and all Omics information (d), with pleiotropic effects for meat quality and carcass traits. Tenderness – meat tenderness; REA – ribeye area and BFT – backfat thickness.
Discussion
Although functional analysis revealed GO terms shared between the different techniques, few genes were identified by more than one procedure for the same trait. Despite the expected, predictable correlation between expressed mRNA and protein, the complexity of biological regulation and methodological limitations suggest a weak relationship among them, causing a few shared genes to be identified in the analyses38. A range of complex mechanisms regulates protein abundance in particular, such as transcriptional control, mRNA processing and stability, translational regulation, post-translational modifications, protein folding and quality control, degradation pathways, and feedback mechanisms to maintain cellular homeostasis. Consequently, when quantifying mRNA, only the first steps in a long chain of regulatory events are observed. Although gene expression in an organism is generally regulated by the transcription of mRNA and its subsequent translation into proteins, many factors can affect the rate and efficiency of this process. One of the main reasons for the low correlation between mRNA and protein is post-transcriptional regulation. The mRNA can undergo alternative processing, rapid degradation, or inefficient translation, events that result in reduced protein production despite the presence of the mRNA transcripts. Furthermore, post-translational modifications such as phosphorylation, glycosylation, and ubiquitination can influence the stability and functionality of proteins. These modifications affect protein localization, interactions, and functional outcomes, impairing the prediction of protein quantity and function based solely on mRNA levels38,39,40.
While genomic analysis evaluates the relationship between polymorphisms and the trait of interest, transcriptomics and proteomics are a portrait of the growth phase of the animal when the samples were collected, and the relationship may, therefore, not be as evident. Nevertheless, functional analysis of omics data might be fundamental to obtaining significant biological insights from complex sets of data that involve different molecular levels, such as genomics, transcriptomics, and proteomics. These data describe the samples from different perspectives and can complement each other in order to obtain relevant biological pathways associated with certain phenotypes, improving the understanding of the biology of the trait studied41,42.
Meat tenderness
Joint functional enrichment analysis of the omics data showed that the GO term negative regulation of growth (GO:0045926) was significant for meat tenderness. Transcripts belonging to this GO term, with positive effects on growth and development, were more expressed (FRZB, IGFBP5, SEMA6C) in the tender meat group, while cell cycle repressors (RB1) and growth inhibitors (RTN4) were downregulated in this group (Table 2). The FRZB gene is recognized for its role in skeletal development and adipogenesis43, but its specific impact on bovine meat quality remains to be elucidated. The IGFBP5 plays a key role in muscle physiology by regulating growth, differentiation, regeneration, and apoptosis44 by modulating the biological activity of IGF-1 and IGF-2. This can influence muscle fiber size and the balance between muscle and connective tissue, thereby affecting meat tenderness. Supporting this, Wang et al.45 found the IGFBP5 gene to be associated with variations in pork meat quality with other QTLs on chromosome 15. Although direct evidence linking the genes FRZB, IGFBP5, and SEMA6C to meat tenderness in cattle remains unclear, their physiological and metabolic functions in muscle biology suggest that they could affect differences in meat tenderness. Therefore, the overexpression of these genes implies that factors affecting muscle physiology may lead to greater meat tenderness46.
The RB1 gene encodes a protein involved in the regulation of cell growth; in addition, its protein interacts with other proteins to influence cell survival47. In a microarray study of muscle tissue from pigs divergent for tenderness, Hamill et al.17 also found the expression of this gene to be repressed in the tender meat group. Furthermore, using GWAS, Hérault et al.48 identified this gene in a QTL region associated with meat quality and carcass composition traits in commercial pigs. These results suggest that the reduced activity of growth inhibitors has a positive effect on meat tenderness. The link between muscle growth and tenderness is characterized by the double-muscling phenotype, which is the result of an inactivating mutation in the myostatin gene49. Cattle carrying inactivated myostatin exhibit a greater capacity to synthesize muscle proteins and to produce more tender meat50.
The omics data also revealed that the biological process response to heat (GO:0009408) was significantly enriched for meat tenderness. Previous studies have linked the regulation of small heat shock proteins (HSPs) to various meat quality attributes, including tenderness. The antiapoptotic and chaperone functions of small HSPs suggest their involvement in the eating quality of meat51. The DNAJA4, HSP90AB1, and HSPA1 transcripts belonging to the term were downregulated in relation to the tender meat group, suggesting that their expression is negatively associated with meat tenderness in Nellore animals51,52,53,54. Additionally, higher protein expression of HSP90AB1 was observed in the low REA group. Although HSP90AB1 may play an important role in the maintenance of cellular function and protection against cellular stress, there is no evidence that this protein is directly involved in muscle growth.
The DNAJA4 gene encodes a chaperone belonging to the 40-kDa heat shock protein (Hsp40) family, which is a co-chaperone of the 70-kDa heat shock protein (Hsp70). The Hsp40/Hsp70 complex plays a role in preserving cellular functions and repairing damaged protein structures, and it also exhibits antiapoptotic activity55,56. The specific function of Hsp40 is likely responsible for the observed negative association of DNAJA4 gene expression with meat tenderness in the Nellore cattle studied. This result aligns with previous findings of Bernard et al.53, who reported lower expression of the DNAJA1 gene, which belongs to the same family as the DNAJA4 gene, in the tender meat group of Charolais cattle. Similarly, Malheiros et al.57 identified the downregulation of the DNAJA1 gene in the group with moderately tender meat. The transcripts encoding Hsp90-beta (HSP90AB1) and Hsp70 (HSPA1A) were also downregulated in relation to the tender meat group. Heat shock proteins (HSPs) exert an antiapoptotic function and can be activated to protect cellular proteins against denaturation18,58. Thus, decreasing expression levels of Hsp90 and Hsp70 probably affects the protection of myofibrillar proteins and favors myofibrillar proteolysis and consequent meat tenderness57,59.
The biological term associated with the cellular component organization or biogenesis (GO:0071840), which was significantly enriched for meat tenderness, plays a key role at the cellular level. This process leads to the assembly, arrangement, or disassembly of cellular components and is aligned with a series of events that occur during the transformation of muscle into meat. This biological process is consistent with biochemical processes that occur during the conversion of muscle to meat. Meat tenderization process involves the breakdown of the structural components of myofibrils through proteolysis, resulting in a decrease in toughness and a gradual increase in meat tenderness60. The CSRP3 transcript, which belongs to the term and was downregulated in relation to the tender meat group, plays an important role in muscle structure maintenance, including myofibril size and sarcomere length61. Therefore, the downregulation of this transcript in the tender meat group probably favors myofibril proteolysis and consequent tenderness.
The CRYAB (αB-crystalline) gene and protein were identified as downregulated in relation to the tender meat group both by transcriptomic analysis and by proteomic analysis. This protein, a member of the small HSP family, plays a fundamental role in the protection of intermediate filaments, stabilizing and protecting target proteins such as desmin62. The hypothesis is that the higher expression of CRYAB in tough meat would prevent the degradation of intermediate filaments and consequently reduce meat tenderness.
Further considering differentially expressed genes and proteins, the NEXN transcript was also identified by both transcriptomic and proteomic analyses, with the transcript being downregulated in the tender meat group in both analyses. The NEXN gene has an essential role in maintaining the Z line and sarcomere integrity and is directly involved in the cytoskeleton organization and apoptotic signaling pathway63. The process of transforming muscle to meat is known to be mediated by a combination of intrinsic and extrinsic factors. The degradation of Z-line proteins catalyzed by proteolytic enzymes contributes to the weakening of the myofibrillar structure and, consequently, to meat tenderness64. Thus, the higher expression of this gene in tough meat appears to inhibit somehow the activity of enzymes responsible for the degradation of muscle fibers and the consequent decrease in tenderness.
Joint functional enrichment for meat tenderness
Joint functional enrichment analysis identified the AUTS2 and CAST genes and the upregulated RDX and PSMD7 proteins as key components (Table S1). The AUTS2 gene has been associated with brain development and central nervous system functioning. Studies suggest the involvement of this gene in diverse processes, including the formation of synapses, the regulation of expression of other genes, and the organization of the cerebral cortex65. We found no evidence of the gene’s action related to meat quality traits.
The CAST (calpastatin) gene plays a fundamental role in beef quality, particularly tenderness66. Calpastatin is a natural inhibitor of the calpain enzyme, which is responsible for the breakdown of muscle proteins during the aging process of meat67. The activity of calpain is essential for meat tenderness since it breaks down muscle proteins into smaller fragments, rendering the meat more tender and juicy68. High expression of the CAST gene leads to a reduction in calpain activity, which can result in meat with a tougher texture. On the other hand, calpain activity is increased when CAST expression is low, resulting in more tender meat60,64. Previous studies have associated the CAST gene with lower meat tenderness in cattle69,70,71. The CAST protein was also upregulated in the high-marbling group. Polymorphisms of the CAST gene have been found to be linked with meat quality characteristics like tenderness, color, and intramuscular fat (IMF) content in pigs72. However, additional biological or functional evidence is required to validate the genetic influence of the calpain system on marbling trait.
The RDX (radixin) gene encodes a protein of the actin-binding protein family. These proteins play an important role in maintaining the structural and functional integrity of cells by connecting the plasma membrane to the actin cytoskeleton73. Evaluating differences in gene expression between pork muscles with different tenderness levels, Hamill et al.17 found the RDX gene to be more expressed in samples related to greater meat tenderness, highlighting the importance of pathways related to cellular organization for increasing meat tenderness.
The PSMD7 gene encodes a subunit of the 26S proteasome, a large protein complex present in the cytoplasm of eukaryotic cells that plays an important role in the degradation of intracellular proteins. The PSMD7 subunit is a non-ATPase that is part of the 19S particle of the 26S proteasome, which is responsible for recognizing and unfolding damaged or unnecessary proteins for degradation by the 20S particle of the proteasome74. Members of the ubiquitin–proteasome play a key role during the transformation of muscle to meat and are involved in proteolysis, leading to the degradation of myofibrillar proteins in muscle cells75 and probably favoring meat tenderness.
Ribeye area
The functional enrichment analysis of omics data for REA identified the cytoskeleton organization (GO:0007010) as a significant term. Cytoskeletal organization is a cellular process that involves the assembly, arrangement, or disassembly of cytoskeletal structures aligned with the muscle development process. Complex gene interactions influence muscle development and growth in animals, muscle growth occurs in two distinct phases, hyperplasia (increase in cell number) during embryonic development and hypertrophy (increase in cell size), during the postnatal phase. Both processes are important for the growth of muscle mass and are regulated by genetic factors, genes and hormones related to growth factors, as well as environmental factors76,77,78.
The CFL1 (cofilin 1) protein, which belongs to the term, was downregulated in relation to the high REA group. Cofilin is an intracellular actin-modulating protein that regulates the dynamic balance between actin depolymerization and polymerization, preserving cytoskeletal function79. In addition to directly cleaving actin, CFL1 also affects gene expression and cell proliferation and plays an important role in the maintenance of cellular homeostasis80. Previous studies on mice have shown that expression of the CFL1 gene tends to decrease as muscle development progresses, while expression of the CFL2 gene tends to increase. Thus, CFL1 is almost undetectable in adult skeletal muscle, which mainly expresses CFL281. Furthermore, comparative functional analysis of the cofilin family during the differentiation of bovine myoblasts revealed a tendency towards a gradual decrease in the expression of the CFL1 gene as muscle development progresses82.
The GO term apoptotic process (GO:0006915) was also considered significant for REA. The processes of protein synthesis and degradation, also called protein turnover, affect muscle growth rates and can thus alter carcass traits in beef cattle83,84,85. In the case of muscle hypertrophy, there is a positive protein balance, i.e., protein synthesis exceeds degradation, which results in protein deposition and consequent muscle growth77. Together, these processes lead to differences in muscle deposition, which directly affects REA. The BAG6 transcript, which was upregulated in relation to the high REA group, encodes an antiapoptotic ubiquitin-like protein86. According to Gomes et al.85, genes encoding proteins of the ubiquitin–proteasome complex, which are related to processes of protein turnover, can influence improvements of economically important traits such as REA in Nellore cattle.
The BCL2L13 protein was downregulated in relation to the high REA group. This protein is a member of the Bcl-2 family that plays a key role in the regulation of apoptosis87. The first pro-apoptotic activity of BCL2L13 was reported in HEK cells (derived from human embryonic kidney cells). Overexpression of BCL2L13 induced apoptosis in HEK cells, while apoptosis inhibitory proteins effectively blocked cell death88. Furthermore, studies on mice demonstrated that increased transcription of the BCL2L13 gene led to greater loss of muscle mass89.
The PRDX5 protein was upregulated in relation to the high REA group, while the gene was associated with BFT. Peroxiredoxins (PRDXs) are members of a highly conserved family of peroxidases that play a role in the maintenance of intracellular reactive oxygen species (ROS) homeostasis. Members of this family are expressed in most organisms and are involved in various biological processes, such as cellular protection against ROS90. As individuals age, oxidative damage to tissues tends to increase, particularly in highly oxidative tissues such as skeletal muscle91. Oxidative damage, in turn, leads to the induction of genes that neutralize the effects of oxidative stress, such as PRDX5, which has antioxidant activity. Oxidative stress can accelerate muscle loss and atrophy, with a consequent reduction in muscle mass92. The RIPK2 protein was also upregulated in relation to the high REA group. RIPK2 encodes a member of the receptor-interacting protein (RIP) family of serine/threonine protein kinases. Its function is related to the modulation of the innate and adaptive immune responses. The gene has been associated with growth traits and meat production in sheep93, representing a potential biomarker for growth traits.
The term regulation of proteolysis (GO:0030162) was also significant for the trait. It is defined as any process that modulates the frequency, rate, or extent of hydrolysis of a bond or peptide bonds within a protein. Together with the number and size of muscle cells and the rate of protein turnover or protein renewal, this process is essential for muscle growth since the final muscle protein content is the result of a balance between the amount of protein synthesized and the amount of protein degraded94.
The UBE2O protein, which belongs to the same term, was upregulated in relation to the high REA group. The ubiquitin-conjugating enzyme (E2) is a critical component of the ubiquitin–proteasome system that controls protein degradation95. Specific roles in different biochemical processes other than ubiquitination have been reported, including growth and differentiation in genes belonging to the same family as the UBE2O protein96, supporting the gene’s positive influence on greater REA.
Joint functional enrichment for ribeye area
Joint functional enrichment analysis identified the FBXW7 transcript upregulated in the high REA group and the CDK7 gene associated with the trait. The FBXW7 transcript encodes a protein that acts as a substrate recruiter of E3 ubiquitin ligase. The function of this protein is to regulate the degradation of key proteins involved in cell cycle regulation, as well as in the regulation of cell signaling and development. The FBXW7 protein is responsible for directing target proteins to the ubiquitin-dependent proteasomal degradation pathway, consequently reducing the concentration of these proteins in the cell97. Gomes et al.85 suggested genes that encode ubiquitin’s to be associated with growth and carcass traits in Nellore cattle. In a GWAS of Nellore cattle, Fernandes Júnior et al.98 also found GO terms related to protein turnover for carcass traits such as REA. The findings suggest an important role of ubiquitins in muscle growth and, consequently, in the development of REA.
The CDK7 gene encodes cyclin-dependent protein kinase 7 (CDK7), an enzyme that plays an important role in the regulation of the cell cycle and gene transcription99. The cell cycle is fundamental for the proliferation of myoblasts and consequent muscle growth. Thus, the CDK family may be indirectly related to muscle growth since it is involved in the regulation of the cell cycle and gene transcription, fundamental processes for the development and maintenance of muscle tissue100.
Marbling
Functional analysis indicated the term cytoskeleton organization (GO:0007010) to be significant for the trait. Most actin-binding proteins (TMP2, XIRP2, and ACTR1B), microtubule-forming proteins (TUBB4A, MAST2, and TPG2), and structural proteins (RAC1) belonging to the term were downregulated in relation to the high marbling group, suggesting cellular rearrangement to provide space for the adipocyte. Decreased expression of actin, tubulin, and other cytoskeletal proteins are events that precede adipocyte differentiation101,102. The differentiation of adipocytes causes a significant change in cell shape, which results in epithelial remodeling of the extracellular matrix and rearrangements of the actin cytoskeleton103,104. Studies have shown that changes in the organization of the cytoskeleton and cell junctions actively participate in marbling deposition during the development of chickens105 and cattle106. The results agree with those of Taye et al.107, who evaluated meat quality parameters, such as marbling deposition, in Ankole cattle. They observed that the organization of cellular components, specifically the GO terms related to actin cytoskeleton organization (GO:0030036), regulates adipocytes.
Furthermore, our results revealed differentially abundant genes and structural proteins between groups. There was decreased expression of myosin7b (MYH7B) and myosin heavy chain 11 (MYH11) in the high marbling group, while the expression of myosin L1 (MYL1) and myosin L6 (MYL6) was increased. Reduced abundance of structural proteins in animals with high marbling content has also been reported in other transcriptomic and proteomic studies108,109,110. Within this context, other authors observed greater myogenesis in Angus cattle compared to Wagyu animals. This finding can be explained by the fact that myogenesis tends to be lower in animals with high marbling since the deposition of intramuscular fat is associated with less proliferation of muscle cells and greater accumulation of adipocytes111.
The MYLK2 gene, which is involved in myosin light chain binding, is predominantly expressed in skeletal muscle fibers and makes up the main myofibrillar proteins in muscle cells112. The kinase encoded by this gene is responsible for the phosphorylation of the myosin light chain and plays an important role in the transport of glucose in adipocytes113. Within this context, considering the importance of glucose in the synthesis of fatty acids, glycerol, and ATP in adipocytes, MYLK2 may influence intramuscular fat deposition. In a transcriptome study, Silva et al.20 found alternatively spliced transcripts of the MYLK2 gene to be differentially expressed in Nellore animals grouped for high intramuscular fat. These findings confirm the occurrence of changes in muscle structure during adipogenesis and support the theory that adipogenesis and myogenesis are competitive processes110.
The GO term negative regulation of catalytic activity (GO:0043086) was also identified as significant by functional enrichment analysis. It is defined as any process that interrupts or reduces the activity of an enzyme. The ARL6IP1 gene enriched to the term, which was downregulated in relation to the high marbling group, encodes an antiapoptotic protein located in the endoplasmic reticulum (ER) that is involved in the formation of ER tubules114. A previous study identified higher methylation and lower expression levels of this gene in obese women, indicating an association between the gene and fat deposition115.
The FASN (fatty acid synthase) gene, which was upregulated in relation to the marbling group, encodes an enzyme involved in fat deposition and fatty acid composition in cattle116,117. Polymorphisms in the FASN gene have been shown to alter milk fat content in dairy cattle118, as well as the composition of fatty acids in bovine subcutaneous and intramuscular fat119. In view of its role in mammalian adipogenesis, the FASN gene is a potential marker in cattle selection and breeding aimed at increasing intramuscular fat deposition in order to improve meat or carcass quality traits based on subcutaneous fat120.
The ACAA2 (acetyl-CoA acyltransferase 2) gene, which was also upregulated in relation to the marbling group, is essential for fatty acid synthesis and is involved in the activation of long-chain fatty acids and their degradation via beta-oxidation121. ACAA2 is expressed in the subcutaneous adipose tissue of beef cattle and is directly involved in adipogenesis122,123.
Joint functional enrichment for marbling
Joint functional enrichment analysis identified upregulated genes and proteins related to the trait, such as CAND1, ACTN4, FGFR2, and NCOR2. The A7MBJ5 protein, encoded by the CAND1 gene, plays an important role in the regulation of the activity of multiprotein complexes involved in cellular protein degradation. Dubiel et al.124 investigated the role of the CAND1 gene in the regulation of adipogenesis and lipid metabolism. The authors showed that CAND1 is expressed at high levels in mature adipose cells when compared to immature cells and that suppression of CAND1 protein expression in adipose cells reduces lipid accumulation and the expression of adipogenesis-related genes. ACTN4, which was also upregulated, is an important structural protein for cytoskeleton organization and the maintenance of cellular integrity in different tissues. Hackl et al.125 demonstrated upregulation of the gene during adipogenesis in mice. In a comparative analysis of the perirenal fat transcriptome of lambs, Suárez-Vega et al.126 also identified expression of the gene related to intramuscular fat in these animals.
The FGFR2 (fibroblast growth factor receptor 2) gene encodes a membrane receptor that binds to specific fibroblast growth factors (FGFs) and activates a series of signaling pathways. The FGFs are important for the growth, development, and repair of tissues from different organs of the body. In a gene co-expression analysis to characterize genes related to marbling in Hanwoo cattle, Lim et al.127 also found the FGFR2 gene to be related to a higher marbling score.
The NCOR2 gene encodes nuclear receptor corepressor 2. This protein is a transcription factor that binds to specific DNA regions and regulates the expression of various genes, inhibiting or activating their transcription. Although the role of NCOR2 in adipocyte function is unclear, a recent study suggests that it may decrease the insulin sensitivity of adipocytes and also reduce the expression of leptin, a protein hormone produced mainly by adipose cells, limiting the capacity of adipocytes to expand with increasing calorie intake128.
Backfat thickness
Neuronal genes are intimately linked to fat accumulation129,130. GWAS of pigs has identified neuronal genes related to BFT129. Although the different analyses performed in the present study for this trait did not detect shared genes, the GO term neuron development (GO:0048666), as well as genes and proteins (CAMK2B, CTNNA2, FRYL, L1CAM, LOC525592, MARK2, NFASC, RAB17, RERE, SMO, SRGAP2, TIAM1, TRIM46, and UBA6) related to neuron development, were identified by functional enrichment analysis. Genes related to actin cytoskeleton organization (FRYL and CTNNA2) and microtubule formation (MARK2 and TRI46) are considered neuronal genes and are classified in the GO term neuron development (GO:0048666).
These genes not only exert a neuronal function but are also involved in actin cytoskeleton organization and cell morphogenesis. Interestingly, these neuronal genes have been associated with brain function, nerves, and disease. Thus, some of the genes associated with BFT also exert a neuronal function. Although neuronal genes are not directly involved in fat deposition, the process of embryogenesis and the expression of certain genes during this process can influence the development of adipose cells131. During embryogenesis, stem cells differentiate into different types of cells, including adipose cells. The expression of neuronal genes during this process may play a role in the differentiation and maturation of these cells131. However, more studies are needed to elucidate how BFT expression and neuronal genes are related to one another.
Enrichment analysis also revealed genes and proteins involved in the GO term cytoskeleton organization (GO:0007010). Most genes, actin-binding proteins, and structural proteins (ACATA1, MARK2, MYOM2, NRAP, and TPM4) were associated (considering the GWAS analysis) or downregulated in relation to the high BFT group. According to the literature, decreased expression of actin, tubulin, integrin, and other cytoskeletal proteins are early events in adipocyte differentiation101,102. A previous study found that, in cells undergoing adipose differentiation, actin synthesis decreased by 90%, while the decrease in tubulin synthesis was greater than 95%, suggesting that biosynthetic events specific to adipocyte differentiation may be influenced by alterations in the cytoskeleton101.
Three shared genes were identified from transcriptomic and proteomic results. The HNRNPM transcript was downregulated in proteomic analysis and upregulated in transcriptomic analysis. CAMK2B was found to be upregulated in both analyses, while INTS3 was downregulated in proteomic analysis and upregulated in transcriptomic analysis. HNRNPM belongs to the subfamily of heterogeneous nuclear ribonucleoproteins (hnRNPs), which are RNA-binding proteins that form complexes with heterogeneous nuclear RNA (hnRNA). These proteins are associated with pre-mRNAs in the nucleus and are thought to influence pre-mRNA processing, as well as other aspects of mRNA metabolism and transport132. The CAMK2B gene is directly involved in the calcium signaling pathway. Cui et al.133 suggested that the calcium ion participates in the regulation of lipid metabolism and regulates fat deposition in adipose tissue in mice. The INTS3 transcript encodes a protein that forms a complex with other proteins, mediating genome stability and the response to DNA damage. INTS3 is believed to be involved in the regulation of the expression of various genes, including those that control cell proliferation, cell differentiation, and cell survival134.
Joint functional enrichment for backfat thickness
Joint functional enrichment analysis to identify the most relevant nodes within the network revealed genes and proteins such as the CTNNA2 gene discussed earlier, as well as the CAND1 and CAMK2D genes and the PAPPA protein, which were upregulated in relation to the high BFT group. The CAND1 gene encodes a negative regulator of the ubiquitin signaling pathway. Dubiel et al.124 investigated the role of the CAND1 gene in the regulation of adipogenesis and lipid metabolism. The results indicated that CAND1 is expressed at higher levels in mature adipose cells compared to immature cells. Furthermore, the suppression of CAND1 protein expression in adipose cells led to a reduction in lipid accumulation and the expression of adipogenesis-related genes. These findings may explain why the CAND1 protein was more expressed in both the high marbling group and the high BFT group.
CAMK2D is an enzyme that participates in important cellular processes, including the regulation of energy metabolism and cell signaling. A study on mice found that inactivation of the CAMK2B gene led to a significant reduction in body fat mass and blood lipid levels135. However, it is important to note that the relationship between the CAMK2B gene and fat deposition is not yet fully understood, and further studies are needed to elucidate the role of this gene in lipid metabolism.
PAPPA is a proteolytic enzyme involved in various biological processes, including hormonal regulation, tissue remodeling, and fetal development. In addition to its role in pregnancy, PAPPA is also involved in the regulation of the activity of IGF, a growth factor that regulates the growth and development of different cells and tissues. PAPPA cleaves an IGF inhibitory protein so that the growth factor can be released and execute its regulatory function136. The IGF system is involved in adipose tissue development, although its roles are not yet well defined137. In addition, elevated PAPPA levels have been implicated in the accumulation of visceral fat in mice138.
Pleiotropy between traits
Transcripts and proteins with pleiotropic effects (Fig. 4) were identified for marbling and BFT (MAST2, FAM207A, MYOM2, NFIX, UXS1, PRSS23, MOG, PAPPA, ENPP1, LZTS2, GPR179, CAND1, UBA6, COX10, and HNRNPM). This is consistent with the fact that body fat deposition is regulated by similar mechanisms involving various metabolic pathways and genes, which explains the genetic correlation (0.29 ± 0.08) between marbling and BFT139. Fat deposition in mammals occurs in specific body regions such as the abdominal cavity (internal fat), intermuscular spaces (between muscle groups), subcutaneous layers, and intramuscular tissues (marbling)140. Internal fat, particularly around the internal organs, is the first to be deposited, followed by intermuscular fat, subcutaneous fat, and, finally, intramuscular fat. However, nutritional mechanisms can promote greater and earlier deposition of intramuscular fat141. For marbling and BFT, the FAM207A, MYOM2, and UXS1 transcripts and proteins were upregulated in animals with the high marbling and BFT. The FAM207A transcript encodes a protein that could be involved in the regulation of cell proliferation and the development of some diseases. A previous study identified this gene by considering alternative splicing events during adipogenesis in human stem cells142.
The MYOM2 gene encodes M-protein, which is found in heart and skeletal muscle cells. M-protein plays an important role in the organization and stability of actin and myosin filaments, the main contractile proteins in muscle. MYOM2 has been associated with marbling and fat deposition in studies on cattle143. UXS1 gene encodes an enzyme called UDP-glucuronate decarboxylase, which is involved in the synthesis of ascorbic acid (vitamin C) in mammals. A previous study has associated the gene with greater intramuscular fat deposition144. PAPPA, which was downregulated in relation to the high marbling and high BFT groups, is an enzyme that cleaves IGF-binding proteins in blood plasma, releasing IGFs to interact with their receptors. Growth hormones like IGF are known to exert effects on fat deposition in animals. In addition, elevated PAPPA levels have been implicated in the accumulation of visceral fat in mice138.
Among the proteins with pleiotropic effects on the traits studied, MGA and TAGLN were identified for marbling and tenderness, explaining the genetic correlation (0.15 ± 0.15) observed by Arikawa et al.139. However, while MGA was more expressed in the tender meat and high marbling groups, TAGLN was more expressed in the tough meat and low marbling groups. MGA is a transcriptional regulator involved in the regulation of genes that control the cell cycle, cell proliferation, apoptosis, and cell differentiation145. The TAGLN protein regulates the contraction of smooth muscle cells present in organs such as the gastrointestinal tract and blood vessels146. TAGLN is also involved in the regulation of cell migration, cell adhesion, and cytoskeleton remodeling. These proteins participate in basal cell functions, but there is no evidence that they are associated with meat quality traits.
The SCAPER and FGG proteins exerted pleiotropic effects on meat tenderness and BFT. While FGG was upregulated in relation to the tender meat and high BFT groups, SCAPER was more expressed in the low BFT and tender meat groups. Located in the ER, SCAPER plays a fundamental role in the regulation of cell division and cell cycle progression. Studies suggest that SCAPER is involved in the regulation of G1/S transition during the cell cycle, which is considered a checkpoint ensuring that cells are ready to progress to DNA replication and cell division147. The FGG (fibrinogen gamma chain) is one of the three subunits that make up the fibrinogen molecule, a plasma protein important for blood clotting. Studies suggest that fibrinogen may also play important roles in other biological processes, such as the immune response, angiogenesis (formation of new blood vessels), and the regulation of platelet function148. SCAPER and FGG participate in basal processes within cells, but we found no studies in the literature that associate these proteins with meat quality traits.
Considering marbling and REA, two proteins with pleiotropic effects were identified: F1N3L7, encoded by the DOCK7 gene, and E3W9A2, encoded by the CLIP1 gene. Both proteins showed higher expression in the high marbling and low REA groups. DOCK7 is involved in many biological processes, including the regulation of cell migration, wound healing, and immune response. Its involvement in the formation of neuronal synapses, which are important for the communication between nerve cells in the brain, has also been reported149. There is no evidence associating the function of DOCK7 with marbling deposition or REA. On the other hand, the CLIP1 gene encodes a protein involved in the regulation of cytoskeleton organization150. The CLIP1 protein contains a CAP-Gly domain that is capable of binding to microtubules. The latter play important roles in cell division, intracellular transport of molecules, and maintenance of cell shape and structure151. We found no evidence associating the function of the gene with REA or marbling. However, a previous study reported an interaction between the CLIP1 gene and mTOR, a protein kinase that regulates cell growth, proliferation, and survival in response to environmental and nutritional stimuli152. The mTOR protein is important for the regulation of metabolism, protein synthesis, autophagy, and cell differentiation153. Also, the mTOR signaling pathway is involved in adipogenesis and has been previously associated with marbling scores in the same Nellore population used in the present study5.
The results suggest that the identified genes and proteins may have pleiotropic effects through multiple biological pathways that impact meat quality traits. However, to confirm this pleiotropic effect, future research should prioritize functional validation. The investigation of pleiotropy becomes even more significant when integrating multiple omics layers, such as genomic, transcriptomic, and proteomic data. This integrated approach can help uncover shared functional connections between different traits, increasing the chances of finding genetic variants that affect gene expression, protein levels, and metabolite profiles. This suggests that some genes may have multifaceted complex biological effects. To support the pleiotropy hypothesis, systems biology methodologies, such as co-expression network analysis and molecular pathway mapping, can be used to identify functional modules linked to multiple traits. Moreover, advanced integrative frameworks, such as matrix factorization and component-based integration techniques, provide robust tools for discovering molecular signatures indicative of pleiotropic activity. Nonetheless, definitive evidence of pleiotropy requires not only the convergence of multi-omics data but also functional validation.
Conclusions
Understanding the molecular mechanisms related to meat quality and carcass traits is fundamental for explaining phenotypic variability. In summary, this study provided a broad view of the biological pathways involved in carcass and meat quality traits of Nellore cattle. Genes related to growth, cell cycle regulation, and HSPs were associated with meat tenderness, while transcribed genes and proteins related to cytoskeleton organization and apoptotic processes were associated with REA. For marbling, proteins related to actin binding and microtubule formation, as well as to fatty acid synthesis and composition, were identified. Finally, genes associated with ubiquitin signaling, regulation of energy metabolism, and tissue remodeling were identified as being important for BFT. These results provide a better explanation of the complex metabolic pathways involved in these traits and indicate target pathways for future studies.
Data availability
The phenotypic information that supports this study’s findings comes from commercial breeding programs. Restrictions are applied to the availability of these data, which were used under license for the current study and are not publicly available (https://gensys.com.br). However, data are available by contacting Dr. Lucia G. Albuquerque (galvao.albuquerque@unesp.br), and the 20 RNA-seq samples are available in NCBI site (https://www.ncbi.nlm.nih.gov/sra/?term=PRJNA780472).
References
Fernandes Júnior, G. A. et al. Current applications and perspectives of genomic selection in Bos indicus (Nellore) cattle. Livest. Sci. 263, 105001 (2022).
FernandesJúnior, G. A. et al. Sustainable intensification of beef production in the tropics: The role of genetically improving sexual precocity of heifers. Animals (Basel) 12, 1–25 (2022).
Silva, R. P. et al. Genomic prediction ability for carcass composition indicator traits in Nellore cattle. Livest. Sci. 245, 104421 (2021).
Magalhães, A. F. B. et al. Genomic selection for meat quality traits in Nelore cattle. Meat Sci. 148, 32–37 (2019).
Arikawa, L. M. et al. Genome-wide scans identify biological and metabolic pathways regulating carcass and meat quality traits in beef cattle. Meat Sci. 209, 109402 (2024).
Leal-Gutiérrez, J. D. et al. Structural equation modeling and whole-genome scans uncover chromosome regions and enriched pathways for carcass and meat quality in beef. Front. Genet. 9, 1–13 (2018).
Salatta, B. M. et al. Differentially expressed messenger RNA isoforms in beef cattle skeletal muscle with different fatty acid profiles. Meat Sci. 222, 109751 (2025).
Fonseca, P. A. de S., Caldwell, T., Mandell, I., Wood, K. & Cánovas, A. Genome-wide association study for meat tenderness in beef cattle identifies patterns of the genetic contribution in different post-mortem stages. Meat Sci. 186, 108733 (2022).
Yu, H. et al. Genome-wide analysis reveals genomic diversity and signatures of selection in Qinchuan beef cattle. BMC Genom. 25, 1–11 (2024).
Ideker, T., Galitski, T. & Hood, L. A new approach to decoding life: Systems biology. Annu. Rev. Genom. Hum. Genet. 2, 343–372 (2001).
Rodosthenous, T., Shahrezaei, V. & Evangelou, M. Integrating multi-OMICS data through sparse canonical correlation analysis for the prediction of complex traits: A comparison study. Bioinformatics 36, 4616–4625 (2020).
Fonseca, P. A. de S. et al. Combining multi-OMICs information to identify key-regulator genes for pleiotropic effect on fertility and production traits in beef cattle. PLoS ONE 13, e0205295 (2018).
Arora, D. et al. Integration of multi-omics approaches for functional characterization of muscle related selective sweep genes in Nanchukmacdon. Sci. Rep. 11, 7219 (2021).
Fonseca, L. F. S. et al. Differences in global gene expression in muscle tissue of Nellore cattle with divergent meat tenderness. BMC Genom. 18 (2017).
He, H., Chen, S., Liang, W. & Liu, X. Genome-wide proteomics analysis on longissimus muscles in Qinchuan beef cattle. Anim. Genet. 48, 131–140 (2017).
Li, J. et al. Integrative analyses of genomic and metabolomic data reveal genetic mechanisms associated with carcass merit traits in beef cattle. Sci. Rep. 12, 1–12 (2022).
Hamill, R. M. et al. Functional analysis of muscle gene expression profiles associated with tenderness and intramuscular fat content in pork. Meat Sci. 92, 440–450 (2012).
D’Alessandro, A. & Zolla, L. Meat science: From proteomics to integrated omics towards system biology. J. Proteom. 78, 558–577 (2013).
Srivastava, S. et al. Haplotype-based genome-wide association study and identification of candidate genes associated with carcass traits in Hanwoo Cattle. Genes 11, 551 (2020).
Silva, D. B. S. et al. Spliced genes in muscle from Nelore Cattle and their association with carcass and meat quality. Sci. Rep. 10, 1–13 (2020).
Wheeler, T. L., Shackelford, S. D. & Koohmaraie, M. Shear Force Procedures for Meat Tenderness Measurement. Animal Research (Clay Center, NE: Roman L. Hruska U. S. Marc. USDA, 2005).
USDA. Method for Grid Assessment of Beef Carcass Ribeye Area. (2011).
United States Departament of Agriculture (USDA). United States Standards for Grades of Slaughter Cattle. (2000).
Misztal, I. et al. Manual for BLUPF90 family of programs. (University of Georgia, 2018).
Wingett, S. W. & Andrews, S. FastQ screen: A tool for multi-genome mapping and quality control. F1000Research 7, 1338 (2018).
Didion, J. P., Martin, M. & Collins, F. S. Atropos: specific, sensitive, and speedy trimming of sequencing reads. PeerJ 5, e3720 (2017).
Cantu, V. A., Sadural, J. & Edwards, R. PRINSEQ++, a multi-threaded tool for fast and efficient quality control and preprocessing of sequencing datasets. PeerJ Prepr. 7, e27553v1 (2019).
Dobin, A. & Gingeras, T. R. Mapping RNA‐seq Reads with STAR. Curr. Protoc. Bioinf. 51, 11.14.1–11.14.19 (2015).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 1–21 (2014).
Haynes, W. Benjamini–Hochberg Method. Encycl. Syst. Biol. 78–78 (2013) https://doi.org/10.1007/978-1-4419-9863-7_1215.
Bradford, M. M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 (1976).
Rauch, A. et al. Computational proteomics analysis system (CPAS): An extensible, open-source analytic system for evaluating and publishing proteomic data and high throughput biological experiments. J. Proteome Res. 5, 112–121 (2006).
Gregori, J., Sánchez, À. & Villanueva, J. msmsEDA & msmsTests: Label-free differential expression by spectral counts. in Statistical Analysis of Proteomic Data vol. 2426 197–242 (Methods in molecular biology, 2023).
Zhang, X., Lourenco, D., Aguilar, I., Legarra, A. & Misztal, I. Weighting strategies for single-step genomic BLUP: An iterative approach for accurate calculation of GEBV and GWAS. Front. Genet. 7, 1–14 (2016).
Bindea, G. et al. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 25, 1091–1093 (2009).
Sherman, B. T. et al. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 50, W216–W221 (2022).
Maier, T., Güell, M. & Serrano, L. Correlation of mRNA and protein in complex biological samples. FEBS Lett. 583, 3966–3973 (2009).
Gygi, S. P., Rochon, Y., Franza, B. R. & Aebersold, R. Correlation between protein and mRNA abundance in yeast. Mol. Cell. Biol. 19, 1720–1730 (1999).
Payne, S. H. The utility of protein and mRNA correlation. Trends Biochem. Sci. 40, 1–3 (2015).
Fontanillo, C., Nogales-Cadenas, R., Pascual-Montano, A. & de Las Rivas, J. Functional analysis beyond enrichment: Non-redundant reciprocal linkage of genes and biological terms. PLoS ONE 6 (2011).
Paczkowska, M. et al. Integrative pathway enrichment analysis of multivariate omics data. Nat. Commun. 11, 1–16 (2020).
Claire D’Andre, H. et al. Identification and characterization of genes that control fat deposition in chickens. J. Anim. Sci. Biotechnol. 4, 43 (2013).
Beattie, J., Allan, G. J., Lochrie, J. D. & Flint, D. J. Insulin-like growth factor-binding protein-5 (IGFBP-5): A critical member of the IGF axis. Biochem. J. 395, 1–19 (2006).
Wang, W., Ouyang, K., Shangguan, X. & Xu, M. Association of porcine IGF binding protein-5 gene with meat quality. Biochem. Genet. 48, 257–265 (2010).
Koohmaraie, M., Kent, M. P., Shackelford, S. D., Veiseth, E. & Wheeler, T. L. Meat tenderness and muscle growth: Is there any relationship?. Meat Sci. 62, 345–352 (2002).
Herwig, S. & Strauss, M. The retinoblastoma protein: A master regulator of cell cycle, differentiation and apoptosis. Eur. J. Biochem. 246, 581–601 (1997).
Hérault, F., Damon, M., Cherel, P. & Le Roy, P. Combined GWAS and LDLA approaches to improve genome-wide quantitative trait loci detection affecting carcass and meat quality traits in pig. Meat Sci. 135, 148–158 (2018).
Grobet, L. et al. A deletion in the bovine myostatin gene causes the double-muscled phenotype in cattle. Nat. Genet. 17, 71–74 (1997).
Wheeler, T. L., Shackelford, S. D., Casas, E., Cundiff, L. V. & Koohmaraie, M. The effects of Piedmontese inheritance and myostatin genotype on the palatability of longissimus thoracis, gluteus medius, semimembranosus, and biceps femoris. J. Anim. Sci. 79, 3069–3074 (2001).
Lomiwes, D., Farouk, M. M., Wiklund, E. & Young, O. A. Small heat shock proteins and their role in meat tenderness: A review. Meat Sci. 96, 26–40 (2014).
De Souza Rodrigues, R. T. et al. Differences in beef quality between Angus (Bos taurus taurus) and Nellore (Bos taurus indicus) cattle through a proteomic and phosphoproteomic approach. PLoS ONE 12, 1–21 (2017).
Bernard, C. et al. New indicators of beef sensory quality revealed by expression of specific genes. J. Agric. Food Chem. 55, 5229–5237 (2007).
Rosa, A. F. et al. Proteome changes of beef in Nellore cattle with different genotypes for tenderness. Meat Sci. 138, 1–9 (2018).
Picard, B. et al. Skeletal muscle proteomics in livestock production. Brief. Funct. Genom. Proteom. 9, 259–278 (2010).
Guillemin, N., Bonnet, M., Jurie, C. & Picard, B. Functional analysis of beef tenderness. J. Proteom. 75, 352–365 (2011).
Malheiros, J. M. et al. Association of CAST2, HSP90AA1, DNAJA1 and HSPB1 genes with meat tenderness in Nellore cattle. Meat Sci. 138, 49–52 (2018).
Zhang, L. et al. Genome-wide association studies for growth and meat production traits in sheep. PLoS ONE 8 (2013).
Baldassini, W. A. et al. Meat quality traits of Nellore bulls according to different degrees of backfat thickness: A multivariate approach. Anim. Prod. Sci. 57, 363 (2017).
Koohmaraie, M. Biochemical factors regulating the toughening and tenderization processes of meat. Meat Sci. 43, 193–201 (1996).
Barash, I. A., Mathew, L., Lahey, M., Greaser, M. L. & Lieber, R. L. Muscle LIM protein plays both structural and functional roles in skeletal muscle. Am. J. Physiol. Cell Physiol. 289, 1312–1320 (2005).
Wang, X. et al. αB-crystallin modulates protein aggregation of abnormal desmin. Circ. Res. 93, 998–1005 (2003).
Hassel, D. et al. Nexilin mutations destabilize cardiac Z-disks and lead to dilated cardiomyopathy. Nat. Med. 15, 1281–1288 (2009).
Bhat, Z. F., Morton, J. D., Mason, S. L. & Bekhit, A. E. D. A. Role of calpain system in meat tenderness: A review. Food Sci. Hum. Wellness 7, 196–204 (2018).
Oksenberg, N. & Ahituv, N. The role of AUTS2 in neurodevelopment and human evolution. Trends Genet. 29, 600–608 (2013).
Goll, D. E., Thompson, V. F., Taylor, R. G. & Christiansen, J. A. Role of the calpain system in muscle growth. Biochimie 74, 225–237 (1992).
Casas, E. et al. Effects of calpastatin and μ-calpain markers in beef cattle on tenderness traits. J. Anim. Sci. 84, 520–525 (2006).
Dransfield, E. Modelling post-mortem tenderisation-IV: Role of calpains and calpastatin in conditioning. Meat Sci. 34, 217–234 (1993).
Pinto, L. F. et al. Association of SNPs on CAPN1 and CAST genes with tenderness in Nellore cattle. Genet. Mol. Res. 9, 1431–1442 (2010).
Schenkel, F. S. et al. Association of a single nucleotide polymorphism in the calpastatin gene with carcass and meat quality traits of beef cattle. J. Anim. Sci. 84, 291–299 (2006).
Magalhães, A. F. B. et al. Genome-wide association study of meat quality traits in Nellore cattle. PLoS ONE 11, 1–12 (2016).
Ropka-Molik, K. et al. Association of calpastatin gene polymorphisms and meat quality traits in pig. Meat Sci. 97, 143–150 (2014).
Cetinkaya, A. et al. Radixin relocalization and nonmuscle α -actinin expression are features of remodeling cardiomyocytes in adult patients with dilated cardiomyopathy. Dis. Markers 2020 (2020).
Zhao, Y. et al. Deubiquitinase PSMD7 regulates cell fate and is associated with disease progression in breast cancer. Am. J. Transl. Res. 12, 5433–5448 (2020).
Sekikawa, M., Seno, K. & Mikami, M. Degradation of ubiquitin in beef during storage. Meat Sci. 48, 201–204 (1998).
dos Santos Silva, D. B. et al. Transcriptome profiling of muscle in Nelore cattle phenotypically divergent for the ribeye muscle area. Genomics 112, 1257–1263 (2020).
Oksbjerg, N., Gondret, F. & Vestergaard, M. Basic principles of muscle development and growth in meat-producing mammals as affected by the insulin-like growth factor (IGF) system. Domest. Anim. Endocrinol. 27, 219–240 (2004).
Silva-Vignato, B. et al. Comparative muscle transcriptome associated with carcass traits of Nellore cattle. BMC Genom. 18, 1–13 (2017).
Gressin, L., Guillotin, A., Guérin, C., Blanchoin, L. & Michelot, A. Architecture dependence of actin filament network disassembly. Curr. Biol. 25, 1437–1447 (2015).
Thirion, C. et al. Characterization of human muscle type cofilin (CFL2) in normal and regenerating muscle. Eur. J. Biochem. 268, 3473–3482 (2001).
Mohri, K. et al. Expression of cofilin isoforms during development of mouse striated muscles. J. Muscle Res. Cell Motil. 21, 49–57 (2000).
Sun, Y. et al. Functional and comparative analysis of two subtypes of cofilin family on cattle myoblasts differentiation. Agriculture 12, 1420 (2022).
Fanzani, A., Conraads, V. M., Penna, F. & Martinet, W. Molecular and cellular mechanisms of skeletal muscle atrophy: An update. J. Cachexia. Sarcopenia Muscle 3, 163–179 (2012).
Maltin, C., Balcerzak, D., Tilley, R. & Delday, M. Determinants of meat quality: tenderness. Proc. Nutr. Soc. 62, 337–347 (2003).
Gomes, R. C. et al. Protein synthesis and degradation gene SNPs related to feed intake, feed efficiency, growth, and ultrasound carcass traits in Nellore cattle. Genet. Mol. Res. 12, 2923–2936 (2013).
Minami, R. et al. BAG-6 is essential for selective elimination of defective proteasomal substrates. J. Cell Biol. 190, 637–650 (2010).
Meng, F. et al. BCL2L13: Physiological and pathological meanings. Cell. Mol. Life Sci. 78, 2419–2428 (2021).
Kataoka, T. et al. Bcl-rambo, a novel Bcl-2 homologue that induces apoptosis via its unique C-terminal extension. J. Biol. Chem. 276, 19548–19554 (2001).
Cohen, T. V. et al. Defective skeletal muscle growth in lamin A/C-deficient mice is rescued by loss of lap2α. Hum. Mol. Genet. 22, 2852–2869 (2013).
Lee, Y. J. Knockout mouse models for peroxiredoxins. Antioxidants 9, 1–19 (2020).
Mecocci, P. et al. Age-dependent increases in oxidative damage to DNA, lipids, and proteins in human skeletal muscle. Free Radic. Biol. Med. 26, 303–308 (1999).
Leite, L. E. de A. et al. Envelhecimento, estresse oxidativo e sarcopenia: uma abordagem sistêmica. Rev. Bras. Geriatr. e Gerontol. 15, 365–380 (2012).
Zhang, H., Wang, Z., Wang, S. & Li, H. Retraction: Progress of genome wide association study in domestic animals. J. Anim. Sci. Biotechnol. 4, 1–10 (2013).
Goldberg, A. L. Protein synthesis during work-induced growth of skeletal muscle. J. Cell Biol. 36, 653–658 (1968).
Nascimento, R. M. P., Otto, P. A., De Brouwer, A. P. M. & Vianna-Morgante, A. M. UBE2A, which encodes a ubiquitin-conjugating enzyme, is mutated in a novel X-linked mental retardation syndrome. Am. J. Hum. Genet. 79, 549–555 (2006).
Contino, G. et al. Expression analysis of the gene encoding for the U-box-type ubiquitin ligase UBE4A in human tissues. Gene 328, 69–74 (2004).
Yeh, C., Bellon, M. & Nicot, C. FBXW7 : A critical tumor suppressor of human cancers. 1–19 (2018).
Fernandes Júnior, G. A. et al. Genome scan for postmortem carcass traits in Nellore cattle1. J. Anim. Sci. 94, 4087–4095 (2016).
Diab, S., Yu, M. & Wang, S. CDK7 Inhibitors in cancer therapy: The sweet smell of success? (2020) https://doi.org/10.1021/acs.jmedchem.9b01985.
Sharma, M., Langley, B., Bass, J. & Kambadur, R. Myostatin in muscle growth and repair. Exerc. Sport Sci. Rev. 29, 155–158 (2001).
Spiegelman, B. M. & Farmer, S. R. Decreases in tubulin and actin gene expression prior to morphological differentiation of 3T3 Adipocytes. Cell 29, 53–60 (1982).
Rodríguez Fernández, J. L. & Ben-Ze’ev, A. Regulation of fibronectin, integrin and cytoskeleton expression in differentiating adipocytes: Inhibition by extracellular matrix and polylysine. Differentiation 42, 65–74 (1989).
Lin, D., Chun, T. H. & Kang, L. Adipose extracellular matrix remodelling in obesity and insulin resistance. Biochem. Pharmacol. 119, 8–16 (2016).
Bonnet, M., Cassar-Malek, I., Chilliard, Y. & Picard, B. Ontogenesis of muscle and adipose tissues and their interactions in ruminants and other species. Animal 4, 1093–1109 (2010).
Cui, H. et al. Identification of differentially expressed genes and pathways for abdominal fat deposition in ovariectomized and sham-operated chickens. Genes (Basel). 10, (2019).
Poleti, M. D. et al. Longissimus dorsi muscle label-free quantitative proteomic reveals biological mechanisms associated with intramuscular fat deposition. J. Proteom. 179, 30–41 (2018).
Taye, M. et al. Whole genome scan reveals the genetic signature of African Ankole cattle breed and potential for higher quality beef. BMC Genet. 18, 1–14 (2017).
Mao, Y. et al. Beef quality with different intramuscular fat content and proteomic analysis using isobaric tag for relative and absolute quantitation of differentially expressed proteins. Meat Sci. 118, 96–102 (2016).
Zhang, Q. et al. Differentially expressed proteins during fat accumulation in bovine skeletal muscle. Meat Sci. 86, 814–820 (2010).
Chen, D., Li, W., Du, M. & Cao, B. Adipogenesis, fibrogenesis and myogenesis related gene expression in longissimus muscle of high and low marbling beef cattle. Livest. Sci. 229, 188–193 (2019).
Duarte, M. S. et al. Enhancement of adipogenesis and fibrogenesis in skeletal muscle of Wagyu compared with Angus cattle. J. Anim. Sci. 91, 2938–2946 (2013).
Janin, A., Bessière, F., Chauveau, S., Chevalier, P. & Millat, G. First identification of homozygous truncating CSRP3 variants in two unrelated cases with hypertrophic cardiomyopathy. Gene 676, 110–116 (2018).
Young, O. C. et al. Implication of phosphorylation of the myosin II regulatory light chain in insulin-stimulated GLUT4 translocation in 3T3-F442A adipocytes. Exp. Mol. Med. 38, 180–189 (2006).
Fowler, P. C. & O’Sullivan, N. C. ER-shaping proteins are required for ER and mitochondrial network organization in motor neurons. Hum. Mol. Genet. 25, 2827–2837 (2016).
Nicoletti, C. F. et al. Altered pathways in methylome and transcriptome longitudinal analysis of normal weight and bariatric surgery women. Sci. Rep. 10, 1–7 (2020).
Roy, R., Taourit, S., Zaragoza, P., Eggen, A. & Rodellar, C. Genomic structure and alternative transcript of bovine fatty acid synthase gene (FASN): Comparative analysis of the FASNgene between monogastric and ruminant species. Cytogenet. Genome Res. 111, 65–73 (2005).
Zhang, S., Knight, T. J., Reecy, J. M. & Beitz, D. C. DNA polymorphisms in bovine fatty acid synthase are associated with beef fatty acid composition. Anim. Genet. 39, 62–70 (2008).
Morris, C. A. et al. Fatty acid synthase effects on bovine adipose fat and milk fat. Mamm. Genome 18, 64–74 (2007).
Abe, T. et al. Novel mutations of the FASN gene and their effect on fatty acid composition in japanese black beef. Biochem. Genet. 47, 397–411 (2009).
Hillgartner, F. B., Salati, L. M. & Goodridge, A. G. Physiological and molecular mechanisms involved in nutritional regulation of fatty acid synthesis. Physiol. Rev. 75, 47–76 (1995).
Ladeira, M. M. et al. Nutrigenomics and beef quality: A review about lipogenesis. Int. J. Mol. Sci. 17, 1–21 (2016).
Wang, Z. et al. Identification of genes related to growth and lipid deposition from transcriptome profiles of pig muscle tissue. PLoS ONE 10, 1–21 (2015).
Jin, W. et al. Transcriptome analysis of subcutaneous adipose tissues in beef cattle using 3’ digital gene expression-tag profiling. J. Anim. Sci. 90, 171–183 (2012).
Dubiel, D., Gierisch, M. E., Huang, X., Dubiel, W. & Naumann, M. CAND1-dependent control of cullin 1-RING Ub ligases is essential for adipogenesis. Biochim. Biophys. Acta Mol. Cell Res. 1833, 1078–1084 (2013).
Hackl, H. et al. Molecular processes during fat cell development revealed by gene expression profiling and functional annotation. Genome Biol. 6, 1–23 (2005).
Suárez-Vega, A. et al. Early adipose deposits in sheep: comparative analysis of the perirenal fat transcriptome of Assaf and Churra suckling lambs. Anim. Genet. 49, 605–617 (2018).
Lim, D. et al. Gene co-expression analysis to characterize genes related to marbling trait in Hanwoo (Korean) cattle. Asian-Australasian J. Anim. Sci. 26, 19–29 (2013).
Sutanto, M. M. et al. The silencing mediator of retinoid and thyroid hormone receptors (SMRT) regulates adipose tissue accumulation and adipocyte insulin sensitivity in vivo. J. Biol. Chem. 285, 18485–18495 (2010).
Lee, K. T. et al. Neuronal genes for subcutaneous fat thickness in human and pig are identified by local genomic sequencing and combined SNP association study. PLoS ONE 6, (2011).
Lee, Y.-S. & Shin, D. Genome-wide association studies associated with backfat thickness in landrace and Yorkshire pigs. Genomics Inform. 16, 59–64 (2018).
Farmer, S. R. Transcriptional control of adipocyte formation. Cell Metab. 4, 263–273 (2006).
Ho, J. S. Y. et al. HNRNPM controls circRNA biogenesis and splicing fidelity to sustain cancer cell fitness. Elife 10, 1–32 (2021).
Cui, H. et al. High-salt intake negatively regulates fat deposition in mouse. Sci. Rep. 7, 2053 (2017).
Skaar, J. R. et al. INTS3 controls the hSSB1-mediated DNA damage response. J. Cell Biol. 187, 25–32 (2009).
Anderson, K. A. et al. Hypothalamic CaMKK2 contributes to the regulation of energy balance. Cell Metab. 7, 377–388 (2008).
Monget, P. & Oxvig, C. PAPP-A and the IGF system. Ann. Endocrinol. (Paris) 77, 90–96 (2016).
Baxter, R. C. & Twigg, S. M. Actions of IGF binding proteins and related proteins in adipose tissue. Trends Endocrinol. Metab. 20, 499–505 (2009).
Conover, C. A., Harstad, S. L., Tchkonia, T. & Kirkland, J. L. Preferential impact of pregnancy-associated plasma protein-A deficiency on visceral fat in mice on high-fat diet. Am. J. Physiol. Metab. 305, E1145–E1153 (2013).
Arikawa, L. M. et al. Genetic parameter estimates for carcass and meat quality traits and their genetic associations with sexual precocity indicator traits in Nellore cattle. J. Anim. Breed. Genet. 0, 1–13 (2025).
Hocquette, J. F. et al. Intramuscular fat content in meat-producing animals: Development, genetic and nutritional control, and identification of putative markers. Animal 4, 303–319 (2010).
Schumacher, M., DelCurto-Wyffels, H., Thomson, J. & Boles, J. Fat deposition and fat effects on meat quality—A review. Animals (Basel) 12, 1550 (2022).
Yi, X., Yang, Y., Wu, P., Xu, X. & Li, W. Alternative splicing events during adipogenesis from hMSCs. J. Cell. Physiol. 235, 304–316 (2020).
Bazile, J., Picard, B., Chambon, C., Valais, A. & Bonnet, M. Pathways and biomarkers of marbling and carcass fat deposition in bovine revealed by a combination of gel-based and gel-free proteomic analyses. Meat Sci. 156, 146–155 (2019).
dos Santos Silva, D. B. et al. Prediction of hub genes associated with intramuscular fat content in Nelore cattle. BMC Genom. 20, 1–12 (2019).
De Paoli, L. et al. MGA, a suppressor of MYC, is recurrently inactivated in high risk chronic lymphocytic leukemia. Leuk. Lymphoma 54, 1087–1090 (2013).
Assinder, S. J., Stanton, J. L. & Prasad, P. D. Transgelin: An actin-binding protein and tumour suppressor. Int. J. Biochem. Cell Biol. 41, 482–486 (2009).
Fasham, J. et al. Delineating the expanding phenotype associated with SCAPER gene mutation. Am. J. Med. Genet. Part A 179, 1665–1671 (2019).
Cooper, A. V., Standeven, K. F. & Ariëns, R. A. S. Fibrinogen gamma-chain splice variant γ′ alters fibrin formation and structure. Blood 102, 535–540 (2003).
Lopes, L. S. et al. Características de carcaça e cortes comerciais de tourinhos Red Norte e Nelore terminados em confinamento. Rev. Bras. Zootec. 41, 970–977 (2012).
Larti, F. et al. A defect in the CLIP1 gene (CLIP-170) can cause autosomal recessive intellectual disability. Eur. J. Hum. Genet. 23, 331–336 (2015).
Izumi, H. et al. The CLIP1–LTK fusion is an oncogenic driver in non-small-cell lung cancer. Nature 600, 319–323 (2021).
Choi, J. H. et al. The FKBP12-rapamycin-associated protein (FRAP) is a CLIP-170 kinase. EMBO Rep. 3, 988–994 (2002).
Aoki, M. S., Miyabara, E. H., Soares, A. G., Saito, E. T. & Moriscot, A. S. mTOR pathway inhibition attenuates skeletal muscle growth induced by stretching. Cell Tissue Res. 324, 149–156 (2006).
Acknowledgements
The authors acknowledge the commercial breeding programs participating in the Nellore Data Alliance (https://gensys.com.br) for providing the data used in this research and the São Paulo Research Foundation (FAPESP) for the financial support.
Funding
This research was funded by São Paulo Research Foundation (FAPESP) grant number #2009/16118-5, #2017/10630-2, #2018/20026-8 and #2022/11852-7. Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brazil (CAPES), finance code 001.
Author information
Authors and Affiliations
Contributions
Conceptualization: G.B.F., L.F.S.F. and L.F.M.M.; methodology: G.B.F, L.F.S.F. and D.B.S.S; resources: L.G.A.; data curation: G.B.F., B.M.S. and L.F.S.F.; writing original draft preparation: G.B.F.; writing review and editing: L.F.M.M.; visualization: L.F.M.M., L.M.A and P.I.S..; supervision: L.G.A, L.F.M.M and L.F.S.F; project administration: L.G.A. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
The animal procedures and data sampling presented in this study were approved and performed following the Animal Care and Ethical Committee recommendations of the São Paulo State University (UNESP), School of Agricultural and Veterinary Science (protocol number 18.340/16). All the data sampling was performed following the CEUA/ FCAV-UNESP guidelines and regulations in agreement to the Regulations for the Administration of Affairs Concerning Experimental Animals (Ministry of Science and Technology, Brazil). In addition, we confirmed the statement that the study was conducted following the ARRIVE guidelines.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Frezarim, G.B., Mota, L.F.M., Fonseca, L.F.S. et al. Multi-omics integration identifies molecular markers and biological pathways for carcass and meat quality traits in Nellore cattle. Sci Rep 15, 10467 (2025). https://doi.org/10.1038/s41598-025-93714-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-93714-x
