
Abstract
Prime editing (PE) is a promising gene editing method that exploits a reverse transcriptase fused to a Cas9, whose single guide RNA (sgRNA) is extended with a reverse transcriptase template containing the desired DNA modifications. Its efficiency and specificity are inconsistent, requiring extensive optimization. To address this, we propose prime editing with prolonged editing window (proPE), which uses a second non-cleaving sgRNA to target the reverse transcriptase template near the edit site. ProPE requires less optimization than PE and extends PE’s potential for allele-specific modifications. By overcoming five limitations of PE, proPE significantly increases overall editing efficiency 6.2-fold up to 29.3% for low-performing edits (<5% with PE) and broadens its applicability to modifications beyond the typical PE range, encompassing a major portion of human pathogenic single nucleotide polymorphisms. With these enhanced properties, proPE holds considerable promise for improved gene editing, including disease modelling and therapeutic intervention.

Similar content being viewed by others

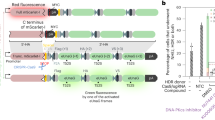
Main
Prime editing (PE), which requires neither the generation of DNA double-strand breaks nor the use of donor DNA1, reduces the production of unwanted modifications compared with nuclease-based gene editing methods. However, PE exhibits low efficiency and requires extended optimization, primarily of the 3′ extension of the prime editing guide RNA (pegRNA) that provides the primer and template for the reverse transcription2. The prime editor complex employs a nickase Streptococcus pyogenes Cas9 (SpCas9)-reverse transcriptase fusion protein (prime editor protein) with a pegRNA and proceeds in five steps (Fig. 1). In the first step (i), the prime editor protein–pegRNA complex binds the DNA and cleaves the non-target DNA strand. In the second step (ii), the released 3′ DNA end hybridizes to the 3′ primer binding site (PBS) of the pegRNA and the reverse transcriptase extends the non-target DNA strand along the reverse transcriptase template (RTT) part of the pegRNA. In the next step (iii), the DNA strand containing the edit (shown in turquoise) forms a 3′ DNA flap that equilibrates with the 5′ flap of the original DNA. Ideally, the 5′ flap is cleaved by an endogenous flap endonuclease and the DNA strand containing the edit is ligated in the fourth step (iv). The edit may become permanent in the last step of the process (v) if the resulting mismatch in the original DNA strand is repaired or when the cell undergoes replication.

Schematic representation of the PE process depicting the steps (i–v), where potential bottlenecks (A–E) may inhibit PE efficiency that are eliminated by the proposed corrective mechanisms of proPE (A*–E*). nCas9, nickase Cas9.
Several approaches have been developed that have effectively increased the efficiency of PE for a number of targets2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21. Other approaches focus on increasing its flexibility: smaller prime editors that can be packed into an adeno-associated virus have been created22,23,24,25,26,27,28,29,30,31. PE variants with altered protospacer adjacent motif (PAM) specificities32,33,34,35 may also increase the flexibility of the editing; however, these variants have been reported to have considerably lower editing activity than PE2,3,36. Attempts have also been made to reduce the formation of unfavourable structures in the pegRNA, although diminishing the efficiency. However, by using circularized RNA forms, that is, prime editing template RNA (petRNA), editing efficiencies could be restored23, although the overall efficiency of classical PE was not demonstrated23,37.
Here we introduce prime editing with prolonged editing window (proPE), a PE tool in which the reverse transcriptase (RT) templating function is targeted independently of the nicking PE complex at the vicinity of the nick site to ensure efficient editing. We have identified five bottlenecks that hinder traditional PE, but their effects are reduced in proPE (Fig. 1). Thus, proPE may substantially enhance editing efficiency when PE is compromised. Another major advantage of proPE derives from its need for two target sites, which further reduces the already low off-target effects of PE and allows allele- and gene-specific (for homologous genes) editing even in cases where PE is not sufficiently specific or effective.
Results
The steps where proPE can outperform PE
ProPE relies on two distinct single guide RNAs (sgRNAs), namely, essential nicking guide RNA (engRNA) and template providing guide RNA (tpgRNA). The engRNA is a conventional sgRNA that is used by the prime editor protein to nick the DNA, releasing the non-target strand. The tpgRNA, which contains the PBS and RTT sequences, facilitates binding to a near target sequence, presenting the PBS and RTT sequences in the vicinity of the nicked DNA strand. The tpgRNA harbours a truncated spacer (11–15 nucleotides), which makes the SpCas9 inactive, that is, it prevents the prime editor protein from nicking the DNA but allows it to bind to its target sequence38.
To provide a conceptual framework for the experiments that follow, we first outline the specific steps in the PE pathway where overall efficiency can be limited and proPE can confer improvements through distinct, hypothesized mechanisms (Fig. 1). For clarity, we summarize these bottlenecks (A–E) and mechanisms (A*–E*) below; supporting evidence for each will be presented in the subsequent results.
Regarding A and A*, the first step of the PE process (i) could be inhibited by the PBS–spacer interaction within the pegRNA as the PBS is complementary to the spacer. In contrast, no such intramolecular interaction occurs when using proPE as the spacer and PBS are on different sgRNAs, allowing routine editing with 17-nucleotide PBS. For B and B*, in the second step of the process (ii), proPE is less susceptible to the inhibitory effects of PBS-degraded tpg/pegRNAs than classical PE likely because the prime editor complex is inactive and uses a short spacer, ensuring a more dynamic exchange of the tpgRNAs on the target DNA39,40,41,42, therefore providing a greater chance of delivering an intact PBS within the appropriate time period during which the nicked non-target strand is available. For C and C*, in the third step of the process (iii), if the new DNA strand is either not fully synthesized or partially degraded, it may reduce the efficiency of PE as the absence of either a sufficiently long right homology arm or the edit diminishes editing efficiency. As the RNaseH domain of the reverse transcriptase is known to digest the RNA template after DNA synthesis30,43, the DNA strand can only be completed to full length via a new RTT after the prime editor protein with the degraded pegRNA has dissociated and a new pegRNA–prime editor complex has bound to the DNA. Using proPE, the inactive prime editing complex harbouring a tpgRNA with a short spacer ensures the faster exchange of tpgRNAs than pegRNAs and thus a more efficient completion of the truncated new DNA strand. The efficiency-enhancing effect of proPE via this mechanism may be more pronounced for edits further away from the nick site as longer DNA strands are more susceptible to degradation during flap equilibration44 and partial digestion is more likely to leave a truncated flap long enough for extension. For D and D*, in the fourth step of the process (iv), the prime editor complex might also re-bind to the DNA strand after the new strand has been created. This could inhibit the exchange of the two flaps and the subsequent DNA repair processes, and thus the incorporation of the edits. The re-binding can easily be reduced using proPE by adjusting the amount of engRNA to an optimal level without decreasing the amount of the RTT-PBS template. For E and E*, in the final step of the process (v), repeated nicking of the edited DNA (re-nicking) can be a frequent cause of low PE efficiency. Re-nicking can be reduced in proPE using a lower but still sufficient amount of engRNA. Apart from increasing the efficiency of editing where target re-nicking is a factor, using an optimal amount of nicking complex can also increase the editing specificity. We discuss bottlenecks D and E together as it is difficult to experimentally separate the effects of re-binding and re-nicking, although the latter is likely to reduce PE efficiency to a higher extent.
In the first part of this study, we used the recently introduced plasmid-based PE activity reporter (PEAR)45 to determine the conditions and parameters where proPE can be used effectively. In the second part, we investigated the efficiency, specificity and potential applications of proPE on genomic targets using amplicon deep sequencing of human cell lines.
First, we performed a proof-of-principle experiment with proPE on a genomic target and showed that editing is only possible in the presence of both tpgRNA and engRNA, even beyond the efficiency of PE. Applying engRNA with a non-targeting tpgRNA (tpgRNA containing a non-targeting truncated spacer with the targeting RTT-PBS) showed negligible editing (Fig. 2a and Extended Data Fig. 1a).

a, Amplicon sequencing results of PE3 using tpg/pegRNAs without the tevopreQ1 extension. The ‘eng fraction’ refers to the fraction of transfected engRNA plasmid (see Supplementary Methods), and T and NT indicate targeting and non-targeting tpgRNA, respectively. del, deletion; w/o, without. b, The effect of the amount of engRNA and tpgRNA plasmids was examined at 50% tpgRNA (top) and 1.24% engRNA (bottom) in the total RNA plasmid fraction, respectively. PE2 and proPE2 were used with tpg/pegRNAs without the tevopreQ1 extension. c, PEAR results showing the range of spacer lengths of tpgRNAs for achieving effective proPE editing. nt, nucleotide. PE2 and proPE2 were used for the PEAR-GFP targets, while PE3 and proPE3 were used for the PEAR-mScarlet targets. The tpg/pegRNAs were used without the tevopreQ1 extension. d, PE and proPE editing was assessed in the presence of increasing amounts of degraded RNAs, PBS-less (left plots), (RTT-PBS)-less (right plots), targeting the same site. PE2 and proPE2 were used for the PEAR-GFP target (top plots), while PE3 and proPE3 were used for the PEAR-mScarlet target (bottom plots). The editing values are normalized to the corresponding values from experiments where non-targeting degraded RNAs were co-expressed. The tevopreQ1 extension was used. Lines are fitted to the data points. The statistical significance between the slopes of the fitted lines was assessed using the non-linear module of GraphPad with the two-tailed extra sum-of-squares F-test. The parameters for the linear equations can be found in Source Data Fig. 2. See Supplementary Fig. 1 for further details on d. Each data point in a–c is presented as the mean ± SD of triplicates; in d, data points represent triplicate means normalized to the mean of the non-target control, with the ± SD including error propagation. In b–d, the sample names (PEAR-GFP-C1(S2) and so on) refer to given proPE or PE combinations, the sequences of which are provided in Supplementary Table 6.
Characterization of proPE using the PEAR assay
The PEAR plasmid contains an intron-interrupted sequence of a fluorescent protein (green fluorescent protein (GFP) or mScarlet) that regains activity when its acceptor splice site is restored (see Extended Data Fig. 1d for a schematic of the PEAR system). The efficiency observed with PEAR plasmids is reflective of the efficiency observed at genomic sites (Extended Data Fig. 1b,c). The PEAR system is preferable to endogenous target testing because it allows the effect of a single parameter to be systematically studied while leaving other parameters of PE unchanged. We used the PEAR system to demonstrate the enhancing effects of proPE, to investigate the mechanisms by which proPE improves editing and to identify the parameter ranges in which proPE is effective.
Decreasing engRNA levels could improve editing efficiency
The following experiment addresses bottlenecks D and E. When increasing the amount of tpgRNA, editing reaches saturation; however, when increasing the amount of engRNA, it reaches a peak and then declines (Fig. 2b). This suggests that the low efficiency of PE may sometimes be a result of either too little or too much nicking activity. In proPE, this nicking activity can be adjusted for each edit without changing the amount of the RTT-PBS sequence. To exploit this advantage of proPE over PE, we routinely tested two or three engRNA coding plasmid quantities in parallel transfections to find the most efficient condition. For clarity, only the results of the most efficient condition are shown; the results of all conditions are provided in an Extended Data figure or Supplementary figure as indicated in the corresponding figure legends.
Degraded tpgRNAs cause less inhibition than degraded pegRNAs
When investigating the spacer length of tpgRNA that enables efficient proPE activity, first we observed that decreasing the spacer length to 15 nucleotides resulted in the SpCas9 nuclease no longer generating indels46 (Extended Data Fig. 1e). Effective proPE editing was achieved with spacer lengths of 10–15 nucleotides, although editing was detectable even with spacers as short as 5 nucleotides (Fig. 2c and Extended Data Fig. 1f).
The following experiments address bottleneck B. Editing is considerably inhibited by degraded pegRNAs4; however, we observed significantly less inhibition of proPE than of PE (Fig. 2d and Supplementary Fig. 1). We proposed that a shortened spacer length, which ensures that SpCas9 becomes inactive and can only bind DNA, offers additional advantages. Contrary to the H840A nickase version, which remains bound to its target for extended periods, similarly to active SpCas9 (ref. 41), inactive SpCas9 does not establish a stable post-cleavage conformation effectively41. Furthermore, when an inactive SpCas9 complex has a shorter spacer, it dissociates from its target more quickly than with a 20-nucleotide spacer, resulting in an even shorter dwell time39,40,42, although the magnitude of this difference is smaller than the difference between the dissociation rates of active and inactive complexes41,42. We proposed that using the prime editor protein with a tpgRNA (which forms an inactive complex due to the short spacer) also results in shorter dwell times, facilitating a more efficient re-priming of reverse transcription (B* in Fig. 1).
To provide support for this more dynamic exchange interpretation, we examined a prime editor protein harbouring a dead SpCas9 and tpgRNAs with either 12- or 20-nucleotide spacers, where the essential nicking was performed by a Staphylococcus aureus Cas9 (SaCas9) prime editor31 (Extended Data Fig. 1g). However, the difference between the dwell times observed here is expected to be substantially reduced compared with that between PE (with an active SpCas9) and proPE (with SpCas9 that is inactivated by tpgRNA). The tpgRNAs with the 20-nucleotide spacers, which have longer dwell times, caused greater inhibition than those with 12-nucleotide spacers (Extended Data Fig. 1g).
Working distance between the engRNA and tpgRNA target sites
Using the same engRNAs and tpgRNAs, the engRNA–tpgRNA target distance at which proPE is more effective than PE in these examples was found to range from 0 to ~30 nucleotides for the trans-oriented targets and from ~6 to ~30 nucleotides for the cis-oriented targets (Extended Data Fig. 2a and Supplementary Fig. 2a). These distances translate to ~70 nucleotides between the two PAMs in the trans orientation and to ~44 nucleotides in the cis orientation.
To assess the extent to which the requirement for a second PAM within a certain distance imposes a constraint, we analysed the ClinVar database (version: 20240502, Pathogenic mutations)47 and found that 98.6% of the closest targets to a pathogenic single nucleotide polymorphism (SNP) have at least one PAM suitably positioned and oriented for tpgRNAs. This supports the notion that the need for two PAMs does not limit the practical utility of our approach.
Characterization of proPE with respect to PBS and RTT length
Using the PEAR system, we observed that proPE requires less RTT optimization and is equally effective with various types of prime editor, such as PE2, PE3 (ref. 1) and PE5 (ref. 8), using tpg/pegRNAs or engineered tpg/pegRNAs (etpg/epegRNAs; with the tevopreQ1 extension4) and PEmax (ref. 8) variants (Extended Data Fig. 2b,c and Supplementary Fig. 2b,c). We also found that decreasing the complementarity between the spacer and the PBS can increase PE efficiency, suggesting an inhibitory effect of their interaction (Extended Data Fig. 2d).
Although the PEAR assay is a very versatile and powerful system, it does not provide any information on the indel background. To obtain this information, we further characterized proPE mainly through amplicon sequencing of edited endogenous targets.
The following experiment addresses bottleneck A. We first compared the efficiency of 6 modifications generated with both 17-nucleotide or shorter (8–15-nucleotide) PBS sequences. While PE was substantially less efficient with the 17-nucleotide PBSs, proPE showed reduced sensitivity to variations in the length of the PBS (Fig. 3a and Extended Data Fig. 3), demonstrating its ability to overcome the inhibition of PE caused by intramolecular spacer–PBS interactions (bottleneck A). These experiments therefore suggest that proPE requires less extensive optimization of the RTT and PBS than PE to achieve efficient editing.

Amplicon sequencing results of PE5max editing and the corresponding proPE5max editing of genomic targets. a, ProPE is markedly less sensitive to PBS length than PE. b,c, The efficiency (b) and specificity (edit/indel ratio) (c) of proPE were not significantly different from PE when tested on 13 within-target edits. d, The editing efficiency of PE decreases as the distance between the editing position and the nicked position increases. The tevopreQ1 extension for tpg/pegRNAs was used in the experiments presented in d,f–h. e, Schematic showing the numbering of the modification position and target sequence. f,g, ProPE is substantially more effective (f) and specific (g) than PE for target-distal edits. The light-grey horizontal lines indicate the ratio where the editing efficiencies and specificities of PE and proPE are the same. h, ProPE shows a significantly higher re-elongation propensity for all three distinct A+B edits, as indicated in the figure. Further details are provided in Extended Data Fig. 3 for a–c, in Extended Data Fig. 4a–c for d, f and g, and in Extended Data Fig. 4d,e for h. For datasets with a normal distribution, statistical significance was assessed by a one-way analysis of variance (ANOVA) (a), a repeated measures (RM) one-way ANOVA (d) or a two-tailed unpaired t-test (b) and means are shown (a,b,d). For datasets with a non-normal distribution, statistical significance was assessed by a two-tailed Mann–Whitney test (c) and a Friedman test with a two-tailed Dunn’s test (f,g) and medians are shown for each dataset (c,f,g). Multiple unpaired t-tests were applied in h. P values are indicated at the top of the graphs (a–d,f,g) or in the plots (h). Each data point is presented as the mean of triplicates (a–d,f,g) or the mean ± SD of triplicates (h). Sample sizes were N = 6 (a), 13 (b,c) and 8 (d,f,g).
Characterization of proPE for the position of the edit
PE and proPE are defined by the combination of the specific pegRNA and second nicking sgRNA and by the combination of engRNA, tpgRNA and the second nicking sgRNA, respectively. We compared the generation of 13 previously described modifications using the RTT and the PBS lengths described in previous studies1,4,8,45 and tpg/pegRNAs without the tevopreQ1 extension and testing a few additional second nicking sgRNAs, resulting in 50 proPE combinations. For each combination, three different amounts of engRNA coding plasmid were transfected (Extended Data Fig. 3). The most efficient proPE and PE combinations for the 13 edits are not significantly different (Fig. 3b,c). This apparent inconsistency with the PEAR experiments, where proPE was more efficient, can be explained if the exact position of the edit is taken into account. The 13 edits were based on the literature1,4,8,45, and all but one were located within the target at one of the positions 1–3 (seed region) or at positions 5 and 6 (PAM region), which can drastically reduce the inhibitory effect of re-nicking (bottleneck E). Also, within-target edits require only a short length for the new DNA strand, making it less prone to degradation or failed synthesis, and even when these do occur, it may become too short for re-elongation (decreasing the effect of bottleneck C and the potential of proPE to overcome bottleneck C). Our investigation using the PEAR plasmids was not restricted to edits within the target.
To investigate the effect of the position of the edit, we designed edits at three different distance groups from the nick site for each of the eight targets with the second nicking sgRNAs and PBS lengths used in previous studies1,4,8,45, while the RTT necessarily differed for the edits at different positions. The first group contained edits within the PE target (positions 1–3, 5 and 6), called within-target edits, the second group in target-proximal positions (positions 4 and 7–10) and the third group in target-distal positions (>10). Figure 3d,e and Extended Data Fig. 4a–c show that PE is significantly weaker at the target-proximal edit positions (mean 17.6%) than at the within-target positions (mean 39.2%); the PE activity decreases even further with increasing the distance from the nick to the target-distal positions (mean 9%), in line with the literature48. In contrast, proPE is less sensitive to the distance of the edit from the essential nick, improving the efficiency of target-distal edits, with median proPE/PE ratios of 6.9 and 7.0 for editing and specificity, respectively (Fig. 3f,g and Extended Data Fig. 4a–c). This may be related to the fact that more inhibitory mechanisms of PE are at play in target-distal edits compared with within-target and target-proximal edits. It is also noteworthy that this increase is achieved with shorter PBSs and by using the tevopreQ1 extension, which tend to reduce the effect of the proposed corrective mechanisms A* (‘no intramolecular spacer–PBS interaction’) and B* (‘more efficient RT priming’), respectively, and emphasizes the role of mechanisms C* (more efficient DNA re-elongation) (Fig. 3h), D* and E* (decreased re-binding and re-nicking) (Fig. 2b).
Effect of proPE on target-distal edits
The following experiment addresses bottleneck C. Flaps produced during PE are likely to be subject to frequent digestion, similarly to other flaps in the cell49,50,51. Such truncated flaps negatively impact the editing process, as demonstrated in a recent study that showed that the co-expression of a flap nuclease resulted in reduced editing efficiency, predominantly in the case of longer flaps44. Truncated flaps may also form due to failed synthesis, especially of longer flaps. The inhibitory effect of truncated flaps during PE can be observed through the lower editing efficiency for long DNA flaps without strong secondary structure, as the absence of a strong secondary structure makes them more susceptible to nuclease digestion44. The digestion of the flap during PE helps to explain the different inhibitory effects of the two types of degraded pegRNA used in our study. Specifically, the longer PBS-less pegRNAs exhibited weaker inhibition than the shorter (RTT-PBS)-less pegRNAs (Fig. 4a,b). This difference probably arises because only PBS-less pegRNAs containing a complementary RTT sequence can repair truncated flaps through re-elongation (Fig. 4c). An earlier study using pegRNAs with shorter flaps, which are less susceptible to degradation, supports this interpretation as there was no difference in the inhibitory effects of the two types of degraded pegRNA4. Altogether, these results strongly suggest that truncated flaps contribute to the frequent low efficiency of PE, particularly for target-distal edits.

DNA flaps harbouring target-distal edits seem to be 3′-truncated and subsequently re-elongated by pegRNAs containing only the RTTs. a,b, PEAR results for PE2 editing of PEAR-GFP targets (a) and PE3 editing of PEAR-mScarlet targets (b). The tevopreQ1 extension was used. PBS-less pegRNAs are less inhibitory than (RTT-PBS)-less pegRNAs. PE was assessed in the presence of increasing amounts of degraded RNAs missing either the PBS or (RTT-PBS) targeting the same site. The editing values are normalized to the corresponding values from experiments where non-targeting degraded RNAs were co-expressed. The error bars (± SD) include error propagation. The dashed lines are fits to the data points. The statistical significance between the slopes of the fitted lines was assessed using the non-linear module of GraphPad with the two-tailed extra sum-of-squares F-test. The parameters for the linear equations can be found in Source Data Fig. 4. The sample names (PEAR-GFP-C1(S3) and so on) refer to PE combinations, the sequences of which are provided in Supplementary Table 6. See Fig. 2d and Supplementary Fig. 1 for further details on these data. c, Schematic illustrating our interpretation of the results shown in a and b.
ProPE effectively reduces the effect of this bottleneck, as shown in Fig. 3h and Extended Data Fig. 4d,e. In an experimental model, where editing depends exclusively on the re-elongation of truncated flaps, proPE demonstrated higher editing efficiency than PE. As discussed earlier, bottleneck C exerts a stronger inhibitory effect for target-distal edits. Therefore, we anticipated the enhancing effect of proPE to be more pronounced at these positions. We also expected this effect to be further enhanced by the ability of proPE to more efficiently re-elongate truncated flaps derived from longer flaps as they remain long enough more often for effective repair through re-elongation. As predicted, in Fig. 5a, proPE showed substantially greater enhancement for target-distal edits. In this experiment, all of the other potential bottlenecks covered in this article (bottlenecks A, B, D and E) were controlled, ensuring appropriate comparison of the effect of different flap lengths in target-proximal and target-distal edit pairs. These results are consistent with the interpretation that the repair of truncated flaps (mechanism C*) is a critical factor in the improved editing efficiency of proPE at target-distal positions. We speculate that the corrective mechanism of proPE for bottleneck C is the same as for bottleneck B. Re-elongation requires the replacement of the degraded tpgRNA–PE protein complex with a new one harbouring an intact RTT, which is facilitated by the reduced dwell time observed in Extended Data Fig. 1g and Fig. 2d.

Amplicon sequencing results of editing genomic targets. a, The editing efficiency of PE decreases as the edit position is moved into the target-distal group, while this difference is less pronounced with proPE. Comparison of PE5max and proPE5max editing for target-proximal and target-distal edits of the same targets using the same tpg/pegRNA with the tevopreQ1 extension with differences only in the RTT sequence containing the edit. This design ensures that, apart from bottleneck C (truncation and re-elongation of the flap), differences between target-proximal and target-distal edits in the effect of bottlenecks are minimized and non-systematic. Editing data were extracted from Fig. 3d,f for better comparison. b,c, Re-evaluation of amplicon sequencing results in HEK293 cells showing editing ratios between samples with different amounts of engRNA plasmid (1:40 and 1:1 engRNA fractions). ProPE tends to be more efficient with the lower amount of engRNA when the corresponding PE exhibits high indel rates (b) or when it is applied to out-of-target edits (c). The light-grey lines in b and c indicate the ratio where the proPE editing with 1:40 and 1:1 engRNA fractions are the same. Data were obtained from the experiments shown in Fig. 7b (b) and Figs. 3, 6a,b and 7b,d (c). For the dataset with a normal distribution, statistical significance was assessed by a two-tailed unpaired t-test and means are shown (a). For the dataset with a non-normal distribution, statistical significance was assessed by a two-tailed Mann–Whitney test (P < 0.0001) and medians are shown (b). Each data point is presented as the mean of triplicates (a–c). P values are indicated at the top of the graphs (a,b). Percentages indicate the fraction of the samples where proPE editing is more efficient (ratio >1) or less efficient (ratio <1) using 1:40 compared with 1:1 engRNA fractions (c).
Effect of proPE on out-of-target edits
The following experiments address bottlenecks D and E. Re-binding and re-nicking by the prime editor complex is expected to negatively impact editing efficiency for out-of-target edits (positions 4, >6). This effect can be reduced with proPE by fine-tuning the amount of engRNA, which can be adjusted irrespective of the amount of RTT for reverse transcription (Figs. 2a,b, 5b,c and 6a). Interestingly, sometimes proPE can function with very small amounts of engRNA plasmid, so we investigated whether such small amounts of engRNA plasmid could indeed cleave the target, yielding detectable indels using the SpCas9 nuclease. Extended Data Fig. 5a,b shows that proPE can yield edits as efficiently as regular PE and that indels can be detected with SpCas9 nuclease even with a substantially reduced 1:80 engRNA fraction (0.4 ng engRNA coding plasmid). The higher proPE efficiency with reduced engRNA levels is observed more often when either the PE produces a higher indel background (Fig. 5b) or in the case of out-of-target edits (Fig. 5c). In both scenarios, re-nicking is expected to be more frequent compared with low-indel-performing PE or within-target edits, respectively. These findings demonstrate that the higher efficiency of proPE observed with a reduced amount of engRNAs is associated with excessive re-nicking and re-binding during editing, highlighting the importance of bottlenecks D and E in the reduced PE and enhanced proPE efficiencies.

Amplicon sequencing results of PE5max editing and the corresponding proPE5max editing of genomic targets. For tpg/pegRNAs, the tevopreQ1 extension was used. a, Different editing efficiency, indel and specificity (edit/indel ratio) patterns are evident with decreasing engRNA fractions when installing edits to HBB, HEK4 site (at the DNMT3B gene), HEXA and PRNP. b,c, ProPE effectively increases the editing efficiency (b) and specificity (edit/indel ratio) (c) of low-performing edits (<5% with PE) for target-distal edits. d–g, Results of editing using proPE in HEK293 cells compared with U2OS and K562 cells (d,e) and HuES cells (f,g), showing an increase in both editing efficiency (d,f) and specificity (edit/indel ratio) (e,g) in all tested cell lines compared with PE. Further details are provided in Extended Data Fig. 5e–g for b and c, in Extended Data Fig. 6 for d and e, and in Extended Data Fig. 7a–c for f and g. The results with the HEK293 cells are extracted from Fig. 3d,f,g for better comparison. Each data point is presented as the mean (b–g) and the mean ± SD (a) of triplicates. Statistical significance was assessed by a two-tailed unpaired t-test for HEK293 (d,f), K562 (e) and HuES (g) cells and by a two-tailed Mann–Whitney test for HEK293 (b,c,e,g), U2OS (d,e), K562 (d) and HuES (f) cells. Medians are shown in the plots (b–g). P values and fold change between proPE and PE are indicated at the top of the plots and either directly above or next to the data points, respectively (b–g). Sample sizes were N = 10 for b,c,f,g and N = 8 for d,e.
Although isolating a single bottleneck experimentally is challenging, it is evident that the resolution of bottlenecks A and B offered by proPE may offer advantages for edits at any position, while for bottlenecks D and E, it provides distinct benefits, particularly with out-of-target edits. In addressing bottleneck C, proPE demonstrates a pronounced advantage for target-distal edits.
ProPE enhances efficiency most for low-performing edits
We next tested 15 additional edits at target-distal positions with a standard 13-nucleotide PBS and the same 8 eng/pegRNA targets used before. For these edits, proPE increased median editing efficiency from 1.4% to 5.5%, while median specificity increased 1.5-fold (Extended Data Fig. 5c–g). ProPE increased editing efficiency to a greater extent for edits where PE worked with low efficiency, presumably, due to a combination of the inhibitory factors shown in Fig. 1. We consider 5% to be the efficiency limit above which single cell cloning attempts are feasible. When we selected edits with a PE editing efficiency of less than 5% (median 0.7%), proPE improved the efficiency up to 14.9%, increasing the median by a factor of 6.5 (Fig. 6b and Extended Data Fig. 5e), while the specificity of the editing increased by a factor of 7.3 (Fig. 6c and Extended Data Fig. 5f,g).
As PE is particularly efficient in HEK293 cells, probably because of its compromised mismatch repair, we explored the applicability of proPE in other cell lines that are less amenable to PE, including human embryonic stem (HuES) cells, as stem cells are notoriously difficult to engineer. The efficiency of proPE relative to PE was assessed by generating eight edits in U2OS and K562 cells (Fig. 6d,e and Extended Data Fig. 6) and ten edits in HuES cells (Fig. 6f,g and Extended Data Fig. 7a–c). ProPE increased the median of the editing efficiency by 5.4-, 9.0- and 4.4-fold (Fig. 6d,f and Extended Data Figs. 6a and 7a) and the median specificity by 2.1-, 2.3- and 1.7-fold (Fig. 6e,g and Extended Data Figs. 6b,c and 7b,c) in the U2OS, K562 and HuES cell lines, respectively. While the optimal engRNA fraction varied with the edits and targets in the HEK293 cells, in the U2OS and K562 cells the fraction of 1:1 proved to be the best for most of these edits, and in the HuES cell line the optimal fraction was 1:40.
ProPE enables access to the majority of human pathogenic SNPs
Enabling PE to be used to generate or correct the majority of human pathogenic SNPs in cell lines4,8 of choice would immensely facilitate the study of pathogenic mechanisms in vivo22,31,52,53,54,55 as well as aid the development of future therapies. Most of the studies on PE have focused on within-target edits1,3,4,8,23,44,56,57,58,59,60,61; however, our results show (Fig. 3d) that the editing efficiency for positions outside the target decreases sharply with distance from the nick site. This phenomenon is also evident in the very recent data of Yu et al.48, who did not restrict their investigations to within-target edits. More than half of the pathogenic SNPs lie outside the target sequence (Fig. 7a), where PE tends to show reduced activity with increasing distance. To better evaluate the potential of proPE to make such modifications, we generated edits that lie outside the target in clinically relevant genes of the human cytochrome P450 superfamily (CYP). These enzymes are involved in many processes, including drug metabolism and the synthesis of cholesterol, steroids and other lipids62. Sequence variations in the CYP genes of patients are often responsible for different individual responses to drug treatment63. We generated a number of naturally occurring variations in the CYP1A1, CYP1A2 and CYP2B6 genes and tested 62 different PE combinations for which no previous editing information was available. We used both PE4max and PE5max editors and 13- and 17-nucleotide PBSs as well as tpg/pegRNAs with the tevopreQ1 extension. Consistent with the literature3,48,64, PE was less efficient with the 17-nucleotide PBS, whereas proPE, as we have demonstrated in Fig. 3a, did not show a decrease in editing efficiency (Fig. 7b,c and Supplementary Fig. 3). The experiment also confirmed the result obtained with the PEAR plasmid system (Extended Data Fig. 2c), that proPE causes a similar increase in editing efficiency regardless of the introduction of a second nick (Extended Data Figs. 7d,e and Supplementary Fig. 3). Overall, the PE efficiency did not reach 5% in 53 PE combinations (the median efficiency was 1%). In none of these 53 combinations did PE exceed the corresponding proPE combination, while proPE increased the editing efficiency for these modifications, with a median proPE/PE ratio of 5.13, up to 20%, and also the specificity, with a median proPE/PE ratio of 3.46 (Extended Data Figs. 7f,g and Supplementary Fig. 3).

a, The distance distribution of human pathogenic mutations from the closest PE target. Less than half of the pathogenic SNPs in the ClinVar database are located in within-target positions that are generally amenable to effective modification. b–f, Amplicon sequencing results of PE5max and proPE5max editing (b–f) as well as PE4max and proPE4max editing (b,c,f) genomic targets. For tpg/pegRNAs, the tevopreQ1 extension was used; f also contains editing results without using the tevopreQ1 extension. Panels b and c show that proPE is less sensitive to PBS length than PE, indicating that minimal PBS optimization is required. ProPE increases the editing efficiency (b) and specificity (edit/indel ratio) (c) for out-of-target edits of clinically relevant CYP genes regardless of the PBS length (N = 31). In d and e, proPE shows potential in terms of efficiency (d) and specificity (edit/indel ratio) (e) for editing pathogenic mutations that are not accessible in the effective editing window of PE. Panel f shows that proPE has a strong enhancing effect on the editing efficiency and specificity of low-performing edits (<5% with PE). The light-grey line indicates the ratio where the editing efficiencies and specificities of PE and proPE are the same (N = 76). The sequences of RNAs targeting T1 to T10 target sites are provided in Supplementary Table 3 (d,e). Medians are shown in b, c and f. Each data point is presented as the mean (b–f) and the mean ± SD (d,e). Statistical significance was assessed by a Kruskal–Wallis test with two-tailed Dunn’s test. P values and fold change between proPE and PE are indicated above the plots and next to the data points, respectively (b,c). Further information on b and c can be found in Supplementary Fig. 3, on d and e in Extended Data Fig. 8a–c and on f in Extended Data Fig. 8d,e.
In HEK293 cells, we also compared the ability of proPE and PE to generate seven human pathogenic SNPs located in the RYR2 and SCN5A genes, both of which cause cardiovascular diseases, and in the KRT12 gene, which causes inheritable corneal dystrophy. These edits are ≥10 nucleotides from the nearest target sites. For each disease mutation, PE was compared with two proPE combinations using different tpgRNA targets. For two mutations, no detectable editing was observed with either PE or proPE, probably due to the presence of polyT stretches within the RTT. For the other five disease mutations, in seven out of ten attempts, editing efficiency was increased by proPE, with proPE/PE ratios between 1.5- and 7.1 (Fig. 7d,e and Extended Data Fig. 8a–c).
The experiments on genomic targets confirm the results obtained with the PEAR system and show that edits that can only be created with low efficiency using PE benefit the most from the use of proPE. In total, 130 different genomic PE4max and PE5max combinations were tested in HEK293 cells in this study, of which 59% (76 cases) did not reach the 5% efficiency level with PE. For these combinations, the median of the proPE/PE ratio was 6.0, improving the editing efficiency up to 29.3% (Fig. 7f and Extended Data Fig. 8d). Several previous approaches that increased the efficiency of PE were also associated with a concomitant increase in indel modifications; proPE was able to install these 76 edits with an increase in specificity with a median of the proPE/PE ratio of 3.8-fold (Fig. 7f and Extended Data Fig. 8e).
PAM-flexible SpCas9 variants35 could provide further advantages for SpCas9-based gene editors and also further increase the availability of pathogenic mutations for PE. However, when comparing the activities of PE and proPE with those of SpRY-PE and SpRY-proPE on eight edits, we observed that both SpRY-PE and SpRY-proPE showed lower overall efficiency than standard PE and proPE (Extended Data Fig. 8f and Supplementary Fig. 4), consistent with the literature36.
Recent approaches may provide benefits similar to proPE. We compared the activities of split PE (sPE) with petRNA23 and two of the highest performing split pegRNA prime editor (SnPE) variants37 (SnPE-5′-MS2 and SnPE-5′-BoxB) with proPE across nine edits and found that proPE consistently exhibited higher activity than split PE with petRNA and both SnPEs (Extended Data Fig. 9a,b).
Recent studies have shown that PE can be successfully applied using adeno-associated virus (AAV) delivery by splitting the editor into two AAV vectors. To determine whether proPE is compatible with AAV delivery, we adapted the v3em PE3-AAV vectors developed by Davis et al.26 to co-express the engRNA, tpgRNA and second nicking sgRNA. We tested these AAV vectors and viruses by transfecting and transducing HEK293 cells, respectively, to introduce the G127V mutation into the prion protein, which confers resistance to Creutzfeldt–Jakob disease. The results demonstrated substantial editing efficiency with both AAV vectors and viruses (Extended Data Fig. 9c), indicating that proPE can be adapted to AAV delivery systems, making it a promising candidate for potential clinical applications.
ProPE improves allele-specific editing and lowers off-target edits
PE works with relatively few off-target edits and off-target indels65,66. As proPE requires the presence of two target sites, whereas PE requires only one, proPE is expected to work on even fewer off-target sites. As a proof of concept, we demonstrated this using the HEK4 site at the DNMT3B gene to introduce a G-to-T edit into position 2, which, based on the literature, could be introduced by PE only with off-target editing60,67,68. Figure 8a,b shows that the use of proPE substantially increases the specificity with respect to off-target editing, on-target indels and off-target indels by 7.0-, 5.0- and 4.7-fold, respectively, while maintaining the on-target editing of PE.

Amplicon sequencing results of PE5max/proPE5max (a,b) and PE4max/proPE4max (d,e) editing of genomic targets. For tpg/pegRNAs, the tevopreQ1 extension was used in d and e. a,b, The off-target modifications produced by PE are reduced by proPE when installing HEK4 2G>T edit, as measured by the metrics of both the ratio of on-target and off-target edits (a) and the ratio of on-target edits and either on-target indels or off-target indels (b). c, Sequence of the nearby region of two homologous genes showing the position to be altered and target sites used. A1 and A2 indicate the target (CYP1A1) and non-target (CYP1A2) genes, respectively. The T1 engRNA target was used with either the T2 tpgRNA target (in cis orientation) or the T3 tpgRNA target (in trans orientation) for proPE editing. T2 can be used by PE for gene-specific editing but not T3 due to its opposite orientation, therefore no PE control is presented for trans editing. Target PAMs are indicated by underlining. d,e, Editing and specificity results for two alternative approaches to efficient gene-specific proPE editing for a modification where PE fails to provide efficient gene-specific editing. The specificity of PE and proPE editing is shown by the metrics of the ratio of the edits at target and non-target sites (d) and the ratios of edit at target sites and indels at target or non-target sites (e). Each data point is presented as the mean ± SD (a,b,d,e). See Extended Data Fig. 10 for further details of the data in d and e.
ProPE has enormous potential for generating clinically relevant models in an allele-specific manner or, in the case of homologous gene families, in a gene-specific manner. PE can also be allele- or gene-specific if the sequence stretch that distinguishes the two alleles/genes is located in the PAM or seed region of the PE target. The unique properties of proPE allow allele- and gene-specific editing in situations where PE cannot be used due to the lack of such a target with the proper orientation or within the appropriate distance. This unique feature is demonstrated by the following two examples. In both cases, with PE4max, we targeted the 196A>G mutation (rs35035798), resulting in a Met-to-Val amino acid change within a region of the CYP1A1 gene that is almost identical to the corresponding sequence of the CYP1A2 gene (Fig. 8c). There is a target site (T1) in which the mutation is located at the seventh editing position. Although PE can achieve an editing efficiency of 13% from T1 (Fig. 8d and Extended Data Fig. 10), due to the lack of sequential variation in the target site between the two genes, T1 cannot be used by PE in a gene-specific manner. In contrast, proPE demonstrated similar efficiency in a gene-specific manner using T1 as the engRNA target in combination with tpgRNA targets (T2 and T3) lying on gene-specific sequence stretches.
In the first case, we used a tpgRNA target (T2) that is found only in the CYP1A1 gene and not in CYP1A2, but is 49 nucleotides away from the mutation, a distance at which PE is no longer effective (it achieves only 1.9% editing; Fig. 8d and Extended Data Fig. 10). There is no other target between the position of the mutation and the T2 target that would be suitable for generating the mutation in a gene-specific manner using PE. Applying a tpgRNA directed to the cis-oriented T2 target with proPE4max, an editing efficiency of 14% was achieved with a highly increased specificity compared with PE for target site edit/non-target site edit (4.8-fold), target site edit/target site indel (3.7-fold) and for target site edit/non-target site indel (7.4-fold), shown in Fig. 8d,e and Extended Data Fig. 10.
In the second example, we used the T3 target for tpgRNA, which is in a trans orientation (that is, on the opposite strand of the DNA) and is therefore not suitable for PE. However, with proPE4max, 18.51% editing could be achieved in a gene-specific manner, with a highly increased specificity compared with PE for target site edit/non-target site edit (4.0-fold), target site edit/target site indel (3.0-fold) and target site edit/non-target site indel (9.7-fold), shown in Fig. 8d,e and Extended Data Fig. 10.
Although an even higher level of editing could be achieved by using proPE5max, and the gene specificity of the on- and off-target edits is not reduced compared with proPE4max, proPE5max is less advantageous because there is a significantly higher background of on- and off-target indels characteristic of the use of a second nicking sgRNA in PE3- and PE5-type approaches (Extended Data Fig. 10).
These examples demonstrate how proPE can effectively extend the effective editing distance from the gene-specific sequence stretch and how we can exploit gene-specific targets in a PE-incompatible orientation to achieve significant increases in specificity when performing allele- or gene-specific editing.
To facilitate the application of proPE, an online design tool, proPE planner, is available via http://prope.welker-group.hu. The planner facilitates the selection and design of engRNA–tpgRNA spacer pairs and suggests RTT and PBS length based on rational design rules.
Conclusions
CYP genes and their enzymes play a critical role in drug metabolism, toxin processing and hormone synthesis. Genetic variations in these genes can affect an individual’s response to drugs, highlighting the importance of studying CYP gene polymorphisms for personalized medicine and optimizing drug therapies. However, current allele-specific gene editing approaches face challenges due to the high sequence identity between homologous CYP genes, as the difficulty of allele-specific editing lies in the rarity of allele variations at the appropriate positions. To make precise modifications, the sequence variation that distinguishes the alleles must both disrupt the target (that is, affect the PAM and/or seed region) and be in close proximity to the modification site. Typically, a distance of not greater than about 15 nucleotides is required for base editing, PE and using single-stranded homologous recombination donor DNA. Our experiments show that proPE extends this distance to over 50 nucleotides for allele-specific editing, highlighting the prominent potential of proPE to advance our understanding of genetic variations and their impact in homologous genes such as the CYP gene family.
Previous studies1,3,4,8,23,44,56,57,58,59,60,61 that focused on increasing PE efficiency may not have recognized the rapid decline in efficiency for edits further away from the target, presumably because they examined only a few edits located beyond 10 nucleotides (nine out of thousands reported in the references surveyed1,3,4,8,44,56,61). We have identified bottlenecks that markedly reduce the efficiency for out-of-target edits and shown that proPE is substantially more effective for these edits, which encompass half of all pathogenic human SNPs.
One of the greatest challenges in biomedical genome engineering is the editing of stem cells, which are very difficult to edit. We were able to make the same edits in HuES cells using PE5max with epegRNAs with approximately 20-fold lower efficiency than in HEK293 cells. Thus, the large increase in efficiency achieved with proPE for low-performing edits may be particularly important in facilitating the generation of individual clones of induced pluripotent stem cells.
In summary, by increasing editing efficiency above a critical level (>5%), proPE may make it practical to generate disease models for the majority of pathogenic SNPs and may also considerably contribute to expanding the potential of PE to enable its use in therapeutic interventions.
Methods
Plasmid construction
Oligonucleotides used in this study
All sequences of the spacers and RTT-PBSs of the RNAs used in this study are listed in Supplementary Tables 2 and 3 (for PEAR and next-generation sequencing (NGS), respectively), while the RNA-coding sequences for the sPE-petRNA and SnPE systems are provided in Supplementary Table 4. The sequences of all other linkers and PCR primers used in this study can be found in Supplementary Table 1. All oligonucleotides were purchased from Sigma-Aldrich except the oligonucleotides used for cloning the plasmid constructs in AAV production, which were kindly provided by Gy. Ferenc, Laboratory of Nucleic Acid Synthesis, Institute of Plant Biology, HUN-REN BRC, Szeged, Hungary.
RNA-expressing plasmids
To monitor transfection efficiency, the RNA-expressing plasmids contained an mCherry expression cassette for the GFP-PEAR experiments and a TagBFP (blue fluorescent protein derived from Entacmaea quadricolor) expression cassette for the mScarlet-PEAR and NGS experiments. The construction of the RNA cloning plasmids is detailed below for each RNA type. Spacer coding linkers were inserted into the RNA cloning plasmids between BpiI sites using 3 U of the BpiI enzyme, 2 U of T4 DNA ligase, 500 μM ATP, 1× Green buffer, 50 ng vector and 0.25 μM of each oligonucleotide. For RTT-PBS cloning, the unique linker was inserted into either the RNA cloning plasmid or the spacer-containing RNA cloning plasmids between Esp3I sites using 3 U of the Esp3I enzyme, 2 U of T4 DNA ligase, 500 μM ATP, 1× Tango buffer, 1 mM dithiothreitol, 50 ng vector and 0.25 μM of each oligonucleotide.
Cloning plasmids for engRNA-expressing and second nicking RNA-expressing plasmids were generated as follows. Cloning plasmids for SpCas9 sgRNA were created by Simon et al.45: pAT9658-sgRNA-mCherry and pAT9679-sgRNA-BFP. The SaCas9 engRNA cloning plasmid (pSLK20330-Sa-sgRNA-mCherry) was constructed by assembling linkers encoding the SaCas9 sgRNA scaffold into the BpiI- and Esp3I-digested pDAS12069-U6-pegRNA-mCherry using an NEB HiFi assembly kit.
Cloning plasmid for tpg/pegRNA-expressing plasmids was generated as follows. tpg/pegRNA cloning plasmids without the tevopreQ1 extension were created by Simon et al.45; pDAS12069-U6-pegRNA-mCherry and pDAS12222-U6-pegRNA-BFP. For tpg/pegRNA cloning plasmids with a tevopreQ1 extension4, a linker containing an exchangeable cassette followed by the tevopreQ1 extension was cloned between the Esp3I sites in pDAS12222-U6-pegRNA-BFP and pDAS12069-U6-pegRNA-mCherry plasmids, resulting in pSLK7824-U6-pegRNA-epeg-BFP and pSLK7822-U6-pegRNA-epeg-mCherry, respectively.
Cloning plasmid for petRNA expressing plasmids was generated as follows. A cloning vector for petRNA23 with the same backbone as the tpg/pegRNA vector, pSLK20322-U6-petRNA-BFP, was constructed using an NEB HiFi assembly kit. PCR was employed to amplify the essential 5′ and 3′ components of the circular RNA (ribozyme, ligation arm, MS2 and flanking sequences) from the petRNA cloning plasmid (acquired from Addgene, 181802). The fragments were subsequently assembled into BpiI- and Esp3I-digested pDAS12222-U6-pegRNA-BFP.
Cloning plasmids for 5′-MS2 and 5′-BoxB RNA-expressing plasmids were generated as follows. To generate plasmids containing a 5′-MS2 or 5′-BoxB sequence along with the tevopreQ1 extension for subsequent cloning, the constructs pSLK20323-U6-MS2-epeg-BFP and pSLK20324-U6-BoxB-epeg-BFP were constructed using an NEB HiFi assembly kit. A linker containing either the MS2 or BoxB sequence, as well as the sequence of an exchangeable cassette followed by the tevopreQ1 extension4, were inserted into the BpiI- and Eco32I-digested pSLK7824-U6-pegRNA-epeg-BFP.
PEAR plasmids
The PEAR-mScarlet plasmid (Addgene no. 162991) used in this study and the pAT9624-BEAR-cloning plasmid (162986) were created by Tálas et al.69. PEAR-GFP plasmids with different eng-tpg target distances and with the SaCas9-engRNA target site were constructed from the pAT9624-BEAR-cloning plasmid (162986)69. The linkers coding the targets were cloned into pAT9624 plasmid between Esp3I sites; the linkers can be found in Supplementary Table 1.
Prime editor-expressing plasmids
The following plasmids were obtained from the non-profit plasmid distribution service Addgene: pCMV-PE2 (132775), created by Anzalone et al.1, pCMV-PEmax-P2A-hMLH1dn (174828), created by Chen and co-workers4, and pCMV-SaCas9-PE (169851), created by Liu et al.31. Several prime editor-expressing plasmids were constructed using an NEB HiFi assembly kit, as described below.
PE2max-expressing plasmid was generated as follows. A PCR fragment synthesized from pCMV-PEmax-P2A-hMLH1dn was used to insert the prime editor coding sequence into NotI- and MssI-linearized pCMV-PE2 (Addgene no. 132775).
tdMCP-PE4max-expressing and N22-PE4max-expressing plasmids were generated as follows. Linkers were used to insert the tdMCP and N22 coding sequences into the pCMV-PEmax-P2A-hMLH1dn vector linearized using SpCas9 in vitro.
nCas9max-expressing plasmid for sPE was generated as follows. nCas9max was created by removing the RT coding sequence from pCMV-PEmax-P2A-hMLH1dn. The plasmid was digested with EcoRI, overlapping segments were created by PCR from the same plasmid and these fragments were assembled.
MCP-M-MLV-RTmax-expressing plasmid for sPE was generated as follows. RTmax was amplified via PCR from pCMV-PEmax-P2A-hMLH1dn, while the plasmid backbone and MS2 coat protein (MCP) were amplified from the MCP-M-MLV-RT vector (created by Liu et al.23 and acquired from Addgene, 181799). Then the PCR fragments were assembled.
PEmax (SpRY-Cas9)-expressing plasmid was generated as follows. SpRY-Cas9 was amplified via PCR, while an upstream fragment was amplified from the PE2max plasmid to generate an overlapping fragment. The two fragments were assembled into NotI- and CpoI-digested PE2max.
Dead SpCas9 prime editor-expressing plasmid was generated as follows. Two regions of the nSpCas9 coding sequence from the pCMV-PEmax-P2A-hMLH1dn vector were PCR-amplified with primers overlapping and containing the D10A mutation in their flanking regions. The resulting fragments were assembled into NotI- and BamHI-digested pCMV-PEmax-P2A-hMLH1dn.
AAV plasmids
p601m-AAV-v3em-Nterm-PE2max (198734) and p601m-AAV-v3em-Cterm-PE2max-∆RNaseH-dualU6 (198735), created by Davis et al.26, were acquired from the non-profit plasmid distribution service Addgene.
The p601m-AAV-v3em-Cterm-PE2max-∆RNaseH-dualU6 construct was used to create plasmids containing the carboxy-terminal part of the prime editor, the tpgRNA expression cassette and a second expression cassette either for engRNA or the second nicking sgRNA. The constructs were assembled using an NEB HiFi assembly kit. For both constructs, PCR was used to generate overlapping fragments for the assembly by amplifying the U6 promoter from AAV-v3em-Cterm-PE2max-∆RNaseH-dualU6. PCR was used to amplify the tpgRNA coding sequence from the ‘tpg_PRNP(1,3-11)_6 G to T (G127V)_RTT23-PBS12_epeg’-expressing plasmid, the second nicking sgRNA coding sequences from the ‘2nd nick_PRNP(4)’-expressing plasmid and the engRNA coding sequence from the ‘eng_PRNP(1)’-expressing plasmid. The two constructs were created by assembling the corresponding fragments with the NotI- and HindIII-digested p601m-AAV-v3em-Cterm-PE2max-∆RNaseH-dualU6.
The sequences of all of the plasmid constructs were confirmed by Sanger sequencing (Microsynth).
Cell culturing and transfection
The HEK293T (CRL-3216), U2OS (HTB-96) and K562 (CCL-243) cell lines were obtained from ATCC. The HuES9 human embryonic stem cell line, provided by D. Melton (Harvard University), was used with approvals from the NIH (Approval number: NIHhESC-09-0022) and the Health Care Research Council, Human Reproduction Committee in Hungary (Approval number: 6681/2012-EHR). The HuES9 cell line was originally described by Cowan et al.70. The cell line carrying the mScarlet-PEAR sequence (HEK-BEAR-mScarlet) was created as described earlier by Tálas et al.69 and were handled using the same protocol applied to HEK293 cells. Short tandem repeat (STR) profiling was used by the respective suppliers to authenticate the HEK293T, U2OS and K562 cell lines. The HuES cell line was previously characterized by the originating laboratory based on morphological features, growth characteristics and expression of molecular markers of undifferentiated pluripotent human stem cells. HEK-BEAR-mScarlet cells were authenticated on the basis of their morphological features and growth characteristics. Cell lines were regularly tested negative for mycoplasma.
HEK293 and U2OS cells were grown in DMEM, and K562 cells were grown in RPMI 1640; both media were supplemented with 10% heat-inactivated fetal bovine serum (FBS) with 100 U ml−1 penicillin and 100 μg ml−1 streptomycin. Cells were cultured at 37 °C in a humidified atmosphere of 5% CO2. HuES9 cells were maintained on Geltrex-coated plates in mTeSR1 medium (Stemcell Technologies) at 37 °C under 5% CO2. HuES cells were passaged every 2–3 days using StemPro Accutase Cell Dissociation Reagent and placed in mTeSR1 supplemented with 10 µM Rho-associated protein kinase (ROCK) inhibitor (Selleckchem, Y-27632-2HCl) for the first 24 h.
Transfections were performed in triplicate. Transfected cells were analysed by flow cytometry 3 days post-transfection, either for PEAR experiments or to assess transfection efficiency, followed by genomic DNA purification.
HEK293 cells were seeded on 48-well plates 1 day before transfection at a density of 3 × 104 to 5 × 104 cells per well. For all experiments, the total DNA was mixed with 0.9 μl turbofect reagent diluted in 50 μl serum-free DMEM and added to the cells after incubation for 20 min at room temperature.
3 × 105 U2OS and 5 × 105 K562 cells were nucleofected in each well using an Amaxa 4D-Nucleofector (Lonza) according to the manufacturer’s protocol. For U2OS cells, the SE Cell Line 4D-Nucleofector X Kit (programme DN-100) was used, and for K562 cells, the SF Cell Line 4D-Nucleofector X Kit (programme FF-120) was used.
For the nucleofection of HuES9 cells, 2 × 105 cells were mixed with 20 µl homemade electroporation buffer (as described by Vriend et al.71) and then electroporated with an Amaxa 4D-Nucleofector using the CA-137 programme. Transfected cells were plated on Geltrex-coated 48-well plates in 500 μl CEPT-supplemented mTeSR1 for the first 24 h and then the media was changed. For further details of the transfection experiments, see Supplementary Methods.
Transduction of HEK293T cells with AAV
AAV viruses were purchased from Creative Cell. HEK293 cells were seeded on 48-well plates at a density of 3 × 104 cells per well 1 day before transduction. Cells were transduced with a total of 1.43 × 106 MOI of AAV using a ratio of 1:0.84:0.16 of the amino-terminal PE2max, carboxy-terminal PE2max with the engRNA and tpgRNA, and carboxy-terminal PE2max with the second nicking sgRNA and tpgRNA coding sequences, respectively. Genomic DNA was extracted 3 days post-transduction.
Inhibition assay
An assay used in the study by Nelson et al.4 was exploited here (Figs. 2 and 4a,b and Extended Data Fig. 1g) in the PEAR reporter system. They used PBS-less RNAs or (RTT-PBS)-less RNAs, mimicking degraded RNAs, to show that they can reduce the accessibility of the target for intact pegRNAs, thereby inhibiting PE4. We examined the inhibitory effect of these degradation-mimicking tpg/pegRNAs (referred to as degraded RNAs) by replacing intact RNAs with an increasing amount of degraded RNAs. To account for the effect of the decreasing amount of intact RNA, in control experiments, an increasing amount of non-targeting RNAs, which do not compete for target binding, was added. The inhibitory effects, shown in the figures (Figs. 2d and 4a,b and Extended Data Fig. 1g), are calculated by normalizing the editing result of targeting inhibitory RNAs to the average of the corresponding non-targeting results.
Re-elongation assay
To directly confirm that proPE is more effective at re-elongating incomplete newly synthesized DNA strands than PE, we designed experiments (Fig. 3h and Extended Data Fig. 4d,e) that rely on re-elongation to create both a within-target edit (A) and a distal edit (B). The first tpg/pegRNA(A) installs the within-target edit, while the second ΔPBS-tpg/pegRNA(B), which lacks a PBS, installs the distal edit. Double edits containing both the A and B modifications can be formed only through the generation of a new DNA strand along the RTT containing edit A, followed by the incorporation of modification B by extending the A-containing short DNA strand along the B-containing RTT. ΔPBS-tpg/pegRNA(B) cannot install the edit when applied alone.
Flow cytometry
Flow cytometry analysis was carried out using an Attune NxT Acoustic Focusing Cytometer (Applied Biosystems by Life Technologies). As a rule, signals from a set target minimum of 10,000 viable single cells were acquired by gating based on the side and forward light-scattering parameters. BFP, GFP, mCherry and mScarlet signals were detected using a 405 nm (for BFP), 488 nm (for GFP) and 561 nm (for mCherry and mScarlet) diode laser for excitation and 440/50 nm (BFP), 530/30 nm (GFP), 620/15 nm (mCherry) and 585/16 nm (mScarlet) filters for emission. The flow cytometry gating strategy is illustrated in Supplementary Fig. 5. In the PEAR experiments, the percentage of GFP or mScarlet positive cells was calculated as the proportion of GFP+mCherry or mScarlet+BFP double positive cells within the mCherry or BFP positive cell population, respectively. mCherry and BFP were used as indicators of the efficiency of transfection. Attune Cytometric software (v.4.2) was used for data analysis.
Genomic DNA purification and genomic PCR
After flow cytometry analysis, genomic DNA was extracted using the Puregene DNA purification protocol (Gentra Systems). Amplicons for next-generation sequencing were generated from the genomic DNA samples using two rounds of PCR to attach Illumina handles. The first-step PCR primers used to amplify the target genomic sequences are listed in Supplementary Table 5 and the indexing of the samples can be found in Supplementary Table 7. PCR was conducted in an S1000 Thermal Cycler (Bio-Rad) or PCRmax Alpha AC2 Thermal Cycler using Q5 high-fidelity polymerase supplemented with Q5 buffer and 150 ng of genomic DNA in a total volume of 50 μl. The thermal cycling profile of the PCR was as follows: 98 °C for 30 s; 35 × (denaturation: 98 °C for 20 s; annealing: see Supplementary Table 5, 30 s; elongation: 72 °C, see Supplementary Table 5); 72 °C for 5 min. i5 and i7 Illumina adapters were added in a second PCR reaction using Q5 high-fidelity polymerase with supplied Q5 buffer and 1 µl of first step PCR product in a total volume of 50 μl. The thermal cycling profile of the PCR was as follows: 98 °C for 30 s; 35 × (98 °C for 20 s; 67 °C for 30 s; 72 °C for 20 s); 72 °C for 2 min. Amplicons were purified by agarose gel electrophoresis. Samples were quantified with Qubit dsDNA HS assay kit and pooled.
The samples used for PCR to observe the on- and off-target modifications in Fig. 8a,b were obtained from the experiment shown in Fig. 3b,c.
Next-generation sequencing, indel and editing frequency analysis
Samples were sequenced on NextSeq (Illumina), resulting in 2 × 150 base-pair (bp) pair-end reads (by Deltabio). Reads were aligned to the reference sequence using BBMap. Primer dimers found among the aligned reads of the FANCF and PRNP amplicons, were removed from further analysis.
Indels at eng/pegRNA and second nicking sgRNA target sites were computationally counted from the aligned reads. Indels without mismatches were searched at ±2 bp around the cut sites. For each sample, indel frequency was determined as (number of reads with indels either at the eng/pegRNA target site or at the second nicking sgRNA target site)/(number of total reads). The frequency of precise edits generated by PE was determined as the percentage of (sequencing reads with the desired modification without indels)/(number of total reads). For intended insertions or deletions in the window of ±2 bp around the cut sites generated by editing, the frequency of precise edits was determined as the percentage of (all sequencing reads with the desired modification)/(number of total reads). For these samples, the indel background was calculated from reads containing indels without considering the desired indel edits. Reads with the intended modifications were identified by searching for a sequence stretch containing the desired edit, flanked by 5 matching nucleotides on both the 5’ and 3’ sides of the edit. We used the modified sequence stretch if the corresponding WT sequence was found in less than 68% of the reads derived from the empty cells, indicating a high sequencing error in the region. In these cases, we modified the sequencing stretch by allowing any type of nucleotide at the position with high sequencing error and an additional nucleotide was added to the particular sequence, keeping 5 matching nucleotides on both the 5’ and 3’ side of the edit. BBMap 38.08, samtools 1.8, BioPython 1.71 and PySam 0.13 software packages were used to analyse the NGS data. The average edit or indel value of the empty cells was subtracted from the value of each independent sample in a triplicate. When specificity values were calculated for the samples and the values were less than 0.3%, then they were arbitrarily set to 0.3% to avoid unrealistic specificity values. The average of the three processed values of a triplicate was then calculated. Specificity values were calculated by dividing the edit values by the indel values for each sample of a triplicate and then taking the average of these ratios.
For the experiments shown in Fig. 6d–g and experiments on CYP genes, this procedure was modified as detailed below.
For Fig. 6d–g, HuES and U2OS cell lines have an SNP (11T>C) in the PRNP locus, in contrast to the HEK293 and K562 cell lines, which causes a mismatch in the RTTs used in this experiment. The desired edit was examined independently from the presence of the SNP.
For each CYP gene, gene-specific primers were used and gene-specific reads were identified on the basis of sequence differences between the two genes. Reads derived from non-gene-specific primer annealing and mixed PCR products due to template switching were excluded by exploiting two gene-specific motifs located at different positions of the amplicon.
Deep sequencing data have been submitted to the National Center for Biotechnology Information Sequence Read Archive under accession number PRJNA1283297.
Statistics and reproducibility
No statistical methods were used to predetermine sample sizes, but our sample sizes are consistent with those commonly reported in PE studies1,4,23. No data were excluded from the analyses. No randomization was necessary for the experimental design. Controlling for covariates was not applicable in this study as it was based on controlled experimental conditions rather than observational data. All key variables were systematically manipulated or held constant, minimizing potential confounding effects. The homogeneity of variances was tested using the Brown–Forsythe test and normality of residuals was tested using the D’Agostino–Pearson omnibus (K2) test. For datasets with a normal distribution, statistical significance was assessed by a two-tailed unpaired t-test to compare two groups. To compare more than two groups, one-way ANOVA or RM one-way ANOVA (in the case of a paired comparison) was used, followed by Šídák’s multiple comparisons test (when comparing only selected groups), Tukey’s post hoc test (when comparing every group with each other group) or Dunnett’s test (when comparing every group with a control group). In cases where the data did not follow a normal distribution, significance was assessed using a two-tailed Mann–Whitney test for comparison between two groups. For comparisons involving more than two groups, the Kruskal–Wallis test was used, or the Friedman test in the case of paired comparisons, both followed by Dunn’s test. Statistical tests were performed using GraphPad Prism 9.2. Means (for data with normal distribution) and medians (for data with non-normal distribution) of each group are shown in each graph. The Investigators were not blinded to allocation during experiments and outcome assessment.
In Fig. 2d, for each normalized dataset, a straight-line model was fitted by non-linear regression using the least-squares fitting method. The null hypothesis that the best-fit slope is the same for all datasets was tested with an extra sum-of-squares F-test. Error bars include error propagation.
In Extended Data Fig. 1g, values were normalized to the corresponding non-targeted controls before calculating the mean. Statistical significance was calculated between the parameters of the fitted curves by using the non-linear module of GraphPad with the two-tailed extra sum-of-squares F-test. A centred second-order polynomial (quadratic) model was used for the fitting in GraphPad. This model provides a better fitting to these points than the linear model, for which the parameters also resulted in significant differences between the 12- and 20-nucleotide spacers.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data are available in the Article and from the corresponding author upon request. The deep sequencing data have been submitted to the NCBI Sequence Read Archive and are available via accession number PRJNA1283297. The ClinVar dataset version 20240502 was used. Source data are provided with this paper.
Code availability
The code for evaluating PE efficiency is available in the Supplementary Code file.
References
Anzalone, A. V. et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157 (2019).
Chen, P. J. & Liu, D. R. Prime editing for precise and highly versatile genome manipulation. Nat. Rev. Genet. 24, 161–177 (2023).
Lin, Q. et al. High-efficiency prime editing with optimized, paired pegRNAs in plants. Nat. Biotechnol. 39, 923–927 (2021).
Nelson, J. W. et al. Engineered pegRNAs improve prime editing efficiency. Nat. Biotechnol. 40, 402–410 (2022).
Zhang, G. et al. Enhancement of prime editing via xrRNA motif-joined pegRNA. Nat. Commun. 13, 1856 (2022).
Liu, Y. et al. Enhancing prime editing by Csy4-mediated processing of pegRNA. Cell Res. 31, 1134–1136 (2021).
Li, X. et al. Enhancing prime editing efficiency by modified pegRNA with RNA G-quadruplexes. J. Mol. Cell Biol. 14, mjac022 (2022).
Chen, P. J. et al. Enhanced prime editing systems by manipulating cellular determinants of editing outcomes. Cell 184, 5635–5652.e29 (2021).
Ferreira da Silva, J. et al. Prime editing efficiency and fidelity are enhanced in the absence of mismatch repair. Nat. Commun. 13, 760 (2022).
Li, X. et al. Highly efficient prime editing by introducing same-sense mutations in pegRNA or stabilizing its structure. Nat. Commun. 13, 1669 (2022).
Zhuang, Y. et al. Increasing the efficiency and precision of prime editing with guide RNA pairs. Nat. Chem. Biol. 18, 29–37 (2022).
Kweon, J. et al. Targeted genomic translocations and inversions generated using a paired prime editing strategy. Mol. Ther. 31, 249–259 (2023).
Wang, J. et al. Efficient targeted insertion of large DNA fragments without DNA donors. Nat. Methods 19, 331–340 (2022).
Tao, R. et al. Bi-PE: bi-directional priming improves CRISPR/Cas9 prime editing in mammalian cells. Nucleic Acids Res. 50, 6423–6434 (2022).
Jiang, T., Zhang, X.-O., Weng, Z. & Xue, W. Deletion and replacement of long genomic sequences using prime editing. Nat. Biotechnol. 40, 227–234 (2022).
Choi, J. et al. Precise genomic deletions using paired prime editing. Nat. Biotechnol. 40, 218–226 (2022).
Anzalone, A. V. et al. Programmable deletion, replacement, integration and inversion of large DNA sequences with twin prime editing. Nat. Biotechnol. 40, 731–740 (2022).
Tao, R. et al. WT-PE: prime editing with nuclease wild-type Cas9 enables versatile large-scale genome editing. Signal Transduct. Target Ther. 7, 108 (2022).
Sun, C. et al. Precise integration of large DNA sequences in plant genomes using PrimeRoot editors. Nat. Biotechnol. 42, 316–327 (2024).
Ponnienselvan, K. et al. Reducing the inherent auto-inhibitory interaction within the pegRNA enhances prime editing efficiency. Nucleic Acids Res. 51, 6966–6980 (2023).
Zheng, C. et al. Template-jumping prime editing enables large insertion and exon rewriting in vivo. Nat. Commun. 14, 3369 (2023).
Böck, D. et al. In vivo prime editing of a metabolic liver disease in mice. Sci. Transl. Med. 14, eabl9238 (2022).
Liu, B. et al. A split prime editor with untethered reverse transcriptase and circular RNA template. Nat. Biotechnol. 40, 1388–1393 (2022).
Grünewald, J. et al. Engineered CRISPR prime editors with compact, untethered reverse transcriptases. Nat. Biotechnol. 41, 337–343 (2023).
Zhi, S. et al. Dual-AAV delivering split prime editor system for in vivo genome editing. Mol. Ther. 30, 283–294 (2022).
Davis, J. R. et al. Efficient prime editing in mouse brain, liver and heart with dual AAVs. Nat. Biotechnol. 42, 253–264 (2024).
She, K. et al. Dual-AAV split prime editor corrects the mutation and phenotype in mice with inherited retinal degeneration. Signal Transduct. Target. Ther. 8, 57 (2023).
Zheng, C. et al. A flexible split prime editor using truncated reverse transcriptase improves dual-AAV delivery in mouse liver. Mol. Ther. 30, 1343–1351 (2022).
Gao, Z. et al. A truncated reverse transcriptase enhances prime editing by split AAV vectors. Mol. Ther. 30, 2942–2951 (2022).
Zong, Y. et al. An engineered prime editor with enhanced editing efficiency in plants. Nat. Biotechnol. 40, 1394–1402 (2022).
Liu, P. et al. Improved prime editors enable pathogenic allele correction and cancer modelling in adult mice. Nat. Commun. 12, 2121 (2021).
Kleinstiver, B. P. et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature 523, 481–485 (2015).
Hu, J. H. et al. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 556, 57–63 (2018).
Nishimasu, H. et al. Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science 361, 1259–1262 (2018).
Walton, R. T., Christie, K. A., Whittaker, M. N. & Kleinstiver, B. P. Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science 368, 290–296 (2020).
Jang, H. et al. Application of prime editing to the correction of mutations and phenotypes in adult mice with liver and eye diseases. Nat. Biomed. Eng. 6, 181–194 (2021).
Feng, Y. et al. Enhancing prime editing efficiency and flexibility with tethered and split pegRNAs. Protein Cell 14, 304–308 (2023).
Dahlman, J. E. et al. Orthogonal gene knockout and activation with a catalytically active Cas9 nuclease. Nat. Biotechnol. 33, 1159–1161 (2015).
Dagdas, Y. S., Chen, J. S., Sternberg, S. H., Doudna, J. A. & Yildiz, A. A conformational checkpoint between DNA binding and cleavage by CRISPR-Cas9. Sci. Adv. 3, eaao0027 (2017).
Ma, H. et al. CRISPR-Cas9 nuclear dynamics and target recognition in living cells. J. Cell Biol. 214, 529–537 (2016).
Aldag, P. et al. Probing the stability of the SpCas9–DNA complex after cleavage. Nucleic Acids Res. 49, 12411–12421 (2021).
Josephs, E. A. et al. Structure and specificity of the RNA-guided endonuclease Cas9 during DNA interrogation, target binding and cleavage. Nucleic Acids Res. 43, 8924–8941 (2015).
Das, D. & Georgiadis, M. M. The crystal structure of the monomeric reverse transcriptase from Moloney murine leukemia virus. Structure 12, 819–829 (2004).
Koeppel, J. et al. Prediction of prime editing insertion efficiencies using sequence features and DNA repair determinants. Nat. Biotechnol. 41, 1446–1456 (2023).
Simon, D. A. et al. PEAR, a flexible fluorescent reporter for the identification and enrichment of successfully prime edited cells. eLife 11, e69504 (2022).
Fu, Y., Sander, J. D., Reyon, D., Cascio, V. M. & Joung, J. K. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol. 32, 279–284 (2014).
Landrum, M. J. et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 42, D980–D985 (2014).
Yu, G. et al. Prediction of efficiencies for diverse prime editing systems in multiple cell types. Cell 186, 2256–2272.e23 (2023).
Liu, Y., Kao, H.-I. & Bambara, R. A. Flap endonuclease 1: a central component of DNA metabolism. Annu. Rev. Biochem. 73, 589–615 (2004).
Suck, D. DNA recognition by structure-selective nucleases. Biopolymers 44, 405–421 (1997).
Perrino, F. W., Harvey, S., McMillin, S. & Hollis, T. The human TREX2 3′ → 5′-exonuclease structure suggests a mechanism for efficient nonprocessive DNA catalysis. J. Biol. Chem. 280, 15212–15218 (2005).
Newby, G. A. & Liu, D. R. In vivo somatic cell base editing and prime editing. Mol. Ther. 29, 3107–3124 (2021).
Li, C. et al. In vivo HSC prime editing rescues sickle cell disease in a mouse model. Blood 141, 2085–2099 (2023).
Jo, D. H., Bae, S., Kim, H. H., Kim, J.-S. & Kim, J. H. In vivo application of base and prime editing to treat inherited retinal diseases. Prog. Retin. Eye Res. 94, 101132 (2023).
Hillen, A. E. J. et al. In vivo targeting of a variant causing vanishing white matter using CRISPR/Cas9. Mol. Ther. Methods Clin. Dev. 25, 17–25 (2022).
Lin, Q. et al. Prime genome editing in rice and wheat. Nat. Biotechnol. 38, 582–585 (2020).
Lee, J. et al. Prime editing with genuine Cas9 nickases minimizes unwanted indels. Nat. Commun. 14, 1786 (2023).
Xue, C. et al. Tuning plant phenotypes by precise, graded downregulation of gene expression. Nat. Biotechnol. 41, 1758–1764 (2023).
Chen, R. et al. Enhancement of a prime editing system via optimal recruitment of the pioneer transcription factor P65. Nat. Commun. 14, 257 (2023).
Li, X. et al. Development of a versatile nuclease prime editor with upgraded precision. Nat. Commun. 14, 305 (2023).
Petri, K. et al. CRISPR prime editing with ribonucleoprotein complexes in zebrafish and primary human cells. Nat. Biotechnol. 40, 189–193 (2022).
Nelson, D. R. et al. Comparison of cytochrome P450 (CYP) genes from the mouse and human genomes, including nomenclature recommendations for genes, pseudogenes and alternative-splice variants. Pharmacogenetics 14, 1–18 (2004).
Ingelman-Sundberg, M. Genetic susceptibility to adverse effects of drugs and environmental toxicants: the role of the CYP family of enzymes. Mutat. Res. 482, 11–19 (2001).
Kim, H. K. et al. Predicting the efficiency of prime editing guide RNAs in human cells. Nat. Biotechnol. 39, 198–206 (2021).
Liang, S.-Q. et al. Genome-wide profiling of prime editor off-target sites in vitro and in vivo using PE-tag. Nat. Methods 20, 898–907 (2023).
Jin, S. et al. Genome-wide specificity of prime editors in plants. Nat. Biotechnol. 39, 1292–1299 (2021).
Kim, D. Y., Moon, S. B., Ko, J.-H., Kim, Y.-S. & Kim, D. Unbiased investigation of specificities of prime editing systems in human cells. Nucleic Acids Res. 48, 10576–10589 (2020).
Kwon, J. et al. TAPE-seq is a cell-based method for predicting genome-wide off-target effects of prime editor. Nat. Commun. 13, 7975 (2022).
Tálas, A. et al. BEAR reveals that increased fidelity variants can successfully reduce the mismatch tolerance of adenine but not cytosine base editors. Nat. Commun. 12, 6353 (2021).
Cowan, C. A. et al. Derivation of embryonic stem-cell lines from human blastocysts. N. Engl. J. Med. 350, 1353–1356 (2004).
Vriend, L. E. M., Jasin, M. & Krawczyk, P. M. in Methods in Enzymology (eds Doudna, J. A. & Sontheimer, E. J.) Vol. 546, 175–191 (Elsevier, 2014).
Acknowledgements
We thank V. Faragó and T. N. Plósz for their help with the illustration and graphical design, R. Csoma for creating the online design tool for proPE, H. N. Stabb, P. Symmons and V. L. Vegi for proofreading the paper and for their helpful comments, and V. Csonka, V. Faragó, V. Karl, I. Szűcsné Pulinka, J. Szűcs, K. Jakab and D. Szeregnyei for their excellent technical assistance. The project was supported by the Hungarian Scientific Research Fund (OTKA; grant nos. K128188, K134968, and K142322 to E.W.) and National Research, Development and Innovation Office of Hungary (grant nos. 2020-1.1.2-PIACI-KFI-2021-00181 to E.W. and 2023-1.1.2-gyorsítósáv-2024-00002). Project no. RRF-2.3.1-21-2022-00015 has been implemented with the support provided by the European Union. Support was provided by the Ministry of Culture and Innovation of Hungary through the National Research, Development and Innovation Fund (project no. KDP-12-10/PALY-2022 to D.A.S.), financed under the KDP-2021 funding scheme.
Author information
Authors and Affiliations
Contributions
S.L.K., D.A.S., Z. Bartos, Z. Biczók, É.V., K.H., P.I.K. and A.T. performed the experiments, processed the data and presented the results. Z.L. analysed the ClinVar dataset. S.L.K. and E.W. designed the experiments and interpreted the results. S.L.K. and D.A.S. analysed the NGS data. E.W. and S.L.K. wrote the paper with input from the other authors.
Corresponding author
Ethics declarations
Competing interests
S.L.K., D.A.S., Z. Bartos, Z. Biczók, É.V., K.H., P.I.K., A.T., Z.L. and E.W. are listed as inventors on a patent application (no. P2500218) filed in Hungary in 2025 by Pannon Plazmid and Biospirál-2006.
Peer review
Peer review information
Nature Catalysis thanks Hanhui Ma, Francisco Sanchez-Rivera and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Characterisation of proPE.
Results of PE3/proPE3 (a-c) and PE2/proPE2 (f) editing without the tevporeQ1 extension (a-c,e,f). a, Indel background of PE and proPE editing on a genomic target shown in Fig. 2a. b,c, The fluorescence readout of the editing obtained by editing the PEAR plasmid correlates with the editing of the PEAR sequence in the genomic context. ‘eng fraction’ refers to the fraction of transfected engRNA plasmid (see Supplementary Methods), ‘T’ and ‘NT’ indicate the targeting and non-targeting tpgRNA, respectively (a-c). d, Schematic representation of the PEAR system45. e, Amplicon sequencing results of SpCas9 nuclease with different spacer length. K- indicates the condition with dead SpCas9. Statistical significance was assessed by a one-way ANOVA and p-values are indicated above the bars. f, ProPE editing is shown on PEAR plasmids using two tpgRNAs with truncated spacers either with or without an upended mismatching 5’ ‘A’ or ‘G’ nucleotide. g, ProPE editing was assessed in the presence of increasing amount of degraded RNAs with 12-nt-long and with 20-nt-long spacers complexed with an inactive prime editor, directed to the target site. TevopreQ1 extension was used. The editing values were normalized to the corresponding values from experiments where non-targeting degraded RNAs were co-expressed before calculating the mean. Statistical significance was calculated between the parameters of the fitted curves by using the nonlinear module of GraphPad with two-tailed extra sum-of-squares F-test. Centred second order polynomial (quadratic) model was used for the fitting in GraphPad. The sample names (PEAR-GFP-C1(S2),…etc) refer to proPE combinations, for which the sequences are provided in Supplementary Table 6. Data are presented as mean values ± SD (a-c,e,f,g).
Extended Data Fig. 2 Working parameter ranges of proPE on PEAR plasmids.
a,b, Fluorescence readouts of PE2 and the corresponding proPE2 editing on PEAR plasmids. tpg/pegRNAs were used without the tevopreQ1 extension. a, The engRNA-tpgRNA target distance at which proPE is more effective, ranges from 0 or ~6 nt for trans-oriented and cis-oriented targets, respectively, to ~30 nt for both, which translates to distances of ~70 nucleotides between the two PAMs in the trans orientation and ~44 nucleotides in the cis orientation. The distances are the only difference between the various conditions within a graph. The light grey line indicates where the editing efficiency of PE control and proPE are equal. b, RTT length has less impact on the efficiency of proPE than of PE and shows little difference between short and long engRNA-tpgRNA target distances. The experiment was performed on cis-oriented targets. c, Comparison of various PE types with the corresponding proPE with/without the tevopreQ1 extension (N = 12). For data sets with a non-normal distribution statistical significance was assessed by a two-tailed Mann-Whitney test (between PE2 and proPE2), the other data sets are normally distributed and therefore a two-tailed unpaired t-test was applied. P-values and fold change between proPE and PE are indicated on the top and directly above the dots, respectively, and medians are shown on the graph (c). d, Fluorescence readouts of PE2 and amplicon sequencing results of the indel formation using a nuclease-sgRNA complex with mismatching spacers on the genome integrated PEAR plasmid. “No MM” refers to the absence of mismatches in the spacer sequence, the position and type of nucleotide changes in the spacer are indicated in the figure. Data are presented as mean values (a-d) ± SD (a,b,d). Further information on panels a, b and c are available in Supplementary figure 2a–c, respectively.
Extended Data Fig. 3 Installing 13 previously described within-target modifications.
a,b, Amplicon sequencing results of PE5max and the corresponding proPE5max editing using tpg/pegRNAs without the tevopreQ1 extension. Data are presented as mean values ± SD. The dashed line shows the 0.3% threshold value. This figure supplements Fig. 3a–c.
Extended Data Fig. 4 ProPE on target-distal edits.
Extended Data Fig. 5 Amplicon sequencing results of target-distal edits.
a,b, PE5max/proPE5max (a) and SpCas9 nuclease (b) editing on 3 genomic targets using tpg/pegRNA with a tevopreQ1 extension. ‘eng fractions’ refers to different amounts of engRNA plasmid. c-g, Data are from the same experiment as shown in Fig. 6b, c. All information is provided in the main figure. Data are presented as mean values (a-g) ± SD (a,b,e-g). The dashed line shows the 0.3% threshold value (f).
Extended Data Fig. 6 Editing various cell lines.
Amplicon sequencing results of PE5max and the corresponding proPE5max editing on the 8 genomic targets (shown also in Fig. 6d, e) in HEK293, U2OS and K562 cell lines using tpg/pegRNAs with tevopreQ1 extension. In a, the editing efficiency, b, the indels formed and c, specificity are shown. Note the logarithmic scale of the ordinate in b and c. For each modification, three engRNA fractions were used. ProPE caused an increase in editing efficiency (a) and specificity (c) in all three cell lines. The indel forming effect of proPE decreased with lower engRNA fractions (b). Data are presented as mean values ± SD. The dashed line shows the 0.3% threshold value (b).
Extended Data Fig. 7 Amplicon sequencing results.
PE4max/proPE4max (d-g) and PE5max/proPE5max (a-g) editing results are shown using tpg/pegRNAs with tevopreQ1 extension (a-g). Data in panels a-c supplement Fig. 6f,g. d,e, ProPE increases the editing efficiency (d) and specificity (e) for both PE4max (N = 12) and PE5max (N = 19) editing types using 13-nt-long PBSs in the CYP genes. f,g, ProPE has a strong enhancing effect on editing efficiency and specificity on low-performing edits on the CYP genes. (N = 53). Data are presented as mean values (a-g) ± SD (a-c). The dashed line shows the 0.3% threshold value (b). Medians are shown (d-g), statistical significance was assessed by a two-tailed Mann-Whitney test (d-f), p-values and fold change between proPE and PE are indicated on the top and next to the dots, respectively (d-f). See Extended Data Fig. 10 for further details of the data in d-g.
Extended Data Fig. 8 ProPE editing on genomic targets.
Amplicon sequencing results of prime editing using tevopreQ1 extension, panels d,e also contain editing results using tpg/pegRNAs without the tevopreQ1 extension. a-c, Individual edit (a), indel background (b) and specificity (c) results of PE5max and corresponding proPE5max editings are shown for seven pathogenic mutations not accessible in the effective editing window of PE. d,e, 130 different genomic PE4max and PE5max editings were tested in HEK293 cells in this study, of which 59% (76 editings) did not reach the 5% efficiency level. For these edits, editing efficiency (d) and specificity (e) are shown. ProPE increased editing efficiency with up to 29.3%, N = 76. f, Comparison of four prime editors as indicated in the figure, SPRY editor variants demonstrated lower editing efficiency when compared to PE4max/PE5max or proPE4max/proPE5max, presented as aggregates. Data are presented as mean values (a-f) ± SD (a-c). Medians are shown (d-f), statistical significance was assessed by a two-tailed Mann-Whitney test, p-values and fold change between proPE and PE are indicated on the top and next to the dots, respectively, N = 76 (d,e). Panels a-c supplement Fig. 7d, e and panels d,e supplement Fig. 7f. Further information on panel f can be found in Supplementary figure 4.
Extended Data Fig. 9 ProPE appears to outperform other split pegRNA approaches and is compatible with AAV-based delivery.
Amplicon sequencing results of prime editing using the tevopreQ1 extension except for sPE-petRNA (a,b) for which a circular RNA is used. a,b, Comparison of the activity of split PE with petRNA23 and two SnPE variants37 (SnPE-5’-MS2, SnPE-5’-BoxB) with proPE and PE on nine edits using the PE4max/PE5max system, presented as aggregates (a) and as individual edits (b), using an identical amount of nicking sgRNA coding plasmid for the corresponding PE variants. ProPE shows a consistently higher activity than the other PE variants. c, ProPE is compatible with AAV delivery. Comparison of proPE delivery systems of plasmid transfection with either our regular conditions or with AAV plasmids and AAV transduction. The results of the regular plasmid transfection from Extended Data Fig. 4a were used as a reference here. Statistical significance was assessed by a Kruskal-Wallis test with two-tailed Dunn’s test (a). Data (a-c) are presented as mean values ± SD (b,c).
Extended Data Fig. 10 Gene-specific editing can be achieved by proPE.
Amplicon sequencing results of PE and proPE using the tevopreQ1 extension. We present the results of two approaches for efficient gene-specific proPE editing, still applicable when PE fails due to the large distance between the desired modification and sequence variations in homologous genes that are amenable to targeting. The actual layouts for these edits and targets are presented in Fig. 8c. Although a higher level of editing can be achieved using proPE5max the gene specificity in terms of the ratio of on-target and off-target edit is not improved compared to proPE4max, while proPE5max produces a significantly higher background of on- and off-target indels. All data are presented as mean values ± SD. Data supplement Fig. 8d, e.
Supplementary information
Supplementary Information
Supplementary Methods, Note 1, Figs. 1–5, Supplementary Methods Table 1 and references.
Supplementary Tables 1–7
Sequences of RNAs and IDs for SRA assigned to the figure numbers, sequences.
Supplementary Data 1
Source data for Supplementary Figs. 1–4.
Supplementary Code
Code for the genomic PE evaluation.
Source data
Source Data Fig. 2
Source data.
Source Data Fig. 3
Source data.
Source Data Fig. 4
Source data.
Source Data Fig. 5
Source data.
Source Data Fig. 6
Source data.
Source Data Fig. 7
Source data.
Source Data Fig. 8
Source data.
Source Data Extended Data Fig. 1
Source data.
Source Data Extended Data Fig. 2
Source data.
Source Data Extended Data Fig. 3
Source data.
Source Data Extended Data Fig. 4
Source data.
Source Data Extended Data Fig. 5
Source data.
Source Data Extended Data Fig. 6
Source data.
Source Data Extended Data Fig. 7
Source data.
Source Data Extended Data Fig. 8
Source data.
Source Data Extended Data Fig. 9
Source data.
Source Data Extended Data Fig. 10
Source data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Krausz, S.L., Simon, D.A., Bartos, Z. et al. ProPE expands the prime editing window and enhances gene editing efficiency where prime editing is inefficient. Nat Catal (2025). https://doi.org/10.1038/s41929-025-01406-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41929-025-01406-6
