
Abstract
Gastric cancer (GC) remains a major global health challenge, characterized by a complex tumor microenvironment (TME) that significantly influences disease progression and therapeutic outcomes. This study focuses on TREM2+ lipid-associated macrophages (LAM) and cancer-associated fibroblasts (CAFs) in modulating the GC microenvironment. Utilizing advanced single-cell RNA sequencing and bulk RNA analyses, we elucidated the interactive mechanisms through which CAFs enhance the immunosuppressive capabilities of TREM2+ LAMs via the CXCL12-CXCR4 signaling axis. Our findings reveal that this interaction facilitates tumor proliferation and inhibits apoptotic processes in GC cells. In vitro experiments confirmed the modulation of this pathway significantly affects tumor cell viability and invasiveness, underscoring the critical roles of these cellular interactions in promoting GC progression. These insights present TREM2+ LAMs and CAFs as potential therapeutic targets, offering new avenues for improving outcomes in GC treatment.

Similar content being viewed by others

Introduction
Gastric cancer (GC) ranks as the fifth deadliest cancer worldwide, and its pathogenesis and treatment strategies have long been focal points in the field of cancer research1,2,3. The intricate tumor microenvironment (TME) is comprised of various cell types, including Cancer-associated fibroblasts (CAFs) and lipid-associated macrophages (LAMs), which interact to influence tumor development and metastasis4,5. CAFs, a prominent cell type in the TME, can impact tumor growth by secreting cytokines and chemokines, as they play a major role in CAF function, as demonstrated by the CXCL12, altering the matrix composition, and facilitating tumor immune evasion6,7,8. On the other hand, TREM2+LAMs, a type of macrophage with immunosuppressive functions, have recently been implicated in promoting tumor growth and immune evasion in various cancers9.
With the advancement of single-cell RNA sequencing and bulk RNA sequencing (bulk RNA-seq) technologies, researchers can now delve deeply into the cellular heterogeneity and complex interactions within the TME, which is crucial for understanding the functions of CAFs and TREM2+LAMs in GC development. These high-throughput sequencing technologies enable the identification of distinct cellular subpopulations within tumors and their dynamic changes during disease progression, offering a novel perspective for unraveling the molecular mechanisms of cellular interactions in the TME10.
Furthermore, the interactions between CAFs and tumor-associated macrophages (TAMs) are increasingly recognized for their roles in tumor progression11. Particularly, the communication between CAFs and TREM2+LAMs via the CXCL12/CXCR4 signaling axis is considered a key factor in regulating the tumor immune microenvironment and promoting GC progression12,13. Therefore, a comprehensive investigation into the specific mechanisms of this signaling axis in GC holds significant importance for discovering new therapeutic targets14,15,16.
This study aims to elucidate the molecular mechanisms of CAFs and TREM2+LAMs in the growth and metastasis of GC. Utilizing single-cell and bulk RNA-seq technologies in conjunction with The Cancer Genome Atlas (TCGA), we conducted a systematic molecular profiling and functional analysis of CAFs and TAMs in GC. Through cell communication analysis, we revealed the interactions between CAFs and TREM2+LAMs and how they collaborate to promote GC development via the CXCL12/CXCR4 pathway. This study not only deepens our understanding of the GC TME but also presents new research directions and potential drug targets for GC treatment. The scientific and clinical significance of these findings provides fresh insights and approaches for shaping future treatment strategies for GC.
Results
Identification of six cell subtypes in GC tissue through scRNA-seq
In order to investigate cellular interactions in GC, we collected adjacent normal tissue and tumor tissue of GC from three patients for scRNA-seq analysis. The data was integrated using the Seurat package. Initial examination of the scRNA-seq data included an assessment of the number of genes per cell (nFeature_RNA), total mRNA molecules (nCount_RNA), and percentage of mitochondrial genes (percent.mt). The majority of cells exhibited nFeature_RNA <5000, nCount_RNA <20,000, and percent.mt <20% (Fig. S1A). Cells with nFeature_RNA between 200 and 5000 percent.mt less than 20% were retained after filtering out low-quality cells, resulting in an expression matrix consisting of 20,989 genes and 48,874 cells. Correlation analysis of the filtered data revealed a coefficient of –0.15 between nCount_RNA and percent.mt, and a coefficient of 0.94 between nCount_RNA and nFeature_RNA (Fig. S1B), indicating good data quality post-filtering for subsequent analysis.
Subsequent analysis of the filtered cells involved the selection of highly variable genes based on gene expression variance, with the top 2000 variable genes chosen for downstream analysis (Fig. S1C). Using the CellCycleScoring function, cell cycle phases were computed (Fig. S1D), followed by data normalization. PCA was then conducted for linear dimension reduction utilizing the selected highly variable genes. Principal gene expression heatmaps for PC_1 – PC_6 were generated (Fig. S1E), along with cell distribution visualization on PC_1 and PC_2 (Fig. S1F), revealing potential batch effects among samples. Batch correction was performed using the harmony package to minimize batch effects and enhance precise cell clustering (Fig. S1G). Subsequently, an ElbowPlot was used to rank the standard deviations of PCs, demonstrating that PC_1-PC_15 sufficiently encapsulated the information from the selected highly variable genes with significant analytical meaning (Fig. S1H). Post-correction results indicated successful elimination of batch effects (Fig. 1A).

A Distribution of cells after Harmony batch correction in PC_1 and PC_2, where each point represents a cell; B clustree package displaying clustering at different resolutions; C Visualization of UMAP clustering results depicting the aggregation and distribution of cells from normal samples and GC samples (Normal, N = 3; GC, N = 3), with red representing GC samples and blue representing normal samples; D Visualization of UMAP clustering results illustrating the aggregation and distribution of cells from different source samples, with each color representing a cluster; E Visualization of cell clustering based on UMAP, with each color representing a unique cell subpopulation and unique color borders indicating the distribution range of each subgroup; F Group visualization of cell annotation results based on UMAP clustering; G Expression levels of six cell marker genes in various cell subgroups, where deeper blue indicates higher average expression levels.
Further nonlinear dimension reduction was carried out using the UMAP algorithm on the top 15 PCs, and clustering across various resolutions was visualized using the clustree package (Fig. 1B). The clustering resolution of 0.4 was selected based on the clustering results, resulting in the identification of 14 cell clusters (Fig. 1C, D). Marker gene expression for each cell cluster was acquired through clustering, and annotation of the 14 cell clusters was performed using the singleR package integrated with literature resources, revealing six cell types: Epithelial cells, B cells, T cells, Fibroblasts, Endothelial cells, and Macrophages (Fig. 1E, F). Consistent results were obtained using the t-SNE algorithm (Fig. S2A, B). Furthermore, UMAP expression plots of the marker genes for these six cell types were presented, with EPCAM representing Epithelial cells, MZB1 for B cells, CD3D for T cells, COL3A1 for Fibroblasts, EMCN for Endothelial cells, and C1QB for Macrophages (Figs. 1G, and S2C).
The findings suggest that GC and normal samples can be classified into 14 clusters, encompassing six cell subtypes, with Epithelial cells, B cells, and Fibroblasts being the most prevalent cell types.
scRNA-seq analysis reveals the key role of TREM2+ LAM in GC
Numerous studies have demonstrated that one of the most crucial metabolic features of cancer cells is lipid metabolic reprogramming to adapt to the TME. Lipid metabolism plays a significant role in the initiation and progression of GC, whereby aberrant lipid metabolism can enhance drug resistance, promote cell proliferation and migration, and support immune suppression functions17,18,19,20.
To explore the association between lipid metabolic pathways and GC, we curated a gene set related to lipid metabolic processes from the Genecards database. Ranking the genes based on their Relevance score, we identified the top 20 key genes (Fig. S3A), revealing that APOE, ranked second, is recognized as a marker gene for macrophages. Studies have suggested that APOE plays a crucial role in modulating macrophage function, especially in lipid metabolism, immune regulation, and macrophage responses associated with diseases21,22,23. We hypothesized a close association between macrophages and lipid metabolism. Further annotation analysis of macrophages validated this hypothesis (Fig. S3B), leading us to believe that LAM may play a critical role in the formation and progression of GC.
Subsequently, using Seurat, we calculated the proportions of different cell types in individual samples (Fig. S3C) and analyzed the differences in cell composition between the tumor-adjacent normal tissues and GC tissues through t-tests. The results indicated a significant increase in the content of fibroblasts and macrophages in GC tissues compared to tumor-adjacent normal tissues (Fig. S3D). Furthermore, we performed differential analysis of gene expression in LAM between normal and GC samples, selecting 94 significantly upregulated genes and 60 significantly downregulated genes based on criteria of |log2FC| > 0.5 and P-value < 0.05 (Fig. S3E).
Moreover, we unexpectedly discovered that TREM2, the most significantly upregulated gene, has received widespread attention in recent years for its involvement in neurodegenerative diseases and various tumors. Multiple studies have confirmed that the expression of TREM2 in LAM serves as a crucial signaling hub in the occurrence, development, and immune escape of tumors24,25,26. Our single-cell transcriptome data further revealed the specific high expression of TREM2 in a subset of macrophages (Fig. S3F, G). Immunofluorescence staining further validated the specific expression of TREM2 in LAM at the protein level, co-expressed with CD68 and APOE (Fig. S3H, I), where CD68 is a macrophage marker protein27, and APOE is a lipid metabolism-related protein. Through RT-qPCR and Western blot experiments, we examined the mRNA and protein expression levels of TREM2 in the tumor adjacent to normal tissues and GC tissues of patients (Fig. S3J). Both differential analysis of single-cell data and cellular experiments in this study consistently indicated a significantly higher level of TREM2 mRNA and protein expression in GC tissues than in tumor-adjacent normal tissues, which is in line with previous research28.
In summary, compared to tumor-adjacent normal tissue samples, the content of fibroblasts and macrophages increases in GC samples. These macrophages are closely associated with lipid metabolism and specifically express TREM2. TREM2+ LAM may play a crucial role in the progression and immune escape of GC.
Increased TREM2 expression associated with high macrophage infiltration density as revealed by joint TCGA analysis
To investigate the expression characteristics of TREM2 in tumor tissues, we initially performed immunofluorescence staining on both the adjacent normal tissues and tumor tissues of five GC patients. Our findings further revealed a co-expression of TREM2 with the total macrophage marker CD68 as well as the M2 macrophage marker CD163 in GC tissues, while its co-expression with the M1 macrophage marker CD86 was less pronounced. These observations suggest that TREM2 may potentially influence macrophage polarization during GC progression and could play an anti-inflammatory role (Fig. S4A, B).
Further analysis in the TCGA public database explored the association between TREM2 expression and clinical outcomes in patients. Compared to normal gastric tissues, the upregulation of TREM2 expression in GC was validated in TCGA-STAD (Fig. S4C). Additionally, we observed a significant difference in TREM2 expression in GC based on tumor stage (T1 to T4) (Fig. S4D), indicating a possible association between TREM2 and the progression and invasiveness of GC. To assess the potential value of TREM2 in the diagnosis and prognosis prediction of GC, ROC curve analysis was conducted, showing slightly higher prognostic AUC values within three years, yet the diagnostic AUC value exceeding 0.9, suggesting that TREM2 could serve as a highly effective biomarker, especially in the early identification of GC patients. However, its efficacy as a sole predictive factor is limited, and it might require a combination with other genes or biomarkers to enhance predictive accuracy (Fig. S4E, F).
To further understand the relationship between TREM2 expression and immune cell characteristics, we employed the ssGSEA method to conduct Pearson correlation analysis on enrichment scores of 24 immune-related cells. The results indicated a strong positive correlation between TREM2 expression levels and macrophages, monocytes and natural killer cells (Fig. S4G). Moreover, using the CIBERSORT method, we performed a deconvolution analysis of immune cell infiltration abundance, revealing a higher proportion of activated B cells, macrophages, and monocytes in TCGA-STAD tumor samples with high TREM2 expression (Fig. S4H).
In conclusion, the elevated expression of TREM2 in GC is closely associated with high macrophage infiltration density. These findings not only underscore the central role of TREM2 in the progression of GC but also imply that TREM2+ LAM may be linked to the growth and invasiveness of GC tumors.
scRNA-seq analysis reveals critical interactions between CAFs and TREM2+LAM in GC via the Cxcl12-Cxcr4 axis
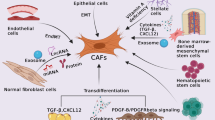
CAFs are a primary component of the TME, influencing cancer hallmarks and impacting tumor growth, invasion, and metastasis29. Through scRNA-seq analysis, we identified fibroblast subpopulations within tumor tissues. Given their distinctiveness within the TME and expression of known CAF markers such as α-SMA (ACTA2), vimentin (VIM), COL1A1, and COL1A2, among others (Fig. 2A), we defined these CAFs.

A Expression levels of CAFs marker genes α-SMA (ACTA2), vimentin (VIM), COL1A1, and COL1A2 in various subgroups of cells in mouse single-cell transcriptome data. Darker shades of red indicate higher expression levels. B Volcano plot showing differential gene expression in CAFs, with red dots representing significantly upregulated genes, blue dots representing significantly downregulated genes, and gray dots indicating genes with no expression difference. C, D Cell lineage trajectory visualization based on cell types and clusters. E, F Differential intercellular communication maps between adjacent normal tissue samples and GC tissue samples (Normal, N = 3; GC, N = 3), with line thickness and color intensity representing the strength of ligand-receptor interactions. G Expression levels of CXCL12 and CXCR4 in various subgroups of cells in mouse single-cell transcriptome data, with deeper shades of blue and red indicating higher expression levels. H, I Expression levels of CXCL12 in CAFs and CXCR4 in TREM2+ LAM. J, K mRNA and protein expression levels of CXCL12 and CXCR4 in mouse adjacent normal tissue and GC tissue samples (Normal, N = 3; GC, N = 3) detected by RT-qPCR and Western blot. Quantitative data are presented as mean ± standard deviation, with intergroup comparisons done using unpaired t-tests. * denotes P < 0.05 for comparison between two groups, ** denotes P < 0.01, *** denotes P < 0.001.
To further explore functional disparities of CAFs between normal and GC samples, differential analysis was conducted on fibroblasts. Applying |log2FC| > 0.585 and P value < 0.05 as selection criteria, we obtained 94 significantly upregulated genes and 71 significantly downregulated genes (Fig. 2B). Gene enrichment analysis revealed that upregulated genes were mainly enriched in biological processes such as response to peptide, regulation of humoral immune response, and cell-substrate junction, whereas downregulated genes were primarily enriched in processes like cytoplasmic translation, peptide binding, and peptide antigen assembly with MHC class II protein complex (Fig. S5A, B). KEGG analysis showed that upregulated genes were predominantly enriched in signaling pathways like Apoptosis and Lysosome, while downregulated genes were enriched in pathways such as Ribosome and Leishmaniasis (Fig. S5A, B).
In order to identify key cell types within the TME, pseudo-temporal analysis was conducted using the “monocle” package. Based on the expression trends of sorted genes, cells were organized and trajectories constructed (Figs. 2C, D, and S5C). The analysis indicated that epithelial cells in the GC microenvironment exhibited a trajectory pointing towards CAFs and TREM2+ LAM cell types, emphasizing the importance of CAFs and TREM2+ LAM in the TME, particularly in their potential roles in cell state transitions and immune modulation processes.
Subsequently, to delve into intercellular communication mechanisms, we employed the R package “CellCall” to analyze pathway activities between different cells. Through chord diagrams and ligand-receptor heatmaps (Figs. 2E, F, and S5D, E), intercellular interactions were visually observed. Among these numerous interactions, particular attention was given to the communication between CAFs and TREM2+ LAM. Compared to adjacent normal tissue, the GC group exhibited a Cxcl12/Cxcr4 interaction between CAFs and TREM2+ LAM, a phenomenon not observed in the normal group.
Further identification of CXCL12/CXCR4 expression in various cell subgroups (Fig. 2G) showed that CXCL12 was primarily expressed in CAFs, while CXCR4 was predominantly expressed in T cells, TREM2+ LAM, and CAFs. These findings further support the notion that the interaction between CAFs and TREM2+ LAM appears to be mediated through the CXCL12-CXCR4 axis.
CXCR4 (C-X-C chemokine receptor type 4) and its ligand CXCL12 (C-X-C motif chemokine 12) are significantly upregulated in various malignant tumors, including GC. This signaling axis not only enhances tumor cell survival, proliferation, angiogenesis, and metastatic capabilities but also plays a pivotal role in the interaction between tumor cells and their microenvironment30,31. Analyzing single-cell transcriptome data from mice, we similarly observed elevated expression of CXCL12 and CXCR4 in CAFs and TREM2+ LAM within GC tissues compared to adjacent normal tissues (Fig. 2H–K).
In conclusion, CAFs and TREM2+ LAM in GC likely engage in critical interactions through the CXCL12-CXCR4 axis, thereby promoting tumor progression and immune escape.
Bulk mRNA sequencing validates the regulatory mechanism of CAFs in the GC microenvironment through the CXCL12-CXCR4 axis
By conducting mRNA sequencing on tumor-adjacent normal tissues and tumor tissues of three GC (GC) patients, a comprehensive exploration of the TME in GC was furthered. The mRNA dataset analyzed a total of 5251 genes, filtering for 106 significantly upregulated genes and 84 significantly downregulated genes based on criteria of |log2FC| > 1.5 and P-value < 0.05 (Fig. 3A). Functional enrichment analysis was performed on the upregulated genes. Gene Ontology (GO) analysis revealed enhanced molecular functions related to extracellular matrix organization, extracellular matrix structural components, and collagen metabolism (Fig. 3B). Additionally, the Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis showed significant upregulation of ECM-receptor interaction and focal adhesion pathways. Moreover, the TNF signaling pathway, PI3K-Akt signaling pathway, IL-17 signaling pathway, and other tumor-related pathways were markedly activated (Fig. 3C). Conversely, downregulated genes were associated with processes like hormone metabolism and humoral immune response and were involved in pathways such as glycolysis and tyrosine metabolism (Fig. S6A, B).

A Volcano plot of gene expression differences in mRNA sequencing data between adjacent normal tissue samples and GC tissue samples (Normal, N = 3; GC, N = 3), with red points indicating significantly upregulated genes, blue points indicating significantly downregulated genes, and gray points indicating genes with no expression difference; B, C Bubble chart and bar graph of GO/KEGG enrichment analysis for highly expressed genes in the differential analysis, where the color of circles and bars represents the significance of enrichment, and the size of circles represents the number of enriched genes; D Gene Set Enrichment Analysis (GSEA) results showing the peaks and valleys represent the degree of dispersion between data; E EPIC analysis heatmap of cell types in adjacent normal tissue samples and GC tissue samples (Normal, N = 3; GC, N = 3); F Violin plot depicting the differential expression of various cell types adjacent to the tumor tissue. Quantitative data are presented as mean ± standard deviation and intergroup comparisons were performed using unpaired t-tests. * indicates P < 0.05 for comparison between two groups, ** indicates P < 0.01 for comparison between two groups; * denotes significance level.
By integrating the significantly upregulated 106 genes in GC and their expression in adjacent normal tissues, gene set enrichment analysis was performed (Figs. 3D, and S6C). Leukocyte transendothelial migration, chemokine receptor binding chemokine pathway, cytokine-cytokine receptor interaction, and cancer-related pathways were all significantly upregulated in GC tissues. Additionally, activation of the CXCR4 pathway was observed in the cancer tissues, which is consistent with our previous findings.
Subsequently, using the EPIC tool, infiltrating cell types in these three pairs of cancer and adjacent normal tissues were analyzed. The overall cell type profile is illustrated (Fig. S6D, E). The results indicated a significant increase in the number of CAFs and a slight growth in macrophages in GC tissues compared to normal tissues (Fig. 3E, F).
These findings suggest that in the GC microenvironment, the increased abundance of CAFs, possibly through activation of the CXCR4 pathway, further enhances the survival, proliferation, and metastatic capabilities of GC cells, thereby driving tumor progression and immune evasion.
High expression of CXCL12/CXCR4 in the GC microenvironment correlates with abundant infiltration of CAFs and macrophages and poor prognosis
To investigate the expression characteristics of CXCL12 and CXCR4 in tumor tissues, we initially performed immunohistochemical staining and H&E staining on cancer-adjacent to normal tissues and tumor tissues from 5 GC patients. The results revealed that in cancer-adjacent normal tissues, CXCL12 and CXCR4 showed weak or no staining, whereas, in tumor tissues, CXCL12 and CXCR4 were moderate to strongly stained in the cell membrane and/or cytoplasm (Fig. 4A). The percentages of CXCL12/CXCR4, positive expression in tumor tissues, were 60.00% and 62.86%, while in normal tissues, the expressions of CXCL12 and CXCR4 were 25.71% and 20.00%, respectively, demonstrating a significant difference in the expression rates of CXCL12/CXCR4 in GC compared to normal tissues (Table S1). These findings suggest that CXCL12 and CXCR4 may interact in the occurrence and metastasis of GC.

A Representative immunohistochemical staining and H&E staining images of CXCL12 and CXCR4 expression in adjacent normal tissues and tumor tissues of GC patients (Normal, N = 5; GC, N = 5) (scale bar: 50 μm, 100 μm); B Correlation bar graphs of CXCL12 and CXCR4 with various infiltrating cell types in the GC microenvironment, where the shade of circle color represents significance level, and the size of the circle represents correlation strength; C Differential expression of CXCL12 and CXCR4 in the mRNA sequencing dataset (Normal, N = 3; GC, N = 3); D, E Differential expression of CXCL12 and CXCR4 in all (left) or paired (right) GC samples and adjacent normal tissues (Normal, N = 32; GC, N = 375) from the TCGA-STAD dataset; F Expression differences of CXCL12 and CXCR4 in GC tumors at different T stages (T1 to T4) from the TCGA-STAD dataset; G Correlation analysis of high and low CXCL12 and CXCR4 expression with overall survival in the TCGA-STAD dataset; H Receiver Operating Characteristic (ROC) curve analysis of CXCR4 in the TCGA-STAD dataset, with the area under the curve (AUC) quantifying its predictive performance. Numerical data is presented as mean ± standard deviation and intergroup differences were analyzed using unpaired t-tests. * indicates P < 0.05, ** indicates P < 0.01, *** indicates P < 0.001 for comparisons between groups, with * denoting statistical significance.
Further exploration of the role of CXCL12/CXCR4 in the TME included analyzing their correlation with various infiltrating cell types. The results indicated a strong positive correlation of CXCL12 and CXCR4 with CAFs and macrophages in the GC microenvironment (Fig. 4B), implying that CAFs may be the primary effector cells of the CXCL12/CXCR4 signaling axis.
Combining mRNA sequencing data, we observed a significant upregulation of both CXCL12 and CXCR4 in GC compared to cancer-adjacent normal tissues (Fig. 4C). Consultation of the TCGA database revealed a similar significant upregulation of CXCR4 in GC, while CXCL12 exhibited a downward trend (Fig. 4D, E). These disparities may arise from differences in sample sources, tumor molecular subtypes, cell lineage characteristics, or other experimental conditions32. Additionally, the correlation analysis of CXCR4 with T tumor size and overall survival period (OS) indicated that its high expression is associated with tumor progression and deterioration (Fig. 4F, G). The ROC curve analysis results also highlighted the good prognostic diagnostic utility of CXCR4 (Fig. 4H).
In conclusion, in the GC microenvironment, CAFs exhibit high expression of CXCR4 and are linked to the CXCL12/CXCR4 signaling pathway. This suggests that CAFs may play a crucial role in regulating the tumor immune microenvironment through this signaling pathway.
TREM2+ LAM promotes GC immune escape and progression in the TME
Studies have identified fatty acid-binding protein (FABP) and fatty acid synthase (FASN) as crucial intracellular mediators in various tumor tissues, with elevated levels of FABP4, FABP5, and FASN associated with promoting tumor proliferation and invasion33,34,35,36. Macrophages can influence cellular lipid metabolism processes37. Previous research has shown that TREM2 is predominantly expressed on the surface of TAMs, suggesting its potential role in immune suppression effects38. However, there is a lack of studies exploring the expression of TREM2 on the surface of macrophages and its regulatory mechanisms on macrophage phenotype and function in vitro.
Initially, we assessed the correct phenotypes of induced macrophage subtypes. RT-qPCR results demonstrated that M2-polarized macrophages exhibited higher expression of the immunosuppressive molecules ARG1 and MRC1 compared to M0 and M1 macrophages (Fig. S7A). Conversely, M1 macrophages showed significantly increased gene expression levels of the pro-inflammatory factors IL-6 and TNF-α compared to M0 and M2 macrophages (Fig. S7B). Flow cytometry revealed a marked increase in the surface expression of the co-stimulatory molecules CD80 and CD86 on M1 macrophages relative to M0 and M2 macrophages, indicative of their M1 phenotype. In contrast, the expression of the specific marker CD206 was significantly higher on the surface of M2 macrophages compared to M0 and M1 macrophages (Fig. S7C). Furthermore, Western blot results confirmed a substantial upregulation of ARG1 protein expression in M2 macrophages compared to the other two macrophage subtypes (Fig. S7D). In summary, the phenotypes of in vitro induced M0, M1, and M2 macrophages were appropriate for subsequent experiments.
Subsequently, we investigated the expression of TREM2 and lipid deposition in different macrophage subtypes in vitro. RT-qPCR results revealed that M2 macrophages exhibited higher expression of TREM2 compared to M0 and M1 macrophages (Fig. 5A). Additionally, there was a significant upregulation of the genes FABP4, FABP5, and FASN in M2 macrophages (Fig. 5B). Flow cytometry analysis of TREM2 surface expression in various subtypes corroborated the gene expression levels (Fig. 5C). Western blot analysis further showed that the protein expression levels of TREM2, FABP4, and FABP5 were highest in M2 macrophages (Fig. 5D).

A, B mRNA expression levels of TREM2, FABP4, FABP5, and FASN in each subtype of macrophages; C Flow cytometry analysis of the proportion of surface TREM2 in each subtype of macrophages; D Western blot analysis of protein expression levels of TREM2, FABP4, and FABP5 in each subtype of macrophages; E, F Immunofluorescence analysis of TREM2 expression and lipid deposition (blue: nucleus; red: TREM2; green: BODIPY 493/503. Scale bar: 25 μm); G ELISA analysis of IL-10 and Arg1 expression levels in cells; H RT-qPCR analysis of IL-10 and Arg1 expression in cells. Quantitative data are presented as mean ± standard deviation, with intergroup comparisons made using unpaired t-tests. * denotes P < 0.05, ** denotes P < 0.01, *** denotes P < 0.001, and “ns” indicates no statistically significant difference between groups. * denotes statistical significance. Cell experiments were replicated three times.
Moreover, immunofluorescence staining confirmed the expression of TREM2 in different macrophage subtypes, utilizing the emerging BODIPY 493/503 lipid tracer to detect intracellular lipid deposition. The results demonstrated elevated red fluorescence, representing high TREM2 expression in M2 macrophages compared to M0 and M1 macrophages (Fig. 5E). Furthermore, the green fluorescence indicative of lipid deposition was higher in M2 macrophages (Fig. 5F). Additionally, RT-PCR and ELISA analysis showed that the expression levels of IL-10 and Arg1 in M2 macrophages were significantly increased in the oe-TREM2 group compared to the oe-NC group, with statistically significant differences (Fig. 5G, H).
The findings suggest that the elevated expression of TREM2 in M2 macrophages is associated with its immunosuppressive function, indicating a potential role in promoting tumor cell proliferation and invasion through the regulation of lipid metabolism pathways. To further elucidate the immunosuppressive regulatory role of TREM2, we employed CRISPR/Cas9 gene editing to generate TREM2 knockout M2 macrophages (TREM2-WT as wild-type control) and overexpressed TREM2 in M2 macrophages using lentiviral vectors (oe-TREM2, with oe-NC as control). RT-qPCR and Western blot were utilized to evaluate TREM2 expression levels, selecting clones with complete TREM2 knockout for expansion (Fig. S8A, B). Furthermore, the transfection efficiency of TREM2 was validated using lentiviral vectors through RT-qPCR and Western blot analysis (Fig. S8C, D).
Different groups of M2-type macrophages were co-cultured separately with T cells (6T-CEM). Flow cytometry analysis identified the proportions of CD3+ T cells (representing all T cells), CD4+ T cells (helper T cells), and CD8+ T cells (cytotoxic T cells). The results revealed a significant increase in the proportions of CD3+ and CD8+ T cells in the TREM2-KO group compared to the TREM2-WT group, while the proportion of CD4+ T cells significantly decreased. Conversely, compared to the oe-NC group, the oe-TREM2 group showed a significant decrease in the proportions of CD3+ and CD8+ T cells and a significant increase in CD4+ T cells (Fig. 6A). Furthermore, the impact on T cell activation status was examined. CD44 is a cell adhesion molecule associated with T cell activation and memory status, while CD69 is an early activation marker for T cells. The results indicated that relative to the TREM2-WT group, the TREM2-KO group exhibited a notable increase in the proportions of T cell CD44 and CD69, whereas the oe-TREM2 group showed a significant decrease in these proportions compared to the oe-NC group (Fig. 6B).

A Flow cytometry analysis to assess the effect of each group on T cell differentiation; B Flow cytometry analysis to evaluate the impact of each group on T cell activation; C RT-qPCR to examine changes in lipid metabolism molecules in different cell groups; D, E Assessment of cell viability and proliferation ability using CCK8 and plate colony experiments; F–H Scratch assays and Transwell assays to measure changes in cell migration and invasion abilities among different groups; I Flow cytometry analysis of apoptotic events in each group, with data on early and late apoptotic cells shown in the respective histograms. Quantitative data are presented as mean ± standard deviation and inter-group comparisons were made using unpaired t-tests. * denotes P < 0.05 for comparison between two groups, ** indicates P < 0.01, *** indicates P < 0.001, and ns represents no statistical difference between the two groups, with * indicating significance. Cell experiments were conducted in triplicate.
These findings suggest a regulatory role of TREM2 in M2-type macrophages regarding T cell subpopulations and activation states, hinting at a potential key role for TREM2 in tumor immunosuppression.
Subsequently, different groups of M2-type macrophages were co-cultured separately with GC cells (MKN-45), and mRNA expression levels of key lipid metabolism molecules, FABP4, FABP5, and FASN, were assessed via RT-qPCR. The results indicated lipid metabolism inhibition in GC cells in the TREM2-KO group compared to the TREM2-WT group, while lipid synthesis activation was observed in the oe-TREM2 group compared to the oe-NC group (Fig. 6C).
In addition, cell viability and proliferation capabilities were assessed through CCK8 and colony formation experiments, cell migration and invasion abilities were evaluated via wound healing and Transwell assays, and flow cytometry was employed to detect cell apoptosis. The results demonstrated that compared to the TREM2-WT group, cells in the TREM2-KO group exhibited significantly reduced viability, proliferation, and migration capabilities, along with a notable increase in apoptosis levels. Conversely, when compared to the oe-NC group, cells in the oe-TREM2 group displayed significantly increased viability, proliferation, and migration capabilities, coupled with a significant decrease in apoptosis levels (Fig. 6D–I).
In conclusion, the manifestation of LAMs’ immunosuppressive effects is TREM2-dependent. Within the GC microenvironment, TREM2+ LAMs suppress anti-tumor immune responses and enhance GC cell vitality, proliferation, migration, and invasion capabilities through lipid metabolism modulation while concurrently inhibiting apoptosis.
CAFs and TREM2+ LAM promote the proliferation, migration, and invasion of GC Cells through the CXCL12/CXCR4 axis
To confirm the successful induction of TAMs and CAFs in cell experiments (Fig. S9B), we measured the concentrations of cytokines and chemotactic factors in the cell culture supernatants using ELISA. The results indicated significantly elevated levels of the anti-inflammatory cytokine IL-10 and the M2 macrophage marker CD206 in TAMs compared to M0 cells. Similarly, CAFs exhibited significantly increased levels of pro-inflammatory cytokine IL-6 and chemokine CXCL12 compared to MRC-5 cells. This successful induction of TAMs and CAFs is depicted in Fig. 7A.

A ELISA was performed to detect the concentrations of the cytokines IL-6 and CD206, as well as the chemokine CXCL12 in the cell culture supernatant after M0 differentiation into TAM and MRC-5 into CAF induction; B Typical morphological changes of M0 macrophages co-cultured with CAFs were observed after H&E staining (scale bar: 25 μm); C RT-qPCR was conducted to assess the mRNA expression levels of IL-6 and CD206, markers for M1 and M2 macrophages, respectively; D ELISA was used to measure the concentrations of CXCL12 and CXCR4 in the cell culture supernatant between the CXCL12-neutralized group and the control group; E, F Cell viability and proliferation changes were examined by CCK-8 and colony formation assays; G–I Migration and invasion abilities of the different groups were evaluated by scratch assay and Transwell assay; J Flow cytometry analysis depicted the occurrence of apoptosis in each group, with statistical data on early and late apoptosis presented in the corresponding graphs. Data are expressed as mean ± standard deviation and intergroup comparisons were made using unpaired t-tests. * indicates P < 0.05, ** indicates P < 0.01, and ns denotes no statistical difference between the two groups; * represents the significance level. Cell experiments were repeated three times.
To mimic the crosstalk among GC cells, CAFs, and TAMs, THP-1-induced M0 cells were treated with CAF-derived conditioned media, resulting in the generation of TAMs associated with CAFs (Fig. S9C). The polarization of macrophages is characterized by distinct morphological changes39. Employing hematoxylin and eosin, staining revealed that CAFs induced an elongated morphology in M0 cells (Fig. 7B). Furthermore, RT-qPCR analysis confirmed that CAFs significantly induced M0 macrophages toward an M2-like phenotype (TAM) by upregulating polarization-associated markers (Fig. 7C). These findings suggest that CAFs may influence their interaction with tumor cells by modulating the polarization state of macrophages.
Further exploring the role of the CXCL12/CXCR4 signaling axis between CAFs and TAMs, we neutralized CXCL12 in MRC5 cells using a CXCL12 antibody. ELISA results showed a significant reduction in the levels of CXCL12 and CXCR4 in the TAM+CAF+CXCL12 Ab group compared to the TAM+CAF+IgG Ab group, indicating effective blockade of this signaling axis (Fig. 7D).
Based on the discovery that TAMs in the GC microenvironment exhibit characteristics related to TREM2+ and lipid metabolism, we coined the term TREM2+ LAM to maintain coherence in the article. To further investigate the interactions between CAFs and TREM2+ LAM in an in vitro co-culture system, GC cells were co-cultured with ordinary DMEM, TREM2+LAM, TREM2+LAM+CAF, and TREM2+LAM+CAF treated with CXCL12 Ab. Cell viability and proliferation capability were assessed using CCK8 and colony formation assays, while wound healing and Transwell assays were employed to evaluate cell migration and invasion. Flow cytometry was utilized to assess cell apoptosis. The results showed that TREM2+LAM significantly promoted the vitality, proliferation, and migration abilities of GC cells while reducing their apoptosis levels. These effects were further enhanced when interacting with CAFs. However, upon blocking the CXCL12/CXCR4 axis, the viability, proliferation, and migration abilities of GC cells significantly decreased while their apoptosis levels increased (Fig. 7E–J).
In conclusion, the interplay between CAFs and TREM2+LAM in the GC microenvironment plays a critical role in immune evasion and progression of GC, providing evidence for targeting the CXCL12/CXCR4 signaling axis as a potential therapeutic strategy.
LAFs and TREM2+LAM regulate macrophage polarization through the CXCL12/CXCR4 axis to promote pulmonary metastasis of GC
To gain a deeper understanding of the critical role of the CXCL12/CXCR4 axis in vivo, we intravenously injected co-cultured cells into a mouse model of GC. As illustrated in Fig. 8A, tumors in the TREM2+LAM group exhibited faster growth compared to the control group, with this effect further enhanced in the presence of CAFs. However, the growth trend was significantly inhibited when a blocker of the CXCL12/CXCR4 axis was employed. Tumor weight notably increased in the TREM2+LAM group compared to the control, and this increase was further amplified in the CAFs+TREM2+LAM group. Treatment with a CXCL12/CXCR4 signaling axis inhibitor led to a significant reduction in tumor weight (Fig. 8B, C). Immunohistochemistry was then used to assess the expression of T-cell markers CD3 and CD8, revealing lower expression of CD3 and CD8 in the TREM2+LAM group compared to the control. The addition of CAFs resulted in a further decline in the expression of these markers. However, following treatment with a CXCL12/CXCR4 inhibitor, their expression levels significantly increased (Fig. 8D). Western blot analysis further confirmed the protein expression of Trem2, Cxcl12, and Cxcr4, showing an increase in the expression of these proteins in the TREM2+LAM group compared to the control, which was further elevated with the addition of CAFs. However, treatment with a CXCL12/CXCR4 inhibitor led to a significant decrease in the expression of these proteins (Fig. 8E).

A–C Comparative analysis and quantitative assessment of tumor size and weight in xenograft tumors from each group (n = 8); D Evaluation of CD3 and CD8 expression in each group by immunohistochemical staining (scale bar: 50 μm); E Protein expression levels of Trem2, Cxcl12, and Cxcr4 in each group analyzed by Western blot; F Effects of GC metastasis on lung tissue in each group and quantitative analysis of metastatic nodules; G Histological changes in lung metastatic nodules evaluated by H&E staining and immunohistochemical staining for CD31 and CD206 (scale bar: 100 μm). Data are presented as mean ± standard deviation and intergroup comparisons were performed using unpaired t-tests. * denotes P < 0.05 for comparison between two groups, ** denotes P < 0.01, *** denotes P < 0.001, and ns indicates no statistical difference between the two groups. Statistical significance is indicated by *. Each group consists of 8 nude mice.
Considering the lungs as a common distant metastatic site for GC, particular attention was focused on the metastasis in the lungs. In the TREM2+LAM group, there was a significant increase in the number of pulmonary metastatic nodules, which was more pronounced in the presence of CAFs. Blocking the CXCL12/CXCR4 signaling axis effectively mitigated the effect on metastasis (Fig. 8F). Consistent with these observations, metastatic nodules were detected in the HE-stained lung tissues, with minimal observation of metastatic nodules in the presence of CXCL12 Ab (Fig. 8G). Moreover, the expression of tumor growth and metastasis proteins CD31 and M2-type macrophage marker protein CD206 was significantly upregulated in the xenograft tumors of the TREM2+LAM and CAFs+TREM2+LAM groups, and this upregulation was reversed by CXCL12 Ab (Fig. 8G).
These findings collectively indicate that LAFs and TREM2+LAM regulate macrophage polarization through the CXCL12/CXCR4 axis, enhancing immunosuppression and thereby promoting the progression and pulmonary metastasis of GC.
Discussion
This study utilized scRNA-seq and bulk RNA-seq technologies to unveil the crucial role of CAFs in the development of GC, particularly in the immunosuppression mediated by TREM2+LAM and the construction of the TME. Unlike previous studies that regarded CAFs as a singular promoting factor, this research discovered that CAFs specifically act on TREM2+LAM through the CXCL12/CXCR4 pathway, fostering the polarization of M2 macrophages and thereby enhancing immunosuppression, distinguishing itself from existing research on the role of CAFs in other tumors40,41. Moreover, this impact of CAFs may also be linked to the migratory and invasive capabilities of tumor cells, providing a novel perspective on exploring CAFs as potential targets for cancer therapy42,43,44.
This paper presents, in detail for the first time, the immunosuppressive role of TREM2+LAM in GC and elucidates its mechanisms in the GC microenvironment. Compared to prior studies on TREM2+LAM in other types of tumors, our findings suggest that TREM2+LAM may play a more complex and specific role in GC. Through a combination of animal experiments and in vitro cell studies, this research further confirms the involvement of TREM2+LAM in GC progression, offering potential directions for targeted therapies45,46,47.
While the CXCL12/CXCR4 pathway has been reported to modulate the TME and the behavior of tumor cells in various cancers, this study emphasizes its particular role in regulating TREM2+LAM in GC, distinguishing it from previous research. This novel finding not only enhances our understanding of the mechanisms underlying GC progression but also suggests that the CXCL12/CXCR4 pathway could serve as a therapeutic target specific to the unique microenvironment of GC48,49,50.
This study further complicates our understanding of the TME. Through cell trajectory and cell communication analyses, it reveals the interactions between CAFs and TREM2+LAM, demonstrating that the construction of the TME is a multifaceted and dynamic process51,52,53. The complexity of the TME necessitates the adoption of a comprehensive approach when investigating therapeutic strategies, considering the holistic ecological context of the tumor54,55. A significant aspect of this study, compared to existing research, is the utilization of integrated analysis techniques such as scRNA-seq and bulk RNA-seq, enabling the exploration of relationships between cell subpopulations at the single-cell level56,57,58. Additionally, this study validated its findings using the TCGA database, enhancing the credibility and relevance of the research results.
In conclusion, our study preliminarily suggests that the interaction between CAFs and TREM2+LAM in GC promotes immune escape, growth, and metastasis through the CXCL12-CXCR4 pathway (Fig. 9). This research provides new insights into the pathogenesis of GC, particularly focusing on the interactions among different cellular subgroups within the TME. The interaction between CAFs and TREM2+LAM and their critical roles in GC offer new avenues for research and potential drug targets. Interventions targeting the CXCL12-CXCR4 pathway may offer more effective treatment strategies for GC patients.

The tumor microenvironment (TME) is illustrated as a dynamic interface involving cancer-associated fibroblasts (CAFs), lipid-associated macrophages (TREM2+LAM), CD8+ T cells, and tumor cells. CAFs secrete CXCL12, which interacts with CXCR4 expressed on TREM2+LAM, promoting their immunosuppressive phenotype via lipid metabolism-related factors (FABP4/5). TREM2+LAM express CD206 and inhibit CD8+ T cell activity by reducing TNF-α secretion, creating an immunosuppressive microenvironment. This interaction enhances tumor proliferation, migration, invasion, and resistance to apoptosis. Metastatic dissemination to secondary sites, such as the lungs, is facilitated by these TME-driven processes. The diagram emphasizes the CXCL12-CXCR4 axis as a critical mediator in CAF and TREM2+LAM crosstalk.
The scientific significance of this study lies in the in-depth exploration of the role of CAFs and TREM2+LAM in GC development. Through the use of scRNA-seq and bulk RNA-seq technologies, this study, for the first time, reveals at the molecular level how CAFs induce immunosuppression in TREM2+LAM via the CXCL12/CXCR4 pathway, thus advancing a new mechanism promoting GC progression. This discovery contributes to our understanding of the complexity of the TME in GC, particularly in terms of immune regulation7,59,60. Furthermore, this study has important clinical implications, offering new biomarkers and potential therapeutic targets for the diagnosis, treatment, and prognosis assessment of GC.
On a clinical level, this study proposes targeted treatment strategies against the pathways related to CAFs and TREM2+LAM, which may facilitate the development of new drugs and innovative treatment approaches. Given the roles of CAFs and TREM2+LAM in tumor immune escape, therapies directed at them could enhance the treatment outcomes of GC patients, especially those resistant to conventional treatments. However, extensive clinical trials are needed for the safe and effective validation of these findings.
Nevertheless, this study has some limitations. While scRNA-seq and bulk RNA-seq can provide abundant molecular information, further bioinformatics analysis and experimental verification are required. In addition, although in vitro experiments and animal models can mimic human diseases, they may not fully replicate the complexity of the human body’s internal environment. Moreover, the complexity of the TME implies that CAFs and TREM2+LAM may have other undiscovered mechanisms of action.
Future research prospects should encompass a more extensive analysis of clinical samples to validate the universality and clinical relevance of our findings. Furthermore, inhibitors targeting the CXCL12/CXCR4 pathway have shown promise in other types of cancers, suggesting potential exploration of these inhibitors in GC treatment. Lastly, deeper investigation into the differences between CAFs and TREM2+LAM in different types of GC may reveal more precise therapeutic targets, thereby advancing personalized medicine.
In conclusion, our study provides a detailed analysis of the roles played by CAFs and TREM2+ LAMs within the GC TME. The interactions between these cell types through the CXCL12-CXCR4 signaling pathway are shown to critically enhance the immunosuppressive environment, facilitating tumor growth and metastasis. Our findings underscore the pivotal role of these interactions in modulating immune responses and suggest that disrupting this axis could offer a novel therapeutic strategy for GC.
We have demonstrated that the CXCL12-CXCR4 pathway is not only a crucial mediator of communication between CAFs and TREM2+ LAMs but also a potential target for therapeutic intervention. Targeting this pathway could disrupt the supportive tumor network, potentially leading to improved responses to existing treatments and better clinical outcomes. This study contributes significantly to the understanding of GC’s complexity and highlights the importance of targeting specific molecular interactions within the TME as a promising approach in cancer therapy. Future research should focus on developing targeted therapies that can inhibit these critical interactions, with the potential to transform the therapeutic landscape for GC patients.
Materials and methods
Clinical sample collection
Our study collected cancer tissue samples from 35 patients undergoing GC therapy in our hospital between January 2022 and December 2022. Tissue samples taken 2 cm from the cancerous lesion were chosen as controls. The clinical information of these patients is detailed in Table S2. Postoperative specimens were examined by two pathologists who diagnosed them as GC based on morphology and immunohistochemical results, including the expression of markers such as HER2 and Ki-67. None of the patients had received any anti-tumor treatments such as radiotherapy or chemotherapy before surgery, and they had no significant medical history of other diseases before the operation. After tissue resection, specimens were immediately frozen in liquid nitrogen and stored at –80 °C. All patients had read and signed informed consent forms before surgery. The experiment was approved by the ethics committee and strictly followed the Helsinki Declaration61,62.
scRNA-seq
Three samples, each of tumor and adjacent normal tissue, from GC patients were collected. Clinical details of these patients are provided in Table S2. Samples 4, 5, and 6 were used for scRNA-seq extraction and sequencing. The tissue samples were cut into small pieces and then digested in RPMI-1640 at 37 °C for 30 min using 1 mg/mL of Type IV collagenase (17104019, Thermo Fisher, USA) and 50 µg/mL of DNase I (10104159001, Roche, Germany). Following digestion, the tissue underwent filtration through a 70 µm cell strainer to obtain a single-cell suspension, followed by treatment with trypsin. Single cells were captured using the C1 Single-Cell Auto Prep System (Fluidigm, Inc., South San Francisco, USA). Once captured, the cells were lysed within the chip to release mRNA, followed by reverse transcription to generate cDNA. The lysed and reverse-transcribed cDNA underwent pre-amplification within a microfluidic chip for subsequent sequencing. The amplified cDNA was used for library construction and subjected to scRNA-seq on the HiSeq 4000 Illumina platform (parameters: paired-end reads, read lengths of 2 × 75 bp, ~20,000 reads per cell)63.
Data analysis was conducted using the “Seurat” package in R software. Data were quality-controlled based on the criteria of 200 < nFeature_RNA < 5000 and percentage of mitochondrial genes <20, with the top 2000 highly variable genes selected for further analysis64.
To reduce the dimensionality of the scRNA-seq dataset, we performed principal component analysis (PCA) based on the top 2000 highly variable genes. The top 20 principal components (PCs) were chosen for downstream analysis using the Elbowplot function in Seurat. Cell subpopulations were identified using the FindClusters function with a default resolution setting res=1. Subsequently, nonlinear dimensionality reduction was performed on the scRNA-seq data using the t-SNE algorithm. Markers for various cell subpopulations were selected using the Seurat package, and cell annotations were performed using the “SingleR” package65.
Cell-cell communication analysis was carried out using the “CellChat” package in R. Differentially expressed genes (DEGs) within the scRNA-seq dataset were identified using the “Limma” package in R. DEGs between normal and GC samples were filtered based on the criteria |logFC| > 0.5 and adjusted P-value < 0.0566.
Bulk RNA extraction and sequencing
Three samples each of tumor tissue were collected from GC patients with GC and adjacent normal tissue. The clinical information of these patients is presented in Table S2. Samples 1, 2, and 3 underwent bulk RNA extraction and sequencing using Trizol reagent (15596026, Invitrogen, Carlsbad, California, USA) for total RNA isolation. The concentration and purity of RNA samples were determined using an Agilent 2100 bioanalyzer (G2939B, Agilent, USA). Total RNA samples meeting the following criteria were selected for subsequent experiments: RNA Integrity Number (RIN) ≥ 7.0 and 28S:18S ratio ≥ 1.5. Sequencing libraries were generated and sequenced by CapitalBio Technology in Beijing, China. Each sample utilized 5 μg of RNA. In brief, the Ribo-Zero™ magnetic bead kit (MRZB12424, Epicentre Technologies Corp, USA) was employed to deplete ribosomal RNA (rRNA) from total RNA. The NEB Next Ultra RNA Library Prep Kit (#E7775S, NEB, USA) was used for Illumina library construction. Subsequently, RNA was fragmented into ~300 base pair (bp) fragments, followed by first-strand cDNA synthesis using reverse transcriptase primers and random primers, and second-strand cDNA synthesis in the presence of dUTP Mix (10×) as a buffer. The cDNA fragments underwent end repair, including polyadenylation and sequencing adapter ligation. After connecting Illumina sequencing adapters, the second cDNA strand was digested using the USER enzyme (#M5508, NEB, USA) to construct strand-specific libraries. The library DNA was then amplified, purified, and enriched by PCR. Subsequent library identification was performed using the Agilent 2100 bioanalyzer, and quantification was carried out using the KAPA Library Quantification Kit (KK4844, KAPA Biosystems). Finally, paired-end sequencing was conducted on the NextSeq CN500 (Illumina) sequencer, achieving an average sequencing depth 50x67,68.
Quality control of sequencing data and analysis of differential gene expression
The quality of paired-end reads of the raw sequencing data was assessed using FastQC software (v0.11.8). Preprocessing of the raw data was conducted with Cutadapt software (1.18) to remove Illumina sequencing adapters and poly(A) tail sequences. Reads with an N content exceeding 5% were filtered out using a Perl script. Reads with a base quality of over 20 and covering at least 70% were extracted using the FASTX Toolkit software (0.0.13). The BBMap software was employed to repair the paired-end sequences. Subsequently, the aligned, high-quality reads fragments were mapped to the hg38 reference genome provided by Gencode using the hisat2 software (0.7.12). Based on high-throughput bulk RNA-seq data, differential gene expression analysis was performed using the R software “Limma” package with thresholds of |log2FC| > 2 and P-value < 0.05 to identify DEGs. The “heatmap” package in R was used to visualize the expression of DEGs, the “ggplot2” package for generating volcano plots of differential genes, and the “clusterProfiler” package for GO and KEGG enrichment analysis. Furthermore, the abundance of immune and cancer cells in malignant tumors was evaluated using EPIC database methodology69,70.
Data download and analysis of public databases
The RNA sequencing data and corresponding clinical pathology data were downloaded from the TCGA project for GC (TCGA-STAD). The gene expression levels for all samples were extracted and quantified using Transcripts Per Million (TPM) as the standardization method. Subsequently, the TPM data of each sample were uniformly integrated to ensure consistency across the dataset. We retrieved the gene set related to the “Lipid Metabolic Process” pathway from the GeneCards database (https://www.genecards.org/). The top 20 genes ranked by Relevance score were visualized using GraphPad software71.
Construction and cultivation of in vitro cell models
Human GC cell line MKN-45 (CL-0292), human embryonic lung fibroblast cell line MRC-5 (CL-0161), human monocyte cell line THP-1 (CL-0233), and human T-cell leukemia cell line 6T-CEM (CL-0008) were all purchased from Procell Life Science & Technology Co., Ltd (Wuhan, China). These cells were cultured in DMEM medium (11965092, Gibco, USA) containing 10% FBS, 10 μg/mL streptomycin, and 100 μ/mL penicillin. THP-1 cells were cultured in RPMI-1640 medium (A1049101, Gibco, USA) supplemented with 10% FBS and 1% penicillin/streptomycin. HEK293T cell line was obtained from ATCC (CRL-3216) and cultured in DMEM medium (11965092, Gibco, USA) with 10% FBS, 10 μg/mL streptomycin, and 100 U/mL penicillin. All cells were maintained in a humidified incubator at 37 °C with 5% CO2 (Heracell™ Vios 160i CO2 Incubator, Thermo Scientific™, Germany) and passaged when reaching 80–90% confluency72.
When the density of THP-1 monocytes reached 1.0 × 105 cells/cm2, 50 nM phorbol-12-myristate-13-acetate (PMA; HY-18739, MedChemExpress, USA) was added, and the cells were incubated in cell culture chambers with transwell inserts (0.4 μm pore size; CLS3412, Corning, USA) for 48 h to establish a co-culture system. This setup allows for the investigation of paracrine signaling and cellular interactions between different cell types, which can influence the differentiation of monocytes into M0 macrophages and better mimic the in vivo microenvironment. Subsequently, the macrophages were treated with LPS (100 ng/mL, HY-P7025A, MedChemExpress) and IL-4 (20 ng/mL, HY-P70445, MedChemExpress) for 24 h each to polarize into M1 and M2 macrophages, respectively (Fig. S9A), for downstream experiments73,74,75.
Construction of TREM2-KO cells using CRISPR/Cas9 technology
TREM2-KO cells were generated using the CRISPR/Cas9 technology with the following sgRNAs: TREM2-sgRNA1: 5′-AGTAAGATGAGCAGCCGGAG-3′ (PAM: AGG); TREM2-sgRNA2: 5′-CATAGGGGCAAGACACCTGC-3′ (PAM: AGG). The sgRNAs were inserted into the Lenti-CRISPR v2 vector containing the Streptococcus pyogenes Cas9 nuclease gene (HanHeng Biotech (Shanghai) Co., Ltd, Shanghai). M2 macrophages were transduced with the lentiviral Lenti-CRISPR v2 vector, and the CRISPR/Cas9 editing system was utilized to generate TREM2-KO cells. Cells were selected using 4 μg/mL puromycin (A1113803, Gibco, USA) post transfection with sgRNA plasmids and donor sequences. Single cells surviving the limiting dilution were screened through RT-qPCR and Western blot to identify TREM2-KO cells, which were further confirmed by DNA sequencing and stored at –80 °C for future use. Cell groups included: (1) TREM2-WT group: M2 macrophages with wild-type TREM2; (2) TREM2-KO group: M2 macrophages with knocked-out TREM276,77.
Construction of overexpression lentiviral vectors and cell transfection
Packaged virus and target vectors were co-transfected into HEK293T cells using Lipofectamine 2000 (11668-500, Invitrogen, USA) until cell confluence reached 80–90%. After 48 h of cell culture, the supernatant was collected, filtered and centrifuged to remove cell debris, yielding virus particles in the supernatant. The virus was harvested at the logarithmic growth phase, and the virus titer was determined. The overexpression lentivirus, constructed and packaged by Genechem (Shanghai), contained the LV-TREM2 gene for overexpression. M2-type macrophages were grown to the logarithmic phase, dissociated with trypsin into a cell suspension of 5 × 104 cells/mL, and then seeded in 6-well plates with 2 mL per well. Prior to establishing the in vitro cell model, different lentiviruses (MOI = 10, virus titer 1 × 108 TU/mL) were added to the cell culture medium and incubated for 48 h. Stable cell lines were selected using 2 μg/mL puromycin (UC0E03, Sigma-Aldrich, Germany) over two weeks.
The cell transfection groups were as follows: (1) oe-NC group: M2-type macrophages were transfected with lentivirus constructed using oe-NC vector; (2) oe-TREM2 group: M2-type macrophages were transfected with lentivirus constructed using oe-TREM2 vector. After 48 h of transfection, RNA and protein levels were examined to confirm knockdown efficiency. Plasmids involved in the study were designed and synthesized by Guangzhou Ruibo Biotechnology Co., Ltd78.
Preparation of conditioned medium (CM)
The preparation steps for CM are as follows: 1. Cultivate MKN-45 and MRC-5 cells until they reach 80% confluency. 2. Rinse the cell culture dishes twice with PBS. 3. Add 10 mL of DMEM containing 1% FBS and incubate at 37 °C for 48 h. 4. Collect the medium, centrifuge to remove cell debris, and harvest the supernatant. 5. Store CM at –80 °C to prevent repeated freeze-thaw cycles. 6. When treating specific cells, mix CM with the complete medium in a 2:1 ratio and incubate at 37 °C for 48 h79. TAMs are obtained by treating M0 cells with CM derived from MKN-45, while CAFs are obtained by treating MRC-5 cells with CM from MKN-45. Subsequently, TAMs are produced by treating M0 cells with CM derived from CAFs (using CM generated from CAFs to treat TAMs) (Fig. S9B). For experiments involving blockade of the CXCL12/CXCR4 signaling axis, 300 μg/mL of CXCL12 antibody (AF-310-NA, R&D Systems, USA) or IgG (1-001-A, R&D Systems, USA) as a control is added to CAF-CM and incubated for 1 h. The treated medium is then used to stimulate M0 cells to obtain TAMs with CXCL12 Ab (TAMs treated with CXCL12 neutralizing antibody based on CAF-CM) and TAMs with IgG Ab (TAMs treated with IgG neutralizing antibody on the basis of CAF-CM) (Fig. S9C). Subsequently, CM from M0, TAMs, TAMs with CAF, as well as TAMs with CXCL12 Ab and TAMs with IgG Ab is further collected following the same procedure (Fig. S9D)79,80,81,82.
Cell coculture
Transwell cell culture inserts (0.4 μm, CLS3412, Corning, USA) were utilized for coculture assays. In investigating the effect of TREM2 on T cells, T cells (6T-CEM) were seeded in the upper chamber of Transwell inserts (105 cells per well), while TREM2-WT, TREM2-KO, oe-NC, or oe-TREM2 M2 cells were seeded in the lower chamber of the Transwell inserts (5 × 104 cells per well). For evaluating the impact of TREM2 on GC cells, GC cells (MKN-45) were seeded in the upper chamber of Transwell inserts (105 cells per well), and TREM2-WT, TREM2-KO, oe-NC, or oe-TREM2 M2 cells were seeded in the lower chamber of the Transwell inserts (5 × 104 cells per well). Cells were harvested for subsequent analysis after 48 h. The groups were categorized as follows: (1) TREM2-WT: coculture of wild-type M2 macrophages with 6T-CEM/MKN-45 cells; (2) TREM2-KO: coculture of M2 macrophages lacking TREM2 with 6T-CEM/MKN-45 cells; (3) oe-NC: coculture of M2 macrophages transfected with empty vector with 6T-CEM/MKN-45 cells; (4) oe-TREM2: coculture of M2 macrophages transfected with TREM2 plasmid with 6T-CEM/MKN-45 cells.
In investigating the crosstalk between GC cells, TAMs, and CAFs, GC cells (MKN-45) were seeded in the upper chamber of Transwell inserts (105 cells per well), while normal DMEM, TAMs, TAMs + CAFs, and TAMs + CAFs + CXCL12 Ab conditioned media were seeded in the lower chamber of the Transwell inserts (5 × 104 cells per well). Cells were collected for subsequent analysis after 48 h. The groups were categorized as follows: (1) Control: coculture of MKN-45 with normal DMEM, serving as the control group; (2) TREM2+LAM: coculture of MKN-45 with TAM-CM; (3) TREM2+LAM+CAF: coculture of MKN-45 with TAM+CAF-CM; (4) TREM2+LAM+CAF+CXCL12 Ab: coculture of MKN-45 with TAM+CAF+CXCL12 Ab-CM83,84.
Hematoxylin and Eosin (H&E) staining
After air-drying the slide containing the tissue slice at room temperature, the slide was fixed at room temperature for 30 s, washed with 1× PBS for 2 s (or rinsed with running water for 30 s in the case of FFPE tissue sections), followed by staining with hematoxylin for 60 s. Subsequently, the slide was washed with 1× PBS for 10 s and underwent differentiation with 1% hydrochloric acid alcohol solution for 3 s, followed by another 2-s wash with 1× PBS. Eosin staining was performed for 3 min, followed by consecutive dehydration with 70%, 80%, 95% ethanol, and absolute ethanol for 5 min each85.
Immunohistochemistry staining
Following fixation, tissue sections were dehydrated in a graded alcohol series, embedded in paraffin, and cut into 5 µm-thick slices. The slices were baked at 60 °C for 20 min, deparaffinized in xylene, with three changes of xylene, each for 15 min, hydrated in absolute alcohol for 5 min, followed by another change of absolute alcohol and 5 min of hydration, then sequentially hydrated in 70% and 95% alcohol for 10 min each. Each slice was treated with 3% H2O2 and left to incubate at room temperature for 10 min to block endogenous peroxidases. Subsequently, antigen retrieval was performed by heating the sections in a citrate buffer in a microwave for 3 min, followed by incubation at room temperature for 10 min; PBS washes were repeated thrice. Normal goat serum blocking solution (C-0005, Haoran Biotechnology, Shanghai, China) was applied onto tissue sections and incubated at room temperature for 20 min. The sections were then incubated overnight at 4 °C with a mixture of primary antibodies: rabbit anti-CD3 (ab16669, 1:1000, Abcam), rabbit anti-CD8 (ab217344, 1:1000, Abcam), rabbit anti-CD31 (ab182981, 1:1000, Abcam), rabbit anti-CD206 (ab64693, 1:1000, Abcam), rabbit anti-CXCL12 (ab155090, 1:1000, Abcam), rabbit anti-CXCR4 (ab181020, 1:1000, Abcam). The next day, the sections were incubated with goat anti-rabbit IgG (ab6721, 1:1000, Abcam) secondary antibody for 30 min, followed by 30 min of incubation at 37 °C with SABC (SA0041, Solarbio, Beijing, China) and development with a DAB chromogenic agent (DA1010, Solarbio, Beijing, China) for 6 min. Counterstaining was performed with hematoxylin (C0107, Beyotime, Shanghai, China) for 30 s. Dehydration was done by sequential immersion in 70%, 80%, 90%, and 95% ethanol for 2 min each, followed by two changes of absolute ethanol for 5 min each, and finally mounted in neutral resin for observation under a light microscope (BX63, Olympus, Japan). The experiment was repeated three times. PBS was used instead of primary antibody for negative control. Five different fields of view were selected for quantitative analysis of IHC images. The number of cells with brown staining signals in each field was counted as positive cells, and the total number of cells in each field was recorded. The percentage of positive cells was calculated as follows: positive cell percentage = (number of brown-stained cells/total cell count) × 100%86.
Immunofluorescence staining
Cell-containing slides were fixed in 4% PFA (catalog number: P0099, manufacturer: Biyuntian) for 30 min, followed by blocking with 0.3% Triton X-100 and 5% BSA for 1 h For single staining, primary antibodies used were rabbit anti-TREM2 (K009576P, 1:500, Solarbio, Beijing), rabbit anti-CD68 (ZRB1427, 1:500, Merck, USA), and rabbit anti-APOE (ab227993, 1:500, Abcam, UK) targeting Human and Mouse reactivity. These primary antibodies were diluted in a blocking buffer and incubated overnight at 4 °C. The secondary antibody mix was prepared by diluting Alexa Fluor® 568-conjugated goat anti-rabbit antibody (ab175471, Abcam, UK), Alexa Fluor® 488-conjugated goat anti-rabbit antibody (ab150077, Abcam, UK), and Alexa Fluor® 647-conjugated goat anti-rabbit antibody (ab6940, Abcam, UK) at 1:1000 ratio. Incubation was carried out at room temperature for 1 h, followed by DAPI staining.
For double staining, primary antibodies used were mouse anti-CD68 (ab955, 1:50, Abcam, UK), rabbit anti-CD86 (ab239075, 1:100, Abcam, UK), mouse anti-CD163 (ab156769, 1:100, Abcam, UK), and goat anti-TREM2 (ab223684, 10 µg/mL, Abcam, UK) targeting Human reactivity. Primary antibodies were diluted according to recommended ratios, and Trem2 primary antibody was mixed in a 1:1 volume ratio with CD68, CD86, or CD163 primary antibodies. Cell-containing slides were incubated overnight at 4 °C in the diluted primary antibody mixture. After washing three times in PBS, the secondary antibody mix was prepared at a 1:500 dilution, consisting of Alexa Fluor® 488-conjugated goat anti-rabbit antibody (ab150077, Abcam, UK), Alexa Fluor® 488-conjugated goat anti-mouse antibody (ab150113, Abcam, UK), and donkey anti-goat Alexa Fluor® 647-conjugated antibody (ab150135, Abcam, UK). Incubation was carried out at room temperature for 1 h. Details of antibodies are shown in Table S3.
Finally, slides or tissue sections were washed three times in PBS. Images were captured using confocal laser scanning microscopy (CLSM; LSM 510 META, Carl Zeiss AG). Five random fields were selected for analysis, and the ImageJ software was utilized to count the number of fluorescently labeled cells (positive cells) and total cells stained with DAPI in each field. The percentage of positive cells was calculated as follows: Positivity rate = (number of positive cells/total number of cells) × 100%87.
Detection of cell lipid deposition using BODIPY staining
Cultured cells were seeded at a density of 1 × 106 cells on confocal culture dishes and allowed to reach 60% confluency. Subsequently, they were fixed in 95% anhydrous ethanol for 15 min and washed with cold PBS. Approximately 500 μL of paraformaldehyde was added to each well and incubated at room temperature for 10–15 min. After two washes with PBS and removal of excess liquid, the cells were stained with BODIPY 493/503 (1 μg/mL, HY-D1614, MedChemExpress) in the dark for 15 min at room temperature. Following two washes with PBS, the cells were co-incubated with DAPI at room temperature for 15 min and then washed. Subsequently, the cells were observed under a confocal laser scanning microscope (LSM 700; Carl Zeiss, Oberkochen, Germany) within 30 min of slide sealing. Five random fields of view were selected for lipid deposition analysis using ImageJ software to quantify the number of cells emitting fluorescent signals (positive cells) and the total number of DAPI-stained cells. The percentage of positive cells was calculated as the ratio of positive cell count to total cell count multiplied by 100%88.
RT-qPCR detection of target gene expression
Total RNA was extracted using a Trizol reagent following the manufacturer’s instructions. The RNA was then reverse-transcribed to cDNA using the PrimeScript RT reagent Kit. The synthesized cDNA was subjected to RT-qPCR analysis using the Fast SYBR Green PCR kit, with each well set up with three replicates. GAPDH was used as the internal control, and relative expression levels were calculated using the 2–ΔΔCt method. The experiment was repeated three times. The primer sequences used for RT-qPCR in our study are shown in Table S4 and were synthesized by Takara89.
Western blot
The RIPA lysis buffer containing PMSF (P0013B, Beyotime, Shanghai, China) was added for tissue and cell extraction to extract total proteins. The protein extraction utilized a kit (P0028, Beyotime, Shanghai) following the manufacturer’s instructions. The supernatant was quantified using the BCA assay kit (P0011, Beyotime, Shanghai) to determine the total protein concentration in each sample. Protein concentration was adjusted to 1 μg/μL, with each sample volume set at 100 μL. Subsequently, samples were boiled for 10 min to denature the proteins and stored at –80 °C for later use.
An 8%–12% SDS gel was prepared based on the size of the target protein bands. Equal amounts of protein samples were loaded into each lane using a micropipette for electrophoresis separation. Proteins on the gel were then transferred onto a PVDF membrane (1620177, BIO-RAD, USA). The membrane was blocked with 5% BSA at room temperature for 1 h. Rabbit anti-TREM2 (ab305103, 1:1000, Abcam), rabbit anti-TREM2 (ab209814, 1:1000, Abcam), rabbit anti-CXCL12 (ab155090, 1:1000, Abcam), rabbit anti-CXCR4 (ab181020, 1:1000, Abcam), rabbit anti-FABP5 (ab255276, 1:1000, Abcam), and rabbit anti-FABP4 (ab92501, 1:1000, Abcam) were added and incubated overnight at 4 °C. Subsequently, the membrane was washed thrice with 1× TBST buffer at room temperature for 5 min each time. HRP-conjugated goat anti-rabbit IgG (ab6721, 1:1000, Abcam) secondary antibody was added and incubated at room temperature for 1 h. The membrane was washed thrice with 1× TBST buffer at room temperature for 5 min each time. The membrane was then immersed in ECL reagent (1705062, BIO-RAD, USA) and incubated at room temperature for 1 min. Excess liquid was removed, and the membrane was covered with plastic wrap. Band exposure was performed using the Image Quant LAS 4000C gel imaging system (GE Healthcare, USA). Cell total proteins were normalized using GAPDH (ab181602, 1:10,000, Abcam) or Vinculin (ab129002, 1:10,000, Abcam) as internal controls. The relative protein expression levels were calculated by comparing the grayscale values of the target bands with the reference bands to detect the expression levels of various proteins90. Each experiment was repeated three times. All the Uncropped and unedited blot/gel images can be found at Figs. S10–S25.
Enzyme-linked immunosorbent assay (ELISA)
An ELISA was conducted following the manufacturer’s instructions. In brief, MKN-45 human GC cells were seeded at an appropriate concentration of 2 × 104 to 1 × 105 cells in a 96-well plate and incubated for 24 h. Subsequently, the cell culture medium was removed and replaced with an equal volume of serum-free DMEM. After 24 h, the supernatant was collected and filtered through a 0.45 μm filter to remove any suspended cells. The secretion of IL-10 (88-7106-88, Invitrogen, USA), IL-6 (EH254RB, Invitrogen, USA), CD206 (EH329RB, Invitrogen, USA), and CXCL12 (EHCXCL12A, Invitrogen, USA) in the cell culture supernatant was quantified using a human ELISA kit80.
CCK-8 and cell proliferation detection in plate cloning
The CCK-8 assay kit (Beyotime, C0037, Shanghai, China) was utilized to assess cell proliferation. MKN-45 human GC cells from different growth phases were taken, and their concentration was adjusted to 5 × 104 cells/mL. Subsequently, they were seeded into a 96-well culture plate, with each well receiving 100 μL of cell suspension. The plate was then placed in an incubator for 48 h, followed by the rapid removal of the supernatant. Next, 10 μL of CCK-8 solution was added to each well, and the plate was incubated at 37 °C for 2 h. The absorbance at 450 nm (A) was measured using a Multiskan FC microplate reader (Thermo Fisher, 51119080, USA). Three replicate wells were set up for each group, and the average was calculated.
For the plate cloning experiment, cells in the exponential growth phase were taken and passaged using standard digestion methods to achieve a cell suspension with over 95% single cells. The cells were counted and diluted to an appropriate concentration with a culture medium, and then 5 mL of cell suspension containing 100 cells per dish was seeded into a culture dish (diameter 60 mm). The dish was gently rocked in a crisscross manner to disperse the cells evenly. The dish was then incubated at 37 °C with 5% CO2 for 2–3 weeks. Upon visible clone formation in the dish, the culture was terminated, the medium was discarded, and the dish was rinsed twice with PBS and air-dried. The cells were fixed with methanol for 15 min, air-dried after methanol removal, stained with Giemsa solution for 10 min, rinsed slowly under running water to remove excess dye, and air-dried. Clones containing more than 10 cells were counted with the naked eye or under a microscope (low magnification). The clone formation rate was calculated using the formula: Clone formation rate = (number of clones/number of seeded cells) × 100%91,92.
Wound healing experiment
The processed test MKN-45 human GC cells were separately seeded in a 6-well plate with a cell density of 70–90%. A clear visible scratch was made in the plate using a 200 μL pipette tip. The cells were washed to remove detached cells, followed by the addition of a fresh culture medium. Incubation took place at 37 °C and 5% CO2, with the initial scratch situation recorded at 0 h. Observation and photography were conducted using an inverted microscope (CKX53, Olympus, Japan). At 24 h, the scratch was observed and photographed again using the same microscope. The width of the scratch was measured using image processing software (ImageJ) to calculate the distance of cell migration93.
Detection of MKN-45 human GC cell migration and invasion through transwell assay
ECM gel (E1270, Sigma-Aldrich, Germany) was added to the upper chamber of a 24-well Transwell plate (8 μm) and then incubated at 37 °C for 30 min to allow gel solidification. Cells treated under different conditions were collected and resuspended in a serum-free culture medium to 105 cells/mL. Subsequently, 200 μL of cell suspension (2 × 104 cells/well) was added to each upper chamber, while 800 μL of culture medium containing 20% FBS was added to the lower chamber. After incubation at 37 °C for 24 h, the Transwell plate was removed, washed twice with PBS, fixed with formaldehyde for 10 min, and then rinsed thrice with water. The cells were stained with 0.1% crystal violet, incubated at room temperature for 30 min, and washed twice with PBS, and the cells on the upper surface were wiped off with a cotton ball. The invaded cells were photographed using an inverted microscope (CKX53, Olympus, Japan). Cell counting and analysis of the invasion capability of cancer cells were performed using ImageJ software. For the migration assay, the Transwell plate was used without ECM gel coating, while all other steps were consistent with the invasion assay. At least four random microscopic fields were selected for cell counting in each experiment94,95.
Flow cytometry analysis
After centrifugation and washing with PBS, the corresponding antibodies were added to the respective cell samples: FITC anti-CD68 (333805, 1:500, BioLegend, USA), PE anti-CD3 (ab22268, 1:1000, Abcam, Shanghai, China), FITC anti-TREM2 (ab236286, 1:1000, Abcam, Shanghai, China), FITC anti-CD44 (ab27285, 1:1000, Abcam, Shanghai, China), and FITC anti-CD69 (310903, 1:1000, BioLegend, USA). The samples were then incubated at 4 °C in the dark for 30 min at 1500 rpm, followed by centrifugation for 5 min to identify macrophage populations, T cells, TREM2, CD44, and CD69. Subsequently, 100 μL of fixative solution was added intracellularly, followed by a 10-min staining at 4 °C and a centrifugation at 1500 rpm for 5 min to discard the supernatant. Then, 100 μL of cell lysis buffer was added, and the samples were incubated at 4 °C for 10 min. Following a PBS wash, the following antibodies were added: FITC anti-CD86 (ab77276, 1:500, Abcam, Shanghai, China), FITC anti-CD80 (ab62635, 1:500, Abcam, Shanghai, China), FITC anti-CD206 (C068C2, 1:300, Elabscience, USA), FITC anti-CD4 (ab269349, 1:200, Abcam, Shanghai, China), and FITC anti-CD8 (ab22504, 1:500, Abcam, Shanghai, China). The samples were then incubated in the dark at 4 °C for 30 min. Cells were centrifuged at 1500 rpm for 5 min to remove unbound antibodies, resuspended in 100 µL PBS, and analyzed using the FACScan flow cytometry system (Becton Dickinson, San Diego, CA, USA)96,97.
Flow cytometry was employed to detect cell apoptosis. In brief, tumor cells (1 × 105/well) were collected and washed in chilled PBS. The cells were then stained with 5 μL of Annexin-V and 5 μL of Propidium Iodide (PI) in the dark for 15 min using the detection kit (APOAF-20TST, Sigma-Aldrich, USA). Subsequently, the pellet was resuspended in 400 μL of binding buffer provided in the kit. The stained cells were analyzed using a flow cytometer98. Cells in the upper right quadrant with the phenotype Annexin V+PI+ represent late apoptotic cells; cells in the lower right quadrant with the phenotype Annexin V+PI- represent early apoptotic cells; cells in the upper left quadrant with the phenotype Annexin V-PI+ represent necrotic cells; and cells in the lower left quadrant with the phenotype Annexin V-PI- represent live cells99.
Establishment of the C57BL/6J nude model
The experimental procedures and animal usage protocols have been approved by the Institutional Animal Ethics Committee (Approval No: SCUAH-2022026). A total of 40 eight-week-old male C57BL/6J nude mice (219, Vital River Laboratory Animal Technology Co., Ltd, Beijing, China) were housed. The mice, weighing 20–24 g, were maintained under non-pathogenic conditions (26–28 °C, 50–65% humidity).
Forty nude mice were selected, and 1 × 107 human GC cell line MKN-45 cells were subcutaneously injected into the dorsal subcutaneous tissue of each mouse. After 2 weeks of inoculation, the mice with established subcutaneous tumor tissues were randomly divided into 5 groups (each comprising 8 mice): 1. GC model group: for subsequent scRNA-seq studies, the mice were kept, and the tumor tissue was aseptically excised along with adjacent normal tissue. 2. Control group: intravenous tail injection of 1 × 107 MKN-45 cells for assessing the interactions between GC cells, TAMs, and CAFs. 3. TREM2+LAM group: tail vein injection of 1 × 107 MKN-45 cells treated with TAM-CM. 4. TREM2+LAM+CAF group: tail vein injection of 1 × 107 MKN-45 cells treated with TAM+CAF-CM. 5. TREM2+LAM+CAF+CXCL12 Ab group: tail vein injection of 1 × 107 MKN-45 cells treated with TAM+CAF+CXCL12 Ab-CM.
Upon the appearance of nodules resembling tumors subcutaneously, the tumor measurements were recorded every 4 days using a vernier caliper to determine the long and short diameters and subsequently calculate the volume. On the 32nd day, the mice were euthanized, and the tumors, along with lung tissues, were dissected, photographed, and weighed using an electronic balance. The visible lung surface tumors were counted, and the experimental data were recorded100. The tumors were stored in cryovials and preserved at –80 °C in the freezer.
Statistical analysis
All data were processed using SPSS 22.0 statistical software (SPSS, Inc., Chicago, IL, USA) and GraphPad Prism 7.0. Continuous variables were expressed as mean ± standard deviation (Mean ± SD), and comparisons between two groups were conducted using independent t-tests. One-way analysis of variance (ANOVA) was employed for comparisons among multiple groups. The homogeneity of variances was assessed using the Levene test. If homogeneity was met, Dunnett’s t and least significant difference (LSD) post-hoc tests were used for pairwise comparisons. If variances were unequal, Dunnett’s T3 test was applied. A significance level of P < 0.05 indicated statistical significance in the comparisons between the two groups.
Data availability
The sequencing data generated in this study have been deposited in the NCBI repository under BioProject ID PRJNA1181929 with the following accession numbers for each dataset: Single-cell RNA sequencing data: Control group: SRR31372926, SRR31372925, SRR31372922; Disease group: SRR31372921, SRR31372920, SRR31372919. Bulk RNA sequencing data: Control group: SRR31372918, SRR31372917, SRR31372916; Disease group: SRR31372915, SRR31372924, SRR31372923.
References
Smyth, E. C., Nilsson, M., Grabsch, H. I., van Grieken, N. C. & Lordick, F. Gastric cancer. Lancet 396, 635–648 (2020).
Machlowska, J., Baj, J., Sitarz, M., Maciejewski, R. & Sitarz, R. Gastric cancer: epidemiology, risk factors, classification, genomic characteristics and treatment strategies. Int. J. Mol. Sci. 21, 4012 (2020).
Sexton, R. E., Al Hallak, M. N., Diab, M. & Azmi, A. S. Gastric cancer: a comprehensive review of current and future treatment strategies. Cancer Metastasis Rev. 39, 1179–1203 (2020).
Timperi, E., Gueguen, P. & Molgora, M. et al. Lipid-associated macrophages are induced by cancer-associated fibroblasts and mediate immune suppression in breast cancer. Cancer Res. 82, 3291–3306 (2022).
Tan, Z., Kan, C. & Sun, M. et al. Mapping breast cancer microenvironment through single-cell omics. Front. Immunol. 13, 868813 (2022).
Mao, X., Xu, J. & Wang, W. et al. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: new findings and future perspectives. Mol. Cancer 20, 131 (2021).
Li, X., Sun, Z. & Peng, G. et al. Single-cell RNA sequencing reveals a pro-invasive cancer-associated fibroblast subgroup associated with poor clinical outcomes in patients with gastric cancer. Theranostics 12, 620–638 (2022).
Wu, F., Yang, J. & Liu, J. et al. Signaling pathways in cancer-associated fibroblasts and targeted therapy for cancer. Signal Transduct. Target Ther. 6, 218 (2021).
Zhou, L., Wang, M. & Guo, H. et al. Integrated analysis highlights the immunosuppressive role of TREM2+ macrophages in hepatocellular carcinoma. Front. Immunol. 13, 848367 (2022).
Purroy, N. & Wu, C. J. Coevolution of leukemia and host immune cells in chronic lymphocytic leukemia. Cold Spring Harb. Perspect. Med. 7, a026740 (2017).
Zhao, Z., Li, W. & Zhu, L. et al. Construction and verification of a fibroblast-related prognostic signature model for colon cancer. Front. Genet. 13, 908957 (2022).
Zhang, F., Cui, J. Y. & Gao, H. F. et al. Cancer-associated fibroblasts induce epithelial-mesenchymal transition and cisplatin resistance in ovarian cancer via CXCL12/CXCR4 axis. Future Oncol. 16, 2619–2633 (2020).
Qin, Y., Wang, F. & Ni, H. et al. Cancer-associated fibroblasts in gastric cancer affect malignant progression via the CXCL12-CXCR4 axis. J. Cancer 12, 3011–3023 (2021).
Wang, Y., Chen, H. & Liu, W. et al. MCM6 is a critical transcriptional target of YAP to promote gastric tumorigenesis and serves as a therapeutic target. Theranostics 12, 6509–6526 (2022).
Shi, W., Zhang, G. & Ma, Z. et al. Hyperactivation of HER2-SHCBP1-PLK1 axis promotes tumor cell mitosis and impairs trastuzumab sensitivity to gastric cancer. Nat. Commun. 12, 2812 (2021).
Qu, X., Liu, B. & Wang, L. et al. Loss of cancer-associated fibroblast-derived exosomal DACT3-AS1 promotes malignant transformation and ferroptosis-mediated oxaliplatin resistance in gastric cancer. Drug Resist. Update 68, 100936 (2023).
Ugel, S., De Sanctis, F., Mandruzzato, S. & Bronte, V. Tumor-induced myeloid deviation: when myeloid-derived suppressor cells meet tumor-associated macrophages. J. Clin. Invest. 125, 3365–3376 (2015).
Kuo, C. Y. & Ann, D. K. When fats commit crimes: fatty acid metabolism, cancer stemness and therapeutic resistance. Cancer Commun. 38, 47 (2018).
Johnson, P. I. & Napier, T. C. GABA- and glutamate-evoked responses in the rat ventral pallidum are modulated by dopamine. Eur. J. Neurosci. 9, 1397–1406 (1997).
Cui, M. Y., Yi, X., Zhu, D. X. & Wu, J. Aberrant lipid metabolism reprogramming and immune microenvironment for gastric cancer: a literature review. Transl. Cancer Res. 10, 3829–3842 (2021).
Stansbury, C. M., Dotson, G. A., Pugh, H., Rehemtulla, A., Rajapakse, I. & Muir, L. A. A lipid-associated macrophage lineage rewires the spatial landscape of adipose tissue in early obesity. JCI Insight 8, e171701 (2023).
Rodriguez-Duarte, J., Galliussi, G. & Dapueto, R. et al. A novel nitroalkene-α-tocopherol analogue inhibits inflammation and ameliorates atherosclerosis in Apo E knockout mice. Br. J. Pharmacol. 176, 757–772 (2019).
Tokuda, T., Calero, M. & Matsubara, E. et al. Lipidation of apolipoprotein E influences its isoform-specific interaction with Alzheimer’s amyloid beta peptides. Biochem. J. 348, 359–365 (2000).
Qiu, H., Shao, Z. & Wen, X. et al. TREM2: keeping pace with immune checkpoint inhibitors in cancer immunotherapy. Front. Immunol. 12, 716710 (2021).
Hsieh, C. L., Koike, M. & Spusta, S. C. et al. A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J. Neurochem. 109, 1144–1156 (2009).
Kawabori, M., Kacimi, R. & Kauppinen, T. et al. Triggering receptor expressed on myeloid cells 2 (TREM2) deficiency attenuates phagocytic activities of microglia and exacerbates ischemic damage in experimental stroke. J. Neurosci. 35, 3384–3396 (2015).
Ferrisse, T. M., de Oliveira, A. B. & Palaçon, M. P. et al. The role of CD68+ and CD163+ macrophages in immunopathogenesis of oral lichen planus and oral lichenoid lesions. Immunobiology 226, 152072 (2021).
Zhang, X., Wang, W., Li, P., Wang, X. & Ni, K. High TREM2 expression correlates with poor prognosis in gastric cancer. Hum. Pathol. 72, 91–99 (2018).
Asif, P. J., Longobardi, C., Hahne, M. & Medema, J. P. The role of cancer-associated fibroblasts in cancer invasion and metastasis. Cancers 13, 4720 (2021).
Domanska, U. M., Kruizinga, R. C. & Nagengast, W. B. et al. A review on CXCR4/CXCL12 axis in oncology: no place to hide. Eur. J. Cancer 49, 219–230 (2013).
Lombardi, L., Tavano, F., Morelli, F., Latiano, T. P., Di Sebastiano, P. & Maiello, E. Chemokine receptor CXCR4: role in gastrointestinal cancer. Crit. Rev. Oncol. Hematol. 88, 696–705 (2013).
Xue, L. J., Mao, X. B., Ren, L. L. & Chu, X. Y. Inhibition of CXCL12/CXCR4 axis as a potential targeted therapy of advanced gastric carcinoma. Cancer Med. 6, 1424–1436 (2017).
Sun, Y., Ren, D. & Yang, C. et al. TRIM15 promotes the invasion and metastasis of pancreatic cancer cells by mediating APOA1 ubiquitination and degradation. Biochim Biophys. Acta Mol. Basis Dis. 1867, 166213 (2021).
Visweswaran, M., Arfuso, F., Warrier, S. & Dharmarajan, A. Aberrant lipid metabolism as an emerging therapeutic strategy to target cancer stem cells. Stem Cells 38, 6–14 (2020).
Biancur, D. E. & Kimmelman, A. C. The plasticity of pancreatic cancer metabolism in tumor progression and therapeutic resistance. Biochim. Biophys. Acta Rev. Cancer 1870, 67–75 (2018).
Cheng, C., Geng, F., Cheng, X. & Guo, D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun. 38, 27 (2018).
Zhang X., Fan L., Wu J. et al. Macrophage p38α promotes nutritional steatohepatitis through M1 polarization. J. Hepatol. 71, 163–174 (2019).
Binnewies, M., Pollack, J. L. & Rudolph, J. et al. Targeting TREM2 on tumor-associated macrophages enhances immunotherapy. Cell Rep. 37, 109844 (2021).
Dahlem, C., Siow, W. X. & Lopatniuk, M. et al. Thioholgamide A, a new anti-proliferative anti-tumor agent, modulates macrophage polarization and metabolism. Cancers 12, 1288 (2020).
Qin, R., Ren, W. & Ya, G. et al. Role of chemokines in the crosstalk between tumor and tumor-associated macrophages. Clin. Exp. Med. 23, 1359–1373 (2023).
Song, H., Lou, C. & Ma, J. et al. Single-cell transcriptome analysis reveals changes of tumor immune microenvironment in oral squamous cell carcinoma after chemotherapy. Front. Cell Dev. Biol. 10, 914120 (2022).
Sobierajska, K., Ciszewski, W. M., Sacewicz-Hofman, I. & Niewiarowska, J. Endothelial cells in the tumor microenvironment. Adv. Exp. Med. Biol. 1234, 71–86 (2020).
Neophytou, C. M., Panagi, M., Stylianopoulos, T. & Papageorgis, P. The role of tumor microenvironment in cancer metastasis: molecular mechanisms and therapeutic opportunities. Cancers 13, 2053 (2021).
Sakamoto, H., Koma, Y. I. & Higashino, N. et al. PAI-1 derived from cancer-associated fibroblasts in esophageal squamous cell carcinoma promotes the invasion of cancer cells and the migration of macrophages. Lab Invest. 101, 353–368 (2021).
Hu, S., Zhang, C. & Ni, L. et al. Stabilization of HIF-1α alleviates osteoarthritis via enhancing mitophagy. Cell Death Dis. 11, 481 (2020).
Huang, L., Rong, Y. & Tang, X. et al. Engineered exosomes as an in situ DC-primed vaccine to boost antitumor immunity in breast cancer. Mol. Cancer 21, 45 (2022).
Ojosnegros, S., Seriola, A., Godeau, A. L. & Veiga, A. Embryo implantation in the laboratory: an update on current techniques. Hum. Reprod. Update 27, 501–530 (2021).
Ouyang, S., Li, H. & Lou, L. et al. Inhibition of STAT3-ferroptosis negative regulatory axis suppresses tumor growth and alleviates chemoresistance in gastric cancer. Redox Biol. 52, 102317 (2022).
Shen, D. D., Pang, J. R. & Bi, Y. P. et al. LSD1 deletion decreases exosomal PD-L1 and restores T-cell response in gastric cancer. Mol. Cancer 21, 75 (2022).
Norberg, S. M. & Hinrichs, C. S. Engineered T cell therapy for viral and non-viral epithelial cancers. Cancer Cell 41, 58–69 (2023).
Gao, Y., Wang, H. & Chen, S. et al. Single-cell N6-methyladenosine regulator patterns guide intercellular communication of tumor microenvironment that contribute to colorectal cancer progression and immunotherapy. J. Transl. Med. 20, 197 (2022).
Chen, P., Wang, Y. & Li, J. et al. Diversity and intratumoral heterogeneity in human gallbladder cancer progression revealed by single-cell RNA sequencing. Clin. Transl. Med. 11, e462 (2021).
Li, X., Zhao, S. & Bian, X. et al. Signatures of EMT, immunosuppression, and inflammation in primary and recurrent human cutaneous squamous cell carcinoma at single-cell resolution. Theranostics 12, 7532–7549 (2022).
Maiorino, L., Daßler-Plenker, J., Sun, L. & Egeblad, M. Innate immunity and cancer pathophysiology. Annu. Rev. Pathol. 17, 425–457 (2022).
Chyuan, I. T., Chu, C. L. & Hsu, P. N. Targeting the tumor microenvironment for improving therapeutic effectiveness in cancer immunotherapy: focusing on immune checkpoint inhibitors and combination therapies. Cancers 13, 1188 (2021).
Zhang, Z., Wang, Z. X. & Chen, Y. X. et al. Integrated analysis of single-cell and bulk RNA sequencing data reveals a pan-cancer stemness signature predicting immunotherapy response. Genome Med. 14, 45 (2022).
Zheng, H., Liu, H., Ge, Y. & Wang, X. Integrated single-cell and bulk RNA sequencing analysis identifies a cancer associated fibroblast-related signature for predicting prognosis and therapeutic responses in colorectal cancer. Cancer Cell Int. 21, 552 (2021).
Bao, X., Shi, R. & Zhao, T. et al. Integrated analysis of single-cell RNA-seq and bulk RNA-seq unravels tumour heterogeneity plus M2-like tumour-associated macrophage infiltration and aggressiveness in TNBC. Cancer Immunol. Immunother. 70, 189–202 (2021).
Shi, T., Zhang, Y. & Wang, Y. et al. DKK1 promotes tumor immune evasion and impedes Anti-PD-1 treatment by inducing immunosuppressive macrophages in gastric cancer. Cancer Immunol. Res. 10, 1506–1524 (2022).
Chen, B., Scurrah, C. R. & McKinley, E. T. et al. Differential pre-malignant programs and microenvironment chart distinct paths to malignancy in human colorectal polyps. Cell 184, 6262–6280.e26 (2021).
Steele, N. G., Carpenter, E. S. & Kemp, S. B. et al. Multimodal mapping of the tumor and peripheral blood immune landscape in human pancreatic cancer. Nat. Cancer 1, 1097–1112 (2020).
Carpenter, E. S., Elhossiny, A. M. & Kadiyala, P. et al. Analysis of donor pancreata defines the transcriptomic signature and microenvironment of early neoplastic lesions. Cancer Discov. 13, 1324–1345 (2023).
Keefe, F., Monzón-Sandoval, J., Rosser, A. E., Webber, C. & Li, M. Single-cell transcriptomics reveals conserved regulatory networks in human and mouse interneuron development. Int J. Mol. Sci. 24, 8122 (2023).
Hao, Y., Hao, S. & Andersen-Nissen, E. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29 (2021).
Ma, L., Hernandez, M. O. & Zhao, Y. et al. Tumor cell biodiversity drives microenvironmental reprogramming in liver cancer. Cancer Cell 36, 418–430.e6 (2019).
Ritchie, M. E., Phipson, B. & Wu, D. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Arunachalam, D., Ramanathan, S. M. & Menon, A. et al. Expression of immune response genes in human corneal epithelial cells interacting with Aspergillus flavus conidia. BMC Genomics 23, 5 (2022).
Linkner, T. R., Ambrus, V. & Kunkli, B. et al. Cellular proteo-transcriptomic changes in the immediate early-phase of lentiviral transduction. Microorganisms 9, 2207 (2021).
Li, K. P., Yu, T., Hwang, J. D., Yeah, K. S. & Winefordner, J. D. Estimation of rate constants using statistical moments of spatially resolved signal profiles for the elucidation of analyte transformation mechanisms in an inductively coupled plasma. Anal. Chem. 60, 1599–1605 (1988).
Racle, J. & Gfeller, D. EPIC: a tool to estimate the proportions of different cell types from bulk gene expression data. Methods Mol. Biol. 2120, 233–248 (2020).
Zhu, X., Zou, W. & Meng, X. et al. Elaiophylin inhibits tumorigenesis of human uveal melanoma by suppressing mitophagy and inducing oxidative stress via modulating SIRT1/FoxO3a signaling. Front Oncol. 12, 788496 (2022).
Lu, T., Li, J., Qian, S., Cao, H., Ning, C. & Liu, X. Enhanced osteogenic and selective antibacterial activities on micro-/nano-structured carbon fiber reinforced polyetheretherketone. J. Mater. Chem. B 4, 2944–2953 (2016).
He, L., Jhong, J. H. & Chen, Q. et al. Global characterization of macrophage polarization mechanisms and identification of M2-type polarization inhibitors. Cell Rep. 37, 109955 (2021).
Wang, P., Wang, H. & Huang, Q. et al. Exosomes from M1-polarized macrophages enhance paclitaxel antitumor activity by activating macrophages-mediated inflammation. Theranostics 9, 1714–1727 (2019).
Nakamura, R., Bing, R., Gartling, G. J. & Branski, R. C. Macrophages alter inflammatory and fibrotic gene expression in human vocal fold fibroblasts. Exp. Cell Res. 419, 113301 (2022).
Zeng, X., Yao, B. & Liu, J. et al. The SMARCA4R1157W mutation facilitates chromatin remodeling and confers PRMT1/SMARCA4 inhibitors sensitivity in colorectal cancer. NPJ Precis Oncol. 7, 28 (2023).
Nishiyama, T., Zhang, Y. & Cui, M. et al. Precise genomic editing of pathogenic mutations in RBM20 rescues dilated cardiomyopathy. Sci. Transl. Med. 14, eade1633 (2022).
Zhang, Z., Wang, J. & Chen, Y. et al. Activin a promotes myofibroblast differentiation of endometrial mesenchymal stem cells via STAT3-dependent Smad/CTGF pathway. Cell Commun. Signal. 17, 45 (2019).
Lee, S. G., Woo, S. M. & Seo, S. U. et al. Cathepsin D promotes polarization of tumor-associated macrophages and metastasis through TGFBI-CCL20 signaling. Exp. Mol. Med. 56, 383–394 (2024).
Li, Y., Shen, Z. & Chai, Z. et al. Targeting MS4A4A on tumour-associated macrophages restores CD8+ T-cell-mediated antitumour immunity. Gut 72, 2307–2320 (2023).
Chen, S., Morine, Y. & Tokuda, K. et al. Cancer‑associated fibroblast‑induced M2‑polarized macrophages promote hepatocellular carcinoma progression via the plasminogen activator inhibitor‑1 pathway. Int. J. Oncol. 59, 59 (2021).
Wu, J. Y., Huang, T. W. & Hsieh, Y. T. et al. Cancer-derived succinate promotes macrophage polarization and cancer metastasis via succinate receptor. Mol. Cell. 77, 213–227.e5 (2020).
Yang, L., Shao, X., Jia, S., Zhang, Q. & Jin, Z. Interleukin-35 dampens CD8+ T cells activity in patients with non-viral hepatitis-related hepatocellular carcinoma. Front. Immunol. 10, 1032 (2019).
Jiang, G., Shi, L. & Zheng, X. et al. Androgen receptor affects the response to immune checkpoint therapy by suppressing PD-L1 in hepatocellular carcinoma. Aging 12, 11466–11484 (2020).
Yang, M., Chen, C., Wang, K., Chen, Y. & Xia, J. Astilbin influences the progression of osteoarthritis in rats by down-regulation of PGE-2 expression via the NF-κB pathway. Ann. Transl. Med. 8, 766 (2020).
Wang, X. & Wu, Y. Protective effects of autophagy inhibitor 3-methyladenine on ischemia-reperfusion-induced retinal injury. Int Ophthalmol. 40, 1095–1101 (2020).
Chen, G., Li, X. & Zhu, H. et al. Transplanting neurofibromatosis-1 gene knockout neural stem cells improve functional recovery in rats with spinal cord injury by enhancing the mTORC2 pathway. Exp. Mol. Med. 54, 1766–1777 (2022).
Warnke, I., Goralczyk, R., Fuhrer, E. & Schwager, J. Dietary constituents reduce lipid accumulation in murine C3H10 T1/2 adipocytes: A novel fluorescent method to quantify fat droplets. Nutr. Metab. 8, 30 (2011).
Garikipati, V. N. S., Verma, S. K., Cheng, Z. et al. Circular RNA CircFndc3b modulates cardiac repair after myocardial infarction via FUS/VEGF-A axis. Nat. Commun. 11, 2234 (2019).
Wu, Q. & Yi, X. Down-regulation of long noncoding RNA MALAT1 protects hippocampal neurons against excessive autophagy and apoptosis via the pi3k/akt signaling pathway in rats with epilepsy. J. Mol. Neurosci. 65, 234–245 (2018).
Zhang, Z., Li, W., Jiang, D., Liu, C. & Lai, Z. MicroRNA-139-5p inhibits cell viability, migration and invasion and suppresses tumor growth by targeting HDGF in non-small cell lung cancer. Oncol. Lett. 19, 1806–1814 (2020).
Guo, S. C., Tao, S. C., Yin, W. J., Qi, X., Yuan, T. & Zhang, C. Q. Exosomes derived from platelet-rich plasma promote the re-epithelization of chronic cutaneous wounds via activation of YAP in a diabetic rat model. Theranostics 7, 81–96 (2017).
Cheung, E. C., DeNicola, G. M. & Nixon, C. et al. Dynamic ROS control by TIGAR regulates the initiation and progression of pancreatic cancer. Cancer Cell. 37, 168–182.e4 (2020).
Li, H., Liang, J. & Wang, J. et al. Mex3a promotes oncogenesis through the RAP1/MAPK signaling pathway in colorectal cancer and is inhibited by hsa-miR-6887-3p. Cancer Commun. 41, 472–491 (2021).
Wu, Y., Zhu, B. & Yan, Y. et al. Long non-coding RNA SNHG1 stimulates ovarian cancer progression by modulating expression of miR-454 and ZEB1. Mol. Oncol. 15, 1584–1596 (2021).
He, W., Zhang, H. & Han, F. et al. CD155T/TIGIT signaling regulates CD8+ T-cell metabolism and promotes tumor progression in human gastric cancer. Cancer Res. 77, 6375–6388 (2017).
Tu, J., Chen, W. & Fang, Y. et al. PU.1 promotes development of rheumatoid arthritis via repressing FLT3 in macrophages and fibroblast-like synoviocytes. Ann. Rheum. Dis. 82, 198–211 (2023).
Tong, D., Zhao, Y., Tang, Y., Ma, J., Wang, Z. & Li, C. Circ-Usp10 promotes microglial activation and induces neuronal death by targeting miRNA-152-5p/CD84. Bioengineered 12, 10812–10822 (2021).
Zhou, K. X., Huang, S. & Hu, L. P. et al. Increased nuclear transporter KPNA2 contributes to tumor immune evasion by enhancing PD-L1 expression in PDAC. J. Immunol. Res. 2021, 6694392 (2021).
Zhang, Z., Fan, Y. & Xie, F. et al. Breast cancer metastasis suppressor OTUD1 deubiquitinates SMAD7. Nat. Commun. 8, 2116 (2017).
Acknowledgements
This study was supported by Ceyuan Puhui (Ningbo) Biotechnology Co.Ltd (Project number 6010223002) and Medical research project of China Medical and Health Development Foundation (Project number JJ-2024-0546). We are deeply grateful to all participants, staff, and the supporting institutions, notably the Cancer Center and the Tumor Research and Therapy Center at Shandong Provincial Hospital affiliated with Shandong First Medical University, for providing essential facilities and resources. This research was adhered to ethical standards approved under protocol number KYLL-2023-405 by the Second Hospital Cheeloo College of Medicine, Shandong University. We thank the Vascular Intervention Department of the First Affiliated Hospital of Ningbo University for technical support. Our appreciation extends to Qianqian Cao, who conceived and designed the research; Dianshui Sun, who performed the experiments; Can Tu, who interpreted the results; Jihua Wang for data analysis; Runjia Fu for Fig. preparation; Rumei Gong for drafting the manuscript; Yueying Xiao and Qin Liu for critical revisions; and Xiaomei Li for approving the final manuscript. Special thanks to the reviewers and editors of the International Journal of Biological Macromolecules for their valuable feedback, which greatly enhanced this manuscript.
Author information
Authors and Affiliations
Contributions
Q.C. conceived and designed research. D.S. performed experiments. C.T. interpreted results of experiments. J.W. analyzed data. R.F. prepared Fig.s. R.G. drafted the paper. Y.X. and Q.L. edited and revised the manuscript. X.L. read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
This study was conducted in strict accordance with the guidelines and regulations of the Second Hospital, Cheeloo College of Medicine, Shandong University. Ethical approval was granted under the protocol number KYLL-2023-405. All research procedures involving animals and human subjects were conducted in compliance with ethical standards and informed consent was obtained from all human participants involved in the study. The welfare and ethical treatment of animals and human subjects were of paramount concern throughout the course of this research.
Peer review
Peer review information
Communications Biology thanks Luciana Marti, and the other, anonymous, reviewer for their contribution to the peer review of this work. Primary Handling Editors: Jin-Min Nam and Mengtan Xing.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cao, Q., Sun, D., Tu, C. et al. Defining gastric cancer ecology: the crucial roles of TREM2+ macrophages and fibroblasts in tumor microenvironments. Commun Biol 8, 514 (2025). https://doi.org/10.1038/s42003-025-07512-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-025-07512-2
