
Abstract
Acute lung injury (ALI), a frequent and severe complication of sepsis, is associated with significant mortality rates. Previous studies indicated that GLS2 plays a key role in promoting ferroptosis. However, its underlying mechanisms remain unclear. Here we show, there were elevated ferroptosis and increased expression levels of protein arginine methyltransferase 1 (PRMT1), early growth response 1 (EGR1), and glutaminase 2 (GLS2) in both in vitro and in vivo ALI models. Additionally, EGR1 was found to induce the transcription of GLS2, thereby promoting ferroptosis. We also discovered that the protein level of EGR1 was increased through enhanced stability, facilitated by PRMT1-mediated arginine methylation, and reduced ubiquitination degradation regulated by neural precursor cell expressed developmentally down-regulated protein 4 like (NEDD4L). The in vivo results confirmed that the knockdown of PRMT1 suppressed ferroptosis via the EGR1/GLS2 axis. Our findings suggest that PRMT1-mediated stabilization of EGR1 promoted sepsis induced ALI via GLS2, highlighting the therapeutic potential of targeting PRMT1 or EGR1 in the treatment of sepsis-induced ALI.

Similar content being viewed by others


Introduction
Sepsis, a life-threatening organ dysfunction, is among the leading causes of mortality in intensive care units1, and acute lung injury (ALI) is a severe respiratory condition characterized by diffuse non-cardiogenic pulmonary edema due to alveolar damage, leading to severe hypoxemia and respiratory distress, often with irreversible outcomes2. Sepsis is identified as the leading cause of ALI attributable to the lungs’ heightened susceptibility to intense immune responses during multiorgan dysfunction stages3. The understanding of ALI pathogenesis remains incomplete, and currently, there is no effective targeted therapy available4. This gap highlights the importance of research into its pathogenic mechanisms and potential treatments. In the past, the pathogenesis of ALI has been associated with processes such as inflammation, coagulation, oxidative stress, and tissue repair5. Recent studies have identified ferroptosis, an iron-dependent form of cell death, as a contributing factor in several models of ALI, including those induced by lipopolysaccharide (LPS), intestinal ischemia-reperfusion, oleic acid, and acute radiation6. Notably, ferroptosis inhibitors, such as ferrostatin-1 and lipoxstatin-1, including the inhibitor of apoptosis-stimulating protein of p53 (iASPP), have been shown to offer protective effects against ALI by inhibiting ferroptosis7. This discovery underscores a promising area of research for developing therapeutic strategies.
Glutaminase 2 (GLS2), a key glutaminase, plays a crucial role in regulating glutamine metabolism8. It facilitates mitochondrial respiration and augments ATP production in cells by catalyzing the conversion of glutamine into glutamate and α-ketoglutarate9. Several studies have shown that GLS2 can promote ferroptosis10. Suzuki et al. reported that GLS2 promotes ferroptosis through glutaminolysis facilitation, contributing to hepatocellular carcinoma suppression10. Furthermore, tumor cells harboring the S47 variant demonstrate reduced GLS2 levels and display resistance to ferroptosis11. Therefore, targeting GLS2 is pivotal in mitigating sepsis-induced ALI. However, its underlying mechanism remains unclear.
Analysis of the GEO database revealed that early growth response 1 (EGR1) is at the intersection of differential genes for sepsis-induced ALI and those for ferroptosis. EGR1, which is expressed in the initial phases of multiple diseases, induces pathological signals from the extracellular environment, thereby accelerating disease development12. Additionally, as a member of the zinc finger transcription factor family, EGR1 can mediate many downstream target genes12. Thus, EGR1 is involved in regulating cell proliferation, apoptosis, invasion, and angiogenesis13. In terms of respiratory disease, EGR1 is elevated in several of them, including ALI12. However, whether EGR1 induces ferroptosis in sepsis-induced ALI is not yet clear. Our preliminary data from the JASPAR database (https://jaspar.elixir.no/) indicate that EGR1 can bind to the promoter of GLS2. Therefore, we speculated that EGR1 may transcriptionally regulate GLS2, driving ferroptosis and aggravating the process of sepsis-induced ALI.
Post-translational modifications of proteins are key factors causing proteome diversity and dynamic cellular equilibrium14. Arginine methylation, a prevalent form of post-translational modification, entails the addition of methyl groups to the guanidino groups of arginine residues within substrate proteins15. The enzymes catalyzing arginine methylation are known as protein arginine methyltransferases (PRMTs)16. The PRMT family consists of 11 members, which influence substrate protein stability, localization, activity, or interaction by catalyzing the methylation of a variety of substrate proteins and are involved in regulating various physiological and pathological processes17. Further analysis revealed that EGR1 can be modified by arginine methylation. Additionally, studies have suggested that inhibiting protein arginine methyltransferase 1 (PRMT1) can alleviate sepsis-induced acute kidney injury18. However, whether PRMT1 can methylate EGR1 arginine and regulate its protein remains to be elucidated.
The steady state of the protein is normally maintained by a dynamic process of protein stabilization and degradation19. Ubiquitination modification promotes protein degradation and is also an important protein regulatory mechanism within the cell20. The prediction of EGR1 ubiquitination from our preliminary data showed that EGR1 was regulated by neural precursor cell expressed developmentally down-regulated protein 4 like (NEDD4L), NEDD4-like E3 Ubiquitin Protein Ligase. Previous studies also have indicated that the expression of NEDD4L decreases in sepsis models induced by LPS21.
Based on the findings above, this study hypothesized that EGR1 was influenced on one hand by PRMT1-mediated arginine methylation, enhancing its stability, and on the other hand ubiquitin-mediated degradation was reduced by inhibiting NEDD4L, leading to an increase in EGR1 protein. This promoted the transcriptional regulation of GLS2, leading to ferroptosis and exacerbating the process of sepsis-induced ALI.
Results
Increased ferroptosis and upregulation of PRMT1, EGR1, and GLS2 expression in sepsis-related ALI models in vivo and in vitro
To investigate the role of ferroptosis in sepsis-induced ALI, a mouse model of ALI was established via CLP, a widely recognized method to induce sepsis in mice. Fer-1 was subsequently administered to inhibit ferroptosis. As depicted in Fig. 1a, the survival rate of mice subjected to CLP was markedly lower than that in the sham group. In contrast, treatment with Fer-1 led to an improvement in survival rates. Moreover, lung injury severity and lung wet-to-dry weight ratios were significantly increased in the sepsis-induced mice compared to the control group (Fig. 1b–d). The administration of Fer-1 results in a reduction of both lung injury severity and wet-to-dry weight ratios. In the CLP group, mitochondrial morphology exhibited thickening of the double membrane and reduced volume. The administration of Fer-1 mitigated changes in mitochondrial morphology, characterized by the reduction in mitochondrial membrane thickening and volume loss (Fig. 1e). This was associated with increased levels of MDA, 4-HNE, and acyl-CoA synthetase long-chain family member 4 (ACSL4), and decreased levels of GSH, glutathione peroxidase 4 (GPX4), and solute carrier family 7 member 11 (SLC7A11), which were reversed by Fer-1 (Fig. 1f–h and Fig. S1a). In the sepsis-affected mice, there was an increased expression of PRMT1, EGR1, and GLS2, which was not altered by the addition of Fer-1 (Fig. 1i).

The cecal ligation and puncture (CLP) procedure was used to establish a sepsis-induced ALI model. Additionally, ferrostatin-1(Fer-1) was employed to inhibit ferroptosis in sepsis-induced ALI. a Survival curves of mice were analyzed using the Kaplan-Meier method. b Hematoxylin & eosin (HE) staining was performed to examine pathological changes in lung tissue. Scale bar = 100 µm. c Lung injury scores were compared among the Sham group, CLP group, and CLP + Fer-1 group. d Lung weight was measured and the wet-to-dry ratio was calculated in each group. e Changes in mitochondrial morphology were observed using electron microscopy. Scale bar = 500 nm. f–h Levels of malondialdehyde (MDA), 4-hydroxynonenal (4-HNE), and glutathione (GSH) were measured. i The expression levels of PRMT1, EGR1, and GLS2 were detected by Western blot. N = 8 mice, *p < 0.05, **p < 0.01, ***p < 0.001.
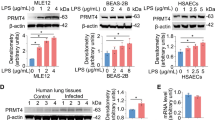
Next, the in vitro ALI model was created by treating the mouse lung epithelial cell line, MLE-12, with LPS. After LPS treatment, the viability of MLE-12 cells was significantly reduced (Fig. 2a). However, the inhibitory effect of LPS on MLE-12 cell viability was attenuated following Fer-1 treatment. Additionally, the addition of Fer-1 counteracted the LPS-induced decrease in GSH levels and increases in MDA, Fe2+, and lipid ROS levels (Fig. 2b–e). Furthermore, Fer-1 treatment attenuated the inhibitory effects of LPS on GPX4 and SLC7A11 and reduced the promotional effects of LPS on ACSL4 levels in MLE-12 cells (Fig. S1b). Similar to the in vivo data above, LPS treatment increased the expression levels of PRMT1, EGR1, and GLS2 (Fig. 2f). After Fer-1 treatment, no significant change in these proteins was observed.

MLE-12 cells were used to construct an in vitro sepsis-related ALI model by treating them without (control) or with LPS. Afterward, the cells were additionally treated with Fer-1 (LPS+Fer-1). a Cell viability was assessed using the CCK-8 assay. b–e The expression levels of malondialdehyde (MDA), the concentration of Fe2+, glutathione (GSH), and lipid ROS were measured. f The expression levels of PRMT1, EGR1, and GLS2 were detected by Western blot. Mean ± SD, n = 3 independent experiments, and box plots represent median with minimum and maximum whiskers. *p < 0.05, **p < 0.01, ***p < 0.001.
In summary, ferroptosis was upregulated in sepsis-associated ALI, accompanied by an increase in the expression of PRMT1, EGR1, and GLS2.
Knockdown of EGR1 suppressed ferroptosis in sepsis-related ALI
To confirm the regulatory effect of EGR1 on ferroptosis in ALI, EGR1 was knocked down in MLE-12 cells by transfecting them with shEGR1 following LPS treatment. The successful knockdown of EGR1 was validated by Western blot analysis (Fig. 3a). The depletion of EGR1 significantly reduced GLS2 expression in MLE-12 cells (Fig. 3a). Furthermore, the silencing of EGR1 suppressed the LPS-induced upregulation of EGR1 and GLS2 expression in MLE-12 cells (Fig. 3b). Transfection with shEGR1 rescued the decrease in MLE-12 cell viability induced by LPS treatment (Fig. 3c). The increased levels of MDA, Fe2+, and lipid ROS, as well as the decreased level of GSH induced by LPS treatment, were reversed after EGR1 knockdown (Fig. 3d–g). In addition, the silencing of EGR1 counteracted the inhibitory effects of LPS on GPX4 and SLC7A11 and decreased the promotional effects of LPS on ACSL4 levels in MLE-12 cells (Fig. S1c).

a EGR1 was knocked down in MLE-12 cells by transfecting them with shEGR1 or shNC. The expression levels of EGR1 and GLS2 were detected by Western blot. b–g MLE-12 cells were treated without or with LPS. Afterward, EGR1 was knocked down in those LPS-treated cells by transfecting them with shEGR1 or shNC. b The expression levels of EGR1 and GLS2 were detected by Western blot. c Cell viability was assessed using the CCK-8 assay. d–g The expression levels of malondialdehyde (MDA), the concentration of Fe2+, glutathione (GSH), and lipid ROS were measured. Mean ± SD, n = 3 independent experiments, and box plots represent median with minimum and maximum whiskers. *p < 0.05, **p < 0.01, ***p < 0.001.
Taken together, these results suggest that the downregulation of EGR1 significantly inhibited ferroptosis in sepsis-related ALI.
EGR1 promoted ferroptosis by transcriptionally regulating GLS2 expression
Next, we explored the downstream mechanism of EGR1-mediated ferroptosis in ALI. It was predicted by JASPAR that EGR1 potentially bound to the -1471/-1461 region of the GLS2 promoter (Fig. 4a). Furthermore, as shown by ChIP results, the binding of EGR1 to the GLS2 promoter was significantly increased in LPS-treated MLE-12 cells (Fig. 4b). The silencing of EGR1 reduced the enrichment of the GLS2 promoter in complexes precipitated with the EGR1 antibody (Fig. 4c). Additionally, results from a dual-luciferase reporter assay revealed that the depletion of EGR1 significantly decreased the luciferase activity in 293 T cells transfected with the GLS2-wt luciferase reporter vector, while it had minimal effect on the luciferase activity of cells transfected with GLS2-mut luciferase vectors (Fig. 4d). The overexpression of EGR1 led to an increase in both EGR1 and GLS2 expression (Fig. 4e). To validate the role of GLS2 in EGR1-regulated ferroptosis in ALI, we knocked down EGR1 and overexpressed GLS2 in MLE-12 cells after LPS treatment. The overexpression of GLS2 reversed the inhibitory effect of EGR1 depletion on GLS2 expression in MLE-12 cells (Fig. 4f). Overexpression of GLS2 suppressed the protective effect of EGR1 knockdown on the viability of LPS-treated MLE-12 cells (Fig. 4g). Furthermore, GLS2 overexpression rescued the EGR1-depletion-induced decrease in MDA, Fe2+, and lipid ROS levels, and the increase in GSH level in LPS-treated MLE-12 cells (Fig. 4h–k). Collectively, these findings suggest that EGR1 enhances the transcription of GLS2, thereby inducing ferroptosis in lung epithelial cells.

a JASPAR predicts the binding between EGR1 with the GLS2 promoter region. b ChIP assay was conducted to detect the binding of EGR1 to the GLS2 promoter region in LPS-treated or not-treated MLE-12 cells. c ChIP assay was used to examine the binding of EGR1 to the GLS2 promoter region in the MLE-12 cells transfected with shNC or shEGR1. d Dual-luciferase reporter assay was performed to evaluate the binding of EGR1 to the GLS2 promoter region in 293 T cells co-transfected with shNC or shEGR1 as well as GLS2-wt or GLS2-mut. e EGR1 was overexpressed in MLE-12 cells by transfecting them with EGR1-overexpressing vectors, and the expression levels of EGR1, and GLS2 were detected by Western blot. f–k MLE-12 cells were treated without or with LPS. Afterward, those LPS-treated cells were transfected with shEGR1 or shNC or GLS2 overexpression vectors or shEGR1 plus GLS2 overexpression vectors. f The expression levels of EGR1 and GLS2 were detected by Western blot. g Cell viability was assessed using the CCK-8 assay. h–k The expression levels of malondialdehyde (MDA), the concentration of Fe2+, glutathione (GSH), and lipid ROS were measured. Mean ± SD, n = 3 independent experiments, and box plots represent median with minimum and maximum whiskers. *p < 0.05, **p < 0.01, ***p < 0.001.
PRMT1-depletion inhibited ferroptosis
Protein post-translational modifications are essential for generating proteomic diversity and maintaining cellular homeostasis. One common modification is arginine methylation, which is catalyzed by a group of enzymes known as PRMTs22. The PRMT family consists of nine key members (PRMT1-9)22, with PRMT8 uniquely expressed primarily in brain tissue23, leading us to focus on the remaining PRMTs. The GPS-MSP (https://msp.biocuckoo.org/wsresult.php) predicted that EGR1 was predominantly dimethylated (Fig. S2a). The PRMT family is classified into three types based on the type of arginine methylation they catalyze: Type I (PRMT1, 2, 3, 4, 6, and 8), which catalyzes both monomethylation and asymmetric dimethylation; Type II (PRMT5 and 9), responsible for monomethylation and symmetric dimethylation; and Type III (PRMT7), which catalyzes only monomethylation24. As a result, PRMT7 was excluded from further investigation. In an in vitro sepsis model, we analyzed the expression levels of PRMT1/2/3/4/5/6/9 using Western blot (Fig. S2b). Following LPS stimulation, PRMT3 and PRMT9 expression remained unchanged, while PRMT1/2/4/5/6 showed increased expression. Furthermore, Co-IP experiments demonstrated that only PRMT1 interacted with EGR1 (Fig. S2c), prompting us to focus on PRMT1 to investigate its role in regulating EGR1. To further investigate the role of PRMT1 in the regulation of ALI, we performed a knockdown of PRMT1 in MLE-12 cells treated with LPS. The efficiency of the transfection using shPRMT1 was validated by Western blot analysis (Fig. 5a). The knockdown of PRMT1 significantly reduced the expression levels of EGR1 and GLS2 (Fig. 5a). Moreover, the depletion of PRMT1 abrogated the enhancing effect of LPS on the expression of PRMT1, EGR1, and GLS2 in MLE-12 cells (Fig. 5b). The inhibitory effect of LPS on the viability of MLE-12 cells was restored following the knockdown of PRMT1 (Fig. 5c). The increase in MDA, Fe2+, and lipid ROS, along with the decrease in GSH levels induced by LPS, were reversed upon PRMT1 knockdown (Fig. 5d–g). Furthermore, PRMT1 depletion restored the LPS-induced decrease in GPX4 and SLC7A11 expression and reversed the LPS-induced increase in ACSL4 levels in MLE-12 cells (Fig. S1d). In conclusion, the downregulation of PRMT1 mitigated ferroptosis in ALI.

a PRMT1 was knocked down in MLE-12 cells by transfecting them with shPRMT1 or shNC. The expression levels of PRMT1, EGR1, and GLS2 were detected by Western blot. b–g MLE-12 cells were treated without or with LPS. Afterward, those LPS-treated cells were transfected with shPRMT1 or shNC. b The expression levels of PRMT1, EGR1, and GLS2 were detected by Western blot. c Cell viability was assessed using the CCK-8 assay. d–g The expression levels of malondialdehyde (MDA), the concentration of Fe2+, glutathione (GSH), and lipid ROS were measured. Mean ± SD, n = 3 independent experiments, and box plots represent median with minimum and maximum whiskers. *p < 0.05, **p < 0.01, ***p < 0.001.
PRMT1-mediated arginine methylation modification maintained EGR1 stability
Ubibrower (http://ubibrowser.bio-it.cn/ubibrowser_v3/) was utilized to predict the binding of E3 ligases with EGR1. The E3 ligases with the highest predicted credibility, including SALL2, BTRC, FANCL, SMURF1, BAZ1B, RAPSN, ITCH, FBXW11, and NEDD4L, were selected to examine their potential interaction with EGR1 (Fig. 6a). After LPS treatment, only ITCH and NEDD4L were significantly downregulated in MLE-12 cells (Fig. S3a), and thus they were chosen for the Co-IP experiment to further validate their interaction with EGR1. The data confirmed that Itch did not interact with EGR1, whereas NEDD4L did (Fig. S3b). Therefore, NEDD4L was chosen for further study. The expression of NEDD4L was decreased in MLE-12 cells treated with LPS (Fig. 6b). Subsequently, NEDD4L was overexpressed in MLE-12 cells by transfecting them with NEDD4L-overexpressing vectors, resulting in a remarkable increase in NEDD4L protein expression post-transfection (Fig. 6c), confirming the success of the NEDD4L overexpression. EGR1 was immunoprecipitated by exogenous SFB-Flag-NEDD4L in 293 T cells and by endogenous NEDD4L protein in MLE-12 cells (Fig. 6d). The overexpression of NEDD4L led to a significant reduction in EGR1 levels (Fig. 6e), an effect that was mitigated by the proteasome inhibitor MG132. Moreover, the overexpression of NEDD4L not only accelerated the degradation of EGR1 but also increased its ubiquitination (Fig. 6f, g). Next, we examined whether PRMT1 was involved in mediating EGR1. The PRMT1 and EGR1 both exogenously and endogenously interacted in 293 T cells or MLE-12 cells, respectively (Fig. 6h, i). Moreover, PTMR1, ADMA, and EGR1 were co-localized in MLE-12 cells (Fig. 6j), suggesting that PTMR1 may play a role in regulating EGR1 arginine methylation. Experiments involving the cleavage of EGR1 into fragments spanning amino acids 1-150 (1-150 aa) and 151-533aa demonstrated PRMT1’s interaction with these truncated forms (Fig. 6k). Mutations at R133 or R278 slightly reduced the ADMA modification of EGR1 by PRMT1 (Fig. 6l), whereas simultaneous mutations at these residues significantly decreased it. These findings emphasize the critical role of PRMT1-mediated ADMA modification in maintaining EGR1 stability. Next, we sought to investigate the function of PRMT1-mediated arginine methylation of EGR1. Co-IP assay showed that PRMT1 knockdown abated the interaction between EGR1 and ADMA but strengthened the interaction between EGR1 and MMA (Fig. 6m). Interestingly, the interaction between NEDD4L and EGR1 was enhanced by PRMT1 deficiency (Fig. 6n) but attenuated by PRMT1 overexpression (Fig. 6o). And EGR1 ubiquitination was reinforced in PRMT1-knockdown AML-12 cells (Fig. 6p). PRMT1 overexpression significantly increased the protein levels of both PRMT1 and EGR1 while decreasing EGR1 ubiquitination (Fig. S4a). Conversely, the knockdown of NEDD4L reduced NEDD4L protein levels but elevated EGR1 protein expression. Additionally, EGR1 ubiquitination was diminished following NEDD4L depletion (Fig. S4b). Finally, we investigated whether PRMT1 modulated the arginine methylation upon LPS challenge or not. As shown in Figure S4c, LPS treatment resulted in enhanced arginine methylation of EGR1, but this effect was reversed by PRMT1 knockdown. Furthermore, LPS treatment reduced the interaction between NEDD4L and EGR1, an effect that was also reversed by PRMT1 knockdown (Fig. S4d). Collectively, these findings suggested that PRMT1-mediated arginine methylation of EGR1 increased EGR1 stability through repressing NEDD4L-mediated EGR1 ubiquitination in AML-12 cells.

a Ubibrowser was used to predict E3 ligases (neural precursor cell expressed developmentally down-regulated protein 4 like, NEDD4L) that bind to EGR1. b The expression of NEDD4L in LPS-treated or non-treated MLE-12 cells was assessed via Western blot. c The expression of NEDD4L in NEDD4L-overexpressed MLE-12 cells was assessed via Western blot. d In 293 T and MLE-12 cells, co-immunoprecipitation (Co-IP) was used to detect the interaction between exogenous and endogenous NEDD4L and EGR1, respectively. (E-G) NEDD4L was overexpressed in MLE-12 cells. e The expression of EGR1 was detected by Western blot after the addition of MG132. f The degradation of EGR1 was assessed by Western blot after the addition of cycloheximide (CHX). g The ubiquitination-mediated degradation of EGR1 was detected by Co-IP. h, i In 293 T and MLE-12 cells, Co-IP was used to detect the interaction between exogenous and endogenous PRMT1 and EGR1, respectively. j The co-localization of ADMA or PRMT1 with EGR1 was confirmed by laser scanning confocal microscopy. Scale bar = 20 µm. k The interaction of EGR1 fragments and PRMT1 was validated by GST pulldown. l Co-IP experiments detected the arginine methylation modification of EGR1 by PRMT1. m Co-IP was performed to assess the levels of arginine methylation on EGR1 in PRMT1-knockdown MLE-12 cells. n Co-IP was used to examine the interaction between EGR1 and NEDD4L in PRMT1 knockdown MLE-12 cells. o Co-IP was conducted to investigate the binding between EGR1 and NEDD4L in PRMT1 overexpressing 293 T cells. p With the transfection with the HA-Ubiquitin, Co-IP was utilized to detect the ubiquitination-mediated degradation of EGR1 in PRMT1 knockdown MLE-12 cells. Mean ± SD, n = 3 independent experiments, and box plots represent median with minimum and maximum whiskers. *p < 0.05, **p < 0.01, ***p < 0.001.
PRMT1 promoted ferroptosis via EGR1/GLS2 axis
To validate the role of PRMT1 in ferroptosis, PRMT1 was knocked down while EGR1 was overexpressed in MLE-12 cells, followed by LPS treatment. Knockdown of PRMT1 decreased the expression of PRMT1, EGR1, and GLS2 (Fig. 7a). In contrast, overexpressing EGR1 reversed the effects of knocking down PRMT1 on the changes in EGR1 and GLS2 (Fig. 7a). PRMT1 depletion also inhibited LPS-induced upregulation of EGR1, and GLS2 (Fig. 7b). Nevertheless, EGR1 overexpression had the opposite effect. The transfection of shPRMT1 significantly increased LPS-treated MLE-12 viability (Fig. 7c), whereas the protective effect of PRMT1 knockdown against LPS on MLE-12 viability was reversed following EGR1 overexpression. The knockdown of PRMT1 led to a decrease in MDA, Fe2+, and lipid ROS levels but an increase in GSH levels (Fig. 7d–g). However, the impact of PRMT1 depletion mentioned above was attenuated by the overexpression of EGR1 (Fig. 7d–g). Additionally, PRMT1 depletion reversed the LPS-induced decrease in GPX4 and SLC7A11 expression and the increase in ACSL4 levels in MLE-12 cells, effects which were subsequently counteracted by EGR1 overexpression (Fig. S1e). Collectively, EGR1 overexpression suppressed the inhibitory effect of PRMT1 knockdown on ferroptosis in MLE-12 cells.

a PRMT1 was knocked down and/or EGR1 was overexpressed in MLE-12 cells by transfecting them with shPRMT1 or EGR1 overexpressing vectors or shPRMT1 plus EGR1 overexpressing vectors. The expression levels of PRMT1, EGR1, and GLS2 were detected by Western blot. b–g MLE-12 cells were treated without or with LPS. Afterward, those LPS-treated cells were transfected with shPRMT1 or EGR1 overexpressing vectors or shPRMT1 plus EGR1 overexpressing vectors. b The expression levels of PRMT1, EGR1, and GLS2 were detected by Western blot. c Cell viability was assessed using the CCK-8 assay. d–g The expression levels of malondialdehyde (MDA), the concentration of Fe2+, glutathione (GSH), and lipid ROS were measured. Mean ± SD, n = 3 independent experiments, and box plots represent median with minimum and maximum whiskers. *p < 0.05, **p < 0.01, ***p < 0.001.
Knockdown of PRMT1 attenuated sepsis-related ALI by suppressing ferroptosis via EGR1/GLS2 axis in vivo
In the subsequent phase of our study, we corroborated the in vitro findings using an in vivo model of ALI. The knockdown of PRMT1 or EGR1 significantly enhanced survival rates in mice subjected to CLP (Fig. 8a and Fig. S5a). Furthermore, silencing of PRMT1 or EGR1 markedly reduced lung injury severity and decreased the lung wet-to-dry weight ratios in CLP-treated mice (Fig. 8b–d and Fig. S5b–d). The alterations in mitochondrial morphology induced by CLP, characterized by the thickening of the mitochondrial double membrane and reduced mitochondrial volume, were notably mitigated upon PRMT1 or EGR1 knockdown (Fig. 8e and Fig. S5e). This was accompanied by a reduction in the levels of MDA and 4-HNE, and an increase in GSH levels in the CLP group, indicating a decrease in ROS and lipid peroxidation (Fig. 8f, h and Fig. S5f, h). Consistently, silencing PRMT1 or EGR1 attenuated the decrease in GPX4 and SLC7A11 levels and reduced the increase in ACSL4 levels in CLP mice (Fig. S5i and Fig. S1f). Additionally, the knockdown of PRMT1 inhibited the CLP-induced upregulation of PRMT1, EGR1, and GLS2 expression (Fig. 8i, j). Similar to PRMT1 depletion, knockdown of EGR1 also attenuated the increase of EGR1 and GLS2 levels in the CLP group but had no impact on PRMT1 expression (Fig. S5i). In summary, the downregulation of PRMT1 or EGR1 mitigated ferroptosis via the EGR1/GLS2 axis, thereby alleviating sepsis-related ALI.

a Survival curves of mice from different groups were analyzed using the Kaplan-Meier method. b HE staining was performed to examine pathological changes in lung tissue. Scale bar = 100 µm. c Lung injury scores were compared among Sham, CLP, CLP + shNC, and CLP+shPRMT1 groups. d Lung weight was measured and the wet-to-dry ratio was calculated in each group. e Changes in mitochondrial morphology were observed using electron microscopy. Scale bar = 500 nm. f–h Levels of malondialdehyde (MDA), 4-hydroxynonenal (4-HNE), and glutathione (GSH) were measured. i, j The expression levels of PRMT1, EGR1, and GLS2 were evaluated by IHC staining and Western blot. Scale bar = 50 µm. N = 8 mice, *p < 0.05, **p < 0.01, ***p < 0.001.
Discussion
ALI, a common and serious complication arising from sepsis, is associated with a high mortality rate25. However, there has been limited advancement in the development of effective pharmacological and ventilatory treatments for sepsis-induced ALI over the past several decades26. Understanding the molecular mechanisms driving ALI is critical for identifying therapeutic strategies. Recent studies have suggested that ferroptosis, a regulated form of cell death, plays a significant role in ALI progression, but its upstream regulatory mechanisms remain unclear. Our study offers insights into these mechanisms, highlighting the role of PRMT1, EGR1, and GLS2 in mediating ferroptosis in sepsis-induced ALI. In this present study, we observed an increase in ferroptosis and elevated expression levels of PRMT1, EGR1, and GLS2 in both in vitro and in vivo ALI models. Furthermore, we demonstrated that EGR1 activated the transcription of GLS2 and promoted ferroptosis. We also found that PRMT1 contributed to the stability of EGR1 through arginine methylation mediation. Moreover, the interaction with NEDD4L was found to reduce EGR1 ubiquitination degradation. Both contributed to the increased level of EGR1 protein. Finally, the regulatory mechanism was validated in the in vivo ALI model. This study may provide a theoretical foundation for employing PRMT1 in the treatment of sepsis-induced ALI.
Ferroptosis plays a significant role in the development of ALI caused by LPS, with GPX4 and SLC7A11 serving as important biomarkers for monitoring this process27. The use of the ferroptosis inhibitor has been shown to effectively mitigate histological damage to the lungs and offer therapeutic benefits27. Electroacupuncture (EA) stimulation at the Zusanli (ST36) point has been found to counteract LPS-induced ferroptosis in alveolar epithelial cells by activating the α7nAchR, which in turn reduces lung inflammation and lessens the severity of LPS-induced ALI28. Additionally, other compounds exhibit protective effects through similar mechanisms. For instance, Puerarin contributes to lung protection by lowering iron levels in lung epithelial cells and reducing GPX4 and GSH expression and synthesis, which helps prevent epithelial cell damage due to ferroptosis29. Moreover, itraconate acts to prevent macrophage ferroptosis through the Nrf2 pathway, while hydrogen sulfide helps to mitigate ferroptosis and inhibit mTOR signaling in the context of sepsis-induced ALI30. Consistently, we demonstrated that ferroptosis was increased in both LPS-induced lung epithelial cells and CLP-induced ALI mouse model. The treatment with ferroptosis inhibitor, Fer-1, significantly reduced ferroptosis in ALI and increased mouse survival rate. GLS2, a key mediator of glutaminolysis, is a well-accepted ferroptosis promoter in many diseases11. For example, in GLS2 depletion mice or human hepatic adenocarcinoma cells, ferroptosis was significantly suppressed10. Jennis M. et al. established a mouse model that expressed a variant of p53 lacking the ability to express GLS211. These mice exhibit a deficiency in their capacity to undergo ferroptosis11. Mechanistically, GLS2 induces ferroptosis by enhancing the conversion of glutamate to α-ketoglutarate, thereby increasing the production of lipid ROS10. We also observed that the levels of PRMT1, EGR1, and GLS2 were elevated in both in vitro and in vivo ALI models, suggesting that EGR1 and PRMT1 might play a role in GLS2-mediated ferroptosis. The addition of Fer-1 did not alter the expression levels of PRMT1, EGR1, or GLS2. Fer-1 scavenges ROS, reduces lipid peroxide accumulation, inhibits lipid peroxidase activity, and protects mitochondria31. It likely functions downstream of GLS2 or through alternative pathways involving PRMT1 and EGR1, which explains why Fer-1 does not impact the expression of PRMT1, EGR1, or GLS2.
Additionally, we demonstrated that EGR1, which is located in nuclear and acts as a critical transcription factor, transpiration regulated the expression level of GLS2. The knockdown of EGR1 markedly reduced GLS2 protein levels and thereby inhibited ferroptosis. This regulatory role of EGR1 in ferroptosis has also been observed in other disease contexts. For instance, Fan et al. demonstrated that EGR1 promoted ferroptosis in acute myocardial infarction via the GPX4/SLC7A11 pathway32. This regulatory mechanism aligns with our findings, further supporting our conclusion. Similarly, another study found that silencing EGR1 in a model of intervertebral disc disease decreased ferroptosis and cartilage degeneration33. In addition, Additionally, α-Hederin was shown to activate the nuclear translocation of EGR1, directly inhibit miR-96-5p, promote DNA damage-inducible transcript 3 (DDIT3)/activating transcription factor 3 (ATF3)-mediated ferroptosis, and reverse cisplatin resistance in non-small cell lung cancer34. Interestingly, previous research also showed the EGR1 level was increased in ALI and may act as a key proinflammatory regulator35. Nevertheless, the link between EGR1 and ferroptosis in ALI had not been previously established. To our knowledge, our study is the first to demonstrate that EGR1 facilitates ferroptosis in sepsis-induced lung injury by modulating GLS2.
Post-translational modifications of proteins are key factors causing proteome diversity and cellular dynamic equilibrium, with arginine methylation modification being one of the common types of post-translational modifications36. PRMT1, the critical arginine methyltransferase in mammalian cells, is essential for normal embryogenesis, cell cycle progression, cellular viability, and signal transduction37. Previous studies indicated that PRMT1 was closely involved in regulating several diseases, including acute kidney injury and ALI38. It was reported by Nakayama et al. that the accumulation of ADMA and elevated expression of PRMT1 in the kidney correlated with the loss of renal capillaries and necrosis of tubular cells38. It was also documented that PRMT1 expression was increased in ALI and the upregulated PRMT1 might contribute to apoptosis induced by oxidative stress39. In line with these findings, this study showed that PRMT1 expression was abnormally enhanced in sepsis-related ALI. Furthermore, we demonstrated that PRMT1 regulated the arginine methylation of EGR1 and thereby enhanced its protein stability. On the other hand, EGR1 ubiquitination was regulated by NEDD4L, and reduced NEDD4L level in ALI decreased EGR1 protein degradation. Together, these factors led to an increase in EGR1 protein levels, promoting sepsis-induced ALI. A similar regulated mechanism was reported in previous studies as well. For example, PRMT1 mediated MYCN methylation and its protein stability in neuroblastoma40. It was also documented that PRMT1 was involved in modulating ATF4 stability via R239 methylation in cardiomyocytes41. Thus, our study highlights the role of PRMT1 in stabilizing key regulatory proteins through arginine methylation, contributing to the pathogenesis of sepsis-induced ALI by enhancing EGR1 stability.
In summary, this study identified a regulatory axis in which PRMT1-driven arginine methylation and NEDD4L-mediated ubiquitination and regulate EGR1 stability, promoting GLS2 transcription and ferroptosis in sepsis-induced ALI. By establishing the PRMT1/EGR1/GLS2 axis as a key regulatory pathway in ferroptosis, this study provides a potential therapeutic target for managing sepsis-induced ALI. Targeting PRMT1 or EGR1 may offer a approach for modulating ferroptosis, reducing lung damage, and improving patient outcomes.
While our findings provide valuable insights into the molecular mechanisms underlying sepsis-induced ALI, it has several limitations. First, we did not assess the broader systemic inflammatory and immune responses, which are critical drivers of sepsis pathology. Including data on inflammatory cytokines and immune cell populations would provide a more comprehensive understanding of how PRMT1 influences the overall immune response in sepsis. Second, although we focused on ferroptosis markers, the potential interactions between ferroptosis and other cell death mechanisms, such as apoptosis or necroptosis, were not explored in this study. Lastly, our findings were derived from in vitro and mouse models, which may not fully replicate the complexity of human sepsis. Further studies using human samples or clinical trials will be necessary to confirm the translatability of our results to clinical practice.
Materials and methods
Animal experiments
Male C57BL/6 J mice, aged 9–10 weeks and weighing between 25 and 29 grams, were procured from the Hunan Slake Jingda Animal Center (Changsha, Hunan). These mice were kept in a specific pathogen-free environment, subject to a 12-hour light-dark cycle, and were given unrestricted access to both food and water. The animal studies conducted as part of this research received approval from Hainan Medical University (No. HYLL-2021-392). Moreover, all experimental procedures were performed in strict adherence to established guidelines. We have complied with all relevant ethical regulations for animal use.
Thirty mice were divided into different groups evenly, the sham group, the cecal ligation and puncture (CLP) group, the CLP+ Ferrostatin-1 (Fer-1) group, the CLP+shNC group, the CLP+shPRMT1 group. The CLP procedure was carried out as previously described42. In brief, mice from the CLP group or CLP+Fer-1 group were anesthetized using isoflurane administered through a vaporizer (Matrx; Midmark Corp., Dayton, OH, USA). A midline incision of 1 cm was made to expose the cecum with care taken to avoid injuring the surrounding blood vessels. The cecum was then securely tied off with a 3/0 silk suture at its midpoint and punctured twice with a 21-gauge needle approximately 0.5 cm from the distal end, allowing a small amount of fecal matter to be expelled to confirm the openings were clear. Afterward, the cecum was returned to its original position within the abdomen, which was then closed in two layers. Mice designated for the sham group underwent the same surgical steps excluding the CLP. Following the procedure, all animals were given a subcutaneous injection of 1 mL of saline at 37 °C for fluid replenishment. Additionally, mice from the CLP+Fer-1 group were intraperitoneally injected with Fer-1 (1 mg/kg; Cat. No.: HY-100579, MedChemExpress) every 12 h for 48 h for 2 consecutive days. For the CLP + shNC group and CLP + shPRMT1 group, shNC-transfected MLE-12 cells (0.5 × 106 cells/kg), shPRMT1-transfected MLE-12 cells (0.5 × 106 cells/kg) were injected through the tail vein. The survival rate of the mice from each group was recorded for 72 h post CLP and then all mice were euthanized. Following euthanasia, the blood samples and the lungs were collected, and the left upper lobe was fixed in formalin pending further histological examination. The left lower lobe was extracted, weighed, and then dried overnight in a heated oven. The right upper lobe was designated for transmission electron microscopy analysis.
Hematoxylin and Eosin (HE) staining
The lung specimens were fixed in 3.7% formaldehyde solution, embedded in paraffin, and sliced into sections of 5 micrometers thickness. These slices underwent a short staining process with hematoxylin (Sigma–Aldrich, St. Louis, MO) for three minutes, and were subsequently washed with flowing tap water for one minute. Following this, eosin (Sigma–Aldrich) was applied for a brief period of 45 s to achieve staining. The stained sections were then prepared for observation, and photographs were taken using a DMi8 optical microscope (Leica, Shanghai, China).
Lung injury score and wet-to-dry ratio measurement
The lung wet-to-dry weight ratio was determined by dividing the wet lung weight by its dry weight. After measuring the wet weight (W) of the tissues, they were dried at a temperature of 65 °C for 48 h to determine their dry weight (D). The semiquantitative assessment (lung injury score) of ALI relied on evaluating factors such as inflammatory cell infiltration, widening of the interstitium, edema, and hemorrhage43,44. A pathologist, who was not aware of the experimental conditions, assigned scores to each characteristic, ranging from 0 (normal) to 4 (very severe).
Immunohistochemistry (IHC)
Lung tissues were subjected to antigen retrieval by heating in a microwave oven in 10 mmol/L citrate buffer for 3 min. Then, sections were incubated with primary antibodies against PRMT1 (ab190892, 1: 500, Abcam), EGR1 (ab300449, 1:100, Abcam), and GLS2 (ab190892, 1:100, Abcam). After incubating with poly-peroxidase-anti-mouse/rabbit IgG, the immunohistochemical reaction was visualized through staining with DAB, followed by counterstaining with hematoxylin-based reagents which were subsequently analyzed under a microscope.
Transmission electron microscopy
Fresh lung samples, measuring 1 mm × 1 mm × 1 mm, were promptly placed in tubes pre-filled with a fixative solution for 4 h. Subsequently, the heart tissues were processed through infiltration, dehydration, and embedding procedures throughout the night. Observations were made using a transmission electron microscope (HITACHI, Tokyo, Japan). The mitochondrial ultrastructural damage was evaluated based on the Flameng score.
Western blot
Protein extraction was performed with radio-immunoprecipitation assay (RIPA) buffer that included protease inhibitors, and the mixture was kept at 4 °C for 30 min (Beyotime Inc., Hiamen, Jiangsu, China). The concentration of proteins was quantified using the BCA protein assay kit (10741395, Thermo Fisher Scientific, Waltham, MA, USA). After quantification, 30 μg of protein from each sample was subjected to SDS-PAGE for separation and then transferred to polyvinylidene fluoride (PVDF) membranes (Merck Millipore, MA, USA). The membranes were blocked and washed with PBS before being incubated with primary antibodies against specific proteins: PRMT1 (ab243146, 1:500, Abcam, Cambridge, UK), EGR1 (ab300449, 1:100, Abcam), GLS2 (ab113509, 1:1000, Abcam), neural precursor cell expressed developmentally down-regulated protein 4 like (NEDD4L, ab245522, 1:2000, Abcam), pan-asymmetric dimethylarginine (ADMA, #13522, CST), pan-methylmalonic acid (MMA, #8015, CST), and β-actin (ab5694, 1:1000, Abcam). Following further PBS washes, the membranes were incubated with secondary antibodies (ab7090, 1:500, Abcam or ab150165, 1:500, Abcam). Protein bands were then detected using an enhanced chemiluminescence (ECL) kit (WBULS0100, Merck Millipore).
Detection of Malondialdehyde (MDA), 4-Hydroxynonenal (4-HNE), Fe2+, and Glutathione (GSH)
Blood samples were centrifuged at 2200 × g for 10 min 4 °C to separate serum, which was then promptly frozen at −80 °C for later analysis. For cell preparation, MLE-12 cells (2 × 106 cells) were washed with PBS and subsequently trypsinized. The levels of MDA, 4-HNE, Fe2+ and GSH in the samples were measured using specific assay kits: Lipid Peroxidation (MDA) Assay Kit (Cat. No.: ab118970, Abcam), Lipid Peroxidation (4-HNE) Assay Kit (Cat. No.: ab238538, Abcam), Iron Assay Kit (Cat. No.: ab83366, Abcam), and Glutathione Assay Kit (Cat. No.: CS0260-1KT, Sigma-Aldrich) following the instructions provided by each manufacturer.
Cell culture
The MLE-12 cell line (Cat. No.: CRL-2110), murine lung epithelial type II cells, was purchased from American Type Culture Collection (USA) and grown in DMEM supplemented with 10% heat-inactivated fetal bovine serum (FBS, Gibco), 100 units/mL of penicillin, and 100 units/mL of streptomycin, and maintained at a temperature of 37 °C. MLE-12 cells were authenticated by STR profiling and tested for mycoplasma contamination.
Cell counting kit-8 (CCK-8) assay
To assess the proliferation of MLE-12 cells, the Cell Counting Kit-8 (CCK-8) assay (Cat. No.: ab228554, Abcam) was utilized. The cells were seeded into a 96-well plate at a density of 5000 cells per well. Forty-eight hours later, 10 µL of CCK-8 solution was introduced into each well. After allowing the cells to incubate for 4 h, the absorbance was recorded using a microplate reader (Thermo Fisher Scientific).
Cell transfection and treatment
Genepharma (Shanghai, China) customized short hairpin RNAs (shRNAs) aimed at targeting EGR1 (shEGR1, targeting sequences: 5’-TCTGACATCGCTCTGAATAAT-3’) and PRMT1 (shPRMT1, targeting sequences: 5’-CCATTGAGGACCGACAATATA-3’), along with a control shRNA (shNC) were inserted into pCD513B-U6 plasmids to create shRNA vectors. To induce overexpression of GLS2 or NEDD4L, their full sequences were replicated using PCR and then inserted into pcDNA3.1 plasmid, which was supplied by SBI (Mountain View, CA, USA). Plasmids without inserts served as negative controls. The vectors were introduced into MLE-12 cells using Lipofectamine 3000 from Invitrogen (Cat. No.: L3000015, Gaithersburg, MD, USA), for 48 h, with a leukocyte activation cocktail (Cat. No.: 554056, BD Bioscience, Heidelberg, Germany) added for the last 5 h to activate the cells.
To assess ferroptosis’s impact on ALI, MLE-12 cells transfected with either shNC or shPRMT1 were maintained until they reached approximately 80% confluence and seeded at 1 × 105 cells/mL density in the cultured plate. Cells were treated with 10 µg/mL LPS (Cat. No.: L2880, Sigma) for 4 h and/or ferroptosis inhibitor Ferrostatin-1 (Fer-1, 10 μM) for 12 h. For cycloheximide (CHX) exposure, NEDD4L overexpressed MLE-12 cells were seeded onto 15-cm culture dishes (Corning Inc., Corning, NY, USA) at a density of 5 × 105 cells/mL, underwent treatment with CHX (100 μg/mL, Cat. No.: HY-12320, MedChemExpress)/DMSO at various intervals (0, 15, 30, 60, 120, and 240 min), after which they were collected for further study.
For MG132 treatment, NEDD4L overexpressed MLE-12 cells were seeded onto 15-cm culture dishes (Corning Inc.) at a density of 5 × 105 cells/mL, underwent treatment with 10 ng/mL MG132 (Cat. No.: 474790, Calbiochem, San Diego, CA, USA) dissolved in DMSO for 48 h. Afterward, the cells were collected for subsequent analysis.
293 T cells (5 × 106 cells per well) received transfections with either shPRMT1 or shNC using Lipofectamine 2000. After 48 h, the lentivirus-containing supernatant was gathered and filtered. This lentiviral supernatant was then applied to MLE-12 cells (5 × 106 cells per well). The cells underwent selection with 2.5 µg/mL puromycin beginning two days after transduction. After 12 days, the puromycin was discontinued, and the cells were maintained in a standard medium until recovery was complete.
BODIPY™ 665/676 dye detected lipid ROS expression
Cells treated with the previously described method45 were seeded in 12-well plates at a density of 1 × 105 cells per well and allowed to adhere for around 12 h. Subsequently, the medium was replaced with one containing DMSO as a control or 5 μM of C11-BODIPY (Cat. No.: D3861, Life Technologies, Waltham, CA, USA), followed by incubation for an additional 20 min at 37 °C. After incubation, the cells were washed with PBS and collected via trypsinization, then washed again with PBS. The cells were resuspended in 400 μL PBS, filtered through a 35 μm nylon mesh filter, and subjected to flow cytometry analysis using a BD LSRII system equipped with a 488 nm laser for excitation (BD Biosciences GmbH).
Chromatin immunoprecipitation (ChIP) assay
MLE-12 cells were collected and lysed using RIP lysis buffer from Merck Millipore. Following lysis, cells underwent cross-linking and sonication. The lysates were then mixed with 900 μL of ChIP Dilution Buffer, 20 μL of 50× Protease Inhibitor Cocktail, and 60 μL of Protein A Agarose/Salmon Sperm DNA beads. Post-incubation, lysates were centrifuged to separate the supernatant, which was carefully transferred to a tube. To this supernatant, 1 μL of either EGR1 antibody (ab300449, 1:100, Abcam) or IgG antibody (ab205718, 1:1000, Abcam) was added, and the mixture was incubated overnight at 4 °C for immunoprecipitation. Following immunoprecipitation and washes, 1 μL of RNase A was introduced to each sample and incubated at 37 °C for an hour to eliminate RNA. Subsequently, 10 μL of 0.5 M EDTA, 20 μL of 1 M Tris-HCl, and 2 μL of 10 mg/mL proteinase K were added to the samples, which were then incubated at 45 °C for two hours. Finally, the DNA was extracted, and its concentration was determined through agarose gel electrophoresis.
Dual-luciferase reporter assay
To explore the influence of EGR1 on the GLS2 promoter’s activity, the GLS2 promoter along with its mutant variants (GLS2-wt, GLS2-mut) were amplified and then inserted into a psiCHECK2 vector (Promega, Madison, WI, USA). 293 T cells underwent co-transfection with either GLS2-wt or GLS2-mut in combination with shNC or shEGR1, using the Lipofectamine 3000 transfection agent (Invitrogen). After a 48-hour incubation period, the Dual-Luciferase Reporter Assay Kit (Promega) was utilized to measure luciferase activity, providing insight into the regulatory effects of EGR1 on the GLS2 promoter.
Co-immunoprecipitation (Co-IP) assay
PRMT1 or EGR1 (1–150) or EGR1 (151–533) cDNA ORF coding plasmids (Origene, Shanghai, China) were subcloned into a pcDNA3.1 vector, using primers designed to append a 6×His-tag to their C-terminal end. MLE-12 cells were treated above or transfected with His-tagged PRMT1 or His-tagged EGR1 (1–150) or His-tagged EGR1 (151–533). MLE-12 cells were initially rinsed twice using PBS, then treated with RIP lysis buffer from Merck Millipore. The harvested cell lysates were subjected to sonication on ice using an IP buffer and then centrifuged at 12,000 rpm for 10 min. From this process, 30 μL of the supernatant was set aside as the ‘Input’ sample. Furthermore, 420 μL of the supernatant was used for overnight immunoprecipitation at 4°C with antibodies against NEDD4L (13690-1-AP, Proteintech, 1:500, Wuhan, Hubei, China), EGR1 antibody (ab300449, 1:100, Abcam), 6xHis (ab9108, 1:500, Abcam), Ubiquitin antibody (ab264179, 1:1000, Abcam) or a non-specific control IgG antibody (ab171870, 1:1000, Abcam). Protein A agarose beads were added to this mixture and incubated for 1 hour at 4 °C. Following four washes with IP buffer and brief centrifugation to remove the supernatant, 30 μL of 2X SDS lysate was added to the pellets and incubated for 10 minutes to elute the protein complexes. These protein complexes were then analyzed through Western blot to detect the presence and enrichment of ADMA, MMA, NEDD4L, and ubiquitin within the complex. NEDD4L having an N-terminal SFB-tag (Flag peptide is part of the SFB tag) was cloned into pcDNA3.1 and transfected into 293 T cells (Origene). Forty-eight hours after transfection, the supernatant was collected. The samples were first cleared of non-specific binders by incubating with protein G-plus beads (sc-2002, Santa Cruz, CA, USA) for 30 min at 4 °C. The beads were then separated from the supernatant by centrifugation. The cleared supernatants were subsequently incubated with an anti-Flag tag antibody (F3165, 1:500, Sigma-Aldrich) for 1 h at 4 °C on a rotating platform. After this period, protein G-plus beads were added, and the mixture was incubated overnight at 4 °C with rotation. The next day, the beads were washed four times with ice-cold 0.2% digitonin wash buffer (150 mM NaCl, 50 mM Tris, pH 7.4) and then twice with ice-cold PBS. For immunoblot analysis, the beads were eluted by boiling them in a twofold concentrated protein sample buffer for 3 min.
Immunofluorescence co-localization analysis
The hepatocytes were fixed in 4% paraformaldehyde for 30 min, incubated with 0.3% Triton X-100, and then immersed in 5% BSA for 1 h. Thereafter, cells were incubated with primary antibodies against PRMT1 (ab190892, 1:80, Abcam), EGR1(ab300449, 1:100, Abcam), asymmetric dimethylarginine (ADMA) (sc-57624, 1:50, Santa Cruz, USA) at 4 °C overnight, followed by incubation with corresponding secondary antibodies for 1 h. Subsequently, the nuclei were stained with DAPI. The stained cells were observed using a laser scanning confocal microscope (Leica).
GST-pull down assay
Site-specific mutations were introduced into EGR1 at positions R133 and R278, changing them to K, either singly or in combination, through PCR-based, oligonucleotide-directed mutagenesis. The mutant cDNA ORF coding plasmids of PRMT1 or EGR1, sourced from Origene, were then integrated into a pcDNA3.1 vector using primers that added an HA-tag or Flag-tag at their C-terminal end. MLE-12 cells underwent the treatment or were transfected with Flag-tagged EGR1 or HA-tagged PRMT1. The EGR1 mutants, fused with GST, were purified using a previously described method46. After purification, these proteins were mixed with Glutathione Sepharose 4B (GE Healthcare) and incubated for three hours at 4 °C. Subsequently, the resins were rinsed four times using a wash buffer composed of 20 mM potassium phosphate buffer (pH 6.0), 100 mM KCl, 0.1 mM EDTA, 10 mM DTT, and 0.5% Triton X-100. Following the washes, the samples were subjected to SDS polyacrylamide gel electrophoresis and stained with Coomassie Brilliant Blue for analysis.
Immunohistochemical (IHC) staining
Lung tissues were preserved in 3.7% formalin, embedded in paraffin, and cut into sections 5 µm in thickness. IHC was conducted on sections 5 µm thick from these paraffin blocks, following a previously established method47. The sections underwent incubation with primary antibodies, specifically PRMT1 (ab73246, 1:250, Abcam), EGR1 (ab300449, 1:100, Abcam), and GLS2 (ab113509, 1:500, Abcam), for one hour at ambient temperature. This was followed by the application of secondary antibodies. Hematoxylin was used for counterstaining before the images were taken with a Leica confocal microscope.
Statistics and reproducibility
All experiments were performed in at least three biological replicates, and each biological replicate contained three technical replicates. Data analysis was performed with SPSS 20.0 software (IBM, Armonk, NY, USA). Findings are displayed as mean ± standard deviation (SD), based on at least three separate trials. Normality distribution of data was evaluated using the Shapiro-Wilk’s test. Detailed sample sizes are described in the figure legends. To compare two groups, unpaired Student’s t-tests were applied, and for analyzes involving more than two groups, a one-way analysis of variance (ANOVA) with subsequent Tukey post hoc test was conducted. A P-value of less than 0.05 was considered statistically significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Source data for the graphs are available as Supplementary Data file. The Animal experiments, co-immunoprecipitation (Co-IP) assay, RNA extraction and quantitative real-time polymerase chain reaction (qPCR), Western blot methods, all of the uncropped images in Western blot were shown in Supplementary information document. To be specific, that the expression of ferroptosis-related markers GPX4, SLC7A11, and ACSL4 under different treatments in in vivo and in vitro sepsis models were shown in Figure S1, and that arginine methylation prediction of EGR1 and PRMT expression/binding in an in vitro sepsis model were presented in Figure S2, that the NEDD4L targeted EGR1 was showed in Figure S3. Besides, that PRMT1 regulated arginine methylation modification of EGR1 was presented in Figure S4, and that knockdown of EGR1 attenuated sepsis-related ALI by suppressing ferroptosis via EGR1/GLS2 axis in vivo were included in Figure S5. Finally, the uncropped and unedited Western blot/gel images were included in Figure S6. The primers used for RT-qPCR were shown in Table S1. The other data generated and/or analyzed during the current study are available from the corresponding author on reasonable request. All data generated or analyzed during this study are included in this published article. Source data can be found in the Supplementary Data file.
Abbreviations
- 4-HNE:
-
4-hydroxynonenal
- ALI:
-
acute lung injury
- ATF:
-
activating rranscription factor
- ADMA:
-
asymmetric dimethylarginine
- ANOVA:
-
one-way analysis of variance
- CCK-8:
-
cell counting kit-8
- Co-IP:
-
co-immunoprecipitation
- ChIP:
-
chromatin immunoprecipitation
- CHX:
-
cycloheximide
- CLP:
-
cecal ligation and puncture
- CCK-8:
-
cell counting kit-8
- DDIT3:
-
DNA damage inducible transcript 3
- EA:
-
electroacupuncture
- ECL:
-
enhanced chemiluminescence
- EGR1:
-
early growth response 1
- FBS:
-
fetal bovine serum
- Fer-1:
-
Ferrostatin-1
- GPX4:
-
glutathione peroxidase 4
- GSH:
-
glutathione
- GLS2:
-
glutaminase 2
- HE:
-
hematoxylin & eosin
- IHC staining:
-
immunohistochemical staining,
- LPS:
-
lipopolysaccharide
- MDA:
-
malondialdehyde
- MMA:
-
methylmalonic acid
- MYCN:
-
proto-oncogene BHLH transcription factor
- NEDD4L:
-
neural precursor cell expressed developmentally down-regulated protein 4 like
- Nrf2:
-
nuclear factor erythroid 2 related factor 2
- PBS:
-
phosphate-buffered saline
- PVDF:
-
polyvinylidene fluoride
- RIP:
-
immunoprecipitation
- RIPA:
-
radio-immunoprecipitation assay
- PRMT1:
-
protein arginine methyltransferase 1
- ROS:
-
reactive oxygen species
- shRNAs:
-
short hairpin RNAs
- SLC7A11:
-
solute carrier family 7 member 11
- α7nAchR:
-
alpha 7 nicotinic acetylcholine receptor
- αKG:
-
alpha ketoglutarate
References
Min, L., Longhui, H., Qiao, K., Chujun, R. & Xiaoran, L. miR-122-3p Alleviates LPS-Induced Pyroptosis of Macrophages via Targeting NLRP1. Ann Clin Lab Sci. 53 (2023).
Luh, S. P. & Chiang, C. H. Acute lung injury/acute respiratory distress syndrome (ALI/ARDS): the mechanism, present strategies and future perspectives of therapies. J Zhejiang Univ Sci B 8, 60–69 (2007).
Sun, B. et al. Acute lung injury caused by sepsis: how does it happen? Front Med. (Lausanne) 10, 1289194 (2023).
Sharp, C., Millar, A. B. & Medford, A. R. Advances in understanding of the pathogenesis of acute respiratory distress syndrome. Respiration 89, 420–434 (2015).
Bezerra, F. S. et al. Oxidative Stress and Inflammation in Acute and Chronic Lung Injuries. Antioxidants (Basel) 12, https://doi.org/10.3390/antiox12030548 (2023).
Yin, X. et al. Ferroptosis, a New Insight Into Acute Lung Injury. Front Pharmacol. 12, 709538 (2021).
Zheng, Y. et al. Ferroptosis, pyroptosis and necroptosis in acute respiratory distress syndrome. Cell Death Discov. 9, 91 (2023).
Suzuki, S. et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci USA. 107, 7461–7466 (2010).
Chen, J., Cui, L., Lu, S. & Xu, S. Amino acid metabolism in tumor biology and therapy. Cell Death & Disease 15, 42 (2024).
Suzuki, S. et al. GLS2 Is a Tumor Suppressor and a Regulator of Ferroptosis in Hepatocellular Carcinoma. Cancer Res. 82, 3209–3222 (2022).
Jennis, M. et al. An African-specific polymorphism in the TP53 gene impairs p53 tumor suppressor function in a mouse model. Genes Dev 30, 918–930 (2016).
Zou, K. & Zeng, Z. Role of early growth response 1 in inflammation-associated lung diseases. Am J Physiol Lung Cell Mol Physiol. 325, L143–l154 (2023).
Wang, B. et al. The Role of the Transcription Factor EGR1 in Cancer. Front Oncol 11, 642547 (2021).
Tak, I.-u.-R. et al. In Protein Modificomics (eds Dar, T. A. & Singh, L. R.) 1-35 (Academic Press, 2019).
Bryant, J. P., Heiss, J. & Banasavadi-Siddegowda, Y. K. Arginine Methylation in Brain Tumors: Tumor Biology and Therapeutic Strategies. Cells 10, https://doi.org/10.3390/cells10010124 (2021).
Tewary, S. K., Zheng, Y. G. & Ho, M. C. Protein arginine methyltransferases: insights into the enzyme structure and mechanism at the atomic level. Cell Mol Life Sci. 76, 2917–2932 (2019).
Litt, M., Qiu, Y. & Huang, S. Histone arginine methylations: their roles in chromatin dynamics and transcriptional regulation. Biosci Rep. 29, 131–141 (2009).
Zhu, Y. et al. Inhibition of PRMT1 alleviates sepsis-induced acute kidney injury in mice by blocking the TGF-β1 and IL-6 trans-signaling pathways. FEBS Open Bio 13, 1859–1873 (2023).
Ross, A. B., Langer, J. D. & Jovanovic, M. Proteome turnover in the spotlight: approaches, applications, and perspectives. Mol Cell Proteomics 20, 100016 (2021).
Suresh, B., Lee, J., Kim, K. S. & Ramakrishna, S. The importance of ubiquitination and deubiquitination in cellular reprogramming. Stem Cells Int 2016, 6705927 (2016).
Liu, Q., Zhang, S., Sun, Z., Guo, X. & Zhou, H. E3 ubiquitin ligase Nedd4 is a key negative regulator for non-canonical inflammasome activation. Cell Death Differ 26, 2386–2399 (2019).
Zhong, Q. et al. Protein posttranslational modifications in health and diseases: functions, regulatory mechanisms, and therapeutic implications. MedComm (2020) 4, e261 (2023).
Dong, R., Li, X. & Lai, K. O. Activity and Function of the PRMT8 Protein Arginine Methyltransferase in Neurons. Life (Basel) 11, https://doi.org/10.3390/life11111132 (2021).
Hwang, J. W., Cho, Y., Bae, G. U., Kim, S. N. & Kim, Y. K. Protein arginine methyltransferases: promising targets for cancer therapy. Exp Mol Med. 53, 788–808 (2021).
Liang, G., Wang, W. & He, Z. Sepsis associated with acute lung injury over the period 2012-2021: a bibliometric analysis. Front Physiol 14, 1079736 (2023).
Sadowitz, B., Roy, S., Gatto, L. A., Habashi, N. & Nieman, G. Lung injury induced by sepsis: lessons learned from large animal models and future directions for treatment. Expert Rev Anti Infect Ther 9, 1169–1178 (2011).
Liu, P. et al. Ferrostatin-1 alleviates lipopolysaccharide-induced acute lung injury via inhibiting ferroptosis. Cell Mol Biol Lett. 25, 10 (2020).
Zhang, Y. et al. Electroacupuncture alleviates LPS-induced ARDS through α7 nicotinic acetylcholine receptor-mediated inhibition of ferroptosis. Front Immunol 13, 832432 (2022).
Xu, B., Wang, H. & Chen, Z. Puerarin inhibits ferroptosis and inflammation of lung injury caused by sepsis in LPS induced lung epithelial cells. Front Pediatr 9, 706327 (2021).
He, R. et al. Itaconate inhibits ferroptosis of macrophage via Nrf2 pathways against sepsis-induced acute lung injury. Cell Death Discov. 8, 43 (2022).
Merkel, M. et al. Mitochondrial Reactive Oxygen Species Formation Determines ACSL4/LPCAT2-Mediated Ferroptosis. Antioxidants (Basel) 12, https://doi.org/10.3390/antiox12081590 (2023).
Fan, K. et al. The Egr-1/miR-15a-5p/GPX4 axis regulates ferroptosis in acute myocardial infarction. Eur J Pharmacol. 909, 174403 (2021).
Zhang, J. et al. EGR1 knockdown confers protection against ferroptosis and ameliorates intervertebral disc cartilage degeneration by inactivating the MAP3K14/NF-κB axis. Genomics 115, 110683 (2023).
Han, S. et al. α-Hederin promotes ferroptosis and reverses cisplatin chemoresistance in non-small cell lung cancer. Aging (Albany NY) 16, 1298–1317 (2024).
Hoetzel, A. et al. Carbon monoxide protects against ventilator-induced lung injury via PPAR-gamma and inhibition of Egr-1. Am J Respir Crit Care Med. 177, 1223–1232 (2008).
Santos, A. L. & Lindner, A. B. Protein posttranslational modifications: roles in aging and age-related disease. Oxid Med Cell Longev 2017, 5716409 (2017).
Infantino, S. et al. Arginine methylation catalyzed by PRMT1 is required for B cell activation and differentiation. Nat Commun 8, 891 (2017).
Nakayama, Y. et al. Asymmetric dimethylarginine accumulates in the kidney during ischemia/reperfusion injury. Kidney Int 85, 570–578 (2014).
Lim, S. K. et al. Activation of PRMT1 and PRMT5 mediates hypoxia- and ischemia-induced apoptosis in human lung epithelial cells and the lung of miniature pigs: the role of p38 and JNK mitogen-activated protein kinases. Biochem Biophys Res Commun. 440, 707–713 (2013).
Eberhardt, A. et al. Protein arginine methyltransferase 1 is a novel regulator of MYCN in neuroblastoma. Oncotarget 7, 63629–63639 (2016).
Jeong, M. H. et al. Correction: PRMT1 suppresses ATF4-mediated endoplasmic reticulum response in cardiomyocytes. Cell Death Dis. 11, 203 (2020).
Toscano, M. G., Ganea, D. & Gamero, A. M. Cecal ligation puncture procedure. J Vis Exp. https://doi.org/10.3791/2860 (2011).
Atabai, K. & Matthay, M. A. The pulmonary physician in critical care. 5: acute lung injury and the acute respiratory distress syndrome: definitions and epidemiology. Thorax 57, 452–458 (2002).
Li, C. et al. LncRNA XIST knockdown alleviates LPS-induced acute lung injury by inactivation of XIST/miR-132-3p/MAPK14 pathway : XIST promotes ALI via miR-132-3p/MAPK14 axis. Mol Cell Biochem. 476, 4217–4229 (2021).
Lange, C., Lehmann, C., Mahler, M. & Bednarski, P. J. Comparison of Cellular Death Pathways after mTHPC-mediated Photodynamic Therapy (PDT) in Five Human Cancer Cell Lines. Cancers (Basel) 11, https://doi.org/10.3390/cancers11050702 (2019).
Akira, S., Uematsu, S. & Takeuchi, O. Pathogen recognition and innate immunity. Cell 124, 783–801 (2006).
Magaki, S., Hojat, S. A., Wei, B., So, A. & Yong, W. H. An Introduction to the Performance of Immunohistochemistry. Methods Mol Biol. 1897, 289–298 (2019).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No.: 82060357 and 81960351).
Author information
Authors and Affiliations
Contributions
Min Li and Xiaoran Liu designed this study. Min Li, Longhui Hu, Qiao Ke, Zhao Li, Chujun Ruan and Hanjing Lu collected the materials and performed the experiments. Min Li and Xiaoran Liu analyzed the data and wrote the manuscript. Xiaoran Liu revised the manuscript. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests
Ethics approval and consent to participate
The animal studies conducted as part of this research received approval from Hainan Medical University (No. HYLL-2021-392). Moreover, all experimental procedures were performed in strict adherence to established guidelines.
Peer review
Peer review information
Communications Biology thanks Qingtao Meng, Yongbo Huang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Christina Karlsson Rosenthal.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, M., Hu, L., Ke, Q. et al. Arginine methyltransferase PRMT1 promotes ferroptosis through EGR1/GLS2 axis in sepsis-related acute lung injury. Commun Biol 8, 159 (2025). https://doi.org/10.1038/s42003-025-07531-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-07531-z
This article is cited by
-
Identification and validation of a novel ferroptosis-related gene signature associated with inherited retinal degeneration in Rd10 mice
Scientific Reports (2026)
-
Identification of Potential Key Ferroptosis-Related Genes in EV71-Infected Cells Through Bioinformatics Analysis and Experimental Study
Current Microbiology (2026)
-
Arginine methylation in cancer: mechanisms and therapeutic implications
Biomarker Research (2025)
-
Identification of shared important genes associated with ferroptosis across different etiologies of acute lung injury
Scientific Reports (2025)
