
Abstract
Background
Retinitis pigmentosa (RP) is a leading cause of blindness affecting 2 million people worldwide. Mutations in cyclic nucleotide-gated channel alpha 1 (CNGA1) account for 2–8% of autosomal recessive RP with no available treatment. Here we further evaluate our previously developed Cnga1-/- mouse model. We aim to present detailed phenotype data and assess the therapeutic efficacy of gene augmentation in this model.
Methods
The retinal function and structure of Cnga1-/- mice were accessed from postnatal month 1 to 6. AAV8-hRHO-mCnga1 was constructed and delivered into the subretinal space of 2-week-old Cnga1-/- mice. Retinal function, photoreceptor survival, and vision-guided behavior were evaluated following treatment.
Results
Here we show that Cnga1-/- mice have a similar phenotype to human patients, characterized by an early loss of rod-mediated retinal function and progressive photoreceptor degeneration, which is nearly complete by 6 months of age. Gene augmentation therapy results in robust expression of correctly localized CNGA1 protein, sustained rescue of retinal function and long-term preservation of photoreceptors for at least 9 months. Treated mice also show improved performance in a vision-guided behavior test. RNA-seq reveals upregulation of genes associated with phototransduction.
Conclusions
To the best of our knowledge, this work demonstrates for the first time that a gene augmentation approach can restore vision and preserves photoreceptors in an animal model of CNGA1-RP. These findings pave the way for future development of gene augmentation therapy for patients with CNGA1-RP.
Plain language summary
Retinitis pigmentosa (RP) is the most common inherited eye disease. It causes progressive vision loss and eventual blindness. Changes in a part of a person’s DNA called the CNGA1 gene are a known cause of RP, but no treatments are currently available. In this study, we used a mouse model lacking the Cnga1 gene to investigate disease features and determine whether adding the gene back into the mice, a treatment called gene therapy, could be a potential treatment. Healthy copies of the Cnga1 gene were delivered into the eye. The treatment restored eye function and improved vision-guided behavior in treated mice. Our findings suggest that gene therapy is a promising treatment approach for people with RP caused by changes in the CNGA1 gene.
Similar content being viewed by others

Introduction
In the plasma membrane of the photoreceptor outer segments, cyclic nucleotide-gated (CNG) channels serve as the final target of phototransduction, converting light-induced changes in the second messenger cGMP into electrical signals1,2,3. Mutations in CNGA1, which encodes the alpha subunit of the rod photoreceptor CNG channel, are associated with autosomal recessive retinitis pigmentosa (RP)4.
RP is the most common inherited retinal disease, characterized by progressive rod-cone photoreceptor degeneration, affecting approximately 1 in 4000 people worldwide5. To date, 89 genes have been identified to be associated with non-syndromic RP (RetNet, accessed on May 20, 2025). CNGA1-RP accounts for 2–8% cases of autosomal recessive RP4,5,6,7,8,9,10, for which there is currently no treatment. Natural history report of CNGA1-RP is still unavailable. However, case reports9,10,11,12,13,14,15,16, as well as our ongoing observation (NCT03691168), suggest that patients with CNGA1-RP exhibit night blindness from early childhood. The rate of photoreceptor degeneration is moderate, and many patients were deemed legally blind by middle age9,10,11,12,13,14,15,16. This disconnection between early-onset clinical symptoms and the relatively slow progression of structural loss offers a potentially wide time window for therapeutic intervention.
Gene augmentation therapy holds great promise in treating inherited retinal diseases, as highlighted by Luxturna, the first FDA-approved gene therapy for RPE65-related retinal degeneration17. However, the lack of a suitable disease model poses an obstacle to the development of therapeutics for CNGA1-RP18,19. Recently, we generated the first Cnga1 knockout (Cnga1-/-) mouse by introducing a 65 bp frame-shift deletion in exon 220. We found that in Cnga1-/- mice, down-regulation of the PI3K-AKT-mTOR pathway triggered the rod photoreceptor death at postnatal day 9, which was exacerbated by the secondary activation of chaperone-mediated autophagy20. The retinal phenotype was not described in detail in our previous publication (Liu et al.20), e.g., the electroretinogram was recorded at a single flash intensity of 0.5 log cd s m−2.
In this study, we have two goals with the Cnga1-/- mouse model: (i) to characterize the longitudinal changes of retinal phenotype in detail, and (ii) to evaluate the efficacy of gene augmentation therapy. We show that the mouse resembles a typical human CNGA1-RP phenotype, and introducing a normal copy of Cnga1 into the rod photoreceptors result in sustained restoration of rod-mediated function and long-term preservation of retinal structure, as well as improvement in vision-guided behavior. This work provides preclinical proof-of-concept for AAV-mediated gene augmentation therapy for patients with CNGA1-RP.
Methods
Ethics Statement
The present study did not contain human data. The Institutional Review Board of Shanghai General Hospital has reviewed and approved the experiments described in this study. All animal procedures were conducted in adherence to the guidelines of the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Visual Research.
Animals
C57BL/6J mice and Cnga1-/- mice were bred and housed at the animal facility of Shanghai General Hospital, where they were maintained under a 12 h/12 h light/dark cycle unless interruptions were required for experiments. The housing temperature ranged from 20–23 °C, and the humidity fluctuated between 45%-55%. The mice were group-housed (3–5 per cage) in individually ventilated cages, with free access to water and a normal chow diet. The health status of mice was regularly monitored by institutional staff. The ratio of males to females was approximately 1:1 in all experiments. The age, strain, and number of mice used for each experiment are stated in the corresponding figure legends. Sample sizes for mouse experiments were based on prior experience of suitable n numbers and a minimum of n of 3 for each condition21. Mice were randomly assigned to treatment and control groups using a computer-generated randomization sequence. At the end of the study, the mice were humanely euthanized by carbon dioxide exposure. Different cohorts of mice were used for OCT imaging and electroretinography experiments to avoid repeated anesthesia. The researchers were not blinded to group allocation as the subsequent experiments were carried by the same researchers from the beginning.
In vivo OCT imaging
OCT imaging was performed using Micron IV System (Phoenix Research Laboratories, Pleasanton, CA, USA) as previously described20. After induction of anesthesia, pupils were dilated with 0.5% tropicamide and 0.5% phenylephrine hydrochloride (Mydrin-P, Santen), and ocular surfaces were kept hydrated with 0.3% hypromellose gel (GenTeal, Alcon). The mice were carefully positioned to ensure that the optic nerve head (ONH) was centered in the image. B-scan images crossing the ONH were captured for analysis. The thickness of the retinal ONL (photoreceptor nuclei) was quantified using Insight software (Phoenix Research Laboratories, Pleasanton, CA, USA). For the natural history study, the ONL thickness was determined from captured images at four positions, each ∼500 μm away from the ONH in the superior, inferior, nasal, and temporal regions of the retina, and the values were averaged for analysis. For the gene augmentation study, considering that the AAV vectors delivered by subretinal injection reached only a fraction of the retina, we selected the temporal position ∼500 μm from the ONH for analysis, based on the delivery route (please see below ‘Subretinal injection’ section).
Electroretinography
Full-field ERGs were recorded using the Celeris Rodent Electrophysiology System (Diagnosys LLC, Lowell, MA, USA)21. Mice were dark-adapted for at least 12 hours prior to testing. Under dim red light, animals were anesthetized with an intraperitoneal injection of pentobarbital sodium (50 mg/kg), and pupils were dilated using a mixture of 0.5% tropicamide and 0.5% phenylephrine hydrochloride (Mydrin-P, Santen). Corneal surfaces were protected and electrically coupled with 0.3% hypromellose gel (GenTeal, Alcon).
Scotopic ERG responses were elicited using white light flashes at increasing intensities (−2.0, −1.5, −1.0, −0.5, 0, 0.5, and 1.0 log cd s m−2). For photopic ERG testing, animals were first light-adapted for 10 minutes under a background light of 1.5 log cd s m−2. Subsequent flashes of −1.0, 0, 1, and 2 log cd s m−2 were delivered under the same background illumination. Data were analyzed with the Espion software V6 (Diagnosys LLC, Lowell, MA, USA).
AAV generation for in vivo delivery
Murine Cnga1, driven by a human Rhodopsin promoter (primer forward 5’-AGC CCA GAT CTT CCC CAC CT-3’ and reverse 5’-GAC TCT GGG TTC TGA CCC CC-3’), was inserted into the pAAV-MCS vector (Stratagene, CA, USA). The construct for AAV packaging was sequenced prior to packaging using Sanger sequencing. AAV vectors were packaged by co-transfecting a triple-plasmid system into HEK293T cells (ATCC), purified by a discontinuous gradient of iodixanol via ultracentrifugation, and titered by quantitative PCR (qPCR) using SYBR Green technology.
Subretinal injection
Subretinal injection was performed under a surgical microscope (Eder Medical Technology, Shanghai, CN) as previously described21. Two-week-old Cnga1-/- mice were anesthetized by intraperitoneal injection of pentobarbital sodium (50 mg/kg), and their pupils were dilated with 0.5% tropicamide and 0.5% phenylephrine hydrochloride (Mydrin-P, Santen). An incision was made through the sclera behind the iris at the nasal side using a 32-gauge needle. A NanoFil syringe with a 36-gauge blunt-end needle (World Precision Instruments, Sarasota, FL, USA) was then inserted through the incision and advanced to the subretinal space of the temporal side. One microliter of viral vector suspension (1 × 1012 vg/ml) was slowly injected, leading to the formation of a visible bleb. Following injection, mice were placed on a heating pad for recovery, and the injected eyes were treated with 0.3% tobramycin and 0.1% dexamethasone (TobraDex, Alcon) for prevention of infection. The eyes with complications from the injection procedure, such as hemorrhage or damage to the lens, were excluded from the study and subsequent analysis.
Immunohistochemistry
Following enucleation, mouse eyes were immersed in 4% paraformaldehyde (PFA) in PBS for 10 min, followed by the removal of the cornea and lens under a dissecting microscope. Eyecups were then fixed in 4% PFA for an additional 60 min, washed with PBS three times for 5 min each, and were dehydrated sequentially with 10%, 20%, and 30% sucrose in PBS for 30 min each. After dehydration, eyecups were flash-frozen in O.C.T. (catalog 4583, Sakura) for cryosectioning at 12 μm thickness (CM3050S, Leica). For immunostaining, the retinal sections were washed with PBS for 5 min and were blocked with 5% goat serum in 0.3% Triton X-100 in PBS. After blocking, retinal sections were incubated at 4 °C overnight with mouse anti-CNGA1 antibody (1:200, catalog ab253296, Abcam), rabbit anti-cGMP antibody (1:200, catalog ab169753, Abcam), or rabbit anti-cone arrestin antibody (1:500, catalog AB15282, Millipore). Retinal sections were washed with PBS 3 times for 10 min each and incubated with secondary antibodies, including goat anti-mouse IgG antibody (1:1000, catalog A11005, Thermo Fisher) and goat anti-rabbit IgG antibody (1:1000, catalog A11008, Thermo Fisher). Nuclei were stained with DAPI (1:2000, catalog D1306, Thermo Fisher). The slides were then mounted with Fluoromount-G medium (catalog 0100-01, Southern Biotech) and imaged with the Leica SP8 Lightning System.
Histology
The eyes were fixed in 4% PFA for 24 h and embedded in paraffin, followed by sectioning at 5 μm through the optic disk. Staining was performed with a commercial hematoxylin and eosin kit (catalog G1003, Servicebio). Slides were imaged using an Olympus BX53 microscope.
RNA-Seq and real-time qPCR
Total RNA of retinas was extracted using the TRIzol reagent (Invitrogen, CA, USA) according to the manufacturer’s protocol. The transcriptome sequencing was conducted by OE Biotech Co., Ltd. (Shanghai, China) using an Illumina Novaseq 6000 platform. Raw data were deposited in the Sequence Read Archive database (accession number PRJNA1214527). Gene Set Enrichment Analysis (GSEA) was performed using GSEA software. All reactions were performed on an ABI Prism 7900HT Sequence Detection System (Applied Biosystems) using SYBR green (RR420A, Takara, Japan). The associated primers were listed in Supplementary Table 1.
Vision-guided behavior testing
The Morris water maze test was modified to assess vision-dependent behavior as previously described21,22. In brief, mice were given a maximum of 60 seconds to locate a visible circular platform (0.01 m2, 0.5 cm above the water level) within a circular pool (1.44 m2, white plastic) filled with water at a temperature of 23 ± 2 °C. The water was made opaque by the addition of nontoxic white pigment (titanium dioxide, JiangHuTaiBai). No intentional cues, apart from the platform, were provided. To evaluate rod-mediated vision, mice were dark-adapted for at least 12 h before testing, and the experiment was performed under very dim light (≤ 0.01 lux at the water level, measured with MAVOLUX 5032B, Gossen, GER).
The experiment was performed on three consecutive days. Each mouse was tested two blocks of four trials per day. During each trial, the mouse was placed in the water from one of four equally spaced starting locations. The starting location was changed in a pseudo-random manner, whereas the platform was kept in a constant location. One trial ended if the mouse climbed onto the platform or if the mouse did not find the platform after 60 seconds. In the latter case, the mouse would be gently placed onto the platform. The mouse was left on the platform during the inter-trial interval for 20 seconds. After each block, the mouse was towel-dried and transferred to its home cage under warm air. The inter-block interval was approximately two hours. Each mouse completed 24 trials (6 blocks) in total during the three-day period. Behavior data were automatically recorded and analyzed with the EthoVision XT tracking system (Noldus, Wageningen, Netherlands). The main outcomes included the time required to reach the platform and the total path length. The latency of mice who did not find the platform within the time limit was recorded as the maximum (60 s) for data analysis.
Statistics and reproducibility
Statistical analysis was performed using GraphPad Prism (9.5.0) software. The statistical tests and the sample sizes used for each experiment are stated in the corresponding figure legends. The experiments were performed in at least three biologically independent animals, and similar results were observed.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Results
Cnga1-/- mice show a progressive loss of photoreceptor cells
We first monitored the photoreceptors over time, as they are the primarily affected cells of RP, and they are the target cells for Cnga1 gene augmentation therapy. Using in vivo optical coherence tomography (OCT) imaging, we observed a progressive, almost linear thinning of the outer nuclear layer (ONL, representing photoreceptor nuclei) in Cnga1-/- mice. The thickness of ONL reduced to 46% of the wild-type at the age of 3 months and became hardly discernible by the age of 6 months (Fig. 1A, B). An important sign detected by OCT was a ‘diffusely-hyperreflective’ appearance and the gradual loss of the IS/OS band (representing the junction of photoreceptor inner and outer segments) from one month postnatal onward. This sign is frequently observed in patients with RP23,24,25,26, indicating an unhealthy status of the photoreceptor cells. Hematoxylin and eosin staining demonstrated that only 1–2 rows of ONL remained by 6 months of age (Supplementary Fig. 1). With the progression of rod photoreceptor degeneration, we found that cone morphology was also compromised in Cnga1-/- mice (Fig. 1C). This is consistent with what was observed in human patients with RP5.

A Representative OCT images of wild-type and Cnga1-/- mice. Asterisks mark the photoreceptor nuclei layer. Scale bars, 100 μm. B Age-related thinning of the photoreceptor nuclei layer of Cnga1-/- mice compared with wild-type mice, as measured by OCT images. Mean ± SD. n = 6 eyes for both group at each time point. C Representative confocal images of immunohistochemistry with CNGA1 (red) and cone arrestin (CAR, a cone marker, green) show morphologically affected but still-persisting cones after advanced thinning of the photoreceptor nuclei layer in Cnga1-/- mice. Scale bar, 10 μm.
Cnga1-/- mice show a lack of rod-driven response to light
We next investigated the effect of CNGA1 deficiency on retinal function using full-field ERGs. This technique assesses the electrical response of the retina to a flash stimulus, with its two main components being the a-wave, a negative potential reflecting photoreceptor activity, and the subsequent positive b-wave, reflecting the activity of downstream retinal interneurons27,28.
We recorded scotopic ERGs (rod-dominated) in dark-adapted mice using a series of flash stimuli (Fig. 2). Three standard stimulus intensities were incorporated as per the recommendation of the International Society for Clinical Electrophysiology of Vision (ISCEV)29: −2 log cd s m−2 for a rod-specific response, and 0.5 and 1 log cd s m−2 as a ‘standard’ and a ‘strong’ flash, respectively, both having mixed rod-cone contribution. For wild-type mice at the age of one month, the b-wave was clearly visible starting at the intensity of −2 log cd s m−2 (Fig. 2A). With increasing stimulus intensities, the a-wave emerged and the amplitudes of both a- and b-waves increased steadily (Fig. 2A). In contrast, the Cnga1-/- mice showed no response to the rod-specific flash at −2 log cd s m−2 (Fig. 2A). With higher light intensities, the ERG waveforms lacked a distinct a-wave and typical oscillations on the peak of b-wave (Fig. 2A), both indicators of rod system activity27,28. These observations suggest that the rod system is not functional in the Cnga1-/- mouse. Indeed, the scotopic ERG waveforms of Cnga1-/- mice are similar to those found in the mouse models of pure cone function, such as the Rho-/- mouse30. The b-wave amplitudes of Cnga1-/- mice significantly declined with age (Fig. 2B, C), which is in keeping with the photoreceptor degeneration (Fig. 1C, D). By 6 months of age, no discernible ERG signals were left in Cnga1-/- mice, whereas the wild-type mice showed only an age-related decrease in amplitudes31 (Fig. 2B, C).

A Representative scotopic (dark-adapted) ERG waveforms in response to a series of flashes. Stimulus intensities are indicated in the left panel. B Quantification of b-wave amplitudes of Cnga1-/- mice compared to wild-type mice at 1 month of age (left), 3 months of age (middle) and 6 months of age (right). Mean ± SEM. C Changes in b-wave amplitudes with age at ISCEV standard flashes of −2 log cd s m−2 (left), 0.5 log cd s m−2 (middle), and 1 log cd s m−2 (right). Mean ± SEM. For the wild-type group, n = 10 eyes at each time point. For the Cnga1-/- group, n = 12 eyes at month 1 and 3; n = 10 eyes at month 2, 4, 5; n = 8 eyes at month 6.
To directly compare the cone function between the wild-type and Cnga1-/- mouse, we further performed photopic (light-adapted) ERGs (Fig. 3). For wild-type mice, b-waves were evident when the stimulus intensity rose to 1 log cd s m−2, and a slight a-wave was visible at the highest stimulus intensity. No marked difference was found in the shape of the waveforms between wild-type mice and Cnga1-/- mice (Fig. 3A). However, the b-wave amplitudes in Cnga1-/- mice were significantly larger than those in wild-type mice during the first two months postnatally (Fig. 3B, C). Likewise, this ‘supernormal’ phenomenon has been reported in the Rho-/- mouse30. The underlying mechanism might be attributed to cone degeneration32,33,34 and warrants further investigation.

A Representative photopic (light-adapted) ERG waveforms in response to a series of flashes. Stimulus intensities are indicated in the left panel. B Quantification of b-wave amplitudes of Cnga1-/- mice compared to wild-type mice at 1 month (left), 3 months (middle) and 6 months (right) of age. Mean ± SEM. C Changes in b-wave amplitudes with age at stimulus intensities of 1.0 log cd s m−2 (left) and 2.0 log cd s m−2 (right). Mean ± SEM. For the wild-type group, n = 10 eyes at each time point. For the Cnga1-/- group, n = 12 eyes at month 1 and 3; n = 10 eyes at month 2, 4, 5; n = 8 eyes at month 6.
In summary, the ERG analysis indicates a lack of rod-driven response in the Cnga1-/- mouse, and reveals a progressive loss of cone-mediated function with an initial ‘supernormal’ phase.
Gene augmentation therapy restores functional rod CNG channels, reverses cGMP accumulation, and rescues rod function
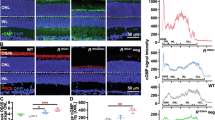
In retina, CNGA1 is selectively expressed in rod photoreceptors, forming the CNG channel with CNGB1 in a 3:1 stoichiometry1. The CNGA1 subunits confer the principal channel properties, whereas the CNGB1 subunit mainly contributes to outer segments localization1,35. Given the above information, we employed an adeno-associated virus (AAV) vector serotype 8 (for photoreceptor tropism36) to deliver murine Cnga1 under the control of a human rhodopsin promoter (for rod-specific expression; AAV8-hRHO-mCnga1). We first conducted a pilot dose-escalation study comparing three doses of AAV8-hRHO-mCnga1 (108, 109, and 1010 vg/eye). Efficacy was evaluated using a −2 log cd s m−2 flash stimulus, a rod-specific intensity recommended by ISCEV29, which elicits a negligible response in Cnga1⁻/⁻ mice (Fig. 2). The 109 vg/eye dose produced the most robust functional rescue and was therefore selected for subsequent experiments (Supplementary Fig. 2). Approximately 109 genome copies of AAV8-hRHO-mCnga1 were injected into the subretinal space of 2-week-old Cnga1-/- mice (Fig. 4A). Immunohistochemical assay of retinas 2.5 months post-injection showed that the introduced Cnga1 was successfully expressed and accurately localized to the rod outer segments (Fig. 4B), suggesting the formation of the rod CNG channel complex. The correct localization of CNGA1 was further confirmed by co-staining with rhodopsin (Supplementary Fig. 3). A major feature of impaired CNG channel function is the abnormal concentration of cGMP1, which triggers photoreceptor death2,37. We found that this toxic accumulation was reversed in the treated retina, whereas cGMP levels remained high in untreated eyes (Fig. 4B).

A Map of gene expression cassette for subretinal delivery and the experimental timeline. The data shown in (B-E) are from mice at 3 months of age. B Representative confocal images showing the expression of correctly localized CNGA1 protein (red) and lack of cGMP accumulation in treated retina. Scale bar, 10 μm. C Representative scotopic (dark-adapted) ERG waveforms in response to a series of flashes from wild-type mouse (left), untreated Cnga1-/- mouse (middle) and treated Cnga1-/- mouse (right). D Quantification of a-wave amplitudes of untreated and treated Cnga1-/- mice. Mean ± SEM. **P < 0.01, ***P < 0.001; two-way ANOVA with Šídák’s multiple comparisons. P values are 0.0001 at −0.5, 0.0011 at 0, and <0.0001 at higher light intensities. E Quantification of b-wave amplitudes of untreated and treated Cnga1-/- mice. Mean ± SEM. ***P < 0.001; two-way ANOVA with Šídák’s multiple comparisons. P values are 0.0006 at −2, 0.0003 at −1.5, 0.0001 at −1, and <0.0001 at higher light intensities. Treated Cnga1-/-, n = 10 eyes; untreated Cnga1-/-, n = 10 eyes.
Further evidence that CNGA1, together with CNGB1, had formed functional CNG channels was provided by scotopic ERG analysis. Figure 4C shows the representative ERG waveforms of wild-type (left), untreated Cnga1-/- (middle), and treated Cnga1-/- (right) eyes 2.5 months post-treatment. At the ISCEV rod-specific flash of −2 log cd s m−2, the treated Cnga1-/- eye exhibited a clear positive b-wave, while the untreated Cnga1-/- eye did not show a response (Fig. 4C). Furthermore, a slight recovery of a-wave was observed starting from −1 log cd s m−2 upward (Fig. 4C). The amplitudes of both a- and b-waves in treated Cnga1-/- eyes showed a clear and statistically significant improvement across all tested stimulus intensity (Fig. 4D, E). Together, these scotopic ERG findings validated the functional recovery of the rod CNG channel following gene augmentation therapy with AAV8-hRHO-mCnga1.
Gene augmentation therapy prolongs photoreceptor survival in Cnga1-/- mice
Patients with CNGA1-RP are characterized by progressive photoreceptor degeneration, which is also observed in the Cnga1-/- mouse model (Fig. 1 and Supplementary Fig. 1). We therefore followed treated Cnga1-/- mice to evaluate whether the gene augmentation therapy could halt or delay the photoreceptor death, thereby enabling long-term functional rescue. In vivo OCT images crossing the optic nerve head revealed that subretinal delivery of AAV8-hRHO-mCnga1 led to a significant preservation of photoreceptor nuclei in the ONL within the treated region compared with the opposite untreated area of the same eye or in the eyes of untreated Cnga1-/- mice (Fig. 5A). This structural rescue detected by OCT was evident at the age of 3 months and persisted up to the end of the experiment at the age of 9 months (Fig. 5A, B).

A Representative OCT images from treated Cnga1-/- mice at 3 months (top), 6 months (middle), and 9 months (bottom) of age showing clear preservation of photoreceptor layer thickness within the treated area. Asterisks mark the photoreceptor nuclei layer. ONH, optic nerve head. Scale bars, 100 μm. B Quantification of the photoreceptor layer thickness in treated Cnga1-/- retinae compared with untreated Cnga1-/- and wild-type retinae, as measured by OCT images. Mean ± SEM. ***P < 0.001; two-way ANOVA with Tukey’s multiple comparisons. P value is <0.0001. n = 6 for each group at each time point. C Representative eye section from a 9-month-old treated Cnga1-/- mouse. GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer. Scale bars, 200 μm in the middle panel and 50 μm in the magnified panels.
A representative hematoxylin and eosin staining of a 9-month-old treated retina is shown in Fig. 5C. It is, again, evident that the gene augmentation therapy resulted in robust protection of photoreceptors, confirming the OCT results. The ONL was 6-7 rows in the treated area, which in striking contrast to the untreated area in the same eye, where only a discontinuous row of ONL remained. Moreover, the inner and outer segments of photoreceptors were also preserved, as demonstrated in the magnified fragments (Fig. 5C). In Cnga1-/- mice, rod degeneration leads to secondary cone degeneration (Fig. 1C). To evaluate treatment effects on cones, we immunolabeled retinal sections with anti-cone arrestin antibody. As expected, cone photoreceptors were preserved in treated region, whereas degeneration continued in untreated areas (Supplementary Fig. 4).
We also evaluated the functional status of preserved photoreceptors at the age of 9 months (Fig. 6). Scotopic and photopic ERG recordings in the treated Cnga1-/- mice revealed clear preservation of the light responses, indicating sustained functional rescue of both rod and cone photoreceptors (Fig. 6).

A Representative scotopic (dark-adapted) ERG waveforms in response to a series of flashes from wild-type (left), untreated Cnga1-/- (middle), and treated Cnga1-/- mice (right) at 9 months of age. B Quantification of a-wave amplitudes of untreated and treated Cnga1-/- mice at 9 months of age. Mean ± SEM. **P < 0.01; two-way ANOVA with Šídák’s multiple comparisons. P values are 0.008 at 0, 0.002 at 0.5, and 0.0034 at 1 light intensities. C Quantification of scotopic b-wave amplitudes of untreated and treated Cnga1-/- mice at 9 months of age. Mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001; two-way ANOVA with Šídák’s multiple comparisons. P values are 0.0208 at −1.5, 0.0067 at −1, 0.0017 at −0.5, 0.0003 at 0, and <0.0001 at higher light intensities. The numbers of eyes were as follows: treated Cnga1-/-, n = 10; untreated Cnga1-/-, n = 4. D Representative photopic (light-adapted) ERG waveforms in response to a series of flashes from wild-type (left), untreated Cnga1-/- (middle), and treated Cnga1-/- mice (right) at 9 months of age. E Quantification of photopic b-wave amplitudes of untreated and treated Cnga1-/- mice at 9 months of age. Mean ± SEM. ***P < 0.001; two-way ANOVA with Šídák’s multiple comparisons. P values are 0.0807 at −1, 0.0567 at 0, and <0.0001 at higher light intensities.
Gene augmentation therapy improves vision-guided behavior in Cnga1-/- mice
Having demonstrated that our gene augmentation strategy restored retinal function and prolonged photoreceptor survival in Cnga1-/- mice, we were curious as to whether the improvement at the retinal level eventually translated to the improvement in the behavioral level. To answer this question, we utilized the water maze test under dim-light conditions. In this vision-guided behavior assay, 9-month-old mice were trained to locate a visible platform over three consecutive days (D1-D3). The time it took for the mice to find the platform was recorded as “escape latency” with a time limit set at one minute.
The representative swimming paths on D3 are illustrated in Fig. 7A. During the three-day period, the learning curves of wild-type and treated Cnga1-/- mice closely paralleled each other (Fig. 7B, C, D). The success rate of both groups rapidly approached nearly 100% on D2, accompanied by a gradual decrease in escape latency (Fig. 7B). In contrast, untreated Cnga1-/- mice often struggled to reach the platform within one minute, and therefore the escape latency showed no significant change over time (Fig. 7B, C).

A Representative swimming route on day 3. B Quantification of the success rate to locate the platform within 1 minute from day 1 to day 3. **P < 0.01, two-way ANOVA with Tukey’s multiple comparisons. Asterisks indicate a significant difference between treated and untreated Cnga1-/- mice at day 3. P value is 0.0026. C Quantification of the escape latency from day 1 to day 3. ***P < 0.001, two-way ANOVA with Tukey’s multiple comparisons. Asterisks indicate a significant difference between treated and untreated Cnga1-/- mice at day 3. P value is <0.0001. D Quantification of the total path length from day 1 to day 3. ***P < 0.001, two-way ANOVA with Tukey’s multiple comparisons. Asterisks indicate a significant difference between treated and untreated Cnga1-/- mice on day 3. P value is <0.0001. The numbers of mice were as follows: wild-type, n = 4; untreated Cnga1-/-, n = 4; treated Cnga1-/-, n = 5. Each mouse completed 8 trials per day.
We also analyzed the path length (Fig. 7D) and identified a pattern similar to that observed in escape latency. Over the training period, the path length of wild-type and treated Cnga1-/- mice decreased progressively, whereas it remained high for untreated Cnga1-/- mice. Together, these observations indicate that the restored retinal function is processed properly in treated Cnga1-/- mice, resulting in improved vision-guided behavior.
Gene augmentation therapy restores the expression of rod phototransduction-related genes in Cnga1-/- mice
To evaluate the transcriptional rescue effects of gene augmentation therapy on photoreceptors, we conducted RNA sequencing (RNA-seq) analysis of retinas from 3-month-old wild-type, untreated Cnga1-/-, and treated Cnga1-/- mice. As expected, untreated Cnga1-/- mice exhibited significant downregulation of rod-specific genes compared to wild-type controls (Fig. 8A). Gene Set Enrichment Analysis (GSEA) revealed substantial repression of gene sets associated with phototransduction and photoreceptor cell maintenance in untreated retinas (Fig. 8C). In contrast, gene augmentation therapy restored the expression of rod-specific genes (Fig. 8B), and re-enriched phototransduction and photoreceptor cell maintenance pathways (Fig. 8D). We validated these findings using quantitative real-time PCR (qPCR). Treated mice showed significant mRNA recovery of Cnga1 and other key rod phototransduction genes, including Rho, Pde6a, Pde6b, Pde6g, Cngb1, Gucy2e, Gucy2f, Guca1a, and Guca1b (Fig. 8E). Collectively, these results demonstrate that gene augmentation therapy successfully preserves the transcriptional program of rod phototransduction in Cnga1-/- mice.

A Volcano plot of differentially expressed genes between wild type and untreated retinas (|log2(fold change) |)> 0.58 and p < 0.05. B Volcano plot of differentially expressed genes between untreated and treated retinas (|log2(fold change) |)> 0.58 and p < 0.05. C GSEA revealed significant downregulation of phototransduction and photoreceptor cell maintenance pathways in untreated versus wild-type retinas. D GSEA showed upregulation of phototransduction and photoreceptor cell maintenance pathways in treated versus untreated retinas. E Relative expression levels of rod phototransduction-related genes in wild type, untreated, and treated retinas, as measured by real-time qPCR (n = 4). mRNA levels were normalized to Gapdh expression and presented as Mean ± SEM. Statistical significance determined by one-way ANOVA with Tukey’s post hoc test.
Discussion
The rod CNG channel is composed of three CNGA1 and one CNGB1 subunit1,35. Biallelic mutations in either CNGA1 or CNGB1 result in progressive rod-cone degeneration known as RP18,19. We are interested in CNGA1-RP for two reasons. First, the prevalence of CNGA1-RP shows dramatically higher in Asian countries8,10 compared to other regions4,5,6,7,9,15 (appropriately 4 times). Second, successful proof-of-concept studies for AAV-based gene augmentation therapy have been reported in animal models of CNGB1-RP22,38,39,40. Considering the close relationship of CNGA1 and CNGB1, we hypothesized that AAV-mediated expression of CNGA1 would be a feasible approach for the treatment of CNGA1-RP. To test this hypothesis, we previously developed a Cnga1-/- mouse model20. In this study, we first performed a detailed phenotypic analysis of this model with OCT and ERG techniques. Cnga1-/- mice exhibited a progressive loss of photoreceptors, which was almost complete by the age of 6 months. Scotopic ERGs indicated a loss of rod-mediated function, as expected, since the CNG channel is the final target of the light-activated signaling pathway. Interestingly, we observed a ‘supernormal’ period of cone function in Cnga1-/- mice during the first two months postnatal. Nonetheless, with the progression of cone degeneration, both scotopic and photopic ERG responses deteriorated. By 6 months of age, ERG responses diminished to a negligible level. Subsequently, we initiated gene augmentation therapy based on the understanding of the above natural history data. We treated Cnga1-/- mice at postnatal day 14, a timepoint prior to major loss of rod photoreceptors, and followed the treated mice to the end-stage of disease at 9 months of age. The therapeutic vector (AAV8-hRHO-mCnga1) delivered to the subretinal space of Cnga1-/- mice restored rod-mediated retinal function and vision-guided behavior for at least 8.5 months post-treatment. Immunohistochemistry showed that CNGA1 was successfully expressed and was correctly trafficked to the outer segments, where the native rod CNG channel is located. In vivo OCT imaging as well as ex vivo histological analysis demonstrated long-term preservation of retinal structure. To our knowledge, the present study is the first report of phenotypic rescue with AAV-based gene augmentation therapy in an animal model of CNGA1-RP.
The availability of suitable disease models has been instrumental in translating any promising treatments to the clinic. Apart from our Cnga1-/- mouse model, four other models were reported in the literature. The first CNGA1-RP mouse model was generated by overexpression of an 890 bp Cnga1 antisense mRNA more than two decades ago41. This transgenic mouse showed slight photoreceptor degeneration at 12 months of age41. However, functional analysis has not been reported so far. In 2015, a spontaneously occurring dog model was identified with 4-bp deletion in exon 9 (c.1752_1755delAACT) of Cnga1 gene42, a detailed description of the retinal phenotype is still pending. More recently, a mouse model was re-derived from the sperm samples of ENU (N-ethyl-N-nitrosourea) mutagenesis repository43. The ENU mouse carries a c.1526 A > G mutation in Cnga1, resulting in Y509C in the cyclic nucleotide-binding domain of CNGA1 protein. This Y509C mutation leads to a complete loss of CNGA1 protein expression43. Consequently, rod-driven ERG responses were diminished in the Cnga1Y509C/Y509C mouse, a phenotype similar to that observed in our Cnga1-/- mouse. However, the progression rate of photoreceptor degeneration in Cnga1Y509C/Y509C mice is much slower than that in Cnga1-/- mice. At 6 months of age, most photoreceptor nuclei were lost in Cnga1-/- mice, whereas 40% photoreceptors remained in Cnga1Y509C/Y509C mice43. The underlying reason for the difference in progression rate is unclear and warrants further investigation. Considering that CNGA1 functions with CNGB1 as a heterotetramer, we recently developed a patient-induced pluripotent stem cell (iPSC) line to facilitate the testing of species-matched gene augmentation therapy44. Together, these disease models provide us with valuable platforms for understanding the disease mechanisms and tools to evaluate therapeutic strategies for CNGA1-RP.
This work demonstrates a proof-of-concept for the treatment of CNGA1-RP using AAV-based gene augmentation therapy. Further efforts are needed before this treatment can be translated into the clinic. Firstly, the natural history of patients with CNGA1-RP should be systematically evaluated. This will not only allow for a more precise comparison of human phenotypes with those of animal models but also may provide suitable biomarkers of disease progression as well as potential outcome measures for future clinical trials45. Furthermore, further optimization of the vector, both in terms of the vector construct and optimal dose, is warranted. Finally, it will be essential to understand the efficacy of therapy applied at different disease stages46,47,48,49. In the current study, we treated Cnga1-/- mice at an early stage of the disease to assess the maximal potential of gene augmentation therapy; however, patients with RP are often diagnosed at a later stage of the disease. It is, therefore, important to determine the therapeutic time window in future studies. With these further efforts, we believe that gene augmentation therapy will, for the first time, provide a therapeutic intervention for patients with CNGA1-RP.
Data availability
The numerical data plotted (source data) in the graphs are in Supplementary Data. Raw data of RNA-seq were deposited in the Sequence Read Archive database (accession number PRJNA1214527). All other data are available from the corresponding author on reasonable request.
References
Kaupp, U. B. & Seifert, R. Cyclic nucleotide-gated ion channels. Physiol. Rev. 82, 769–824 (2002).
Power, M. et al. Cellular mechanisms of hereditary photoreceptor degeneration - Focus on cGMP. Prog. Retin. Eye Res. 74, 100772 (2020).
Arshavsky, V. Y., Lamb, T. D. & Pugh, E. N. Jr G proteins and phototransduction. Annu. Rev. Physiol. 64, 153–187 (2002).
Dryja, T. P. et al. Mutations in the gene encoding the alpha subunit of the rod cGMP-gated channel in autosomal recessive retinitis pigmentosa. Proc. Natl. Acad. Sci. USA 92, 10177–10181 (1995).
Hartong, D. T., Berson, E. L. & Dryja, T. P. Retinitis pigmentosa. Lancet 368, 1795–1809 (2006).
Chen, X. et al. Targeted sequencing of 179 genes associated with hereditary retinal dystrophies and 10 candidate genes identifies novel and known mutations in patients with various retinal diseases. Invest. Ophthalmol. Vis. Sci. 54, 2186–2197 (2013).
Hanany, M., Rivolta, C. & Sharon, D. Worldwide carrier frequency and genetic prevalence of autosomal recessive inherited retinal diseases. Proc. Natl. Acad. Sci. USA 117, 2710–2716 (2020).
Gao, F. J. et al. Genetic anD CLINICAL FINDINGS IN A LARGE COHORT OF CHINESE PATIENTS WITH SUSPECTEd Retinitis Pigmentosa. Ophthalmology 126, 1549–1556 (2019).
Paloma, E. et al. Novel homozygous mutation in the alpha subunit of the rod cGMP gated channel (CNGA1) in two Spanish sibs affected with autosomal recessive retinitis pigmentosa. J. Med. Genet. 39, E66 (2002).
Katagiri, S. et al. Whole exome analysis identifies frequent CNGA1 mutations in Japanese population with autosomal recessive retinitis pigmentosa. PLoS One 9, e108721 (2014).
Zhang, Q. et al. Autosomal recessive retinitis pigmentosa in a Pakistani family mapped to CNGA1 with identification of a novel mutation. Mol. Vis. 10, 884–889 (2004).
Ye, Z. et al. Case Report: The First Reported Concurrence of Wilson Disease and Bilateral Retinitis Pigmentosa. Front. Med. 9, 877752 (2022).
Gao, Q. et al. A novel CNGA1 GENE MUtation (c.G622A) of autosomal recessive Retinitis Pigmentosa leads to the CNGA1 protein reduction on membrane. Biochem. Genet. 57, 540–554 (2019).
Wang, M., Gan, D., Huang, X. & Xu, G. Novel compound heterozygous mutations in CNGA1in a Chinese family affected with autosomal recessive retinitis pigmentosa by targeted sequencing. BMC Ophthalmol. 16, 101 (2016).
Jin, X. et al. Novel compound heterozygous mutation in the CNGA1 gene underlie autosomal recessive retinitis pigmentosa in a Chinese family. Biosci. Rep. 36, e00289 (2016).
Saito, K. et al. A case of retinitis pigmentosa homozygous for a rare CNGA1 causal variant. Sci. Rep. 11, 4681 (2021).
Russell, S. et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet 390, 849–860 (2017).
Gerhardt, M. J., Priglinger, S. G., Biel, M. & Michalakis, S. Biology, pathobiology and gene therapy of CNG channel-related retinopathies. Biomedicines 11 https://doi.org/10.3390/biomedicines11020269 (2023).
Gerhardt, M. J., Petersen-Jones, S. M. & Michalakis, S. CNG channel-related retinitis pigmentosa. Vis. Res. 208, 108232 (2023).
Liu, Y. et al. Retinal degeneration in mice lacking the cyclic nucleotide-gated channel subunit CNGA1. FASEB J. 35, e21859 (2021).
Wu, Y. et al. AAV-mediated base-editing therapy ameliorates the disease phenotypes in a mouse model of retinitis pigmentosa. Nat. Commun. 14, 4923 (2023).
Koch, S. et al. Gene therapy restores vision and delays degeneration in the CNGB1(-/-) mouse model of retinitis pigmentosa. Hum. Mol. Genet. 21, 4486–4496 (2012).
Aizawa, S. et al. Correlation between visual function and photoreceptor inner/outer segment junction in patients with retinitis pigmentosa. Eye 23, 304–308 (2009).
Tao, L. W., Wu, Z., Guymer, R. H. & Luu, C. D. Ellipsoid zone on optical coherence tomography: a review. Clin. Exp. Ophthalmol. 44, 422–430 (2016).
Gong, Y., Chen, L. J., Pang, C. P. & Chen, H. Ellipsoid zone optical intensity reduction as an early biomarker for retinitis pigmentosa. Acta Ophthalmol. 99, e215–e221 (2021).
Oh, J. K., Nuzbrokh, Y., Lima de Carvalho, J. R. Jr., Ryu, J. & Tsang, S. H. Optical coherence tomography in the evaluation of retinitis pigmentosa. Ophthalm. Genet. 41, 413–419 (2020).
Tanimoto, N. et al. Vision tests in the mouse: Functional phenotyping with electroretinography. Front Biosci. (Landmark Ed.) 14, 2730–2737 (2009).
Benchorin, G., Calton, M. A., Beaulieu, M. O. & Vollrath, D. Assessment of murine retinal function by Electroretinography. Bio Protoc. 7, https://doi.org/10.21769/BioProtoc.2218 (2017).
Robson, A. G. et al. ISCEV Standard for full-field clinical electroretinography (2022 update). Doc. Ophthalmol. 144, 165–177 (2022).
Jaissle, G. B. et al. Evaluation of the rhodopsin knockout mouse as a model of pure cone function. Invest. Ophthalmol. Vis. Sci. 42, 506–513 (2001).
Park, J. C. et al. Effects of normal aging on the mouse retina assessed by full-field flash and flicker electroretinography. Sci. Rep. 13, 8860 (2023).
Yagasaki, K., Miyake, Y., Litao, R. E. & Ichikawa, K. Two cases of retinal degeneration with an unusual form of electroretinogram. Doc. Ophthalmol. 63, 73–82 (1986).
Alexander, K. R. & Fishman, G. A. Supernormal scotopic ERG in cone dystrophy. Br. J. Ophthalmol. 68, 69–78 (1984).
Phillips, M. J. et al. Retinal function and structure in Ant1-deficient mice. Invest. Ophthalmol. Vis. Sci. 51, 6744–6752 (2010).
Hüttl, S. et al. Impaired channel targeting and retinal degeneration in mice lacking the cyclic nucleotide-gated channel subunit CNGB1. J. Neurosci. 25, 130–138 (2005).
Pupo, A. et al. AAV vectors: The Rubik’s cube of human gene therapy. Mol. Ther. 30, 3515–3541 (2022).
Li, S., Ma, H., Yang, F. & Ding, X. cGMP Signaling in Photoreceptor Degeneration. Int. J. Mol. Sci. 24 https://doi.org/10.3390/ijms241311200 (2023).
Petersen-Jones, S. M. et al. Patients and animal models of CNGβ1-deficient retinitis pigmentosa support gene augmentation approach. J. Clin. Invest. 128, 190–206 (2018).
Wagner, J. E. et al. In vivo potency testing of subretinal rAAV5.hCNGB1 gene therapy in the Cngb1 Knockout Mouse Model of Retinitis Pigmentosa. Hum. Gene Ther. 32, 1158–1170 (2021).
Occelli, L. M. et al. Development of a translatable gene augmentation therapy for CNGB1-retinitis pigmentosa. Mol. Ther. https://doi.org/10.1016/j.ymthe.2023.04.005 (2023).
Leconte, L. & Barnstable, C. J. Impairment of rod cGMP-gated channel alpha-subunit expression leads to photoreceptor and bipolar cell degeneration. Invest. Ophthalmol. Vis. Sci. 41, 917–926 (2000).
Wiik, A. C. et al. Progressive retinal atrophy in Shetland sheepdog is associated with a mutation in the CNGA1 gene. Anim. Genet. 46, 515–521 (2015).
Kandaswamy, S. et al. Mutations within the cGMP-binding domain of CNGA1 causing autosomal recessive retinitis pigmentosa in human and animal model. Cell Death Discov. 8, 387 (2022).
Wu, Y. et al. Establishment of the induced pluripotent stem cell line SJTUGHi002-A from a CNGA1-related recessive retinitis pigmentosa patient. Stem Cell Res. 76, 103334 (2024).
Ayala, A., Cheetham, J., Durham, T. & Maguire, M. The Importance of Natural History Studies in Inherited Retinal Diseases. Cold Spring Harb. Perspect. Med. 13, https://doi.org/10.1101/cshperspect.a041297 (2023).
Koch, S. F. et al. Halting progressive neurodegeneration in advanced retinitis pigmentosa. J. Clin. Invest. 125, 3704–3713 (2015).
Koch, S. F. et al. Genetic rescue models refute nonautonomous rod cell death in retinitis pigmentosa. Proc. Natl. Acad. Sci. USA 114, 5259–5264 (2017).
Kajtna, J., Tsang, S. H. & Koch, S. F. Late-stage rescue of visually guided behavior in the context of a significantly remodeled retinitis pigmentosa mouse model. Cell. Mol. Life Sci. 79, 148 (2022).
Scalabrino, M. L. et al. Late gene therapy limits the restoration of retinal function in a mouse model of retinitis pigmentosa. Nat. Commun. 14, 8256 (2023).
Acknowledgements
This work was supported by the Shanghai Sailing Program (24YF2734000 to Yidong Wu), Natural Science Foundation of Shanghai (24ZR1458800 to Xiaoling Wan), National Natural Science Foundation of China (U22A20311 and 82388101 to Xiaodong Sun), and Science and Technology Commission of Shanghai Municipality (23J41900200 to Xiaodong Sun).
Author information
Authors and Affiliations
Contributions
Yi.W., X.W., F.W., and X.S. conceived and designed the research. Yi.W., Tia.Z., Yaz.W., and Tin.Z. designed, performed, and analyzed the in vivo experiments. C.H. performed immunohistochemistry experiments. Yaf.W., Y.L., Z.C., and J.C. performed RNA sequencing. T.L., J.S., H.J., and L.Z. contributed to data collection and interpretation. Yi.W. and X.W. wrote the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
F.W. is an equity owner of Innostellar Biotherapeutics, a company developing human gene therapies. The remaining authors declare no competing interests.
Peer review
Peer review information
Communications Medicine thanks Michelle McClements and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wu, Y., Zhao, T., Wang, Y. et al. Gene augmentation therapy restores vision and preserves photoreceptors in a mouse model of CNGA1-retinitis pigmentosa. Commun Med 5, 384 (2025). https://doi.org/10.1038/s43856-025-01108-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s43856-025-01108-x
