
Abstract
Immunity by vaccination can protect human against heterologous viruses. However, protective abilities of artificial vaccines are still weaker than natural infections. Here we develop a kinetically engineered vaccine (KE-VAC) that mimics the multidimensional immunomodulation in natural infections via dynamic activation of antigen presenting cells with masked TLR7/8 agonist and sustained supplies of antigens and adjuvants to lymph nodes, leading to follicular helper T and germinal centre B cell activation in vaccinated mice. KE-VAC demonstrates superior efficacy than traditional alum and mRNA vaccines, achieving a 100% survival rate with increased neutralizing antibodies titers and polyfunctional CD8+ T cells, recognizing heterologous SARS-CoV-2 variants, and inducing broad and long-term protection against multiple strains of influenza viruses. Prime/boost vaccination with KE-VAC also protect aged ferrets from severe fever with thrombocytopenia syndrome virus infection, with no virus detected in any organs at day 6 p.i. The efficacy of KE-VAC across various pathogens thus highlights its potential as an effective vaccine against emerging infectious risks.
Similar content being viewed by others


Introduction
As the COVID-19 pandemic, which poses a global threat, approaches its end, countries worldwide are gradually lowering their risk levels, signalling the beginning of the endemic era. However, the recent resurgence of highly pathogenic avian influenza underscores the need for innovative and safe vaccines over diseases that threaten humanity1,2,3. During the COVID-19 pandemic, various types of new vaccines, including mRNA vaccines, received emergency approval and entered the vaccine market, spurring research activity in vaccine and adjuvant development. Despite this progress, most of these newly developed vaccines exhibit significantly weaker protective capabilities than the natural immune responses observed in patients who have recovered from natural infections4,5. For example, when the immune capacities of individuals vaccinated against COVID-19 were compared with those of individuals who were infected and subsequently recovered, significant differences in long-term protection, protection against variants, and T-cell responses were reported6,7.
In this study, we focused on understanding why artificial vaccines and natural infections differ in their protective abilities, and on the basis of this insight, we aimed to develop a new vaccine‒adjuvant system that could provide a protective immune profile similar to those of recipients who have recovered from natural infections with healthy state. Artificial vaccines typically use a single antigen or an adjuvant such as alum, which provides a stable humoral response but is often insufficient to induce a robust T-cell response. This inadequacy may result in a lack of early resistance to pathogens following vaccination or infection7,8,9,10,11. In contrast, natural pathogens contain not only antigenic information but also various pathogen-associated molecular patterns (PAMPs) that trigger complex T-cell-related immune responses12,13,14,15,16. Additionally, while vaccination delivers an immunogen only at the time of administration, pathogens undergo a gradual replication process after invading the body, continuously stimulating the immune system. Research has shown that when antigens or adjuvants are gradually supplied, they enhance the GC immune response17,18,19,20,21,22. For an example, utilising specially designed osmotic pumps for slow and sustained immunisation has demonstrated significant effectiveness in GC activation22. These findings suggest that a vaccine platform that mimics the dynamic stimulation observed in natural infections could improve both humoral and T-cell-related responses, leading to effective, long-term protection.
To overcome the limitations of conventional vaccines, we develop a kinetically engineered vaccine (KE-VAC) that mimics the multidimensional immune stimulation observed in natural infections. KE-VAC achieves prolonged antigen presentation and sustained immune activation by utilizing a masked TLR7/8 agonist and a continuous antigen-adjuvant delivery system. This design enhances germinal center activation, contributing to long-term immune protection, while also strengthening T-cell responses, which play a crucial role in early and rapid pathogen clearance. By modulating immune responses at both the cellular and tissue levels, KE-VAC effectively orchestrates humoral and cellular immunity, ensuring both early defence and durable immune memory. These findings highlight the potential of KE-VAC as an effective vaccine platform capable of providing broad and sustained protection against emerging infectious diseases.
Results
Designing kinetically engineered vaccine mimicking natural infection
To develop a vaccine-adjuvant platform that mimics the characteristics of natural infection and increases protective immunity, we designed a kinetically engineered vaccine (KE-VAC) capable of controlling multidimensional immune responses through the design of masked TLR7/8 agonist (m-TLR7/8a) as a synthetic PAMP-based molecular adjuvant. m-TLR7/8a allowed delayed activation of TLR7/8a by endolysosome responsive cleavage to adjust kinetics of immune response at the cellular level and the recapitulated the latent proliferation and gradual stimulating immune system of invading pathogen at tissue level by FDA-approved alum (Fig. 1). At the cellular level, most of the conventional synthetic PAMPs available in vaccines facilitated APCs activation and antigen presentation, but they also induce cellular exhaustion23,24. As the strategy for the kinetic immune modulation of antigen presenting cells (APCs) that can achieve robust innate and adaptive immune responses without inducing exhausted immune cells, we designed m-TLR7/8a that remained in an inactive state based on the transient chemical masking of putative active site in TLR7/8a (C4 amine)) and recovered activity via a chemical linker that was cleavable by gamma-interferon-inducible lysosomal thiol reductase (GILT) within the endolysosome microenvironment (Fig. 1, Supplementary Fig. 1 and 2)25. Cholesterol was additionally chosen as a transient shielding blocker because it could enable facile incorporation of m-TLR7/8a into nanoliposomes with a high encapsulation efficacy. Additionally, nanoliposomes (m-TLR7/8a) has been engineered to be negatively charged to facilitate interactions with alum and various antigens (such as recombinant proteins or inactivated viruses) (Fig. 1). The nanoliposome (m-TLR7/8a) in KE-VAC can provide timely and sustained stimulation of APCs and prolong antigen presentation on immune complexes and interaction time with TFH cells to promote GC-related immune responses with delayed exhaustion in addition to the significant enhancement of T-cell responses26,27. At the cellular level, when nanoliposome (m-TLR7/8a) entered an immune cell, the masking site of m-TLR7/8a was cleaved by GILT (Supplementary Fig. 3a)28. During the cleavage process, the activity of TLR7/8a was gradually recovered from nanoliposome (m-TLR7/8a), inducing prolonged stimulation of APCs (Supplementary Fig. 3b). Delayed TLR7/8 signaling helps prevent exhaustion in APCs, enabling them to respond to stimuli over a longer period (Supplementary Fig. 3c-e). The extended activity of APCs facilitated prolonged antigen presentation, which, in turn, allows sustained interactions between APCs and TFH cells, thereby initiating robust germinal centre immune responses at the cellular level24,29.

Chemical synthetic process of m-TLR7/8a and simple description of nanoliposome (m-TLR7/8a) and KE-VAC.
Selection of vaccination strategy for durable immune responses
To investigate the impact of different immunisation strategies mimicking natural infection and conventional artificial vaccination on the immune response, mice were vaccinated with the same total doses of antigen or adjuvant via three different methods: increasing (Inc), splitting (Spt), and bolus (Bol). Blood was collected and analysed after the first cycle, and the popliteal lymph nodes (pLNs), spleen, and blood were collected and analysed after the second cycle (Supplementary Fig. 4a). Serum anti-OVA IgG, IgG1, and IgG2c end-point titers were compared on days 14 and 28, revealing that the Bol group presented no titer values on day 14, whereas both the Inc and Spt groups presented high titers at that time. Even on the 28th day postvaccination, both the Inc and Spt groups maintained higher titers than the Bol group did (Supplementary Fig. 4b). Analysis of TFH cells and GC B-cells, which are involved in durable humoral immune responses in pLNs and spleens, revealed significantly greater values in the Inc and Spt groups than in the Bol group (Supplementary Fig. 4c–e). The Inc group generally demonstrated higher values than the Spt group did, but statistical significance was observed only for pLN GC B-cells. Furthermore, analysis of central memory T (TCM) cells in the spleen revealed higher levels of CD4+ TCMs in the Inc and Spt groups than in the Bol group, with the Spt group exhibiting the highest levels of CD8+ TCMs. The results for the Inc and Spt groups suggested that sustained stimulation with the antigen and adjuvant significantly impacted durable immune responses in lymphoid organs. Consequently, the development of a vaccine platform with controlled delivery kinetics of antigens and adjuvants holds promise for eliciting a durable immune response.
Designing a kinetically engineered nanovaccine platform
To identify optimal immunostimulatory adjuvant candidates for promoting the GC-related immune response, TFH, GC B-, and plasma cell induction, we screened various adjuvant candidates by coculturing DCs, T-cells, and B-cells (Fig. 2a). Among the PAMPs tested, nanoliposome (m-TLR7/8a), which was developed on the basis of a novel TLR7/8 agonist (m-TLR7/8a), exhibited superior efficacy in inducing TFH and plasma cell differentiation and IL-21 secretion than did the well-known TLR7/8 agonist (R848) and even nanoliposome formulation of R848 (Fig. 2b, c and Supplementary Fig. 5). DC antigen presentation after antigen and adjuvant treatment was greatly prolonged in the nanoliposome (m-TLR7/8a) group (Fig. 2d). Additionally, the nanoliposome (m-TLR7/8a)-treated DCs exhibited extended secretion of GC-related cytokines (IL-23), Th1 response-related cytokines (IL-12p70), and activation-related cytokines (IL-6) (Fig. 2e).

a Schematic illustration of the coculture method for mimicking the GC response. BMDC, Naïve T cell, and B cell were harvested from C57BL/6 mice and co-culture for analysis. b TFH and plasma cells were characterised from cocultured cells by flow cytometry (n = 3). c IL-21 secretion was evaluated in the coculture supernatant by ELISA (n = 3). d Antigen presentation by BMDCs were characterized by flow cytometry and (e) IL-23 (left), IL-12p70 (middle), and IL-6 (right) cytokine secretion from activated BMDCs depend on time after stimulation were detected by ELISA (n = 5). f Releasing mechanism of nanoliposome (m-TLR7/8a) from KE-VAC. g The ability of alum to capture liposomes and antigens was characterised with fluorescence-labelled liposomes and antigens (n = 3). h The interaction of nanoliposome (m-TLR7/8a) with alum was observed via TEM (scale bar = 200 nm). i The colocalization of antigens and liposomes with alum was observed via confocal microscopy with fluorescence-labelled liposomes and antigens. j The antigen and nanoliposome (m-TLR7/8a) released from KE-VAC were evaluated via a Transwell assay (n = 3). k FITC-labelled antigens and liposomes accumulated in pLNs after i.m. injection with or without alum (n = 3). Data presented as mean ± SD. Statistical significance was analysed via one-way ANOVA in (b-f). p values: ns, not significant; *p < 0.05; ** p < 0.01; ***p < 0.001; ****p < 0.0001).
Prior results (Supplementary Fig. 4c-e) demonstrated that a continuous supply of adjuvants and antigens fostered a sustained immune response. We recapitulated these phenomena and engineered a vaccine platform with a kinetic modulator enabling the continuous supply of nanoliposome (m-TLR7/8a) and antigens. Alum, the most widely used FDA-approved adjuvant, was selected as the most suitable candidate for this purpose (Fig. 2f). Owing to its positively charged surface, we hypothesised that alum could interact with negatively charged antigens and nanoliposome (m-TLR7/8a). Indeed, alterations in pH enabled the modulation of alum’s charge to facilitate interactions with the fluorescence-labelled antigen (AF647-OVA) and DiD-liposome (as a model of nanoliposome (m-TLR7/8a)). Dissociation studies using DiD-liposomes confirmed the effective binding of the two substances when alum was positively charged, with subsequent release upon attaining a negative charge state (Supplementary Fig. 6). Subsequent centrifugation of the fluorescence-labelled material resulted in nearly 100% adsorption efficiency at the intended concentration (Fig. 2g). Transmission electron microscopy (TEM) examination of KE-VAC, which is composed of an antigen and nanoliposome (m-TLR7/8a) adsorbed to alum, confirmed the proximity of nanoliposome (m-TLR7/8a) to alum (Fig. 2h). Confocal microscopy further confirmed the colocalization of alum, the antigen, and nanoliposome (m-TLR7/8a) (Fig. 2i). Analysis of the material released through a Transwell system revealed a nearly linear release profile over approximately 2 weeks (Fig. 2j). By using in vivo imaging to confirm the delivery efficiency of the antigen and nanoliposome (m-TLR7/8a) to LNs with and without alum, we observed significant differences. Without alum, the intramuscularly injected antigen and nanoliposome (m-TLR7/8a) were delivered to the pLNs, peaking in delivery efficiency the day after injection but then diminishing sharply and becoming barely detectable by 7 days after injection. Interestingly, when alum was included, the amount of antigen and nanoliposome (m-TLR7/8a) in the pLNs increased gradually, reaching its peak at 7 days after injection, and remained detectable even at 14 days after injection (Fig. 2k).
KE-VAC potentiate a broad and long-lasting protective immune response
In murine models, KE-VAC, formulated with the model antigen OVA, was administered twice at 2-week intervals (Fig. 3a). One week following the second inoculation, pLNs and spleens adjacent to the inoculation site were harvested for immune cell analysis. Compared with those inoculated with alum or antigen alone, KE-VAC (G4) resulted in significant increases in the numbers of TFH cells, GC B-cells, and plasma B-cells (Fig. 3b, c). Noteworthy variations were also observed in memory T-cells associated with the cellular memory response, with a marked increase in the numbers of both CD4+ and CD8+ TCMs upon KE-VAC inoculation (Fig. 3d).

a Timeline detailing the immunisation schedule and doses and blood and organ collection. The components were intramuscularly injected into mice twice, with a 2-week interval. The doses were as follows: OVA, 10 µg; Alum, 100 µg; and m-TLR7/8a in nanoliposome, 20 µg (n = 5). (b–d) Antigen (OVA) and Immune cells related to the GC and memory response were analysed 7 days after boosting. TFH and GC B-cells were characterised in the (b) pLNs and (c) spleen. d CD4+ and CD8+ TCMs were analysed in the spleen by flow cytometry. e Timeline detailing the immunisation schedule and doses and blood and organ collection. The components were intramuscularly injected into mice twice, with a 2-week interval. The doses were as follows: OVA, 10 µg; Alum, 100 µg; and m-TLR7/8a in nanoliposome, 20 µg. Blood was collected every 7 days, and the spleen was harvested every 14 days for analysis. f–j Several parameters related to humoral immunity and the GC response were observed over 49 days after priming (n = 3). f Serum anti-OVA IgG (left), IgG1 (middle), and IgG2c (right) antibody titers were evaluated at the endpoint of dilution. (g) TFH cells, (h) GC B-cells, (i) plasma B-cells, and (j) memory B-cells were characterised in the spleen during this period by flow cytometry. Data presented as mean ± SD. Statistical significance was analysed via one-way ANOVA for (b-d) and (g-j). p values: ns not significant; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001).
To ascertain the contribution of activated immune cells to the long-term immune response, KE-VAC was administered following the same schedule as the mRNA lipid nanoparticle (mLNP) vaccine, with analyses conducted at weekly intervals (Fig. 3e). The serum anti-OVA IgG, IgG1, and IgG2c titers decreased in both the mLNP and KE-VAC groups beginning on the 28th day after priming. While mLNP-induced antibody titers sharply decreased and were undetectable by the 49th day, the mice inoculated with KE-VAC maintained high antibody titers even on day 49 (Fig. 3f). Although both vaccines initially induced increases in the numbers of TFH and GC B-cells, KE-VAC inoculation maintained elevated levels beyond the 21st day post inoculation, suggesting a sustained and robust immune response (Fig. 3g, h). Additionally, KE-VAC uniquely stimulated significant and prolonged expansion of plasma B-cells and memory B-cells, in contrast with the minimal changes observed in mLNP-treated groups (Fig. 3i, j). This sustained increase highlights the potency and durability of KE-VAC as a vaccine platform, underscoring its potential for eliciting long-lasting immune protection. These findings underscore the potential of KE-VAC, which is designed for continuous supply and kinetic stimulation of antigen and nanoliposome (m-TLR7/8a), as a vaccine platform to elicit a prolonged and robust immune response by inducing TFH cells and GC B-cells.
The long-term and sustained efficacy of KE-VAC can be attributed to its modulation effects at both the macroscopic and cellular levels. To elucidate the effect of different kinetic factors provided by KE-VAC on the vaccine efficacy, we have assessed the immune responses at different kinetic immune modulation condition at cellular and macroscopic levels. Compared to single shot + R848 injection, KE-VAC induced significantly higher GC immune responses in both the spleen and LNs by day 7, following an initial response observed at day 3 post-vaccination. The GC immune responses were dominant in KE-VAC-treated group compared with those in nanoliposome (m-TLR7/8a)-treated group, suggesting that macroscopic kinetic modulation played a significant role (Supplementary Fig. 7). The anti-OVA IgG titers in nanoliposome (m-TLR7/8a)-treated group were higher than those of nanoliposome (R848)-treated group, demonstrating that the different cellular-level kinetic control can lead to different immune responses, even with same macroscopic kinetic modulation condition (Supplementary Fig. 8).
Robust and broad protective immunity against multiple SARS-CoV-2 variants
To evaluate the efficacy of our KE-VAC platform in combating natural diseases, we inoculated C57BL/6 mice with KE-VAC containing the wild-type (WT) SARS-CoV-2 hexapro antigen, which includes six proline substitutions stabilizing the spike protein in its prefusion form and increasing immunogencity30. Following priming, the serum collected on day 28 was subjected to a serum neutralizing assay against SARS-CoV-2 transfected pseudovirus (Supplementary Fig. 9a). Analysis of the anti-spike endpoint titers of IgG, IgG1, and IgG2c in the day 28 serum revealed significantly higher titers in the KE-VAC (hexapro) group than in the control group (Supplementary Fig. 9b). Additionally, the neutralizing assay assessed titers against seven mutations, including WT and variants Alpha, Beta, N501Y + D614G, Gamma, Kappa, and Delta. The results revealed high neutralizing titer values against the WT SARS-CoV-2 and all tested variant strains in the serum collected from mice inoculated with KE-VAC (hexapro) (Supplementary Fig. 9c).
To investigate the immune response induced by KE-VAC with spike protein antigen against SARS-CoV-2, BALB/c mice were immunised with antigens as stabilized omicron-spike trimers either alone or in combination with alum or KE-VAC (spike) on days 0 and 21. Fourteen days after the last immunisation, we collected serum and spleen samples to assess both humoral and cellular immune responses. Additionally, a subset of mice was challenged with 100LD50 of a mouse-adapted SARS-CoV-2 strain (Wuhan strain) to determine vaccine efficacy (Fig. 4a). KE-VAC (spike) group induced a 100% survival rate and a minimal decrease in body weight (4.8%) at 2 days post infection (dpi), demonstrating robust protection against SARS-CoV-2 infection. Although the group inoculated with KE-VAC group presented significantly lower lung viral titers than did the spike-alone group at 3 dpi, the viral load was comparable to that in the spike protein with alum group. Notably, at 5 dpi, the KE-VAC group exhibited clear viral clearance from the lungs. However, although the alum group also achieved 100% survival, these mice experienced a greater decrease in body weight (9.3%) at 3 dpi, and infectious viruses were detected in the lungs until 7 dpi (Fig. 4b, c).

a Timeline detailing the immunisation schedule, blood collection intervals, and SARS-CoV-2 challenge protocol. BALB/c mice were administered the spike protein (BA.2) alone or in combination with alum or KE-VAC on days 0 and 21. The components were intramuscularly injected into mice twice, with a 3-week interval. The doses were as follows: omicron spike protein, 3 µg; Alum, 100 µg; and m-TLR7/8a in nanoliposome, 20 µg. b Following viral infection, the infectious virus was titrated from the lung homogenates of the mice (n = 3). c Virus-infected mice were monitored for body weight (n = 5) and survival rate (n = 12) from 5 to 7 dpi. d ELISA was performed to determine antibody titers of spike-specific IgG (n = 19). e A serum neutralization assay was performed against live SARS-CoV-2 virus variants: Omicron BA.2, WT, alpha, beta, and delta strains, and the results are presented as GMTs (n = 18 to 20). For the measurement of GC immune cells and antigen-specific CD8+ T-cells, splenocytes were collected from the immunised mice. TFH cells (f) and GC B-cells (g) were characterised via FACS (n = 5). h ELISPOT was performed to determine antigen-specific T cell in splenocyte of immunised mice (n = 5). i Total polyfunctional CD8+ T-cells were analysed via FACS in the presence of monensin for 12 h, and each population of triple- or double-cytokine-positive CD8+ cells were analysed (n = 5). Data presented as mean ± SD. Statistical significance was analysed via one-way ANOVA/Bonferroni in (b–i). p values: ns, not significant; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001).
Analysis of antigen-specific immune responses revealed significantly higher spike-specific IgG titers in both the alum and KE-VAC (spike) groups than in the spike-alone group (Fig. 4d). Importantly, the KE-VAC (spike) significantly increased the serum neutralizing titers against SARS-CoV-2 virus of both homologous and heterologous virus strains (Fig. 4e). Increased formation of GCs was observed, highlighted by increased populations of TFH cells and GC B-cells in the spleen, indicating a superior humoral immune response (Fig. 4f, g). Regarding the role of CD8+ cytotoxic T lymphocytes (CTLs) in viral clearance, the KE-VAC (spike) group increased significantly more IFN-γ+ spot-forming units (SFUs) in splenocytes of immunised mice upon stimulation with the homologous variant than those of both the spike-alone and spike-alum groups (Fig. 4h and Supplementary Fig. 10 and 11). However, SFUs by stimulating heterologous variants were comparable across all groups. Flow cytometry analysis revealed an increased proportion of polyfunctional CD8+ T-cells in the KE-VAC group (triple or double positive for IFN-γ, IL-2 and TNF-α), indicating an enhanced CTL response capable of rapid viral clearance by killing virus infected cells (Fig. 4i). Furthermore, multiple cytokine secretion by CD8+ T-cells was induced against multiple SARS-CoV-2 variants by KE-VAC (Supplementary Fig. 12). These results suggest that the KE-VAC (spike) platform is highly effective in enhancing cross-protection and CTL responses against multiple variants, culminating in the rapid elimination of infectious viruses from the lungs.
Protective immunity against various subtypes of influenza viruses
As current influenza vaccines using variable surface antigens are not suitable for providing effective protection against unpredictable influenza pandemic outbreaks, many approaches have been investigated to develop new vaccines that offer broad cross-protection as well as convenient preparation and administration. As influenza matrix protein 2 (M2) which is well conserved across influenza strains and the stalk domain (HA2) of hemagglutinin (HA) which is another potent conserved region have been considered as attractive targets for inducing cross-protection against influenza A subtypes31,32. Considering these observations, we hypothesized that a fusion vaccine based on sM2 and HA2 (sM2HA2) would confer significant cross-protection. However, the drawback of these M2- and HA2-based vaccines is their low immunogenicity which necessitates the use of appropriate adjuvants to enhance their efficacy33,34. To overcome the low immunogenicity of the sM2HA2 antigen and enhance its protective ability against influenza virus, we integrated the sM2HA2 antigen into our KE-VAC platform and evaluated its efficacy. The groups of mice were intramuscularly (i.m.) immunised on days 0 and 14 with PBS alone (control), sM2HA2 alone, sM2HA2 with alum, nanoliposome (m-TLR7/8a), or KE-VAC (sM2HA2). To evaluate the effects inoculation of KE-VAC, IgG levels in the serum were measured via ELISA after the plates were coated with sM2HA2 protein or sM2 or HA2 peptides (Fig. 5a)35. The serum IgG responses specific to sM2HA2, sM2, and HA2 were elicited in the sM2HA2-immunised mice with the KE-VAC (sM2HA2) at markedly higher levels than those observed in the sM2HA2 alone, sM2HA2 with alum, or nanoliposome (m-TLR7/8a) groups (Supplementary Fig. 13 and 14). However, sM2HA2 alone did not significantly affect the level of IgG specific to sM2HA2, sM2, or HA2. Moreover, IgG isotypes specific to sM2 or HA2 were assessed to validate the above results. Similar to the previous results, sM2HA2 alone did not significantly affect the levels of IgG1 and IgG2a isotypes, whereas compared with the sM2HA2-immunised group and the other two adjuvant groups, the KE-VAC (sM2HA2) group significantly increased the robust levels of IgG1 and IgG2a (Supplementary Fig. 14). These results indicate that the enhanced humoral immune responses observed in the sM2HA2 group were primarily attributed to the combination with KE-VAC (sM2HA2).

a Timeline detailing the immunisation schedule, blood collection intervals, and virus challenge protocol. The components were intramuscularly injected into mice twice, with a 2-week interval. The doses were as follows: sM2HA2 antigen, 15 µg; Alum, 100 µg; and m-TLR7/8a in nanoliposome, 20 µg. (b and c) Cytokine production by splenocytes after vaccination (n = 5 for PBS and Mitogen, and n = 10 for sM2HA2). (b) IFN-γ and (c) IL-4 production in splenocytes after vaccination was confirmed by ELISPOT. (d) Following viral infection, the infectious virus was titrated from mouse lung homogenates (n = 6). (e) Survival rates and body weight changes were observed in infected mice after infection with influenza variants H1N1, H5N2, H7N3, H9N2, and H3N2 (n = 7). (f) Haematoxylin and eosin (H&E) staining was conducted on H1N1- and H5N2-infected mouse lungs. The yellow arrows indicate infiltrated immune cells. (g) Timeline detailing the immunisation schedule, blood and spleen collection, and virus challenge protocol for confirming long-term protection. The virus challenge and analysis conducted 6 months after immunisation. (h) The ant-sM2HA2 IgG concentration in the serum was evaluated, and (i) a representative value at a 1:100 dilution was used (n = 7). (j and k) Cytokine production by splenocytes 6 months after vaccination (n = 6 for PBS and Mitogen, and n = 12 for sM2HA2). (j) IFN-γ and (k) IL-4 production in splenocytes after vaccination was confirmed by ELISPOT. (l) Survival rates and (m) body weight changes were observed in the mice after virus challenge (n = 7). Data presented as mean ± SD. Statistical significance was analysed via two-way ANOVA in (b, c, j and k) and one-way ANOVA in (i), at 7 days post injection in.(e, m). p values: ns not significant; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001).
Furthermore, an ELISPOT assay was conducted to investigate the efficacy of KE-VAC in inducing a cell-mediated immune response specific to sM2HA2, sM2, and HA2 in combination with the sM2HA2 antigen. After 21 days of initial immunisation, splenocytes were harvested from the mice and subjected to IFN-γ and IL-4 ELISPOT assays. IFN-γ is a characteristic Th1 cytokine produced by cytotoxic T lymphocytes, while IL-4 functions as a Th2 cytokine36. Compared with those from mice immunised with sM2HA2 alone or sM2HA2 with alum or nanoliposome (m-TLR7/8a), the splenocytes from mice immunised with KE-VAC (sM2HA2) presented significantly greater numbers of sM2HA2-, sM2- and HA2-specific IFN-γ-secreting splenocytes and IL-4-secreting splenocytes (Fig. 5b, c, and Supplementary Fig. 15). However, recombinant sM2HA2 alone did not elicit notable levels of either IFN-γ- or IL-4-producing splenocytes in response to stimulation with sM2HA2, sM2, or HA2. These findings suggest that, compared with the well-known adjuvant alum and nanoliposome (m-TLR7/8a), immunising with KE-VAC (sM2HA2) is superior in leading to a stronger cell-mediated response to antigens. Additionally, KE-VAC can intensify cell-mediated immune responses induced by sM2HA2.
Based on earlier findings (Fig. 5a–c), the vaccination of KE-VAC (sM2HA2) elicits both humoral and cell-mediated immunity. Next, we investigated the ability of KE-VAC (sM2HA2) to confer protection against various lethal influenza subtypes. For this purpose, vaccinated animals were challenged with 10 LD50 of mouse-adapted influenza subtype A viruses, including H1N1, H5N2, H7N3, H9N2, and H3N2. To investigate whether immunisation with the influenza vaccine or adjuvants could prevent viral replication, lung samples from vaccinated mice were collected at 3 and 5 dpi after challenge with H1N1 or H5N2 to measure viral titers. The mice immunised with KE-VAC (sM2HA2) presented significantly lower virus titers than those immunised with sM2HA2 alone or other alum- or nanoliposome (m-TLR7/8a)-adjuvanted mice did, which suggests the potent capacity of KE-VAC (sM2HA2) for better lung viral clearance (Fig. 5d). Surprisingly, the KE-VAC (sM2HA2) inoculated group of mice showed almost complete virus clearance at 5 dpi.
Furthermore, the survival rate and body weight loss of the mice were observed for 13 days post challenge. The results revealed that none of the mice in the control group survived lethal influenza infection (Fig. 5e). In contrast, the infected mice in the groups vaccinated with sM2HA2 alone presented significant body weight loss and 100% mortality rates, except for those infected with H1N1 and H3N2. Only 0–20% of the mice survived lethal infection with H1N1 and H3N2. However, the group that was immunised with sM2HA2 plus alum or nanoliposome (m-TLR7/8a) demonstrated 60–80% survival ability. Interestingly, KE-VAC (sM2HA2)-immunised mice exhibited 100% protection against lethal challenge with H1N1, H5N2, H7N3, H9N2, and H3N2. GC formation is a key feature of inducible bronchus-associated lymphoid tissue in response to influenza A virus infection37. Histopathological examination of lung tissue sections from vaccinated and challenged mice was conducted to assess the extent of immune cell infiltration. Therefore, the lung sections of control mice and sM2HA2-immunised mice analysed on day 5 after challenge revealed dense peribronchial structures containing infiltrating immune cells, as indicated by the yellow arrows in Fig. 5f and Supplementary Fig. 16. In contrast, the challenged mice that received the KE-VAC (sM2HA2) lacked these structures, similar to the control groups. Furthermore, even though the antigen was low immunogenic sM2HA2, KE-VAC showed a specific increase in cross-variant protective neutralizing antibodies against H1N1, H5N2, H9N2, and H7N3 (Supplementary Fig. 17). These findings suggest that combining sM2HA2 as an universal antigen with conserved domain with KE-VAC that can induce sufficient Ab-specific humoral and T cell-mediated immunity may be a promising approach for the cross-protection against various lethal influenza subtypes.
Long-lasting immunity and protection against influenza virus infection
Long-lasting immunity is mediated primarily by memory B-cells, a subset of B lymphocytes generated during the initial immune response to an antigen. The duration of the immune response and protection are crucial factors in determining the effectiveness of a vaccine38. Therefore, to determine long-lasting immunogenicity, the serum IgG level was detected via ELISA, followed by the collection of serum samples from the mice after 6 months of priming (Fig. 5g). Compared with the other groups, the KE-VAC (sM2HA2) group presented significantly higher levels of IgG specific to sM2HA2 (Fig. 5h, i), sM2 (Supplementary Fig. 18a) and HA2 (Supplementary Fig. 18b), even six months after the first immunisation. When the IgG isotypes were considered, the IgG1 and IgG2a levels specific to sM2 and HA2 (Supplementary Fig. 18c-f) were also significantly greater in the KE-VAC (sM2HA2) group. However, sM2HA2 inoculation alone did not induce any considerable level of serum IgG or any IgG isotypes. In addition, the long-term cell-mediated immune response was also examined by isolating splenocytes six months after the first vaccination and conducting an ELISPOT assay. Interestingly, mice vaccinated with KE-VAC presented more antigen-specific IFN-γ- and IL-4-secreting cells than the other groups did (Fig. 5j, k). The results demonstrated that the KE-VAC (sM2HA2) elicits long-lasting humoral and cell-mediated immune responses, which persist for at least six months following the first immunisation (Supplementary Fig. 19). To evaluate the protective efficacy against H1N1 after 6 months of final vaccination, the mice were challenged with H1N1, and their body weights and survival rates were monitored for 13 days. As shown in Fig. 5m, all the mice in the control group and sM2HA2 only group presented more than 20% body weight loss by 8 and 10 days after the challenge, respectively. However, alum or nanoliposome (m-TLR7/8a)-immunised mice demonstrated approximately 80% protection against lethal infection. In contrast, the KE-VAC (sM2HA2) group showed 100% survival even 6 months after vaccination (Fig. 5l), and body weights started to recover after 7 dpi (Fig. 5m). These results indicated that KE-VAC (sM2HA2) could confer long-term protection against influenza A viruses, and the findings of this study suggest the potential utility of integrating sM2HA2 and KE-VAC in the development of a novel and improved influenza vaccine.
KE-VAC induces protective immunity against SFTSV in ferrets
To assess the efficacy of adjuvanted SFTSV vaccines, groups of ferrets (n = 6/group) were intramuscularly administered an inactivated SFTSV vaccine with or without the TLR7/8a adjuvant (KE-VAC). The experiments utilized an aged ferret model, which mimics the immune response of an immunocompromised person39,40. The study included a mock-PBS group (G1), an inactivated CB1/2014 SFTSV vaccine (30 μg) alone group (G2), and a KE-VAC (inactivated SFTSV) group (G3). These were administered twice with a 2-week interval (Fig. 6a). Sera were collected 2 weeks after each priming and boosting immunisation to evaluate neutralizing antibody production, with subsequent collections at 2-week intervals up to 30 weeks.

a Timeline detailing the immunisation schedule, blood collection intervals, and SFTSV challenge protocol. Ferrets were administered a mock-PBS, an inactivated SFTSV vaccine alone, or an inactivated SFTSV with Alum/nanoliposome (KE-VAC). The components were intramuscularly injected into ferret twice, with a 2-week interval. The doses were as follows: Inactivated SFTSV, 30 µg; Alum, 100 µg; and m-TLR7/8a in nanoliposome, 20 µg. b Changes in the serum neutralizing titers against live virus of ferrets were tracked at 2-week intervals for 30 weeks after immunisation (n = 5). c and d IFN-γ production in PBMCs immediately before the challenge was confirmed by an ELISPOT assay (c), with a representative image shown in (d) (n = 2). e On days 2, 4 and 6 after the SFTSV challenge, organ tissues from three ferrets per group were collected, and the viral load was quantified via qRT‒PCR (n = 3). f-h Health monitoring of ferrets after challenge included measuring changes in body weight (f), body temperature (g), and platelet count (h) until 6 dpi (n = 6). i Serum neutralizing titers were measured at various time points after the challenge (n = 6). Data presented as mean ± SD. Statistical significance was analysed via unpaired two-tailed t test in (e and g). p values: ns not significant; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001).
The data revealed robust induction of long-term neutralizing antibody activities for up to 16–18 weeks after priming. At 2 weeks after the first immunisation, the G2 group exhibited serum neutralizing titers of 40-160, peaking at 6 weeks with a serum neutralizing titers of 320. Meanwhile, the G3 group, which received the SFTSV vaccine with KE-VAC (inactivated SFTSV), displayed serum neutralizing titers of 80-160, peaking at 4 weeks post-priming, compared to the mock-PBS (G1) group (Fig. 6b). In both G2 and G3 groups, serum neutralizing titers gradually declined after reaching their peak, falling below the detection limit at 18 weeks and 20 weeks after priming, respectively (Fig. 6b). At 30 weeks post-priming, we assessed the ability to induce antigen-specific T-cell responses in ferrets from the SFTSV vaccine alone (G2) or KE-VAC (G3) groups using an IFN-γ ELISPOT assay (Fig. 6c, d). Results showed that KE-VAC (G3) group had significantly higher SFTSV-specific T-cell responses compared to the SFTSV vaccine alone (G2) group. (Fig. 6c, d).
To evaluate protective efficacy, ferrets from the mock-PBS (G1), SFTSV vaccine alone (G2), and KE-VAC (G3) groups were challenged with SFTSV (CB1/2014) at a dose of 106.0 TCID50. All ferrets were monitored for clinical signs of infection, body weight and temperature changes, platelet counts, serum neutralizing, and viral titers until 6 days post-infection (dpi). After the challenge, the G1 group, showed an increasing viral load in the serum, while viral RNA was detected only at 2 dpi in the G2 and G3 groups (Fig. 6e). Additionally, viral RNA was detected in all tissues collected at 4 and 6 dpi in G1 group, whereas in the G2 group, viral RNA was detected in PBMC, spleen, and bone marrow only at 4 dpi (Fig. 6e). In the G3 group, viral RNA was not detected in any tissues, indicating more rapid virus clearance compared to the SFTSV vaccine alone (G2) group (Fig. 6e). Both G2 and G3 groups showed no significant changes in body weight, temperature, or platelet counts, whereas mock-PBS (G1) ferrets experienced high fevers, 20% weight losses, and severe thrombocytopenia from day 4 after the SFTSV challenge (Fig. 6f–h). Furthermore, both G2 and G3 groups exhibited neutralizing activity until 6 dpi, with no significant difference in serum neutralizing tirers between the two groups (Fig. 6i).
Taken together, although the inactivated SFTSV vaccine alone can induce robust neutralizing antibodies, the KE-VAC platform significantly enhances the SFTSV-specific immune response. This enhancement provides sufficient protection against the lethal SFTSV challenge and enables rapid virus clearance even after long-term vaccination.
Discussion
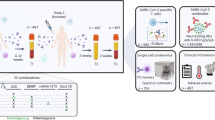
Here, we developed a kinetically engineered vaccine (KE-VAC) that (1) allows the presentation of antigen information for a long period and sustained immune stimulation with m-TLR7/8a without exhausting the APCs; (2) gradually supplies the antigen and adjuvant to the lymph nodes (LNs) to promote TFH, GC B-, and T-cell differentiation; (3) strongly induces both germinal centre and T-cell-related immune responses for long-term and broad protection (Fig. 7). The development of KE-VAC represents a significant advancement in vaccine adjuvants based on inspiration from natural infections. In natural infections, a more robust immune response often occurs due to two factors: local lung immunity resulting from the virus’s mucosal entry route and sustained viral replication that continuously stimulates the immune system, generating a persistent response41,42,43,44.

a Multiscale spatiotemporal immune-modulated KE-VAC by alum-assisted nanoliposome (m-TLR7/8a). b Summary of the therapeutic effect of KE-VAC in infectious diseases.
In the future research, combining this continuous stimulation with the capacity to induce local immunity through mucosal vaccination would likely yield even better outcomes.41,42,43,44 Since KE-VAC is an intramuscular vaccine containing alum, our approach focuses on providing continuous systemic support by mimicking viral replication kinetics, and we successfully achieved this through modulation at the cellular and tissue levels. Through a dual modulation approach at both the cellular and tissue levels, KE-VAC effectively orchestrates cytotoxic T lymphocyte (CTL) and GC immune responses, enhancing the vaccine’s capacity for prolonged protection against infectious diseases. Notably, our platform has demonstrated superior efficacy compared with traditional vaccine methods and mRNA vaccines, showing extended immunity against SARS-CoV-2, influenza, and SFTS viruses in animal models.
To design a novel vaccine platform, we synthesised nanoliposome (m-TLR7/8a), which could be slowly activated in immune cells and prolong its action and antigen presentation ability at the cellular level without exhaustion of APCs. The nanoliposome (m-TLR7/8a) can provide sustained stimulation of APCs and prolong antigen presentation on immune complexes and interaction time with TFH cells to promote GC-related immune responses with delayed exhaustion in addition to the significant enhancement of T-cell responses. Additionally, the engineered KE-VAC system facilitated the interaction with negatively charged nanoliposomes (m-TLR7/8a) and various antigens (such as recombinant proteins or inactivated viruses), subsequently released antigens and nanoliposomes (m-TLR7/8a) into the body, enhancing TFH cell, short- and long-term IgG, GC B-cell, and plasma B-cell responses at the macroscopic level inspired by the natural infection profile. This sustained response is achieved through multidimensional modulation at macroscopic and cellular levels, as KE-VAC induces a gradual increase in GC B cells post-vaccination, surpassing the effects of individual alum or nanoliposome (m-TLR7/8a) treatments. Moreover, by incorporating modulation at the cellular level, KE-VAC significantly enhances IgG production compared to systems lacking this feature (Supplementary Figs. 7 and 8). Our KE-VAC suggested here should be differentiation from the previous research that used osmotic pumps and hydrogels to supply antigens and adjuvants in a gradual manner in that KE-VAC was designed based on FDA-approved alum and novel masked TLR7/8a to modulate immune response kinetically at both the cellular and tissue levels17,18,19,20,21,22. The engineered KE-VAC system, which releases antigens and nanoliposomes (m-TLR7/8a) into the body, could enhance TFH cell, short- and long-term IgG, GC B-cell, and plasma B-cell responses at the macroscopic level inspired by the natural infection profile (Fig. 7a). Owing to the harmonised cellular and tissue modulation capabilities of KE-VAC, the cellular immune response, which is crucial for early and rapid defence, is significantly enhanced compared with that of conventional vaccines. Additionally, the GC response greatly strengthens the humoral immune response, which is vital for long-lasting protection (Fig. 7a).
To evaluate the efficacy of KE-VAC against natural virus infection in animal models, we tested it against three viruses: SARS-CoV-2, influenza, and SFTS. In the evaluation of immune function in the SARS-CoV-2 model, in which mice were vaccinated with the Omicron spike protein and KE-VAC, a 100% survival rate was achieved, alongside a significant increase in NAbs and polyfunctional CD8+ T-cells for the original strain and heterologous variants (wild type, alpha, beta, and delta). For influenza, we used sM2HA2, a universal antigen that targets conserved domains, and injected it into mice to induce a response against various strains. This resulted in a 100% survival rate for mice challenged with each of five strains (H1N1, H5N2, H7N3, H9N2, and H3N2). Furthermore, a virus challenge conducted 192 days after the initial vaccination resulted in high secretion levels of IFN-γ and IL-4. Through the immune responses induced by KE-VAC, mice recovered their weight rapidly after viral infection, returning to their baseline weight within two weeks whereas groups treated with other vaccine modalities showed persistent weight deficits (Fig. 5e, m). For SFTS, we tested KE-VAC in an aged ferret model, which exhibits an immune response similar to that of an immunocompromised person. A virus challenge was conducted 30 weeks after the initial vaccination. KE-VAC demonstrated high protection efficiency, with no virus detected in the serum, PBMCs, or any of four organs. In particular, during the early stages of infection, KE-VAC facilitated rapid recovery by promoting high viral clearance in immune-active sites such as the spleen, PBMC, and bone marrow (Fig. 6e). This finding indicates a robust and lasting immune response against SFTSV long after inoculation. Vaccination with KE-VAC is expected to provide recipients with an immune profile similar to those of patients who have recovered from natural infections, an outcome not typically achieved with conventional vaccines (Fig. 7b). Moreover, KE-VAC can offer enhanced protection while maintaining safety, unlike natural infections (Fig. 7b). In practical application in animal studies, KE-VAC demonstrated superior and long-lasting protective effects and significantly improved survival rates in healthy states compared with existing vaccine methods across models of three diseases: SARS-CoV-2, severe fever with thrombocytopenia syndrome virus (SFTSV), and influenza virus (Fig. 7b).
Through challenge and analysis of three distinct disease models, we observed that KE-VAC facilitates rapid recovery from initial infections via T cell responses and, from a long-term perspective, offers disease prevention through a durable immune response mediated by germinal centre activation. Notably, in experiments with influenza and SARS-CoV-2, KE-VAC demonstrated the ability to elicit cross-reactive neutralizing responses against multiple heterologous viral variants, which represents a significant advantage for its potential application as a practical vaccine (Fig. 7b). Moreover, the adaptability of KE-VAC to diverse antigens confers a notable advantage, facilitating swift responses during disease outbreaks. This inherent versatility positions KE-VAC as a potent candidate for combating emerging infectious threats. The widespread efficacy of KE-VAC across various pathogens and vaccination schedules highlights its potential as a versatile tool in disease control efforts and the mitigation of emerging infectious risks.
Methods
Materials and reagents
Alhydrogel adjuvant 2% (alum, cat. no. vac-alu-50) and Adju-Phos adjuvant (AdjuPhos, cat. no. vac-phos-250) were purchased from InvivoGen (San Diego, CA, USA). Ovalbumin from chicken egg white (OVA, cat. no. A5503) and cholesterol (Chol, cat. no. C3045) were purchased from Sigma‒Aldrich (St. Louis, MO, USA). We purchased 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC, cat. no. 850375 C) and 1,2-dipalmitoyl-sn-glycero-3-phospho-(1’-rac-glycerol) (DPPG, cat. no.840485 C) from Avanti Polar Lipids (Birmingham, AL, USA). Resiquimod (R848, cat. no. orb304065) was purchased from Biorbyt (Cambridge, UK). CPG-ODN (ODN1668, cat. no. tlrl-1668) and poly I:C (cat. no. tlrl-pic) was purchased from Invitrogen (USA). Red blood cell lysis buffer (cat. no. 420301) was purchased from BioLegend (San Diego, CA, USA). Recombinant mouse GM-CSF (cat. no. 415) was purchased from R&D Systems (Minneapolis, MN, USA). Phosphate-buffered saline (PBS, cat. no. SH30256) was purchased from Cytiva (GE Healthcare, South Logan, UT, USA), and 10X Dulbecco’s phosphate-buffered saline (DPBS, cat. no. LB001-01) was purchased from Welgene (Gyeongsan, Korea).
Animals, viruses, and antibodies
The KE-VAC mouse study conducted in this research was ethically reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of the Sungkyunkwan University School of Medicine (Protocol Nos: SKKUIACUC2020-06-17-1 and SKKUIACUC2022-05-09-1). This committee is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC International) and adheres to the Institute of Laboratory Animal Resources (ILAR) guidelines. 315 stocks of C57BL/6 (Orient Bio, C57BL/6NCrlOri) and 120 stocks of BALB/C mice (Orient Bio, BALB/cAnNCrlOri) (6- to 8-week-old females) were procured from Orient Bio (Korea) and 45 stocks of C57BL/6 mice (DBL, C57BL/6NTac) (6- to 8-week-old females) were procured from DBL (Korea). All the animals were housed in individually ventilated cages under controlled humidity (30–70%) and temperature (21–26 °C) conditions and were subjected to a 12-h light‒dark cycle. The mouse study for influenza was performed in strict compliance with the National Institutes of Health’s (NIH) Guide for the Care and Use of Laboratory Animals. The Institutional Animal Care and Use Committee (IACUC) of Chungnam National University approved all the mouse procedures (Approval Number: 202307A-CNU-113). 340 stocks of BALB/C mice (Hanil Laboratory Animal Center, BALB/cAnNHsd) (6- to 8-week-old females) were procured from Hanil Laboratory (Korea). All efforts were made to minimise animal suffering. The ferret study was approved by the Medical Research Institute, a member of the Laboratory Animal Research Center of Chungbuk National University (LARC) and was conducted in strict accordance with the relevant policies regarding animal handling mandated under the Guidelines for Animal Use and Care of the Korea Center for Disease Control (K-CDC). Viruses were handled in an enhanced biosafety level 3 (BSL3) containment laboratory as approved by the Korean Centers for Disease Control and Prevention (KCDC-14-3-07). 18 stocks of 3-year-old male ferrets were purchased from ID bio (Korea).
All mice obtained from Orient Bio, DBL, and Hanil Laboratory were supplied and separately bred in SPF (Specific Pathogen-Free) facilities, including both experimental and control groups. Ferrets were supplied from Laboratory animal facilities, and all individuals, including those in the experimental and control groups, were separately bred in a barriered environment.
Detailed information concerning the antibodies used in this study, including the fluorescence antibody type, manufacturer, clone, and catalogue number, is provided in Supplementary Table 1.
For the SARS-CoV-2 experiment, SARS-CoV-2 spike-stabilized trimers from B.1.1.529 (Omicron variant, cat. no. REC32008) were purchased from the Native Antigen Company. SARS-CoV-2 variants (Wuhan strain; hCoV-19/South Korea/NMC-02/2020, Alpha (B.1.1.7); hCoV-19/South Korea/NMC-nCoV-07/2021, Beta (B.1.351); hCoV-19/South Korea/NMC-nCoV-08/2021, Delta (B.1.617.2); hCoV-19/South Korea/NMC-nCoV-11/2021, BA.2/omicron (B.1.1.529); hCoV-19/South Korea/CBNU-nCoV-55/2021); and mouse-adapted SARS-CoV-2 (originating from the Wuhan strain) were used for the serum neutralization assay and viral challenge experiment, respectively. The viruses were propagated in a Vero-E6 monolayer (ATCC no. CRL-1586) via DMEM (Gibco, cat.no. 11995073) for 1 h at 37 °C in 5% CO2. After incubation, the medium was changed to 2% FBS-supplemented DMEM (Gibco). Three days after infection, the viruses were harvested from the infected Vero-E6 cells, centrifuged at 1,000 × g for 20 min, and then stored at −80 °C until use. All the experiments were performed under BSL3 conditions. For inactivation of the virion, SARS-CoV-2 in the culture supernatant was treated with β-propiolactone for 72 h at 4 °C. The inactivated virions were purified via ultracentrifugation at 100,000 × g for 2 h in a 30% sucrose density gradient. BCA protein assay kits (Thermo Fisher, cat.no 23227) were used for the quantification of purified virions.
For the influenza experiment, five mouse-adapted low-pathogenic strains—AI A/Puerto Rico/8/34 (H1N1), A/Aquatic bird/Korea/W81/2005 (H5N2), A/Aquatic bird/Korea/W44/2005 (H7N3), A/Chicken/Korea/116/2004 (H9N2), and A/Philippines/2/2008 (H3N2)—were used. All influenza viruses were propagated in pathogen free 10 to 12-day-old embryonated chicken eggs and titrated by determining TCID50 values on Madin-Darby Canine kidney epithelial cells (MDCK; ATCC no. CCL-34)45. These five mouse-adapted low-pathogenic strains were then stored at −80 °C and used to infect BALB/c (Hanil Laboratory Animal Center, BALB/cAnNHsd) mice for the challenge experiments.
For the SFTS virus experiment, the SFTS virus (CB1/2014) was passaged onto confluent monolayers of Vero E6 cells (ATCC no. CRL-1586) in DMEM (Gibco, cat. no. 11995073) supplemented with 2% FBS (Gibco, cat. no. 10082-147), penicillin (100 U ml−1), and streptomycin (100 μg ml−1; Gibco, cat. no. 15140122) and incubated at 37 °C with 5% CO2. The cell culture supernatant was collected at 5 dpi and stored at −80 °C as the working virus stock for this study. Viral infectivity titers were determined through an immunostaining assay, with the TCID50 calculated via an in-house-generated monoclonal NP antibody against SFTSV via an immunofluorescence assay46.
BMDC culture for in vitro experiment and activation assay
In all in vitro experiment, mouse BMDC was adapted for experiment. Mouse BMDCs were produced from the bone marrow of female C57BL/6 mice (Orient Bio, C57BL/6NCrlOri) aged 6 to 8 weeks. The femurs and tibias were extracted, and the bone marrow was flushed out using RPMI 1640 medium (Gibco, cat. no. A1049101) and a 26-gauge syringe. To eliminate red blood cells (RBCs), RBC lysis buffer (BioLegend) was used. After several washes, the cells were suspended in RPMI medium (24 mL) supplemented with mGM-CSF (20 ng mL−1; R&D systems, cat. no. 415) and seeded at a concentration of 2.5 × 106 cells per well in a 6-well culture plate. On the second day, the medium containing mGM-CSF (20 ng mL−1) was replaced after thorough washing with PBS to remove nonadherent cells. On the fourth day, fresh medium containing mGM-CSF (20 ng mL−1) was added. On the sixth day, the cells had differentiated into immature BMDCs, which were utilized for subsequent experiments.
Cell lines
To titrate influenza viruses, Madin-Darby Canine kidney epithelial cells (MDCK; ATCC no. CCL-34) was adopted in DMEM (Gibco, cat. no. 11995073, USA) supplemented with 10% FBS (Gibco, cat. no. 10082-147, USA), penicillin (100 U ml−1), and streptomycin (100 μg ml−1; Gibco, cat. no. 15140122) and incubated at 37 °C with 5% CO2 condition. Vero-E6 monolayer (ATCC no. CRL-1586) was adopted for the propagation of SARS-CoV-2 and SFTS viruses with DMEM (Gibco, cat. no. 11995073, USA) supplemented with 2% FBS (Gibco, cat. no. 10082-147), penicillin (100 U ml−1), and streptomycin (100 μg ml−1; Gibco, cat. no. 15140122) and incubated at 37 °C with 5% CO2 condition. To quantify EC50 value of TLR 7/8 agonist, HEK-BLUE hTLR8 cell line (Invitrogen, cat. no. hkb-htlr8) was purchased and cultured with DMEM (Gibco, cat. no. 11995073, USA) supplemented with 10% FBS (Gibco, cat. no. 10082-147, USA), penicillin (100 U ml−1), streptomycin (100 μg ml−1; Gibco, cat. no. 15140122), and normocin (100 μg ml−1; Invitrogen, cat. no. ant-nr-05) and incubated at 37 °C with 5% CO2 condition
BMDC, naïve T cell, and B cell co-culture
To co-culture DCs, T cells, and B cells, we prepared BMDCs using the aforementioned method. Spleens were harvested from 6-week-old C57BL/6 mice (Orient Bio, C57BL/6NCrlOri), and T cells (Naive CD4 + T Cell Isolation Kit, mouse, BD cat. no. 130-104-453) and B cells (B Cell Isolation Kit, mouse, BD cat. no. 130-090-862) were isolated using microbeads. A total of 2 × 104 BMDCs were seeded into a 96-well cell culture plate with RPMI-1640 media (Gibco, cat. no. A1049101) supplemented by 10% FBS (Gibco, cat. no.26140079), penicillin (100 U ml−1) and incubated at 37 °C with 5% CO2. The cells were treated with OVA at a concentration of 10 µg mL−1 and each adjuvant at a concentration of 1 µg mL−1. After 24 h, 4 × 104 T cells and B cells were added to each well. Following an additional 24-hour incubation, single cells were analysed using flow cytometry, and cytokine levels were quantified using ELISA.
Enzyme-linked immunosorbent assay (ELISA) for cytokine measurement
Prepared BMDCs were treated with the indicated immune stimulants and incubated for indicated time. The cell culture supernatants were centrifuged at 10,000 × g for 10 min at 4 °C, after which the supernatants were collected. All the collected supernatants were analysed with mouse IL-12p70 (BD, cat. no. 555256), IL-6 (BD, cat. no. 555240), TNF-α ELISA kits (BD, cat. no. 560478), IL-21 (Thermo Fisher, cat. no. BMS6021) and IL-23 ELISA kits (Thermo Fisher, cat. no. BMS6017) following the manufacturer’s instructions.
Preparation of nanoliposome (m-TLR7/8a)
Nanoliposome (m-TLR7/8a), which is a nanosized liposome, was fabricated via a thin-film hydration method followed by tip sonication. DOPC, DPPG, Chol, and m-TLR7/8a (60:10:15:15) were dissolved in chloroform. The synthetic scheme and characterization of chemical structure for m-TLR7/8a are described in Supplementary Fig. 1 and 2. The organic solvent was removed via rotary evaporation at room temperature (RT) for 30 min or more. The thin film was hydrated with PBS for 2 h, and the solution was sonicated by tip sonication in an ice bath. Liposomes were then extruded through a 0.2-μm pore size filter membrane via a Mini Extruder (Avanti polar lipids). The hydrodynamic size and zeta potential of the filtered solution were analysed via dynamic light scattering (ELS-Z electrophoretic light scattering photometer, ELSZ-2000 series, Otsuka, Osaka, Japan). The encapsulated m-TLR7/8a was quantified via ultraviolet‒visible light (UV‒vis) spectrometry (UV-1800, SHIMADZU, Kyoto, Japan). The m-TLR7/8a containing nanoliposome was sonicated (Branson Ultrasonics™) in ethanol condition and calculated by serially diluted UV absorption standard curve. (Supplementary Fig. 20b)
Preparation of nanoliposome (R848)
Nanoliposome (R848), which is a nanosized liposome, was fabricated via a thin-film hydration method followed by tip sonication. DOPC, DPPG, and Chol (60:10:30) were dissolved in chloroform. In this step, R848 also dissolved in chloroform. The organic solvent was removed via rotary evaporation at room temperature (RT) for 30 min or more. The thin film was hydrated with PBS for 2 h, and the solution was sonicated by tip sonication in an ice bath. Liposomes were then extruded through a 0.2-μm pore size filter membrane via a Mini Extruder (Avanti polar lipids). After the fabrication, the nanoliposome (R848) concentrated by 30 kDa MWCO centrifugal filter (Amicon® Ultra Centrifugal Filter). The encapsulated R848 was quantified via ultraviolet‒visible light (UV‒vis) spectrometry (UV-1800, SHIMADZU, Kyoto, Japan). The R848 containing liposome was sonicated (Branson Ultrasonics™) in ethanol condition and calculated by serially diluted UV absorption standard curve. (Supplementary Fig. 20a)
Preparation of mLNP (RNA)
LNP containing target CleanCap OVA mRNA (TriLink cat. no. L-7210-1000) was synthesized using microfluidic device (The NanoAssemblr Ignite Precision Nanosystems). 100 mg mL−1 of ALC-0315 (Ionizable lipid, cat. no. HY-138170, MCE), 50 mg mL−1 of ALC-0159 (PEGylated lipid, MCE, cat. no. HY-138300), 10 mg mL−1 of DSPC (1,2-distearoyl-sn-glycero-3-phosphocholine, helper lipid, Avanti Polar Lipids, cat. no. 850365P-1g,), and 10 mg mL−1of cholesterol (sterol lipid, Sigma-Aldrich, cat. no. C8667-1G) was dissolved in ethanol (organic phase) with 43/5/9/20 weight ratio and 0.1 mg mL−1 of mRNA was dissolved in pH4 buffer (100 mM sodium citrate, aqueous phase). Using microfluidic device, organic phase and aqueous phase was mixed with 3: 1 volume ratio. Synthesized LNP was dialyzed using Slide-A-Lyzer™ Dialysis Cassettes, 10 K MWCO (Thermo Fisher, cat. no. 66380) in 1X PBS overnight and stored in 4 °C before use.
GILT-dependent m-TLR7/8a cleavage
Nanoliposome (m-TLR7/8a) (79.5 mM, 100 μl) was capped in 12-14 kDa MWCO dialysis membrane (Spectra/Por™) and prepared in a 50 ml tube. Cysteine (1 μM or 200 nM, 50 μl, Sigma-Aldrich) was dissolved in PBS with or without GILT (2.5 μg, recombinant human IFI30, RayBiotech) and added to the tube. After 2, 4, 6, 24, 48, and 72 h, the outer PBS were collected, and released amount of R848 was analysed by UV-vis.
HEK-BLUE hTLR8 reporter cell and Quanti-BLUE assay
Pre-cultured HEK-BLUE hTLR8 reporter cell (Invitrogen) prepared in 96-well cell culture plate. R848 and nanoliposome (m-TLR7/8a) were gradually treated and incubated for 24 h. The cell supernatant was collected and mixed with Quanti-BLUE solution (Invitrogen) in 96-well plate. The optical density (OD) of mixed solution were detected at 650 nm.
In vitro alum dissociation, interaction, and release profile
All alum used in the study was Alhydrogel purchased from InvivoGen. The alum was placed in buffers of various pH values ranging from 1 to 13. HCl was used for the 1-6 pH range buffer, and NaOH was used to prepare the 8-13 pH range buffer. After being placed in buffer, the samples were rotated for 24 h, and the zeta potential of alum was analysed via dynamic light scattering (DLS, ELS-Z electrophoretic light scattering photometer) for charge transition analysis. Proteins were conjugated to Alexa Fluor 647 (AF647) via NHS labelling (Invitrogen, cat. no. O34784), and liposomes were generated with DiD fluorescence (Invitrogen, cat. no. D7757) (liposome (DiD)) mixed with alum and rotated at room temperature (r.t.) for 30 min to enable adsorption. The samples were subsequently centrifuged at 10,000 × g for 10 min to remove pellet alum-adhered protein and liposomes, and the supernatant was replaced with various pH buffers for dissociation, unless otherwise noted, in PBS. All replaced supernatants were analysed for fluorescence using a SpectraMax M5 (Molecular Devices, San Jose, CA, USA), and the results were normalized to samples that contained no alum. The release profile was measured via the same method but at the indicated time points of 0.5, 1, 3, 6, and 12 h and 1, 3, 7, and 14 days after mixing and rotation at 37 °C.
DC exhaustion after sustained stimulation
To evaluate the effect of sustained stimulation on DC exhaustion, 1 × 106 BMDCs were seeded into 6-well plates. The DCs were subjected to the following conditions: (1) simultaneous treatment with OVA (20 µg mL−1) and R848 (1 µg mL−1), (2) OVA (20 µg mL−1) and sequential treatment with R848 at 0, 4, and 8 h, and (3) OVA (20 µg mL−1) treatment with nanoliposomes containing masked TLR7/8 agonist (m-TLR7/8a) instead of R848. At 12 h after the initial treatment, an additional dose of R848 (1 µg mL−1) and OVA (20 µg mL−1) was applied to the DCs. After 24 h, cell supernatants were collected and analysed using ELISA to measure cytokine levels.
Model antigen (OVA) vaccination and analysis
For the model antigen (OVA) vaccination experiment, six-week-old female C57BL/6 mice (Orient Bio, C57BL/6NCrlOri) were purchased from Orient Bio. C57BL/6 female mice were treated with KE-VAC or the single components. The mice were intramuscularly administered 10 μg of OVA, 100 μg of alum, nanoliposome (m-TLR7/8a) containing 79.5 nmol of m-TLR7/8a, or combination of them on days 0 and 14. In an experiment comparing KE-VAC with the mLNP, 5 μg for OVA mRNA containing LNP was immunised. The blood, spleen, and pLN of the mice were harvested from immunised mice and analysed.
Model study for evaluating the effects of sustained antigen and adjuvant vaccination
For the sustained vaccination experiment, six-week-old female C57BL/6 mice (Orient Bio, C57BL/6NCrlOri) were purchased from Orient Bio. C57BL/6 female mice were treated with KE-VAC or the single components. The mice were intramuscularly administered components as 3 different patterns: Increasing (Inc), Splitting (Spt), and Bolus (Bol). The mics in Inc groups inoculated by 0.15, 0.3, 0.6, 1.23, 2.51, 5.07, 10.14 µg of OVA with 0.2, 0.4, 0.8, 1.5, 3.1, 6.3, 12,7 µg of R848 at day 0, 2, 4, 6, 8, 10, 12. The mice in Spt groups administrated 2.86 µg of OVA and 3.6 µg of R848 for 7 times with 2 days interval. The mice in Bol groups inoculated by 25 µg of OVA and 20 µg of R848 at once. The 14-day vaccination cycle was repeated 2 time for all groups. During the vaccination, the blood was collected from mice and analysed at 14-day post injection. After the vaccination, the mouse pLN, spleen, and blood were harvested from the mice and analysed at 28-day post 1st injection.
SARS-CoV-2 (omicron spike) vaccination, viral challenge and analysis
For the SARS-CoV-2 vaccination and viral challenge experiment, 6-week-old female BALB/c mice (Orient Bio, BALB/cAnNCrlOri) were purchased from OrientBio (Korea). All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of the Institute for Basic Science (Approval number: IBS-2023-043) and performed under the guidelines. The mice were intramuscularly administered 0.5 μg of SARS-CoV-2 spike-stabilised trimer protein alone or in combination with 100 μg of alum or 79.5 nmol of m-TLR7/8a on days 0 and 21. Sera and splenocytes were collected on day 35. Fourteen days after the final vaccination, the mice were challenged intranasally with 100 LD50 of mouse-adapted SARS-CoV-2, and the survival rate and body weight were monitored for up to seven days. Mice that lost more than 25% of their body weight reached the experimental endpoint and then use inhalation of CO2 gas for euthanasia. To determine the infectious viral titers in the virus-infected lungs, the lungs of the challenged mice were harvested and frozen at −80 °C at 3, 5, and 7 days post infection. Homogenates were prepared from the frozen tissues via a TissueLyser II (Qiagen, Venlo, Netherlands) and centrifuged at 15000 × g for 10 min. Vero-E6 cells (ATCC no. CRL-1586) were treated with 10-fold serially diluted homogenates for 1 h at 37 °C. The homogenates were removed and incubated with DMEM supplemented with 2% FBS for 72 h at 37 °C. Cytopathic effects (CPEs) were monitored three days post infection, and the viral titers were calculated via the Reed–Muench method and expressed as the 50% tissue culture infective dose (TCID50).
SARS-CoV-2 vaccination and IgG assay
C57BL/6 female mice (DBL, C57BL/6NTac) aged 6 weeks were immunized twice with the KE-VAC or components: m-TLR7/8a in nanoliposome (m-TLR7/8a), (79.5 μmol), HexaPro antigen (priming) (1 μg); and HexaPro antigen (priming) (5 μg). One hundred microlitres of blood from immunized mice was collected on days 14 and 28 and then centrifuged at 10,000 × g for 10 min at 4 °C to separate the serum. For the IgG, IgG1, and IgG2c ELISAs, a 96-well plate was coated overnight with recombinant SARS-CoV-2 spike protein (2 μg mL−1). The plate was subsequently washed two times with 0.05% (v/v) Tween-20 and blocked for 3 h at 37 °C using a 5% (w/v) skim milk solution. Serum samples were loaded onto the plate and incubated for 2 h at 37 °C. To specifically detect IgG, IgG1, and IgG2c, secondary antibodies, including goat anti-mouse IgG (H + L)-HRP, rat anti-mouse IgG1-HRP, and goat anti-mouse IgG2c-HRP (SouthernBiotech), were added to each plate at a 1:6000 dilution. After 1 h of incubation at 37 °C, the plate was washed, and the proteins were detected using a TMB solution. The absorbance of the plate at 450 nm was measured using a microplate reader (VersaMax).
Production of SARS-CoV-2 Spike pseudovirus
Plasmids encoding the SARS-CoV-2 spike protein (Wuhan-Hu-1) were purchased from Sino Biological (pCMV3-SARS-CoV-2 Spike, VG40589-UT). The SARS-CoV-2 spike mutants were generated by site-directed mutagenesis or artificially synthesized DNA from the SARS-CoV-2 spike protein. SARS-CoV-2 pseudoviruses were produced by cotransfecting 293 T cells with pMDLg/pRRE (Addgene plasmid, cat. no. 12251), pRSV-Rev (Addgene plasmid, cat. no. 12253), pCDH-CMV-Nluc-copGFP-Puro (Addgene plasmid, cat. no. 73037), and plasmids encoding either version of the SARS-CoV-2 spike by using polyetherimide. Sixty hours post-transfection, culture supernatants containing SARS-CoV-2 spike pseudoviruses were harvested and filtered (0.45 μm pore size, S2HVU01RE, Millipore, California, USA), after which the copy numbers of the pseudoviruses were quantified via qPCR.
Quantification of viral copy numbers by quantitative reverse transcription PCR (qRT-PCR)
The RNA used in the experiment was extracted using TRIzol reagent (Thermo Fisher, cat. no. 15596018) or the RNeasy kit (Qiagen, cat. no. 74104), and cDNA synthesis was performed using SuperiorScript III Reverse Transcriptase (Enzynomics) with a specific primer set targeting the M segment of SFTSV. The forward primer used was SFTSV-M-F: AATTCACATTTGAGGGTAGTT, and the reverse primer used was SFTSV-M-R: TATCCAAGGAGGATGACAATAAT. Real-time PCR was performed using SYBR Green Supermix (Bio-Rad Laboratories) and the CFX96 real-time PCR detection system (Bio-Rad). The experimentally determined copy numbers were calculated as a ratio based on the standard control.
Influenza vaccination, viral challenge and analysis
For the influenza vaccination and viral challenge experiment, of 5-week-old female BALB/c (Hanil Laboratory Animal Center, BALB/cAnNHsd) mice were purchased from Hanil Laboratory Animal Center (Jeonju, Korea), and housed in a temperature- and light-controlled SPF environment and allowed free access to food and water. The mice were separated into five groups (n = 7 per each group). The i.m. vaccinations were administered to four groups of mice in each set two times at two-week intervals using 15 µl of sM2HA2 (15 µg) alone, Alum with sM2HA2, Nanoliposome with sM2HA2 or KE-VAC. One group from each set was exposed to the PBS (Sigma‒Aldrich, cat. no. 56064 C) treatment as a control. Before being inoculated, all mice were anaesthetised with ether, and the investigation was performed in a biosafety level (BSL)-2 facility. Serum samples were collected on days 21 and 180 (long-lasting) to assess immune responses. Mice that lost more than 25% of their body weight reached the experimental endpoint and then use inhalation of CO2 gas for euthanasia. The sera were separated by centrifugation and stored at -20 °C for further analysis. Spleens were collected on day 21 and day 180 to isolates splenocytes to analyse antigen-specific T-cell responses. For the viral challenge, the mice were infected intranasally one week after the last vaccination, with 10 MLD50 of the mouse-adapted low-pathogenic AI A/Puerto Rico/8/34(H1N1), A/Aquatic bird/Korea/W81/2005 (H5N2), A/Aquatic bird/Korea/W44/2005 (H7N3), A/Chicken/Korea/116/2004 (H9N2), and A/Philippines/2/2008 (H3N2) viruses in 20 μL of PBS. The mice were observed at predetermined intervals to assess survival and weight loss for 13 days. Mice exhibiting more than 20% body weight loss were considered as an experimental endpoint and were humanely euthanized using CO2 inhalation. Six mice of each group were euthanized at 3- and 5-days post-infection (dpi) to evaluate viral titers in the lungs.
Viral titres and histopathological analysis of lungs
Viral titers in the lungs were measured using 50% tissue culture infectious dose (TCID50) tests. Concisely, to eliminate cellular debris, the lung tissues were homogenized in PBS containing an antibiotic and antimycotic solution (Gibco) and centrifuged at 12,000 × g. The confluent MDCK cells were exposed to ten-fold serial dilutions of samples for one hour at 37 °C in a humid environment. The infected cells were kept for 72 h with an overlay medium containing L-1-tosylamide-2-phenylethyl chloromethyl ketone (TPCK) trypsin (Sigma-Aldrich, cat. no. 4370285). Hemagglutination assay (HA) was conducted following the observation of a cytopathic effect (CPE). The virus titers were determined using the Reed and Muench method and expressed as log10 TCID50 / lung tissue. Lung tissues were preserved in 10% formalin-containing neutral buffer as soon as they were collected, and samples were kept in a shaker for two days to prepare histopathology samples. Subsequently, the middle portion of the tissues was used to embed the samples in paraffin wax. Using a microtome, the slices were cut to a thickness of 4-6 mm, placed on slides, and stained with hematoxylin and eosin (H&E). Histological alterations were evaluated using light microscopy.
SFTSV vaccination, viral challenge and analysis
For the SFTSV vaccination and challenge, eighteen 3-year-old male ferrets were purchased from ID bio (Korea). In immunisation experiments on ferrets, ferrets were confirmed to have no antibodies against SFTSV. All procedures, including immunisation, blood collections and euthanasia, were performed under anaesthesia using xylazine (UNIBIOtech, cat. no. 326R02, Gyeonggido, Korea) and alfaxalone (Jurox, cat. no. 520635, Rutherford, Australia). The naïve aged ferrets were i.m. immunised with 300 μL PBS (G1, n = 6) or 30 µg of inactivated SFTSV (G2, n = 6) alone or in combination with 20 µg of TLR7/8a (G3, n = 6). Vaccines were administered two times (prime and boost) at 2-week intervals, and sera were collected at 2-week intervals until 30 weeks after each first immunisation. The ferrets were subsequently challenged i.m. with SFTSV (CB1/2014) at 106.0 TCID50 mL−1 at 30 weeks after the first immunisation. Clinical signs of infection (viral load, platelet counts, body weight, and body temperature) were monitored at 0, 2, 4, and 6 days post challenge. Haematological parameters were analysed via EDTA-treated whole-blood samples from infected animals using a Celltac haematology analyser (MEK-6550J/K, Nihon Kohden, Tokyo, Japan). Sera were collected from each ferret at 2-day intervals after infection, and peripheral virus titers were determined. Three animals per group were analysed at days 4 and 6 to collect tissue samples (liver, spleen, kidney, bone marrow, and PBMCs) with individual scissors. The viral loads in the collected tissues and sera were quantified via qRT‒PCR. The ferrets were housed in an ABSL3 facility within Chungbuk National University (Cheongju-si, South Korea) with a 12-h light/dark cycle and access to water and food. All animal care was performed strictly according to the animal care guidelines and experimental protocols approved by the Institutional Animal Care and Use Committee (IACUC) (approval number CBNUA-2130-23-02) of Chungbuk National University.
In vivo flow cytometry analysis
For preparing single-cell suspensions, the spleens and pLNs were disrupted mechanically and placed in medium containing collagenase D (1 mg mL−1; Sigma Aldrich). The mixtures were incubated in a shaking incubator at 37 °C for 30 min. The resulting cell solutions were filtered through 70-μm cell strainers and washed twice with PBS.
Single cells from the spleens or pLNs were stained with viability stain (BD) to segregate live cells and then stained with antibodies specific for T-cells (CD3, CD4, and CD8), memory T-cells (CD44 and CD62L), TFH cells (PD-1, ICOS, and CXCR5), GC B-cells (CD19, CD95 and GL-7), and plasma cells (CD20 and CD138).
To analyse antigen-specific multifunctional CD8+ T cells, intracellular staining was performed. Single cells obtained from the spleen and LN were stimulated by an S peptide pool or inactivated SARS-CoV-2 variants with 1.0 µg mL−1 GolgiPlug™ (BD cat. no 555029). The stimulated cells were washed twice with ice-cold PBS and then stained for surface markers with antibodies against anti-mouse CD3, CD4, and CD8. After surface staining, the cells were washed and suspended in a fixation/permeabilization solution, incubating for 20 min at 4 °C. The fixed cells were subsequently washed twice with BD Perm/Wash buffer (BD Biosciences) and stained with antibodies specific for cytokine-producing CD8+ T cells, including anti-mouse IL-2, TNF-α, and IFN-γ, for 30 min at room temperature (RT). After intracellular staining, the cells were washed twice with BD Perm/Wash buffer and resuspended in buffer.
Flow cytometry data were analysed via a BD FACSCanto II (Sungkyunkwan University Cooperative Center for Research Facilities), FACSymphony A3 (Korea Virus Research Institute) and quantified via FlowJo V 10. Detailed information on the antibodies and gating strategies used are provided in Supplementary Table 1 and Supplementary Figs. 21-25.
Serum anti-OVA IgG antibody assay
100 μL of blood from immunised mice was collected and then centrifuged at 10,000 × g for 10 min at 4 °C to separate the serum. For the IgG, IgG1, and IgG2c ELISAs, a 96-well plate (Immunoplate, SPL) was coated overnight with recombinant OVA protein (2 μg mL−1). The plate was subsequently washed two times with 0.05% (v/v) Tween-20 and blocked for 3 h at 37 °C using a 5% (w/v) skim milk solution. Serum samples were loaded onto the plate and incubated for 2 h at 37 °C. To specifically detect IgG, IgG1, and IgG2c, secondary antibodies, including goat anti-mouse IgG (H + L)-HRP, rat anti-mouse IgG1-HRP, and goat anti-mouse IgG2c-HRP (SouthernBiotech), were added to each plate at a 1:6000 dilution. After 1 h of incubation at 37 °C, the plate was washed, and the proteins were detected using a TMB solution. The absorbance of the plate at 450 nm was measured using a microplate reader (VersaMax).
Serum anti-sM2HA2, sM2 and HA2 IgG antibody assay
Serum IgG levels and the IgG1 and IgG2a isotypes were measured using ELISA. Overnight at 4 °C, 200 ng/well of sM2, HA2 peptide, or sM2HA2 protein was coated on 96-well ELISA plates (Costar, cat. no. 2592) in coating buffer (pH 9.6). The plates were blocked with 200 µL of 10% skim milk (BD, cat. no. 232100) after being washed with PBS containing 0.05% Tween 20 (ChemCruz, cat. no. 5C-2911313). To detect IgG, IgG1, and IgG2a, mouse sera diluted into a series of dilutions (1:50 to 1:1600) in PBS containing 2% skim milk were dispensed into designated wells and incubated at 37 °C for 2 h, followed by the addition of 1:3000 diluted horseradish peroxidase-conjugated goat anti-mouse IgG (GeneTex, cat. no. GTX213111-01), IgG1 (Invitrogen, cat. no. a10551), and IgG2a (Invitrogen, cat. no. a10685). The plates were incubated for 2 h at 37 °C. Afterwards, 100 microlitres of a substrate mixture containing tetramethylbenzidine (TMB; BD, cat. no. 555214) was added, and the reaction was allowed to continue for 10 min. Using an ELISA auto reader (Molecular Devices), the optical density was measured at 450 nm following the addition of the stop solution (2 NH2SO4).
Enzyme-Linked Immunospot (ELISPOT) assay specific to SARS-CoV-2 virus
To measure the T-cell immune response, the frequency of IFN-γ-producing T-cells was evaluated via mouse IFN-γ ELISPOT kits (BD Bioscience, cat. no. 551849, USA). Briefly, the splenocytes were plated at 5 × 105 cells/well onto purified IFN-γ Ab-coated ELISPOT plates and stimulated with 1 μg mL−1 S peptide pool (Sino Biological, China) and 1 μg mL−1 BPL-inactivated SARS-CoV-2 variants for 60 h at 37 °C. The SFUs were enumerated via an ELISPOT plate reader (iSpot SPECTRUM System, AID Diagnostika, USA). For measurement of the levels of antigen-specific B-cell responses, enzyme-linked immunosorbent assay (ELISA) plates (Thermo Fisher) were coated with 0.5 μg mL−1 SARS-CoV-2 spike-stabilized trimers, washed with PBS-T, and blocked with 5% skim milk in PBS-T. The sera from each group were added to the antigen-coated plates, followed by incubation with HRP-anti-mouse IgG (Abcam, cat. no. ab97023, USA). The plates were washed and developed with the chromogenic tetramethylbenzidine substrate (Merck), and the reactions were terminated with 2 N H2SO4. The absorbance was measured at 450 nm using an Infinite M plex microplate reader (Tecan).
ELISPOT assay specific to sM2HA2, sM2 and HA2
Mouse IFN-γ (BD, cat. no. 551083) and IL-4 ELISPOT kits (BD, cat. no. 551017) were used to conduct interferon (IFN)-γ and interleukin (IL)-4 ELISPOT assays on splenocytes. Anti-mouse IFN-γ and IL-4 capture antibodies (5 µg mL−1) were coated on BD ELISPOT 96-well plates and incubated overnight at 4 °C in PBS. The plates were blocked with a solution containing full RPMI 1640 medium (HyClone, cat. no. SH30027.01) supplemented with 10% foetal bovine serum (Gibco, cat. no. 17904731) after the antibodies were discarded, after which they were incubated for 2 h at room temperature. Following aseptic isolation of splenocytes, 1 × 106 cells/well, sM2HA2 recombinant protein, sM2 or HA2 peptide (1 μg/well), medium alone (negative control) or 0.5 µg mL−1 phytohemagglutinin (positive control; Gibco, cat. no. 10576-015) were added to the media. Following a 24-h incubation period at 37 °C with 5% CO2, the plates were subjected to sequential treatments with streptavidin-horseradish peroxidase, biotinylated anti-mouse IFN-γ and IL-4 antibodies, and substrate solution (BD cat. no. 551951). Ultimately, an ImmunoScan Entry analyser (Cellular Technology, Shaker Heights, USA) was used to count the spots.
Serum neutralization against SARS-CoV-2 pseudovirus (pseudotyped SARS-CoV-2)
Transfected 293 T cells expressing ACE2 were generated by transducing 293 T cells with an ACE2 expression lentiviral vector (Addgene plasmid, cat. no. 145839). Single-cell clones were derived by limiting dilution from the bulk populations. 293T-ACE2 cells were seeded at a density of 2 × 104 cells per well in 96-well luminometer-compatible tissue culture plates (SPL, Korea) 24 h before infection. For the neutralization assay, pseudoviruses (1.8 × 107 copies) were mixed with diluted serum ranging from 50 to 984150-fold. Then, the mixture was added to 96-well 293T-ACE2 cells. After 24 h of incubation, the inoculum was replaced with fresh medium. Luciferase activity was measured 72 h after infection. Briefly, cells were lysed with 40 μL per well of Passive Lysis Buffer (Promega, cat. no. E1910). The luciferase activity in the lysates was measured using the Nano-Glo Luciferase Assay System (Promega, Wisconsin, USA). Specifically, 40 μL of substrate in Nano-Glo buffer was mixed with 40 μL of cell lysate and incubated for 3 min at RT. NanoLuc luciferase activity was measured using a GloMax Navigator Microplate Luminometer (Promega, Wisconsin, USA) using an integration time of 300 ms integration time.
Serum neutralization against SARS-CoV-2 virus
To examine neutralizing antibodies against SARS-CoV-2 variants in vaccinated mice, the collected sera were inactivated by heating at 56 °C for 30 min. Initial 1:10 serum dilutions were prepared in serum-free DMEM (Gibco), followed by twofold serial dilutions to achieve final serum dilutions ranging from 1:10 to 1:12,800. For each well, serially diluted sera were mixed with 100 TCID50 of SARS-CoV-2 and incubated at 37 °C for 1 h to neutralize the infectious virus. The serum-virus mixtures were then inoculated onto Vero-E6 cell (ATCC) monolayers, incubated for 1 h at 37 °C in 5% CO, and subsequently replaced with 2% FBS-supplemented DMEM (Gibco). After three days, the neutralization of infectious virus was assessed based on cytopathic effect (CPE).
Serum neutralization against Influenzas virus
The microneutralization assay was conducted to assess the levels of H1N1, H5N2, H9N2 and H7N3-specific neutralizing antibodies in sera of immunised mice. Briefly, Serum was treated with a receptor-destroying enzyme (Denka Seiken, Japan) and subsequently inactivated at 56 °C for 30 min in FBS-free DMEM (HyClone, USA, cat. no. SH30243.01). A volume of 50 μl of 2-fold serial dilution of inactivated serum was added to 96-well microtiter plates and mixed with 50 μl of 100 TCID50 of H1N1, H5N2, H9N2, and H7N3 viruses. This mixture was then incubated at 37 °C for 1 h, after which 100 μl of 5 × 103 of MDCK cells suspension were added to each well. The plates were incubated at 37 °C with 5% CO2 for 4 days. The virus-induced cytopathic effect was evaluated, and neutralizing antibody titers were determined by the highest serum dilution where the cytopathic effect remained observable.
Serum neutraliation against SFTS virus
To evaluate the presence of neutralizing antibodies in the blood of vaccinated ferrets, serum samples from each time point were collected, with each serum then heat-inactivated at 56 °C for 30 min. Serum dilutions were prepared in serum-free DMEM (Gibco), starting with a 1:10 initial dilution, and further diluted twofold to achieve a final range of 1:10 to 1:2560. For each well, serially diluted sera were incubated at 37 °C for 1 h with 100 TCID50 SFTSV (CB1/2014) to neutralize the infectious virus. The serum-virus mixtures were then inoculated onto Vero-E6 (ATCC) monolayer cells, incubated for 1 h at 37 °C with 5% CO, and subsequently replaced with DMEM (Gibco) supplemented with 2% FBS. After 5 days, neutralization of the infectious virus was confirmed by immunochromogenic staining of Vero-E6 cells using an anti-SFTSV NP antibody.
Statistics and reproducibility
All the results are presented as the mean ± standard deviation (SD). A two-tailed unpaired t test was used to compare the two groups. One-way ANOVA (or two-way ANOVA) with Tukey’s multiple comparisons test was used to analyse multiple groups of data. The log-rank (Mantel–Cox) test was used for survival data. All the statistical analyses were performed via GraphPad Prism 8 and Microsoft Excel 2022. P values (n.s.: not significant, *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001) were used to indicate statistical significance.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data are included in the Supplementary Information or available from the authors, as are unique reagents used in this Article. The raw numbers for charts and graphs are available in the Source Data file whenever possible. Source data are provided with this paper.
References
Nguyen, T.-Q. et al. Emergence and interstate spread of highly pathogenic avian influenza A(H5N1) in dairy cattle. bioRxiv 2024.05.01.591751 https://doi.org/10.1101/2024.05.01.591751. (2024)
Reardon, S. Bird flu in US cows: where will it end? Nature 629, 515–516 (2024).
Burrough, E. R. et al. Early Release - Highly Pathogenic Avian Influenza A(H5N1) Clade 2.3.4.4b Virus Infection in Domestic Dairy Cattle and Cats, United States, 2024 - Volume 30, Number 7—July 2024 - Emerging Infectious Diseases journal - CDC. Emerg Infect Dis 30, (2024).
Chemaitelly, H. et al. Protection from previous natural infection compared with mRNA vaccination against SARS-CoV-2 infection and severe COVID-19 in Qatar: a retrospective cohort study. Lancet Microbe 3, e944–e955 (2022).
Chemaitelly, H. et al. Protection of prior natural infection compared to mRNA vaccination against SARS-CoV-2 infection and severe COVID-19 in Qatar. medRxiv 2022.03.17.22272529 https://doi.org/10.1101/2022.03.17.22272529. (2022)
Moss, P. The T cell immune response against SARS-CoV-2. Nat Immunol 23, 186–193 (2022).
Hall, V. et al. Protection against SARS-CoV-2 after Covid-19 Vaccination and Previous Infection. N. Engl. J. Med. 386, 1207–1220 (2022).
Mosmann, T.R., McMichael, A.J., LeVert, A. et al. Opportunities and challenges for T cell-based influenza vaccines. Nat Rev Immunol 24, 736–752 (2024).
Wherry, E. J. & Barouch, D. H. T cell immunity to COVID-19 vaccines. Science (1979) 377, 821–822 (2022).
Pulendran, B. Learning immunology from the yellow fever vaccine: innate immunity to systems vaccinology. Nat. Rev. Immunol. 2009 9:10 9, 741–747 (2009).
Zinkernagel, R. M. On natural and artificial vaccinations. Ann. Rev. Immunol 21, 515–546 (2003).
Medzhitov, R. Recognition of microorganisms and activation of the immune response. Nat. 2007 449:7164 449, 819–826 (2007).
Tang, D., Kang, R., Coyne, C. B., Zeh, H. J. & Lotze, M. T. PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol. Rev. 249, 158 (2012).
Bhagchandani, S., Johnson, J. A. & Irvine, D. J. Evolution of Toll-like receptor 7/8 agonist therapeutics and their delivery approaches: From antiviral formulations to vaccine adjuvants. Adv. Drug Deliv. Rev. 175, 113803 (2021).
Ugolini, M. et al. Recognition of microbial viability via TLR8 drives TFH cell differentiation and vaccine responses. Nat. Immunol. 19, 386–396 (2018).
Dowling, D. J. Recent Advances in the Discovery and Delivery of TLR7/8 Agonists as Vaccine Adjuvants. Immunohorizons 2, 185–197 (2018).
Tam, H. H. et al. Sustained antigen availability during germinal center initiation enhances antibody responses to vaccination. Proc. Natl Acad. Sci. USA 113, E6639–E6648 (2016).
Roth, G. A. et al. Injectable hydrogels for sustained codelivery of subunit vaccines enhance humoral immunity. ACS Cent. Sci. 6, 1800–1812 (2020).
Yin, Q. et al. A TLR7-nanoparticle adjuvant promotes a broad immune response against heterologous strains of influenza and SARS-CoV-2. Nat. Mater. 22, 380–390 (2023).
Irvine, D. J., Aung, A. & Silva, M. Controlling timing and location in vaccines. Adv. Drug Deliv. Rev. 158, 91–115 (2020).
Ou, B. S., Saouaf, O. M., Baillet, J. & Appel, E. A. Sustained delivery approaches to improving adaptive immune responses. Adv. Drug Deliv. Rev. 187, 114401 (2022).
Cirelli, K. M. et al. Slow Delivery Immunization Enhances HIV Neutralizing Antibody and Germinal Center Responses via Modulation of Immunodominance. Cell 177, 1153–1171.e28 (2019).
Langenkamp, A., Messi, M., Lanzavecchia, A. & Sallusto, F. Kinetics of dendritic cell activation: impact on priming of TH1, TH2 and nonpolarized T cells. Nat. Immunol. 1, 311–316 (2000).
Abdi, K., Singh, N. J. & Matzinger, P. Lipopolysaccharide-Activated Dendritic Cells: “Exhausted” or Alert and Waiting? J. Immunol. 188, 5981–5989 (2012).
Jin, S. M. et al. A nanoadjuvant that dynamically coordinates innate immune stimuli activation enhances cancer immunotherapy and reduces immune cell exhaustion. Nat. Nanotechnol. 18, 390–402 (2023).
Benson, R. A. et al. Antigen presentation kinetics control T cell/dendritic cell interactions and follicular helper T cell generation in vivo. Elife 4, (2015).
Martínez-Riaño, A. et al. Long-term retention of antigens in germinal centers is controlled by the spatial organization of the follicular dendritic cell network. Nat. Immunol. 24, 1281–1294 (2023).
Arunachalam, B., Phan, U. T., Geuze, H. J. & Cresswell, P. Enzymatic reduction of disulfide bonds in lysosomes: Characterization of a gamma-interferon-inducible lysosomal thiol reductase (GILT). Proc. Natl Acad. Sci. USA 97, 745–750 (2000).
Trefzer, A. et al. Dynamic adoption of anergy by antigen-exhausted CD4+ T cells. Cell Rep. 34, 108748 (2021).
Seephetdee, C. et al. Mice Immunized with the Vaccine Candidate HexaPro Spike Produce Neutralizing Antibodies against SARS-CoV-2. Vaccines 9, 498 (2021).
Neirynck, S. et al. A universal influenza A vaccine based on the extracellular domain of the M2 protein. Nat. Med. 5, 1157–1163 (1999).
Lee, J. S. et al. The highly conserved HA2 protein of the influenza a virus induces a cross protective immune response. J. Virol. Methods 194, 280–288 (2013).
Noh, H. J. et al. Programming of Influenza Vaccine Broadness and Persistence by Mucoadhesive Polymer-Based Adjuvant Systems. J. Immunol. 195, 2472–2482 (2015).
Chowdhury, M. Y. E. et al. Mucosal vaccination of conserved sM2, HA2 and cholera toxin subunit A1 (CTA1) fusion protein with poly gamma-glutamate/chitosan nanoparticles (PC NPs) induces protection against divergent influenza subtypes. Vet. Microbiol 201, 240–251 (2017).
Moyer, T. J. et al. Engineered immunogen binding to alum adjuvant enhances humoral immunity. Nat. Med 26, 430–440 (2020).
Albanesi, C. et al. Interleukin-17 is Produced by Both Th1 and Th2 Lymphocytes, and Modulates Interferon-γ- and Interleukin-4-Induced Activation of Human Keratinocytes. J. Invest. Dermatol. 115, 81–87 (2000).
Tan, H. X. et al. Inducible bronchus-associated lymphoid tissues (IBALT) serve as sites of B cell selection and maturation following influenza infection in mice. Front Immunol. 10, 611 (2019).
Nichol, K. L., Nordin, J. D., Nelson, D. B., Mullooly, J. P. & Hak, E. Effectiveness of influenza vaccine in the community-dwelling elderly. N. Engl. J. Med 357, 1373–1381 (2007).
Park, S. J. et al. Ferret animal model of severe fever with thrombocytopenia syndrome phlebovirus for human lethal infection and pathogenesis. Nat. Microbiol. 4, 438–446 (2018).
Kim, Y. I. L. et al. Age-dependent pathogenic characteristics of SARS-CoV-2 infection in ferrets. Nat. Commun. 13, 1–13 (2022).
Barman, S., Soni, D., Brook, B., Nanishi, E. & Dowling, D. J. Precision Vaccine Development: Cues From Natural Immunity. Front Immunol. 12, 662218 (2022).
Neutra, M. R. & Kozlowski, P. A. Mucosal vaccines: the promise and the challenge. Nat. Rev. Immunol. 6, 148–158 (2006).
Laidlaw, B. J. & Ellebedy, A. H. The germinal centre B cell response to SARS-CoV-2. Nat. Rev. Immunol. 22, 7–18 (2021).
Cromer, D. et al. Prospects for durable immune control of SARS-CoV-2 and prevention of reinfection. Nat. Rev. Immunol. 21, 395–404 (2021).
Eisfeld, A. J., Neumann, G. & Kawaoka, Y. Influenza A virus isolation, culture and identification. Nat. Protoc. 9, 2663–2681 (2014).
Yu, M. A. et al. Evaluation of two different enzyme-linked immunosorbent assay for severe fever with thrombocytopenia syndrome virus diagnosis. Clin. Exp. Vaccin. Res. 7, 82–86 (2018).
Acknowledgements
This work was supported by National Research Foundation (NRF) grants funded by the Korean government (grant numbers NRF-2020R1A2C3006888, NRF-2022M3A9J3072298, and RS-2023-00218648), Republic of Korea. (Prof. Yong Taik Lim). This work was supported by National Research Foundation (NRF) grants funded by the Korean government (grant No. NRF-2021R1A6A1A03045495), Republic of Korea. (Prof. Jong-Soo Lee). This work was supported by Institute for Basic Science (grant No. IBS-R801-D1), Republic of Korea. (Director Young Ki Choi).
Author information
Authors and Affiliations
Contributions
S.N.L., J.S.L., Y.K.C., and Y.T.L. conceived and designed the trial. S.N.L., Y.I.K., J.K., D.K.H., R.E., S.H.P., J.H., J.G., Y.S., M.-H.L., and Y.W.N. performed the experiments and analysed the data. S.N.L., Y.I.K., J.K., D.K.H., J.S.L., Y.K.C., and Y.T.L. wrote the manuscript. J.S.L., Y.K.C., and Y.T.L. edited the manuscript and supervised the project. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lee, S.N., Kim, YI., Kim, J. et al. Kinetically activating nanovaccine mimicking multidimensional immunomodulation of natural infection for broad protection against heterologous viruses in animal models. Nat Commun 16, 2914 (2025). https://doi.org/10.1038/s41467-025-58006-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-58006-y
