
Abstract
Type II-D Cas9 proteins (Cas9d) are more compact than typical Type II-A/B/C Cas9s. Here, we demonstrate that NsCas9d from Nitrospirae bacterium RBG_13_39_12 derived from a metagenomic assembly exhibits robust dsDNA cleavage activity comparable to SpCas9 in vitro. Unlike typical Cas9 enzymes that generate blunt ends, NsCas9d produces 3-nucleotide staggered overhangs. Our high-resolution cryo-EM structure of the NsCas9d-sgRNA-dsDNA complex in its catalytic state reveals the target and non-target DNA strands positioned within the HNH and RuvC catalytic pockets, respectively. NsCas9d recognizes the 5′-NRG-3′ protospacer adjacent motif (PAM), with 5′-NGG-3′ showing the highest cleavage efficiency. Its sgRNA structure, resembling the 5′ end of IscB ωRNA, along with structural features shared with other Cas9 variants, suggests that Cas9d are hypothesized to resemble evolutionary intermediates between other Cas9 sub-types and IscB. These findings deepen our understanding of Cas9 evolution and mechanisms, highlighting NsCas9d as a promising genome-editing tool due to its compact size, DNA cleavage pattern, and efficient PAM recognition.
Similar content being viewed by others

Introduction
Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-Cas systems are RNA-guided immune mechanisms in bacteria and archaea, and they have been adapted as powerful genome-editing tools1,2,3,4. Cas9, the hallmark protein of Type II CRISPR-Cas systems, is an RNA-guided endonuclease that features two nuclease domains, HNH and RuvC5. Through binding to CRISPR RNA (crRNA) and trans-activating crRNA (tracrRNA) or chimeric single-guide RNA (sgRNA), Cas9 recognizes DNA sequences containing a protospacer adjacent motif (PAM) and complementary target regions, enabling programmable DNA cleavage, forming the basis of Cas9-based genome editing6.
Numerous Cas9 proteins have been characterized, with Streptococcus pyogenes Cas9 (SpCas9) being the most widely used due to its high cleavage efficiency and robust genome-editing performance3,6. However, despite the availability of various Cas9 orthologs, their large size remains a significant limitation. For instance, SpCas9 (1368 amino acids), Staphylococcus aureus Cas9 (SaCas9, 1053 amino acids), and even smaller orthologs like Campylobacter jejuni Cas9 (CjCas9, 984 amino acids) exceed the packaging capacity of adeno-associated viruses (AAVs), which is limited to ~4.7 kb7,8,9,10. This highlights the ongoing need for more compact Cas9 variants that maintain high cleavage activity for efficient gene-editing applications.
Cas9 orthologs are believed to share a common evolutionary history with IsrB/IscB proteins, with notable divergences in the acquisition of HNH domains, insertion of REC-like domains, and the loss of the PLMP domain11. These evolutionary changes were accompanied by modifications to their RNA components and CRISPR arrays, including acquisitions, truncations, and decoupling events. Recently identified Type II-D Cas9 proteins represent an evolutionary intermediate between IsrB/IscB and canonical Cas9s11,12. This subtype includes smaller Cas9 proteins (~700 amino acids), offering significant potential for genome-editing applications. Notably, Cas9d proteins have demonstrated nuclease-mediated genome editing and base editing activity at specific target sites in mammalian cells12,13.
NsCas9d, a representative of Type II-D Cas9, was identified from the metagenome of the Nitrospirae bacterium RBG_13_39_1211,14. Due to its evolutionary importance and potential as a genome-editing tool, further structural studies on Type II-D Cas9 are essential. Recently, structures of Cas9d-MG34-1 were reported, but they lack complete RuvC and HNH domains, limiting mechanistic insight13,15. This study aims to address this gap through the structural and biochemical characterization of NsCas9d, providing insights into its mechanism and evolutionary development.
In this study, we present the cryo-EM structure of the NsCas9d-sgRNA-DNA complex in its catalytic state. In this structure, the HNH domain is positioned in an active state, with its catalytic residue H387 facing the target strand cleavage site. Five nucleotides from the non-target strand are positioned in the RuvC catalytic pocket. Interestingly, the sgRNA architecture differs from that of Cas9s in Type II-A/B/C systems but assembles similarly to the 5′ region of ωRNA bound to IscB. We also demonstrate that NsCas9d recognizes both NGG and NAG PAM sequences. Our findings expand the understanding of the Type II-D CRISPR-Cas system, introduce the Type II-D Cas9 subtype, and highlight the untapped potential within the diversity of CRISPR systems.
Results
NsCas9d cleaves target DNA with minimal 20-nt complementarity
To explore the diversity of Cas9 proteins, we selected 162 orthologs to construct a phylogenetic tree. The Type II-D Cas9s formed a distinct clade, showing a closer evolutionary relationship with Type II-C Cas9s. Notably, NsCas9d, identified from the metagenome of the Nitrospirae bacterium RBG_13_39_1211,14, differs significantly from other Type II-D variants like Cas9_665 and Cas9_1261 (Fig. 1a). Previous phylogenetic analysis further reveals that NsCas9d is more closely related to canonical Cas9s, whereas Cas9_665 and Cas9_1261 share a closer evolutionary affinity with IscB, suggesting that NsCas9d may represent an ancestral form of Type II-A/B/C Cas9s11,12.

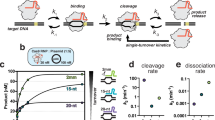
a Phylogenetic representation of the diversity provided by Cas9 orthologs. Subtype II-A, B, C, D systems are colored green, purple, orange and red respectively. Several typical Cas9s are indicated by black dots. b In vitro cleavage activity of NsCas9d. dsDNA substrates of varying base pairing with guide of sgRNA were used to explore the minimal heteroduplex length requisite for sustained high cleavage activity. c Depletion assay to identify functional PAM for NsCas9d. Log relative frequency of each base at every position relative to 3′ end of target sequence from libraries before and after cleavage. d In vitro cleavage activity of NsCas9d. The dsDNA targets have different sequences at the PAM region. e In vitro cleavage activity of several Cas9 proteins. Source data are provided as a Source Data file.
In the Nitrospirae genome, we identified four nearly identical repeat sequences, with ~37 nucleotides found between the repeats (Supplementary Fig. 1a). To design the sgRNA for NsCas9d, we used Repeat-1 to identify a complementary sequence within the tracrRNA, which allowed us to determine the anti-repeat sequence. The sgRNA scaffold for NsCas9d was created by fusing the 3′ end of a truncated direct repeat with the 5′ end of the corresponding tracrRNA via a 4-nt GAAA linker. This resulted in a 173-nt sgRNA with a 37-nt guide sequence. The sgRNA associated with NsCas9d is longer than those typically found in Type II-A/B/C Cas9s but shorter than the ωRNA that binds to IscB11,16,17.
We co-expressed the NsCas9d and its sgRNA in Escherichia coli BL21(DE3) cells, and purified the NsCas9d-sgRNA binary complex. To determine the minimal guide-target base-pair requirement for DNA cleavage by NsCas9d, we prepared linear plasmid DNA with varying base-pair lengths (16-37 nt) matching the sgRNA guide sequence and conducted DNA cleavage assays (Fig. 1b and Supplementary Fig. 1b). Our results demonstrated that NsCas9d efficiently cleaved DNA when the target sequence formed a 20-nt or longer duplex with the sgRNA. Cleavage activity was significantly reduced with 18 base-pair heteroduplexes and completely abolished with 16 base pairs. These findings indicate that a 20-nt guide-target duplex is essential for NsCas9d activity. Subsequently, we designed a 20-nt sgRNA guide region and conducted additional assays using this optimized sgRNA.
NsCas9d recognizes 5′-NRG-3′ PAM sequence
To identify the PAM sequence recognized by NsCas9d, we conducted a PAM depletion assay. A 20-nt protospacer with an 8-bp randomized sequence was inserted into the pUC19 plasmid, generating a Protospacer-PAM(8N) plasmid library. After linearizing the library with HindIII enzyme, the NsCas9d-sgRNA complex was used to cleave the DNA. Deep sequencing was performed on the DNA before and after cleavage, and the nucleotide frequency at the 8N PAM positions was analyzed to identify the depleted functional PAM sequences. The results revealed that cleavage primarily occurred at the 5′-NGG-3′ PAM sequence (Fig. 1c).
To further validate the recognition of the 5′-NGG-3′ PAM, we conducted an in vitro linear plasmid DNA cleavage assay using substrates with variations at positions [−1*] to [−4*]. Our results showed that G at positions [−2*] and [−3*] optimized NsCas9d cleavage efficiency. Additionally, the combination of AG at positions [−2*] and [−3*] also produced substantial cleavage activity (Fig. 1d). In contrast, variations at these positions significantly reduced or completely abolished cleavage. These findings confirm that NsCas9d specifically recognizes the 5′-NRG-3′ PAM sequence, where R is either A or G, with the highest cleavage efficiency observed for the 5′-NGG-3′ PAM. All target DNA used in subsequent assays contained the 5′-NGG-3′ PAM.
NsCas9d exhibits comparable DNA cleavage activity to SpCas9
We next compared the DNA cleavage activity of NsCas9d with other well-characterized Cas9 orthologs, including SpCas9, SaCas9, NmCas9 and HpCas9. In vitro cleavage assays revealed that NsCas9d exhibited activity comparable to SpCas9, moderately higher activity than SaCas9, and significantly greater activity than NmCas9 and HpCas9 (Fig. 1e).
Structure of the NsCas9d-sgRNA-dsDNA complex in catalytic state
Wild-type NsCas9d (Fig. 2a) was co-expressed with this 156-nt sgRNA containing a 20-nt guide sequence in E. coli, and the NsCas9d-sgRNA binary complex was purified. To reconstitute the NsCas9d-sgRNA-DNA ternary complex, we mixed the purified NsCas9d-sgRNA complex with a 41-bp dsDNA target, which contains a 5′-GGG-3′ PAM sequence and is complementary to the guide strand. The complex was formed at a molar ratio of 1:1.1, resulting in the NsCas9d-sgRNA-DNA ternary complex. To facilitate DNA binding, both strands of the dsDNA were identical in the guide-complementary region (Fig. 2b).

a Architectural domains of NsCas9d. BH, bridge helix. WED wedge, PI PAM-interacting. b Schematic representation of the sgRNA and dsDNA used for cryo-EM. TS, target strand. NTS non-target strand. c Cartoon representations of NsCas9d-sgRNA-dsDNA complex in a catalytic state. Domains of NsCas9d are colored according to the scheme used in (a). d Overall structure of NsCas9d-sgRNA-dsDNA complex, while NsCas9d in surface style and nucleic acid in cartoon style.
We determined the cryo-EM structure of NsCas9d-sgRNA-dsDNA ternary complex at a 2.86 Å resolution (Supplementary Fig. 2 and Supplementary Table 1). Except for 30 amino acids in the REC1 domain (E109-Q138), 41 amino acids in the REC2 domain (P215-D255), and 11 nt in the non-target strand (C[10*]-A[17*], C[2*]-A[4*]), which are disordered, all other amino acids and nucleotides were resolved. In contrast to the recently published structures of Cas9d-MG34-1, where the HNH domain is completely disordered and most of the RuvC and PI domains are disordered, the NsCas9d structure shows more ordered features13,15.The NsCas9d structure revealed a bilobed architecture consisting of the REC lobe and NUC lobe, similar to other Cas9 proteins (Fig. 2a, c).
NsCas9d consists of 762 amino acids, shorter than the Cas9 proteins found in type II-A/B/C systems. Despite its smaller size, NsCas9d retains the key domains found in larger Cas9 proteins, albeit with shorter versions of those domains (Supplementary Fig. 3a). Notably, the REC lobe of NsCas9d is particularly compact, with only 168 amino acids compared to 624 in SpCas9 and 354 in NmCas9. This compact structure includes the REC1 and REC2 domains, which are connected by a 13-aa flexible loop, creating greater separation than observed in SpCas9 or NmCas9 (Supplementary Fig. 3b). This increased separation likely enhances the flexibility of the REC2 domain, leading to disorder in 41 residues within this region. Additionally, the RuvC domain in NsCas9d is also more compact, ~120 amino acids smaller than those in other Cas9 proteins (Supplementary Fig. 3b).
The sgRNA consists of a guide region (nucleotides 1-20), a repeat region (G21-U39), and the tracrRNA (G40-A156) (Fig. 3a). In the NsCas9d-sgRNA-DNA ternary complex, the full R-loop is formed. The guide segment of the sgRNA forms a 20-bp RNA-DNA heteroduplex with partial TS (C1-C20), adopting A-form and located in the central tunnel between REC and NUC lobes. The non-target strand is positioned in the RuvC binding channel, with 5 nucleotides observed in the RuvC catalytic pocket. The remaining nucleotides within the target strand reform the duplex with the non-target strand, with 5 base pairs resolved (Fig. 2c, d). Notably, both DNA strands remain intact, suggesting that NsCas9d is in a catalytic pre-cleavage state. This observation aligns with the finding that structural rearrangement of the HNH domain into its catalytic state is essential for activation of the RuvC domain18.

a Schematics of the sgRNA depicting secondary structure. b The interactions of N659 and upstream portion of the repeat: anti-repeat duplex. c In vitro cleavage activity of NsCas9d with sgRNAs which have different length of repeat: anti-repeat duplex via truncation. Source data are provided as a Source Data file. d, e Cartoon representation of the tracrRNA scaffold of NsCas9d and the ωRNA of IscB (PDB: 7UTN), colored according to the scheme used in (a). f The interaction of sgRNA with BH and the loop connecting two REC domains. The protein part is represented in the form of surface electrostatic potential. g The stacking interactions of the flip within sgRNA and residues of NsCas9d. h Cartoon representation of the pseudoknot structure.
Catalytic state of NsCas9d
The HNH domain of Cas9 is known for its high flexibility, which often results in DNA-bound Cas9 structures adopting an inactive conformation where the HNH domain is positioned away from the target strand cleavage site. However, in our DNA-bound NsCas9d complex, the cryo-EM density map clearly reveals that the HNH domain is positioned adjacent to the TS cleavage site (Fig. 2c). This configuration closely resembles the catalytic state observed in NmCas918, indicating that NsCas9d is also captured in its catalytic state. This structural arrangement suggests that NsCas9d adopts a catalytically competent conformation upon target DNA binding, providing insights into the activation mechanism of Type II-D Cas9 orthologs.
NsCas9d sgRNA architecture resembles the 5′-end of ωRNA
The sgRNA of NsCas9d (Fig. 3a) features repeat and anti-repeat regions (A44-A62) forming an A-form-like duplex with a G21-A62 mismatch, 17 Watson-Crick base pairs, and a wobble pair (G27·U56) (Supplementary Fig. 5a). This structure, known as the P1 stem loop, occupies a binding site similar to that observed in other Cas9 proteins and IscB8,16,18,19. Its upstream segment, near the guide RNA, is stabilized through interactions with the WED and REC1 domains (Supplementary Fig. 4). Specifically, the N659 side chain of the WED domain forms hydrogen bonds with the bases of A61 and the ribose of A62 (Fig. 3b). In contrast, the downstream portion of the P1 stem loop extends beyond the NsCas9d protein, showing no direct interaction, which suggests a limited role in RNA-guided DNA cleavage.
To evaluate the functional relevance of the downstream segment, we conducted DNA cleavage assays using sgRNAs with truncated P1 stem loops (Supplementary Fig. 5b). Truncations of up to 10 base pairs (A30-U39 and A44-U53) did not affect cleavage efficiency, indicating that the truncated 136-nt sgRNA is as effective as the full-length version in guiding NsCas9d-mediated DNA cleavage (Fig. 3c). Notably, this 136-nt sgRNA is comparable in length to those used by SpCas9 and other Cas9 orthologs, while NsCas9d is ~300 amino acids shorter.
Beyond the anti-repeat region, the sgRNA adopts a compact tertiary structure comprising J1, J2, P2, P3 and a pseudoknot (PK) (Fig. 3d). Positioned at the PI-WED interface and contacting the REC lobe (Fig. 2d), this structured region is stabilized by interactions with the Arg-rich bridge helix (BH) and the loop connecting the REC domains, forming an extensive contact network (Fig. 3f). J1 connects to the P1 stem loop, while J1 and J2 align antiparallel to P3, with P2 bridging them. The 3′ end of the sgRNA flips between nucleotides 145 and 146, stabilized by stacking interactions with Q50 and R47 (Fig. 3g). This flipping promotes base pairing between nucleotides 146-149 and the loop in P3, resulting in pseudoknot formation (Fig. 3h).
Interestingly, the overall fold of this sgRNA region diverges from those of sgRNAs in other Cas9 proteins from type II-A/B/C systems (Supplementary Fig. 5c)8,18,19. However, the sgRNA structure is strikingly similar to the 5′ portion of ωRNA bound to IscB (Supplementary Fig. 5d). Compared to the NsCas9d sgRNA, ωRNA spans over 200 nt, featuring a significantly longer single-stranded linker and an extended P5 stem at its 3′ end (Supplementary Fig. 5d). Apart from these differences, the remaining regions of ωRNA exhibit high similarity to the NsCas9d sgRNA, with the P4 region of ωRNA being just 4 bp longer than that of NsCas9d sgRNA (Fig. 3e).
5′-NRG-3′ PAM recognition
The target double-stranded DNA (dsDNA) containing the 5′-NGG-3′ protospacer adjacent motif (PAM) is positioned within a positively charged cleft formed by the PI and WED domains (Fig. 4a, Supplementary Fig. 6a). These domains enclose the DNA, precisely aligning with the minor groove of the PAM. The side chain of K662 forms critical interactions with the base pair at position (−2) from the minor groove via water molecules. Additionally, the side-chain of N664 and the carbonyl oxygen of G647 establish hydrogen bonds with the bases of G(−2*) and G(−3*), respectively, further stabilizing the PAM interaction (Fig. 4b). Moreover, K728 engages with the base of G(−2*) from the major groove through a water-mediated hydrogen bond, while R711 and N667 interact with the phosphate backbone of the non-target strand, contributing to the overall stability of the DNA-protein complex.

a Binding of the PAM duplex with NsCas9d. NsCas9d was shown in cartoon representation with the coloring of the domains and residues referenced from Fig. 2a. b Interactions of residues and the PAM duplex. Residues are color-coded by domains in Fig. 2a. c Effects on in vitro cleavage of mutations on the PAM-interacting residues. Data are represented as mean ± SD (n = 3 independent experiments). Source data are provided as a Source Data file.
To elucidate the mechanism underlying NsCas9d’s recognition of the 5′-NAG-3′ PAM, we performed a simulation that substituted the GC base pair at position (−2) with an AT pair. Despite this substitution, residues K662 and N446 and K728 maintained their interactions with the same base pair (Supplementary Fig. 6b). These conserved interactions suggest that NsCas9d retains critical base contacts, thereby sustaining its cleavage activity with the NAG PAM sequence.
To evaluate the functional significance of these PAM-interacting residues, we introduced alanine substitutions at key positions and performed DNA cleavage assays. The K662A mutation nearly abolished DNA cleavage activity, underscoring its essential role in PAM recognition. Similarly, the equivalent residue K649 in Cas9d-MG34-1 is crucial for PAM recognition13,15. Furthermore, alanine substitutions at R711 and N667 significantly impaired NsCas9d’s cleavage efficiency (Fig. 4c), emphasizing the importance of these residues in stabilizing the DNA-protein interface and facilitating efficient target cleavage.
NsCas9d cleaves dsDNA, generating staggered ends
We captured the stable, catalytically active conformation of the HNH domain, with the side chain of H387 oriented toward the phosphate group between C(3) and A(4) on the target strand (TS) (Fig. 5a). This positioning defines the TS cleavage site, which was validated by Sanger sequencing of the cleavage products (Fig. 5c), consistent with previously reported Cas9 cleavage site6,18,20,21.

a, b Residues and interactions around cleavage sites in TS and NTS. c Sequence of supercoiled plasmid used as substrates when detecting exact cleavage sites and Sanger-sequencing traces. The cleavage sites are indicated by red isosceles triangles. The red isosceles triangles in Sanger-sequencing traces show the termini of the polymerases’ catalytic reaction during sequencing.
For the non-target strand (NTS), a 5-nt fragment was observed in the cryo-EM density map near the conserved catalytic residue D10 of the RuvC domain (Fig. 5b). Due to incomplete electron density, the precise nucleotide sequence of this fragment could not be resolved. To determine the precise cleavage site, we performed Sanger sequencing of the supercoiled plasmid DNA cleavage product, which confirmed the NTS cleavage site between nucleotides (6*) and (7*) (Fig. 5c). Therefore, NsCas9d generates staggered DNA ends with a 3-nt 5′-overhang, in contrast to other reported Cas9s that typically produce blunt ends when cleaving dsDNA19,20. However, this finding is consistent with the results from previous studies on other Type II-D Cas9s12,15.
Additionally, a cation specificity assay revealed that NsCas9d requires cations for cleavage, with Mg2+, Mn2+, and Co2+ acting as effective cofactors, while Zn2+, Ca2+, Cu2+, Cd2+, and Ni2+ showed no activity. This cation preference mirrors that of other Cas9 orthologs (Supplementary Fig. 7a)18,22,23,24.
Effect of mismatch position on NsCas9d cleavage
The length of the heteroduplex between the target strand (TS) and guide strand has minimal impact on cleavage activity within the 20–37 bp range (Fig. 1b). However, single mismatches within a 20 bp heteroduplex significantly reduce cleavage efficiency. Mismatches at positions 18–19 and 13–14 cause a moderate reduction in activity, decreasing NsCas9d cleavage by ~50% (Supplementary Fig. 7b). In contrast, mismatches at most other positions severely impair cleavage, while U:T mismatches at positions 6 and 11 have only a minor effect. To further investigate the influence of mismatch position and type on cleavage efficiency, we introduced various substitutions at positions 6 and 11. Replacing the wild-type A with T, C, or G at these positions produced distinct cleavage outcomes (Supplementary Fig. 7c), suggesting a dependency on both the mismatch position and nucleotide type. These findings indicate that both the position and the type of mismatch affect DNA cleavage by NsCas9d.
NsCas9d as an active nuclease and genome-editing tool in mammalian cells
NsCas9d, with its compact size and two endonuclease domains, exhibits DNA cleavage activity comparable to SpCas9 when paired with 136–156 nt sgRNAs in vitro (Fig. 1e). To evaluate its potential as a genome-editing tool in mammalian cells, we synthesized a human codon-optimized version of NsCas9d and cloned it into the PX459 plasmid, along with an sgRNA targeting the EMX1.1 site of the EMX1 gene25. After 52 h of incubation with HEK293T cells, genomic DNA was extracted, and T7E1 cleavage assays were performed to assess in vivo cleavage efficiency26. While NsCas9d successfully edited the target gene, its editing efficiency was notably lower than that of SpCas9 (Fig. 6). These results demonstrate the functional potential of NsCas9d for genome editing but highlight the need for further optimization to improve its efficiency in mammalian cells.

Schematic for the Cas9-sgRNA expression plasmids. Editing efficiency of NsCas9d in HEK293T cells comparing with SpCas9 was detected by evaluating the cleavage efficiency of the targeted region amplicons using the T7E1 endonuclease. Source data are provided as a Source Data file.
Discussion
NsCas9d, one of the smallest Cas9 orthologs, exhibits robust DNA cleavage activity and high specificity for its PAM sequence. In this study, we captured the pre-cleavage catalytic state of NsCas9d in complex with sgRNA and target DNA, showing that the HNH domain is correctly positioned at the scissile site on the target strand. We also confirmed that NsCas9d specifically recognizes the NRG PAM sequence, which is crucial for its cleavage mechanism.
Unlike most Cas9 proteins, which generate blunt ends or a few 1-nt 5′-staggered ends27, NsCas9d produces 3-nt staggered DNA ends. The HNH domain cleaves the target strand between the 3rd and 4th nucleotides, while the RuvC domain cleaves the non-target strand between the 6th and 7th nucleotides, generating a 3-nt 5′-overhang. In addition, staggered DNA ends were also observed in other Type II-D Cas9s12,15, suggesting that staggered cleavage is a characteristic feature of Cas9d enzymes. This cleavage pattern distinguishes Cas9d from other Cas9 variants and resembles the cleavage products generated by Cas1228. Notably, this staggered cleavage can facilitate more predictable and efficient DNA repair processes, particularly in applications like gene insertions, as well as improve the efficiency of non-homologous end joining (NHEJ)-based gene insertions into mammalian genomes, thereby enhancing editing efficiency29,30.
Type II-D CRISPR-Cas systems represent a distinct phylogenetic cluster, characterized by compact Cas9 proteins (600–970 amino acids) that are smaller than those in type II-A (~1200–1400 amino acids) and type II-C (~1000–1100 amino acids) systems, but larger than the ancestral IsrB/IscB (~300–500 amino acids). Unlike IsrB, which lacks the HNH nuclease and α-helical recognition (REC) lobe31, type II-D Cas9 retains these critical domains. The sgRNA associated with NsCas9d is ~130–150 nucleotides long, smaller than the ~200–400-nt ωRNA in IsrB/IscB but larger than the ~100-nt sgRNAs in conventional Cas9 proteins such as SpCas9 and SaCas9. While IsrB/IscB’s smaller size facilitates AAV packaging, their lower DNA cleavage efficiency limits their usage. In contrast, NsCas9d’s compact size, simplified sgRNA, and high cleavage activity make it more suitable for efficient packaging into a single AAV vector, offering a superior alternative for genome editing applications10.
The sgRNA associated with NsCas9d adopts the scaffold structure, distinct from those in other type II CRISPR-Cas systems. It contains a pseudoknot, a feature rarely observed in type II systems, except in CjCas98. Additionally, the P2 stem-loop disrupts the double-stranded pairing between RNA stem regions J2 and P3, forming an alpine-butterfly-knot conformation, also seen in IscB’s ωRNA16. The sgRNA bound to NsCas9d is a shorter, simplified version of the ωRNA found in IscB. This simplification reflects an evolutionary trend toward protein-dominated complexes, with ~64% of the NsCas9d complex consisting of protein, higher than IscB (<50%) but lower than other type II systems (>70%) (Supplementary Fig. 8). This positions NsCas9d as an intermediate between typical Cas9s and IsrB, underscoring the evolutionary significance of type II-D CRISPR-Cas systems.
NsCas9d recognizes the 5′-NRG-3′ PAM sequence similarly to SpCas9, but through a distinct mechanism. The interactions between the guanine bases within the PAM and the corresponding residues (K662, K728, N664, and G647) are weaker in NsCas9d compared to the stronger interactions observed in SpCas9 with the NGG PAM32. This disparity likely contributes to reduced DNA unwinding efficiency in NsCas9d, which may explain its comparatively lower DNA cleavage activity in vivo. Structural optimization of these interactions could enhance NsCas9’s DNA cleavage activity, improving its potential for efficient genome editing.
In conclusion, NsCas9d is a promising, compact genome-editing tool with efficient PAM recognition and robust catalytic activity. Its features, including a distinct sgRNA scaffold and the ability to generate 3-nt staggered DNA ends, set it apart from other Cas9 variants. While its in vivo DNA cleavage efficiency is lower than SpCas9, targeted optimization could enhance its activity, making NsCas9d an excellent candidate for applications requiring small Cas9 proteins, particularly in AAV packaging and genome editing.
Methods
NsCas9d identification and Cas9s’ expression and purification
The protein sequence of NsCas9d was obtained from a scaffold of Nitrospirae bacterium RBG_13_39_12 genome as previous reported11,14. The gene encoding full-length NsCas9d (residues 1-762) was codon-optimized for Escherichia coli and synthesized from GenScript. NsCas9d was cloned into an expression vector pET28a-Sumo between the BamHI and XhoI restriction sites with a His6-sumo tag at the N terminus. For NsCas9d-sgRNA co-expression, the gene encoding His6-Sumo tagged NsCas9d was cloned into the first multiple cloning site (MSC-1) of pRSFDuet vector. The crRNA and tracrRNA were combined to a single guide RNA (sgRNA) via a GAAA linker. The sequence encoding sgRNA was cloned into the MCS-2 of pRSFDuet vector with a hepatitis delta virus (HDV) ribozyme at the 3′ end. E. coli BL21 (DE3) cells were transformed with pRSFDuet-NsCas9d-sgRNA to overexpress NsCas9d-sgRNA complex.
The E. coli cells were cultured at 37 °C in LB medium supplemented with 50 μg/mL kanamycin. After OD600 reached 0.6–0.8, protein expression was induced with 0.1 mM isopropyl-β-D-1-thiogalactopyranoside (IPTG) for 14 h at 18 °C. Cells were collected by centrifugation and resuspended in buffer A (20 mM Tris-HCl, pH 7.5, 500 mM NaCl) for NsCas9d-sgRNA complex purification. Collected cells were lysed by sonication, and then high speed centrifuged. After removing cell debris, the supernatant was loaded onto Ni2+-charged Sepharose resin column (GE HealthCare). The complex was eluted with lysis buffer supplemented with 300 mM imidazole. To remove the His6-Sumo tag, the purified protein or complex were mixed with His-tagged ubiquitin-like protein 1 (Ulp1) protease for 2 h at 4 °C while dialyzing against buffer B (20 mM Tris-HCl, pH 7.5, 200 mM NaCl). The free His6-Sumo tag was removed by Ni2+-charged Sepharose resin column equilibrated with buffer B. The flow-through collections were further purified by a Heparin column (GE Healthcare). The bound NsCas9d-sgRNA complex were eluted with a linear gradient of 0.2–1 M NaCl with 20 mM Tris-HCl, pH 7.5. Protein concentrations in co-expressed complex were determined using the Bradford assay (Bio-Rad). NsCas9d-sgRNA mutants were constructed using a PCR-based method, the plasmid encoding wild-type full-length NsCas9d-sgRNA complex as the template. NsCas9d-sgRNA mutants were purified with an identical protocol.
The expression and purification protocols for SpCas9, SaCas9, NmCas9 and HpCas9 closely follow the aforementioned procedures. However, these four proteins were not co-expressed with their respective sgRNAs, which were obtained from in vitro transcription with T7 RNA polymerase.
Phylogenetic tree construction
To compare type II-D Cas9s with other usual Cas9 orthologs, a phylogenetic tree was constructed by combining 159 representatives type II-A, -B, -C Cas9 orthologs along with three type II-D Cas9 orthologs. Multiple sequence alignment of total 162 Cas9s was performed using MUSCLE and then the result was analyzed using IQ-TREE 2.2.2.7 with 1000 Ultrafast bootstrap replicates and other default parameters33,34.
In vitro cleavage assay
The target DNA plasmid was generated by cloning target and PAM sequence into the pUC19 vector. The linearized target plasmid for in vitro cleavage assay was generated by HindIII digestion. For determining the cleavage sites on the target strand (TS) and the non-target strand (NTS), the supercoiled pUC19 plasmid was used as substrate. NsCas9d-sgRNA was incubated with substrate dsDNA at 37 °C for 10 min in 10 μL reaction buffer (20 mM Tris-HCl, pH 7.5, 100 mM KCl, 5 mM MgCl2, 1 mM DTT and 5% glycerol) unless stated otherwise. For metal ion assays, the MgCl2 was replaced with other divalent cations or EDTA at a concentration of 5 mM. The reactions were stopped by the addition of 6× gel loading dye (New England Biolabs) and analyzed by 1% agarose gel electrophoresis and ethidium bromide staining. To compare in vitro activity of NsCas9d and its mutants, assays were performed at least three times, followed by quantification of gel bands using ImageJ (v1.51 d)35.
PAM depletion assay
The dsDNA oligonucleotides containing 8N randomized nucleotides downstream of a 20 nt target sequence were cloned into pUC19 to generate a plasmid library. The HindIII-linearized plasmid library was cleaved in vitro by NsCas9d-sgRNA complex and resolved by 1% agarose gel. The uncleaved substrate was purified with a Gel Extraction Kit (Omega Bio-tek) and then PCR amplified. The amplified DNA library was pair-end (150 bp) sequenced on a NOVAseq 6000 System (Illumina). The 8N nucleotide PAM regions were extracted from resulting reads and used to calculate the fractions of reads matching each one-base derivative relative to the total number of reads as described36. For a given PAM, enrichment was calculated as the log2-transformed ratio of the fold depletion.
Cryo-EM sample preparation and data collection
The NsCas9d-sgRNA-dsDNA ternary complex was reconstituted by incubating the co-expressed NsCas9d-sgRNA20 binary complex and the full-length 41 bp dsDNA with a 20 nt bubble on the target region, at a molar ratio of 1:1.1 on ice for 30 min. The NsCas9d-sgRNA-dsDNA ternary complex was purified via size-exclusion chromatography (Superdex 200 Increase 10/300, GE Healthcare), equilibrated with buffer B (20 mM Tris-HCl, pH 7.5, 200 mM NaCl). Au 300 mesh R1.2/1.3 grids (Nanodim) were glow-charged in O2-Ar condition for 60 s. The purified samples (~1.2 mg/mL, 3 μL) were applied to the grids in a Vitrobot Mark IV (Thermo Fisher Scientific) at 4 °C under 100% humidity conditions followed by a waiting time of 10 s and a blotting time of 3 s. The grids were then plunge-frozen in liquid ethane and stored in liquid nitrogen.
The cryo-EM data were collected using a 300 kV Titan Krios microscope (Thermo Fisher Scientific) equipped with a K2 Summit direct electron detector (Gatan) and a GIF-Quantum energy filter. Movie stacks were dose-fractioned into 32 frames, yielding a total dose of 60 e−/Å2 and binned to a pixel size of 0.82 Å (0.41 Å per super-resolution pixel) at a nominal magnification of 16,500. The data were automatically collected using SerialEM, with a nominal defocus set range of −1.2 to −1.5 μm.
A total of 3149 movie stacks were imported into CryoSPARC (v4.5.1) with collection parameters and then aligned into single-frame micrographs by performing patch motion correction37. Contrast transfer function (CTF) parameters of the micrographs were estimated by patch CTF estimation. An initial dataset of 3,430,206 particles were extracted and screened by rounds of 2D classification and hetero refinements. The selected 795,352 particles were then used for 3D classification with a focus mask of REC domain as an input parameter. One of four classes displayed better density of REC2 domain, therefore this class containing 215,731 particles was subjected to non-uniform refinement. A 2.86 Å map was generated after refinement, according to the gold standard Fourier shell correlation (GSFSC) 0.143 criterion.
The ternary complex model was manually built using WinCOOT (v0.9.6) with an initial NsCas9d structural model generated by AlphaFold3 as reference38,39. The clear densities for most nucleotides allowed de novo construction of sgRNA and dsDNA. The complex model was refined by Phenix.real_space_refine with the secondary structure restraints and evaluated by Phenix.validation_cryoem incorporating MolProbity40. All model graphics and cryo-EM density map figures were prepared using UCSF ChimeraX (v1.7.1) and PyMOL (v2.5.5)41,42.
Gene editing activity in mammalian cells
The expression sequence of NsCas9d protein was synthesized at GenScript with codon-optimized. NsCas9d protein expression sequence and its sgRNA expression sequence targeting the EMX1.1 site of the EMX1 gene were cloned into the PX459 plasmid as previously described protocol25. Additionally, the protein expression sequence of SpCas9 and its corresponding sgRNA expression sequence were cloned into the same site within the PX459 plasmid. The guide sequence of SpCas9 sgRNA specifically targets the EMX1.1 locus within the EMX1 gene. Genomic DNA was extracted after 52 h transfection in HEK293T cells. Primers were designed for the upstream and downstream regions of the EMX1.1 locus to amplify the targeted region using Q5 Hot start High-Fidelity DNA Polymerase (New England Biolabs). PCR product was extraction from agarose gel, and the recovered DNA was annealed at 95 °C for 5 min. The annealed sample was then incubated with T7E1 at 37 °C for 15 min26. The cleavage result of the targeted region DNA fragment by T7E1 was assessed by agarose gel electrophoresis.
Statistics and reproducibility
All biochemical assays were repeated at least three times with robust reproducibility.
Data availability
The atomic coordinate generated in this study has been deposited in the Protein Data Bank (PDB) under the accession code 9LGI (NsCas9d-sgRNA-dsDNA). The corresponding cryo-EM data has been deposited into the Electron Microscopy Data Bank (EMDB) with accession code EMD-63066. Four previously published atomic coordinates were used in Fig. 3e and Supplementary Fig. 3b, 5c, d: 7UTN (IscB-ωRNA-dsDNA), 5F9R (SpCas9-sgRNA-dsDNA), 6JE4 (NmCas9-sgRNA-dsDNA-AcrIIC3) and 5X2G (CjCas9-sgRNA-dsDNA). The source data underlying Figs. 1, 3c, 4c, 6 and Supplementary Figs 7, 8 are provided as a Source Data file. Source data are provided with this paper.
References
Barrangou, R. et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712 (2007).
Gasiunas, G., Barrangou, R., Horvath, P. & Siksnys, V. Cas9–crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 109, E2579–E2586 (2012).
Cong, L. et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013).
Mali, P. et al. RNA-guided human genome engineering via Cas9. Science 339, 823–826 (2013).
Chylinski, K., Makarova, K. S., Charpentier, E. & Koonin, E. V. Classification and evolution of type II CRISPR-Cas systems. Nucleic Acids Res. 42, 6091–6105 (2014).
Jinek, M. et al. A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012).
Nishimasu, H. et al. Crystal structure of Staphylococcus aureus Cas9. Cell 162, 1113–1126 (2015).
Yamada, M. et al. Crystal structure of the minimal Cas9 from Campylobacter jejuni reveals the molecular diversity in the CRISPR-Cas9 systems. Mol. Cell 65, e1103 (2017).
Wang, D., Tai, P. W. L. & Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 18, 358–378 (2019).
Li, C. & Samulski, R. J. Engineering adeno-associated virus vectors for gene therapy. Nat. Rev. Genet. 21, 255–272 (2020).
Altae-Tran, H. et al. The widespread IS200/IS605 transposon family encodes diverse programmable RNA-guided endonucleases. Science 374, 57–65 (2021).
Aliaga Goltsman, D. S. et al. Compact Cas9d and HEARO enzymes for genome editing discovered from uncultivated microbes. Nat. Commun. 13, 7602 (2022).
Ocampo, R. F. et al. DNA targeting by compact Cas9d and its resurrected ancestor. Nat. Commun. 16, 457 (2025).
Anantharaman, K. et al. Thousands of microbial genomes shed light on interconnected biogeochemical processes in an aquifer system. Nat. Commun. 7, 13219 (2016).
Yang, J. et al. Insights into the compact CRISPR–Cas9d system. Nat. Commun. 16, 2462 (2025).
Schuler, G., Hu, C. & Ke, A. Structural basis for RNA-guided DNA cleavage by IscB-ωRNA and mechanistic comparison with Cas9. Science 376, 1476–1481 (2022).
Kato, K. et al. Structure of the IscB–ωRNA ribonucleoprotein complex, the likely ancestor of CRISPR-Cas9. Nat. Commun. 13, 6719 (2022).
Sun, W. et al. Structures of Neisseria meningitidis Cas9 Complexes in Catalytically Poised and Anti-CRISPR-Inhibited States. Mol. Cell 76, e935 (2019).
Jiang, F. et al. Structures of a CRISPR-Cas9 R-loop complex primed for DNA cleavage. Science 351, 867–871 (2016).
Gasiunas, G. et al. A catalogue of biochemically diverse CRISPR-Cas9 orthologs. Nat. Commun. 11, 5512 (2020).
Zhang, Y. et al. Catalytic-state structure and engineering of Streptococcus thermophilus Cas9. Nat. Catal. 3, 813–823 (2020).
Gong, S., Yu, H. H., Johnson, K. A. & Taylor, D. W. DNA unwinding is the primary determinant of CRISPR-Cas9 activity. Cell Rep. 22, 359–371 (2018).
Nierzwicki et al. Principles of target DNA cleavage and the role of Mg2+ in the catalysis of CRISPR–Cas9. Nat. Catal. 5, 912–922 (2022).
Das, A. et al. Coupled catalytic states and the role of metal coordination in Cas9. Nat. Catal. 6, 969–977 (2023).
Ran, F. A. et al. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 (2013).
Sentmanat, M. F., Peters, S. T., Florian, C. P., Connelly, J. P. & Pruett-Miller, S. M. A survey of validation strategies for CRISPR-Cas9 editing. Sci. Rep. 8, 888 (2018).
Zuo, Z. & Liu, J. Cas9-catalyzed DNA cleavage generates staggered ends: evidence from molecular dynamics simulations. Sci. Rep. 6, 37584 (2016).
Zetsche, B. et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163, 759–771 (2015).
Swarts, D. C. & Jinek, M. Cas9 versus Cas12a/Cpf1: structure–function comparisons and implications for genome editing. WIREs RNA 9, e1481 (2018).
Shou, J., Li, J., Liu, Y. & Wu, Q. Precise and predictable CRISPR chromosomal rearrangements reveal principles of Cas9-mediated nucleotide insertion. Mol. Cell 71, e494 (2018).
Hirano, S. et al. Structure of the OMEGA nickase IsrB in complex with ωRNA and target DNA. Nature 610, 575–581 (2022).
Anders, C., Niewoehner, O., Duerst, A. & Jinek, M. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature 513, 569–573 (2014).
Edgar, R. C. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 5, 113 (2004).
Minh, B. Q. et al. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534 (2020).
Schneider, C. A., Rasband, W. S. & Eliceiri, K. W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012).
Esvelt, K. M. et al. Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nat. Methods 10, 1116–1121 (2013).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D. 66, 486–501 (2010).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. Sect. D. 75, 861–877 (2019).
Meng, E. C. et al. UCSF ChimeraX: tools for structure building and analysis. Protein Sci. 32, e4792 (2023).
Schrödinger, L. & DeLano, W. PyMOL. Retrieved from http://www.pymol.org/pymol (2020).
Acknowledgements
We would like to thank Professor Yongming Wang from the School of Life Sciences, Fudan University for sharing plasmid library containing 8N randomized nucleotides. We thank staff of the Center for Biological Imaging, Core Facilities for Protein Science at the Institute of Biophysics (IBP), Chinese Academy of Sciences (CAS). We thank the grants from National Key R&D Program of China (2023YFC3403400, 2023YFA0915000), Beijing Municipal Science & Technology Commission (Z231100007223004), the Natural Science Foundation of China (32330055, 31930065, 22121003, 32071198, 32071444), Beijing Natural Science Foundation (5232022), the Chinese Academy of Sciences (XDB0570000 and XDB37010202).
Author information
Authors and Affiliations
Contributions
K.W. and Y.W. conceived the study. K.W. expressed, purified NsCas9d-sgRNA binary complex, and made the sample of NsCas9d-sgRNA-dsDNA ternary complex for cryo-EM structure determination. K.W. and X.Y. collected the cryo-EM data. K.W., J.W. and X.Y. built and refined the structural model. K.W., W.S. and G.S. performed the biochemical assays. Y.W. and K.W. wrote the manuscript. Y.W. supervised all the structural and biochemical studies.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Ailong Ke, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, K., Wang, J., Yang, X. et al. Structural insights into Type II-D Cas9 and its robust cleavage activity. Nat Commun 16, 7396 (2025). https://doi.org/10.1038/s41467-025-62128-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-62128-8
