
Abstract
Trimethylamine N-oxide (TMAO) contributes to cardio-metabolic diseases, with hepatic flavin-containing monooxygenase 3 (FMO3) recognized as its primary source. Here we demonstrate that elevated adipocyte FMO3 and its derived TMAO trigger white adipose tissue (WAT) dysfunction and its related metabolic disorders in ageing. In adipocytes, ageing or p53 activation upregulates FMO3 and TMAO levels. Adipocyte-specific ablation of FMO3 attenuates TMAO accumulation in WAT and circulation, leading to enhanced glucose metabolism and energy and lipid homeostasis in ageing and obese mice. These improvements are associated with reduced senescence, fibrosis and inflammation in WAT. Proteomics analysis identified TMAO-interacting proteins involved in inflammasome activation in adipocytes and macrophages. Mechanistically, TMAO binds to the central inflammasome adaptor protein ASC, promoting caspase-1 activation and interleukin-1β production. Our findings uncover a pivotal role for adipocyte FMO3 in modulating TMAO production and WAT dysfunction by promoting inflammasome activation in ageing via an autocrine and paracrine manner.
Similar content being viewed by others
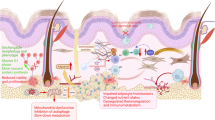

Introduction
Ageing, featured by chronic inflammation and senescence, is a major contributor to cardiometabolic diseases, such as type 2 diabetes1,2,3,4. White adipose tissue (WAT) acts as an endocrine organ to maintain systemic energy and glucose homeostasis. Transcriptomic and proteomic analyses indicate that WAT is the first tissue showing functional decline in ageing5,6,7. WAT is composed of diverse cell populations, including mature white adipocytes that produce bioactive adipokines to communicate and coordinate with the neighboring cells and distal metabolic tissues in control of systemic metabolism under varying nutritional and environmental conditions8,9,10,11,12. Ageing alters composition and functionality as well as the interaction of the adipocytes and the WAT-resident cells. For instance, activation of the NLR family pyrin domain-containing 3 (NLRP3) inflammasome in adipose tissue-resident macrophages (ATM) produces growth differentiation factor-3 and monoamine oxidase A that impair lipolysis in mature adipocytes during ageing13,14. On the other hand, p53-dependent senescence in adipocytes impairs adipogenesis and induces inflammation and insulin resistance in WAT under ageing and obesity15,16,17. Despite these insights, the mechanisms underlying WAT deterioration and its precise contribution to metabolic regulation during ageing remain inadequately understood.
Gut microbiota controls host metabolism by generating an array of metabolites targeting to multiple metabolic tissues18,19,20,21. Flavin-containing monooxygenase 3 (FMO3), a xenobiotic metabolizing enzyme primarily expressed in the liver, converts gut microbiota-produced trimethylamine (TMA) from its nutrient precursors (such as choline, L-carnitine, and betaine) into trimethylamine-N-oxide (TMAO) via hepatic FMO322,23,24. Early human and animal studies showed the important role of this microbiota-host axis in cardiometabolic health22,23,25. In rodent models, knockdown of hepatic FMO3 using anti-sense oligonucleotides or global deletion of FMO3 improves hepatic insulin resistance, hyperlipidemia, obesity and atherosclerosis22,26,27. Dietary treatment with TMAO promotes inflammation in visceral WAT (vWAT) by upregulating the expression of pro-inflammatory cytokines, including MCP-1 and TNF-α of C57BL/6 mice fed with a high-fat diet (HFD)26. Furthermore, Schugar et al demonstrated that global deletion of FMO3 or hepatic downregulation of FMO3 alleviates diet-induced obesity by promoting adaptive thermogenesis and beiging in WAT via TMAO, but whether this effect is adipocyte-autonomous is unknown27. Recently, circulating TMAO level but not its nutrient precursors has been reported to be significantly increased in elderly humans (above 65-year-old) compared with those in young (18–44-year-old) or middle-aged (45–64-year-old) humans and pre-mature ageing rodents28. In addition, TMAO has been shown to accelerate ageing-related vascular and brain diseases by triggering a senescent response in rodents28,29, however, its effects on metabolic homeostasis and its source of production in ageing remain elusive.
Although the liver is considered the main site for TMAO production via FMO3, we here demonstrate that adipocyte FMO3 is the contributor to the elevated TMAO level in ageing. We found that FMO3 and TMAO are abundantly expressed in mature adipocytes of WAT, and their levels are induced in humans and rodents with ageing via a p53-dependent pathway. Adipocyte-specific deletion of FMO3 protects against ageing- or obesity-induced functional decline of WAT, accompanied by improvement of glucose, lipid homeostasis and energy balance in mouse models. Adipocyte FMO3-derived TMAO acts as an autocrine and paracrine factor to trigger inflammasome activation and subsequent IL-1β production in mature adipocytes and ATM. Our proteomics analysis identifies numerous TMAO-binding proteins that participate in inflammatory pathways, particularly inflammasome activation. Our study uncovers how aged adipocytes convert gut microbiota-derived metabolite to elicit adipose tissue dysfunction and systemic dysmetabolism in ageing.
Results
Matured adipocytes synthesize TMAO via FMO3 in mice
The gene expression level of FMO3 in human subcutaneous WAT (sWAT) has been shown to be positively correlated with body mass index27. However, whether FMO3 protein and its metabolic product TMAO can be found in fat depots, and its physiological relevance has never been explored. We first examined expression profiles of FMO3 and TMAO levels across various WAT, BAT, and the liver in 8-week-old male and female C57BL/6 J mice. In line with previous studies30,31, hepatic FMO3 mRNA and protein levels were significantly higher in female mice compared to males (Fig. 1a, b), perhaps due to the negative effect of androgen on FMO3 expression25,31. Notably, Fmo3 mRNA expression in WAT and BAT was approximately 99% lower than in the liver for both sexes (Fig. 1a), yet FMO3 protein expression in these adipose tissues approached ~60% and ~30% of hepatic levels in male and female mice, respectively (Fig. 1b). Interestingly, FMO3 expression in adipose depots did not exhibit sexual dimorphism, indicating that factors other than sex hormones may regulate adipose FMO3 expression. Ex vivo assay showed that both WAT and BAT explants could oxidize deuterated trimethylamine (d9-TMA) to d9-TMAO in a dose-dependent manner, albeit less efficiently than the primary hepatocytes (Fig. 1c–f). We further identified the cellular sources of FMO3 and TMAO production within WAT. Immunoblotting revealed that mature adipocytes expressed FMO3 at higher levels than stromal vascular fraction (SVF) cells isolated from sWAT of 8-week-old C57BL/6 mice (Fig. 1g). Moreover, FMO3 expression increased during the differentiation of 3T3-L1 preadipocytes into mature adipocytes (Fig. 1h). Both SVF- and 3T3-L1-derived mature adipocytes synthesized d9-TMAO from d9-TMA (Fig. 1i, j); however, this capability was abolished upon Fmo3 knockdown in mature 3T3-L1 adipocytes (Fig. 1k, l). Similar to hepatocytic AML12 cells, FMO3 was abundantly expressed in the microsomes of 3T3-L1 mature adipocytes (Supplementary Fig. 1a). The microsome preparation from the adipocytes was capable of converting d9-TMA into d9-TMAO under incubation conditions of 37 oC and a pH range of 7.4–9.4 (Supplementary Fig. 1b, c). However, this conversion was inhibited by addition of the FMO3 inhibitor 3,3’-diindolylmethane (DIM)32, increasing temperature to 45 oC, or reducing the pH to 6.4 or 5.4 during the incubation (Supplementary Fig. 1b–d).

a–f 8-week-old male and female C57BL/6 mice were used. qPCR (a) and immunoblotting (b) analyses of FMO3 expression in subcutaneous white adipose tissue (sWAT), epididymal white adipose tissue (eWAT), brown adipose tissue (BAT) and the liver. Relative FMO3 expression is normalized to that in eWAT of male mice. Densitometry analysis of FMO3 normalized with HSP90 is shown in (b). a: n = 5 for male; n = 7 for female. b: n = 4. c–f d9-TMAO production after incubating eWAT, sWAT and BAT explants and primary hepatocytes (HCs), with d9-TMA (200 μM or 500 μM) for 48 h. Levels in tissue lysates were normalized to total protein concentration. sWAT: n = 5 (c and e); n = 6 (d and f). eWAT: n = 4 (c and e, males), and n = 6 in remaining panels. BAT: n = 4 (f, males), and n = 6 in other panels. Hepatocytes: n = 3. g Immunoblotting analysis of FMO3. n = 4. h 3T3-L1 cells were differentiated into mature adipocytes, followed by immunoblotting analysis of FMO3, adiponectin (APN; adipocyte marker) and HSP90. The day before differentiation is day 0. Equal amounts of liver lysate were loaded as the positive control of FMO3. n = 2. i, j Mature adipocytes or stromal vascular fraction (SVF) from eWAT (i), and undifferentiated 3T3-L1 fibroblasts (preadipocytes) or differentiated 3T3-L1 adipocytes (j) were incubated with 100 μM d9-TMA for 24 h, followed by d9-TMAO measurement in the cell lysate. i: n = 4. j: n = 3. k, l 3T3-L1 mature adipocytes transfected with siRNA against Fmo3 (siFmo3) or scramble control (siScramble) for 48 h, were subjected to immunoblotting analysis of FMO3 (k) and d9-TMAO detection as indicated (l). n = 3. m, n Matured adipocytes differentiated from SVF of sWAT of 6-week-old adipocyte-FMO3 knock-out (KO) mice and their wild-type (WT) littermates were used. m Immunoblotting and respective densitometry analyses of FMO3 normalized with β-actin. n = 4. n d9-TMAO levels measured as indicated. n = 4. Data represented as mean ± SEM. Significance was calculated using one-way ANOVA for (a–f) and two-tailed Student’s t-test for (g, h, k, l–n), with Welch’s correction for (i and j).
To validate these findings in vivo, we generated an adipocyte-specific FMO3 knockout (Adipo-FMO3 KO) mouse model using the Cre-loxP recombination system (Supplementary Fig. 2a-b). Adipose tissue and liver samples were harvested from 6-week-old male Adipo-FMO3 KO mice and wild-type (WT) controls for analysis. Immunoblotting analysis showed no difference in FMO3 protein in the livers of male Adipo-FMO3 KO mice and WT controls (Supplementary Fig. 2c). Immunofluorescence staining revealed a robust expression of FMO3 in perilipin-positive adipocytes within eWAT and sWAT of WT mice, which was markedly diminished in male Adipo-FMO3 KO counterparts (Supplementary Fig. 2d-e). In line with this, adipocytes fractionated from sWAT and eWAT of male Adipo-FMO3 KO mice displayed reduced FMO3 protein levels relative to WT controls, whereas the SVF showed no FMO3 expression (Supplementary Fig. 2f, g). This reduction in FMO3 was associated with decreased production of d9-TMAO from d9-TMA in sWAT and eWAT explants (Supplementary Fig. 2h, i). Additionally, mature adipocytes derived from the SVF of Adipo-FMO3 KO mice exhibited lower Fmo3 mRNA (Supplementary Fig. 2j) and protein expression (Fig. 1m), as well as diminished d9-TMAO synthesis (Fig. 1n), compared to WT controls. Notably, the deletion of FMO3 did not affect the expression of adipogenic markers, such as adiponectin (Adipoq) and peroxisome proliferator-activated receptor gamma (Pparg2), nor the expression of Fmo2 (Supplementary Fig. 2j). In BAT, Fmo3 expression (Supplementary Fig. 2k) and d9-TMAO production (Supplementary Fig. 2h) were also significantly reduced in male Adipo-FMO3 KO mice compared to WT controls. Intraperitoneal administration of d9-TMA in male WT mice led to a rapid increase in circulating d9-TMAO levels, an effect that was partially attenuated by adipocyte-specific FMO3 deletion (Supplementary Fig. 2l). We observed similar reduction of FMO3 expression in SVF-adipocytes and d9-TMAO biosynthesis from d9-TMA in WAT and BAT depots but not in the liver from female Adipo-FMO3 KO mice compared to those from WT controls (Supplementary Fig. 3). These results indicate that adipose tissues are also able to produce TMAO via FMO3.
FMO3 and its derived TMAO are increased in WAT during ageing
As FMO protein family has been implicated in ageing33,34,35, we hypothesised that FMO3 expression changes in the adipose tissue and other tissues during ageing. We re-analysed the publicly available RNAseq dataset containing Fmo3 mRNA expression in multiple tissues of 1-month-old and extremely old (27-month-old) C57BL/6JN mice6. This analysis showed that Fmo3 mRNA expression was increased in BAT, eWAT and mesenteric WAT (mWAT), but not in other examined tissues, including the liver, brain, heart, etc, of 27-month-old mice (Fig. 2a). To confirm the change of FMO3 expression at the protein level and examine TMAO abundance, we isolated sWAT, eWAT, BAT and the liver from 12-week-old (so-called “young” hereafter) and 2-year-old male C57BL/6 J (so-called “old” hereafter) mice15. Immunoblotting analysis revealed that FMO3 protein was induced in eWAT and sWAT but not in the liver of the old mice (Fig. 2b-d). These changes were accompanied by the elevation of circulating and adipose TMAO levels (Fig. 2e–g), as determined by LC-MS/MS. On the other hand, no change was observed in the level of circulating choline (Fig. 2e), the precursor of TMAO. The increase of FMO3 expression in WAT and TMAO levels in circulation were also observed in the aged female C57BL6/J mice (Supplementary Fig. 4). The transcriptome dataset GSE175495 revealed an upregulation of FMO3 mRNA levels in sWAT from the aged human subjects (aged 60-85 years) compared to those from younger subjects (aged 20–35 years)36 (Fig. 2h). Weighted Gene Co-expression Network Analysis (WGCNA)37 of this dataset identified 13 gene modules in which the brown module was significantly and positively associated with age (Supplementary Fig. 5a-e). Importantly, the brown module contained FMO3 gene. The co-expressed genes together with FMO3 were functionally related to immune cell activation, as revealed by Gene-ontology-biological process (GO-BP) analysis (Supplementary Fig. 5f). We further performed differential gene expression analysis with low FMO3 as a reference group, followed by Gene Set Enrichment Analysis (GSEA). To get a better differential resolution, we only included the top 9 and bottom 9 samples from each group (so-called low FMO3 and high FMO3 expression groups)38 (Supplementary Fig. 6a, b). The GSEA results demonstrated that high FMO3 expression group were positively correlated with inflammatory responses and negatively correlated with adipose tissue functions, including adipogenesis, cholesterol metabolism, and fatty acid oxidation (Supplementary Fig. 6c-d).

a FMO3 expression in various tissues of 1-month-old (so-called “Young”, n = 6), 27-months-old (so-called “Old”, n = 4) and between these two time points from RNA-sequencing data of Mus musculus with accession number GSE132040. Liver: n = 17 for 0–6 M, n = 12 for 7–12 M and 13-18 M, and n = 14 for 19–27 M. b–g 3-month-old (Young) and 24-month-old (Old) male C57BL/6 J mice were used. b–d Immunoblotting analysis of FMO3 expression in eWAT, sWAT, BAT and liver. Densitometry analysis of FMO3 normalized with HSP90 is shown in (d). n = 4 for sWAT, eWAT, and BAT. n = 3 for liver. e Circulating levels of TMAO and choline. n = 5 for young. n = 4 for old. f TMAO level in the liver, eWAT and BAT as indicated. TMAO level is normalized with total protein concentration. n = 5 for liver and BAT. n = 3 for young and n = 5 for old in eWAT. g sWAT explants were incubated with 200 μM d9-TMA for 48 h, followed by d9-TMAO detection in tissue lysate. d9-TMAO level is normalized with total protein concentration. n = 3. h FMO3 expression in sWAT of 20–35 years old (so-called “Young”) and 60-85 years old (so-called “Old Aged”) from RNA-sequencing data of human subjects with accession number GSE175495. n = 12. i, j “Young” is defined as ages 16-46 years old (n = 6) and “Old Aged” as age ≥ 60 years old (n = 8) obtained from non-diabetes patients with nonfunctional adrenal adenoma who underwent adrenalectomy or partial adrenalectomy. i TMAO level in human sWAT. n = 14. j Pearson correlation analysis of TMAO level in sWAT and age. n = 14. k FMO3 expression in sWAT of humans after 1 and 2 years of calorie restriction. The BioProject accession number is PRJNA1018321. n = 6. All samples are biologically independent replicates, except for figure k for which samples used are repetitive measures of biological samples. Data are represented as mean ± SEM. Statistical significance was assessed using one-way ANOVA (a), one-way repeated (k) measures ANOVA, Student’s t-test (d–f with Welch’s correction for h), and Mann-Whitney U test (i). Abbreviations: Mesenteric white adipose tissue (mWAT).
To examine TMAO abundance in WAT in ageing, we collected sWAT from human subjects with non-functional adrenal adenoma who underwent adrenalectomy or partial adrenalectomy. The subjects were grouped according to their age into young (aged 16–46-year-old) and aged (age ≥60-year-old). LC-MS/MS analysis showed that the TMAO level was higher in sWAT isolated from the aged subjects and was positively associated with age (Fig. 2i-j). Calorie restriction is known to alleviate ageing-induced adipose tissue dysfunction. We examined whether calorie restriction alters FMO3 expression in sWAT using the RNAseq dataset of human sWAT from humans who underwent an average of 14% sustained calorie restriction39. FMO3 expression was reduced after 1 and 2 years of calorie restriction (Fig. 2k). On the contrary, expression of other FMO protein members were not altered by calorie restriction (Supplementary Fig. 7).
p53 activation induces FMO3 expression and TMAO production in mature adipocytes and WAT
To investigate the mechanism underlying the above changes in FMO3 level in ageing, we predicted which transcription factor(s) control the FMO3 promoter activity using Jaspar database. Apart from hypoxia-inducible factor 1 alpha (HIF-1α) as recently reported40, we found two putative p53 responsive elements (RE) in the promoter and intron 1 of the human FMO3 gene (Fig. 3a). p53 is activated in adipocytes during ageing, which triggers senescence, inflammation, and insulin resistance15,16. We treated 3T3-L1 adipocytes with two different p53 activators, namely doxorubicin (a chemotherapy drug with DNA-damaging and senescence-inducing properties) (Fig. 3b–e) and nutlin-3a (an MDM2 inhibitor that blocks the MDM2-p53 interaction and upregulates p53 expression and senescence) (Supplementary Fig. 8a, b) for 24 h17. Treatment with nutlin-3a increased mRNA levels of Fmo3 by 7-fold and protein levels by 5.5-fold, accompanied by induction of the senescent markers, including p53 and Cdkn1a (Supplementary Fig. 8a, b). Treatment with doxorubicin not only upregulated the senescent markers but also FMO3 expression and d9-TMA to d9-TMAO conversion in 3T3-L1 mature adipocytes (Fig. 3a–e and Supplementary Fig. 9a, b). Likewise, FMO3 and TMAO could be detected in human adipocytes derived from adipose-derived mesenchymal stem cells and SGSB cells, and their levels were also upregulated by doxorubicin or nutlin-3a treatment (Supplementary Figs. 8c–g and 9c–h). The doxorubicin-induced TMAO production in mature adipocytes was largely abrogated by genetic deletion of FMO3 (Fig. 3f) or treatment with DIM (Supplementary Fig. 9e).

a A diagram showing two putative p53 responsive elements (RE) on the human FMO3 gene identified by JASPAR. Upper panel: consensus p53 RE suggested by JASPAR. Lower panel: The first and second putative p53 RE sites are located on the promoter and intron 1 of FMO3 gene as indicated. The number of nucleotides is relative to the ATG start codon. b–d 3T3-L1 adipocytes treated with DMSO as vehicle or doxorubicin (10 μM) for 24 h were subjected to qPCR analysis (b) and immunoblotting analysis (c). The bar chart in (d is the densitometry analysis of FMO3, p53 and p21 normalized with HSP90. n = 3 for vehicle. n = 4 in (b) and n = 3 in (c) for doxorubicin. e, f 3T3-L1 adipocytes (e) or SVF-derived adipocytes from Adipo-FMO3-KO mice or WT controls (f) were treated with vehicle (Veh) or doxorubicin (Doxo; 10 μM) for 24 h, followed by 200 μM d9-TMA for 16 h. d9-TMAO production in cell lysate or conditioned medium (e, f) as indicated. n = 4 for vehicle, doxorubicin and d9-TMA in (e, f). n = 5 in panel e for doxorubicin ± d9-TMA. n = 4 in the cell lysate and n = 3 in the cell culture medium of panel f for doxorubicin ± d9-TMA. g–k 16-week-old male C57BL/6 mice injected with a single dose of 2 mg and 10 mg of doxorubicin per kg of body weight or 1X PBS (Veh) were used. The day before doxorubicin injection is defined as day 0. qPCR analysis of Fmo3, p53 and Cdkn1a mRNA expression are normalized with 36b4 and 18 s in sWAT (g) and eWAT (h). g: n = 5 for vehicle and 2 mg doxorubicin injection, and n = 4 for 10 mg doxorubicin injection for Cdkn1a and p53; for Fmo3, n = 5 for vehicle and 10 mg doxorubicin injection, and n = 4 for 2 mg doxorubicin injection. h: n = 4 for Cdkn1a and p53; for Fmo3, n = 4 for vehicle and 10 mg doxorubicin injection, and n = 5 for 2 mg doxorubicin injection. TMAO level in eWAT and sWAT at day 10 (i) and in serum at day 0 and day 10 (j). i: n = 4 for eWAT. n = 4 for vehicle and 10 mg doxorubicin, and n = 5 for 2 mg doxorubicin for sWAT. j: n = 5. k qPCR analysis of Fmo3 gene expression normalized with 18 s in liver. n = 5. l 3T3-L1 adipocytes were co-transfected with a vector expressing luciferase (pGL3) under the control of Fmo3 promoter or intron-1 region containing p53 RE or no promoter (Basic) for 12 h, followed by treatment with 10 μM nutlin-3a for 24 h. n = 4. m 3T3-L1 adipocytes were co-transfected with plasmids expressing GFP or GFP-tagged p53 together with indicated luciferase vectors for 48 h, followed by measurement of luciferase activity. n = 3. The data are expressed as fold change over pGL3-Basic in the cells treated with Veh (l) or transfected with GFP (m). Data expressed as mean ± SEM, analysed by two-tailed Student’s t-test or one-way ANOVA.
To confirm the effect of doxorubicin on FMO3 and TMAO induction in vivo, we subjected 12-week-old male and female mice with a single dose injection of doxorubicin (2 mg/kg or 10 mg/kg) as previously described17,41. After 10 days, the senescent markers and FMO3 (Fig. 3g, h and Supplementary Fig. 10a, b and g) and its derived TMAO (Fig. 3i and Supplementary Fig. 10c) in sWAT and gonadal WAT (gWAT) were upregulated in the mice injected with doxorubicin compared to those injected with vehicle in both genders. These changes were accompanied by an increase in circulating TMAO level (Fig. 3j and Supplementary Fig. 10d). However, doxorubicin treatment exerted no obvious effect on Fmo3 mRNA or protein levels and TMAO levels in the liver (Fig. 3k and Supplementary Fig. 10c and e, f). To examine whether p53 directly controls FMO3 activity, we cloned the human FMO3 promoter and intron 1 region consisting of p53 RE into a pGL3-luciferase vector. The luciferase assay showed that overexpression of p53 or treatment with nutlin-3a enhanced luciferase expression controlled by the promoter but not the intron 1 of FMO3 in 3T3-L1 cells (Fig. 3l-m).
Adipocyte-specific deletion of FMO3 alleviates ageing-induced adipose tissue dysfunction and metabolic diseases
Next, we investigated whether adipocyte-specific deletion of FMO3 protects against age-related adipose tissue dysfunctions and its associated metabolic disorders. Circulating levels of TMAO were comparable between male Adipo-FMO3 KO and WT mice at 3 months and 1-year of age (Fig. 4a). However, significantly lower TMAO level, by 1.5-fold, was observed in male Adipo-FMO3 KO mice at around 2 years of age, indicating that elevated circulating TMAO level in ageing is mainly because of increased adipocyte FMO3-derived TMAO (Fig. 4a). Circulating levels of glucose, insulin, homeostatic model assessment for insulin resistance (HOMA-IR), glucose tolerance and insulin sensitivity were similar between male Adipo-FMO3 KO mice and their WT littermates at 1 year of age (Supplementary Fig. 11a–e). However, adipocyte-specific deletion of FMO3 markedly improved glucose homeostasis in mice aged ~18–20-month-old (Fig. 4b–i), which age is analogous to 60–70-years in human42. Furthermore, energy expenditure (Fig. 4j), oxygen consumption and locomotor activity (Fig. 4k-m) were significantly elevated in 22-month-old male Adipo-FMO3 KO mice, when compared to their respective WT controls. 22-month-old male Adipo-FMO3 KO mice also had a modest reduction of respiratory exchange ratio (RER) (Fig. 4n), reflecting a better ability in lipid utilisation. At 1-year-old, male Adipo-FMO3 KO mice displayed a higher energy expenditure and RER but no change in locomotor activity or oxygen consumption (Supplementary Fig. 11f–i). Dyslipidaemia, characterised by hypercholesterolemia and hypertriglyceridemia, is commonly observed in the elderly. Male Adipo-FMO3 KO mice displayed improved triglyceride clearance and lower levels of low-density lipoprotein cholesterol (LDL-C) relative to WT controls when they were around 1-year-old and 2-year-old (Fig. 4o & Supplementary Fig. 11j-k). Additionally, there was a tendency towards increased life expectancy in male Adipo-FMO3 KO mice compared to their WT counterparts, yet a larger sample size and longer monitoring duration are required to confirm the change (Fig. 4p). Loss of muscle mass is a well-documented phenomenon in ageing. We found an increase in lean mass in male Adipo-FMO3 KO mice at age of 18-20-month-old, when compared to their respective WT control groups (Supplementary Fig. 12).

6 to 8-week-old (so-called “Young”), 48 to 50-week-old (so-called “Middle-aged) and 85 to 105-week-old (so-called “Old”) male WT and KO mice on standard chow diet were used. a Serum levels of TMAO. n = 5. Circulating glucose (b) and serum insulin levels (d) during IPGTT in the 8-week-old and 99-week-old mice; AUC of glucose level during GTT in (c). b, c n = 6 for young and n = 7 for old animals. d n = 6 for WT and n = 4 for KO animals. e, f Circulating glucose level (e) during ITT in the 7-week-old and 87-week-old mice. The bar chart on (f) represents the AUC of glucose during ITT. n = 7 for young and n = 5 for old animals. Circulating glucose (g), insulin (h) and calculated HOMA-IR (i) after fasting for 6 h. n = 7 for young animals. n = 5 for WT and n = 7 KO for old animals. Energy expenditure (j), locomotor activity and its AUC (k, l) oxygen consumption rate (m) and respiratory exchange ratio (RER) (n) measured in the 95-week-old mice using metabolic cage. WT: n = 6. KO: n = 4 in (j and m), and n = 6 in (k and n). o Serum total cholesterol (TC), low-density lipoprotein (LDL) and high-density lipoprotein (HDL) in 105-week-old mice after fasting for 6 h. n = 5 for WT. n = 8 for KO. p Survival rates were compared using log rank test for the trend up to 105 weeks. n = 9. Data are represented as mean ± SEM. Statistical data were analysed by two-tailed Student’s t-test or one-way ANOVA. Significant P-values: # WT-young v/s WT-old; $ KO-young v/s WT-old; * WT-old v/s KO-old.
Next, we examined whether the amelioration of systemic metabolism is due to the alleviation of adipose tissue dysfunction during ageing. We harvested eWAT from 2-year-old male Adipo-FMO3 KO mice and WT controls for RNA sequencing (RNAseq) analysis. Differential gene expression analysis revealed 267 upregulated and 265 downregulated genes in male Adipo-FMO3 KO mice when compared to the controls. GSEA demonstrated that the pathways related to p53-related senescence, interferon-α/γ, inflammatory responses and NF-κB were downregulated, whereas those related to adipogenesis, and cholesterol homeostasis were upregulated by adipocyte-specific deletion of FMO3 (Fig. 5a and Supplementary Fig. 13a–e). This data indeed overlaps with the GSEA analysis of human sWAT with high and low FMO3 expression (Supplementary Fig. 6c, d). Notably, senescence, inflammation and fibrosis are recognised as hallmark features of WAT dysfunction in ageing43, and therefore were further evaluated in subsequent experiments. QPCR analysis confirmed reduction of the gene related to inflammatory cytokines and chemokine (such as Il1b, Il18, Cxcl9, Ccl5, F4/80, Nos2), senescent genes (such as p53 and Cdkn1a) and fibrotic genes (Col1a1, Col3a1, Col4a1, Col6a3 and Tgfb) (Fig. 5b and Supplementary Fig. 13f). Histological analysis corroborated these gene expression data, showing decreased immune cell infiltration (Fig. 5c), pro-inflammatory M1 macrophages (identified by F4/80+iNOS+ positive staining) (Fig. 5d), and lower collagen deposition (revealed by Sirius red staining) (Supplementary Fig. 13g) in eWAT of male Adipo-FMO3 KO mice. In line with the improvement of fibrosis and inflammation, a reduction in adipocyte hypertrophy was observed in the eWAT of male Adipo-FMO3 KO mice (Supplementary Fig. 13h). To further substantiate the observed senescence amelioration, we evaluated the expression of the DNA damage marker (phosphorylation of H2AX at Ser 139 [pH2AX]) and p53 in adipocytes via immunofluorescent staining. This analysis showed that pH2AX and p53 intensity were less pronounced in adipocytes of male Adipo-FMO3 KO mice than that in the WT controls (Fig. 5e, f). Immunoblotting analysis also indicated a significant reduction of p53 and its downstream target p21, which is responsible for cell cycle arrest and senescence-associated secretory phenotype (SASP)2, in the eWAT of male Adipo-FMO3 KO mice (Fig. 5g). Given inflammasome activation is observed in aged WAT1 and reduction of Il1b and Il18 in eWAT of aged male Adipo-FMO3 KO mice, we further evaluated the key inflammasome molecules in eWAT of our animal model. Mature IL-1β, the active form of caspase-1 and NLRP3 proteins were downregulated in eWAT of male Adipo-FMO3 KO mice (Fig. 5g). Circulating levels of inflammasome cytokines, including IL-1β and IL-18 were diminished in male Adipo-FMO3 KO mice, whereas MCP-1 and adiponectin remained unchanged (Fig. 5h–k). In addition, circulating leptin level was also reduced in male Adipo-FMO3-KO mice (Fig. 5l).

eWAT and serum isolated from 105-week-old male WT and KO mice on standard chow diet were used. a Gene set enrichment analysis (GSEA) plot of WT vs KO mice. Inflammatory response and p53 pathways are downregulated in KO. n = 4 for WT. n = 5 for KO. b qPCR analysis of genes related to inflammation and p53 pathways. n = 5 for WT. n = 6 for KO. c H&E staining. Immune cell clusters are pointed by red arrows. The lower panel is the quantification of the number of immune cells per 103 μm2 of area. Scale bar: 200 µm. n = 5. d Immunofluorescence staining for the macrophage marker F4/80 and iNOS. Bar charts on the right of panel d show the quantification of iNOS level in F4/80+ cells and the number of F4/80+ cells. Scale bar: 200 µm. n = 5. e, f Immunofluorescence staining for cellular senescence marker p53 (e) and DNA damage marker phospho-γ-H2AX (p-H2AX) (f) together with the adipocyte marker perilipin. The bar charts in (e and f) are quantification of p53 and p-H2AX in perilipin+ adipocytes. Scale bar: 200 µm. n = 4 for WT. n = 5 for KO. g Immunoblotting analysis of the proteins related to p53 pathways (p53 and p21) and inflammasome (pro-IL-1β), cleaved-IL-1β (p17), pro-caspase-1, cleaved caspase-1 (p10) and NLRP3. The bar chart on the right panel is the densitometric quantification for the indicated proteins normalized with β-actin. n = 5 for WT. n = 6 for KO. Circulating levels of IL-1β (h), IL-18 (i), MCP-1 (j), adiponectin (k) and leptin (l) in the 105-week-old mice. n = 5 for WT. n = 6 for KO. All samples are biologically independent replicates. Data are represented as mean ± SEM. Statistical data were analysed by the Mann-Whitney U test for (c) and a two-tailed Student’s t-test for the remaining graphs, with Welch’s correction applied for (d).
We also evaluated the impacts of FMO3 deletion on sWAT and BAT. Despite no change in the number of immune cells and crown-like structure detected by H&E staining, adipocytes in sWAT of male Adipo-FMO3 KO mice were less hypertrophic (Supplementary Fig. 14a-d). Sirius red staining and the QPCR analysis showed that adipocyte deletion of FMO3 also modestly improved fibrosis in sWAT (Supplementary Fig. 14e-f). QPCR and immunoblotting analysis showed that inflammatory macrophage markers, including F4/80, iNOS and IL-1β and inflammasome protein NLRP3 were reduced in sWAT of male Adipo-FMO3 KO mice (Supplementary Fig. 14f-g). The improvement was associated with reduced TMAO level in the sWAT of male Adipo-FMO3 KO mice (Supplementary Fig. 14h). The BAT isolated from male Adipo-FMO3 KO mice displayed less lipid accumulation and higher gene expression of Prmd16 (a key transcriptional factor maintaining brown adipocyte identity and function in adults and ageing44,45) and a lower level of p53, despite no change in other thermogenesis-related genes including Ucp1 and Cidea and inflammatory cytokines including Tnfa and Il1b (Supplementary Fig. 15). Consistent to the findings in the male mice, female Adipo-FMO3 KO mice demonstrated (1) a reduction of TMAO levels in the circulation and WAT; (2) better insulin sensitivity and glucose and lipid homeostasis; (3) improvement of senescence, inflammation and fibrosis as well as reduction of inflammasome proteins and genes in WAT under ageing conditions (Supplementary Figs. 16-18). These findings suggested that adipocyte-specific deletion of FMO3 alleviates ageing-induced adipose tissue dysfunction.
Obesity is known to accelerate ageing and induce metabolic diseases by exacerbating adipose tissue inflammation and senescence2,46. We next examined whether adipocyte-specific deletion of FMO3 prevents high-fat-diet (HFD)-induced metabolic dysfunction. Male Adipo-FMO3 KO mice had lower levels of circulating TMAO, fasting insulin, glucose and IL-1β when compared to WT controls, after the HFD feeding for 24-weeks (Fig. 6a-d). Although there was an increase in oxygen consumption in male Adipo-FMO3 KO mice under room temperature and cold environment, there was no significant difference in body weight, food intake, RER, and locomotor activity among the two groups (Supplementary Fig. 19a-e). Male Adipo-FMO3 KO mice exhibited better glucose tolerance (Fig. 6e) and insulin sensitivity (Fig. 6f) after the HFD feeding for 11 and 12 weeks, respectively. In addition, insulin secretion during GTT (Fig. 6g) and HOMA-IR (Fig. 6h) were reduced in male Adipo-FMO3 KO mice, reflecting improved insulin sensitivity. Similar to the observation in the ageing model, M1 macrophage infiltration (assessed by flow cytometry), senescence (assessed by β-gal staining), crown-like structure (assessed by H&E staining), fibrosis (assessed by Sirius red staining) and inflammasome activation (assessed by mature form of IL-1β and cleavage form of caspase-1) were diminished in eWAT of male Adipo-FMO3 KO mice when compared to those in WT controls (Fig. 6i-n). These changes were associated with ~50% reduction of adipose TMAO level (Fig. 6o). Circulating levels of IL-1β (Fig. 6d), triglyceride, and ALT were reduced in HFD-fed male Adipo-FMO3 KO mice, whereas serum leptin, MCP1, total cholesterol and LDL cholesterol remained unchanged between the two groups (Supplementary Fig. 19f-j). In addition, the insulin-sensitizing and anti-inflammatory adipokine adiponectin was increased in the circulation of male Adipo-FMO3 mice under HFD (Supplementary Fig. 19k).

eWAT and serum isolated from 32-week-old male Adipo-FMO3-WT and KO mice on a high-fat diet (HFD, fed from week 8) were used unless stated otherwise. a Serum levels of TMAO. WT: n = 7 for WT. n = 8 for KO. Circulating insulin (b) and glucose (c) after fasting for 6 h. n = 8. d Circulating level of IL-1β. n = 7 for WT. n = 8 for KO. e Circulating glucose levels during IPGTT in the 19-week-old mice on HFD feeding for 11 weeks. n = 8 for WT. n = 6 for KO. f Circulating glucose levels during ITT in the 20-week-old mice fed on high fat diet for 12 weeks. n = 7 for WT. n = 8 for KO. g Circulating serum insulin levels during IPGTT in the 19-week-old mice after 11 weeks of HFD feeding. n = 5. h Calculated HOMA-IR after fasting for 6 h. n = 7 for WT. n = 10 for KO. i FACS analysis of proportions of M1 and M2 macrophage populations in eWAT. n = 5. j SA-β-gal staining for eWAT. n = 5. k H&E staining. Immune cell clusters are pointed by red arrows. The bar chart on the right of (k) is the quantification of the number of immune cells per 103 μm2 of area. n = 11 for WT. n = 8 for KO. l Sirius Red staining. The bar chart in the right panel is the quantification of fibrous structures that are in red color n = 8 for WT. n = 7 for KO. m, n Immunoblotting analysis of the proteins related to inflammasome pro-IL-1β, cleaved-IL-1β pro-caspase-1, cleaved caspase-1 (p10), NLRP3 and ASC (m). The bar chart in panel n is the densitometric quantification for the indicated proteins. n = 7. o TMAO level in eWAT. TMAO level in tissue lysate is normalized with total protein concentration. n = 4 for WT. n = 5 for KO. All samples are biologically independent replicates. Data are represented as mean ± SEM. Statistical data were analysed by the Mann-Whitney U test for (b) and a two-tailed Student’s t-test for the remaining graphs, with Welch’s correction applied for (a, d and k).
FMO3-derived TMAO induces inflammation and senescence in mature adipocytes and macrophages
Next, we examined whether the anti-inflammatory and -senescent effects of adipocyte-specific FMO3 deletion on WAT are cell-autonomous and TMAO dependent. To this end, we isolated SVF from sWAT of 6–8-week-old male Adipo-FMO3 KO and WT controls, followed by induction of mature adipocyte differentiation. We used SVF from sWAT instead of eWAT because of its well-established higher differentiation potential. To induce senescence, we treated the SVF-derived adipocytes with doxorubicin for 48 h. Treatment with doxorubicin upregulated Fmo3 expression and TMAO production in WT adipocytes, and such upregulation was blocked by FMO3 deletion (Fig. 7a-b). Doxorubicin alone triggered a similar degree of senescence between both FMO3 deficiency and WT adipocytes, which was reflected by β-gal staining (Fig. 7c) and gene expression of p53, Cdkn1a and Cdkn2a as well as the SASP IL-1β (Fig. 7d-g, i, j). Importantly, incubation with TMA potentiated the doxorubicin-induced senescent responses in WT adipocytes, but such potentiating effect was completely abrogated by FMO3 deletion (Fig. 7c-j). Likewise, SVF-derived adipocytes from female Adipo-FMO3 KO mice were also resistant to Doxo+TMA induced TMAO production and senescent response without affecting the apoptotic program (Supplementary Fig. 20). Furthermore, treatment with doxorubicin and TMAO synergistically induced IL-1β secretion and the senescent genes expression in eWAT explants isolated from 12-week-old male C57BL/6 mice (Fig. 7k-l). In addition, the senescent effect of doxorubicin was increased pronouncedly by 1.5-fold in human SGSB adipocytes in the presence of TMA, but such effect of TMA was blocked by the FMO3 inhibitor DIM (Fig. 7m).

a–i SVF-derived mature adipocytes from Adipo-FMO3-KO and WT mice were treated with doxorubicin (10 μM) or DMSO as vehicle (Veh) for 20 h, followed by incubation with d9-TMA (500 μM) for 4 h. a qPCR analysis of Fmo3 mRNA expression normalized with 36b4 and 18 s. n = 7 for WT. n = 8 for KO. b d9-TMAO in the cell lysate (n = 3) and conditioned medium (n = 4) as indicated. d9-TMAO level in cell lysate is normalized with total protein concentration. c SA-β-gal staining. The bar graph shows the quantification of the β-gal-positive area (blue color) and presented as fold change over WT-veh. n = 3. d–h qPCR analysis of genes related to senescence and inflammation. n = 8 for p53, Cdkn1a and Pycard genes. n = 5 for Il1b and Cdkn2a genes. i IL-1β in the conditioned medium measured as indicated. n = 4. j–l eWAT explants from 12-week-old Adipo-FMO3-KO mice or WT control (j) or 12-week-old C57BL/6 mice (k, l) were treated with doxorubicin (10 μM) for 20 h, followed by stimulation with TMA (500 μM) for 4 h. j, k IL-1β level in the conditioned medium. j: n = 3. k: n = 9. l qPCR analysis of the genes related to senescence and normalized with 36b4 and 18 s. n = 8 for vehicle and TMAO. n = 5–8 for doxorubicin and doxorubicin + TMAO. m SGBS mature adipocytes were treated with doxorubicin (1 μM) or DMSO as vehicle (Veh) along with DIM (50 μM) for 20 h, followed by incubation with TMA (500 μM) for 4 h. qPCR analysis of p53, CDKN2A and CDKN1A mRNA expression are normalized with 36B4 and 18S. n = 4. Data are represented as mean ± SEM. Statistical data were analysed by two-tailed Student’s t-test or one-way ANOVA.
To induce inflammation, we treated the SVF-derived adipocytes with lipopolysaccharide (LPS), a gut-derived endotoxin known to trigger adipose inflammation in both obesity and ageing46. mRNA expression of Fmo3 was not altered by LPS and/or TMA stimulation in SVF-adipocytes from both genotypes, but its expression was significantly reduced by 3.7-fold in those from Adipo-FMO3 KO mice, accompanied by diminished activity in converting TMA to TMAO (Supplementary Fig. 21a, b). LPS induced expression of IL-1β, and the chemokine Cxcl10 mRNA expression (Supplementary Fig. 21c, d) and such effect was further enhanced by TMA treatment in WT adipocytes. FMO3 deletion abrogated the potentiating effects of TMA on LPS-induced inflammatory responses in adipocytes (Supplementary Fig. 21e-g). Taken together, these data suggest FMO3 control inflammation and senescence in adipocytes via a TMAO-dependent manner.
TMAO activates inflammasome via binding to ASC
Our in vitro and in vivo data showed that IL-1β, mainly produced by inflammasome activation, is positively regulated by FMO3-TMAO. The inflammasome genes including IL1B, IL18, NLRP3 and PYCARD (apoptosis-associated speck-like protein containing a CARD; also known as ASC) were upregulated in sWAT from aged human individuals and were positively correlated with FMO3 mRNA level (Supplementary Fig. 22).
Inflammasome activation requires two signals. The priming signals (such as LPS) upregulate inflammasome genes, including NLRP3, IL1B and CASP1 via toll-like receptor 4 (TLR4) and NF-κB pathway46. The activation signals such as ATP and nigericin trigger the assembly of inflammasome, the adaptor protein ASC and pro-caspase-1, which lead to cleavage and activation of caspase-1 and subsequent maturation of IL-1β and IL-1847. We next investigated whether TMAO serves as the priming and/or activation signal in macrophages and adipocytes, two major cell types involved in adipose tissue inflammation in ageing13,48.
Similar to the response in endothelial cells49, treatment with TMAO dose-dependently induced IL-1β and IL-18 but not TNF-α secretion in BMDM primed with LPS (Supplementary Fig. 23a-d). The potentiating effect of TMAO on IL-1β was blocked by treatment with the NLRP3 inflammasome inhibitor or caspase-1 inhibitor but not potassium chloride (rescue the potassium efflux during inflammasome activation) or the P2X7 receptor antagonist A438079 (Supplementary Fig. 23e). LPS + TMAO treatment in BMDM modestly enhanced p65 and IKB-α phosphorylation, but it did not exert consistent effects on inflammasome gene expression, including Nlrp3, Casp1, Il1b and Il18 when compared to those treated with LPS alone (Supplementary Fig. 23f-g). On the contrary, the cleaved form of caspase-1 in the supernatant was dramatically markedly induced by LPS + TMAO treatment (Supplementary Fig. 23h). Likewise, LPS + TMAO increased IL-1β production but had no effect on gene expression of CASP1 and PYCARD and a moderate effect on IL-1β in human THP-1 macrophages compared to those treated with LPS alone (Supplementary Fig. 24).
To determine whether FMO3-TMAO also induces inflammasome activation adipocytes, we established three stable 3T3-L1 cell lines overexpressing (1) GFP as control, (2) wild-type (WT) FMO3 and (3) a FMO3-P153L mutant with defective catalytic activity for TMAO production50 (Supplementary Fig. 25a). As expected, overexpression of WT-FMO3 but not FMO3-P153L increased the conversion of d9-TMA into d9-TMAO (Supplementary Fig. 25b). Consistently, overexpression of WT-FMO3 but not FMO3-P153L augmented LPS-induced IL-1β secretion in the presence of TMA in mature adipocytes (Supplementary Fig. 25c). Unlike macrophages, overexpression of WT-FMO3 but not FMO3-P153L mutant significantly upregulated the genes related to inflammasome activation including Nlrp3, Casp1, Il1b and Pycard in 3T3-L1 adipocytes under the LPS or LPS-TMA-stimulated conditions (Supplementary Fig. 25d-g). On the contrary, LPS + TMA induced inflammasome genes (including Casp1, Nlrp3, Il1b and Pycard) and IL-1β secretion were downregulated by deletion of FMO3 in SVF-derived adipocytes (Supplementary Fig. 21c, e-g). Consistently, pharmacological inhibition of FMO3 with DIM or methimazole (MMI) largely abrogated LPS + TMA-induced TMAO production and inflammasome activation (reflected by caspase-1 activity and IL-1β in medium) in 3T3-L1 mature adipocytes (Supplementary Fig. 26). In human SGSB adipocytes, treatment with LPS + TMAO also augmented IL-1β expression (Supplementary Fig. 27). The above findings indicate that TMAO potentiates the effects of LPS-induced inflammasome activation in adipocytes.
Recent studies have shown that metabolites can regulate biological processes through interactions with proteins and TMAO is known as a chemical chaperon that controls protein stability and conformation51,52. PERK has been identified as the receptor for mediating the detrimental effects of TMAO on metabolism53, but our data showed that it is not affected by TMAO treatment in macrophages (Supplementary Fig. 23g). To identify novel TMAO-binding proteins involved in inflammasome, we utilised Limited Proteolysis-small molecule mapping (LiP-SMap)54. We incubated the lysates from macrophages and adipocytes primed with LPS in the presence or absence of TMAO, followed by proteinase digestion. Proteomics analysis revealed that 379 and 194 proteins were resistant to proteinase-K-induced limited proteolysis in the presence of increasing concentrations of TMAO, in the samples from BMDM and adipocytes, respectively (Supplementary Fig. 28a). By comparing these potential TMAO-binding proteins among macrophages and adipocytes, there were 18 proteins in common, including ASC and superoxide dismutase 2 (SOD2) (Supplementary Fig. 28b-c). ASC is an essential component of the NLRP3 inflammasome, whereas SOD2 neutralizes mitochondrial ROS. Interestingly, TMAO has been shown to increase mitochondrial ROS by inhibiting SOD2 for mitochondrial reactive oxygen species production and the subsequent NLRP3 inflammasome activation in endothelial cells49. Ingenuity Pathway Analysis (IPA) of the potential TMAO-binding proteins revealed that these proteins are involved in inflammatory pathways such as non-canonical NF-κB, interleukins in both adipocytes and macrophages, whereas the fibrotic pathway collagen chain trimerization is enriched in adipocytes only (Supplementary Fig. 28d-e). Among the enriched pathways, IL-1 signalling, necroptosis, pyroptosis, CGAS-STING signalling and Parkinson’s signalling pathways are known to link with inflammasome activation.
ASC is the adaptor protein bridging NLRP3 inflammation and pro-caspase-1 via oligomerization. Inhibition of ASC abrogates caspase-1 activation and IL-1β production in macrophages and improves lipogenesis in sWAT55,56,57. Therefore, we further investigated whether and how TMAO induces inflammasome activation and IL-1β production via ASC. Consistent with the LiP-SMap analysis, drug affinity responsive target stability (DARTS) analysis showed that TMAO protected proteinase-induced ASC protein degradation in a dose-dependent manner (Fig. 8a-b), suggesting a direct interaction between ASC and TMAO. Indeed, treatment with TMAO led to an increase of ASC expression but not its mRNA expression in LPS-primed THP-1 macrophages by 2.5-fold change (Fig. 8c-d) or BMDM (Supplementary Fig. 29a-b) by 2.8-fold change. siRNA-mediated silencing of PYCARD completely abrogated TMAO-induced IL-1β production and caspase-1 activation in THP-1 macrophages (Fig. 8e-h) and BMDM primed with LPS (Supplementary Fig. 29c-e). Immunofluorescence staining revealed that ASC expression on F4/80+ macrophage was induced in eWAT of male WT mice when they were old, whereas adipocyte-FMO3 deletion abrogated such an ageing effect (Fig. 8i-j). Similarly, expression of ASC on F4/80+ macrophages in the sections of gWAT and ASC protein in gWAT homogenates were also reduced in aged female Adipo-FMO3 KO mice (Supplementary Fig. 17b, d). In addition, ASC protein expression in eWAT of HFD-fed male Adipo-FMO3 KO mice was also reduced when compared to the WT controls (Fig. 6m, n). These findings suggest that TMAO binds with ASC and stabilises its expression for inflammasome activation in macrophages.

a Lysates from BMDM treated with LPS were incubated with or without proteinase K in the presence of different concentrations of TMAO as indicated for 7 min. The cell lysates were subjected to immunoblotting analysis of ASC and HSP90. b Recombinant ASC proteins were incubated with proteinase K and different concentrations of TMAO for 2 h at 37 °C, followed by immunoblotting analysis (left panel). The ASC recombinant protein was subjected to SDS-PAGE and silver staining to assess purity. c, d Differentiated THP-1 macrophages were primed with LPS (50 ng/mL) for 20 h and incubated with TMAO (500 μM) for 4 h. c qPCR analysis of PYCARD mRNA level normalized with 18S. n = 6. d Immunoblotting analysis of ASC protein. (n = 3). e–h THP-1 macrophages were transfected with siRNA against PYCARD (siPYCARD) or scramble control (siScramble) for 48 h, followed by priming with LPS (50 ng/mL) for 20 h and incubation with TMAO (500 μM) for 4 h. qPCR (e) and immunoblotting (f) analysis of PYCARD mRNA and ASC protein, respectively. Measurement of IL-1β (g; n = 5 for scramble control treated with LPS and TMAO, and n = 4 for the remaining) and caspase-1 activity (h; n = 3) in the conditioned medium. i, j Young and Old male WT and KO mice on STC were used. i Immunofluorescence staining of F4/80 and ASC. The bar chart shows the quantification of ASC level in F4/80+ cells. Scale bar: 200 μm. (n = 5). j Immunofluorescence staining for caspase-1 and F4/80. Bar chart in the right panel shows the quantification of caspase-1 level in F4/80+ cells. Scale bar: 200 μm. (n = 5). Data are represented as mean ± SEM. Statistical data were analysed by two-tailed Student’s t-test, with Welch’s correction for (j).
Discussion
This study uncovers for the first time that p53-induced FMO3 expression in adipocytes is a major contributor to evaluated circulating TMAO in ageing, and its excessive accumulation triggers WAT dysfunction and metabolic disorders (Supplementary Fig. 30). TMAO provokes senescence and inflammasome activation, two major pathogenic events in ageing, in adipose tissue-resident cells, including mature adipocytes and macrophages. These detrimental effects of TMAO contribute to multiple hallmarks of WAT dysfunction, including fibrosis, low-grade chronic inflammation, and adipocyte hypertrophy, which results in glucose intolerance, insulin resistance, dyslipidaemia, and impaired energy balance in ageing. Our mechanistic study further reveals that TMAO binds with a cluster of inflammatory molecules, in particular ASC, which acts as the sensor of TMAO to induce cleavage of caspase-1 and subsequent IL-1β maturation and secretion in macrophages.
NLRP3 inflammasome is activated in WAT in ageing and obesity, leading to systemic elevation of IL-1β and IL-181. These pro-inflammatory cytokines impair insulin secretion and actions, thereby causing metabolic diseases including type 2 diabetes58,59. While inflammasome activation in adipose resident macrophages and B cells impairs lipolysis and energy balance in ageing13,60, its activation in human visceral adipocytes causes inflammation and fibrosis in obesity61. Human elderly with gene signatures showing constitutive inflammasome activation have a higher incidence of vascular dysfunction when compared to those without inflammasome activation62. On the other hand, the anti-ageing interventions, including calorie restriction and exercise, reduce IL-1β and NLRP3 gene expression in sWAT of patients with type 2 diabetes62. In rodents, genetic inactivation of key molecules in the NLRP3 inflammasome components, including NLRP3, ASC, IL-1β and caspase-1 improves WAT dysfunctions, including inflammation, defective lipolysis, fibrosis, lipid oxidation and adipogenesis under obese conditions13,46,58,60. Consistently, we demonstrated that reduced NLRP3 inflammasome activation and IL-1β expression in WAT of Adipo-FMO3 KO mice, which alleviates ageing-associated WAT dysfunction and metabolic disorders. In vitro, inhibition of the inflammasome components, also block the effects of TMAO on macrophages. In human, FMO3 and TMAO levels are also induced in aged sWAT, and FMO3 level positively associates with inflammasome genes and is downregulated by calorie restriction. These findings suggest that the beneficial effects of inhibition of FMO3-TMAO axis in adipocytes on metabolism and adipose tissue functions are closely linked with the reduced inflammasome activity in WAT.
Despite the recent identification of several obesity-associated inflammasome second signal activators58,63,64, the origins of endogenous inflammasome activators and their specific target cells within WAT during ageing remain elusive. In this study, we have demonstrated that adipocyte FMO3 and its derived TMAO are upregulated in the WAT of aged humans and rodents. Suppression of TMAO by deletion of FMO3 in adipocytes abrogated inflammasome activation in WAT of aged mice. We observed a marked reduction of the active form of caspase-1 and ASC in the ATM of aged Adipo-FMO3 KO mice. This suggests that adipocyte-derived TMAO acts as a paracrine factor to induce inflammasome activation in macrophages. On the other hand, we demonstrated that FMO3 and its derived TMAO potentiates IL-1β secretion and inflammasome gene expression in both macrophages and mature adipocytes. Given limited adipose tissues available in aged Adipo-FMO3 KO mice and WT controls, we were unable to assess inflammasome activity in adipocytes using more sensitive and high-throughput methods. In macrophages, the effect of TMAO on second signal activation is dramatic, whereas its effect on the priming step is relatively moderate. Our unbiased proteomics identified ASC as a TMAO-binding protein in both adipocytes and macrophages. Furthermore, we demonstrated that TMAO upregulates Pycard as well as other inflammasome gene levels in adipocytes, whereas TMAO treatment increases protein but not mRNA expression of ASC in macrophages. The differential effects of TMAO on inflammasome genes in adipocytes and macrophages are currently unknown. But ASC expression can be regulated at a transcriptional level by p53 or at a post-translational level, such as autophagy- and ubiquitination-mediated degradation65,66,67. Since TMAO triggers p53 activation and senescence in mature adipocytes, therefore it is possible that TMAO upregulates Pycard expression via p53. On the other hand, TMAO might mainly control ASC protein at a post-translation level in macrophages. Indeed, we also showed that inhibition of ASC or caspase-1 abrogated the effect of TMAO on IL-1β secretion in macrophages, further confirming that TMAO as the second signal for inflammasome activation.
Apart from its effect on inflammation, we also demonstrated that the FMO3-TMAO axis exerts pronounced effects on adipose senescence and fibrosis. Consistent with our study, TMAO triggers senescence in multiple cell types such as neurons, endothelial cells, and smooth muscle cells via Sirt-1 and p53-p21 pathway2,68. In addition, TMAO induces adipocytic progenitor cells into myofibroblasts via PERK upon TGF-β stimulation69. Inflammasome activation in adipocytes and macrophages also induces senescence and fibrosis61, therefore it is possible that FMO3-TMAO directly induces senescence and fibrosis or indirectly via inflammasome. In addition, we showed that TMAO binds and upregulates Pycard expression. Since Pycard induces p53 phosphorylation and p21 expression in cancer cells, and this signalling axis has been recently shown to suppress lipogenesis in adipocytes and sWAT57,67,70. It is tempting to speculate that TMAO induces p53-dependent senescence via ASC in adipocytes. Further investigation on which pathogenic event (senescence, inflammasome and/or fibrosis) is the initiator and their interplay in adipose tissue dysfunction induced by FMO3-TMAO axis in ageing is warranted.
Adipocyte-specific FMO3 KO mice exhibited a higher energy expenditure and lipid utilization in old age and obese conditions. These improvements are associated with a lower level of p53 and a high level of Prmd16 in BAT. Consistent with our findings, global knockout of FMO3 promotes browning in sWAT and energy expenditures in the mice fed with HFD27. In addition, the inactivation of p53 in adipocytes promotes beiging in aged WAT of mouse model via inhibiting mitophagy71. Taken together, adipocyte FMO3 directly regulates thermogenic program in WAT and/or BAT, but the underlying mechanism is currently unknown.
Recent studies have detected FMO3 protein in the extra-hepatic tissues31,72,73. Single-cell sequencing revealed that FMO3 is expressed in multiple cell types on the aorta where it produces TMAO and induces vascular inflammation via multiple pathways72. Here, we showed that mature adipocytes also express FMO3 and TMAO, and this metabolite product pathway is enhanced in ageing and contributes to the increase of systemic TMAO levels. On the contrary, we did not observe any significant difference in hepatic FMO3 expression between young and aged C57BL/6 J mice. Previous studies have shown that hepatic FMO3 expression increases with age in male C57BL/6 N and female C57BL/6 J mice74,75. In hepatocytes, Fmo3 mRNA expression is regulated by multiple factors, including sex hormones, glucagon, insulin and bile acids25,31. The discrepancy between our study and previous studies in hepatic FMO3 expression during ageing may be due to the nutritional state (such as fasting period) prior to sacrifice or variations in microbiota-derived bile acid composition resulting from different housing environments. In addition, hepatic Fmo3 mRNA is downregulated by LPS via TLR4 in female mouse models76. However, in this study, we showed that adipose/adipocyte FMO3 expression is not sexually dimorphic and not controlled by LPS but is mainly regulated by p53. p53 activity is increased in mature adipocytes during ageing and obesity15,16. We showed that p53 activation upregulates FMO3 expression by enhancing its promoter activity and TMAO production, which in turn provokes inflammation and senescence. On the other hand, we show that TMAO also increases p53 expression in adipose tissues and adipocytes, thereby forming a viscous cycle to exacerbate senescence and inflammation in WAT during ageing.
In summary, our study has identified a source of TMAO production in adipocytes, mediated by FMO3. This metabolite biosynthesis pathway plays a crucial role in the age-related activation of the inflammasome and the onset of senescence in WAT, both of which contribute to systemic metabolic dysregulation. We also identified ASC as a sensor for TMAO-induced inflammation activation in macrophages. Although we demonstrated that FMO3 substrate-competitive inhibitors, such as MMI and DIM77,7879,80, can suppress inflammasome activation in mature adipocytes induced by LPS and TMA, these inhibitors may be less specific and less efficient than traditional inhibitors that directly block FMO3 enzymatic activity. Therefore, the development of novel, highly specific FMO3 inhibitors, combined with adipose tissue-targeted drug delivery systems81,82, could represent a promising strategy for treating age-associated metabolic disorders. Importantly, this approach may help avoid the undesirable “fish odour” syndrome caused by TMA accumulation, which can occur when hepatic FMO3 activity is broadly inhibited.
Methods
Animal studies
All animals were kept in groups of five to six per cage and maintained at the Centralized Animal Facility of the Hong Kong Polytechnic University, Hong Kong. The animals were grouped according to their genotypes, sex, and age in the experiments involving Adipo-FMO3-KO mice and their WT controls and young and old mice used for collection of adipose tissue depots and serum. Fmo3floxed/floxed mice carrying two loxP sites on the intron 3 and 5 of Fmo3 gene were generated and confirmed by Shanghai Model Organisms Center. Homozygous Adipo-FMO3 knock-out (Adipo-FMO3 KO) mice were generated by mating Fmo3floxed/floxed mice with Adipo-Cre+ mice on a C57BL6/J background39. FMO3floxed/floxed were used as the wild-type (WT) littermate controls. Genotyping was performed by digesting pinna samples in DirectPCR Lysis Reagent (Catalogue# 102-T, Viagen Biotech) supplemented with 0.5 mg/mL proteinase K (Catalogue# P2308, Sigma-Aldrich) overnight at 55 oC, followed by heat inactivation at 85oC for 1 h before PCR with 2xEs Taq MasterMix (Catalogue# CW0690, CoWin Biosciences). We genotyped the mice for Fmo3flox/flox and Adipo-Cre+ alleles using the primers listed in Supplementary Table 2. Mice were kept at room temperature 22 oC (±0.5) and humidity (60 ± 10%) on a 12 h/12 h light/dark cycle with ad libitum access to water and either fed on a standard chow diet, (STC; 24.7 kcal% protein, 13.2 kcal% fat, and 62.1 kcal% carbohydrates; Catalogue# 5053, PicoLab Rodent Diet 2.0) or 45% high-fat diet, (HFD; 20 kcal% protein, 45 kcal% fat and 35 kcal% carbohydrates; Catalogue# D12451, Research Diets Inc.).
A minispec body composition analyser (Bruker Minispec LF90) was used to determine lean and fat mass. Continuous oxygen consumption, carbon dioxide release, energy expenditure, respiratory exchange ratio (RER), food intake and locomotor activity at room temperature of 22 oC as mentioned in figure legends were recorded by the Promethion metabolic Cage System (Sable Systems International). Mice were acclimated in the system for 1-2 days before the data acquisition and analysis.
For IPGTT, mice were fasted for 16 h, followed by an intraperitoneal injection of D-glucose (Catalogue# 346351, Sigma-Aldrich). For ITT, mice fasted for 6 h, followed by an intraperitoneal injection of human recombinant insulin (Catalogue# 91077 C, Sigma-Aldrich). Blood glucose was measured using a glucometer (ACCU-Check Performa, Roche) and the dosage of D-glucose and recombinant insulin was mentioned in the figure legends. For GSIS, mice fasted for 16 h were intraperitoneally injected with D-glucose injection, followed by tail vein blood collection at time 0, 10, 20 and 30 min. The collected blood was kept at room temperature for a minimum of 30 min, centrifuged at a speed of 4000 rpm for 15 min and supernatant serum was collected. Serum insulin was measured by using ultra-sensitive or normal-range insulin ELISA kits (Mercodia).
For the chemically induced senescent model, 16-week-old male and female C57BL6/J mice were assigned randomly and intraperitoneally injected with a single dose of 2 mg and 10 mg per kg of body weight of doxorubicin (Catalogue# S1208, Selleck Chemicals) or Phosphate-Buffered Saline (PBS) (as control). The animals were sacrificed on day 10 for tissue collection.
For the lipid tolerance test, animals were fasted for 16 h in a clean cage. Oral gavage with olive oil was performed at 10 μL per gm of body weight concentration. Tail vein blood sampling was done for 6 h at 1 h intervals post-gavage. Samples were kept at room temperature for a minimum of 30 min and centrifuged at a speed of 4000 rpm for 15 min for serum collection. Serum triglyceride level was measured according to the manufacturer’s protocol using the triglycerides detection kit (Catalogue #2100-430, Stanbio Laboratory). All animal experimental protocols were approved by the Animal Subjects Ethics Sub-Committee (approval number: 18-19/35-HTI-R-GRF) at the Hong Kong Polytechnic University, Hong Kong.
For euthanasia, animals underwent 6 h of fasting, followed by an intraperitoneal injection of ketamine (10%; Catalogue# 0904088-05, Alfasan International B.V.) and xylazine (2%; Catalogue# 1205117-05, Alfasan International B.V.) combination, administered at a dosage of 12.5 μL per gram of body weight.
Biochemical and immunological assays
Serum insulin (Catalogue# 10-1249-01 or Catalogue# 10-1247-01, Mercodia), leptin (Catalogue# RD291001200R, BioVendor) and adiponectin (Catalogue# 32010, Immunodiagnostics Ltd.) were measured using ELISA kits. Serum cholesterol (Catalogue# 1010, Stanbio Laboratory), LDL (Catalogue# 0710-080, Stanbio Laboratory), HDL (Catalogue# 0590-080, Stanbio Laboratory), triglycerides (Catalogue# 2100, Stanbio Laboratory) and free fatty acid (Catalogue# 11383175001, Roche) were estimated according to the manufacturer’s protocol. To measure serum IL-18 (Catalogue# BMS618-3) and IL-1β (Catalogue# BMS6002), ELISA kits from Invitrogen were used. Levels of IL-1β and MCP-1 in tissue or cell homogenate and cell culture media were quantified using mouse IL-1β/IL-IF2 DuoSet ELISA (Catalogue# DY401, R & D Systems) and mouse MCP-1 DuoSet ELISA (Catalogue# DY479, R & D Systems), respectively. Activity of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were determined using ALT/SGP+ Liqui-UV test (Catalogue# 2930/430, Stanbio Laboratory) and AST/SGOT Liqui-UV test kits (Catalogue# 2930/2920, Stanbio Laboratory). All the above measurements were done according to the manufacturer’s protocol.
Histological analysis and immunofluorescence staining
Post-cull, the tissues were fixed in a 10% neutral buffered saline (Catalogue# 5705, Thermo Fisher Scientific) for 24 h and then subjected to tissue processing in the Excelsior AS Tissue Processor (Thermo Fisher Scientific) and cut into 5-μm sections. The paraffin sections of various tissues were stained with haematoxylin-eosin (Catalogue# G1121, SolarBio; H&E) and Picro Sirius Red (Catalogue# ab246832, Abcam) and analysed with ImageJ software.
For immunofluorescent staining, deparaffinised and rehydrated sections were subjected to antigen retrieval in sodium citrate buffer (0.1 mol/L sodium citrate, 0.1% Tween 20, pH 6.0) at boiling for 20 min. Following, sections were blocked with 5% fetal bovine serum (FBS) in 1x PBS for 1 hour at room temperature, incubated with the primary antibodies (1:80-200 dilution) overnight at 4 °C in antibody buffer (3% BSA and 0.1# triton-X 100 in 1X PBS). The slides were washed with 1X TBST (0.1% Tween-20) for 2 min with rigorous washing, followed by 3 min of resting for three times. Post-washing, the slides were incubated with respective fluorescent dye-conjugated secondary antibodies for 1 hour at room temperature, followed by washing with 0.1% TBST (5 min rigorous washing, 5 min resting, three times). The slides were mounted with Prolong Glass Antifade Mountant (Catalogue# P36980, Invitrogen). Images were acquired with Leica TCS SPE Confocal microscope, and the intensities of positively stained cells were quantified in 7-8 randomly selected fields by the ImageJ software.
Senescence activity measurement
Senescence in the fat explants and differentiated adipocytes were determined using the SA-β Gal staining kit (Catalogue# 9860, Cell Signaling Technology), following the manufacturer’s protocol. Briefly, ~30 mg adipose tissue explants were first washed with 1X PBS in a 96-well plate and then fixed in 300 μL of 1X Fixative Solution at room temperature overnight. For SVF-differentiated mature adipocytes, the cells were rinsed once with 1X PBS and fixed with 0.5 mL of 1X Fixative Solution for 1–2 h at room temperature. Meanwhile, the β-Galactosidase Staining Solution was prepared, maintaining a pH of 6.0 ± 0.1. The solution was prepared by combining 930 μL of 1X Staining solution, 10 μL of 100X Solution A, 10 μL of Solution B, and 50 μL of 20 mg/mL X-gal stock solution. After fixation, the tissue explants or cells were rinsed twice with 1X PBS. Next, 300 μL or 0.5 mL of the β-Galactosidase Staining Solution was added to the tissue explants or cells, respectively, and the plates were sealed with parafilm to prevent evaporation and subsequent crystallisation. The sealed plates were then incubated at 37 °C overnight in a dry incubator without carbon dioxide. While the β-Galactosidase staining solution was still on the plate, the cells were observed under a microscope to check for the development of blue colour. For tissue explants, the colour formation was observed by the naked eye. Once the colour developed, the β-galactosidase staining solution was removed, and the plates were overlaid with 70% glycerol for imaging and long-term storage at 4 °C.
Isolation and differentiation of SVF to matured adipocytes
Fat depots were digested with collagenase type I (Catalogue# 17100017, Thermofisher Scientific; 2 mg/mL) at 37 oC for 60 min, filtered through a cell strainer with a pore size of 100 µm (Catalogue# 3520350, Coring) and centrifuged for 5 min at 500 x g. Cell pellets containing SVF were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (Catalogue# 12800082, Gibco™) with 10% FBS (Catalogue# 10270, ThermoFisher Scientific) and 1 % penicillin-streptomycin (PS, Catalogue# 151401229, ThermoFisher Scientific) until 100 % confluent. Differentiation of white adipocytes from SVF was achieved by treating cells with isobutyl methylxanthine (IBMX) 0.5 mM, Catalogue# 858455, Sigma Aldrich), insulin (10 μg/mL, Catalogue# 12-585-014, Gibco), dexamethasone (0.25 μM, Catalogue# A13449, Thermo Scientific Fisher), and rosiglitazone (100 nM, Catalogue# AC462410010, Fisher Scientific) for 4 days. While differentiating media was replenished after 2 days. Cells were kept in maintenance media (DMEM supplemented with 0.1% insulin) for 2–4 days to achieve complete differentiation. For induction of inflammasome activation, matured adipocytes were primed with 100 ng/mL LPS (Catalogue# L4391, Sigma-Aldrich, USA) for 20 h before treatment with 500 μM TMA (Catalogue# C847, AKSci) or d9-TMA (Catalogue#613843, Sigma Aldrich) or 500 μM TMAO (Catalogue# 317594, Sigma-Aldrich) or 500 μM d9-TMAO (Catalogue# DLM-4779-1, Cambridge Isotope Laboratories) for 4 h. To induce senescence, matured adipocytes were treated with 0.3 μg/mL doxorubicin for 24 h, followed by treatment with 500 μM each of TMA, d9-TMA, TMAO and d9-TMAO for 24 h. Conditioned media was collected for the measurement of pro-inflammatory cytokines and cell lysates were collected for qPCR, western blot, and LC-MS/MS analyses. D9-TMA was added to matured adipocytes derived from the WT and Adipo-FMO3 KO animals sWAT and incubated for 48 h. Media and cells were collected to detect d9-TMAO levels using the LC-MS/MS technique. Also, various pro-inflammatory cytokines IL-1β and MCP-1 both in conditioned media and cell lysates were measured using the commercial kits mentioned in the section “Biochemical and immunological assays”.
Flow cytometry analysis of M1 and M2 macrophages in eWAT
SVF cells from eWAT were resuspended in the FACS buffer (5 mM HPES, 2 mM EDTA, 1% BSA in PBS) and Fc receptor was blocked with 2% rat serum for 20 min at 4 °C. Single-cell suspensions were then stained with a cocktail of fluorophore-conjugated antibodies for 30 min on ice in the dark followed by washing twice with FACS buffer. The following antibodies were used39: CD45-APC-Cy7 (Catalogue# 103116, Bio Legend; 1:100 dilution), F4/80-PE (Catalogue# 111604, Bio Legend), CD11b-BV605 (Catalogue# 563015, BD Horizon), CD11c-FITC (Catalogue# 117306, Bio Legend), CD206-PERCP-Cy5.5 (Catalogue# 141716, Bio Legend), CD3-BV421 (Catalogue# 100227, Bio Legend), and B220-PE-Cy7 (Catalogue# 552772, BD Horizon). Next, these cells were fixed and permeabilised with Transcription Factor Staining Buffer Set (Catalogue# 562574, eBioscience) according to the manufacturer’s instructions. The labelled cell suspensions were finally analysed using BD FACS Aria III Cell Sorter (BD Biosciences) (Supplementary Fig. 31) and Flow Jo software.
Isolation of BMDM, differentiation into macrophages and inflammasome induction
Bone marrow cells were harvested from the tibias and femurs of 8–10-week-old male C57BL/6 J mice. The bone marrow cells were cultured in the complete DMEM media supplemented with L929 cell-conditioned media for 7 days as we previously described83. The L929 cell conditioned media was prepared by culturing L929 mouse fibroblasts (Catalogue# CCL-1 NCTC clone 929, ATCC) in DMEM containing 12% FBS and 1% PS, until the cells reached confluence. The conditioned media was then collected after 3 days of culturing and used to differentiate murine macrophages from the bone marrow. After the 7-day culture period, the differentiated BMDM were washed and replaced with complete DMEM prior to experimental treatments. To trigger inflammasome activation, the BMDM were first primed with 100 ng/mL of LPS or vehicle control for 20 h, followed by a 4-hour stimulation with 500 μM TMAO. Both the conditioned media and the cells were then collected for various analyses. The levels of the pro-inflammatory cytokines IL-1β and MCP-1 were measured in both the conditioned media and the cell lysates. Additionally, qPCR analyses were performed on the samples.
For gene silencing in BMDM, siRNAs targeting Pycard were transfected into BMDM using DharmaFECT 3 (Catalogue# T-2003-03, Dharmacon) as per the manufacturer’s protocol. Briefly, the transfection was conducted simultaneously with BMDM plating (1–1.5 × 10^6 cells/well of 6-well plate). For each well, 5 μL of the transfection agent and a final concentration of 10 nM siRNA were used in a total volume of 0.25 mL of Opti-MEM I (Catalogue# 31985070, Invitrogen) and the final volume made up to 1 mL using complete DMEM. A scramble siRNA (siScramble) was used as a control. The media was changed to 1.5 mL of fresh complete DMEM after 7 h and the cells were incubated for another 36 h in a 5 % CO2 incubator at 37 °C, after which they were lysed to isolate total RNA or protein.
Cell culture and differentiation of 3T3-L1 cells
To differentiate 3T3-L1 preadipocytes into mature adipocytes, the 3T3-L1 preadipocytes cells were cultured in complete DMEM media with 10% FBS. After 48–72 h of reaching 100% confluence, the cells were treated with a differentiation cocktail containing IBMX (0.5 mM, Catalogue# 858455, Sigma Aldrich), insulin (10 μg/mL, Catalogue# 12-585-014, Gibco), dexamethasone (0.25 μM, Catalogue# A13449, Thermo Scientific Fisher), and rosiglitazone (100 nM, Catalogue# AC462410010, Fisher Scientific) for 2 days. On day 2, the cells were refreshed with DMEM supplemented with the same differentiation factors for an additional 2 days. Subsequently, the cells were maintained in complete DMEM supplemented with insulin (1 μg/mL) for 3–4 days to allow full differentiation into mature adipocytes. Following the differentiation process, the cells were treated with vehicle control DMSO and, either with 100 ng/mL of doxorubicin, or 100 ng/mL of nutlin-3a (Catalogue# S8059, Selleck Chemicals) for 24 h. Both the cells and the conditioned media were collected as described in the previous protocol.
Mature adipocytes were transfected with siRNA using the reverse transfection technique. First, the siRNA against Fmo3 or Scramble control and Lipofectamine RNAiMAX (Catalogue# 13778150, Invitrogen) were separately diluted in Opti-MEM, and then mixed by pipetting. This siRNA-RNAiMAX mixture was added to a gelatin-coated 6-well cell culture plate and left to incubate for 25 min at room temperature. The final concentrations were 50 μL/mL for the Lipofectamine RNAiMAX and 400 nM for the siRNA. The final volume of media per well was 1 mL in the 6-well plate. The mature 3T3-L1 adipocytes were transfected on day 6 of differentiation. To prepare the cells, mature adipocytes were trypsinized with 0.25% trypsin for 10–15 min, counted, and spun down for 5 min at 300 x g. The cells were then resuspended in culture media and added to the 6-well plates on top of the pre-incubated siRNA-RNAiMAX mixture. The final number of cells per well was around 0.5 × 10^6 in the 6-well plates. The media was changed after two days, and the cells were harvested four days after the initial transfection.
Differentiation of THP-1 and activation of inflammasome
THP-1 monocytes were cultured in 3 mL of RPMI-1640 media supplemented with 15-20 ng/mL of phorbol-12-myristate 13-acetate (PMA, Catalogue# 10008014, Cayman Chemicals) to induce macrophage differentiation. After 48–72 h of incubation, the differentiated cells were washed with 1X PBS and then incubated in RPMI-1640 media for an additional 1–2 days. Following the rest period, the cells were primed with 100 ng/mL of LPS for 16–20 h. Subsequently, the THP-1 macrophages were treated with 500 μM of TMAO for 4 h. The cells and conditioned media were collected for the evaluation of various pro-inflammatory cytokines using ELISA and qPCR techniques.
For gene silencing in THP-1 macrophages, the cells were seeded at 1.5 × 106 cells/well in a 6-well plate. Simultaneous with cell plating, transfection was performed using 5 μL of Lipofectamine™ 3000 (Catalogue# L3000001, Thermo Fisher Scientific) and a final concentration of 10 nM siRNA in a total volume of 0.25 mL Opti-MEM I, made up to 1 mL with complete RPMI-1640. A Scramble siRNA was used as a control. After 7 h, the media was changed to 1.5 mL of fresh complete media, and the cells were incubated for an additional 36 h at 37 °C in a 5% CO2 incubator before being lysed for total RNA or protein isolation.
Generation of 3T3-L1 stable cell lines
Mouse 3T3-L1 preadipocytes were seeded in 6-well plates and grew until 50% confluence, followed by infection with lentiviral particles carrying GFP, wild-type (WT)FMO3, or the FMO3 (P153L) mutant. The human FMO3 gene and its mutant P153L were inserted into the lentivirus vector pLV4ltr-PGK-ZsGreen(2 A) PURO-CMV (Catalogue# PRCDCMUP, Cellecta) and then packaged into lentivirus particles at a titre of ZsGreen 2.5 × 108 TU/mL, FMO3 3.5 × 108 TU/mL and Mutation-FMO3(P153L) 1.5 × 108 TU/mL. For each experimental group, 800 TU of the respective lentivirus and 2 μL of polybrene (10 mg/mL, Catalogue# TR1003, Sigma Aldrich) were added to the cells and mixed well. After a 4 h infection period, the media was changed to fresh culture media. This process resulted in the generation of stable 3T3-L1 cell lines expressing GFP, WT-FMO3 and FMO3-P153L mutant. These cell lines were maintained in culture media supplemented with puromycin (2 μg/mL, Catalogue# A1113803, Thermo Fisher Scientific) to ensure continued transgene expression. Differentiation of these stable cell lines into mature adipocytes was done as mentioned above. To confirm the expression of FMO3 and FMO3(P153L), western blot analysis was performed. Additionally, the production of TMAO was quantified using LC-MS analysis, as described in the section below.
SGBS cell culture
The Simpson-Golabi-Behmel syndrome (SGBS) cells were used as a model to resemble human adipocytes in vitro84,85. SGBS preadipocytes were cultured in DMEM/Nutrient Mixture F12 (DMEM/F12) (Catalogue# 31330-038, Invitrogen) medium with 10% FBS. The SGBS cells were seeded in 12-well plates and allowed to reach approximately 90% confluence. To induce adipocyte differentiation, the SGBS cells were incubated with DMEM/F12 medium containing 1% penicillin/streptomycin, 33 μM biotin (Catalogue# B-4639, Sigma-Aldrich), 17 μM pantothenate (Catalogue# P-5155, Sigma-Aldrich), 20 nM human insulin (Catalogue# 12585-014, ThermoFisher Scientific), 100 nM cortisol (Catalogue# H-0888, Sigma-Aldrich), 200 pM triiodothyronine (Catalogue# T-6397, Sigma-Aldrich), 10 μg/mL transferrin (Catalogue# T-2252, Sigma-Aldrich), 100 nM rosiglitazone, 25 nM dexamethasone, and 250 μM IBMX but without FBS for four days. The SGBS cells were then maintained in the medium with the same concentrations of biotin, pantothenate, human insulin, cortisol, and triiodothyronine as before for another three days, and this procedure was repeated twice. After that, the differentiated SGBS adipocytes were used for treatment with p53 activators or TMAO as indicated in the figure legends.
Differentiation of human adipocytes from mesenchymal stem cells
Human adipose tissue-derived mesenchymal stem cells (hADMSCs) were established using our previously established protocols86,87. The adipose tissue was washed with 1X PBS containing 1% PS for three times. Subsequently, the tissue was finely minced using a surgical scissor and enzymatically digested with collagenase type I (1 mg/mL) at 37 °C with shaking for 30 min. The digested tissue was incubated with DMEM/F12 medium supplemented with 10% FBS and 1% PS, followed by centrifugation at 300 x g for 5 min. The cell pellet was re-suspended in DMEM/F12 with 10% FBS and 1% PS and cultured at 37 °C. hADMSCs used in this study were from passages 6 to 1088. Ethical approval was obtained for the collection of adipose tissue samples. Experimental protocols were approved by the Institutional Review Board (approval number: UW 21-624) at the Hong Kong University, Hong Kong.
hADMSCs were subjected to adipogenic differentiation using the OriCell® Human Related Stem Cells Adipogenic Differentiation Induction Kit (Catalogue# HUXXC-90031, Cyagen, China) according to the manufacturer’s instructions. In brief, hADMSCs were seeded in a six-well plate at a density of 2 × 104 cells/cm2 and allowed to reach 90% confluence in DMEM/F12 supplemented with 10% FBS and 1% PS. The cells were then incubated with Adipogenic Induction Medium A for 3 days, followed by a switch to Adipogenic Induction Medium B for 1 day. This alternating process was continued for 2 rounds. Daily monitoring of the cells was performed to observe the formation of lipid droplets, and adipogenic differentiation was confirmed through Oil Red O staining.
Assessment of senescent and inflammatory response in WAT explant
Approximately 20–30 mg of WAT was minced and placed in each well of a 96-well plate containing complete DMEM media. To achieve this seeding density, around 150 mg of WAT was coarsely minced in 150 μL of complete DMEM using autoclaved scissors, resulting in a total volume of approximately 300 μL containing about 20 mg of tissue per 40 μL of the minced tissue mixture. Using cut-end pipette tips, 40 μL of the minced tissue mixture was transferred to each well of the 96-well plate, thereby seeding around 20 mg of tissue per well. The tissue samples were then treated with either doxorubicin (100 ng/mL) for 20 h or LPS (100 ng/mL) for 16 h, followed by a 4 h treatment with 500 μM TMA or d9-TMA. Conditioned media was collected for measurement of pro-inflammatory cytokines and LC-MS/MS analyses. Minced tissue samples were collected for qPCR analyses of various genes involved in senescence and inflammation as indicated in the figure legends, and LC-MS/MS analysis.
Measurement of Fmo3 promoter activity by luciferase assay
The luciferase reporter plasmid pGL.3-Basic was inserted with the Fmo3 promoter or Fmo3 intron 1 region containing p53 RE element: (i) the Fmo3-WT-promoter, which spanned from −2000 base pairs to the transcription start site (+1 bp) and (ii) the Fmo3-Intron, which was located between exon 1 and exon 2, spanning from +6664 bp to +8664 bp, respectively. The luciferase reporter plasmids and the p53 expression plasmid or GFP control, and a renilla plasmid (for normalisation of transfection efficiency) were co-transfected into 3T3-L1 adipocytes using Lipofectamine™3000 for 10 h as indicated in the figure legend. After the cells were replaced with fresh media and continued to culture for 24 h, the luciferase activity was measured using a luciferase assay kit (Catalogue# R027, Beyotime), according to the manufacturer’s instructions. Briefly, 250 μL lysis buffer was added to each well of the 6-well plates. The cell lysates were collected after centrifugation at 12,000 rpm for 4 min at 4 °C. Next, 100 μL of the cell lysate supernatant from each sample was added to a 96-well plate, and the same volume of firefly luciferase substrate was mixed in. The luciferase values were then determined using a Varioskan LUX multimode microplate reader (Thermo Fisher Scientific). In cases where the Renilla luciferase was used as an internal reference, an additional step was performed. After measuring the firefly luciferase, 100 μL of the Dual-Lumi™ Renilla luciferase assay (Catalogue# RG088S, Beyotime) working solution was added to each well of the 96-well plate and mixed well. The value obtained from the firefly luciferase assay was then divided by the value from the renilla luciferase assay to obtain a ratio. This ratio was used to compare the degree of activation of the target reporter gene between samples.
Detection of choline, TMA and TMAO by LC-MS/MS
Choline, TMA and TMAO were quantified according to a previous study89. Briefly, labelled standards and internal standards (choline, TMA, TMAO, d9-TMA, d9-TMAO and d8-valine) were individually prepared in ultrapure water at a concentration of 10 mM and stored at −80 °C. Calibration standards were prepared by serial dilutions of the highest concentration standard (62.5 μM, 31.5 μM, 15.625 μM, 7.8 μM, 3.9 μM and 1.95 μM). The internal standard working solution was prepared by mixing the stock solutions of the labelled standards into 50 mM tert-butyl bromoacetate (TBB) to final concentrations of 1 μM for d9-TMA (used as internal standard for TMA), d9-TMAO (used as internal standard for d9-TMAO), and d8-valine (used as internal standard for d9-TMA and d9-TMAO), respectively. 5 μL of serum or conditioned media was directly used, but for tissue samples, around 30 mg of tissue was homogenised with 100 μL of chloroform, methanol, and water (or 50 mM TBB, when TMA derivatisation was achieved for measurement of TMA) prepared at the ratio of 1:8:1 and supernatant post-centrifugation of homogenised samples were used for the analysis. Further, 5 μL of standard, blank and sample was taken in a 1.6 mL microcentrifuge tube. For this, 46 μL of the 1 μM internal standard solution prepared in 50 mM TBB and 70% of ammonium hydroxide was added (we used 30%). The mixture was vortexed briefly and kept at room temperature for 30 min. To this mixture, 20 μL of 1% formic acid was added and subjected to centrifugation at 15,000 x g for 5 min at room temperature and the supernatants were transferred into HPLC vials for analysis. Quantification of choline, TMA and TMAO was performed using an Agilent 6460 Liquid Chromatography-Electrospray Ionisation Triple Quadrupole Mass Spectrometer (LC-MS/MS). Chromatographic separations were performed on an Atlantis Silica HILIC 3μm 4.6 ×50 mm column. The column was heated to 40 °C while maintaining the flow rate at 1 ml/min. The gradient was: 5% A for 0.05 min, to 15% A in 0.35 min, to 20% A in 0.6 min, to 30% A in 1 min, to 45% A in 0.55 min, to 55% A in 0.05 min, at 55% A for 0.9 min, to 5% A in 0.05 min, at 5% A for 1.45 min; “A” 10% acetonitrile/90% water with 10 mM ammonium formate and 0.1% formic acid and “B” was 90% acetonitrile/10% water with 10 mM ammonium formate and 0.1% formic acid. Analytes and their corresponding isotopes were monitored using characteristic precursor-product ion transitions: 104 > 45 for choline, 174 > 59 for TMA, 183 > 68 for d9-TMA, 76 > 58 for TMAO and 85 > 66 for d9-TMAO. The concentrations of each analyte in samples were determined from calibration curves using the peak area ratio of the analyte to its isotope.
Drug affinity responsive target stability (DARTS) analysis
BMDM were stimulated with LPS (100 ng/mL) and IFN-γ (100 ng/mL) for 24 h to increase the expression of inflammatory and inflammasome components. The cells were then harvested by adding 400 μL of freshly prepared lysis buffer (containing protease inhibitor cocktail (HY-K0010, MCE), 0.2% Triton-X-100, 50 mM Tris-HCl, and 10 mM sodium chloride) to the 10 cm cell culture dish, and the lysates were collected into Eppendorf tubes on ice. The cell lysates were centrifuged at 12000 rpm for 5 min, and the supernatants were collected. The protein concentration of the cell lysates was determined using a Bicinchoninic acid (BCA) protein assay kit (Catalogue# 23225, Thermo Fisher Scientific). Equal volumes of protein (100 μg) were aliquoted into eight tubes and incubated with four different concentrations of TMAO (0 μM, 100 μM, 250 μM, and 500 μM), with two tubes per concentration, on ice for 1 hour. One tube per TMAO concentration was randomly selected, and 10 μL of protease (0.5 μg/100 μg protein) prepared in 1× TNC buffer (containing 500 μL of 1 M Tris-HCl (pH 8.0), 100 μL of 5 M NaCl, 100 μL of 1 M CaCl2, and 300 μL of ultrapure water) was added, followed by incubation at room temperature for 7 min. Finally, an equal volume of 5× protein loading buffer was added to each tube, and the samples were boiled in preparation for Western blot analysis.
Limited proteolysis small molecule mapping (LiP-SMap) approach
SVF-derived adipocytes or BMDM treated with LPS (100 ng/mL) were lysed with a lysis buffer (100 mM HEPES pH 7.5, 150 mM KCl, 1 mM MgCl2) were homogenised with the Bertin Technologies Precellys Evolution Homogenizer. The samples were then centrifuged at 20,000 x g for 5 min at 4 °C. Endogenous small molecules including metabolites and nucleic acids were eliminated from the samples by size-exclusion chromatography using Zeba Spin Desalting Columns 7 MWCO (Catalogue# 89890, ThermoFisher Scientific). Protein concentration in each sample was then determined with BCA (Catalogue# 23225, ThermoFisher Scientific). Cell lysates were then aliquoted into tubes that contained 300 µg of whole proteome sample with equal volume and incubated with TMAO (prepared in 100 mM HEPES, pH 7.5) for 10 min at 25 °C. Proteinase K from Tritirachium album (Catalogue# P2308, Sigma Aldrich) was added to all samples at a proteinase K to protein mass ratio of 1:100 and incubated at 25 °C for 5 min. Limited digestion was quenched by incubating the samples at 98 °C for 3 min, followed by the addition of sodium deoxycholate to a final concentration of 5% and another incubation at 98 °C for 3 min before proceeding to the complete digestion.
Samples that had undergone limited proteolysis were further reduced, alkylated, and completely digested using the EasyPep™ Mini MS Sample Prep Kit (Catalogue# A40006, ThermoFisher Scientific) according to the manufacturer’s instructions. In short, the Reduction solution and Alkylation solution from the kit were added sequentially to the reaction mix, followed by heating the samples to 95 °C for 10 min. After the heat incubation, the samples were cooled to room temperature before adding the reconstituted Trypsin/Lys-C Protease mix supplied in the kit. The digestion mix was then incubated at 37 °C with shaking for 3 h and the digestion was stopped with the Digestion Stop Solution. The digested peptide mixtures were transferred and cleaned up using the Peptide Clean-up column provided with the kit and centrifugation. The eluted peptide samples were dried in a vacuum centrifuge and resuspended in 0.1% formic acid and immediately analysed by mass spectrometry.
Peptide samples were analysed with the Thermo Scientific Orbitrap Fusion Lumos Mass Spectrometer equipped with equipped with a nano electrospray ion source and the Ultimate 3000 RSLCnano liquid chromatography system in the University Research Facility in Chemical and Environmental Analysis, The Hong Kong Polytechnic University. Peptides were separated using the Acclaim PepMap C18 analytical columns and Trap Column Cartridges Holders with nanoViper Fittings (ThermoFisher Scientific). Flow rate was set as 300 nL/min and 1 µL of peptide samples was injected for fractionation and analysis. The gradient settings for LC fractionation were as follows: 0.1% formic acid as buffer A, and 0.1% formic acid in 100% acetonitrile as buffer B. The mobile phase gradient started at 2% buffer B until 5 min, then gradually increased to 6% buffer B at 7 min, 20% buffer B at 82 min, 30% buffer B at 90 min, and 90% buffer B at 100 min. The gradient was held until 105 min and re-equilibrated with 2% buffer B at 115 min. The peptides were analysed by an Orbitrap Fusion Lumos Mass Spectrometer using a data-dependent acquisition (DDA) strategy in positive ion mode with a scan range of 400–1500 m/z at a resolution of 60,000 and standard automatic gain control (AGC) target. 2–7 multiple charged ions were used to trigger MS-MS scans, whereas the dynamic exclusion duration was set at 40 s. Selected precursors were fragmented using high-energy collision dissociation (HCD) with normalised collision energy set as 30%. MS/MS was acquired using an Orbitrap as a mass analyser with mass resolution of 7500 and standard AGC target. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository. Identified targets with increasing abundance along with increasing TMAO concentration were selected for further analysis. Pathway enrichment was performed using QIAGEN IPA (QIAGEN Inc., https://digitalinsights.qiagen.com/IPA).
Quantitative RT-PCR
Total RNA was extracted using TRIzol (Catalogue# 15596026, Takara) according to the manufacturer’s instructions. The cDNA was synthesised by reverse transcription of 1 μg RNA using a GoScript Reverse Transcription kit (Catalogue# A2801, Promega) with random universal hexamer primers. Quantitative real-time PCR was performed using SYBR Green (Catalogue# 208056, Qiagen) with gene-specific primers in the ViiA7 Real-time PCR System (Applied Biosystems). Target gene expression level was normalised against various housekeeping genes, including GAPDH, 36B4 and 18S as indicated in figure legends.
RNA sequencing analysis
Total RNA was extracted using the RNeasy mini kit (Catalogue# 74104, Qiagen) following manufacturer’s instructions. After passing quality control, the libraries were generated and sequenced with the DNBseq sequencing platform, BGI Genomics, China. Briefly, Poly-A enriched mRNA was fragmented and used to synthesise first-strand cDNA with random primers. Following, second-strand cDNA was synthesised using dUTP. The cDNA was end-repaired, 3’-adenylated, and adaptors were ligated. The U-labelled second strand was digested with UDG, and the remaining fragments were PCR amplified. The library was assessed for quality, circularised, and amplified into DNA nanoballs that were sequenced on the DNBSEQ platform. Raw data was filtered using SOAPnuke to remove adapters, short/low-quality reads, and high N-content sequences. The cleaned reads were used for further analysis (NCBI Gene Expression Omnibus (GEO) under the accession number GSE294785).
Functional enrichment analysis was performed on differentially expressed genes in the eWAT of 105-week-old male WT and Adipo-Cre- FMO3flox/flox mice using gene set enrichment analysis (GSEA) with the clusterProfiler R package (v4.10.0). The unfiltered differentially expressed gene list was ranked by decreasing test statistics. GSEA calculates an enrichment score (ES) for each MSigDB gene set (https://download.baderlab.org/EM_Genesets/current_release/Mouse/symbol/) by examining the ranked gene list. Positive NES values indicate enrichment at the top of the list, while negative NES values indicate enrichment at the bottom. Enrichment plots were generated using the gseaplot function from the enrichplot R package (v1.22.0).
Isolation of sWAT from human subjects
Subcutaneous adipose tissues were obtained from non-diabetes patients (young: 16–55 years, n = 6; aged: 60–72 years, n = 8) during nephrectomy by the Department of Urology Surgery in Zhongshan Hospital, Fudan University. The Institute’s Ethics Committee (No. Y2023-620) approved the design of the study. Additionally, the ethics approval number provided by The Hong Kong Polytechnic University for the use of human adipose tissue samples is HSEARS20230811002. All participants were fully informed about the study and signed informed consent forms. Briefly, after removing visible blood vessels and connective tissues, each adipose tissue sample was dissected into small pieces (50 mg/sample) for TMAO extraction. Clinical and metabolic characteristics of human subjects are listed in the table.
RNA-seq analysis of FMO3/Fmo3 expression in fat depots in humans and rodents
The mouse bulk RNA-Sequencing data used in this study for comparing the expression of FMO family at different ages was obtained from the Tabula Muris Consortium and deposited in the NCBI GEO under the accession number GSE132040. This dataset consists of transcriptomic data from 17 different mouse tissues collected across 10 time points (1, 3, 6, 9, 12, 15, 18, 21, 24, and 27 months) from both male and female mice. We only re-analysed the data at 1 and 27 months old.
We also re-analysed the RNA-Sequencing data (GSE175495) of sWAT from 12 young (20-35 years old) and 12 aged (60–85 years old) human participants in a previous study36. To identify the genes co-expressed with FMO3, we conducted the weighted gene correlation network analysis. First, the gene co-expression network was created with the weighted gene correlation network analysis (WGCNA) protocol of the R package WGCNA (v.1.72-5)37. WGCNA was performed on the normalised and variance-stabilising transformed (VST) expression data. A soft threshold (power) of 7, a signed topological overlap matrix, and a minimum module size of 30 were selected to construct a scale-free network and module detection. A module–trait correlations were represented as a heatmap with the module eigengenes and FMO3 normalised count. The brown modules displaying the most positive correlation to FMO3 were further analysed. We identified 405 module hub genes based on gene significance (GS) > 0.2 and module membership (MM) > 0.7. We further created a design matrix from the ‘Age variable’ (young and old aged) and ran a linear model with the limFit function of limma package (V 3.58.1). After applying multiple testing correction, we plotted the differential eigengenes within the brown module across the young and old aged samples. The shinyGO (V 0.77) was used to test for the over-representation analysis (ORA) of co-expressed hub genes of the brown. Pathways were sorted by fold enrichment and colour coded by -log10(FDR).
Second, we sorted the data from the same dataset (GSE175495) based on FMO3 expression and split them into two groups- high FMO3 and low FMO3. We included 18 samples with top 9 and bottom 9 FMO3 expression levels and excluded the remaining 4 with intermediate FMO3 expression levels. To this end, differential gene expression (DGE) analysis between the two groups was performed using DESeq2 package. Genes with p-adj ≤ 0.05, |log2FC | ≥ 2 and base mean ≥ 20 were identified as significantly differentially expressed genes (DEG) and visualised by volcano plot. Functional enrichment analysis of DEGs was performed by gene set enrichment analysis (GSEA) using R package clusterProfiler (V 4.10.0) with the unfiltered rank-ordered gene list. The MSigDB gene set collections were downloaded from Bader lab (https://download.baderlab.org/EM_Genesets/current_release/Human/symbol/). The gsea plot function of R package enrichplot (V 1.22.0) was used to generate the plot of running ES scores.
To investigate the effects of calorie restriction on FMO3 expression, we re-analysed RNAseq data of sWAT samples collected in the CALERIE-II study39. Human participants without obesity and health participants were randomly assigned to either a 25% calorie restriction (CR) group or an ad libitum control group for 2 years with samples collected at baseline, 1 year, and 2 years. Abdominal sWAT biopsies from the CR group participants who achieved an average 14% sustained calorie restriction were used for the RNA-Sequencing analysis.
Caspase-1 activity assay
To investigate the activation of caspase-1 in the conditioned medium, a chloroform-methanol protein extraction method was employed, followed by the colorimetric assay using a commercially available kit (Catalogue# C1101, Beyotime Biotechnology) according to the manufacturer’s protocol. Briefly, the collected conditioned medium was mixed with methanol and chloroform at a 4:4:1 ratio, vortexed for 15 s, and then centrifuged at 20,000 x g for 15 min. The upper phase of the mixture was discarded. If the original volume of the conditioned medium was 1 ml, 400 μL of methanol was added to the sample, followed by another centrifugation at 20,000 x g for 15 min. The supernatant was removed, and the protein pellet was dried for 10 min. The dried sample pellets were dissolved in the cell lysis buffer provided in the kit and incubated with the assay buffer containing the caspase-1 substrate Ac-YVAD-pNA (Catalogue# C1101, Beyotime Biotechnology). The absorbance at 405 nm was measured using a microplate reader.
Western blot analysis
Tissues or cells were homogenised in a Radioimmunoprecipitation assay (RIPA) buffer (150 mM NaCl, 50 mM Tris HCl, 2 mM EDTA, 0.1% SDS, 1% NP-40 (pH = 7.4) with protease inhibitors (Catalogue# HY-K0010, MedChemExpress, USA) and phosphatase inhibitors (Catalogue# B15002, Bimake, USA) cocktail. An equal amount of proteins was separated by SDS-PAGE and transferred to a polyvinylidene difluoride (PVDF) membrane (Catalogue # 1620177, Bio-Rad). The membrane was blocked with 10% non-fat milk in TBST and probed with primary antibody overnight at 4 oC. Following, the membrane was washed with TBST and incubated with corresponding secondary antibodies conjugated to horseradish peroxidase for 1 h at room temperature. Specific protein signals were visualised using enhanced chemiluminescence reagents (Catalogue#1705061 & #1705062, Bio-Rad, USA) and the intensity of protein bands was quantified using ImageJ software.
To analyse the maturation of IL-1β and activation of caspase-1 in the conditioned medium, the chloroform-methanol extraction method described earlier was employed. After drying the protein pellets, they were resuspended in SDS loading buffer, boiled for 10 min at 95 °C, and then subjected to immunoblot analysis.
Extraction of microsomes immunoblotting analysis and TMAO production assay
Briefly, adipocytes or AML12 cells were washed with homogenizing medium, which contains Tris-HCl (10 mM), sucrose (255 mM), MgCl2 (2 mM) and phenylmethylsulphonyl fluoride (PMSF, 0.1 mM) at 37 °C. The cells were then resuspended in 2 volumes of the same homogenizing medium. The de-fatted homogenate, obtained by repeatedly suctioning adipocytes with a 29-gauge needle, was centrifuged at 16,000 x g for 15 min. The resulting pellet, which contained plasma membranes, mitochondria, and lysosome, was collected along with the supernatant, which contained the microsomes. The supernatant was centrifuged at 212,000 x g (Optima XE-90 Ultracentrifuge, Beckman Coulter) for 70 min at 4 °C. The pellet containing microsomes was resuspended in 0.1 M potassium phosphate buffer (pH = 7.4) to a final volume of approximately 2 mg protein/mL.
The reaction system for measuring FMO3 enzymatic activity contained 0.50 mg/mL microsomal protein and an NADPH-generating system consisting of 1.3 mM NADP+, 3.3 mM glucose-6-phosphate, 3.3 mM MgCl2, and 0.4 unit/mL glucose-6-phosphate dehydrogenase in 0.1 M potassium phosphate buffer (pH = 5.4, 6.4, 7.4, 8.4 and 9.4). In experiments testing the effect of temperature on FMO3 activity, 0.5 mg/mL of microsomal protein were preheated at 45 °C for 0, 1, 3, 5, or 10 min without an NADPH-generating system. After heat-treatment, the mixtures were cooled on ice for 15 min. After a 5 min preincubation at 37 °C, reactions were started by the addition of substrate d9-TMA (1000 μM) and were allowed to continue at 37 °C for 20 min. The reactions were terminated with the addition of two volumes of methanol. After addition of internal standard (d8-valine) solution to the reaction mixture, protein was precipitated by centrifugation. The supernatant was subjected to liquid chromatography (LC) quantification.
We collected AML12 cells and mature 3T3-L1 adipocytes from six-well plates (each plate as one sample), with three independent replicates for each cell type. The cells were lysed and centrifuged using the aforementioned method to obtain the pellet and supernatant. Subsequently, the supernatant underwent ultracentrifugation to isolate microsomal proteins as above. Immunoblotting was used to detect the content of FMO3 in the cells, with TOM20 serving as a marker protein to distinguish whether the microsomes and the pellets were fractionated.
Statistical analysis
All statistical analyses were performed using GraphPad Prism 7.0. The data were presented as mean ± SEM. The normality of data distribution and homogeneity of variance were assessed using the Shapiro-Wilk and Levene’s tests, respectively, to guide the selection of appropriate parametric or non-parametric statistical methods. For comparisons between two groups, Student’s t-test with Welch’s correction when variance is unequal or Mann-Whitney U-test was used. One-way ANOVA with Bonferroni correction was employed for multiple group comparisons. To assess the relationship between two variables, Pearson co-efficient analysis was conducted. Kaplan-Meier survival rate curves were constructed to evaluate the longevity effects, and a non-parametric log-rank test was used to compare the overall survival distributions between the groups. A p-value < 0.05 was considered statistically significant. Animal experiments were performed using independent samples, and in vitro experiments were repeated at least twice with consistent results.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The RNA-sequencing data generated in this study have been deposited in the NCBI database under accession code GSE294785 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE294785]. The experimental data generated in this study are provided in the Supplementary Information/Source Data file. The sequencing data used in this study are available in the NCBI database under accession codes GSE132040 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE132040], GSE175495 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE175495] and PRJNA1018321 [https://www.ncbi.nlm.nih.gov/sra/PRJNA1018321]. The proteomics data for TMAO-binding proteins is available in ProteomeXchange with accession number PXD066391 [https://www.ebi.ac.uk/pride/archive/projects/PXD066391]. All data supporting the findings described in this manuscript is also available from the corresponding author upon request. Source data are provided with this paper.
References
Youm, Y. H. et al. Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell Metab. 18, 519–532 (2013).
Liu, Z., Wu, K. K. L., Jiang, X., Xu, A. & Cheng, K. K. Y. The role of adipose tissue senescence in obesity- and ageing-related metabolic disorders. Clin. Sci.134, 315–330 (2020).
Palmer, A. K. et al. Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell 18, e12950 (2019).
Islam, M. T. et al. Senolytic drugs, dasatinib and quercetin, attenuate adipose tissue inflammation, and ameliorate metabolic function in old age. Aging Cell 22, e13767 (2023).
Yu, Q. et al. Sample multiplexing for targeted pathway proteomics in aging mice. Proc. Natl. Acad. Sci. USA 117, 9723–9732 (2020).
Tabula Muris, C. A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature 583, 590–595 (2020).
Wu, Y. et al. Dynamics of single-nuclei transcriptomic profiling of adipose tissue from diverse anatomical locations during mouse aging process. Sci. Rep. 14, 16093 (2024).
Zhao, S. et al. Partial leptin reduction as an insulin sensitization and weight loss strategy. Cell Metab. 30, 706–719 e706 (2019).
Matsubara, T. et al. PGRN is a key adipokine mediating high fat diet-induced insulin resistance and obesity through IL-6 in adipose tissue. Cell Metab. 15, 38–50 (2012).
Chen, Q. et al. A brown fat-enriched adipokine Adissp controls adipose thermogenesis and glucose homeostasis. Nat. Commun. 13, 7633 (2022).
Petrus, P. et al. Glutamine links obesity to inflammation in human white adipose tissue. Cell Metab. 31, 375–390 e311 (2020).
Yore, M. M. et al. Discovery of a class of endogenous mammalian lipids with anti-diabetic and anti-inflammatory effects. Cell 159, 318–332 (2014).
Camell, C. D. et al. Inflammasome-driven catecholamine catabolism in macrophages blunts lipolysis during ageing. Nature 550, 119–123 (2017).
He, M. et al. An acetylation switch of the NLRP3 inflammasome regulates aging-associated chronic inflammation and insulin resistance. Cell Metab. 31, 580–591 e585 (2020).
Liu, Z. et al. The dysfunctional MDM2-p53 axis in adipocytes contributes to aging-related metabolic complications by induction of lipodystrophy. Diabetes 67, 2397–2409 (2018).
Minamino, T. et al. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat. Med 15, 1082–1087 (2009).
Vergoni, B. et al. DNA damage and the activation of the p53 pathway mediate alterations in metabolic and secretory functions of adipocytes. Diabetes 65, 3062–3074 (2016).
Yang, H. et al. Gut microbial-derived phenylacetylglutamine accelerates host cellular senescence. Nat. Aging (2025). https://doi.org/10.1038/s43587-024-00795-w
Sun, L. et al. Age-dependent changes in the gut microbiota and serum metabolome correlate with renal function and human aging. Aging Cell 22, e14028 (2023).
Ghosh, T. S., Shanahan, F. & O’Toole, P. W. The gut microbiome as a modulator of healthy ageing. Nat. Rev. Gastroenterol. Hepatol. 19, 565–584 (2022).
Awoyemi, A., Troseid, M., Arnesen, H., Solheim, S. & Seljeflot, I. Markers of metabolic endotoxemia as related to metabolic syndrome in an elderly male population at high cardiovascular risk: a cross-sectional study. Diabetol. Metab. Syndr. 10, 59 (2018).
Koeth, R. A. et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 19, 576–585 (2013).
Wang, Z. et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 472, 57–63 (2011).
Rath, S., Heidrich, B., Pieper, D. H. & Vital, M. Uncovering the trimethylamine-producing bacteria of the human gut microbiota. Microbiome 5, 54 (2017).
Miao, J. et al. Flavin-containing monooxygenase 3 as a potential player in diabetes-associated atherosclerosis. Nat. Commun. 6, 6498 (2015).
Gao, X. et al. Dietary trimethylamine N-oxide exacerbates impaired glucose tolerance in mice fed a high fat diet. J. Biosci. Bioeng. 118, 476–481 (2014).
Schugar, R. C. et al. The TMAO-producing enzyme flavin-containing monooxygenase 3 regulates obesity and the beiging of white adipose tissue. Cell Rep. 19, 2451–2461 (2017).
Li, D. et al. Trimethylamine-N-oxide promotes brain aging and cognitive impairment in mice. Aging Cell 17, e12768 (2018).
Hu, J. et al. Trimethylamine N-oxide promotes abdominal aortic aneurysm formation by aggravating aortic smooth muscle cell senescence in mice. J. Cardiovasc. Transl. Res 15, 1064–1074 (2022).
Greg Falls, J., Yan Cao, D.-Y. R., Levi, P. atriciaE. & Hodgson, E. rnest Regulation of mouse liver flavin-containing monooxygenases 1 and 3 by sex steroids. Arch. Biochem. Biophys. 342, 212–223 (1997).
Bennett, B. J. et al. Trimethylamine-N-oxide, a metabolite associated with atherosclerosis, exhibits complex genetic and dietary regulation. Cell Metab. 17, 49–60 (2013).
Licznerska, B. & Baer-Dubowska, W. Indole-3-carbinol and its role in chronic diseases. Adv. Exp. Med. Biol. 928, 131–154 (2016).
Rossner, R., Kaeberlein, M. & Leiser, S. F. Flavin-containing monooxygenases in aging and disease: Emerging roles for ancient enzymes. J. Biol. Chem. 292, 11138–11146 (2017).
Gonzalez Malagon, S. G. et al. The phenotype of a knockout mouse identifies flavin-containing monooxygenase 5 (FMO5) as a regulator of metabolic ageing. Biochem. Pharm. 96, 267–277 (2015).
Leiser, S. cottF. e. a. Cell nonautonomous activation of flavin-containing monooxygenase promotes longevity and health span. Science 350, 1375–1378 (2015).
Trim, W. V. et al. Divergent immunometabolic changes in adipose tissue and skeletal muscle with ageing in healthy humans. J. Physiol. 600, 921–947 (2022).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinforma. 9, 559 (2008).
Schurch, N. J. et al. How many biological replicates are needed in an RNA-seq experiment and which differential expression tool should you use?. RNA 22, 839–851 (2016).
Spadaro, O. et al. Caloric restriction in humans reveals immunometabolic regulators of health span. Science 375, 671–677 (2022).
Luo, M. et al. Hif-1alpha expression targets the TMA/Fmo3/TMAO axis to participate in gallbladder cholesterol stone formation in individuals living in plateau regions. Biochim. Biophys. Acta Mol. Basis Dis. 1870, 167188 (2024).
Covarrubias, A. J. et al. Senescent cells promote tissue NAD(+) decline during ageing via the activation of CD38(+) macrophages. Nat. Metab. 2, 1265–1283 (2020).
Dutta, S. & Sengupta, P. Men and mice: relating their ages. Life Sci. 152, 244–248 (2016).
Nguyen, T. T. & Corvera, S. Adipose tissue as a linchpin of organismal ageing. Nat. Metab. 6, 793–807 (2024).
Wang, W. et al. A PRDM16-driven metabolic signal from adipocytes regulates precursor cell fate. Cell Metab. 30, 174–189 e175 (2019).
Harms, M. J. et al. Prdm16 is required for the maintenance of brown adipocyte identity and function in adult mice. Cell Metab. 19, 593–604 (2014).
Wu, K. K., Cheung, S. W. & Cheng, K. K. NLRP3 Inflammasome Activation in Adipose Tissues and Its Implications on Metabolic Diseases. Int. J. Mol. Sci. 21 (2020). https://doi.org/10.3390/ijms21114184
Guo, H., Callaway, J. B. & Ting, J. P. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat. Med 21, 677–687 (2015).
Kursawe, R. et al. A role of the inflammasome in the low storage capacity of the abdominal subcutaneous adipose tissue in obese adolescents. Diabetes 65, 610–618 (2016).
Chen, M. L. et al. Trimethylamine-N-oxide induces vascular inflammation by activating the NLRP3 inflammasome through the SIRT3-SOD2-mtROS signaling pathway. J. Am. Heart Assoc. 6 (2017). https://doi.org/10.1161/JAHA.117.006347
Yeung, C. K. e. a. Functional Characterization of Genetic Variants of Human FMO3 Associated with Trimethylaminuria. Arch. Biochem. Biophys. 464, 251–259 (2007).
Figlia, G., Willnow, P. & Teleman, A. A. Metabolites regulate cell signaling and growth via covalent modification of proteins. Dev. Cell 54, 156–170 (2020).
Kumari, K., Singh, K. S., Singh, K., Bakhshi, R. & Singh, L. R. TMAO to the rescue of pathogenic protein variants. Biochim. Biophys. Acta Gen. Subj. 1866, 130214 (2022).
Chen, S. et al. Trimethylamine N-oxide binds and activates PERK to promote metabolic dysfunction. Cell Metab. 30, 1141–1151 e1145 (2019).
Piazza, I. et al. A map of protein-metabolite interactions reveals principles of chemical communication. Cell 172, 358–372 e323 (2018).
Zhang, L. et al. Peli1 facilitates NLRP3 inflammasome activation by mediating ASC ubiquitination. Cell Rep. 37, 109904 (2021).
Soriano-Teruel, P. M. et al. Identification of an ASC oligomerization inhibitor for the treatment of inflammatory diseases. Cell Death Dis. 12, 1155 (2021).
Chen, H. et al. ASC regulates subcutaneous adipose tissue lipogenesis and lipolysis via p53/AMPKalpha axis. Int. J. Mol. Sci. 23 (2022). https://doi.org/10.3390/ijms231710042
Vandanmagsar, B. et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 17, 179–188 (2011).
Arnush, M. et al. IL-1 produced and released endogenously within human islets inhibits B cell function. J. Clin. Investig. 102, 516–526 (1998).
Camell, C. D. et al. Aging induces an Nlrp3 inflammasome-dependent expansion of adipose B cells that impairs metabolic homeostasis. Cell Metab. 30, 1024–1039 e1026 (2019).
Unamuno, X. et al. NLRP3 inflammasome blockade reduces adipose tissue inflammation and extracellular matrix remodeling. Cell Mol. Immunol. 18, 1045–1057 (2021).
Furman, D. et al. Expression of specific inflammasome gene modules stratifies older individuals into two extreme clinical and immunological states. Nat. Med. 23, 174–184 (2017).
Wen, H. et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 12, 408–415 (2011).
Wu, H. et al. Deficiency of mitophagy receptor FUNDC1 impairs mitochondrial quality and aggravates dietary-induced obesity and metabolic syndrome. Autophagy 15, 1882–1898 (2019).
Shi, C.-S. et al. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 13, 255–263 (2012).
Lopez-Castejon, G. Control of the inflammasome by the ubiquitin system. FEBS J. 287, 11–26 (2020).
Ohtsuka, T. et al. ASC is a Bax adaptor and regulates the p53-Bax mitochondrial apoptosis pathway. Nat. Cell Biol. 6, 121–128 (2004).
Zhang, L., Yu, F. & Xia, J. Trimethylamine N-oxide: role in cell senescence and age-related diseases. Eur. J. Nutr. 62, 525–541 (2023).
Kim, S. J. et al. Gut microbe-derived metabolite trimethylamine N-oxide activates PERK to drive fibrogenic mesenchymal differentiation. iScience 25, 104669 (2022).
Drexler, S. K. et al. Tissue-specific opposing functions of the inflammasome adaptor ASC in the regulation of epithelial skin carcinogenesis. Proc. Natl. Acad. Sci. USA 109, 18384–18389 (2012).
Fu, W., Liu, Y., Sun, C. & Yin, H. Transient p53 inhibition sensitizes aged white adipose tissue for beige adipocyte recruitment by blocking mitophagy. FASEB J. 33, 844–856 (2019).
Saaoud, F. et al. Aorta- and liver-generated TMAO enhances trained immunity for increased inflammation via ER stress/mitochondrial ROS/glycolysis pathways. JCI Insight 8 (2023). https://doi.org/10.1172/jci.insight.158183
Wang, J. et al. Deficiency of flavin-containing monooxygenase 3 protects kidney function after ischemia-reperfusion in mice. Commun. Biol. 7, 1054 (2024).
Wei, D. et al. Melatonin relieves hepatic lipid dysmetabolism caused by aging via modifying the secondary bile acid pattern of gut microbes. Cell Mol. Life Sci. 79, 527 (2022).
Brunt, V. E. et al. Suppression of the gut microbiome ameliorates age-related arterial dysfunction and oxidative stress in mice. J. Physiol. 597, 2361–2378 (2019).
Zhang, J. et al. Hepatic flavin-containing monooxygenase gene regulation in different mouse inflammation models. Drug Metab. Dispos. 37, 462–468 (2009).
Collins, H. L. et al. L-Carnitine intake and high trimethylamine N-oxide plasma levels correlate with low aortic lesions in ApoE-/-transgenic mice expressing CETP. Atherosclerosis 244, 29–37 (2016).
Emiliano, A. B., Governale, L., Parks, M. & Cooper, D. S. Shifts in propylthiouracil and methimazole prescribing practices: antithyroid drug use in the United States from 1991 to 2008. J. Clin. Endocrinol. Metab. 95, 2227–2233 (2010).
Zhu, S. et al. TMAO is involved in sleep deprivation-induced cognitive dysfunction through regulating astrocytic cholesterol metabolism via SREBP2. Front. Mol. Neurosci. 17, 1499591 (2024).
Prokopienko, A. J. et al. Metabolic activation of flavin monooxygenase-mediated trimethylamine-N-oxide formation in experimental kidney disease. Sci. Rep. 9, 15901 (2019).
Kolonin, M. G., Saha, P. K., Chan, L., Pasqualini, R. & Arap, W. Reversal of obesity by targeted ablation of adipose tissue. Nat. Med 10, 625–632 (2004).
Wang, L., Jia, Q., He, J. & Li, Y. Adipose tissue-targeting nanomedicines for obesity pharmacotherapy. Trends Endocrinol. Metab. (2025). https://doi.org/10.1016/j.tem.2025.03.010
Wu, K. K. L. et al. The APPL1-Rab5 axis restricts NLRP3 inflammasome activation through early endosomal-dependent mitophagy in macrophages. Nat. Commun. 12, 6637 (2021).
Wabitsch, M. et al. Characterization of a human preadipocyte cell strain with high capacity for adipose differentiation. Int. J. Obes. 25, 8–15 (2001).
Tews, D. et al. 20 Years with SGBS cells—a versatile in vitro model of human adipocyte biology. Int. J. Obes. 46, 1939–1947 (2022).
Zhang, Y. et al. Adult mesenchymal stem cell ageing interplays with depressed mitochondrial Ndufs6. Cell Death Dis. 11, 1075 (2020).
Zhang, C. et al. Topical and intravenous administration of human umbilical cord mesenchymal stem cells in patients with diabetic foot ulcer and peripheral arterial disease: a phase I pilot study with a 3-year follow-up. Stem Cell Res. Ther. 13, 451 (2022).
Sun, S. J. et al. Immunomodulation by systemic administration of human-induced pluripotent stem cell-derived mesenchymal stromal cells to enhance the therapeutic efficacy of cell-based therapy for treatment of myocardial infarction. Theranostics 11, 1641–1654 (2021).
Zhao, X., Zeisel, S. H. & Zhang, S. Rapid LC-MRM-MS assay for simultaneous quantification of choline, betaine, trimethylamine, trimethylamine N-oxide, and creatinine in human plasma and urine. Electrophoresis 36, 2207–2214 (2015).
Acknowledgements
This project was supported by Hong Kong Research Grant Council (RGC) General Research Fund (15105119) and Collaborative Research Fund (C5044-23G), National Natural Science Foundation of China (92357305), Shenzhen Municipal Science and Technology Innovation Commission Basic Research General Programme: 20210324130202006 and PolyU internal funding (P0036848) to Dr. Kenneth King-yip Cheng. Martin Wabitsch received funding by the Federal Ministry of Education and Research (Bundesministerium für Bildung und Forschung, BMBF) as part of the German Center for Child and Adolescent Health (DZKJ) under the funding code 01GL2407A. Juntao Yuan and his research is supported by the Hong Kong Scholars Program.
Author information
Authors and Affiliations
Contributions
T.G. and J.Y. conducted most of the experiments, manage the raw data, prepare the figures, and drafted the manuscript. K.K.L.W. and W.K. performed LiP-SMap and some experiments in macrophages and revised the manuscript. M.Y.M.H. conducted the DARTS analysis and contributed to the revision of the manuscript. M.M.H. performed WGCNA and DGE analyses. B.W. and X.L. collected human samples and conducted data analysis. M.W. provided SGSB cells and methods for SGBS cell cultures and experiments. Y.Z. advised on SGSB cell experiments. Q.L., X.F., and Y.N. provided hADMSCs and conducted related experiments. K.L. conducted flow cytometry analysis. K.K.C. acquired the funding, provided resources, supervised the project, and wrote the manuscript. All authors reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflicts of interest.
Peer review
Peer review information
Nature Communications thanks Andrew Neilson and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ganapathy, T., Yuan, J., Ho, M.Ym. et al. Adipocyte FMO3-derived TMAO induces WAT dysfunction and metabolic disorders by promoting inflammasome activation in ageing. Nat Commun 16, 8873 (2025). https://doi.org/10.1038/s41467-025-63905-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-63905-1
