
Abstract
Tpt1/TRPT1/KptA family proteins are evolutionarily conserved in all three domains of life. In fungi and plants, Tpt1 transfers 2’-PO42- from tRNA splice junction to NAD+, which is the final step of tRNA maturation and is critical for the function of tRNA. In mammals and bacteria, Tpt1-catalyzed reaction leads to 5’-end ADP ribosylation, a reversible chemical modification of nucleic acids. Based on in vivo and in vitro biochemical studies, a two-step catalytic mechanism has been established for Tpt1-catalyzed RNA 2’-PO42- transfer, including (i) the 2’-PO42- attacks NAD+, releasing nicotinamide and forming a 2’-phospho-ADP-ribosylated RNA (2’-p-ADPR-RNA) intermediate; and (ii) transesterification of the ADP-ribose 2”-OH to RNA 2’-PO42-, displacing the 2’-OH RNA and producing ADP-ribose-1”,2”-cyclic phosphate (Appr>P). However, neither 2’-p-ADPR-RNA intermediate nor Appr>P product has been captured in any reported Tpt1 structures. Here, we report a series of crystal structures of T. kodakarensis Tpt1 (TkoTpt1), capturing the key 2’-p-ADPR-RNA intermediate. In addition, our structures also capture the 5’-p-ADPR-DNA intermediate and Appr>P product. Structural analysis and in vitro catalytic assays revealed that TkoTpt1 utilizes similar mechanism in 2’-PO42- and 5’-PO42- transfer. In conclusion, our structures reaffirm the catalytic mechanism of Tpt1-catalyzed phosphate transfer.
Similar content being viewed by others

Introduction
Transfer RNA (tRNA) plays vital roles in living organisms1,2. In addition to end processing and base modification, maturation of many tRNAs also requires splicing of intron3. Although not present in all organisms, tRNA introns are widely distributed in Archaea and Eukarya4,5. tRNA splicing has been investigated in many species6,7,8,9, especially the budding yeast S. cerevisiae10,11. It was found that tRNA splicing in S. cerevisiae includes three distinct stages, namely (i) intron excision by the splicing endonuclease12,13, (ii) ligation of the 5’-half and 3’-half by the ligase Trl114,15, and (iii) removal of 2’-PO42- at splice junction of the ligated tRNA by the enzyme Tpt116,17,18. Depletion of Tpt1 gene leads to the accumulation of multiple ligated tRNA species bearing a splice junction 2’-PO42- and deceased viability of S. cerevisiae, confirming that Tpt1 is essential for both tRNA splicing and vegetative growth of S. cerevisiae19. Besides S. cerevisiae, Tpt1 also plays identical roles in tRNA splicing in plants and other fungal species, such as the top-priority human fungal pathogens C. auris, C. albicans, A. fumigatus, and C. immitis20,21. Due to their high conservation and critical roles, Tpt1 family proteins have been viewed as a potential target for antifungal drug development21.
Tpt1 homologs are evolutionarily conserved in bacterial, archaeal, and eukaryotic species22. The mammalian and bacterial homologs of Tpt1 are known as TRPT1 and KptA, respectively. TRPT1 and KptA family proteins possess in vitro RNA 2’-phosphotransferase activity and can functionally replace Tpt1 in S. cerevisiae22,23, but the tRNAs of mammals and bacteria do not contain intron or utilize different types of enzymes for intron splicing24, which strongly indicates that Tpt1/TRPT1/KptA family proteins may function on other types of substrates. Such a hypothesis has been confirmed by several discoveries in recent years. In 2022, Ohira et al. discovered that many thermophilic bacterial tRNAs are phosphorylated at 2’-position of U47, which confers thermal stability and nuclease resistance to tRNAs. This UP47 modification can be dephosphorylated by archaeal Tpt1 protein, tuning the structural rigidity of tRNAs under extreme environmental conditions25. Besides internal 2’-PO42- of tRNAs, Munir et al. found that Tpt1 homologs from A. pernix (Ape), P. horikoshii (Pho), C. thermocellum (Cth), and A. fulgidus (Afu) can all cap RNA or DNA 5’-phosphate with ADP-ribose26. Munnur et al. demonstrated that such 5’-end ADP-ribosylation activity is conserved for KptA from S. coelicolor and TRPT1 from higher eukaryotes, such as humans27.
The catalytic activity of Tpt1/TRPT1/KptA family proteins is NAD+-dependent. Based on extensive in vivo and in vitro studies16,17,18,28, a two-step catalytic mechanism has been well established for Tpt1 family proteins (Fig. 1a), including (i) the 2’-PO42- of the RNA attacks NAD+, releasing nicotinamide and forming a 2’-phospho-ADP-ribosylated RNA (2’-p-ADPR-RNA) intermediate; and (ii) transesterification of the ADP-ribose 2”-OH to RNA 2’-PO42-, displacing the 2’-OH RNA and producing ADP-ribose-1”,2”-cyclic phosphate (Appr>P)16,29. To verify the catalytic mechanism, structural studies have been carried out for various Tpt1/TRPT1/KptA family proteins, such as Tpt1 from S. cerevisiae, A. pernix, A. thermocellus, P. horikoshii, and R. slithyformis, and TRPT1 from human and mouse30,31,32. Besides NAD+ and RNA binding, these structures also captured some unexpected substrates of Tpt1/TRPT1/KptA family proteins, such as NADP(H)31. However, none of these structures captured the key intermediate or the Appr>P product.

a The established mechanism for the Tpt1-catalyzed RNA 2’-PO42- transfer reaction. b Size-exclusion profile and SDS-PAGE gel analysis of the purified TkoTpt1 protein. c, d In vitro assays showing DNA 5’-PO42- transfer activity of TkoTpt1. Sequence of the DNA is listed at the bottom of the figure. The reaction system contains 0.5 μM TkoTpt1, 0.5 μM substrate DNA, and 2 mM NAD+ in reaction buffer.
To provide direct structural evidences for the catalytic mechanism of Tpt1/TRPT1/KptA family proteins, we performed extensive crystallization trials on T. kodakarensis Tpt1 (TkoTpt1). Here, we report a series of TkoTpt1 structures (Supplementary Tables 1–4), including four structures in the presence of NAD+ and 5’-p-DNA, capturing the reaction either prior to 5’-PO42- transfer, at the intermediate state, or after the completion of the reaction. Four TkoTpt1 structures in complex with 2’-PO42--containing nucleotides were also determined. Structural analysis indicated that Tpt1/TRPT1/KptA family proteins all utilize the conserved mechanism in transferring phosphate from their substrates.
Results
TkoTpt1 has nucleic acid 5’-PO4 2- catalytic activity
TkoTpt1 is composed of 182 amino acids (aa) with a theoretical molecular weight of 21.0 kDa. A previous study showed that TkoTpt1 can dephosphorylate UP47 of thermophilic bacterial tRNAs25, whereas it is unclear whether TkoTpt1 can function on other substrates, such as RNA and DNA with 5’-PO42-. To this end, we first expressed and purified TkoTpt1 protein to homogeneity (Fig. 1b). The protein was eluted at 12.4-mL on the Superdex G75 size-exclusion column, suggesting that TkoTpt1 exists as a monomer in solution. We then carried out in vitro catalytic assays using one DNA with PO42- at the 5’-end and FAM-label at the 3’-end (5’-p-GGTGGCGTGT-FAM-3’) as substrate. As depicted in Fig. 1c, no reaction could occur to the DNA in the absence of TkoTpt1 and/or NAD+ at 37 °C. Although very weak, one product band was observed at 4 °C when both TkoTpt1 and NAD+ were present. At 37 °C, 60 °C, and 80 °C, one additional product was also generated.
To determine the possible identities of the two products, we performed a time-course analysis at 80 °C (Fig. 1d). At the reaction times of 1 min and 2 min, mainly the slow-moving product was generated. At the reaction time of 5 min and 10 min, both products were observed. However, mainly the fast-moving product could be detected at the reaction times of 30 min and 60 min. These observations suggested that TkoTpt1 can work on DNA with 5’-PO42- and the slow-moving and fast-moving bands might correspond to an intermediate and the final product, respectively. Besides the original 5’-p-DNA, we designed three variant DNAs with different 5’-end nucleotides and performed time course analysis. As depicted in Fig. 2, all DNA variants can be catalyzed by TkoTpt1 in vitro. Compared to other DNAs, TkoTpt1 is more efficient in catalyzing the first step reaction on the variant with 5’-end Cytosine (kstep1 = 0.93±0.16 min−1, Table 1). The rate constant is very slow for the variant with Guanosine at 5’-end (kstep1 = 0.39 ± 0.09 min−1).

The reaction system contains 0.25 μM TkoTpt1 protein, 0.5 μM substrate DNA, and 2 mM NAD+ in reaction buffer. Experiments were repeated independently three times with similar results. Source data are provided in the Source Data file. The distributions of the 5′-p-ADPR DNA intermediate and 5′-OH DNA product (expressed as percentage of total DNA) are plotted as a function of reaction time. Each datum in the graphs is the average of three independent experiments ±SEM.
TkoTpt1 can catalyze the conversion from the intermediate to the final product (Fig. 2). At a reaction time of 90 min, about 92% final product was observed for the DNA variant with 5’ Guanosine. Only 26-76% final product were produced for other three variants. Compared to the first step, the rate constant for the second step is much slower (Table 1), which are 2.68 ± 0.29 × 10−2 min−1, 1.34 ± 0.15 × 10−2 min−1, 0.32 ± 0.10 × 10−2 min-1, and 1.44 ± 0.12 × 10−2 min-1 for the DNAs with Guanosine, Thymine, Cytosine, and Adenosine at 5’-ends, respectively. These observations indicated that the identity of DNA 5’-end nucleotide affects both steps of the reaction catalyzed by TkoTpt1. TkoTpt1 can also catalyze the reaction between 5’-p-RNA (5’-p-AGUGGCGUGU-FAM-3’) and NAD+ at 80 °C (Supplementary Fig. 1). Compared to the corresponding DNA, the rate constants for both steps of the RNA are slower (kstep1 = 0.44 ± 0.06 min-1; kstep2 = 0.17± 0.08 × 10−2 min−1).
It is worth mentioning that the 5’-phosphate transfer reaction has been investigated for human TRPT1, ApeTpt11, PhoTpt1, AfuTpt1, and CthTpt1 at 37 °C26,27. Although these proteins can catalyze the reaction between NAD+ and 5’-p-DNA (or 5’-p-RNA), the reaction was limited to the first step and no 5’-OH DNA (or 5’-OH RNA) product from the second step was generated. Production of 5’-OH DNA by TkoTpt1 requires very high temperature (80 °C) and the rate constant is very low (Table 1). Instead of being a biologically relevant property of Tpt1 family enzymes, dephosphorylation of a DNA 5’-phosphate is likely specific to TkoTpt1.
The overall conformation of TkoTpt1 is flexible
Upon confirming the catalytic activity of TkoTpt1, we then performed crystallization trials and solved one unliganded TkoTpt1 structure (Supplementary Table 1). The crystal belongs to C2221 space group; per asymmetric unit contains four TkoTpt1 protomers. As depicted in Supplementary Fig. 2a, each TkoTpt1 protomer is composed of two lobes, the N-terminal lobe (N-lobe, aa 1–86) and the C-terminal lobe (aa 87–182). Since it was involved in NAD+ binding in the Tpt1 and TRPT1 structures31,32, the C-terminal lobe was also named as the NAD+-lobe. Both N-lobe and NAD+-lobe are of α/β fold in nature (Supplementary Fig. 2a–c). The N-lobe contains two anti-parallel β-strands (β1 and β2) and three α-helices (α1-3). The NAD+-lobe is composed of four α-helices (α4-7) and five β-strands (β3-7) that are anti-parallel to each other.
Structural superposition showed that the folding of the N-lobe is conserved among the four TkoTpt1 protomers; the root mean square deviation (RMSD) value between them is only 0.4–0.6 Å (Supplementary Fig. 2d, left panel). Supported by the low RMSD value (0.5–0.7 Å), the folding of the NAD+-lobe is also conserved in the four protomers (Supplementary Fig. 2d, right panel). However, the relative orientation between the N-lobe and the NAD+-lobe is very flexible. When superimposed on the NAD+-lobe, the N-lobe undergoes obvious tilting in the protomers B and D (Supplementary Fig. 2e), suggesting that TkoTpt1 could adopt multiple conformations.
Structural basis for NAD+ and substrate DNA binding by TkoTpt1
In order to reveal the underlying basis for the TkoTpt1-catalyzed reaction, we synthesized one 5’-p-DNA (5’-p-AAAAAAAAAA-3’) and performed co-crystallization trials with TkoTpt1 and NAD+. One complex structure, TkoTpt1/DNA/NAD+, was determined (Fig. 3a). The structure was refined at 2.01-Å resolution (Supplementary Table 2) and resulted in clear density maps for the protein (Supplementary Fig. 1a) and the NAD+ molecule (Fig. 3b and Supplementary Fig. 3b). The conformation of NAD+ is stabilized by extensive interactions with TkoTpt1 NAD+-lobe (Fig. 3c, upper panel), which is virtually identical to that reported in the PhoTpt1 structure (PDB_ID: 8TFZ, Supplementary Fig. 3c)31. In brief, the N1 and N7 atoms of the ADP motif form hydrogen bond (H-bond) interactions with the side chain hydroxyl oxygen of Ser107 and the main chain N atom of Lys114, respectively. In addition, the nucleobase also forms stacking interactions with the side chains of Ile108 and Met116. The 2’-OH and 3’-OH groups of the sugar form three H-bond interactions, including two with the side chain of Thr100 and one with the side chain of His98. The α- and β-phosphate groups form two and one H-bond interactions with the side chains of Arg118 and His137, respectively. The ribose motif packs against His122 and its 2”-OH group H-bonds with the ND1 atom of His122. The nicotinamide motif packs against Thr133 and its O7 atom H-bonds with the main chain N atom of Gly99.

a Overall folding of the TkoTpt1/DNA/NAD+ complex. The N-lobe and NAD+-lobe are shown as cartoon in yellow and green, respectively. b 2Fo-Fc electron density maps of the bound DNA molecule, NAD+, and SO42-. c The detailed interactions between TkoTpt1 and NAD+ (top panel), DNA (middle panel), or SO42- (bottom panel). The Hydrogen-bond interactions are indicated by black dashed lines. d Superposition showing the large conformational change between the N-lobe and NAD+-lobe in the TkoTpt1/DNA/NAD+ complex and the unliganded structure, which is colored in blue.
The 5’-p-DNA is composed of ten nucleotides, but only three showed clear density in the structure (Fig. 3b and Supplementary Fig. 3b). Instead of 5’-end, these nucleotides are all located at the internal region of the DNA. The nucleotide at the catalytic site is numbered as A + 1; and, the two neighboring nucleotides are numbered as A-1 and A + 2, respectively. The backbone of the nucleotides adopts a U-type conformation (Fig. 3c, middle panel). In addition to H-bond interactions with the side chain of Arg118 of the NAD+-lobe, the phosphate group of A + 2 also H-bonds with the side chains of N-lobe residues Arg17 and His18. The conformation of A + 2 is further stabilized by stacking with His18. A-1 is mainly stabilized by stacking with the nucleobase of A + 2. Instead of stacking with other nucleotides, the nucleobase of A + 1 is flipped out and points in the opposite direction. In addition to H-bond interaction with the side chain hydroxyl oxygen of Ser9, the nucleobase of A + 1 forms stacking interactions with the side chains of Ser9, Lys10, and Arg65 of the N-lobe. The conformation and interactions of A + 1 are similar to the RNA splice junction nucleobase in the reported ApeTpt1 structure (PDB_ID: 8TG4, Supplementary Fig. 3d)31.
Our in vitro assays clearly showed that TkoTpt1 can catalyze the reaction between NAD+ and 5’-p-DNA (Figs. 1c, d and 2), but NAD+ remains intact in the TkoTpt1/DNA/NAD+ structure (Fig. 3b). Structural analysis (Fig. 3c, bottom panel) suggested that the reaction might be inhibited by high concentration SO42− (2.0 M) present in the crystallization conditions (Supplementary Table 5). In the structure, one well-defined SO42− is bound between DNA and the NAD+ molecule (Fig. 3b and Supplementary Fig. 3b). In addition to the 3”-OH group of NAD+, the SO42− also H-bonds with the side chains of Arg17, Lys63, Arg65, and Tyr77 of the N-lobe and the side chain of Arg136 of the NAD+-lobe. Superposition with the reported PhoTpt1 structure (PDB_ID: 8TFX, Supplementary Fig. 3e) revealed that the bound SO42− can mimic the 2’-PO42− of the RNA and prevent it from entering the catalytic site. The inhibitory effects of SO42− can be further supported by our in vitro catalytic assays (Supplementary Fig. 4).
The folding of the N-lobe and NAD+-lobe are conserved in the unliganded and the complexed structures of TkoTpt1, but their relative orientations are significantly different. When superposed on the NAD+-lobe, the N-lobe undergoes almost a 90° rotation in the complex structure (Fig. 3d), likely due to the interactions mediated by DNA and SO42− with residues from both lobes of TkoTpt1 (Fig. 3c). The high degree of flexibility of the two lobes has also been observed for a bacterial Tpt1 by solution NMR method previously33.
TkoTpt1 can catalyze the formation of the Appr > P product
To better demonstrate that TkoTpt1 is active in catalyzing the reaction of 5’-p-DNA, we performed further co-crystallization trials for TkoTpt1, 5’-p-DNA, and NAD+. Two structures, TkoTpt1/DNA/Appr>P complex-A and TkoTpt1/DNA/Appr>P complex-B, were solved. Although identical in sample composition, the crystallization conditions for the TkoTpt1/DNA/Appr>P complexes are different from that of TkoTpt1/DNA/NAD+ (Supplementary Table 5). The condition for TkoTpt1/DNA/Appr>P complex-B is free of ammonium sulfate. Although it is present in the condition for TkoTpt1/DNA/Appr>P complex-A, the concentration of ammonium sulfate is low (50 mM). In combination with the in vitro catalytic assays (Supplementary Fig. 4), these observations suggested that the elimination of high concentration ammonium sulfate is the key factor in solving the TkoTpt1/DNA/Appr>P complexes.
The complex-A (Fig. 4a and Supplementary Fig. 5a) was refined at 1.82-Å resolution; its space group and cell parameters are similar to those of the TkoTpt1/DNA/NAD+ structure (Supplementary Table 2). As indicated by the clear density maps (Fig. 4b and Supplementary Fig. 5b), NAD+ in complex-A has been converted into the product Appr>P. The cyclic PO42− forms three H-bond interactions, including one with the side chain of Arg17 and two with the side chain of Arg65 (Fig. 4c). The O atom at the 2”-position of the ribose H-bonds with the side chain of Lys63; the O4 atom H-bonds with the side chain of Arg136. Structural superposition (Supplementary Fig. 5c, d) revealed that the overall conformation of Appr>P is similar to that of ADP-ribose-1”-phosphate captured in the reported A. thermocellus (Ath) Tpt1 structure (PDB_ID: 6E3A)32 and the ApeTpt1 structure (PDB_ID: 8TG3)31. Out of the 10 nucleotides of the DNA, six are ordered in complex-A. However, TkoTpt1 mainly interacts with the three internal nucleotides adopting U-type conformation. Likely, formation of the complex involves the release of the product 5’-OH DNA after the reaction and rebinding of the DNA during the crystallization process.

a Overall folding of the TkoTpt1/DNA/Appr>P complex-A. The bound DNA and Appr>P molecules are shown as sticks with their C-atoms colored in pink and cyan, respectively. b 2Fo-Fc electron density maps of the bound DNA and Appr>P molecules. c The detailed interactions between TkoTpt1 and the cyclic PO4 and ribose of Appr>P. d Superposition showing the conformational changes prior to and after the formation of the Appr>P product. Protein residues, DNA, and Appr>P in TkoTpt1/DNA/Appr>P complex-A are colored as in (c). For the TkoTpt1/DNA/NAD+ complex, the C-atoms of protein residues, DNA, and NAD+ are colored in gray and the sulfur atom of the bound SO42− is colored in green.
TkoTpt1/DNA/Appr>P complex-B was refined at 2.40-Å resolution (Supplementary Table 2). The crystal belongs to P21 space group; per asymmetric unit contains two TkoTpt1/DNA/Appr>P complexes (Supplementary Fig. 6a, b). All DNA nucleotides are ordered. The 5’-OH group of nucleotide A + 1 is not engaged at the product site but is pointing away from the Appr>P phosphate, which is adjacent to C2’ of A + 1 nucleotide. Except A + 1 and A + 2 nucleotides, the remaining nucleotides form non-canonical A:A base pairs between the DNA strands of the two complexes. As indicated by the low RMSD value (0.4 Å), the overall conformations of complex-A and complex-B are similar (Supplementary Fig. 6c); the conformations of Appr>P are virtually identical in the two structures. Superposition with the TkoTpt1/DNA/NAD+ structure showed that Appr>P formation does not cause obvious changes in the overall conformation of TkoTpt1 (Supplementary Fig. 6d), supported by the very low RMSD value (0.2 Å). The conformations of the ADP motifs are conserved, but the ribose undergoes certain conformational changes (Fig. 4d). Compared to that of the intact NAD+, the ribose of Appr>P is rotated and shifted, ensuring the proper interactions between the cyclic PO42- and TkoTpt1. Besides the rotation of the side chain of Arg136, subtle conformational changes can also be observed for the side chain of Lys63 prior to and after the reaction.
5’-p-ADPR-DNA is the key intermediate of TkoTpt1-catalyzed reaction
The terminal 5’-OH groups of the DNAs are not engaged in the product sites of both TkoTpt1/DNA/Appr>P complexes (Fig. 4a and Supplementary Fig. 6a), suggesting that they are off-pathway with respect to the DNA 5’-PO42- removal reaction. However, formation of Appr>P in these structures clearly confirm that TkoTpt1 is capable of transferring PO42− from DNA 5’-end to NAD+. To identify the residues critical for the catalytic activity of TkoTpt1, we designed and purified a series of single point TkoTpt1 mutants, in which the residue responsible for DNA or NAD+ binding is substituted with Ala. Using 5’-p-DNA (5’-p-GGTGGCGTGT-FAM-3’) as substrate, we analyzed the in vitro catalytic activities of these mutants at 80 °C (Fig. 5a). In contrast to the wild-type (WT) TkoTpt1 protein, neither the R17A, H98A, nor R118A mutants produced any product, indicating that Arg17, His98, and Arg118 all play critical roles in the reaction. Compared to WT TkoTpt1, the catalytic activities of the K6A, R7A, and K10A mutants are weakened; in addition to the substrates, significant number of intermediates are remained after 1 h reaction.

a In vitro assays showing DNA 5’-PO42- transfer activities of wild-type and mutant proteins of TkoTpt1. Experiments were repeated independently three times with similar results. Source data are provided in the Source Data file. b Time course analysis of the DNA 5’-PO42- transfer activities of TkoTpt1 K63A mutant. The reaction system contains 0.25 μM K63A mutant, 0.5 μM substrate DNA, and 2 mM NAD+ in reaction buffer. The distributions of the 5′-p-ADPR DNA intermediate and 5′-OH DNA product (expressed as percentage of total DNA) are plotted as a function of reaction time. Each datum in the graphs is the average of three independent experiments ±SEM. c Overall folding of the K63A/5’-p-ADPR-DNA complex. The key 5’-p-ADPR-DNA intermediate is shown as sticks. d 2Fo-Fc electron density maps of the intermediate bound in the K63A/5’-p-ADPR-DNA complex. e The detailed interactions between TkoTpt1 and the phosphate groups of the bound intermediate. f Superposition showing the conformational differences between the intermediate and the product. Protein residues and the intermediate in K63A/5’-p-ADPR-DNA complex are colored as in (e). For TkoTpt1/DNA/Appr>P complex-A, protein residues, DNA, and the Appr>P product are all colored in light blue.
Interestingly, no final product could be generated when the reaction was catalyzed by the H18A, K63A, R65A, Y77A, R136A, or H137A mutants; however, they produced various numbers of intermediates. Compared to other mutants, the K63A mutant of TkoTpt1 is more efficient in intermediate formation (Fig. 5a). To further verify the functional importance of the above residues, we carried out the catalytic assays at 16 °C, which is identical to the crystallization temperature. As depicted in Supplementary Fig. 7, the H18A, K63A, R65A, and Y77A can catalyze the production of the intermediate. Again, the K63A mutant showed the highest potential in catalysis. To further confirm the functional importance of Lys63, we performed a time course analysis for the K63A mutant at 80 °C. As depicted in Fig. 5b, the mutant can catalyze the first step of the rection with a rate constant of 0.83 ± 0.15 min−1. No obvious 5’-OH DNA product could be observed.
To clarify the identity of the intermediate, we performed co-crystallization trials for the K63A mutant, 5’-p-DNA, and NAD+. One complex structure was determined at 2.33-Å resolution (Fig. 5c and Supplementary Fig. 8a, Supplementary Table 2). As supported by the clear density maps (Fig. 5d and Supplementary Fig. 8b), the nicotinamide moiety of NAD+ had been released; and, the 5’-PO42− of the DNA covalently linked with the ribose at the 1” position, representing the key intermediate (5’-p-ADPR-DNA) of the reaction. The structure was termed as K63A/5’-p-ADPR-DNA hereafter. In the structure, the PO42− at the linker region of 5’-p-ADPR-DNA forms two H-bond interactions: one with the side chain of Arg17 and another with the side chain of Arg65 (Fig. 5e). The side chain of Arg136 H-bonds with the β-phosphate of intermediate.
Superposition showed that the overall folding of the K63A/5’-p-ADPR-DNA structure is very similar to that of the TkoTpt1/DNA/NAD+ complex (Supplementary Fig. 8c) and TkoTpt1/DNA/Appr>P complex-A (Supplementary Fig. 8d); the RMSD values between them are only 0.3 Å and 0.2 Å, respectively. However, careful analysis does reveal some conformational differences, especially around the linker region (Fig. 5f). In TkoTpt1/DNA/Appr>P complex-A, the Adenosine in the catalytic site adopts a full cis conformation and its N7 atom forms H-bond interaction with the side chain of Ser9 of TkoTpt1. Compared to the corresponding Adenosine, the sugar of the ADPR-linked Adenosine is tilted and the nucleobase is rotated about 180° in the K63A/5’-p-ADPR-DNA structure; the Adenosine adopts a very different conformation (intermediate between cis and anti) and its N1 atom H-bonds with Ser9 of TkoTpt1.
The orientations of the corresponding PO42− are completely different in K63A/5’-p-ADPR-DNA and TkoTpt1/DNA/Appr>P complex-A, likely due to the release and rebinding of the product DNA in the latter structure. Rather than terminal nucleotide, TkoTpt1 interacted with the internal nucleotides in TkoTpt1/DNA/Appr>P complex-A (Fig. 4a). The conformation of the visualized nucleotide A + 1 might be different from that of the actual product Adenosine residue. For the second step to occur in the DNA 5’-PO42− transfer reaction, the ADPR O2” nucleophile must be properly poised to attack the DNA-5’-ADPR phosphodiester and expel the DNA O5’ via an in-line mechanism. In the K63A/5’-p-ADPR-DNA structure, the O2”-P-O5’ angle is approximately 108°, which is incompatible with the second step of transesterification. Lys63 is highly conserved in TkoTpt1 and the homologous proteins (Supplementary Fig. 9). Although TkoTpt1 K63A mutant could not catalyze the second step of the reaction, the corresponding mutants of ScTpt1, HsTRPt1 and MmTRPT1 are fully functional in previous in vivo assays29,30. Instead of direct role in catalysis, these observations suggested that Lys63 of TkoTpt1 may be involved in structural rearrangement required for the in-line attacking.
Lys63 of TkoTpt1 can be covalently linked with ADPR
The K63A/5’-p-ADPR-DNA structure confirmed that TkoTpt1 is capable of transferring PO42- from DNA 5’-end to NAD+, but no structure has captured 5’-PO42- at the catalytic site prior to the reaction. To close this gap, we performed further crystallization trials. To prevent the reaction, ADP ribose (ADPR) was utilized in the screening process. ADPR is an analog of NAD+; instead of nicotinamide, ADPR has one hydroxyl group attached to the 1” position of the ribose (Fig. 6a). Two structures, TkoTpt1/DNA/ADPR complex-A* (Fig. 6b) and TkoTpt1/DNA/ADPR complex-B*, were determined. The complex-A* was refined at 1.82-Å resolution (Supplementary Fig. 10a and Supplementary Table 3), resulting in clear density maps for 5’-PO42− of the DNA (Fig. 6c). The conformation of 5’-PO42− is stabilized by H-bond interactions with the side chains of Arg17, Arg65, and Tyr77 (Fig. 6d). Superposition with the K63A/5’-p-ADPR-DNA structure confirmed that 5’-PO42− of the DNA is located at the catalytic site in TkoTpt1/DNA/ADPR complex-A* (Fig. 6e).

a The chemical structures of NAD+ and ADPR. b Overall folding of the TkoTpt1/5’-p-DNA/ADPR complex-A*. Lys63 and the bound DNA and ADPR are shown as sticks. c 2Fo-Fc electron density maps of the bound DNA and the covalently linked Lys63 and ADPR. d The detailed interactions between TkoTpt1 and DNA 5’-PO4 and ADPR. e Superposition showing the conformational differences between the intermediate and the covalently linker ADPR. Protein residues, DNA, and the covalently linked ADPR in TkoTpt1/5’-p-DNA/ADPR complex-A* are colored as in panel d. For the K63A/5’-p-ADPR-DNA complex, protein residues and the intermediate are all colored in light blue.
As expected, ADPR did not interact with DNA 5’-PO42- in TkoTpt1/DNA/ADPR complex-A*. However, as supported by the strong density maps, the ribose of ADPR adopts a linear conformation and is covalently linked with the side chain of Lys63 of TkoTpt1 (Fig. 6c and Supplementary Fig. 10b). The OH group at the 4” and 2” positions of the ribose form H-bond interactions with DNA 5’-PO42− and the side chain of Arg136, respectively. Compared to the K63A/5’-p-ADPR-DNA structure, the side chains of Tyr77 and Arg136 are slightly shifted in TkoTpt1/DNA/ADPR complex-A*, facilitating the interactions with DNA 5’-PO42− and ADPR (Fig. 6e).
The side chain of Lys63 of TkoTpt1 is also covalently linked with ADPR ribose in TkoTpt1/DNA/ADPR complex-B* (Supplementary Fig. 10c, d), which was refined at 1.97-Å resolution (Supplementary Table 3). As indicated by the very low RMSD value, the overall folding of TkoTpt1/DNA/ADPR complex-A* and complex-B* are similar (Supplementary Fig. 10e). The conformations of Lys63 and ADPR are identical in the two structures. However, instead of 5’-PO42− of the DNA, one SO42− (present in the crystallization buffer) is captured at the catalytic site in TkoTpt1/DNA/ADPR complex-B*, which further confirming that SO42− can prevent DNA 5’-PO42- from reaching the catalytic site. To further confirm the linkage between ADPR and TkoTpt1, we analyzed the reaction mixture by mass spectra (Supplementary Fig. 11). When incubated at 80 °C for 1 h, low percentage of TkoTpt1-ADPR covalent adduct can be detected. And, majority of TkoTpt1 have been linked by ADPR after heating for 19 h.
RNA 2’-PO4 2- is recognized by the N-lobe of TkoTpt1
The detailed basis for DNA 5’-PO42− transferring by TkoTpt1 have been revealed by the structures presented above, but how TkoTpt1 recognizes RNA 2’-PO42− is unclear. Since no tRNA with 2’-PO42− modification is available, we performed co-crystallization trials for TkoTpt1 and nucleotide with phosphate groups at 2’, 3’, and 5’ positions, mimicking the 2’-phosphorylated nucleotide at the junction site of tRNA. Two complex structures were determined, which were termed as TkoTpt1/2’,3’,5’-p-A (Supplementary Fig. 12a) and TkoTpt1/2’,3’,5’-p-U (Supplementary Fig. 12b). The two structures were refined at 1.95-Å and 1.94-Å (Supplementary Table 4), produced clear density maps for 2’,3’,5’-p-A (Supplementary Fig. 12c) and 2’,3’,5’-p-U (Supplementary Fig. 12d).
In the TkoTpt1/2’,3’,5’-p-A structure (Fig. 7a), the N1 atom of the nucleobase H-bonds with the side chain of Ser9. The conformation of the nucleobase is further stabilized by stacking with the side chains of Lys10 and Arg65. The 5’-PO42− does not form stable interaction with TkoTpt1, but 3’-PO42− H-bonds with the side chains of Arg17 and His18. Like 3’-PO42−, 2’-PO42− also H-bonds with Arg17; in addition, it also forms H-bond interactions with Lys63 and Arg65. In the TkoTpt1/2’,3’,5’-p-U structure (Fig. 7b), the N3 atom of the nucleobase H-bonds with Ser9. Apart from that, the interactions between 2’,3’,5’-p-U and TkoTpt1 are similar to that observed in the TkoTpt1/2’,3’,5’-p-A structure.

a Detailed interactions between TkoTpt1 and 2’,3’,5’-p-A in the TkoTpt1/2’,3’,5’-p-A complex. b Detailed interactions between TkoTpt1 and 2’,3’,5’-p-U in the TkoTpt1/2’,3’,5’-p-U complex. c Detailed interactions between TkoTpt1 K63A mutant and 2’,3’,5’-p-A and ADPR in the K63A/2’,3’,5’-p-A/ADPR complex. d Detailed interactions between TkoTpt1 K63A mutant and the 2’-p-ADPR-RNA intermediate in the K63A/2’-p-ADPR-RNA complex.
As supported by the very low RMSD value (0.3 Å), the overall conformations of TkoTpt1/2’,3’,5’-p-A and TkoTpt1/2’,3’,5’-p-U are very similar (Supplementary Fig. 12e). Due to the compositional difference of the nucleobase, the two nucleotides exhibit slightly different conformations in their sugar, 3’-PO42−, and 5’-PO42−. In contrast, the conformation of 2’-PO42− and its interactions with TkoTpt1 are conserved (Supplementary Fig. 12f). Compared to other complex structures, the relative orientation between the N-lobe and NAD+-lobe in the TkoTpt1/2’,3’,5’-p-A or TkoTpt1/2’,3’,5’-p-U structure is more similar to that of the unliganded TkoTpt1 structure. In such orientation, 2’-PO42− could not form any interaction with the NAD+-lobe. In the reported PhoTpt1 structure (PDB_ID: 8TFX), one 2’,5’-p-A is captured31. Although the relative orientations between the N-lobe and NAD+-lobe are very different, 2’-PO42- of 2’,5’-p-A and 2’,3’,5’-p-A formed similar interactions with the N-lobe in the two structures (Supplementary Fig. 13a, b). Very recently, one structure of PhoTpt1 in complex with an RNA 2’-PO42- splice junction (PDB_ID: 8TG5) has been reported34, which also showed different domain orientation but similar 2’-PO42- recognition with the TkoTpt1/2’,3’,5’-p-A complex (Supplementary Fig. 13c, d). Taken together, these observations confirmed that the N-lobe plays major role in 2’-PO42- recognition.
TkoTpt1 is capable of catalyzing RNA 2’-PO4 2- and NAD+ reaction
TkoTpt1/2’,3’,5’-p-A and TkoTpt1/2’,3’,5’-p-U structures confirm that TkoTpt1 can recognize RNA 2’-PO42-. We then wondered how TkoTpt1 transfers 2’-PO42- from RNA to NAD+. To this end, we solved one K63A/2’,3’,5’-p-A/ADPR complex (Supplementary Fig. 14a). ADPR and TkoTpt1 K63A mutant were utilized to capture the structure prior to reaction. The structure was refined at 1.82-Å resolution (Supplementary Table 4) and produced clear density maps for 2’,3’,5’-p-A and ADPR (Supplementary Fig. 14b). Like the DNA-complexed structure, ADPR and 2’,3’,5’-p-A are next to each other in the K63A/2’,3’,5’-p-A/ADPR complex (Fig. 7c). In addition to N-lobe residues Arg17 and Arg65, 2’-PO42- also H-bonds with Arg136 of the NAD+-lobe; 3’-PO42- forms two H-bond interactions with NAD+-lobe Arg118. As expected, no reaction could occur with ADPR in the K63A/2’,3’,5’-p-A/ADPR complex. The ribose of ADPR forms a closed five-member ring with all OH groups attached to C1”, C2”, and C3” in α configuration. ADPR 1”-OH and 3”-OH form H-bond interactions with 2’-PO42- of 2’,3’,5’-p-A and Arg136 of the protein, respectively.
To better understand how TkoTpt1 transfers RNA 2’-PO42- to NAD+, we solved a structure of TkoTpt1 K63A mutant in the presence of 2’,3’,5’-p-A and NAD+ (Supplementary Fig. 14c) at 1.97-Å resolution (Supplementary Table 4). The structure belongs to P21212 space group; per asymmetric unit contains two TkoTpt1 K63A protomers, protomer A and protomer B. Each protomer binds one 2’,3’,5’-p-A and one NAD+. As indicated by the clear density maps, nicotinamide group of NAD+ has been substituted by 2’-PO42- of 2’,3’,5’-p-A, forming 2’-p-ADPR-RNA intermediate (Supplementary Fig. 14d). The structure was termed as K63A/2’-p-ADPR-RNA complex hereafter. Superposition showed that the Adenine nucleobase could adopt either cis or anti conformation, leading to slight shifting of the sugar; whereas, the conformations of the 2’-PO42- and 3’-PO42- are conserved within the two intermediates (Supplementary Fig. 14e).
The overall folding of the K63A/2’-p-ADPR-RNA complex is very similar to that of the K63A/2’,3’,5’-p-A/ADPR complex (Supplementary Fig. 15a); the RMSD value between them is only 0.5 Å. Like the K63A/2’,3’,5’-p-A/ADPR complex, 2’-PO42- of the intermediate H-bonds with Arg65 in the K63A/2’-p-ADPR-RNA complex (Fig. 7d); 3’-PO42- forms H-bond interactions with Arg17 and Arg118. However, instead of 2’-PO42-, Arg136 H-bonds with the β-phosphate group of the intermediate. The relative orientation between the N-lobe and NAD+-lobe is very different in the K63A/2’-p-ADPR-RNA complex and the TkoTpt1/2’,3’,5’-p-A complex (Supplementary Fig. 15b), but the conformation of 2’,3’,5’-p-A in the latter structure is very similar to that of the intermediate associated with TkoTpt1 K63A protomer A (Supplementary Fig. 15c). The conformation of 2’,3’,5’-p-A of the intermediate associated with TkoTpt1 K63A protomer B is similar to 2’,3’,5’-p-U observed in the TkoTpt1/2’,3’,5’-p-U complex (Supplementary Fig. 15d). Taken together, these observations suggest that TkoTpt1 N-lobe can recognize 2’-PO42- attached to different nucleotides; upon formation of catalytic conformation, TkoTpt1 could transfer 2’-PO42- from all these nucleotides to NAD+.
TkoTpt1 follows similar mechanism in 5’-PO4 2- and 2’-PO4 2- transfer
Both K63A/5’-p-ADPR-DNA and K63A/2’-p-ADPR-RNA complexes represent the intermediate state of the TkoTpt1-catalyzed nucleic acid PO42- transfer reaction. The folding of the N-lobe and NAD+-lobe are conserved in these structures, but the detailed orientations are not identical; when superimposed on the NAD+-lobe, the N-lobe undergoes a significant rotation (Fig. 8a). Structural comparison revealed that the interactions between the intermediate 3’-PO42- and NAD+-lobe Arg118 are conserved (Fig. 8b). To generate the 5’-p-ADPR-DNA intermediate captured in the K63A/5’-p-ADPR-DNA structure, the target nucleoside must undergo large rotation around its linkage to the flanking 3’ phosphodiester to deliver the 5’-PO42- to the catalytic site. In contrast, such rotation is not required for the formation of the 2’-p-ADPR-RNA intermediate, which may lead to the larger ADPR-O2”–P–O2’-RNA’ angles (125˚ and 137˚) for the two intermediates captured in the K63A/2’-p-ADPR-RNA structure. However, further conformational changes are still required for the two 2’-p-ADPR-RNA intermediates to carry out the second step in-line attacking reaction. Again, Lys63 of TkoTpt1 may help in this process.

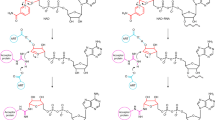
a Superposition of the K63A/5’-p-ADPR-DNA complex and the K63A/2’-p-ADPR-RNA complex. b Conformational comparison of the two intermediates and their interacting residues. c Proposed mechanisms for TkoTpt1-catalyzed nucleic acid 5’-PO42- transfer.
Based on our structural observations, we concluded that TkoTpt1 utilizes a similar mechanism in transferring 5’-PO42- or 2’-PO42- from nucleic acid to NAD+. The mechanism proposed for nucleic acid 5’-PO42- transfer by TkoTpt1 is summarized in Fig. 8c. First, the target 5’-PO42- is recognized and orientated for in-line attacking of NAD+, expelling nicotinamide and generating the 5’-p-ADPR-DNA intermediate. Second, the 2”-OH group of the intermediate attacks the target PO42-, displacing nucleic acid and generating the Appr>P product.
Discussion
In conclusion, we determined a series of TkoTpt1 structures in this work. Besides the unliganded structure, we also determined several structures of TkoTpt1 in complex with NAD+ and 5’-p-DNA either prior to reaction, at the intermediate state, or after the completion of the reaction, which captured the key intermediate for 5’-PO42- transfer (Fig. 5c) and the Appr>P product (Fig. 4a and Supplementary Fig. 6a). The intermediate for RNA 2-PO42- transfer was captured by the K63A/2’-p-ADPR-RNA complex (Fig. 7d). Structural analysis revealed that the orientation between the N-lobe and NAD+-lobe of TkoTpt1 is very flexible (Fig. 3d). The N-lobe is responsible for target PO42- recognition, but transfer of PO42- requires the binding of both target nucleic acid and NAD+ in proper conformation (Supplementary Fig. 15b).
TkoTpt1 utilizes similar mechanism in 5’-PO42- and 2’-PO42- transfer. Previous studies have defined a conserved Arg-His-Arg-Arg tetrad essential for Tpt1 activity in vivo and in vitro23,29. These tetrad residues are conserved in TkoTpt1, including Arg17, His18, Arg65, and Arg118 (Supplementary Fig. 9). Our in vitro assays (Fig. 5a) and structural observations (Figs. 3c, 4c, 5e and 7c, d) suggested that these tetrad residues play similar role in the TkoTpt1-catalyzed PO42- transfer reaction. Our study also revealed the functional importance for some other conserved residues, including Lys63 and His98 (Fig. 5a and Supplementary Fig. 9). Ala substitution of Lys63 prevented TkoTpt1 from catalyzing the second step of the reaction (Figs. 5b, c and 7d), whereas the corresponding mutants of ScTpt1, HsTRPt1 and MmTRPT1 are fully functional29,30. Instead of direct role in catalysis, Lys63 of TkoTpt1 may be involved in structural rearrangement essential for the second step of the reaction. The functional role of Lys63 could be compensated by other residues in the homologous proteins. For the second step of the reaction to occur, the 2”-OH of the intermediate needs to be deprotonated (Figs. 1a, 8c). However, what is responsible for the deprotonation of the intermediate is unclear at present.
KptA, TRPT1, and Tpt1 are homologous to each other. They are evolutionarily conserved in fungi, bacterial, archaeal, and eukaryotic species22. Although it is not required for tRNA intron splicing in all species, Tpt1 catalyzes 2’-PO42- removal from tRNA splice junction, which is the last step of tRNA maturation in fungi and plants19,24. Since the splice junction 2’-PO42- is located one base 3’ of the anticodon, its presence interferes with anticodon recognition and the function of tRNA in protein translation, which might be the main reason for the death of yeast with Tpt1 gene disruption19. Functional substitution of Tpt1 in yeast indicated that bacterial KptA and eukaryotic TRPT1 can all catalyze tRNA 2’-PO42- removal.
The Appr>P product has been identified for over 30 years17,35 and formation of the intermediate has also been observed in many in vitro assays catalyzed by various Tpt1/TRPT1/KptA family proteins16, indicating that they use a similar mechanism in removing PO42- from nucleic acids. In addition to biochemical studies, structural studies have been reported for TRPT1 from mouse and human30. Although NAD+ was included in the crystallization sample, the determined MmTRPT1 structure (PDB_ID: 7YW2) and HsTRPT1 structure (PDB_ID: 7YW3) captured one ADP-ribose-1”-phosphate in their NAD+-lobes. The overall folding of the MmTRPT1 and HsTRPT1 structures is very similar (Supplementary Fig. 16a). Interestingly, although no nucleic acid substrate is present in the MmTRPT1 and HsTRPT1 structures, their folding is very similar to that of the TkoTpt1/DNA/Appr>P complex; the RMSD value between them is only 0.7 Å (Supplementary Fig. 16b).
Compared to the TRPT1 family protein, more structural studies have been performed for the Tpt1 family protein. Besides unliganded structures and complexes with either substrate-mimicking RNA or NAD+, ternary complexes have also been reported for ApeTpt1 and AthTpt131,32. In the ApeTpt1 structure (PDB_ID: 8TG4), one 2’-OH RNA molecule and one ADP-ribose-2”-phosphate molecule were captured (Supplementary Fig. 16c). Like ADP-ribose-1”-phosphate captured by the AthTpt1 structure (Supplementary Fig. 5a), ADP-ribose-2”-phosphate in the ApeTpt1 structure was also produced by Tpt1 proteins during in vivo expression and crystallization process. Structural superposition showed that the overall folding of the ApeTpt1 and AthTpt1 structures is very similar to that of the TkoTpt1/DNA/Appr>P complex (Supplementary Figs. 5c, d and 16c). Sequence alignment revealed that the residues critical for the function of TkoTpt1 are highly conserved in Tpt1/TRPT1/KptA family proteins (Supplementary Fig. 9). Although no intermediate or Appr>P product has been captured by any other Tpt1/TRPT1/KptA family proteins, the sequence and structural similarity indicate that they use the conserved mechanism (Fig. 1a) in transferring 2’-PO42- from their substrates to NAD+. Besides fungal tRNAs, plant tRNAs, and thermophilic bacterial tRNAs, Tpt1/TRPT1/KptA family proteins also participate in 2’-PO42- removal of certain mRNAs, such as HAC1 mRNA during the unfolded protein response in yeast36,37. Based on the structural observations, it has been believed that Tpt1/TRPT1/KptA family proteins can remove 2’-PO42- from NADP(H)31. In the future, it is worth investigating whether Tpt1/TRPT1/KptA family proteins can function on other substrate carrying 2’-PO42-.
Our in vitro assays (Figs. 1c, d and 2) and crystal structures (Fig. 4 and Supplementary Fig. 6a) clearly confirmed that TkoTpt1 is capable of transferring 5’-PO42- from DNA to NAD+, generating 5’-OH DNA and the Appr>P product. Although it is not as efficient as DNA, TkoTpt1 can also transfer 5’-PO42- from RNA to NAD+ (Supplementary Fig. 1). In contrast to TkoTpt1, all the reported Tpt1/TRPT1/KptA family proteins can only catalyze the first step of the reaction in vitro, leading to ADP-ribosylation at the 5’-end of the nucleic acids. Weixler and co-workers found that RNAs in human cells can be ADP-ribosylated, protecting against the cleavage by XRN1 nuclease38. ADP-ribosylation on human genomic DNAs were demonstrated by Tromans-Coia and co-workers39. Based on structural analysis and in vitro assay results, Yang and co-workers suggested that mammalian TRPT1s might employ different residues for nucleic acid ADP-ribosylation and RNA 2’-PO42- transfer30, but no reported structure has ever captured nucleic acids with 5’-PO42- at any reaction state. Our structures have captured both reactions at various states, clearly indicating that Tpt1/TRPT1/KptA family proteins use the same residues for ADP-ribosylation and 2’-PO42- transfer. Unlike TkoTpt1, the structures of many Tpt1/TRPT1/KptA family proteins are relatively rigid; they adopt similar conformations in the presence and absence of substrate and NAD+. The structural rigidity may prevent these proteins from conformational changes required for the complete transfer of the 5’-PO42-. Although TkoTpt1 can catalyze the removal of DNA 5’-PO42- in vivo, it requires high temperature and the reaction rate is very low (Fig. 2). Since DNA 5’-phosphate can be removed by many phosphatases in vivo, the biological function for the TkoTpt1-catalyzed 5’-phosphate removal needs to be further investigated.
In addition to S. cerevisiae, Tpt1 is also conserved in many human fungal pathogens, such as C. auris, C. albicans, A. fumigatus, and C. immitis. Due to their critical roles in tRNA splicing in these species, Tpt1 has been viewed as a potential drug target21. The structures of HsTRPT1/ADP-ribose-1”-phosphate complex and ApeTpt1/ADP-ribose-2”-phosphate complex (Supplementary Figs. 5 and 16) have provided valuable information for Tpt1-specific antifungal drug development. Both ADP-ribose-1”-phosphate and ADP-ribose-2”-phosphate in the reported structures were co-purified with the target protein. In contrast, no such molecule was co-purified with TkoTpt1, allowing the co-crystallization of TkoTpt1 with different combinations of substrate and ligand. The resulting TkoTpt1/DNA/ADPR complexes are interesting, in which the side chain of Lys63 is covalently linked with the ribose of ADPR (Fig. 6b and Supplementary Fig. 10a). Lys63 is highly conserved in Tpt1/TRPT1/KptA family protein (Supplementary Fig. 9). Covalent linking between Lys residue and ADPR has also been observed in the structures of some other proteins, such as the C6orf130 protein associated with the neurodegenerative disease in human40. One plausible mechanism for the formation of the Lys-ADPR adduct is depicted in Supplementary Fig. 17. In brief, the ribose of ADPR is opened, forming an aldehyde form of ADPR ribose sugar. Then, the resulting ADPR sugar interacts with the side chain of Lys63, forming a Schiff base. However, as revealed by mass analysis, the reaction between ADPR and TkoTpt1 is very slow (Supplementary Fig. 11), suggesting that ADPR is not an ideal lead compound for the development of small molecule inhibitors.
Our structures represent the only 5’-p-nucleic acid complexed Tpt1/TRPT1/KptA family protein structures available to date. In addition to the Appr>P product, our structures also captured the key intermediate for nucleic acid ADP-ribosylation and 2’-PO42- transfer. Our studies reaffirm the catalytic mechanism of Tpt1-catalyzed phosphate transfer and advance our understanding on intron splicing of tRNA and ADP-ribosylation of nucleic acid.
Methods
Plasmid construction
Gene containing the codon-optimized cDNA sequence of TkoTpt1 (Supplementary Table 6) was synthesized by Tsingke Biotech company. The target fragments were amplified by polymerase chain reaction (PCR) and recombined into pET28a-His-Sumo vector using ClonExpress II One Step Cloning Kit (Vazyme). Two primers (Primer_F: 5′-AGAGAACAGATTGGTGGATCCATGAAACCGGAACGTAAACGTG-3′ and Primer_R: 5′-GTGGTGGTGGTGGTGCTCGAGCTAAACAGC CAGAGTGATGCAGT-3′) were used in the PCR reaction. The engineered pET28a-His-Sumo- TkoTpt1 vector was transformed into E. coli BL21 (DE3) competent cells and cultured at 37 °C overnight. All TkoTpt1 mutants were constructed by site-directed mutagenesis using the primers listed in Supplementary Table 7. The gene of the wild-type TkoTpt1 was used as a template during the construction. Sequences of all constructs were confirmed by DNA sequencing.
Protein expression and purification
All wild-type TkoTpt1 and the mutant proteins were expressed and purified using the same procedures. Briefly, every 10 mL of E. coli BL21 (DE3) cells transformed with recombined vector was inoculated into 1 L LB medium supplemented with 50 μg/mL kanamycin and cultured at 37 °C. When the OD600 reached 0.6, isopropyl β-D-1-thiogalactopyranoside (IPTG, final concentration 0.2 mM) was added to induce the expression of protein. After culturing at 18 °C for additional 16 h, the cells were harvested by centrifugation at 4 °C, 5400 × g for 15 min.
The cell pellets were resuspended in lysis buffer (20 mM Tris pH 8.0, 500 mM NaCl, 25 mM Imidazole), lysed by ultrahigh-pressure homogenizer, and clarified by centrifugation. The supernatant was loaded onto a HisTrap HP column (5 mL, Cytiva). The protein was eluted by buffer composed of 20 mM Tris pH 8.0, 500 mM NaCl, and 500 mM Imidazole with a linear gradient. The target protein was treated with Ulp1 protease and dialyzed against buffer composed of 20 mM Tris pH 8.0 and 100 mM NaCl. The sample was loaded onto HisTrap HP column again, and the flow-through containing the target protein was collected and loaded onto a HiTrap Heparin column (Cytiva) equilibrated with buffer composed of 20 mM Tris pH 8.0, 100 mM NaCl. The target protein was eluted by buffer composed of 20 mM Tris pH 8.0, 1 M NaCl with a linear gradient. The fractions containing the target protein were collected and then loaded onto a Superdex™ 75 increase10/300 GL column (Cytiva) equilibrated with gel filtration buffer (20 mM Tris pH 8.0, 150 mM NaCl, 1 mM DTT). Peak fractions were collected and analyzed using 4-20% SDS-PAGE gradient gel. The purified protein was concentrated to 10 mg/mL and stored at −80 °C.
Crystallization and X-ray diffraction data collection
For crystallization of the unliganded TkoTpt1 structure, the protein was diluted to 5 mg/mL with gel filtration buffer. To prepare the sample for the TkoTpt1/2’,3’,5’-p-A or the TkoTpt1/2’,3’,5’-p-U complexes, the protein and the nucleotide were mixed to final concentrations of 5 mg/mL and 5 mM, respectively. The samples for the K63A/2’,3’,5’-p-A/ADPR and K63A/2’-p-ADPR-RNA complexes were prepared by mixing TkoTpt1 K63A mutant protein, 2’,3’,5’-p-A nucleotide, and ligand (ADPR or NAD+) with final concentrations of 5 mg/mL, 5 mM, and 5 mM, respectively. To prepare the sample for the 5’-p-DNA complexed structures, TkoTpt1 protein (either wild-type or K63A mutant), DNA (5’-p-AAAAAAAAAA-3’), and ligand (NAD+ or ADPR) were mixed with final concentrations of 5 mg/mL, 0.5 mM, and 5 mM, respectively. All samples were incubated at room temperature for 30 min. The initial crystallization conditions were identified by a robot system at 16 °C using the sitting-drop vapor diffusion method, whereas the hanging-drop vapor diffusion method was utilized for the optimization of all crystals. The detailed crystallization conditions for each individual structure are summarized in Supplementary Table 5.
All crystals were cryoprotected using their mother liquor supplemented with 25% glycerol and snap-frozen in liquid nitrogen. The diffraction data were collected at beamlines BL10U2 at the Shanghai Synchrotron Radiation Facility (SSRF) and beamlines BL18U1 and BL19U1 of National Facility for Protein Science Shanghai (NFPS). Data processing was carried out using autoPROC or xia2_3dii programs developed by the staff of the beamline. The data collection and processing statistics were summarized in Supplementary Tables 1–4.
Structure determination and refinement
The unliganded TkoTpt1 structure was determined by molecular replacement (MR) method using the Phaser program41 from the CCP4 suite42. The isolated N-lobe and NAD+-lobe predicted by AlphaFold2 were used as the search model. The resulting models were refined against the diffraction data using the Refmac5 program of the CCP4 suite. The 2Fo –Fc and Fo –Fc electron density maps were regularly calculated and used as guide for the model adjustment in COOT43. The refined N-lobe and NAD+-lobe of TkoTpt1 were used as the search model for the determination of the TkoTpt1/DNA/NAD+ complex by the MR method. The missing DNA and NAD+ molecules were manually built in COOT. The refined TkoTpt1/DNA/NAD+ complex was used as search model for the determination of all other structures. Final refinement of all structures was performed using the phenix.refine program44. The structural refinement statistics were summarized in Supplementary Tables 1–4.
In vitro phosphate transfer assays
TkoTpt1 (either wild-type or mutant), NAD+, and DNA or RNA substrate (which are all composed of 10 nucleotides and contain a PO4 at the 5’ end and a FAM label at the 3’ end) were mixed in reaction buffer (20 mM Tris pH 8.0, 150 mM NaCl, 1 mM DTT). The final concentrations of the protein, DNA substrate, and NAD+ were 0.25 or 0.5 μM, 0.5 μM, and 2 mM, respectively. The reaction mixtures were incubated at the specified temperature and quenched by the addition of equal volume Formamide loading buffer (90% formamide, 50 mM EDTA). The products were analyzed by electrophoresis through a 15% polyacrylamide gel containing 7 M urea in 90 mM Tris-borate and 2 mM EDTA. Due to the lack of commercially available intermediate standard, no size marker was included in the gel analysis. The gel was visualized using Typhoon FLA-9000 imaging device. The reaction products were quantified by scanning the gels with Image J. The data were displayed by Excel and GraphPad Prism (version 9.5). All experiments were carried out at least three independent experiments.
MALDI-TOF mass analysis
The reaction mixtures containing TkoTpt1 (0.25 mM), ssDNA (5’-AAAAAAAAAA-3’, 0.5 mM) and 2 mM ADPR in reaction buffer (20 mM Tris pH 8.0, 150 mM NaCl) were incubated at 80 °C for 1 or 19 h. These samples and the wild-type TkoTpt1 sample were then used for analysis. MALDI MS analysis was performed on Bruker Ultraflex TOF/TOF MS (Bruker, Bremen, Germany). The mixture of sample and matrix (20 mg/mL Sinapinic acid in 70% ACN and 1% TFA) was dropped onto the sample supports. On-target desalting was performed with 1% TFA. All spectra were obtained in positive reflector mode, and mass spectrometric data analysis was performed using the Bruker FlexAnalysis Software (version 3.3). All experiments were carried out in triplicate.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Structural factors and coordinates have been deposited in the Protein Data Bank under accession codes 9LD4, 9LD6, 9LDF, 9LDG, 9LDA, 9LDD, 9LDC, 9LDH, 9LDI, 9LD3, and 9LDE for the unliganded structure, the TkoTpt1/DNA/NAD+ complex, the TkoTpt1/DNA/Appr>P complex-A, the TkoTpt1/DNA/Appr>P complex-B, the K63A/5’-p-ADPR-DNA complex, the TkoTpt1/DNA/ADPR complex-A* complex, the TkoTpt1/DNA/ADPR complex-B* complex, the TkoTpt1/2’,3’,5’-p-U complex, the TkoTpt1/2’,3’,5’-p-A complex, the K63A/2’,3’,5’-p-A/ADPR complex, and the K63A/2’-p-ADPR-RNA complex, respectively. Accession codes are 6E3A, 7YW2, 7YW3, 8TFZ, 8TFX, 8TG3, 8TG4, and 8TG5 for the previously reported AthTpt1, MmTRPT1, HsTRPT1, PhoTpt1/NAD+, PhoTpt1/2’,5’-p-A, ApeTpt1/ADPR-1”-PO4, ApeTpt1/ADPR-2”-PO4, and PhoTpt1/2’-p-RNA structures, respectively. Source data are provided as a Source Data file. Source data are provided with this paper.
References
Kirchner, S. & Ignatova, Z. Emerging roles of tRNA in adaptive translation, signalling dynamics and disease. Nat. Rev. Genet 16, 98–112 (2015).
Hoagland, M. B., Stephenson, M. L., Scott, J. F., Hecht, L. I. & Zamecnik, P. C. Soluble ribonucleic acid intermediate in protein synthesis. J. Biol. Chem. 231, 241–257 (1958).
Phizicky, E. M. & Hopper, A. K. The life and times of a tRNA. Rna 29, 898–957 (2023).
Chan, P. P. & Lowe, T. M. GtRNAdb 2.0: an expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res. 44, D184–D189 (2016).
Belfort, M. & Weiner, A. Another bridge between kingdoms: tRNA splicing in archaea and eukaryotes. Cell 89, 1003–1006 (1997).
Baldi, M. I., Mattoccia, E., Ciafre, S., Attardi, D. G. & Tocchinivalentini, G. P. Binding and cleavage of pre-transfer RNA by the Xenopus Splicing Endonuclease − 2 separable steps of the intron excision reaction. Cell 47, 965–971 (1986).
Filipowicz, W. & Shatkin, A. J. Origin of splice junction phosphate in transfer-Rnas processed by Hela-Cell extract. Cell 32, 547–557 (1983).
Ogden, R. C. et al. Invitro transcription and processing of a yeast transfer-rna gene containing an intervening sequence. Cell 17, 399–406 (1979).
Knapp, G., Beckmann, J. S., Johnson, P. F., Fuhrman, S. A. & Abelson, J. Transcription and processing of intervening sequences in yeast transfer-RNA genes. Cell 14, 221–236 (1978).
Knapp, G., Ogden, R. C., Peebles, C. L. & Abelson, J. Splicing of yeast transfer-RNA precursors - structure of the reaction intermediates. Cell 18, 37–45 (1979).
Peebles, C. L., Ogden, R. C., Knapp, G. & Abelson, J. Splicing of yeast transfer-RNA Precursors − 2-stage reaction. Cell 18, 27–35 (1979).
Peebles, C. L., Gegenheimer, P. & Abelson, J. Precise excision of intervening sequences from precursor transfer-RNAs by a membrane-associated yeast endonuclease. Cell 32, 525–536 (1983).
Trotta, C. R. et al. The yeast tRNA splicing endonuclease: a tetrameric enzyme with two active site subunits homologous to the archaeal tRNA endonucleases. Cell 89, 849–858 (1997).
Greer, C. L., Peebles, C. L., Gegenheimer, P. & Abelson, J. Mechanism of action of a yeast rna ligase in transfer-RNA splicing. Cell 32, 537–546 (1983).
Phizicky, E. M., Schwartz, R. C. & Abelson, J. Saccharomyces-cerevisiae transfer-RNA ligase - purification of the protein and isolation of the structural gene. J. Biol. Chem. 261, 2978–2986 (1986).
Spinelli, S. L., Kierzek, R., Turner, D. H. & Phizicky, E. M. Transient ADP-ribosylation of a 2′-phosphate implicated in its removal from ligated tRNA during splicing in yeast. J. Biol. Chem. 274, 2637–2644 (1999).
Culver, G. M. et al. An nad derivative produced during transfer-RNA splicing - Adp-ribose 1”-2” cyclic phosphate. Science 261, 206–208 (1993).
Mccraith, S. M. & Phizicky, E. M. An enzyme from Saccharomyces-Cerevisiae uses Nad+ to transfer the splice junction 2’-phosphate from ligated transfer-rna to an acceptor molecule. J. Biol. Chem. 266, 11986–11992 (1991).
Spinelli, S. L., Consaul, S. A. & Phizicky, E. M. A conditional lethal yeast phosphotransferase (tpt1) mutant accumulates tRNAs with a 2′-phosphate and an undermodified base at the splice junction. Rna 3, 1388–1400 (1997).
Organization, W. H. WHO Fungal Priority Pathogens List to Guide Research, Development, and Public Health Action (WHO, 2022).
Dantuluri, S., Schwer, B., Abdullahu, L., Damha, M. J. & Shuman, S. Activity and substrate specificity of Candida, Aspergillus, and Coccidioides Tpt1: essential tRNA splicing enzymes and potential antifungal targets. Rna 27, 616–627 (2021).
Spinelli, S. L., Malik, H. S., Consaul, S. A. & Phizicky, E. M. A functional homolog of a yeast tRNA splicing enzyme is conserved in higher eukaryotes and in Escherichia coli. Proc. Natl. Acad. Sci. USA 95, 14136–14141 (1998).
Munir, A., Abdullahu, L., Damha, M. J. & Shuman, S. Two-step mechanism and step-arrest mutants of NAD+-dependent tRNA 2’-phosphotransferase Tpt1. Rna 24, 1144–1157 (2018).
Gerber, J. L., Köhler, S. & Peschek, J. Eukaryotic tRNA splicing - one goal, two strategies, many players. Biol. Chem. 403, 765–778 (2022).
Ohira, T. et al. Reversible RNA phosphorylation stabilizes tRNA for cellular thermotolerance. Nature 605, 372–379 (2022).
Munir, A., Banerjee, A. & Shuman, S. NAD+-dependent synthesis of a 5′-phospho-ADP-ribosylated RNA/DNA cap by RNA 2′-phosphotransferase Tpt1. Nucleic Acids Res. 46, 9617–9624 (2018).
Munnur, D. et al. Reversible ADP-ribosylation of RNA. Nucleic Acids Res. 47, 5658–5669 (2019).
Mccraith, S. M. & Phizicky, E. M. A highly specific phosphatase from saccharomyces-cerevisiae implicated in transfer-RNA sSplicing. Mol. Cell Biol. 10, 1049–1055 (1990).
Sawaya, R., Schwer, B. & Shuman, S. Structure-function analysis of the yeast NAD+-dependent tRNA 2’-phosphotransferase Tpt1. Rna 11, 107–113 (2005).
Yang, X. Y. et al. Structural and biochemical insights into the molecular mechanism of TRPT1 for nucleic acid ADP-ribosylation. Nucleic Acids Res. 51, 7649–7665 (2023).
Jacewicz, A., Dantuluri, S. & Shuman, S. Structural basis for Tpt1-catalyzed 2′-PO4 transfer from RNA and NADP(H) to NAD. Proc. Natl. Acad. Sci. USA 120, e2312999120 (2023).
Banerjee, A. et al. Structure of tRNA splicing enzyme Tpt1 illuminates the mechanism of RNA 2′-PO4 recognition and ADP-ribosylation. Nat. Commun. 10, 218 (2019).
Alphonse, S., Banerjee, A., Dantuluri, S., Shuman, S. & Ghose, R. NMR solution structures of RNA 2′-phosphotransferase Tpt1 provide insights into NAD+ binding and specificity. Nucleic Acids Res. 49, 9607–9624 (2021).
Jacewicz, A., Damha, M. J. & Shuman, S. Structures of RNA phosphotransferase Tpt1 reveal distinct binding modes for an RNA 2′-PO4 splice junction versus a 5′-PO4 mononucleotide. Rna 31, 916–922 (2025).
Culver, G. M., Consaul, S. A., Tycowski, K. T., Filipowicz, W. & Phizicky, E. M. Transfer-RNA splicing in yeast and Wheat-Germ - a Cyclic Phosphodiesterase implicated in the metabolism of Adp-Ribose 1”,2”-cyclic phosphate. J. Biol. Chem. 269, 24928–24934 (1994).
Cherry, P. D., White, L. K., York, K. & Hesselberth, J. R. Genetic bypass of essential RNA repair enzymes in budding yeast. Rna 24, 313–323 (2018).
White, L. K., Strugar, S. M., MacFadden, A. & Hesselberth, J. R. Nanopore sequencing of internal 2′-PO4 modifications installed by RNA repair. Rna 29, 847–861 (2023).
Weixler, L., Feijs, K. L. H. & Zaja, R. ADP-ribosylation of RNA in mammalian cells is mediated by TRPT1 and multiple PARPs. Nucleic Acids Res. 50, 9426–9441 (2022).
Tromans-Coia, C. et al. TARG1 protects against toxic DNA ADP-ribosylation. Nucleic Acids Res. 49, 10477–10492 (2021).
Sharifi, R. et al. Deficiency of terminal ADP-ribose protein glycohydrolase TARG1/C6orf130 in neurodegenerative disease. Embo J. 32, 1225–1237 (2013).
Bunkoczi, G. et al. Phaser.MRage: automated molecular replacement. Acta Crystallogr D. Biol. Crystallogr. 69, 2276–2286 (2013).
Potterton, E., Briggs, P., Turkenburg, M. & Dodson, E. A graphical user interface to the CCP4 program suite. Acta Crystallogr. D. Biol. Crystallogr. 59, 1131–1137 (2003).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D. 60, 2126–2132 (2004).
Afonine, P. V. et al. Towards automated crystallographic structure refinement with pehnix.refine. Acta Crystallogr. D. 68, 352–367 (2012).
Acknowledgements
We thank the staff members of BL10U2 beamline at Shanghai Synchrotron Radiation Facility (SSRF), the staff members of BL18U1 and BL19U1 beamlines of National Facility for Protein Science Shanghai (NFPS), and the Large-scale Protein Preparation System at the National Facility for Protein Science (NFPS) in Shanghai for providing technical support and assistance in data collection and analysis. We thank Drs. Baixing Wu and Xing Jian for helpful discussion. This study was supported by the National Natural Science Foundation of China (32371252, 32171197, and 32571387, J.H.G.), the Key Research and Development Project of China (2023YFC2307104 and 2024YFE0105100, J.H.G), Natural Science Foundation of Shanghai (25ZR1402332, R.X.C.), and Open Funding Project of the State Key Laboratory of Bioreactor Engineering.
Author information
Authors and Affiliations
Contributions
C.L.C., J.Y., and W.Z.Z. performed crystal screening, diffraction data collection, and structure determination. C.L.C., J.Y., and W.Z.Z. constructed and purified the proteins with the help from, Y.Q.G., Y.X.Z., H.L.L., L.X.L., Z.R.L., C.X.W., G.L.T. and H.H.L. In vitro catalytic assays were performed by C.L.C. and J.Y. with help from Y.Q.Y. and R.X.C. Mass spectrum analysis were performed by J.Y. and J.Q.C. The structures were analyzed by C.L.C., J.Y., W.Z.Z., and J.H.G. The paper was written by C.L.C., J.Y., W.Z.Z., H.H.L., and J.H.G. with input from Q.Y.W., Z.H., and J.B.M. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cao, C., Yang, J., Zhang, W. et al. Crystal structures and snapshots along Tpt1-catalyzed phosphate transfer from nucleic acid to NAD+. Nat Commun 16, 10888 (2025). https://doi.org/10.1038/s41467-025-65881-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-65881-y
