
Abstract
The crosstalk between translation and metabolism is fundamental for cellular plasticity. While most studies focus on translation within canonical coding regions, the roles of non-canonical open reading frames (ORFs) in metabolic regulation and early development remain unclear. Here, we show that selective translation of an upstream ORF in the 5′ untranslated region (UTR) of Lin28b produces an 85-amino acid microprotein, PLUM (pluripotency-associated Lin28b uORF-encoded microprotein). Depletion of PLUM leads to deterministic and synchronized (near 100%) induction of naïve pluripotency and causes embryo implantation defects in vivo. Mechanistically, PLUM depletion dissolves L1td1 condensates and enhances L1td1 binding to pluripotency mRNAs such as Tfcp2l1 and Zfp42, stabilizing them and promoting coordinated gene activation. Concurrently, PLUM loss disrupts P-bodies enriched with a subset of nuclear-encoded mitochondrial mRNA, potentially preventing their degradation. Together, these alterations trigger an early burst of mitochondrial oxidative phosphorylation and synchronized naïve gene expression, accelerating acquisition of the naïve state. Our study identifies the novel uORF-encoded microprotein PLUM as a pluripotency determinant integrating RNA regulation and metabolic remodeling.
Similar content being viewed by others


Introduction
Embryonic development is an intricate and precisely regulated process that can be broadly divided into two stages: the pre-implantation and post-implantation periods. Cells originating from the inner cell mass of the pre-implantation blastocysts exhibit the ability to generate all somatic lineages of the adult organism, maintaining full developmental potential, which is defined as “naïve” pluripotency1,2. Following implantation, these naïve cells undergo transcriptional, epigenetic and metabolic changes to progress to a primed pluripotent state1,3,4. Although naïve and primed pluripotent states are functionally similar, they differ in various aspects, exhibiting distinct developmental potential, with only naïve cells are able to contribute efficiently to chimeras, making them a promising source of generating chimeric organs for heterologous transplantation1,5,6,7. Elucidating the regulatory mechanisms of different pluripotent states should not only lead to a better understanding of cell fate decisions and embryonic development, but also provide important clues for regenerative medicine.
Translational control plays a key role in cell fate decisions and early embryonic development. During oogenesis and the post-fertilization period, when transcription is not activated, translational regulation of maternal mRNA serves as the primary regulatory mechanism of gene expression. This process is essential for early embryonic development8,9,10. Importantly, it has been demonstrated that mRNA and protein levels are poorly correlated in mouse embryonic fibroblasts (MEFs) and pluripotent stem cells (PSCs) derived from the early mammalian embryo, with only 40–70% of variability in protein levels is attributable to mRNA levels11,12, highlighting the importance of translational control in shaping the cellular proteome required for cell fate decisions and early development. Indeed, PSCs have been characterized by low overall protein synthesis rates but high ribosome biogenesis rates, consistent with their prominent nucleoli and high nucleus-to-cytoplasmic ratio13.
The crosstalk between translation and metabolism is fundamental for cellular plasticity14. During PSC differentiation, metabolic remodeling accompanies dynamic changes in ribosome biogenesis and protein synthesis15,16. While translation is a highly energy-consuming process dependent on metabolic pathways, it also directly controls the expression of metabolic genes14. However, previous studies predominantly focused on the translation of known proteins or at developmental periods during which transcriptional input is minimal, with a notable gap in research regarding the translation of non-canonical open reading frames (ORFs), i.e., regions outside of annotated protein coding sequences17,18. The functional significance of selective translation of non-canonical ORFs and their dynamic changes in pluripotency regulation and embryonic development remains poorly understood.
Lin28 is an evolutionarily conserved RNA-binding protein that was first discovered in Caenorhabditis elegans as a critical regulator of developmental timing19. There are two Lin28 paralogs in most vertebrates: Lin28a and Lin28b, which are highly expressed in early development and undifferentiated cells and are involved in translational regulation by binding mRNA and governing microRNA biogenesis20. Lin28 proteins are localized in organelles associated with translational control, such as cellular processing bodies (P-bodies), stress granules and nucleoli21,22. Deficiency in Lin28 proteins leads to severe developmental abnormalities23, while forced expression of Lin28 promotes reprogramming of differentiated cells to pluripotency and enhances tissue growth and repair by regulating metabolic remodeling24,25,26. Moreover, Lin28 proteins may be reactivated in somatic cells to drive tumor growth and progression27,28. Thus, Lin28 proteins have emerged as master regulators of growth and metabolism in development and pathogenesis.
Here, we investigate the translation of non-canonical ORFs across pluripotent and somatic states and show that the selective translation of an upstream ORF (uORF) in the 5′ untranslated region (5′UTR) of Lin28b generates an 85-amino acid microprotein, PLUM, which is critical for the transition between naïve and primed pluripotent states and for metabolic reprogramming during embryo implantation. Loss of PLUM disrupts L1td1 phase-separated condensates, enabling L1td1 bind and stabilize naïve pluripotency transcripts such as Tfcp2l1 and Zfp42, thereby enhancing their translation. Simultaneously, PLUM depletion disrupts P-bodies containing nuclear-encoded mitochondrial mRNAs, preventing their degradation and revealing a previously unrecognized layer of metabolic gene regulation. These two layers of regulation orchestrate an early burst of mitochondrial oxidative phosphorylation and synchronized activation of naïve pluripotency genes, leading to deterministic acquisition of naïve pluripotency. Our results identify a novel uORF-encoded microprotein, PLUM, as a deterministic reprogramming factor in pluripotency transitions and also reveal a mechanism for cell fate regulation through metabolism and cytoplasmic phase separation.
Results
Ribo-seq reveals a uORF-encoded microprotein in Lin28b 5′UTR
We investigated genome-wide translation to identify actively translated noncanonical ORFs by ribosome profiling (Ribo-seq) in pluripotent stem cells (mouse ESCs and EpiSCs) and somatic cells (mouse embryonic fibroblasts, MEFs)29 (Fig. 1a). High-confidence actively translated ORFs were identified based on quantitative assessment of the three-nucleotide periodicity using RiboCode30 (Supplementary Fig. 1a). In addition to 14,730 canonical ORFs (86.36% of the identified ORFs), we detected 2,326 noncanonical ORFs (13.64%), including 2034 uORFs (11.93%, the largest part of noncanonical ORFs), 200 dORFs (1.17%) and 92 lncRNA ORFs (0.54%) (Fig. 1b, c). Nearly half of uORFs use the AUG start codon, but the majority of ORFs that use a non-AUG start codon are uORFs (Supplementary Fig. 1b). To identify noncanonical ORFs with physiological function, we performed differential translation analysis on all identified ORFs. Hundreds of noncanonical ORFs were differentially translated in ESCs, EpiSCs and MEFs, especially uORFs, implying these noncanonical ORFs may be involved in pluripotency regulation (Supplementary Fig. 1c, d).

a Schematic representation of the Ribo-seq procedure in ESCs, EpiSCs and MEFs. b Ribosome profiling reveals widespread translation of non-canonical ORFs in ESCs, EpiSCs and MEFs. c Start codon usage in the identified ORFs. d Scatter plot showing the uORFs with significantly changed translation efficiency in EpiSCs compared with ESCs and MEF. e RPF (Ribosome protection fragment) densities in the 5′UTR and mORF of Lin28b in ESCs, EpiSCs and MEFs. f Amino acid sequence of pluripotency-associated Lin28b uORF-encoded microprotein (PLUM) from different species. g Left: Schematic representation of the expression constructs. Right: immunoblot analysis of translation of uORF in the 5′UTR of Lin28b in HEK293 cells. h Immunoblot analysis of endogenous PLUM expression in ESCs, EpiSCs, MEFs and PLUM overexpressing control (OE PLUM) samples. i Representative unique peptide spectrum and sequence of PLUM identified by LC-MS. j Immunofluorescence of PLUM-Flag in EpiSCs. Scale bar, 5 µm. k Immunoblot analysis of endogenous PLUM in nuclear and cytoplasmic fractions of EpiSCs. The experiments in g, h, j, k were repeated independently three times with similar results. Representative data are shown.
Since uORFs are generally considered to act as cis-regulatory elements to regulate the translation of downstream main ORFs (mORFs)31, we then explored the translational changes of uORFs and corresponding mORFs in different cell types. Among differentially translated uORFs, approximately one-third uORFs were altered without corresponding changes in the translation of their respective mORFs, while the remaining two-thirds changed together with their mORFs. Importantly, although uORFs can function as inhibitory cis-regulatory elements for their corresponding mORFs (Supplementary Fig. 1e), we observed almost no uORFs that changed in the opposite direction of their mORFs when comparing translation changes across different cell types (Supplementary Fig. 1f, g). These suggest that these differentially translated uORFs may have other functions, such as coding for microproteins. Integrating RNA-seq and Ribo-seq, we calculated the translational efficiency (TE) and found uORFs displayed significant differences in TE across different pluripotent and somatic states (Fig. 1d). Among the uORFs with significant changes, we observed a uORF, Lin28b uORF, beginning in the 5′UTR of Lin28b and partially overlapping its coding sequence (CDS) in a different reading frame (Fig. 1d, e). Lin28b uORF begins with the ATG start codon and possesses a partial Kozak sequence, indicating a strong translation capability. The translation of Lin28b uORF was significantly increased in EpiSCs compared to ESCs and MEF (Fig. 1d, e), and because Lin28b is an important pluripotency factor, we selected this noncanonical ORF for further research.
Lin28b uORF was predicted to encode an 85-amino acid microprotein, which is highly conserved among mammals (Fig. 1f). To validate the translation of Lin28b uORF, we generated a series of expression vectors with a C-terminal fusion of 3 × Flag tag to the uORF, including a uORF expression control lacking the 5′UTR preceding the uORF (uORF-Flag), a uORF with the full 5′UTR (UTR-Flag), and a uORF with the full 5′UTR but harboring an ATG to TAA mutation (TAA-Flag) (Fig. 1g). We detected the fusion protein of predicted molecular weight with an anti-Flag antibody after transfection of uORF-Flag and UTR-Flag constructs into HEK293T cells, whereas no signal was observed after transfection of TAA-Flag, indicating Lin28b uORF is capable of being translated to its 85-amino acid microprotein (Fig. 1g).
To further confirm the endogenous existence of this pluripotency-associated Lin28b uORF-encoded microprotein, which we termed PLUM, we produced a custom polyclonal antibody against PLUM using recombinant protein as antigen. We examined the protein level of PLUM in ESC, EpiSC and MEF and found PLUM is highly expressed in EpiSC compared to ESC and MEF, consistent with our Ribo-seq data (Fig. 1h and Supplementary Fig. 1h). Furthermore, the existence of PLUM was confirmed by liquid chromatography-mass spectrometry (LC-MS) (Fig. 1i). In addition, immunofluorescence revealed a punctate cytoplasmic localization of PLUM (Fig. 1j), which was further confirmed by nucleus/cytoplasm fractionation and immunoblotting of the endogenous protein (Fig. 1k). Notably, our custom antibody was not suitable for immunofluorescence of endogenous PLUM, and future strategies such as noncanonical amino acid labeling may enable more precise localization studies. Altogether, these data demonstrate that the selective translation of Lin28b uORF in primed pluripotency generates a novel conserved microprotein PLUM.
PLUM depletion induces deterministic naïve pluripotency
To investigate the functional role of PLUM in PSCs, we conducted a CRISPR/Cas9-mediated knockout (KO) of PLUM in EpiSCs derived from embryonic mice carrying a GFP reporter for naïve pluripotency driven by a cassette lacking the proximal enhancer of Oct432. Frameshift mutations were introduced into the uORF region that does not overlap with the Lin28b mORF (Fig. 2a and Supplementary Fig. 2a), resulting in the loss of PLUM without affecting Lin28b protein level (Fig. 2b and Supplementary Fig. 2b). Because the uORF start codon is essential for its regulation of mORF translation, this strategy eliminates the microprotein produced by uORF translation without altering its regulatory context (Supplementary Fig. 2c–e). PLUM KO EpiSCs had a slightly reduced proliferation rate and more compact clonal morphology (Fig. 2c and Supplementary Fig. 2f), similar to what is observed in naïve ESC1, suggesting that PLUM may be involved in the transition between different pluripotent states.

a Schematic of sgRNAs used to generate PLUM knockout (KO) EpiSCs. b Immunoblot analysis of PLUM in wild type (WT), KO and PLUM overexpressing (OE PLUM) EpiSCs. c Representative phase-contrast images of WT and KO EpiSCs. Scale bar, 250 µm. d Representative phase-contrast and OCT4-GFP images of WT and KO EpiSCs on day 6 of BMP4 induced primed-naïve transition (BiPNT). rESCs, naïve ESC reset from EpiSCs via BiPNT. Scale bar, 250 µm. e Number of Oct4-GFP+ colonies in WT and KO EpiSCs on day 6 of BiPNT. P = 1.156 × 10−6. f Number of Oct4-GFP+ colonies in WT and KO EpiSCs with or without PLUM overexpression on day 6 of BiPNT. Significant differences were detected between KO and WT (P = 6.044 × 10⁻⁷), and KO and KO + PLUM (P = 3.92 × 10⁻⁶). g Flow cytometry analysis of Oct4-GFP+ cells in WT and KO EpiSCs on day 0 to 10 of BiPNT. Significant differences were detected between KO and WT on day 4 (P = 0.0002), day 5 (P = 0.0002), day 6 (P = 6.27 × 10−5), day 7 (P = 6.85 × 10−5), day 8 (P = 0.0001), day 9 (P = 0.0003) and day 10 (P = 2.39 × 10−5). h Representative phase-contrast images of ESCs and ESCs overexpressing PLUM (ESC + PLUM) cultured in Activin A/bFGF medium for 5 days. Scale bar, 250 µm. i RT-qPCR analysis of naïve and primed genes as in (h). Significant differences between KO and WT in naïve genes (Nanog, P = 0.0004; Klf4, P = 0.0012; Essrb, P = 0.0153; Tfcp2l1, P = 0.0009; Dppa5a, P = 0.0008; Sox2, P = 0.0009) and primed genes (Otx2, P = 0.0165; Fgf5, P = 2.65 × 10⁻⁵; Dnmt3b, P = 0.0012). The experiments in (b, c, d, h) were repeated independently three times with similar results. Representative data are shown. Data in (e–g, i) are mean ± s.d., n = 3 biological replicates. P-values were calculated by two-tailed unpaired student’s t test, *P < 0.05, **P < 0.005, ***P < 0.001, ****P < 0.0001. Source data are provided as a Source Data file.
We then examined the effects of PLUM KO on the induction of naïve pluripotency using a chemically defined, BMP4-induced primed-naïve transition (BiPNT) system33 (Fig. 2d). Remarkably, PLUM KO significantly increased the efficiency of PNT, as shown by an increase in the numbers of Oct4-GFP+ colonies (Fig. 2d, e and Supplementary Fig. 3a), up-regulation of naïve pluripotency genes, including Nanog, Klf4, Klf2, Esrrb and Dppa5a, and down-regulation of primed pluripotency genes, including Fgf5, Otx2 and Dnmt3b (Supplementary Fig. 3b). Moreover, overexpression of PLUM reduced efficiency of PNT in WT EpiSCs and was able to completely counteract the effects of PLUM KO, further validating a role for PLUM in safeguarding primed pluripotency (Fig. 2f).
Detection of Oct4-GFP+ cells by flow cytometry and alkaline phosphatase (AP) staining at different time points showed that PLUM KO accelerated the kinetics of PNT (Fig. 2g and Supplementary Fig. 3c). Notably, more than 98% of PLUM KO cells were Oct4-GFP+ at day 10 (Fig. 2g). The resulting PLUM KO colonies (reset naïve state stem cells, rESC) expressed protein markers of naïve pluripotency, exhibited a transcriptional profile similar to that of naïve cells, and were capable of generating chimeric mice (Supplementary Fig. 4a–d). Clonal analysis demonstrated that multiple independent PLUM KO clones displayed consistent pluripotency gene expression and maintained long-term self-renewal with stable AP activity for at least 20 passages (Supplementary Fig. 5a–e). In addition, teratoma formation assays confirmed that these cells retained multilineage differentiation potential (Supplementary Fig. 5f). Together, these findings indicate that the converted cells are homogeneous, stably naïve, and fully pluripotent, and demonstrate that PLUM depletion induces a synchronized and deterministic acquisition of naïve pluripotency.
The transition from primed to a naïve pluripotent state can be achieved by overexpressing key transcription factors34,35. To determine whether PLUM’s function is specific to BMP4-induced condition, we tested PLUM KO in PNT induced by Klf2 or Esrrb and observed a marked increase in Oct4-GFP+ colony numbers compared with WT cells, a phenotype that was reversed by PLUM overexpression (Supplementary Fig. 6a). Importantly, in the BiPNT culture system devoid of BMP4, PLUM KO EpiSCs could be converted into a naïve pluripotent state, whereas WT EpiSCs could not (Supplementary Fig. 6b). Moreover, we confirmed the expression of PLUM in human primed ESCs (Supplementary Fig. 6c) and found PLUM knockout was able to promote the transition from primed to naïve pluripotent state, evidenced by increased alkaline phosphatase-positive (AP+) colonies and elevated naïve gene expression (Supplementary Fig. 6d–f). These data indicate that the function of PLUM in pluripotency transition is independent of culture conditions and is not limited to mice.
Finally, to extend our observations, we evaluated the function of PLUM in the conversion from naïve to primed pluripotent state. Overexpression of PLUM in naïve ESCs cultured with the conversion medium containing Activin A and bFGF resulted in the generation of larger EpiSC-like colonies, significant upregulation of primed genes and downregulation of naïve genes (Fig. 2h, i). Conversely, PLUM KO rESCs showed a delayed exit from the naïve state under primed conditions, which was rescued by PLUM overexpression, supporting a role for PLUM in promoting the naïve-to-primed transition and safeguarding the primed state (Supplementary Fig. 7a, b).
PLUM loss drives coordinated naïve pluripotency activation
To further investigate the mechanism underlying the deterministic naïve transition induced by PLUM loss, we performed a comprehensive time-course transcriptomic analysis across days 0–10 (Fig. 3a). Using WT- and KO-derived rESCs as naïve references, we found that WT cells at day 10 were still transcriptionally distinct from rESCs, whereas KO cells had largely converged to an rESC-like profile by day 8, indicating a fast and more deterministic reprogramming (Fig. 3a). Time-series clustering further revealed that most genes in KO cells fell into two major groups: cluster 2, which showed a sharp increase in the first 3 days and then declined, and cluster 6, which decreased early but rose steadily at later stages (Fig. 3b). Gene ontology (GO) analysis showed that cluster 2 genes were enriched for translation, mitochondrial respiration, and apoptosis, whereas cluster 6 genes were involved in transcription, chromatin remodeling, DNA repair, and embryonic development (Fig. 3c).

a Principal component analysis (PCA) of time-course transcriptomes (day 0–10) from WT and PLUM KO cells compared with rESCs. b Time-series clustering of differentially expressed genes in WT and KO cells. c Gene ontology (GO) enrichment analysis of cluster 2 and cluster 6 genes in KO cells. d Cluster assignments of naïve pluripotency marker genes in WT and KO cells. e Heatmap showing the expression dynamics of representative pluripotency markers during PNT.
Notably, pluripotency markers, which typically display heterogeneous dynamics during cell fate transitions, exhibited divergent cluster assignments in WT cells (clusters 1, 3, 4, 5, and 6), but shifted into a more uniform pattern in KO cells, all mapping to cluster 6 (Fig. 3d). Further analysis revealed that these genes were activated earlier and reached higher levels in KO cells, with the exception of Klf4 and Klf5 (Fig. 3e). These results suggest that PLUM loss not only synchronizes but also accelerates the induction of pluripotency marker genes, thereby accelerating naïve state acquisition.
The strong enrichment of mitochondrial functions in KO cluster 2 was particularly striking (Fig. 3c and Supplementary Fig. 8a). We analyzed genes encoding mitochondrial complexes I-V and found that they exhibited a pronounced early upregulation in KO cells, but not in WT, suggesting an OXPHOS burst during the early phase of PNT in KO cells (Supplementary Fig. 8b). Consistently, Seahorse assays confirmed elevated mitochondrial oxygen consumption rate (OCR) and reduced glycolysis in KO cells, both of which were rescued by PLUM overexpression (Supplementary Fig. 8c, d).
Together, these findings reveal that PLUM loss drives deterministic naïve transition by inducing an early OXPHOS burst and synchronizing the activation of naïve pluripotency genes, thereby accelerating the acquisition of the naïve state.
PLUM interacts with L1td1 to regulate its phase separation
To understand the mechanisms by which PLUM regulates cell fate transitions in PSCs, we conducted immunoprecipitation for PLUM followed by mass spectrometry (IP-MS) to identify its binding partners in EpiSCs. Among the top-ranked candidates, which were rigorously filtered to remove contaminants using CRAPome36, we observed L1td1, an RNA-binding protein highly expressed in PSCs37 (Fig. 4a and Supplementary Fig. 9a). L1td1 has been suggested involved in pluripotency regulation38,39, however, its mechanisms of action remain largely unexplored. Using co-IP, we confirmed that PLUM interacts with L1td1 and found their interaction is resistant to RNase A treatment (Fig. 4b and Supplementary Fig. 9b). In addition, PLUM was successfully pulled down with His-tagged L1td1 in E. coli lysates (Supplementary Fig. 9c, d). These data suggest a direct interaction between PLUM and L1td1.

a Flag-PLUM IP-MS in EpiSCs. Candidate PLUM-interacting proteins are ranked by Mascot score. b L1td1 co-IP with Flag-PLUM in EpiSCs with or without RNase A treatment. c Schematic of L1td1 domain-deletion mutants. d Flag-PLUM co-IP with L1td1 domain-deletion mutants. e Numbers of Oct4-GFP+ colonies in EpiSCs transfected with CTRL, shL1td1-1/2 or L1td1. Significant differences were detected between CTRL and shL1td1-1 (P = 0.0043), shL1td1-2 (P = 0.0105), L1td1 (P = 0.0015). f Numbers of Oct4-GFP+ colonies in PLUM KO EpiSCs transfected with CTRL, shL1td1-1/2 or L1td1. Significant differences were detected between CTRL and shL1td1-1 (P = 0.0002), shL1td1-2 (P = 7.77 × 10−5), L1td1 (P = 0.0017). g Number of Oct4-GFP+ colonies in EpiSCs transfected with control, L1td1 or L1td1 mutant (∆620–782 and ∆620–713). Significant differences were detected between control and L1td1 (P = 0.0012), Δ620–782 (P = 0.0159), Δ620–713 (P = 0.0202). h Left: Representative images of mCherry-L1td1 in WT and KO EpiSCs with or without PLUM overexpression. Scale bar, 5 µm. Right: Number of L1td1 condensates per cell. Significant differences were detected between KO and WT (P = 1.63 × 10⁻¹¹). i Schematic of L1td1 condensates formation through PLUM binding. j Representative images of L1td1-GFP in EpiSCs treated with 1,6-Hexanediol for five minutes. Scale bar: up, 5 µm; down, 1 µm. k FRAP analysis of L1td1-GFP condensates in EpiSCs. Scale bar, 1 µm. Significant differences were detected between 2 s and 60 s (P = 2.53 × 10⁻8). l Representative images of mCherry-L1td1, ∆620–782 and ∆ 620-713 in EpiSCs. Scale bar, 5 µm. The experiments in (b, d, j, l) were repeated independently three times with similar results. Representative data are shown. Data in (e–h, k) are mean ± s.d., n = 3 biological replicates (e–g), n = 20 clones (h), 25 clones (k). P-values were calculated by two-tailed unpaired student’s t test, *P < 0.05, **P < 0.005, ***P < 0.001, ****P < 0.0001. Source data are provided as a Source Data file.
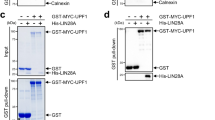
RNA-binding proteins typically possess specific RNA-binding domains (RBD) or RNA-recognition motifs (RRM) that facilitate their functions40. L1td1 is predicted to have three functional RBD-like domains (RBDL), prompting us to investigate their interaction with PLUM. We constructed four L1td1 mutants with different RBDL domain deletions and assessed their interaction with PLUM (Fig. 4c). The simultaneous deletion of RBDL2 and RBDL3 (Δ620–782) abolished the interaction between PLUM and L1td1(Fig. 4d). Consistently, deletion of RBDL2 alone (Δ620-713), but not RBDL3 (Δ718-782), also abolished the interaction, suggesting the RBDL2 domain is critical for PLUM-L1td1 interaction (Fig. 4d).
The interaction between PLUM and the functional RBDL2 domain of L1td1 led us to explore their functional similarities and interplay. We examined the effects of L1td1 overexpression and knockdown on the primed-naïve transition of WT and PLUM KO EpiSCs. L1td1 knockdown promoted PNT in WT EpiSCs, while its overexpression was inhibitory (Fig. 4e and Supplementary Fig. 9e, f), indicating that L1td1 and PLUM may share roles in pluripotency regulation. Intriguingly, the effects of L1td1 manipulation were reversed in PLUM KO EpiSCs (Fig. 4f), suggesting a functional interaction between PLUM and L1td1. To further explore this interaction, we tested the effects of the L1td1 mutant lacking the crucial interaction domain (Δ620–782 and Δ620-713) on PNT. Unlike WT L1td1, the mutants lost their inhibitory effect on PNT and even slightly enhanced it (Fig. 4g).
Next, we assessed whether the cellular localization of L1td1 is affected by PLUM depletion. L1td1 formed punctate condensates in WT EpiSCs but was dispersed in PLUM KO cells (Fig. 4h). Re-expression of PLUM in KO cells restored condensate formation (Fig. 4h), indicating that PLUM-L1td1 binding is required for the formation of L1td1 condensates (Fig. 4i). As RNA-binding proteins often undergo phase separation to exert their functions40, we tested the sensitivity of L1td1 condensates to 1,6-hexanediol (1,6-HD)41, an aliphatic alcohol known to disrupt weak hydrophobic interactions, and found that L1td1 condensates dissolved in 1.5% 1,6-HD (Fig. 4j). Fluorescence recovery after photobleaching (FRAP) experiments revealed partial recovery of L1td1 condensates after bleaching, further supporting their phase separation property (Fig. 4k). Moreover, the L1td1 Δ620–782 and Δ620–713 mutants, which lost interaction with PLUM, failed to form condensates (Fig. 4l).
To assess whether PLUM itself could undergo phase separation, we analyzed its sequence and found no intrinsically disordered regions (Supplementary Fig. 10a). Consistently, purified PLUM protein failed to form condensates across a range of concentrations, salt conditions, or in the presence of RNA (Supplementary Fig. 10b, c). Together, these findings indicate that PLUM does not phase separate independently but instead regulates the phase separation and condensate formation of L1td1, thereby influencing its role in pluripotency.
PLUM modulates L1td1 RNA binding to stabilize Tfcp2l1 mRNA
Given that PLUM regulates the formation of L1td1 condensates in EpiSCs, we sought to address whether PLUM KO alters the RNA-binding capacity of L1td1. We conducted enhanced UV cross-linking immunoprecipitation (eCLIP) with an L1td1 antibody followed by high-throughput sequencing42 (Supplementary Fig. 11a). Global analysis identified 4449 significant L1td1 binding peaks in WT EpiSCs compared to 3457 in PLUM KO cells (Fig. 5a). Most L1td1 peaks were mapped to the CDSs and introns of target transcripts in WT EpiSCs. In PLUM KO, the number of L1td1 binding peaks in CDSs and 5′ UTRs was significantly reduced (Fig. 5a). Interestingly, consensus motif analysis showed that L1td1 preferentially bound to “GAGCGU” sequences in both WT and PLUM KO EpiSCs (Fig. 5b). The mRNAs bound by L1td1 displayed significant differences between WT and PLUM KO cells (Fig. 5c). GO analysis revealed that WT-specific L1td1 binding genes were involved in transcription regulation, protein phosphorylation and cell cycle, while PLUM KO-specific L1td1 binding genes were associated with organ morphogenesis, cell adhesion and gene expression (Supplementary Fig. 11b).

a, b Distribution of L1td1-binding peaks (a) and enriched motifs(b). c Specific and common L1td1 binding genes in WT and KO EpiSCs. d Overlap of specific L1td1 binding genes with cluster 2 genes in KO EpiSCs. e IGV views of L1td1 peaks across Tfcp2l1. f RIP-qPCR of Tfcp2l1 3′UTR-L1td1 interaction in WT and KO EpiSCs with or without PLUM overexpression. Significant differences were detected between L1td1-IP and IgG-IP in KO (P = 8.85 × 10⁻⁵). g RT-qPCR of Tfcp2l1 in WT and KO EpiSCs with or without PLUM overexpression. Significant differences were detected between KO and WT (P = 0.0018). h RT-qPCR of Tfcp2l1 in WT and KO EpiSCs on BiPNT day 5 with or without PLUM overexpression. Significant differences were detected between KO and WT (P = 0.0001). i Immunoblot of Tfcp2l1 as in (h). j Tfcp2l1 binding on TSS of Naïve and OXPHOS genes. k Representative images of PLUM-Flag and DCP1α in EpiSCs. Scale bar, 5 µm. l Representative images and quantification of L1td1 condensate colocalization with P-body in WT and KO EpiSCs with or without PLUM overexpression. Scale bar, 5 µm. Boxes show the interquartile range, line marks the median, and whiskers denote min-max values. Significant differences were detected between KO and WT (P = 1 × 10⁻15). m P-body number per cell in WT and KO EpiSCs with or without PLUM overexpression. Significant differences were detected between KO and WT (P = 3.5 × 10⁻11). n Schematic for P-bodies purification with GFP-LSM14A and qRT-PCR of mRNAs in purified P-body and cytoplasmic fractions. Significant differences were detected between P-body and cytosol in OXPHOS genes (Ndufs8, P = 0.0002; Ndufa3, P = 0.0005; Sdha, P = 0.0243; Uqcr10, P = 0.0007; Atp5d, P = 1.79 × 10⁻5; Atp5g, P = 0.0013), and glycolysis gene Pgk1 (P = 0.0165). The experiments in (i, k) were repeated independently three times with similar results. Representative data are shown. Data in (f–h, l–n) are mean ± s.d., n = 3 biological replicates (f–h, n), n = 20 clones (l, m). P-values were calculated by a two-tailed unpaired student’s t test, *P < 0.05, **P < 0.005, ***P < 0.001, ****P < 0.0001. Source data are provided as a Source Data file.
To elucidate how differential RNA binding of L1td1 contributes to pluripotency regulation, we examined the overlap between KO-specific L1td1 targets and Cluster 6 genes during the PNT process. Among these, we identified several critical regulators of naïve pluripotency, such as Tfcp2l1 and Zfp42 (Fig. 5d). We next focused on Tfcp2l1 for validation and found that PLUM loss significantly increased L1td1 binding to its 3′UTR, accompanied by elevated Tfcp2l1 mRNA levels (Fig. 5e), raising the possibility that PLUM depletion enhances L1td1’s binding to 3′UTR of Tfcp2l1 to modulate RNA levels and pluripotency. To this end, we examined the binding of L1td1 to 3′UTR of Tfcp2l1 by performing RNA immunoprecipitation coupled with quantitative PCR (RIP-qPCR). Notably, the observed increased binding of L1td1 to 3′UTR of Tfcp2l1 was markedly reversed when PLUM was overexpressed in PLUM KO EpiSCs (Fig. 5f).
Given the importance of the 3′UTR and associated RNA-binding proteins in mRNA stability and degradation, we inhibited mRNA transcription by Actinomycin D and examined the degradation of Tfcp2l1 mRNA. The loss of Tfcp2l1 mRNA was slower in PLUM KO EpiSCs, suggesting that PLUM depletion enhances Tfcp2l1 mRNA stability (Fig. 5g). To link PLUM-mediated Tfcp2l1 mRNA stability to pluripotency transition, we measured the RNA and protein levels of Tfcp2l during the PNT process. Both mRNA and protein levels of Tfcp2l1 were increased in PLUM KO cells, which could be reduced by PLUM re-expression (Fig. 5h, i), confirming translational regulation of Tfcp2l1 by PLUM during pluripotency transition.
By analyzing Chromatin immunoprecipitation (ChIP) and RNA-seq data43, we found Tfcp2l1 binding is highly enriched around the transcription start site (TSS) of Naïve pluripotency and mitochondrial OXPHOS genes (Fig. 5j). Further GO analysis showed that genes directly bound and activated by Tfcp2l1 are associated with embryonic development and mitochondrial metabolism (Supplementary Fig. 11c).
To test whether other naïve regulators are similarly affected, we examined Nanog and Zfp42 mRNA stability. PLUM KO had no effect on Nanog but increased Zfp42 stability (Supplementary Fig. 11d), indicating that L1td1 contributes to the post-transcriptional regulation of multiple pluripotency-associated genes. The much longer half-life of Zfp42 suggests limited functional consequences of its stabilization, and we therefore focused on Tfcp2l1 as the most responsive target. A more comprehensive understanding of additional downstream targets will require future investigation. To assess the role of L1td1 domains in Tfcp2l1 regulation, we tested the four L1td1 mutants in WT cells. Both Δ620–782 and Δ620–713, which lost interaction with PLUM, gained the ability to bind Tfcp2l1 mRNA and enhanced its translation, increasing downstream OXPHOS gene expression, whereas Δ718-782 had no effect (Supplementary Fig. 12a–c). These results indicate that L1td1’s interaction with Tfcp2l1 mRNA is independent of RBDL2 or RBDL3, but is modulated by PLUM binding and the formation of L1td1 condensates.
Overall, these results support a model in which PLUM interacts with L1td1 to regulate its phase separation and RNA binding, thereby influencing Tfcp2l1 mRNA stability, mitochondrial OXPHOS, and Naïve gene expression.
PLUM loss reduces P-bodies with nuclear-encoded mt-mRNAs
L1td1 was reported to co-localize with P-bodies and regulate RNA processing38,39. We found partial co-localization of PLUM with P-bodies (marked by Dcp1α) (Fig. 5k). In PLUM KO EpiSCs, the co-localization of L1td1 condensates with P-bodies was significantly reduced, and the overall number of P-bodies was decreased (Fig. 5l, m). Both phenotypes were rescued by re-expression of PLUM (Fig. 5l, m), implying a role for PLUM in the regulation of RNA processing and translation by influencing P-bodies assembly.
P-bodies primarily function to store RNAs and inhibit their translation, thereby regulating the availability and translational activity of RNA within the cell44,45,46. To investigate the potential relationship between reduced P-body numbers and PLUM-mediated metabolic reprogramming and pluripotency, we employed a fluorescence-activated particle sorting method to isolate P-bodies from EpiSCs, and assessed the mRNAs associated with P-bodies, using total cytoplasmic fractions as a control (Fig. 5n). Our results revealed that a subset of nuclear-encoded mitochondrial mRNAs, particularly those encoding components of mitochondrial complexes I and V, was significantly enriched in P-bodies (Fig. 5n). In contrast, we observed no substantial enrichment of mRNAs encoding glycolytic enzymes or naïve proteins (Fig. 5n).
These results indicate that the loss of PLUM disrupts P-bodies that are enriched with specific nuclear-encoded mitochondrial mRNAs, potentially inhibiting the degradation of these mRNAs.
PLUM loss impairs metabolic reprogramming during embryo implantation
To explore the physiological functions of PLUM, we generated PLUM knockout mice using CRISPR/Cas9 system with guide RNA (gRNA) targeting the uORF in the 5′UTR of Lin28b (Fig. 6a and Supplementary Fig. 13a). We found expression of PLUM in brain, heart, liver, kidney, intestine, spleen and testis in WT mice (Supplementary Fig. 13b) and confirmed the knockout of PLUM at the protein level (Fig. 6b). In the process of mouse breeding through mating of heterozygotes, we found that homozygous PLUM knockout mice (PLUM-/-) were not born at the expected Mendelian ratio, showing a decreased birth rate without sex bias (Fig. 6c).

a Schematic of sgRNA used to generate PLUM knockout mice. b Immunoblot of endogenous PLUM in testis of PLUM +/+ and PLUM -/- mice. c Inheritance pattern of the mutated PLUM allele. d Immunoblot of endogenous PLUM in post-implantation embryo and pre- and post-implantation uterus. e Representative images and numbers of PLUM +/+, PLUM -/- and PLUM +/- embryo on implantation. Scale bar, 1 cm. Significant differences were detected between PLUM -/- and PLUM +/+ (P = 0.0004), PLUM +/- (P = 0.0009). f Timeline of embryo transfer. Decidualization was assessed at 6.5 dpc. g Decidua formation efficiency. Significant differences were detected between PLUM -/- and PLUM +/+ embryo (P = 7.79 × 10⁻5). h RT-qPCR analysis of OXPHOS genes in 4.5 dpc and 6.5 dpc embryos of PLUM+/+ and PLUM-/- mice. Significant differences were detected between PLUM-/- and PLUM+/+ in Ndufs8 (P = 0.0004), Ndufa3 (P = 0.0003), Sdha (P = 0.0027), Sdhb (P = 0.0004), Uqcrc1 (P = 0.0002), Uqcr10 (P = 5.78 × 10⁻5), Cox6a (P = 0.0003), Cox6b (P = 0.0009), Atp5d (P = 0.0007) and Atp5g1 (P = 0.0004). i, Representative images of TMRM staining in PLUM +/+ and PLUM -/- embryo. Scale bar, 50 µm. j Procedure of dissecting the epiblasts from 6.5 dpc embryos for ΔΨm and ATP measurement. k, l Flow cytometry analysis of TMRM in the epiblasts of PLUM +/+ and PLUM -/- embryos. Significant differences were observed between PLUM -/- and PLUM +/+ embryo (P = 0.0006). m ATP levels in the epiblasts of PLUM+/+ and PLUM -/- embryos. Significant differences were detected between PLUM -/- and PLUM +/+ embryo (P = 0.0004). The experiments in (b, d, i) were repeated independently three times with similar results. Representative data are shown. Data in (e, g, h, l, m) are mean ± s.d., n = 6 female mice (e, g), n = 3 biological replicates (h), n = 5 and 6 biological replicates (l), n = 4 biological replicates (m). P-values were calculated by two-tailed unpaired student’s t test, **P < 0.005, ***P < 0.001, ****P < 0.0001. Source data are provided as a Source Data file.
Given the critical role of PLUM in regulating naïve and primed pluripotent stem cells, which resemble cells of the pre- and post-implantation embryo, respectively, we speculated that PLUM deficiency might affect embryo implantation. To investigate this, we first analyzed published RNA-seq data47, which showed increased Lin28b mRNA in post-implantation embryos (Supplementary Fig. 13c). We further examined PLUM expression in post-implantation embryos and pre- and post-implantation uteri and found PLUM was highly expressed only in post-implanted embryo, suggesting that PLUM likely affects embryos themselves and not uteri (Fig. 6d). Consistently, immunoblotting revealed no detectable Lin28b protein in uteri from 3.5–6.5 dpc WT and PLUM-/- mice, and comparable Lin28b protein levels in 5.5 and 6.5 dpc post-implantation embryos (Supplementary Fig. 13d, e), ruling out indirect effects through Lin28b.
Next, we assessed implantation sites in WT, PLUM and PLUM+/− 6.5 dpc uteri by Chicago sky blue 6B staining. PLUM-/- uteri had significantly fewer implantation sites compared with WT uteri, indicating defects in implantation of PLUM-/- embryos (Fig. 6e). Importantly, the number of implantation sites in PLUM+/- uteri obtained by crossing male WT mice with female PLUM-/- mice showed no differences compared with WT uteri (Fig. 6e), indicating that the implantation defect was caused by PLUM deficient-embryo but not uteri. To further clarify whether the implantation defect originated from impaired pre-implantation development or implantation capacity, we cultured WT and PLUM-/- 2-cell embryos in vitro and observed no difference in blastocyst formation (Supplementary Fig. 13f). However, when transferred into pseudopregnant WT recipients, PLUM-/- blastocysts exhibited reduced implantation efficiency compared to WT (Fig. 6f, g). Together, these results demonstrate that implantation failure arises PLUM-deficient embryos rather than maternal effects.
Metabolic remodeling is critical for early embryonic development48,49,50. To determine whether PLUM contributes to metabolic remodeling during embryo implantation, we measured expression of mitochondrial OXPHOS genes in pre- and post-implantation embryos. In WT embryos, mitochondrial OXPHOS genes decreased during implantation, whereas in PLUM-/- embryos, these genes did not decrease properly (Fig. 6h). Tetramethylrhodamine methyl ester (TMRM) staining of isolated post-implantation embryos showed elevated mitochondrial membrane potential (ΔΨm) in PLUM-/- post-implantation embryos (Fig. 6i).
To accurately assess mitochondrial function in the epiblasts of post-implantation embryos, we dissected epiblast to analyze ΔΨm and ATP levels (Fig. 6j). The epiblasts of PLUM-/- post-implantation embryos exhibited higher ΔΨm and increased ATP levels compared to those of WT embryos (Fig. 6k–m). Consistent with these metabolic abnormalities, histological analysis revealed delayed implantation and increased apoptosis in 6.5 dpc PLUM-/- embryos, as indicated by enhanced TUNEL staining (Supplementary Fig. 13g), suggesting that the implantation defects may result from elevated apoptosis and delayed embryo development, potentially associated with altered mitochondrial activity or dysregulated reactive oxygen species (ROS) homeostasis.
Together, these data indicate that PLUM deficiency disrupts metabolic remodeling and compromises proper embryo implantation.
Discussion
In this study, we reveal selective translation of non-canonical ORFs in stem cells and identify PLUM, a novel 85-amino acid microprotein encoded by a uORF in the 5′ UTR of Lin28b, acting as a cytoplasmic and metabolic determinant in the regulation of pluripotency and embryonic development (Fig. 7). Mechanistically, we show that PLUM depletion dissolves L1td1 condensates and enhances L1td1 binding to pluripotency-related mRNAs, including Tfcp2l1 and Zfp42, thereby promoting their stabilization and coordinated activation. In addition, PLUM loss disrupts P-bodies enriched with nuclear-encoded mitochondrial mRNA, potentially limiting their degradation. These parallel effects converge on an early burst of mitochondrial OXPHOS and synchronized activation of naïve pluripotency genes, ultimately driving a deterministic transition to the naïve state. Our results identify uORF-encoded microprotein PLUM as a deterministic reprogramming factor and elucidate a novel mode of PLUM-mediated metabolic remodeling and cytoplasmic phase separation in pluripotency regulation, highlighting the importance of dynamic translation of non-canonical ORFs in early development and cell fate decisions.

Proposed model showing Lin28b uORF-encoded microprotein PLUM functions in pluripotency transition and embryonic development.
We demonstrate that the selective translation of a uORF in the 5′UTR of Lin28b produces a novel microprotein, PLUM, which is crucial for pluripotency regulation and embryonic development. Depletion of PLUM enables deterministic induction of naïve pluripotency in mouse EpiSCs, representing a distinct route to naïve pluripotency. PLUM loss triggers an early burst of mitochondrial OXPHOS and a synchronized induction of naïve pluripotency genes, changes that markedly accelerate the acquisition of the naïve state. Mechanistically, loss of PLUM dissolves L1td1 condensates and enhances L1td1 binding to naïve pluripotency mRNA such as Tfcp2l1 and Zfp42, thereby increasing their stability and accelerating coordinated gene activation. L1td1 binding to the Tfcp2l1 3′UTR may protect it from CNOT-mediated degradation, thereby promoting transcript stability. The limited colocalization of L1td1 with P-bodies and the reduction in P-body number upon L1td1 loss suggest that L1td1 may influence mRNA decay by preventing the recruitment of RNA decay factors through modulation of P-body assembly.
PLUM depletion also disrupts P-bodies enriched with nuclear-encoded mitochondrial mRNAs, potentially limiting RNA degradation and revealing a previously unrecognized layer of mitochondrial gene regulation via P-body-associated post-transcriptional control. Tfcp2l1, a central regulator of naïve pluripotency, may further stabilize the naïve network and counteract the naïve-to-primed transition by activating OXPHOS-associated transcriptional programs.
To date, only MBD3 and C/EBPα have been reported to achieve deterministic reprogramming to pluripotency with near 100% efficiency51,52,53. However, these deterministic factors require specific reprogramming systems or ectopic expression of classic Yamanaka factors such as Sox2, Klf4, Oct4 and Myc, with mechanisms typically associated with epigenetic remodeling. In contrast, the PLUM-mediated deterministic induction of naïve pluripotency occurs independently of exogenous transcription factors and BMP4-related mechanisms, instead acting through metabolic remodeling and cytoplasmic phase separation, whereby PLUM loss elicits an early OXPHOS burst that synchronizes naïve pluripotency gene activation. The resulting cells are homogeneous, stably naïve, and fully pluripotent, with germline transmission assays currently in progress to rigorously confirm this capacity. Thus, our study not only identifies uORF-encoded microprotein PLUM as a predominant barrier preventing the establishment of naïve pluripotency, but also reveal the metabolism and cytoplasmic phase separation mechanism underlying deterministic induction of naïve pluripotency.
Importantly, by generating PLUM knockout mice, we reveal that PLUM deficiency causes embryo implantation defects. During implantation, the epiblast undergoes a crucial metabolic transition from OXPHOS to glycolytic metabolism to meet the biosynthetic and energetic demands of rapid proliferation. The elevated ΔΨm and ATP levels observed in PLUM-deficient embryos indicate impaired metabolic remodeling, leading to excessive mitochondrial activity and redox imbalance. Such defects are likely to compromise implantation efficiency, highlighting that PLUM-dependent post-transcriptional regulation of L1td1 and metabolic pathways is essential for coupling metabolic state with developmental competence. Due to the conserved protein sequence of PLUM in mammals and its function in regulating pluripotency of both human and mouse cells, PLUM may also play a crucial role in human embryo implantation, indicating that mutations within the uORF in the 5′UTR of LIN28B should be considered in genomic studies related to abnormal embryonic development. Although PLUM exerts a strong regulatory function, the potential inhibitory effect of the uORF on Lin28b mORF translation should be considered when interpreting PLUM’s impact on pluripotency and early embryonic development.
Early pregnancy loss is a significant clinical issue in human reproduction, affecting over 15% of conceptions54,55, primarily due to genetic abnormalities. Our results suggest that mutations in non-canonical ORF may play a role in early pregnancy loss associated with implantation failure and identify PLUM deficiency as a risk factor for pregnancy failure, which could be considered in the diagnosis and prediction of early pregnancy loss, offering potential applications for in vitro fertilization and assisted reproductive technologies. Furthermore, given the partial overlap between Lin28b uORF and the exon 1 and 2 of Lin28b mORF, particular attention should be paid during data analysis in clinical genome sequencing and in the design of targeted regions for gene knockout studies. In addition, future studies exploring whether PLUM translation is dynamically regulated under stress or altered metabolic conditions will be important to understand its broader physiological and pathological roles.
Methods
Ethics statement
This study complies with all relevant ethical regulations of Guangzhou Institutes of Biomedicine and Health, Chinese Academy of Sciences. All animal experiment protocols were approved and conducted in accordance with the guidelines of the Institutional Animal Care and Use Committee (IACUC, No. 2021059 and 2025065) of the Guangzhou Institutes of Biomedicine and Health, Chinese Academy of Sciences.
Mice
ΔPE-Oct4-GFP transgenic allele-carrying mice (CBA/CaJ×C57BL/6 J) were purchased from The Jackson Laboratory, and 129 Sv/Jae mice were purchased from Beijing Vital River Laboratory. PLUM knockout mice were generated by Biocytogen (Beijing, China) through CRISPR/Cas9-mediated genome engineering. The Cas9 mRNA and guide RNA (gRNA) targeting the uORF in the 5′UTR of Lin28b (ACTGATGCTGAAGATCACTCCGG) were co-injected into C57BL/6 N zygotes, which were transferred to pseudo-pregnant females. The founders were genotyped by sequencing of the PCR products and crossed with wild-type C57BL/6 N mice.
Cell culture
Mouse ESCs, EpiSCs and MEFs were derived from E3.5, E5.5 and E13.5 embryos, respectively, obtained by crossing male homozygous ΔPE-Oct4-GFP transgenic-allele-carrying mice (CBA/CaJ×C57BL/6 J) with 129/Sv female mice3,56,57. Mouse ESCs and iPSCs were cultured on 0.1% gelatin-coated dishes in N2B27 medium supplemented with 2i (1 µM PD0325901 (Selleck) and 3 µM CHIR99021 (Selleck)) and 1,000 U/ml mouse leukemia inhibitory factor (mLIF, made in GIBH). The N2B27 medium comprised 50% Neurobasal (Gibco), 50% DMEM/F12 (HyClone), 1% B27 (Gibco), 0.5% N2 (Gibco), 1% GlutaMAX (Gibco), 1% NEAA (Gibco) and 0.1 mM β-mercaptoethanol (Gibco). EpiSCs were cultured on fetal bovine serum (FBS, Gibco)-coated dishes in N2B27 medium supplemented with 20 ng/ml bFGF (PeproTech), 20 ng/ml activin A (PeproTech) and 1 µM XAV939 (Selleck)). MEFs and HEK 293 T cells were cultured in DMEM\high glucose (Gibco) supplemented with 10% FBS (Aicon).
Plasmids and lentivirus production
The coding sequences of PLUM, L1td1 and its mutants were cloned into the pLVX lentiviral vector. shRNA inserts were cloned into pLKO.1 lentiviral vector. pX459 and lentiCRISPR v2 vectors were purchased from Addgene. The plasmids pLV-Esrrb and pLV-Klf2 were a generous gift from Dr. Jing Liu. Lentivirus were produced from HEK293T cells using polyethylenimine (Polyscience) as previously described58. Cells were infected with the lentiviral supernatant in the presence of polybrene (Beyotime) and selected with puromycin (Selleck) for 3 days.
Induction of mouse EpiSCs to naïve state
To induce mouse EpiSCs to naïve state by BMP433, EpiSCs were dissociated into single cells using Accutase (Sigma) and seeded on FBS-coated 12-well plates at a density of 2 × 104 cells per well. The next day, the medium was replaced with Stage Ⅰ medium, which contains DMEM\high glucose, 1% B27, 0.5% N2, 1% GlutaMAX, 1% NEAA, 0.1 mM β-mercaptoethanol, 50 µg/ml vitamin C (Sigma), 5 mM LiCl (Sigma), 20 ng/ml bFGF, 1 µM CHIR99021, 1000 µ/ml mLIF, 1 µM EPZ5676 (Selleck), 2.5 µM EPZ6438 (Selleck) and 10 ng/ml BMP4 (R&D system). After culturing in Stage Ⅰ medium for 3 days, the medium was replaced with Stage II medium, which is N2B27 medium supplemented with 2i and mLIF, for 3 or 5 days.
To induce mouse EpiSCs to the naïve state by transcription factors, EpiSCs were infected with lentivirus containing Essrb or Klf2 for 10 h with polybrene. 24 h after infection, EpiSCs were selected with puromycin for 3 days and replated on FBS-coated 12-well plates at a density of 2 × 104 cells per well. The next day, the medium was replaced with N2B27 medium supplemented with 2i and mLIF for 5 days.
Induction of mouse ESCs to primed state
Mouse ESCs were seeded on FBS-coated 6-well plates at a density of 5 × 104 cells per well and cultured in N2B27 medium supplemented with 20 ng/ml bFGF, 20 ng/ml activin A, 1 µM XAV939 and 3 µM CHIR99021 for 5 days.
CRISPR-Cas9 medicated gene knockout
To generate CRISPR-Cas9-meditaed PLUM knockout in EpiSCs, gRNAs targeting the uORF in the 5′ UTR of Lin28b were designed on the website (http://crispor.tefor.net/) and cloned into pX459 or lentiCRISPR v2 vectors. For targeting, 5 × 104 EpiSCs were transfected with the plasmids using Lipofectamine Stem (Thermo fisher). 48 h after transfection, EpiSCs were selected with puromycin for 3 days and picked and verified by genomic PCR sequencing.
Implantation localization
7-8 weeks PLUM-deficient and wild-type female mice were superovulated by injection of PMSG (pregnant mare serum gonadotropin, AibeiBio) and human chorionic gonadotropin (hCG, AibeiBio) after 48 h. The day a plug was found designated as E0.5. Implantation sites E6.5 were localized by intravenous injection of 1% Chicago Sky Blue 6B (HARVEYBIO).
Embryo transfer experiments
WT and PLUM KO 2-cell embryos were cultured in BSA-free KSOM medium until 4.0 dpc. Four hours prior to transfer, embryos were pre-equilibrated in fresh BSA-free KSOM medium and then surgically transferred into the uterine horns of pseudopregnant females at 2.5 dpc. Recipient uterine deciduae were collected at 6.5 dpc, and implantation sites were visualized by intravenous injection of 1% Chicago Sky Blue 6B.
Flow cytometry
Cells were dissociated into single cell with 0.25% Trypsin-EDTA and collected by centrifugation. The cell pellet was resuspended with PBS containing 0.1% BSA and filtered with cell strainer (BD biosciences) to remove large clumps of cells. The cells were then analyzed with an Accuri C6 flow cytometer (BD biosciences). Data analysis was performed using FlowJo v10.0.7.
RT-qPCR
Total RNA was extracted with TRIzol (ThermoFisher), and 2 μg RNA was used to generate complementary DNA using HiScript II reverse transcriptase (Vazyme). Transcriptional levels of genes were determined by ChamQ SYBR qPCR Master Mix (Vazyme) and analyzed with a CFX-96 Real-Time system (Bio-Rad). The primers used for RT-qPCR is provided in Supplementary Table 1.
Immunofluorescene
Cells on glass dishes coated with 0.1% gelatin or FBS were fixed with 4% paraformaldehyde for 30 min, permeabilized in 0.5% Trition X-100 for 30 min, and then blocked in 10% FBS for 2 h at room temperature. The cells were incubated with primary antibodies overnight at 4 °C and then with secondary antibodies for 1 hour at room temperature. Next, the cells were stained with DAPI (Sigma) for 10 min and analyzed using a Zeiss LSM 800 confocal microscope. The following primary antibodies were used: anti-Flag (Sigma, F1804, 1:500), anti-Nanog (Bethyl, A300-397A, 1:500), anti-Rex1 (Abcam, ab28141, 1:500) and anti-Dcp1α (Abcam, ab183709, 1:500). The secondary antibodies used were goat anti-rabbit DyLight 488 (Abcam, ab96883, 1:1000) and goat anti-rabbit DyLight 594 (Abcam, ab96885, 1:1000)
Western blot
Whole-cell extracts were obtained using radioimmunoprecipitation (RIPA) buffer (Beyotime) supplemented with protease inhibitor cocktail (Selleck). The samples were separated by 10–12.5% SDS-PAGE, transferred to polyvinylidene difluoride (PVDF) membrane (Millipore), and incubated with antibodies. The following primary antibodies were used: anti-GAPDH (Bioworld, AP2063, 1:5000), anti-Actin (Huabio, ET1702-52, 1:5000), anti-Flag (Sigma, F1804, 1:1000), anti-Lin28b (Cell signaling, 5422S, 1:1000), anti-L1td1 (Proteintech, 21528, 1:1000) and anti-Tfcp2l1 (Proteintech, 14248, 1:1000). The custom polyclonal antibody against PLUM was produced by GenScript (Nanjing, China) using recombinant protein as antigen. The secondary antibodies used were goat anti-rabbit (Kangchen, KC-RB-035, 1:5000) and goat anti-mouse (Kangchen, KC-MM-035, 1:5000). Uncropped and unprocessed scans are provided in the source data file.
Immunoprecipitaion
Cells were lysed in NP-40 lysis buffer (Beyotime) supplemented with protease inhibitor cocktail for 30 min at 4 °C as described previously59. Antibodies anti-Flag (Sigma, F1804, 1:200), anti-L1td1 (Proteintech, 21528, 1:200), normal mouse IgG (Millipore, 12-371, 1:200) or normal rabbit IgG (Cell signaling, 2729, 1:200) were linked to Dynabeads with protein A and G (Thermo fisher) for 4 h at 4 °C in lysis buffer. Lysates were incubated with the washed antibody-linked Dynabeads overnight at 4 °C. After immunoprecipitation, the beads were washed with lysis buffer five times and boiled in SDS buffer for 10 min. The eluents were analyzed by western blot or mass spectrometry.
RNA immunoprecipitaion
RNA immunoprecipitation (RIP) assay was performed with the RNA Immunoprecipitation Kit (Geneseed) following the manufacturer’s instructions. Briefly, cells were lysed by RIP lysis buffer supplemented with protease inhibitor and RNase inhibitor, and incubated at 4 °C with antibody-conjugated Dynabeads overnight. After rigorous washes with RIP lysis buffer, co-precipitated RNA was purified and analyzed by RT-qPCR. The following antibodies were used: anti-L1td1 (Proteintech, 21528, 1:200), anti-HA (Proteintech, 51064, 1:200) and normal rabbit IgG (Cell signaling, 2729, 1:200).
RNA stability assay
Cells were treated with 10µg/ml Actinomycin D (Selleck) for the indicated time, and RNA was extracted at the indicated time points for further RT-qPCR analysis. RNA stability was estimated by the relative amount of target mRNA remaining after inhibiting mRNA transcription with Actinomycin D.
RNA-seq
Total RNA was extracted with TRIzol. Libraries were constructed with the Illumina TruSeq RNA Sample Prep kit following the manufacturer’s instructions. The experiments were performed by the Annoroad Gene Technology (Beijing, China). The clean data were analyzed with STAR software60 and the differentially expressed genes were identified by DESeq261. The Gene Ontology analysis was performed using the DAVID database (https://david.ncifcrf.gov). The P-values represent the modified Fisher’s exact corrected EASE score.
eCLIP-seq and data analysis
A total of 1 × 107 EpiSCs were cross-linked with 254 nm UV light at 400 mJ/cm2, lysed and partial digested with RNase I (Thermo fisher). The lysates were incubated with 10 µg anti-L1td1 (Proteintech, 21528) conjugated to Dynabeads with protein A and G for 4 h, followed by washing, end repair, and 3′ adapter ligation. The protein-RNA complexes were eluted from the beads, resolved by denaturing gel electrophoresis, and transferred onto a nitrocellulose membrane. Library preparation was performed by DIATRE Biotechnology (Shanghai, China) as in the original protocol42. The data were first trimmed adapter by Cutadapt and then mapped against the full mouse genome (mm10) with STAR. PCR duplication removal was performed by Picard (http://broadinstitute.github.io/picard/) and only the read 2 were kept by SAMtools for performing the following clusters identification using CLIPper (https://github.com/YeoLab/clipper). The peak-level input normalization was performed by overlapping the eCLIP reads of the sample and the paired SMInput sample to the CLIPper-identified peaks to get the count number separately to calculate the fold-enrichment, and the enrichment p-value was further obtained using Chi-Square or Fisher Exact Test.
Ribo-seq and data analysis
Ribo-seq were performed as previously described29. Briefly, cells were treated with 100 μg/ml cycloheximide (Sigma) at 37 °C for 10 min and lysed on ice, followed by digestion with DNase I and RNase I. Ribosomes were purified using illustra MicroSpin S400 HR columns (GE healthcare), and the ribosome protected RNA fragments (RPFs) were isolated with the RNA Clean and Concentrator-25 Kit (Zymo) and further purified using magnet beads (Vazyme). Ribosomal profiling libraries were constructed using NEBNext Multiple Small RNA Library Prep Set for Illumina following the manufacturer’s instructions. The 140–160 bp size PCR products were enriched to generate a cDNA library and sequenced using Illumina HiSeqTM X10 by Gene Denovo Biotechnology (Guangzhou, China). After pre-processing, the Cutadapt program was used to trim the adapter in the reads. Next, sequencing reads originating from rRNAs were identified and discarded by aligning the reads to rRNA sequences of mouse mm10 using Bowtie2). The remaining reads were then mapped to the mm10 genome using STAR, and the ORF identification was performed by the RiboCode software.
Translational efficiency (TE) analysis
Raw read counts for RNA-seq and Ribo-seq datasets were quantified using featureCounts, and normalization of raw counts was performed using DESeq2. Count matrices were imported into DESeq2 using DESeqDataSetFromMatrix, and size factors were estimated with estimateSizeFactors to account for library size differences and sequencing depth. Normalized counts were extracted using the counts function with the normalized = TRUE option. TE for each ORF was calculated as the ratio of Ribo-seq normalized counts to RNA-seq normalized counts (TE = Ribo-seq normalized counts / RNA-seq normalized counts).
Mitochondrial membrane potential (∆ψm) analysis
Mouse 6.5 dpc (day post-coitum) embryos were dissected from the decidua. For ∆ψm analysis, embryos were seeded on glass dishes and stained with 25 nm TMRM for 30 min at 37 °C, followed by analysis using a Zeiss LSM 800 confocal microscope. For ∆ψm quantification via flow cytometry, epiblast tissues dissected from embryos were dissociated into single cell with Accutase and then neutralized with DMEM. Cells were stained with 25 nm TMRM for 30 min, resuspended in DPBS, and analyzed with an Accuri C6 flow cytometer (BD biosciences). Data analysis was performed using FlowJo v10.0.7.
Measurement of ATP levels
ATP was measured with the ENLITEN ATP assay kit according to the manufacturer’s instructions (Promega, FF2000). Briefly, ATP was extracted from embryos using 2.5% TCA, then neutralized and diluted to a final concentration of 0.1% using 0.1 M Tris-acetate buffer (pH 7.75). After the addition of an equal volume of rL/L Reagent from the kit, luminescence was measured as readout for ATP levels.
P-body purification
P-body purification was performed as previously described44, with some modifications. Briefly, LSM14A-GFP-expressing cell pellets were lysed on ice for 20 min using a lysis buffer composed of 50 mM Tris (pH 7.4), 1 mM EDTA, 150 mM NaCl, and 0.2% Triton X-100, supplemented with 65 µ/ml of RNaseOut ribonuclease inhibitor (Promega) and protease inhibitor cocktail (Selleck). Following lysis, the samples were centrifuged at 200 × g for 5 min at 4 °C to remove nuclei. To eliminate residual DNA, the supernatant was incubated for 30 min with 4 µ/ml of DNase I (Vazyme). The lysates were then centrifuged at 10,000 × g for 7 minutes at 4 °C, and the resulting pellets were resuspended in 40 μl of lysis buffer containing an additional 80 U of RNaseOut (Promega) to isolate the cytoplasmic fraction. P-bodies were subsequently sorted from this fraction using a MoFlo Astrios flow cytometer, pelleted by centrifugation at 10,000 × g for 7 min at 4 °C, and stored at − 80 °C. An aliquot of the corresponding pre-sorted cytoplasmic fraction was also retained for further analysis.
Statistical analyses
The statistical tests used in this study are indicated in the respective figure legends. In general, data were analyzed by student unpaired t test to determine statistically significant effects (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive in the National Genomics Data Center, China National Center for Bioinformation under CRA014487 and CRA030203. The raw LC-MS data has been deposited to the ProteomeXchange Consortium under dataset identifier PXD065363. The remaining data are available with the article, supplementary information and source data file. Source data are provided in this paper.
References
Weinberger, L., Ayyash, M., Novershtern, N. & Hanna, J. H. Dynamic stem cell states: naive to primed pluripotency in rodents and humans. Nat. Rev. Mol. Cell Biol. 17, 155–169 (2016).
Evans, M. J. & Kaufman, M. H. Establishment in culture of pluripotential cells from mouse embryos. Nature 292, 154–156 (1981).
Tesar, P. J. et al. New cell lines from mouse epiblast share defining features with human embryonic stem cells. Nature 448, 196–199 (2007).
Li, M., Liu, G. H. & Belmonte, I. zpisua J. C. Navigating the epigenetic landscape of pluripotent stem cells. Nat. Rev. Mol. Cell Biol. 13, 524–535 (2012).
Chen, Y. et al. Generation of cynomolgus monkey chimeric fetuses using embryonic stem cells. Cell Stem Cell 17, 116–124 (2015).
Kobayashi, T. et al. Generation of rat pancreas in mouse by interspecific blastocyst injection of pluripotent stem cells. Cell 142, 787–799 (2010).
Schmitz, D. A. & Wu, J. Hidden totipotency in naive human pluripotent stem cell cultures. Life Med. 1, 250–253 (2022).
Li, L., Zheng, P. & Dean, J. Maternal control of early mouse development. Development 137, 859–870 (2010).
Iyyappan, R. et al. The translational oscillation in oocyte and early embryo development. Nucleic Acids Res. 51, 12076–12091 (2023).
Zhang, C., Wang, M., Li, Y. & Zhang, Y. Profiling and functional characterization of maternal mRNA translation during mouse maternal-to-zygotic transition. Sci. Adv. 8, eabj3967 (2022).
Atlasi, Y. et al. The translational landscape of ground state pluripotency. Nat. Commun. 11, 1617 (2020).
Schwanhausser, B. et al. Global quantification of mammalian gene expression control. Nature 473, 337–342 (2011).
Saba, J. A., Liakath-Ali, K., Green, R. & Watt, F. M. Translational control of stem cell function. Nat. Rev. Mol. Cell Biol. 22, 671–690 (2021).
Biffo, S., Ruggero, D. & Santoro, M. M. The crosstalk between metabolism and translation. Cell Metab. 36, 1945–1962 (2024).
Sampath, P. et al. A hierarchical network controls protein translation during murine embryonic stem cell self-renewal and differentiation. Cell Stem Cell 2, 448–460 (2008).
Corsini, N. S. et al. Coordinated Control of mRNA and rRNA Processing Controls Embryonic Stem Cell Pluripotency and Differentiation. Cell Stem Cell 22, 543–558 (2018).
Mudge, J. M. et al. Standardized annotation of translated open reading frames. Nat. Biotechnol. 40, 994–999 (2022).
Chen, J. et al. Pervasive functional translation of noncanonical human open reading frames. Science 367, 1140–1146 (2020).
Ambros, V. & Horvitz, H. R. Heterochronic mutants of the nematode Caenorhabditis elegans. Science 226, 409–416 (1984).
Shyh-Chang, N. & Daley, G. Q. Lin28: primal regulator of growth and metabolism in stem cells. Cell Stem Cell 12, 395–406 (2013).
Balzer, E. & Moss, E. G. Localization of the developmental timing regulator Lin28 to mRNP complexes, P-bodies and stress granules. RNA Biol. 4, 16–25 (2007).
Sun, Z. et al. LIN28 coordinately promotes nucleolar/ribosomal functions and represses the 2C-like transcriptional program in pluripotent stem cells. Protein Cell 13, 490–512 (2022).
Tsialikas, J. & Romer-Seibert, J. LIN28: roles and regulation in development and beyond. Development 142, 2397–2404 (2015).
Zhang, J. et al. LIN28 Regulates stem cell metabolism and conversion to primed pluripotency. Cell Stem Cell 19, 66–80 (2016).
Yu, J. et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 318, 1917–1920 (2007).
Shyh-Chang, N. et al. Lin28 enhances tissue repair by reprogramming cellular metabolism. Cell 155, 778–792 (2013).
Zhou, J., Ng, S. B. & Chng, W. J. LIN28/LIN28B: an emerging oncogenic driver in cancer stem cells. Int. J. Biochem. Cell Biol. 45, 973–978 (2013).
Viswanathan, S. R. et al. Lin28 promotes transformation and is associated with advanced human malignancies. Nat. Genet. 41, 843–848 (2009).
Ingolia, N. T., Ghaemmaghami, S., Newman, J. R. & Weissman, J. S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324, 218–223 (2009).
Xiao, Z. et al. De novo annotation and characterization of the translatome with ribosome profiling data. Nucleic Acids Res. 46, e61 (2018).
Zhang, H., Wang, Y. & Lu, J. Function and evolution of upstream ORFs in eukaryotes. Trends Biochem. Sci. 44, 782–794 (2019).
Yeom, Y. I. et al. Germline regulatory element of Oct-4 specific for the totipotent cycle of embryonal cells. Development 122, 881–894 (1996).
Yu, S. et al. BMP4 resets mouse epiblast stem cells to naive pluripotency through ZBTB7A/B-mediated chromatin remodelling. Nat. Cell Biol. 22, 651–662 (2020).
Yeo, J. C. et al. Klf2 is an essential factor that sustains ground state pluripotency. Cell Stem Cell 14, 864–872 (2014).
Adachi, K. et al. Esrrb Unlocks Silenced Enhancers for Reprogramming to Naive Pluripotency. Cell Stem Cell 23, 266–275 (2018).
Mellacheruvu, D. et al. The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat. Methods 10, 730–736 (2013).
Wong, R. C. et al. L1TD1 is a marker for undifferentiated human embryonic stem cells. PLoS ONE 6, e19355 (2011).
Emani, M. R. et al. The L1TD1 protein interactome reveals the importance of post-transcriptional regulation in human pluripotency. Stem Cell Rep. 4, 519–528 (2015).
Narva, E. et al. RNA-binding protein L1TD1 interacts with LIN28 via RNA and is required for human embryonic stem cell self-renewal and cancer cell proliferation. Stem Cells 30, 452–460 (2012).
Corley, M., Burns, M. C. & Yeo, G. W. How RNA-Binding Proteins Interact with RNA: Molecules and Mechanisms. Mol. Cell 78, 9–29 (2020).
Gao, Y., Li, X., Li, P. & Lin, Y. A brief guideline for studies of phase-separated biomolecular condensates. Nat. Chem. Biol. 18, 1307–1318 (2022).
Van Nostrand, E. L. et al. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat. Methods 13, 508–514 (2016).
Yan, H. et al. Fatty acid oxidation is required for embryonic stem cell survival during metabolic stress. EMBO Rep. 22, e52122 (2021).
Hubstenberger, A. et al. P-Body purification reveals the condensation of repressed mRNA regulons. Mol. Cell 68, 144–157.e145 (2017).
Kodali, S. et al. RNA sequestration in P-bodies sustains myeloid leukaemia. Nat. Cell Biol. 26, 1745–1758 (2024).
Safieddine, A. et al. Cell-cycle-dependent mRNA localization in P-bodies. Mol. Cell 84, 4191–4208 (2024).
Wang, C. et al. Reprogramming of H3K9me3-dependent heterochromatin during mammalian embryo development. Nat. Cell Biol. 20, 620–631 (2018).
Zhang, J. et al. Metabolism in pluripotent stem cells and early mammalian development. Cell Metab. 27, 332–338 (2018).
Sharpley, M. S., Chi, F., Hoeve, J. T. & Banerjee, U. Metabolic plasticity drives development during mammalian embryogenesis. Dev. Cell 56, 2329–2347 (2021).
Todorova, P. K. et al. Amino acid intake strategies define pluripotent cell states. Nat. Metab. 6, 127–140 (2024).
Rais, Y. et al. Deterministic direct reprogramming of somatic cells to pluripotency. Nature 502, 65–70 (2013).
Di Stefano, B. et al. C/EBPalpha poises B cells for rapid reprogramming into induced pluripotent stem cells. Nature 506, 235–239 (2014).
Mor, N. et al. Neutralizing Gatad2a-Chd4-Mbd3/NuRD complex facilitates deterministic induction of naive pluripotency. Cell Stem Cell 23, 412–425 (2018).
Quenby, S. et al. Miscarriage matters: the epidemiological, physical, psychological, and economic costs of early pregnancy loss. Lancet 397, 1658–1667 (2021).
Li, L. et al. Mutations in TUBA4A lead to human zygotic arrest and early developmental failure. Life Med. 2, https://doi.org/10.1093/lifemedi/lnad032 (2023).
Li, L. et al. Glis1 facilitates induction of pluripotency via an epigenome-metabolome-epigenome signalling cascade. Nat. Metab. 2, 882–892 (2020).
Wu, Y. et al. Srebp-1 interacts with c-Myc to enhance somatic cell reprogramming. Stem Cells 34, 83–92 (2016).
Wu, Y. et al. Plin2-mediated lipid droplet mobilization accelerates exit from pluripotency by lipidomic remodeling and histone acetylation. Cell Death Differ. 29, 2316–2331 (2022).
Wu, Y. et al. Phospholipid remodeling is critical for stem cell pluripotency by facilitating mesenchymal-to-epithelial transition. Sci. Adv. 5, eaax7525 (2019).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Acknowledgements
This work was financially supported by the National Key Research and Development Program of China (2024YFA0916400), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB0480000), the National Key Research and Development Program of China (2023YFE0210100, 2022YFE0210100, 2024YFA1802302, 2022YFA1103800, 2022YFE0210600), the National Natural Science Foundation projects of China (32025010, 32488301, 92254301, 92357302, 92157202, 32241002, 32261160376, 32100619, 32170747, 32322022, 32370782, 32371007, 32300608, 32300620, 32471358, 32461160288, 32500669, 82505003, 32500691, 32570905), NSFC/RGC Joint Grant Scheme 2022/2023 (N_CUHK 428/22), Major Project of Guangzhou National Laboratory (GZNL2024A03006, GZNL2024B01003) the Key Research Program, CAS (ZDBS-ZRKJZ-TLC003), CAS Project for Young Scientists in Basic Research (YSBR-075), the International Partnership Program of Chinese Academy of Sciences(188GJHZ2024048GC), Guangdong Province Science and Technology Program (2023B0303000023, 2023A1515030231, 2022A1515110493, 2023B1212060050, 2021B1515020096, 2022A1515110951, 2023B1212120009, 2024A1515010782, 2024B1515040020, 2024A1515030120, 2023TQ07A024, 2024A1515012839, 2022B1212050004, 2024TQ08A297, 2024TQ08A035), Guangzhou Science and Technology Program (202206060002, 2023A04J0414, 2025A04J2106, 2025A04J7110, 2025A04J5485, 2023A04J0863, 2023A04J0727), Shandong Provincial Natural Science Foundation (ZR2025QC1639), Health@InnoHK funding support from the Innovation Technology Commission of the Hong Kong SAR, CAS Youth Innovation Promotion Association (to Y.W. and K.C.), Major Research Project (GIBHMRP25-01) and Basic Research Project of Guangzhou Institutes of Biomedicine and Health, Chinese Academy of Sciences. We acknowledge and thank all members of Dr. Xingguo Liu’ lab for helpful discussion, Dr. Jing Liu for providing pLV-Esrrb and pLV-Klf2 plasmids, Haozhao Liang for technical support in the immunohistochemistry experiments, the core facility and animal center in GIBH for technical help and support.
Author information
Authors and Affiliations
Contributions
Conceptualization: X.L., Y.W., Z.H., and M.Z. Supervision: X.L. Methodology: X.L., Y.W., Z.H., Y.H., Z.R., and M.Z. Investigation: Z.H., Y.W., Y.H., Y. Liu., Y.Li., W.L., Z.R., Y.D., J.Z., Q.W., W.W., G.L., D.Q., and W-Y.C. Visualization: Y.W., Z.H., and Y.H. Funding acquisition: X.L. and Y.W. Data curation: Y.W., Z.H., and Y.H. Formal analysis: Z.H., Y.W., and Y.H. Software: Y.H. Resource: K.C., L.L., G.X., Z.L., and Y.Z. Writing-original draft: X.L., Y.W., and Z.H. Writing-review & editing: X.L., Y.W., Z.H., and Y.H.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hao, Z., Wu, Y., Huang, Y. et al. Microprotein PLUM encoded by Lin28b uORF is a cytoplasmic determinant of pluripotency and embryonic development. Nat Commun 16, 10324 (2025). https://doi.org/10.1038/s41467-025-66297-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-66297-4
