
Abstract
Renal tubulointerstitial abnormalities predict diabetic kidney disease (DKD) progression, and targeting them may prevent DKD. Insulin-like growth factor binding-protein 7 (IGFBP7) is expressed in renal tubular cells and is elevated in both blood and urine during the early stages of human diabetes, serving as a predictor of the rate of disease progression. We showed that tubule- and glomerular-specific IGFBP7 promotes DKD, with tubular-derived IGFBP7 disrupting the renal microenvironment. IGFBP7 impairs mitochondrial bioenergetics in tubular cells, causing lipid accumulation, cell cycle arrest, interstitial inflammation, fibrosis, and glomerulosclerosis. These findings were substantiated by transgenic overexpression and the specific deletion of IGFBP7 in type 1/2 DKD mice. Mechanistically, IGFBP7 interacts with STAT3, promoting its acetylation/dimerization and downregulating mitochondrial bioenergetics. Our study identified levomefolic acid as a novel inhibitor of IGFBP7 and demonstrated its efficacy in mitigating the progression of DKD. Here we showed IGFBP7 is a promising therapeutic target for DKD.
Similar content being viewed by others


Introduction
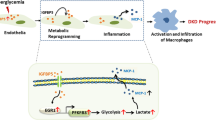
Diabetic kidney disease (DKD) is the leading cause of end-stage kidney disease, affecting approximately 40% of individuals worldwide1,2. Its progression involves multifaceted mechanisms, including immune activation, pro-inflammatory cytokine release, chemokine dysregulation, and renal cell dysfunction in tubular epithelial cells (TECs), podocytes, and endothelial cells2,3. Recent spatial transcriptomic analyses of human kidneys have delineated distinct cellular microenvironments (e.g., tubular, glomerular, and immune-associated regions), highlighting the critical role of cell-specific interactions in DKD pathogenesis4. These advances emphasize the complexity of the molecular pathways driving DKD and underscore the need for targeted therapeutic strategies.
The cellular origins of myofibroblasts in kidney pathology remain debated, with proposed mechanisms including phenotypic transitions from epithelial, endothelial, or immune cells, as well as the recruitment of bone marrow-derived precursors5,6,7,8. These activated mesenchymal cells exhibit characteristic markers such as α-smooth muscle actin and participate in tissue remodeling processes8,9. Emerging evidence implicates multiple signaling pathways—including TGF-β1, Notch, Wnt, and Hedgehog cascades—in driving metabolic dysregulation and cellular reprogramming in diabetic kidneys10,11,12,13. These molecular alterations promote pathological extracellular matrix alterations through mechanisms involving cell cycle regulation and metabolic adaptations14,15. Interestingly, insulin-like growth factor-binding protein 7 (IGFBP7), a ubiquitously expressed 29-kDa secretory protein16,17, functions as a senescence-associated secretory phenotype component with pleiotropic biological effects, particularly through the regulation of G1/S cell cycle progression18,19,20. Clinically, urinary IGFBP7 combined with TIMP-2 has demonstrated predictive value for acute kidney injury risk stratification21,22. Beyond renal pathology, this multifunctional protein shows diagnostic potential for hepatic disorders, where its expression correlates with inflammation and fibrosis in non-alcoholic fatty liver disease23. Emerging evidence suggests that circulating IGFBP7 is a sensitive indicator of early tubular pathology and prognostic marker of proteinuria-associated renal decline in patients with diabetes24,25. These collective observations support the hypothesis that IGFBP7-mediated cellular mechanisms may critically influence DKD progression, warranting a systematic investigation of its pathophysiological role in DKD pathogenesis.
Metabolic reprogramming, a hallmark of cellular adaptation to kidney injury, manifests through a dynamic shift in energy substrate utilization across renal cell populations26,27,28,29. While the regulatory networks driving these metabolic alterations and their contribution to mesenchymal cell expansion remain incompletely characterized, accumulating evidence implicates impaired fatty acid oxidation (FAO) in injured epithelial cells as a critical mediator of pathological tissue remodeling26,30,31,32. This metabolic dysregulation represents a key mechanistic node in DKD progression, necessitating deeper exploration of its therapeutic implications. Intriguingly, evolutionary studies in Drosophila have revealed that tumor-derived IGFBP7 reprograms systemic metabolism by sequestering glucose from adipose tissues to fuel tumor growth and induce host energy depletion33,34. In contrast, contemporary murine models have demonstrated that macrophage-specific IGFBP7 silencing ameliorates hyperglycemia and lipid accumulation in obesity-associated hepatic injury35, highlighting its context-dependent metabolic roles. Our work expands this paradigm to DKD by identifying tubular- and podocyte-derived IGFBP7 as central orchestrators of renal microenvironment dysregulation. Through high-throughput screening of 24,400 compounds, we discovered that levomefolic acid (LA), a clinically utilized nutrient adjuvant, selectively inhibits IGFBP7-bound STAT3 signaling. This intervention normalized aberrant FAO and attenuated cellular injury in both in vitro and preclinical models, suggesting a druggable mechanism. Collectively, these findings suggest that renal IGFBP7 is a master regulator of metabolic maladaptation in DKD patients. The efficacy of LA in counteracting IGFBP7-driven pathology underscores the translational potential of targeting this axis and offers a precise therapeutic strategy for the management of DKD.
Results
Renal H3K4me3-dependent IGFBP7 expression and secretion is higher in multiple mouse models of diabetes and in DKD patients
Western blot analyses demonstrated that IGFBP7 was expressed in various organs of adult mice, including the kidneys, liver, colon, lungs, and muscle (Fig. 1A). Clinical investigations revealed substantial upregulation of IGFBP7 expression in renal biopsies from DKD patients compared with that in controls (Fig. 1B). To further explore the relationship of IGFBP7 in the progression of DKD, human serum samples were collected for detection. The basic characteristics and laboratory diagnostic data of the 92 participants are summarized in Supplementary Table 1. Among these participants, there were no significant differences in age and gender across groups (p greater than 0.05). In contrast, we detected significant disparities between the case and control groups in terms of participants’ BMI, fasting plasma glucose, fasting insulin (FI), and homeostasis model assessment of insulin resistance, as well as renal function parameters including creatinine (Cr), blood urea nitrogen (BUN), and estimated glomerular filtration rate (eGFR) (all p < 0.05); however, no differences in blood pressure (systolic blood pressure and diastolic blood pressure) were observed between the three groups. These findings are consistent with the typical clinical features distinguishing healthy individuals from those with DKD. Importantly, correlation analysis showed that serum IGFBP7 levels were significantly positively correlated with DKD progression and urinary protein excretion (UPE), while significantly negatively correlated with eGFR (Fig. 1C and Supplementary Fig. 1A). ROC curve analysis showed that the area under curve values of IGFBP7 related to eGFR and UPE were both 0.98 (Supplementary Fig. 1B). Taken together, the results suggest that IGFBP7 has a strong ability to identify or correlate these key indicators of DKD (Fig. 1C and Supplementary Fig. 1A, B). To establish an experimental model of DKD, we used mice with streptozotocin (STZ)-induced type 1 diabetes mellitus over a 24-week observation period. Immunofluorescence analysis localized IGFBP7 expression predominantly in the renal tubular compartments, including the proximal tubules, distal tubules, and collecting ducts, in STZ-treated animals (Fig. 1D). Notably, both serum and urinary IGFBP7 concentrations showed strong positive correlations with albuminuria in this model (Fig. 1E, F). Parallel findings were observed in a high-fat diet (HFD)/STZ-induced type 2 diabetes model, in which renal IGFBP7 expression (Fig. 1G), serum/urinary IGFBP7 levels, and albuminuria exhibited significant intercorrelations (Fig. 1H, I). These observations were further validated in db/db mice, a genetic model of type 2 diabetes, confirming the consistent elevation in IGFBP7 levels (Fig. 1J–L and Supplementary Fig. 1C).

A Western blot of IGFBP7 expression in mouse tissues (n = 4, one-way ANOVA with Tukey’s multiple comparisons test). The kidney shows relatively higher IGFBP7 expression compared to other tested tissues (not indicating high basal expression in normal kidneys). B Representative IF images and quantification of LTL+IGFBP7+ expression in DKD renal tissues and normal kidney (n = 6, Spearman correlation analysis, two-tailed). C Correlation analysis of serum IGFBP7 levels and eGFR changes in patients with diabetic kidney disease (restricted cubic spline plot). D IGFBP7 expression in renal tubules in STZ-induced type 1 DKD mice kidney; LTL (proximal), PNA (distal), DBA (collecting ducts). E, F Correlation analysis of serum (E) and urinary (F) IGFBP7 level with urinary albumin/creatinine in STZ-treated type 1 DKD mice (n = 12, Spearman correlation analysis, two-tailed). G IGFBP7 expression in renal tubules in HFD/STZ-induced type 2 DKD mice kidney (n = 9, Spearman correlation analysis, two-tailed). H, I Correlation analysis of serum (H) and urinary (I) IGFBP7 level with urinary albumin/creatinine in HFD/STZ-induced type 2 DKD mice (Control: n = 7; HFD/STZ: n = 8, Spearman correlation analysis, two-tailed). J Immunofluorescence of IGFBP7 in db/db type 2 mice kidney. K, L Serum (K) and urinary (L) IGFBP7 levels in db/db mice and db/m mice (n = 8, two-tailed unpaired Student’s t test). M Relative western blot analysis and quantification of IGFBP7 expression levels in mTEC, MPC5, and SV40 after stimulation with different concentrations of glucose (n = 3). N ELISA of IGFBP7 secretion levels in mTEC, MPC5, and SV40 supernatants with 30 mM glucose (n = 3). O Western blot analysis of H3K4Me1, H3K4Me3, and H3K27Ac in HG-treated HK2 cells (n = 4, one-way ANOVA with Tukey’s multiple comparisons test). P Western blot analysis of IGFBP7 and H3K4Me3 in HK2 cells (n = 4, one-way ANOVA with Tukey’s multiple comparisons test). Q ChIP assay for the binding of H3K4Me3 to IGFBP7 (n = 4, one-way ANOVA with Tukey’s multiple comparisons test). R Schematic diagram of MLL1 and WDR5-mediated H3K4Me3 regulation of IGFBP7 transcription (by FigDraw 2.0). Data represent the mean ± SEM. NC negative control, STZ streptozotocin, HFD high-fat diet, HG high glucose (30 mmol/L), mTEC mouse tubular epithelial cells, MPC5 mouse podocyte clone-5, SV40 mouse glomerular mesangial cells. Source data are provided as a Source Data file.
Given the established role of glomerular pathology in DKD progression2,36, we conducted in vitro investigations using mouse renal TECs (mTECs), podocytes (MPC5), and mesangial cells (SV40). High-glucose conditions induced dose- and time-dependent upregulation of IGFBP7 expression across all cell types, with the maximal induction observed in mTECs (Fig. 1M and Supplementary Fig. 1D). This finding was corroborated using the human renal TEC line HK2 (Supplementary Fig. 1E). Interestingly, we also found that IGFBP7 expression in renal TECs and podocytes was mainly due to glucotoxicity rather than osmotic stress (Supplementary Fig. 1F, G). Notably, we also observed alterations in the secretion levels of IGFBP7, and ELISA results indicated a significant increase in IGFBP7 levels in the mTEC supernatant following high glucose (HG) stimulation (Fig. 1N). Recent studies have identified palmitic acid as a significant pathogenic factor in DKD. Consistent with this finding, our results showed a marked increase in IGFBP7 expression in mTECs after palmitic acid stimulation (Supplementary Fig. 1H).
To investigate why IGFBP7 expression was elevated in DKD, we analyzed its promoter using the UCSC Genome Bioinformatics platform (http://genome.ucsc.edu/). The promoter showed high H3K4 methylation, indicating a role of chromatin methylation in regulation (Supplementary Fig. 2A). The H3K4me3 methyltransferase MLL1, which requires WDR5 for activation, was significantly upregulated in hyperglycemic HK2 cells (Fig. 1O). Pharmacological inhibition of the MLL1/WDR5 interaction using MM-102 and OICR-9429 resulted in dose- and time-dependent reductions in both IGFBP7 expression and global H3K4Me3 levels (Fig. 1P and Supplementary Fig. 2B–E). Silencing MLL1 also alleviated the high glucose-induced increase in IGFBP7 expression (Supplementary Fig. 2F). Chromatin immunoprecipitation (IP) assays demonstrated glucose-dependent enrichment of H3K4Me3 at the IGFBP7 promoter (Fig. 1Q, R). In addition, we have achieved targeted epigenetic modification inhibition of the promoter region of IGFBP7 through single guide RNAs (sgRNAs) associated with dCas9-KRAB (Supplementary Fig. 2G). Notably, when the promoter region is deleted, the expression of IGFBP7 is strongly inhibited, which established an epigenetic mechanism for the upregulation of IGFBP7 in DKD (Supplementary Fig. 2H). Our findings indicate a strong positive correlation between renal IGFBP7 and the DKD process, primarily involving TEC expression and secretion.
Collectively, these findings established IGFBP7 as a responsive biomarker with a strong correlation to albuminuria across multiple experimental models of DKD. These data further identified TECs as the primary source of IGFBP7 production and implicated MLL1/WDR5-mediated histone modifications in its pathological overexpression during DKD progression.
Renal IGFBP7 promotes DKD progression in mouse models
To investigate the role of IGFBP7 in DKD, we generated IGFBP7 knockout (KO) mice and established a type 1 DKD model through STZ administration (Supplementary Fig. 3A). Both wild-type (WT) and IGFBP7 KO mice developed sustained hyperglycemia within 1 week post-injection (Supplementary Fig. 3B). Although diabetic WT mice exhibited significant albuminuria, IGFBP7 deficiency markedly attenuated urinary albumin excretion (Supplementary Fig. 3C). The KO mice also demonstrated attenuation of STZ-induced elevation in serum creatinine levels, although no significant difference was observed in BUN levels between the groups (Supplementary Fig. 3D, E). Histopathological evaluation demonstrated that genetic ablation of IGFBP7 significantly attenuated diabetic renal pathology, including renal tubular dilation, interstitial fibrosis, mitochondrial structural abnormalities, and glomerulosclerosis (Supplementary Fig. 3F–I). Ultrastructural analysis revealed podocyte foot process integrity in diabetic IGFBP7 KO mice, in contrast to the characteristic foot process effacement observed in DKD mice (Supplementary Fig. 3J).
We further established a type 2 DKD model through unilateral nephrectomy combined with a HFD and low-dose STZ administration in IGFBP7 KO mice (Supplementary Fig. 3K)37. Systemic IGFBP7 deletion resulted in a modest reduction in body weight compared with the WT controls (Supplementary Fig. 3L). Intraperitoneal glucose tolerance tests and insulin tolerance tests confirmed the development of impaired glucose tolerance and insulin resistance following HFD feeding, validating the successful induction of type 2 diabetes mellitus (Supplementary Fig. 3M–O). Immunofluorescence analysis revealed significantly attenuated HFD/STZ-induced IGFBP7 expression in KO mice compared with diabetic controls (Supplementary Fig. 3P). Histopathological assessment demonstrated that IGFBP7 deficiency substantially ameliorated renal pathology, with Masson’s trichrome staining showing reduced interstitial fibrosis (Supplementary Fig. 3Q) and electron microscopy revealing a preserved tubular ultrastructure (Supplementary Fig. 3R). Consistent with these findings, glomerular histological analysis indicated mitigation of mesangial matrix expansion and basement membrane thickening in KO mice (Supplementary Fig. 3S, T).
Complementary gain-of-function experiments were conducted using IGFBP7-overexpressing transgenic mice subjected to STZ-induced diabetes (Supplementary Fig. 4A–C). These animals exhibited exacerbated renal injury markers, including amplified fibrosis, mitochondrial structural abnormalities, inflammatory infiltration, and dysregulated lipid accumulation compared with non-transgenic diabetic controls (Supplementary Fig. 4D–G). Our complementary loss-of-function and gain-of-function approaches collectively demonstrated the critical role of renal IGFBP7 in promoting DKD progression in both type 1 and 2 diabetes models.
IGFBP7 deficiency in tubules and podocytes suppresses DKD progression
Initially, we observed that HG levels increased IGFBP7 expression and secretion in renal TECs and podocytes, with more pronounced changes in renal tubular cells. To this end, we injected STZ into flox/flox (FF) mice and IGFBP7 renal tubular epithelial conditionally KO (cKO) mice to induce type 1 DKD (Fig. 2A and Supplementary Fig. 5A, B). Successful model creation was confirmed using blood glucose tests (Supplementary Fig. 5C). ELISA results showed that cKO mice exhibited a significant reduction in STZ-induced urinary albumin increase and a slight reduction in serum creatinine levels, with no significant effect on BUN (Fig. 2B and Supplementary Fig. 5D, E). Notably, cKO mice exhibited reduced renal tubule dilation and interstitial fibrosis, which are key features of DKD, compared with FF mice (Fig. 2C and Supplementary Fig. 5F). We found that IGFBP7 cKO mice showed significantly less severe STZ-induced DKD damage (Supplementary Fig. 5G), with reduced glomerular damage, inflammation, fibrosis, and lipid accumulation in the renal tubules (Supplementary Fig. 5H–J). Mitochondrial damage in DKD mice was also notably decreased in IGFBP7 cKO mice (Supplementary Fig. 5K). However, there was no significant difference in glucose tolerance and insulin resistance between FF and cKO mice (Supplementary Fig. 5L, M).

A Schematic diagram showing induction of type 1 diabetic kidney disease in IGFBP7 renal tubular epithelial cell conditional knockout (cKO) mice. Art was created by FigDraw 2.0. B Urinary albumin/creatinine level of FF and cKO mice. The results showed that knockout IGFBP7 in tubular significantly reduced STZ-induced glomerular dysfunction (n = 8 biological replicates, one-way ANOVA with Tukey’s multiple comparisons test). C PAS and Masson staining of renal tubules. Scale bars, 20 and 50 μm. D Schematic diagram showing induction of type 2 diabetic kidney disease in cKO mice. Art was created by FigDraw 2.0. E Oil red O and immunofluorescence staining of kidney tissues from HFD/STZ-treated FF and cKO mice. F Urinary albumin/creatinine level of FF and cKO mice. The results showed that cKO mice significantly reduced HFD/STZ induced type 2 diabetic kidney disease (n = 6 biological replicates, one-way ANOVA with Tukey’s multiple comparisons test). G PCA plot of the kidney lipidomics analysis of FF + HFD/STZ and cKO+HFD/STZ mice (n = 5). H Heat map showing DEGs related to Phosphatidylcholine (PC) in FF and cKO mouse kidney (n = 5). I In vivo imaging analysis of IGFBP7 cKO mice. IGFBP7 expression in the kidneys was successfully restored by tubule-targeted AAV in cKO mice. J Immunofluorescence results of KIM-1 and αSMA in IGFBP7 cKO mice. K Schematic diagram showing induction of type 2 diabetic kidney disease in IGFBP7 renal podocyte conditional knockout (cKONphs1) mice. Art was created by FigDraw 2.0. L Transmission electron microscopy of glomerular basement membrane thickness and foot process width in FF and cKONphs1 mice. Data represent the mean ± SEM. FF floxflox, cKO conditional knockout in tubular, cKONphs1 conditional knockout in podocyte, PC phosphatidylcholine, LPC lyso phosphatidylcholin, EV empty vector, OE overexpression. Source data are provided as a Source Data file.
Similarly, we established an HFD/STZ-induced type 2 DKD model in IGFBP7 cKO mice (Fig. 2D and Supplementary Fig. 6A). No significant differences in body weight were observed between HFD-fed FF and cKO mice (Supplementary Fig. 6B). Biochemical and histopathological analyses (ELISA, Masson’s trichrome staining, and immunofluorescence) demonstrated attenuated renal injury, inflammation, and fibrosis in cKO mice compared with diabetic controls (Fig. 2F and Supplementary Fig. 6C–E). Ultrastructural and microscopic evaluations further revealed that IGFBP7 cKO mice showed ameliorated glomerular damage and mitochondrial dysfunction in the renal tubular cells (Supplementary Fig. 6F, G). Notably, Oil Red O staining and immunofluorescence analyses confirmed a significant reduction in lipid accumulation in the renal tubules of cKO mice (Fig. 2E). Targeted lipidomic profiling identified substantial reductions in renal lipid metabolites in cKO mice, particularly phosphatidylcholine and phosphatidylethanolamine (Fig. 2G, H and Supplementary Fig. 6H–J). In addition, we further explored the function of IGFBP7 in db/db mice. ELISA, glomerular filtration rate detection, and Masson results further demonstrated that silencing IGFBP7 could significantly alleviate glomerular injury and tubulointerstitial fibrosis in DKD (Supplementary Fig. 7A–D).
To further elucidate the pathogenic role of renal tubular epithelial IGFBP7 in DKD, we used adeno-associated virus (AAV)-mediated IGFBP7 overexpression (Supplementary Fig. 8A). Quantification using ELISA revealed that tubular IGFBP7 overexpression significantly exacerbated STZ-induced urinary albumin excretion (Supplementary Fig. 8B). Histopathological evaluation demonstrated concurrent aggravation of renal interstitial fibrosis and tubular lipid accumulation in the overexpression models, with transmission electron microscopy confirming heightened mitochondrial structural damage characterized by cristae disorganization and matrix swelling (Supplementary Fig. 8C–F). Rescue experiments using IGFBP7 reintroduction in cKO mice recapitulated its disease-promoting effects, exacerbating glomerular lesions and renal fibrosis, as validated by ELISA and Masson’s trichrome staining (Fig. 2I and Supplementary Fig. 8G–J). Immunofluorescence analyses further demonstrated that IGFBP7 overexpression reinstated the expression of the renal injury marker KIM-1 and the fibrosis marker αSMA in STZ-treated cKO mice (Fig. 2J).
Our previous studies have revealed upregulated IGFBP7 expression and enhanced secretory activity in podocytes exposed to chronic hyperglycemic conditions. To elucidate its pathogenic role in DKD, we generated podocyte-specific IGFBP7 cKO (cKONphs1) mice. PCR confirmed the successful KO construction (Supplementary Fig. 9A), and a type 2 DKD model was developed using HFD/STZ in cKONphs1 mice (Fig. 2K). Multi-parameter assessment showed that IGFBP7 cKONphs1 mice attenuated glomerular damage and renal interstitial fibrosis in DKD compared with diabetic controls (Supplementary Fig. 9B, C). However, the renoprotective effects were less pronounced than those observed in tubular epithelial-specific IGFBP7 KO mice. Transmission electron microscopy revealed decreased foot process fusion and basement membrane thickening in cKONphs1 mice (Fig. 2L and Supplementary Fig. 9D). Mechanistic exploration via RNA sequencing identified IGFBP7-overexpressing podocytes as key mediators of pathogenic pathways, particularly through the activation of inflammatory cascades and dysregulation of signaling pathways, such as ferroptosis (Supplementary Fig. 9E, F).
Our in vivo findings demonstrated that IGFBP7 exerts compartment-specific pathogenic effects in DKD, with synergistic contributions from both renal TECs and glomerular podocytes. Notably, tubule-derived IGFBP7 is the predominant driver of disease progression, highlighting the importance of cell type-specific mechanisms in DKD pathogenesis
IGFBP7 inhibits fatty acid metabolism in renal TECs
To elucidate the regulatory role of IGFBP7 in renal TECs, we performed RNA sequencing across four experimental groups: normal control + empty vector (NG + EV), HG + EV, NG + HG, and HG + IGFBP7 overexpression (Supplementary Fig. 10A). KEGG pathway analysis demonstrated that IGFBP7 overexpression significantly suppressed key energy metabolism pathways, particularly the FA metabolism pathway, and concurrently activated inflammation-associated pathways (Fig. 3A and Supplementary Fig. 10B). This finding is particularly relevant given the kidney’s unique metabolic architecture—as an organ enriched with mitochondria–it exhibits compartment-specific substrate preferences: glomeruli primarily utilize glucose for energy production, whereas renal tubules predominantly rely on FAO, a critical energy-generating process for TEC function38,39. Importantly, our data revealed that IGFBP7 overexpression specifically downregulated FAO-related pathways in renal tubular cells (Fig. 3B, C and Supplementary Fig. 10C).

A KEGG enrichment analysis of upregulated (above) and downregulated (down) genes following IGFBP7 overexpression (Padj. Value, false discovery rate). B GSEA plots of fatty acid metabolism gene sets. C KEGG module analysis of fatty acid metabolism. The results showed a significant enrichment in the fatty acid β-oxidation pathway (Padj. Value, false discovery rate). D GSEA plots for oxidative phosphorylation and TCA cycle genes. E Real-time PCR analysis of the mRNA expression of electron transport chain (ETC, Complex I–V) complex genes in BP7-Tg and WT kidney at 8 months of age (n = 6, two-tailed unpaired Student’s t test). F Western blot of PGC-1α in FF versus cKO mice (n = 6, one-way ANOVA with Tukey’s multiple comparisons test). G Real-time monitoring the oxygen consumption rate (OCR) in HK2 with or without IGFBP7 overexpression (BP7-EV (n = 3), BP7-OE (n = 4); biological replicates). H Real-time monitoring the OCR in HG-treated HK2 with or without IGFBP7 knockdown (n = 3). I Relative OCR of HG-treated HK2 with or without IGFBP7 overexpression (n = 12, two-tailed unpaired Student’s t test). J Etomoxir-induced decreases in OCR from HG-treated HK2 (n = 12, two-tailed unpaired Student’s t test). K Schematic of the mechanism by which IGFBP7 induces impaired fatty acid degradation and lipid accumulation in renal tubular epithelial cells; key components and inhibitors involved are shown (by FigDraw 2.0). L Schematic of in vitro co-culture experiments. M Western blot analysis of Wilms’ Tumer 1 (WT-1) and Nephrin protein levels in MPC5 cells under mTEC conditioned media (n = 4, one-way ANOVA with Tukey’s multiple comparisons test). N Western blot analysis of COL-1 and αSMA protein levels in NIH-3T3 cells under mTEC conditioned media (n = 4, one-way ANOVA with Tukey’s multiple comparisons test). O Kidney spheroids from FF and IGFBP7-cKO mice. P Western blotting and quantification of COL-1 and αSMA in FF and cKO mouse kidney spheroids treated with high glucose (n = 3, one-way ANOVA with Tukey’s multiple comparisons test). Data represent the mean ± SEM. n.s. not significant, FF floxflox, cKO conditional knockout in tubular, WT wild type, BP7-Tg IGFBP7 overexpression transgenic, HG high glucose, EV empty vector, OE overexpression. Source data are provided as a Source Data file.
Renal fibrosis is closely linked to metabolic dysfunction in TECs, a process mediated by mitochondrial energy deficits30. FAO serves as the primary driver of mitochondrial energy production, fueling subsequent ATP generation via the tricarboxylic acid (TCA) cycle and oxidative phosphorylation–processes critical for tubular cell homeostasis. RNA sequencing revealed that IGFBP7 overexpression markedly impaired the oxidative phosphorylation, as evidenced by the downregulation of key metabolic genes (Fig. 3D and Supplementary Fig. 10D). Real-time PCR confirmed that IGFBP7 overexpression suppressed oxidative phosphorylation-related gene expression under HG conditions, whereas IGFBP7 silencing restored these effects (Fig. 3E and Supplementary Fig. 10E, F). Collectively, these findings demonstrate that IGFBP7 disrupts mitochondrial bioenergetics by targeting early-stage FAO.
The AMPK signaling pathway, a central energy sensor regulating cellular metabolic balance, promotes mitochondrial biogenesis through PGC-1α activation40,41,42,43. RNA sequencing indicated significant suppression of AMPK signaling under HG and IGFBP7 overexpression conditions, with PGC-1α expression particularly sensitive to IGFBP7-mediated regulation (Fig. 3F and Supplementary Fig. 10G). In DKD mice, PGC-1α downregulation was observed but was attenuated in IGFBP7 conditional KO models (Fig. 3F and Supplementary Fig. 10H). Importantly, silencing IGFBP7 also reversed the high glucose-mediated inhibition of the AMPK signaling pathway and the reduced expression of PGC-1α in primary renal TECs isolated from human donors (Supplementary Fig. 10I). Mechanistically, IGFBP7 inhibited AMPK activation and PGC-1α expression in renal tubular cells under HG stress, thereby compromising mitochondrial energy metabolism (Fig. 3G, H and Supplementary Fig. 10J).
Pharmacological inhibition of mitochondrial FAO using etomoxir demonstrated that IGFBP7 overexpression substantially reduced basal oxygen consumption rates in renal tubular cells, confirming its functional impact on mitochondrial energetics (Fig. 3I, J). This metabolic disruption was corroborated by Oil Red O staining, which showed that IGFBP7 overexpression promoted lipid droplet accumulation in HG-treated TECs; this effect was reversed by IGFBP7 silencing (Supplementary Fig. 10K, L). Collectively, these data established that IGFBP7 drives lipid deposition in glucotoxic environments, primarily by impairing mitochondrial FAO, thereby blocking FA catabolism in renal tubular cells (Fig. 3K).
TEC-derived IGFBP7 promotes the disorder of renal microenvironment induced by HG levels
Notably, both IGFBP7 expression and secretion were significantly upregulated in renal TECs under HG conditions (Fig. 1M, N), indicating its paracrine/autocrine roles in DKD progression. IGFBP7 overexpression exacerbated HG-induced expression of injury/fibrosis markers in tubular cells, while genetic silencing attenuated these pathological changes (Supplementary Fig. 11A–D). In addition, dCAS9-sgRNA-mediated transcriptional repression effectively mitigated the HG-induced fibrosis-like response in renal TECs (Supplementary Fig. 11E). More importantly, we further validated this regulatory effect in human primary renal TECs, which strengthens the translational relevance of our findings (Supplementary Fig. 11F). Furthermore, GSEA revealed a positive correlation between IGFBP7 expression and the activation of NF-kappa B as well as p53 signaling pathways (Supplementary Fig. 11G). We also provided compelling evidence that HG-induced renal tubular epithelial cell injury is mediated by IGFBP7 rather than osmotic pressure, thus clarifying the key mediator underlying HG-associated renal tubular damage (Supplementary Fig. 11H). Furthermore, IGFBP7 potentiated palmitic acid-induced tubular cell injury (Supplementary Fig. 11I), indicating that endogenous IGFBP7 drives tubuloepithelial damage and fibrotic transformation under glucotoxic stress.
To explore the paracrine effects of tubule-derived IGFBP7, we treated podocytes and fibroblasts with conditioned media from IGFBP7-overexpressing tubular cells (Fig. 3L). HG conditions alone reduced podocyte marker expression, which was aggravated by IGFBP7-enriched medium (Fig. 3M). Similarly, HG-conditioned media increased fibrosis markers (COL-1 and αSMA) in NIH-3T3 fibroblasts, with combined HG/IGFBP7 media producing synergistic activation (Fig. 3N). Interestingly, the levels of transforming growth factor-beta 1 (TGF-β1) and connective tissue growth factor (CTGF) were also elevated in the culture supernatant of renal TECs with IGFBP7 overexpression (Supplementary Fig. 11J). More notably, this IGFBP7-overexpressing renal tubular epithelial cell-derived supernatant further enhanced TGF-β1 expression in fibroblasts and podocytes (Supplementary Fig. 11K, L). Direct treatment with recombinant IGFBP7 independently induced fibroblast-to-myofibroblast transition (Supplementary Fig. 11M), confirming its fibrogenic potential.
To establish in vivo relevance, we generated kidney organoids from IGFBP7 tubular cKO mice (Fig. 3O). HG exposure markedly increased the fibrotic indices in FF organoids, whereas cKO-derived organoids exhibited attenuated fibrotic responses (Fig. 3P). These multimodal findings suggest that tubular-derived IGFBP7 is a central mediator of renal microenvironment dysregulation in DKD pathogenesis.
IGFBP7 impedes ubiquitination and subsequent degradation of STAT3 mediated by the E3 ubiquitin ligase MDM2
Gene Ontology enrichment analysis identified protein binding as the predominant molecular function of IGFBP7 (Supplementary Fig. 12A). To delineate the interactome, IP-mass spectrometry of renal TECs from the HG and IGFBP7 overexpression group was performed and 69 overlapping proteins were identified, of which 15 had identified roles in renal pathophysiology (Fig. 4A). STAT3 emerged as the top-ranked IGFBP7-binding partner under glucotoxic stress based on combined bioinformatics and experimental validation (Fig. 4B and Supplementary Fig. 12B, C). Direct interactions were confirmed by co-IP (Co-IP) in HEK-293T cells, supported by surface plasmon resonance, immunofluorescence colocalization, and molecular docking simulations (Fig. 4C–F and Supplementary Fig. 12D). Domain mapping using IGFBP7 truncation constructs (Flag-IGFBP7, -Nter, -Nter-Ka, -Ka-Ig, and -Ig) and STAT3 domain fragments (Flag-STAT3, - CCD, -DBD, and -SH2-TAD) localized the interaction interface to the CCD of STAT3 and the Kazal/Ig C2 domain of IGFBP7 (Fig. 4G, H).

A Venn diagram of the high glucose (HG) model group and IGFBP7 overexpression (OE) group. Filtering the binding strength with differentially expressed protein in high glucose-stimulated mTEC cells with or without IGFBP7, identified STAT3 as a direct target protein of IGFBP7. B Co-IP results of IGFBP7 binding to STAT3 in control and high glucose model groups in renal tubular epithelial cells. C Co-IP results of HA-IGFBP7 binding to Flag-STAT3 in HEK-293T cells. D Surface plasmon resonance (SPR) analysis of the interactions between recombinant STAT3 protein and recombinant IGFBP7 protein. Recombinant IGFBP7 protein concentration: 25, 12.5, 6.25, 3.125, and 1.56 μM. E Immunofluorescence staining of IGFBP7 and STAT3 in HG-treated mTEC and HK2 cells. F Molecular docking of IGFBP7 with STAT3. G Co-IP was performed to determine the interaction between endogenous STAT3 and the functional domains of IGFBP7, including Flag-IGFBP7, Flag-Nter, Flag-Nter-Ka, Flag-Ka-Ig, and Flag-Ig in HG-treated HK2 cells. H Co-IP was performed to determine the interaction between endogenous IGFBP7 and the functional domains of STAT3, including Flag-STAT3, Flag-CCD, Flag-DBD, and Flag-SH2-TAD in HG-treated HK2 cells. I Western blotting analysis of STAT3 levels in HG-treated mTEC and HK2 cells with or without IGFBP7 overexpression (n = 4). J The UbiBrowser assay results indicate that MDM2 is the key E3 ubiquitin ligase for STAT3. K Co-IP assay detects the effect of MDM2 overexpressing on STAT3 ubiquitination level in HK2 cells. L Flag-STAT3, Myc-MDM2, and IGFBP7 OE were co-expressed and applied with MG-132 in HK2 cells. Flag was isolated by IP, and levels were assessed with anti-Myc antibody. M Myc-MDM2, Flag-STAT3, and IGFBP7 OE were co-expressed and applied with MG-132 in HK2 cells. Myc was isolated by IP, and levels were assessed with anti-Flag antibody. Data represent the mean ± SEM. mTEC mouse renal tubular epithelial cells, HK2 human renal tubular epithelial cells, LC-MS/MS liquid chromatography-tandem mass spectrometry, EV empty vector, OE overexpression, Nter IGFBP N-terminal, KA Kazal-like, Ig Ig-like C2-type, CCD Coiled-coil domain, DBD DNA-binding domain, SH2-TAD Src homology 2-transactivation domain. Source data are provided as a Source Data file.
Functional studies demonstrated that IGFBP7 stabilized STAT3 protein levels in renal cells under HG conditions, a phenotype recapitulated in diabetic mice (Fig. 4I and Supplementary Fig. 12E–H). Cycloheximide chase assays revealed that IGFBP7 overexpression extended STAT3 half-life by impairing proteasomal degradation (Supplementary Fig. 12I). Mechanistically, IGFBP7 enhanced the polyubiquitination of STAT3 while reducing its interaction with MDM2, the primary E3 ligase identified by Ubibrowser analysis (Fig. 4J and Supplementary Fig. 12J, K). Co-IP validated MDM2-mediated STAT3 ubiquitination (Fig. 4K) and showed that IGFBP7 competitively disrupted STAT3-MDM2 binding (Fig. 4L, M). This established IGFBP7 as a novel post-translational regulator of STAT3 stability by shielding it from MDM2-dependent ubiquitin-proteasome degradation under diabetic conditions.
Conditional STAT3 KO or knockdown in renal TECs eased DKD
To elucidate the pathogenic role of STAT3 in DKD, we established renal tubule-specific STAT3 KO mice (S3cKO; Supplementary Fig. 13A). In STZ-induced type 1 diabetes-associated DKD, S3cKO mice exhibited significantly attenuated albuminuria compared with their WT littermates (Fig. 5A, B), accompanied by reduced renal interstitial fibrosis (Fig. 5C and Supplementary Fig. 13A).

A Schematic diagram showing induction of type 1 diabetic kidney disease in STAT3 renal tubular epithelial cell conditional knockout (S3cKO) mice. Art was created by FigDraw 2.0. B Urinary albumin/creatinine level of S3FF and S3cKO mice. The results showed that knockout STAT3 in tubular significantly reduced STZ-induced glomerular dysfunction (n = 6 biological replicates, one-way ANOVA with Tukey’s multiple comparisons test). C Masson and immunohistochemical staining results of STZ-treated S3cKO mice. Scale bars, 50 μm. D Schematic illustration of overexpression of STAT3 in IGFBP7 renal tubule conditional knockout (cKO) mice. E Immunofluorescence results of mouse kidney STAT3 expression levels. The results showed that STAT3 was successfully overexpressed in cKO mice. F PAS, Masson, and immunohistochemistry staining in cKO mice with STAT3 overexpression. Scale bars, 50 μm. G Urinary albumin/creatinine level in STZ-treated IGFBP7 cKO mice with STAT3 overexpression (n = 6 biological replicates, one-way ANOVA with Tukey’s multiple comparisons test). H Western blot analysis of NLRP3, COL-1, αSMA, KIM-1, and STAT3. I PAS and Masson staining of IGFBP7 cKO mice kidney. Scale bars, 50 μm. J Urinary albumin/creatinine level in HFD/STZ-treated IGFBP7 cKO mice with STAT3 overexpression (n = 6 biological replicates, one-way ANOVA with Tukey’s multiple comparisons test). K Schematic representation of STAT3 silencing in IGFBP7 overexpression transgenic (Tg) mice. L PAS and Masson staining of IGFBP7 Tg mice kidney (n = 6 biological replicates, two-tailed unpaired Student’s t test). Scale bars, 50 μm. M Urinary albumin/creatinine level in STZ-treated IGFBP7 Tg mice with STAT3 knockdown (n = 6 biological replicates, two-tailed unpaired Student’s t test). Data represent the mean ± SEM. Source data are provided as a Source Data file.
Complementary AAV-mediated STAT3 knockdown in TECs further confirmed these findings. Mice subjected to STZ- or combined HFD/STZ-induced DKD showed blunted disease progression upon STAT3 silencing, as evidenced by improved renal function and histopathology (Supplementary Fig. 13B–E). Consistently, silencing renal TECs STAT3 in db/db mice also alleviated renal tubulointerstitial fibrosis and glomerular injury (Supplementary Fig. 13F–I). In vitro validation using STAT3-silenced HK2 cells demonstrated mitigation of HG-induced cellular injury and lipotoxicity (Supplementary Fig. 14A). In addition, STAT3 silencing in vitro further attenuated the activation of HIF-1α and NF-kappa B signaling pathways (Supplementary Fig. 14B).
IGFBP7 targets STAT3 to promote DKD progression
To delineate the IGFBP7-STAT3 regulatory axis in DKD progression, we performed AAV-mediated STAT3 overexpression in renal tubule-specific IGFBP7 cKO mice, followed by induction of DKD via STZ or combined HFD/STZ (Fig. 5D). Immunofluorescence confirmed robust STAT3 overexpression in tubular epithelia (Fig. 5E), which effectively rescued STZ-induced renal dysfunction, impaired oxidative phosphorylation, and lipid accumulation in cKO mice while augmenting STAT3 phosphorylation (Fig. 5F, G and Supplementary Fig. 15A–C). A comparable phenotypic was observed in the HFD/STZ-induced type 2 DKD model (Fig. 5H–J and Supplementary Fig. 15D, E).
Conversely, AAV-driven STAT3 silencing in IGFBP7 transgenic mice markedly attenuated diabetic renal injury, decreased glomerular filtration rate, and interstitial fibrosis (Fig. 5K–M). Mechanistic consistency was demonstrated in vitro, where STAT3 knockdown suppressed HG-induced inflammatory responses and fibrotic transformation in IGFBP7-overexpressing renal tubular cells (Supplementary Fig. 15F)
IGFBP7 facilitates dimerization of acetylated STAT3, subsequently hindering PGC-1α-driven mitochondrial bioenergetics
STAT3 primarily functions via dimerization in cellular processes44. To assess the potential influence of IGFBP7 on STAT3 dimerization, we performed Co-IP assays. The results demonstrated that Flag-tagged STAT3 and Myc-tagged STAT3 formed dimers in cells, with IGFBP7 overexpression significantly enhancing this interaction (Fig. 6A). A similar enhancement in STAT3 dimerization by IGFBP7 was observed under HG conditions (Supplementary Fig. 16A). While STAT3 can form homodimers and heterodimers with STAT1 or STAT5, IGFBP7 specifically interacted with endogenous STAT3 and STAT1, but not with STAT5, in HK2 cells (Supplementary Fig. 16B). Notably, no direct STAT3-STAT1 or STAT3-STAT5 interactions were detected in HK2 cells, and IGFBP7 did not alter these associations (Supplementary Fig. 16C), suggesting its selective promotion of STAT3 autodimerization.

A Co-IP assay indicating that IGFBP7 promotes the dimerization of STAT3. HK2 cells were transfected with the indicated vectors followed by IP using Flag antibody. The GFP vector worked as an unrelated control protein. B Co-IP assay indicating the promotion of IGFBP7 on the dimerization of phosphorylated (Y705D) or acetylated (K4Q) STAT3 in HK2 cells. The relative quantification of STAT3 dimerization was listed. C Co-IP assay for the interaction of IGFBP7 with the different indicated STAT3 construct (Flag-K4R, Flag-STAT3, and Flag-K4Q) (n = 3 biological replicates, one-way ANOVA with Tukey’s multiple comparisons test). D Co-IP assay of the interaction of SIRT2 with different STAT3 constructs (Flag-STAT3 WT and Flag-STAT3 K685R) in high glucose-treated HK2 cells. E Co-IP assay indicating the inhibition of IGFBP7 on the interaction of SIRT2 with STAT3 in HG-treated HK2 cells. F Real-time monitoring the oxygen consumption rate (OCR) in HG-treated HK2 with or without STAT3 overexpression (HG + S3EV (n = 3), HG + S3OE (n = 4); biological replicates). G Western blot analysis of PGC-1α and pAMPK after STAT3 overexpression in STZ-induced IGFBP7 cKO mice (n = 6 biological replicates, one-way ANOVA with Tukey’s multiple comparisons test). H Real-time monitoring the OCR in IGFBP7 overexpressed HK2 cells with or without STAT3 knockdown (n = 3 biological replicates). I Luciferase reporter assay measured the luciferase activities of the HK2 cells transfected with PGC-1α luciferase plasmids and STAT3 plasmids (n = 4 biological replicates, one-way ANOVA with Tukey’s multiple comparisons test). J Wild-type (WT) and mutant (Mut) PGC-1α were inserted into pmirGLO reporter vectors. K Luciferase reporter assay measured the luciferase activities of PGC-1α-CDS WT or PGC-1α-CDS Mut in HG-treated HK2 cells with or without IGFBP7 knockdown (n = 4 biological replicates, one-way ANOVA with Tukey’s multiple comparisons test). L Luciferase reporter assay measured the luciferase activities of the HG-treated HK2 cells transfected with PGC-1α-Luc, IGFBP7, STAT3, STAT3 K685R (inactivated of STAT3 mutants with lysine 685 reduced acetylation) (n = 4 biological replicates, one-way ANOVA with Tukey’s multiple comparisons test). Data represent the mean ± SEM. n.s. not significant. Source data are provided as a Source Data file.
Given the critical roles of phosphorylation (Y705) and acetylation in stabilizing STAT3 dimers during cytokine/growth factor signaling45,46, we evaluated the effects of IGFBP7 on these post-translational modifications in renal TECs under HG conditions. IGFBP7 overexpression increased global STAT3 phosphorylation and acetylation. However, the phosphorylated (Y705)/total STAT3 ratio remained unchanged, implying that elevated phosphorylation stemmed from increased total STAT3 levels (Supplementary Fig. 16D). In contrast, IGFBP7 markedly enhanced STAT3 acetylation under HG conditions (Supplementary Fig. 16E). To determine the dependency of IGFBP7 on these modifications, we introduced a non-phosphorylatable STAT3 mutant (Y705A) into 293 T cells. Co-IP assays revealed comparable dimerization between the WT and Y705A STAT3 (Supplementary Fig. 16F), indicating phosphorylation-independent dimerization. Intriguingly, while IGFBP7 bound to Y705A STAT3, it failed to promote its dimerization (Supplementary Fig. 16G).
To investigate acetylation dependency, p300 acetyltransferase knockdown via siRNA was used to reduce STAT3 acetylation, which concurrently impaired STAT3 dimerization and IGFBP7-STAT3 binding (Supplementary Fig. 16H). This confirmed the necessity of acetylation in both processes. STAT3 acetylation (K4Q: K601/615/631/685Q) and phosphorylation (Y705D) mimetics were generated. IGFBP7 bound to both mutants and promoted their dimerization (Fig. 6B). However, a dual mutant (Y705D-K4R) abolished IGFBP7-mediated dimerization (Supplementary Fig. 16I), and K4Q enhanced IGFBP7 binding, while K4R abrogated this effect (Fig. 6C). These data establish the specificity of IGFBP7 for acetylated STAT3 in dimerization.
Focusing on K685 acetylation, a key functional site regulated by SIRT deacetylases47,48, we found that SIRT2 interacted with STAT3 under HG conditions (Supplementary Fig. 16J). The STAT3 K685R mutant was resistant to SIRT2-mediated deacetylation (Fig. 6D), confirming that K685 was a SIRT2 target. Importantly, IGFBP7 diminished the SIRT2-STAT3 interaction (Fig. 6E), suggesting that its promotion of STAT3 dimerization involved the inhibition of SIRT2-mediated deacetylation at K685.
To investigate whether IGFBP7 inhibits mitochondrial bioenergetics via STAT3 targeting, we first observed that STAT3 overexpression significantly suppressed mitochondrial bioenergetics, as evidenced by reduced oxygen consumption rates (Fig. 6F). In IGFBP7 cKO mice, impaired AMPK pathway and downregulated PGC-1α in DKD mice were markedly rescued, whereas STAT3 overexpression reversed these effects (Fig. 6G and Supplementary Fig. 16K). Hippocampal metabolic profiling further revealed that STAT3 silencing alleviated IGFBP7 overexpression-induced mitochondrial bioenergetic deficits (Fig. 6H). Notably, STAT3 overexpression robustly inhibited PGC-1α transcriptional activity in renal TECs, as demonstrated by dual luciferase reporter assays (Fig. 6I). To determine the specificity of this regulation, a mutant PGC-1α plasmid (Mut) was generated (Fig. 6J). Silencing IGFBP7 in renal TECs enhanced the transcriptional activity of WT PGC-1α but not of the mutant under HG conditions (Fig. 6K). Furthermore, overexpression of IGFBP7 in cells expressing the STAT3 K685R mutant (a non-acetylatable form) revealed that the K685R mutation itself elevated PGC-1α transcriptional activity. However, IGFBP7 overexpression significantly attenuated this increase (Fig. 6L).
Collectively, these findings demonstrate that IGFBP7 suppresses PGC-1α-mediated mitochondrial bioenergetics in renal TECs under HG conditions by specifically targeting acetylated STAT3 dimerization.
LA acts as an IGFBP7 antagonist to alleviate the progression of DKD
Based on these findings, we explored therapeutic agents targeting IGFBP7 for the treatment of DKD. Using computer-aided drug screening, we identified novel IGFBP7 inhibitors and evaluated their anti-fibrotic effects (Fig. 7A). From a library of 24,327 bioactive compounds (MCE, HY-L001), five candidates–HY-18600A, HY-16659, HY-N0002, HY-14302, and HY-14781 (LA)–showed significant anti-fibrotic activity (Supplementary Fig. 17A, B). Further validation in HG-treated HK2 cells revealed that LA was the most potent compound in reducing fibrosis markers (Supplementary Fig. 18A). Molecular docking based on the IGFBP7 crystal structure highlighted that LA interacted with key residues (MET85, ARG134, SER137, and GLN138) (Fig. 7B). CETSA and SPR assays confirmed LA-IGFBP7 binding (Fig. 7C, D), while Co-IP demonstrated that LA disrupted the IGFBP7-STAT3 interaction (Supplementary Fig. 18B). Additionally, LA suppressed COL-1 and αSMA expression in renal TECs under HG conditions (Fig. 7E) and restored energy metabolism by counteracting acetylated STAT3 upregulation and PGC-1α downregulation, as well as reducing lipid droplet accumulation (Supplementary Fig. 18C, D). Furthermore, we have further clarified that the combined administration of LA with either BAY 11-7082 (a specific inhibitor of the NF-κB pathway) or rapamycin (a well-characterized inhibitor of the mTOR signaling pathway) fails to confer a superior renal protective effect compared with their individual use (Supplementary Fig. 18E, F). Next, we assessed LA functions in two murine DKD models. In STZ-induced type 1 DKD (Fig. 7F), LA (2.5 mg/kg, weekly) administration for 10 weeks attenuated urinary albumin levels (Fig. 7G). Histopathological and western blot analyses confirmed renal fibrosis reduction and marker expression by LA (Fig. 7H and Supplementary Fig. 18G), supported by diminished lipid deposition via Oil Red O staining (Fig. 7I). Similar efficacy was observed in HFD/STZ-induced type 2 DKD mice (Fig. 7J), with LA alleviating glomerular injury and fibrosis (Fig. 7K, L and Supplementary Fig. 18J). More importantly, we further evaluated the effect of LA on glomerular function using transmission electron microscopy. As illustrated in the results, LA significantly attenuated the thickening of the glomerular basement membrane (GBM), the increase in foot process width, and the decrease in foot process number that occur during DKD progression (Supplementary Fig. 18H, I). Of note, LA did not alter the symptoms of STZ-induced hyperglycemia (Supplementary Fig. 18K). To further investigate the functional role and therapeutic efficacy of LA in advanced DKD, we administered LA to 6-month-old db/db mice, followed by a 4-week drug withdrawal period. Results showed that LA effectively mitigated glomerular injury and interstitial fibrosis in mice with advanced DKD. Notably, a slight rebound in pathological manifestations was observed after drug discontinuation, which further underscores the critical therapeutic importance of LA in advanced DKD (Supplementary Fig. 18L–O). Notably, LA administration did not affect liver function, as evidenced by unaltered ALT and AST levels (Supplementary Fig. 19A, B). Furthermore, histological analysis of major vital organs, including the heart, liver, spleen, and lungs, revealed no discernible pathological damage. These observations collectively confirm that LA does not induce significant toxic side effects within the tested dosage range (Supplementary Fig. 19C).

A Workflow of the hybrid virtual screening strategy. B Molecular docking demonstrated that Levomefolic acid (LA) physically bound to the catalytic domain of IGFBP7. C Cellular thermal shift assay (CETSA) analysis showing the stabilisation of IGFBP7 in vitro, with or without LA treatment. D Surface plasmon resonance (SPR) analysis of the interactions between IGFBP7 and LA. Concentration of salmeterol (μmol/L): 16, 8, 4, 2, 1, 0.5. E Immunofluorescence (IF) analysis of COL-1 and αSMA in LA-treated HK2 cells in response to high glucose (HG). F Schematic illustration of treatment of levomefolic acid after STZ-induced type 1 diabetic kidney disease. G Urinary albumin/creatinine level in STZ-induced DKD with LA treatment (n = 6 biological replicates, one-way ANOVA with Tukey’s multiple comparisons test). H PAS and Masson staining in STZ-induced DKD with LA treatment. Scale bars, 50 μm. I Oil red O staining of STZ-induced DKD with LA treatment. Scale bars, 100 μm. J Schematic illustration of treatment of LA after HFD/STZ-induced type 2 diabetic kidney disease. K Urinary albumin/creatinine level in HFD/STZ-induced DKD with LA treatment (n = 6 biological replicates, one-way ANOVA with Tukey’s multiple comparisons test). L PAS and Masson staining in HFD/STZ-induced DKD with LA treatment (n = 6 biological replicates, one-way ANOVA with Tukey’s multiple comparisons test). Scale bars, 50 μm. Data represent the mean ± SEM. Source data are provided as a Source Data file.
In conclusion, our study identified LA as a therapeutic candidate that attenuated DKD progression by targeting IGFBP7.
Discussion
IGFBP7 in renal tubules and podocytes plays a pivotal role in DKD. Our findings demonstrated that cell-specific IGFBP7 acts as a key mediator in DKD by promoting acetylated STAT3 dimerization, which suppresses AMPK signaling-driven mitochondrial bioenergetics. These processes correlate with the activation of mesenchymal transdifferentiation in diabetic kidneys, establishing a mechanistic link between renal interstitial fibrosis and glomerular dysfunction, which are the two hallmark features of DKD. While IGFBP7 is predominantly expressed in the kidney, its functional role in DKD pathogenesis and renal microenvironment regulation remains underexplored (Supplementary Fig. 20, by FigDraw 2.0).
Clinical studies have shown that elevated IGFBP7 levels correlate with DKD progression25. In vitro, IGFBP7 silencing attenuates HG-induced TGF-β1/Smad pathway activation in podocytes49. Previous studies have identified IGFBP7 as an early responder in the proximal tubules during acute kidney injury22. Notably, IGFBP7 mRNA is markedly upregulated in STZ-induced diabetic rat kidneys24, and elevated circulating IGFBP7 levels predict the progression from DKD to end-stage kidney disease25. Our data provide mechanistic insights into the role of IGFBP7 in DKD pathophysiology, showing that its tubular expression is significantly increased in DKD patients and diabetic mouse models, with its levels positively correlated with renal fibrosis and inflammation. During the progression of DKD, distinct types of epigenetic memory—specifically metabolic memory and hypoxia memory—exhibit tight associations with altered expression of disease-relevant genes50. Our study uniquely demonstrates that histone H3 lysine 4 trimethylation (H3K4Me3)—a canonical activating epigenetic modification—mediates the upregulation of IGFBP7: we observed enhanced H3K4Me3 enrichment at the IGFBP7 promoter region in diabetic renal tissues, which directly promotes IGFBP7 transcriptional activation. These findings support the hypothesis that IGFBP7 secretion by proximal renal tubules initiates a cascade of events that play a critical role in the pathophysiology of DKD.
To the best of our knowledge, this is the pioneering study to delineate how IGFBP7 modulates the renal microenvironment to drive DKD progression. We identified IGFBP7 as a stressor in proximal tubular cells, inducing pro-inflammatory and profibrotic responses in both type 1 and 2 diabetic kidneys. Although hyperglycemia is the primary driver of DKD51,52,53, previous research has focused on its effects on apoptosis and inflammation. In contrast, our work highlights HG-induced dysregulation of FA metabolism in renal tubular cells as a key contributor to interstitial fibrosis. Strikingly, IGFBP7 deletion in TECs restored mitochondrial FA metabolism and protected against proteinuria in diabetic DKD mice, despite persistent hyperglycemia. To our knowledge, this is the notable report linking FA regulatory effects of IGFBP7 to DKD pathogenesis.
Renal lipid imbalance is an early and persistent feature of DKD, with disrupted FA uptake/degradation directly contributing to tubular injury32,54. Elevated lipid accumulation and de novo lipogenesis genes are correlated with fibrosis in human kidneys55. For instance, acyl-CoA synthetase short-chain family 2 drives lipogenesis, NADPH depletion, and ROS-mediated NLRP3 pyroptosis in renal tubules55. Mitochondrial FAO deficiency exacerbates lipid overload, triggering myofibroblast activation and fibrogenesis26,54. In this study, we demonstrated that IGFBP7 upregulation in diabetic kidneys exacerbated FAO inhibition, lipid accumulation, and fibrosis. Conversely, IGFBP7 deficiency enhanced FAO and attenuated mesenchymal activation and fibrosis.
RNA sequencing identified IGFBP7 as a “protein-binding” modulator of signaling pathways. STAT3, a key signal transducer activated by pathological stress56,57, undergoes dimerization as the final activation step, which can occur independent of Tyr705 phosphorylation. Unphosphorylated STAT3 regulates specific gene targets45,58. Critically, STAT3 acetylation at K685 (the SH2 domain) and other residues (K49, K87, and K631) governs nuclear localization, transcriptional activity, and dimer stability47,48. Acetylation, particularly at K685, may supersede phosphorylation and regulate STAT3 function59,60. Our study demonstrated that IGFBP7 promoted acetylated STAT3 dimerization by antagonizing SIRT2-mediated deacetylation. Acetylation-driven STAT3 activation disrupts FAO and amplifies fibrotic signaling. Persistent STAT3 activation correlates with poor DKD outcomes61, which is consistent with our observation that IGFBP7 deficiency restored FAO and mitigated fibrosis despite hyperglycemia. Our work suggests that IGFBP7-STAT3 is a pivotal regulator that links defects in FA degradation to DKD pathology. By impairing FAO and amplifying lipid-driven fibrogenesis, IGFBP7 establishes a metabolic-cytokine crosstalk that accelerates DKD progression.
This study further elucidated that PGC-1α acts as a pivotal downstream effector in the IGFBP7-STAT3 pathway, driving renal dysfunction and mitochondrial bioenergetic deficits. Notably, in the progression of chronic kidney disease (CKD), AMPK plays a key role in maintaining energy metabolism homeostasis and autophagy in renal tubular cells62—and as a core regulator of the AMPK signaling pathway, PGC-1α is activated by signals such as cAMP and cytokines to transiently regulate mitochondrial oxidative phosphorylation genes63,64,65. Dysregulated PGC-1α levels impair mitochondrial electron transport chain complexes and bioenergetics, exacerbating renal fibrosis66,67,68,69. In diabetic kidneys, we observed an inverse correlation between PGC-1α expression and IGFBP7 upregulation. IGFBP7 overexpression suppressed PGC-1α mRNA/protein levels in vivo and in vitro, whereas IGFBP7 knockdown restored PGC-1α expression. STAT3 is a negative regulator of PGC-1α70,71,72. For instance, hepatic STAT3 deletion increases PGC-1α expression, inducing insulin resistance73, and STAT3 suppresses PGC-1α via miR-23a upregulation71. Our findings align with these mechanisms, showing that IGFBP7 inhibits PGC-1α via STAT3. Critically, STAT3 silencing reversed mitochondrial dysfunction and renal injury caused by IGFBP7 overexpression, highlighting that the IGFBP7-STAT3-PGC-1α axis is central to DKD progression.
LA, the bioactive form of folic acid74,75,76, is directly utilized without metabolic conversion77. It also supports nucleic acid synthesis, cardiovascular health, and neural development78,79. Although high-dose folic acid induces kidney injury (> 200 mg/kg)80,81, dietary folate is associated with reduced mortality and end-stage kidney disease risk in CKD patients82,83. In this study, we identified LA as a novel small-molecule IGFBP7 inhibitor after screening > 24,000 compounds. LA mitigated IGFBP7-mediated lipotoxicity in vitro and attenuated renal fibrosis and glomerular dysfunction in vivo. Mechanistically, LA disrupted IGFBP7-STAT3 binding and restoreed mitochondrial energy metabolism. Systemic LA administration did not cause major organ toxicity, indicating its therapeutic potential. These findings elucidate the specific mechanisms by which LA ameliorates DKD and paves the way for its future clinical applications.
In conclusion, IGFBP7 expression is upregulated in the early stages of DKD25. This elevated expression in proximal tubules demonstrates persistent upregulation in both human and rodent models. IGFBP7-mediated suppression of FA metabolism in proximal tubular cells induces intracellular lipid deposition, subsequently triggering the activation and secretion of pro-inflammatory and profibrotic cytokines. Severe renal interstitial fibrosis resulting from these cytokine-mediated processes progresses to glomerular sclerosis, ultimately culminating in elevated glomerular albumin filtration rates84. Renal TEC-derived IGFBP7 directly induces glomerular podocyte injury and fibroblast activation. The absence of secondary glomerular pathology in diabetic mice with conditional IGFBP7 KO in renal tubular cells provides additional evidence of a mechanistic connection between tubule-derived IGFBP7 and the development of interstitial inflammation, fibrosis, and glomerulosclerosis. Therefore, the therapeutic targeting of IGFBP7-mediated dysregulation of the renal microenvironment could potentially attenuate DKD progression, particularly in renal fibrotic processes and glomerular injury. Interestingly, LA, a newly recognized IGFBP7 inhibitor, showed promising efficacy in ameliorating diabetic nephropathy in both animal and cellular experiments. Collectively, these findings advance our understanding of the role of IGFBP7 in shaping the pathological renal microenvironment in DKD and highlight a novel therapeutic strategy targeting IGFBP7 to slow disease progression.
Methods
Reagent and materials
Antibodies specific for IGFBP7 (ab171085, ab214262: Abcam; and 19961-1-AP: Proteintech), COL-1 (66761-1-Ig; Proteintech), αSMA (14395-1-AP; Proteintech), STAT3 (10253-2-AP; Proteintech), pSTAT3 (R381552; Zen-Bio), AcSTAT3 (#2523; CST), STAT1 (R25799; Zen-Bio), STAT5 (R381427; Zen-Bio), PGC-1α (R381615; Zen-Bio), pAMPK (AP1441; Abclonal), AMPK (A1229; Abclonal), H3K4Me1 (A22079; Abclonal), H3K4Me3 (A22146; Abclonal), H3K27Ac (A22264; Abclonal), Histone3 (A2348; Abclonal), Plin2 (15294-1-AP; Proteintech), KIM-1 (30948-1-AP; Proteintech), NLRP3 (WL02635; Wanlei), TNFα (60291-1-Ig; Proteintech), γH2AX (R381558; Zen-Bio), WT-1 (ab267377; Abcam), Nephrin (ab216341; Abcam), NFκB1 (10745-1-AP; Proteintech), HSPA5 (R24618; Zen-Bio), SIRT2 (ET1611-72; HuaBio), Flag (20543-1-AP; Proteintech), HA (51064-2-AP; Proteintech), Myc (60003-2-Ig; Proteintech), GFP (M20004; Abmart), MDM2 (66511-1-Ig; Proteintech) and β-actin (bs-10966R; Bioss). Fluorescein-labelled LTL (FL-1321), PNA (FL-1071), and DBA (FL-1031) were obtained from Vector Laboratories (Newark, CA, USA). The IRDye 800-conjugated secondary antibody was obtained from Rockland Immunochemicals (Gilbertsville, PA, USA). PAS, Creatinine Assay, and BUN Assay Kits were obtained from the Nanjing Jiancheng Bioengineering Institute (Nanjing, China). An IGFBP7 ELISA Kit was purchased from Shanghai MLBIO Biotechnology (Shanghai, China). Recombinant mouse IGFBP7 protein was purchased from Novoprotein (CU51; Shanghai, China). STZ and glucose were purchased from Sigma Chemical Company (MO, United States). MM-102, etomoxir, and LA were purchased from MCE (Shanghai, China).
Human renal biopsy samples
Samples of kidney tissue and peripheral blood were collected from both healthy individuals and DKD patients at the First Affiliated Hospital of Anhui Medical University. Patients diagnosed with DKD via renal biopsy participated in the study. Normal controls were obtained from non-tumor kidney tissue of patients who underwent nephrectomy for renal cell carcinoma. Serum levels of proteinuria, creatinine, and blood glucose were used to diagnose DKD patients. The clinical details for all donors are thoroughly outlined in Table S1. This study was approved by Biomedical Ethics Committee of Anhui Medical University (82240036). The studies were conducted with each participant’s informed and written consent, following the guidelines of the Declaration of Helsinki.
Mouse models of diabetic kidney disease
All animal experiments were approved by the Institutional Animal Care and Use Committee of Anhui Medical University (Ethics No. LLSC20221109) and strictly adhered to NIH guidelines for laboratory animal welfare. Male littermate mice (age and group sizes specified in figure legends) were randomly assigned to experimental groups using a blinded protocol for surgical interventions and outcome assessments. Animals were maintained in specific pathogen-free facilities under 12-h light/dark cycles at 24 °C, with ad libitum access to autoclaved water and either standard chow or high-fat diet (HFD). Housing conditions included corncob-bedded cages (3–5 mice/cage) replaced thrice weekly following steam sterilization. Sex selection was based on established metabolic phenotypes, with male C57BL/6 mice chosen due to their heightened susceptibility to diet-induced obesity and glucose intolerance compared to females. Veterinary staff routinely monitored animals, with no clinical signs of distress or pathology requiring intervention observed throughout the study.
For the unilateral nephrectomy and STZ-induced type 2 diabetic mouse studies, 8–10-week old male C57BL/6 mice (IGFBP7 KO, cKOKsp, and cKONphs1 mice) were used. Ksp-Cre mice (Strain NO. T012960) and Nphs1-Cre mice (Strain NO. T016576) purchased from GemPharmatech Co., Ltd. (Jiangsu, China). Eight-week-old male Cre+/IGFBP7fl/fl mice and their littermate Cre+/IGFBP7+/+ mice were fed either a HFD (60 kcal% from fat, Research Diets) or control diet for 4 weeks and then uninephrectomized. After one week recovery period from uninephrectomy, mice were injected low-dose STZ dissolved in 50 mmol/L sodium citrate buffer (pH 4.5) (50 mg per kg body weight intraperitoneally daily for 3 days) after starving for 4 h to induce partial insulin deficiency or injected vehicle for control. Mice continued to be maintained on a HFD or control diets for another 20 weeks (as shown in Fig. 2). Mice received ad libitum HFD feeding for 24 weeks prior to terminal procedures. Systemic biochemical profiling was performed using serum and 24-h urine samples. Following euthanasia, renal tissues were harvested with cortical regions isolated for histopathological evaluation and molecular analyses (protein/RNA extraction).
For the STZ induced type 1 diabetic mouse studies, 8–10-week old male C57BL/6 mice (IGFBP7 KO, cKO, and Tg mice; STAT3 cKO mice) were used. Type 1 Diabetes was induced in 10-week-old male mice on a C57BL/6 background by five consecutive intraperitoneal injections of STZ (50 mg/kg) with 10 mM citrate buffer (pH 4.5), and were maintained for totally 24 weeks.
Glomerular filtration rate
GFR was measured by determining elimination of fluorescein isothiocyanate (FITC)-sinistrin transcutaneously as described85. A fluorescence detector (NICKidney; Mannheim Pharma & Diagnostics GmbH, Germany) was placed on a depilated region of the back and FITC-sinistrin (50 mg/kg body weight) injected intravenously via the tail vein. Data were acquired in conscious mice and GFR calculated using the half-life (t1/2).
Cell culture and treatment
The human tubular epithelial cells (HK2) and mouse tubular epithelial cells (mTEC) lines were provided by Prof. Hui Yao Lan (The Chinese University of Hong Kong, Hong Kong, China), and prepared as previously reported22. Conditionally immortalized mouse podocytes (MPC5) were acquired from the Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences. The cells were maintained in RPMI 1640 supplemented with 10% fetal bovine serum and 20 U/mL murine recombinant interferon-γ (IFN-γ) at 33 °C under permissive conditions. For differentiation induction, MPC5 were transferred to IFN-γ-free medium and cultured at 37 °C for 7 days under non-permissive conditions prior to experimentation. Mouse mesangial cells (SV40) were obtained from the Cell Bank of Chinese Academy of Sciences and propagated in DMEM with 5% serum supplementation. Genetic manipulation of TECs was performed using Lipofectamine 3000 transfection reagent (Invitrogen) according to standardized protocols. siRNA constructs and overexpression plasmids (HanBio, China) were introduced following manufacturer specifications, with scrambled siRNA/plasmid (HanBio, China) serving as negative controls. Briefly, nucleic acid-lipid complexes were prepared by incubating diluted siRNA/plasmid with transfection reagent for 20 min at room temperature, followed by administration to cells cultured in OPTI-MEM medium. Following an incubation period of 6 to 12 h, the medium was replaced with complete culture medium. The selected sequences are listed in Table S2. In this study, the cells were subjected to different treatment conditions: High glucose (HG) (D-glucose at a final concentration of 30 mmol/L in culture medium), medium contained 5.6 mmol/L-glucose as normal glucose (NG), mannitol was added in the medium as the osmotic pressure control for HG.
Mouse kidney organoids
The newly isolated FF and IGFBP7-cKO mouse kidney primary cells were cultured in 24-well ultra-low attachment plates (Corning, NY, USA) using an appropriate medium. Each well was supplemented with 3–4 Matrigel pellets, each measuring 10 μL. Fixed matrix glue pellets were introduced into each well alongside organoid culture medium, and 1 mL of phosphate-buffered saline (PBS) was added to the peripheral wells. The cells were subsequently incubated in a 5% CO2 atmosphere at 37 °C. The development of the kidney organoids was systematically monitored. The procedures related to organoid culture were primarily executed by Anhui Luohua Biotechnology Co., Ltd (Anhui, China). Proteins from the kidney organoids were harvested for subsequent analysis via western blotting. Renal organoids of FF in cKO mice were subjected to high glucose stimulation, and proteins were subsequently extracted from renal organoids for subsequent western blot analysis.
Renal histology and immunohistochemistry
Renal specimens from mice were processed through standard histological protocols involving fixation, graded ethanol dehydration, paraffin infiltration, and embedding to generate 4 μm-thick sections. Tissue sections underwent histomorphological analysis using Periodic Acid-Schiff (PAS) and Masson’s trichrome staining protocols, with microscopic evaluation performed under brightfield illumination. Renal fibrosis quantification followed established methodologies, with immunohistochemical analyses performed according to manufacturer protocols. For detection of the target protein, sections were incubated with primary antibodies overnight at 4 °C, after which they were exposed to secondary antibodies for 60 min at room temperature. Subsequent, the sections were examined using an Olympus IX83 microscope (Olympus, Japan).
Immunofluorescent staining and high-content fluorescence imaging
To ascertain the primary localization of IGFBP7 expression within renal tissues, kidney sections were subjected to staining with LTL, PNA, and DBA. The deparaffinized sections were incubated overnight at 4 °C with primary antibodies, including anti-IGFBP7, anti-COL-1, anti-αSMA, anti-KIM1, anti-Plin2, and anti-STAT3, followed by a 2-h incubation with a secondary antibody. Imaging was conducted using an Olympus IX83 microscope. Additionally, HK2 cells were uniformly seeded into 96-well plates and exposed to high glucose conditions, as well as to 19 screened drug candidates at concentrations of 0.625, 1.25, and 2.5 μM. Fluorescence staining was performed according to previously established protocols. High-throughput fluorescence imaging was carried out utilizing the ImageXpress Micro 4 system (Molecular Devices, San Jose, CA, USA).
RNA sequencing
mTEC cells were collected and divided into four groups: normal glocose + empty vector (NG + EV), high glucose + empty vector group (HG + EV), high glucose + IGFBP7 overexpression (NG + OE), high glucose + overexpression group (HG + OE). MPC5 cells were collected and divided into two groups: high glucose + IGFBP7 empty plasmid vector group (HG + EV) and high glucose + IGFBP7 overexpression group (HG + OE). The two cell types were subjected to RNA-seq sequencing analysis. RNA sequencing was executed by BGI (Huada Genomics). Then used the DESeq2 R package to differential expression analysis, followed by identification of DEGs based on |LogFC| > 1 and p value < 0.05.
Absolute quantitative lipidomics
Renal specimens were cryopreserved at −80 °C for subsequent absolute lipid quantification. Lipidomic profiling was conducted through a commercial analytical platform (Shanghai Luming Bio) employing chromatographic separation with a UHPLC eNexera LC-30A system (Shimadzu) coupled to a Q Exactive Hybrid Quadrupole-Orbitrap mass spectrometer (Thermo Scientific™). Quantitative lipid characterization was achieved through LipidSearch software (v4.2, Thermo Scientific™) using a 13-component isotope-labeled internal standard panel for normalization, enabling precise determination of lipid molecular species concentrations.
Oil red O stain
Oil Red O staining was conducted utilizing an Oil Red O staining kit (Beyotime). Briefly, frozen kidney tissue sections (8 to 10 µm) or cultured cells fixed with 4% paraformaldehyde were air-dried and subsequently washed under running tap water for 5 min. The samples were then rinsed with 60% isopropanol and stained with freshly prepared Oil Red O solution for 15 min. Following this, the samples were washed with 60% isopropanol, and the nuclei were lightly counterstained with hematoxylin, followed by a wash with distilled water.
Glucose tolerance test
The mice underwent a fasting period of 6 h, followed by an intraperitoneal injection of glucose dissolved in PBS. The administered dosage was 2 g/kg body weight for mice on a control diet and 1.2 g/kg body weight for those on a high-fat diet. Blood glucose levels were assessed prior to the injection and subsequently at 15, 30-, 60-, 90-, and 120-min post-injection, utilizing the Roche glucose monitoring system.
Insulin tolerance test
Mice were subjected to a 4-h fasting period prior to receiving an intraperitoneal injection of human insulin (Sigma) dissolved in saline, administered at a dosage of 0.35 U kg−1 body weight for mice on a control diet and 0.6 U kg−1 body weight for those on a high-fat diet. Blood glucose levels were subsequently measured according to the previously outlined methodology.
Measurement of oxygen consumption rate (OCR)
Mitochondrial respiratory function was assessed using an XF24 extracellular flux analyzer (Seahorse Bioscience). Renal TECs (1 × 104 cells/well) were seeded in specialized microplates and subjected to sequential mitochondrial stress testing: basal OCR measurement followed by serial injections of 1 μM oligomycin (ATP synthase inhibition), 0.75 μM FCCP (uncoupling agent), and a 1 μM oligomycin/rotenone cocktail (electron transport chain termination). Acquired data were normalized to total cellular protein content.
Transmission electronic microscopy
The renal cortex was isolated from the medulla and sectioned into cubes measuring 1 mm³. Kidneys from all experimental groups were preserved using Karnovsky’s fixative. The samples were subsequently examined with a Jeol 1010 Transmission Electron Microscope. A total of 60 to 80 micrographs were captured from each sample at varying magnifications (10,000× to 25,000×). These micrographs were assessed for indicators such as mitochondrial swelling, fragmentation, the presence of disorganized cristae, basement membrane thickness, and podocyte foot effacement.
Luciferase reporter assays
The luciferase reporter assays were conducted following established methodologies. STAT3 overexpression plasmids, blank vector plasmids, PGC-1α promoter (PGC-1α-WT), and PGC-1α mutant (PGC-1α-Mut) vector plasmids were procured from HanBio. Luciferase activity was measured using a dual-luciferase assay kit (HanBio) in accordance with the manufacturer’s instructions. Renilla luciferase served as the normalization control.
Cytotoxicity assay
Cellular viability assessments for HK-2 cells exposed to test compounds were conducted with a CCK-8 assay kit (Sigma). Following experimental treatments, 10 μL CCK-8 reagent was added to each well containing 100 μL culture medium, followed by 2-h incubation at 37 °C. Absorbance measurements were performed at 450 nm wavelength using a SpectraMax i3x plate reader (Molecular Devices).
Cellular thermal shift assay
Cellular thermal shift assays were conducted following established methodology86. Briefly, HK-2 cells with or without LA pretreatment were lysed in RIPA buffer and mechanically homogenized (4 °C) with 3 mm zirconium beads. Lysates were normalized for protein concentration using a commercial assay kit (Beyotime) and distributed into six equal aliquots. Thermal denaturation was performed across graded temperatures (8 min/cycle) in a Bio-Rad thermal cycler, followed by three liquid nitrogen snap-freeze/thaw cycles. Post-centrifugation supernatants were subjected to immunoblot analysis.
Chromatin immuno-precipitation (ChIP) assay
Chromatin immunoprecipitation assays were performed with anti-H3K4Me3 antibody and IgG control (4 °C overnight incubation) using the SimpleChIP® Enzymatic Chromatin IP Kit (Cell Signaling Technology #9003). Quantitative analysis of the IGFBP7 promoter region was conducted via SYBR Green-based real-time PCR (7500 Fast System, Applied Biosystems) with Premix Ex Taq reagents (Takara Bio). The following primer pairs specific to the human IGFBP7 gene were used: forward primer, 5′-GAAGAGCCGCTCGCACTG-3′, and reverse primer, 5′-GTCCGAAGAGGAGGAAGAGG-3′.
Western blotting
Protein extracts from renal and cellular specimens were processed following standard extraction protocols for immunoblot analysis22. The samples were incubated with primary antibodies for 12 h at 4 °C, subsequently treated with IRDye 800-conjugated secondary antibody (1:10,000 dilution) for 1.5 h at room temperature. Imaging was conducted using a Li-Cor/Odyssey infrared imaging system (LI-COR Biosciences, Lincoln, NE, USA), and the resulting images were quantified utilizing ImageJ software (NIH, Bethesda, MD, USA).
RNA extraction and real-time PCR
RNA extraction was performed with RNeasy kits (Qiagen) followed by cDNA synthesis using reverse transcriptase reagents (Bio-Rad). Quantitative PCR analysis was carried out on an Opticon2 thermal cycler (Bio-Rad) with SYBR Green detection chemistry, adhering to standardized amplification protocols. Gene expression quantification implemented established RT-qPCR methodologies referenced in previous investigations87. The sequences of the primers utilized are provided in Supplemental Table 2.
Co-immunoprecipitation
Immunoprecipitation analyses were conducted in accordance with previously established protocols22. Cellular/nuclear preparations underwent triple PBS rinses (4 °C) and lysis in NP-40 buffer (1%). Lysates were immunocomplexed with Flag/HA/Myc antibodies (1 μg/antibody) coupled to protein A/G magnetic beads (Bio-linkedin) through 16 h incubation at 4 °C. Thermally eluted proteins (SDS buffer, 95 °C) were separated on 4–20% gradient gels. Western blotting analyses were performed using antibodies specific for Flag, HA, Myc, GFP, STAT1, STAT3, STAT5, and SIRT2.
Identification of IGFBP7-interacting proteins via LC-MS/MS analysis
HK2 cells were subjected to treatment and subsequently categorized into two distinct groups: the high glucose model group (HG) and the IGFBP7 overexpression group (OE). Cell lysates underwent immunoprecipitation utilizing anti-IGFBP7 antibodies in conjunction with protein A/G magnetic beads (Bio-linkedin). Orbitrap-Fusion mass spectrometer (Thermo Scientific) accompanied by Easy-nLC 1000 (Thermo Scientific) was used to identify Co-IP proteins. The online capillary column (150 μm ID × 12 cm bed length, 1.9 μm) was coupled to mass spectrometry. Samples were eluted from the column by a 0–69-min 5–31% step linear gradient of the buffer B (100% ACN, 0.1% FA) to a 5-min isocratic elution at 95% of the buffer B, supplemented with buffer A (100% water, 0.1% FA). The electrospray voltage was 2.1 kV, capillary temperature was 320 °C, and normalized collision energy was 32 eV. HCD energy of 32% was used. The mass range for MS scans was 300–1400 m/z. The mass spectrometer was conducted in standard MS/MS data-dependent acquisition mode. The false positive rate was set to 1 and 5% on peptide level as restrict and relax cutoff, respectively. The LC-MS/MS detection and analysis were performed by Shanghai Luming Biological Technology Co., Ltd. (Shanghai, China).
Virtual screening and molecular docking
The Discovery Studio 2017 R2 software (BIOVIA Software Inc.), developed by Beijing Chuangteng Technology Co., Ltd. (Beijing, China), was utilized for this study. The analysis incorporated commercial databases, including the MCE database, which comprises over 24,000 compounds. The crystal structure of IGFBP7 was obtained from the predicted AlphaFold model (UniProt code: Q16270). The Protein Preparation module was employed to refine the free protein structure by eliminating all co-crystallized water molecules. Additionally, the protein structure underwent energy optimization through the incorporation of missing polar hydrogens and the application of CHARMM force fields. Compounds sourced from TargetMol were filtered based on Lipinski’s rule of five and evaluated for Absorption, Distribution, Metabolism, Excretion, and Toxicityf properties to identify active compounds. Ultimately, a total of 17,681 molecules were identified as active compounds from the database. These molecules were subjected to molecular docking studies using LibDock and CDOCKER, with the docking protocol being validated against the Kazal domain of IGFBP7. The top 89 candidate compounds, exhibiting docking scores ranging from 139.5 to 167.3, were selected for subsequent cytotoxicity and activity testing, leading to the identification of the most active compound.
Surface plasmon resonance assay
The interactions between IGFBP7 and either STAT3 or LA were quantitatively assessed utilizing a Biacore T200 system (Cytiva, Marlborough, MA, USA). Purified recombinant STAT3 or IGFBP7 was immobilized onto a carboxymethylated CM5 sensor chip. Gradient concentrations of IGFBP7 (1.56, 3.12, 6.25, 12.5, and 25 μmol/L) or LA (0.5, 1, 2, 4, 8, and 16 nmol/L) were introduced as analytes. The resulting data were analyzed using Biacore Evaluation Software (T200 Version 2.0).
Statistical analyses
Data analysis was performed with SPSS 23.0 (IBM) and visualization with GraphPad Prism 9.0. Normality assessment employed the Shapiro-Wilk test, with normally distributed data expressed as mean ± SEM. Intergroup comparisons utilized independent t-tests and one-way ANOVA with Tukey’s post hoc correction. A statistical significance threshold was set at P < 0.05.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data associated with this study are present in the paper or the Supplementary Materials. The RNA-seq data are available in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) under BioProject accession PRJNA1220270 and PRJNA1219984. Source Data are provided with this paper.
References
Herrington, W. G. & Haynes, R. Diabetic kidney disease — semaglutide flows into the mainstream. N. Engl. J. Med. 391, 178–179 (2024).
Mohandes, S. et al. Molecular pathways that drive diabetic kidney disease. J. Clin. Investig. 133, e165654 (2023).
Balzer, M. S. et al. Single-cell analysis highlights differences in druggable pathways underlying adaptive or fibrotic kidney regeneration. Nat. Commun. 13, 4018 (2022).
Abedini, A. et al. Single-cell multi-omic and spatial profiling of human kidneys implicates the fibrotic microenvironment in kidney disease progression. Nat. Genet. 56, 1712–1724 (2024).
Lovisa, S. et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat. Med. 21, 998–1009 (2015).
Allison, S. J. Targeting EMT to reverse renal fibrosis. Nat. Rev. Nephrol. 11, 565–565 (2015).
Grande, M. T. et al. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 21, 989–997 (2015).
Kuppe, C. et al. Decoding myofibroblast origins in human kidney fibrosis. Nature 589, 281–286 (2021).
Meng, X., Nikolic-Paterson, D. J. & Lan, H. Y. TGF-β: the master regulator of fibrosis. Nat. Rev. Nephrol. 12, 325–338 (2016).
Meng, X.-M., Tang, P. M.-K., Li, J. & Lan, H. Y. TGF-Î2/Smad signaling in renal fibrosis. Front. Physiol. 6, 82 (2015).
Ramachandran, P. et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 575, 512–518 (2019).
Cohen, C. et al. WNT-dependent interaction between inflammatory fibroblasts and FOLR2+ macrophages promotes fibrosis in chronic kidney disease. Nat. Commun. 15, 743 (2024).
O’Sullivan, E. D. et al. Indian Hedgehog release from TNF-activated renal epithelia drives local and remote organ fibrosis. Sci. Transl. Med. 15, eabn0736 (2023).
Srivastava, S. P. et al. Renal Angptl4 is a key fibrogenic molecule in progressive diabetic kidney disease. Sci. Adv. 10, eadn6068 (2024).
Eun, M. et al. Chromatin accessibility analysis and architectural profiling of human kidneys reveal key cell types and a regulator of diabetic kidney disease. Kidney Int. 105, 150–164 (2024).
Sun, Y. et al. Blockade of the CD93 pathway normalizes tumor vasculature to facilitate drug delivery and immunotherapy. Sci. Transl. Med. 13, eabc8922 (2021).
Li, Q. et al. An IGFBP7hi endothelial cell subset drives T cell extravasation in psoriasis via endothelial glycocalyx degradation. J. Clin. Investig. 133, e160451 (2023).
Jia, H.-M. et al. Cell cycle arrest biomarkers for predicting renal recovery from acute kidney injury: a prospective validation study. Ann. Intensive Care 12, 14 (2022).
Fiorentino, M. et al. Serial measurement of cell-cycle arrest biomarkers [TIMP-2] · [IGFBP7] and risk for progression to death, dialysis, or severe acute kidney injury in patients with septic shock. Am. J. Respir. Crit. Care Med. 202, 1262–1270 (2020).
Shah, A. M. et al. Large scale plasma proteomics identifies novel proteins and protein networks associated with heart failure development. Nat. Commun. 15, 528 (2024).
Molinari, L. et al. Utility of biomarkers for sepsis-associated acute kidney injury staging. JAMA Netw. Open 5, e2212709 (2022).
Yu, J. et al. Insulin-like growth factor binding protein 7 promotes acute kidney injury by alleviating poly ADP ribose polymerase 1 degradation. Kidney Int. 102, 828–844 (2022).
Stanley, T. L. et al. Relationship of IGF-1 and IGF-binding proteins to disease severity and glycemia in nonalcoholic fatty liver disease. J. Clin. Endocrinol. Metab. 106, e520–e533 (2021).
Ashrafi Jigheh, Z. et al. Empagliflozin attenuates renal and urinary markers of tubular epithelial cell injury in streptozotocin-induced diabetic rats. Ind. J. Clin. Biochem. 35, 109–114 (2020).
Januzzi, J. L. et al. Insulin-like growth factor binding protein 7 predicts renal and cardiovascular outcomes in the canagliflozin cardiovascular assessment study. Diabetes Care 44, 210–216 (2021).
Kang, H. M. et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 21, 37–46 (2015).
Tran, M. T. et al. PGC1α drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature 531, 528–532 (2016).
Miguel, V., Shaw, I. W. & Kramann, R. Metabolism at the crossroads of inflammation and fibrosis in chronic kidney disease. Nat. Rev. Nephrol. 21, 39–56 (2025).
Knol, M. G. E., Wulfmeyer, V. C., Müller, R.-U. & Rinschen, M. M. Amino acid metabolism in kidney health and disease. Nat. Rev. Nephrol. 20, 771–788 (2024).
Miguel, V. et al. Renal tubule Cpt1a overexpression protects from kidney fibrosis by restoring mitochondrial homeostasis. J. Clin. Investig. 131, e140695 (2021).
Baek, J., He, C., Afshinnia, F., Michailidis, G. & Pennathur, S. Lipidomic approaches to dissect dysregulated lipid metabolism in kidney disease. Nat. Rev. Nephrol. 18, 38–55 (2022).
Midha, A. D. et al. Organ-specific fuel rewiring in acute and chronic hypoxia redistributes glucose and fatty acid metabolism. Cell Metab. 35, 504–516.e5 (2023).
Kwon, Y. et al. Systemic organ wasting induced by localized expression of the secreted insulin/IGF antagonist ImpL2. Dev. Cell 33, 36–46 (2015).
Figueroa-Clarevega, A. & Bilder, D. Malignant drosophila tumors interrupt insulin signaling to induce cachexia-like wasting. Dev. Cell 33, 47–55 (2015).
Morgantini, C. et al. Liver macrophages regulate systemic metabolism through non-inflammatory factors. Nat. Metab. 1, 445–459 (2019).
Zhao, Y. et al. Podocyte OTUD5 alleviates diabetic kidney disease through deubiquitinating TAK1 and reducing podocyte inflammation and injury. Nat. Commun. 15, 5441 (2024).
Fu, Y. et al. Elevation of JAML promotes diabetic kidney disease by modulating podocyte lipid metabolism. Cell Metab. 32, 1052–1062.e8 (2020).
Forbes, J. M. & Thorburn, D. R. Mitochondrial dysfunction in diabetic kidney disease. Nat. Rev. Nephrol. 14, 291–312 (2018).
Mitrofanova, A., Merscher, S. & Fornoni, A. Kidney lipid dysmetabolism and lipid droplet accumulation in chronic kidney disease. Nat. Rev. Nephrol. 19, 629–645 (2023).
Huynh, C., Ryu, J., Lee, J., Inoki, A. & Inoki, K. Nutrient-sensing mTORC1 and AMPK pathways in chronic kidney diseases. Nat. Rev. Nephrol. 19, 102–122 (2023).
Malik, N. et al. Induction of lysosomal and mitochondrial biogenesis by AMPK phosphorylation of FNIP1. Science 380, eabj5559 (2023).
Qu, Q. et al. Lithocholic acid binds TULP3 to activate sirtuins and AMPK to slow down ageing. Nature https://doi.org/10.1038/s41586-024-08348-2 (2024).
Yanagi, T. et al. ULK1-regulated AMP sensing by AMPK and its application for the treatment of chronic kidney disease. Kidney Int. 106, 887–906 (2024).
Yuan, Z., Guan, Y., Chatterjee, D. & Chin, Y. E. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science 307, 269–273 (2005).
Bi, J. et al. TMEM25 inhibits monomeric EGFR-mediated STAT3 activation in basal state to suppress triple-negative breast cancer progression. Nat. Commun. 14, 2342 (2023).
Li, Y.-J. et al. Fatty acid oxidation protects cancer cells from apoptosis by increasing mitochondrial membrane lipids. Cell Rep. 39, 110870 (2022).
Mi, Y. et al. Loss of fatty acid degradation by astrocytic mitochondria triggers neuroinflammation and neurodegeneration. Nat. Metab. 5, 445–465 (2023).
Ye, Y. et al. SIRT2 counteracts primate cardiac aging via deacetylation of STAT3 that silences CDKN2B. Nat. Aging 3, 1269–1287 (2023).
Cai, X., Wang, L., Wang, X. & Hou, F. Silence of IGFBP7 suppresses apoptosis and epithelial mesenchymal transformation of high glucose induced‑podocytes. Exp. Ther. Med. 16, 1095–1102 (2018).
Mimura, I., Chen, Z. & Natarajan, R. Epigenetic alterations and memory: key players in the development/progression of chronic kidney disease promoted by acute kidney injury and diabetes. Kidney Int. 107, 434–456 (2025).
Zhong, D. et al. Genetic or pharmacologic blockade of mPGES-2 attenuates renal lipotoxicity and diabetic kidney disease by targeting Rev-Erbα/FABP5 signaling. Cell Rep. 43, 114075 (2024).
Mise, K. et al. NDUFS4 regulates cristae remodeling in diabetic kidney disease. Nat. Commun. 15, 1965 (2024).
Elwakiel, A. et al. Factor XII signaling via uPAR-integrin β1 axis promotes tubular senescence in diabetic kidney disease. Nat. Commun. 15, 7963 (2024).
Lee, L. E., Doke, T., Mukhi, D. & Susztak, K. The key role of altered tubule cell lipid metabolism in kidney disease development. Kidney Int. 106, 24–34 (2024).
Mukhi, D. et al. ACSS2 gene variants determine kidney disease risk by controlling de novo lipogenesis in kidney tubules. J. Clin. Investig. 134, e172963 (2024).
Prutsch, N. et al. STAT3 couples activated tyrosine kinase signaling to the oncogenic core transcriptional regulatory circuitry of anaplastic large cell lymphoma. Cell Rep. Med. 5, 101472 (2024).
Xiao, J. et al. L3MBTL3 and STAT3 collaboratively upregulate SNAIL expression to promote metastasis in female breast cancer. Nat. Commun. 16, 231 (2025).
Liu, Y. et al. STAT3 and its targeting inhibitors in osteosarcoma. Cell Prolif. 54, e12974 (2021).
Wang, J. et al. SHF acts as a novel tumor suppressor in glioblastoma multiforme by disrupting STAT3 dimerization. Adv. Sci. 9, 2200169 (2022).
Ma, L. et al. Lysyl oxidase 3 is a dual-specificity enzyme involved in STAT3 deacetylation and deacetylimination modulation. Mol. Cell 65, 296–309 (2017).
Liu, R. et al. Role of transcription factor acetylation in diabetic kidney disease. Diabetes 63, 2440–2453 (2014).
Claude-Taupin, A. et al. The AMPK-sirtuin 1-YAP axis is regulated by fluid flow intensity and controls autophagy flux in kidney epithelial cells. Nat. Commun. 14, 8056 (2023).
Bennett, C. F., Latorre-Muro, P. & Puigserver, P. Mechanisms of mitochondrial respiratory adaptation. Nat. Rev. Mol. Cell Biol. 23, 817–835 (2022).
Zhao, Q. et al. Targeting mitochondria-located circRNA SCAR alleviates NASH via reducing mROS output. Cell 183, 76–93.e22 (2020).
Zhang, Y. et al. ZHX2 emerges as a negative regulator of mitochondrial oxidative phosphorylation during acute liver injury. Nat. Commun. 14, 7527 (2023).
Inoue, S. et al. Short-term cold exposure induces persistent epigenomic memory in brown fat. Cell Metab. 36, 1764–1778.e9 (2024).
Varuzhanyan, G. et al. PGC-1α drives small cell neuroendocrine cancer progression toward an ASCL1-expressing subtype with increased mitochondrial capacity. Proc. Natl. Acad. Sci. USA 121, e2416882121 (2024).
Xu, C. et al. Regulation of pericyte metabolic reprogramming restricts the AKI to CKD transition. Metabolism 145, 155592 (2023).
Huang, Y. et al. IRF1-mediated downregulation of PGC1α contributes to cardiorenal syndrome type 4. Nat. Commun. 11, 4664 (2020).
Nie, Y. et al. STAT3 inhibition of gluconeogenesis is downregulated by SirT1. Nat. Cell Biol. 11, 492–500 (2009).
Wang, B., Hsu, S.-H., Frankel, W., Ghoshal, K. & Jacob, S. T. Stat3-mediated activation of microRNA-23a suppresses gluconeogenesis in hepatocellular carcinoma by down-regulating Glucose-6-phosphatase and peroxisome proliferator-activated receptor gamma, coactivator 1 alpha. Hepatology 56, 186–197 (2012).
Meng, T. et al. Tamoxifen induced cardiac damage via the IL-6/p-STAT3/PGC-1α pathway. Int. Immunopharmacol. 125, 110978 (2023).
Inoue, H. et al. Role of STAT-3 in regulation of hepatic gluconeogenic genes and carbohydrate metabolism in vivo. Nat. Med. 10, 168–174 (2004).
Liu, F., Edelmann, M., Piironen, V. & Kariluoto, S. 5-methyltetrahydrofolate is a crucial factor in determining the bioaccessibility of folate in bread. J. Agric. Food Chem. 70, 13379–13390 (2022).
Griffith, T. M., Chaytor, A. T., Bakker, L. M. & Edwards, D. H. 5-methyltetrahydrofolate and tetrahydrobiopterin can modulate electrotonically mediated endothelium-dependent vascular relaxation. Proc. Natl. Acad. Sci. USA 102, 7008–7013 (2005).
Annibal, A. et al. Regulation of the one carbon folate cycle as a shared metabolic signature of longevity. Nat. Commun. 12, 3486 (2021).
Sullivan, M. R. et al. Methionine synthase is essential for cancer cell proliferation in physiological folate environments. Nat. Metab. 3, 1500–1511 (2021).
Viswanathan, M., Urrutia, R. P., Hudson, K. N., Middleton, J. C. & Kahwati, L. C. Folic acid supplementation to prevent neural tube defects updated evidence report and systematic review for the US Preventive Services Task Force. Obstet. Anesthesia Dig. 44, 63–64 (2024).
An, P. et al. Micronutrient supplementation to reduce cardiovascular risk. J. Am. Coll. Cardiol. 80, 2269–2285 (2022).
Valiño-Rivas, L. et al. Growth differentiation factor-15 preserves Klotho expression in acute kidney injury and kidney fibrosis. Kidney Int. 101, 1200–1215 (2022).
Radhakrishnan, K. et al. Orphan nuclear receptor ERR-γ regulates hepatic FGF23 production in acute kidney injury. Proc. Natl. Acad. Sci. USA 118, e2022841118 (2021).
Xu, X. et al. Efficacy of folic acid therapy on the progression of chronic kidney disease: the renal substudy of the China Stroke Primary Prevention Trial. JAMA Intern. Med. 176, 1443 (2016).
Liu, M. et al. Relationship of dietary intake of food folate and synthetic folic acid intake from fortified foods with all-cause mortality in individuals with chronic kidney disease. Food Funct. 15, 559–568 (2024).
Hasegawa, K. et al. Renal tubular Sirt1 attenuates diabetic albuminuria by epigenetically suppressing Claudin-1 overexpression in podocytes. Nat. Med. 19, 1496–1504 (2013).
Gao, L. et al. STING/ACSL4 axis-dependent ferroptosis and inflammation promote hypertension-associated chronic kidney disease. Mol. Ther. 31, 3084–3103 (2023).
Wang, J. et al. Inhibition of METTL3 attenuates renal injury and inflammation by alleviating TAB3 m6A modifications via IGF2BP2-dependent mechanisms. Sci. Transl. Med. 14, eabk2709 (2022).
Shan, R. et al. Madecassoside alleviates acute kidney injury by regulating JNK-mediated oxidative stress and programmed cell death. Phytomedicine 123, 155252 (2024).
Acknowledgements
We thank the Anhui Medical University First Hospital for provision of tissue samples and the Center for Scientific Research of Anhui Medical University for valuable assistance. This work was financially supported by National Natural Science Foundation of China (No. 82570824 to X.-M.M., 82270738 to X.-M.M., 82500846 to J.-T.Y.), the Natural Science Foundation of Anhui Province (No.2508085QH348 to X.-W.H.), and Anhui Medical University Basic and Clinical Cooperative Research Promotion Plan (2023xkiT009 to X.-M.M.).
Author information
Authors and Affiliations
Contributions
X.-M.M. and J.-T.Y. designed the study. X.-M.M., Y.-Y.J., and J.J. conceptualized and oversaw the project. J.-T.Y., X.-W.H., J.-N.W., and Q.Y. performed most of the experiments. J.-T.Y., X.-Y.L., R.-R.S., and X.-Y.S. performed in vivo studies. J.-T.Y., R.H., Y.-Q.W., and F.Z. performed in vitro studies. T.P. performed protein purification and plasmid construction. J.-T.Y., X.-W.Y., J.W., F.-H.L. J.-G.W., and Y.F. interpreted the results. X.-M.M., J.-T.Y., and X.-W.H. acquired funding to support the project. J.-T.Y., X.-W.H., J.-N.W., and X.-M.M. wrote the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Pedro Geraldes, Joachim Jankowski and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yu, Jt., Hu, Xw., Wang, Jn. et al. Renal insulin-like growth factor binding-protein 7 is a critical promoter of progressive diabetic kidney disease. Nat Commun 17, 30 (2026). https://doi.org/10.1038/s41467-025-66490-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-66490-5
