
Abstract
Leber hereditary optic neuropathy (LHON), a maternally inherited mitochondrial disorder, results from point mutations in mitochondrial DNA (mtDNA), primarily affecting the MT-ND4 gene. To date, no animal model harboring authentic LHON mutations has been available, limiting therapeutic development. However, when we attempted to generate such models using mitochondrial base editors, we found that activity-enhanced DddA11-based cytosine base editors (DdCBEs) induce off-target mtDNA mutations and developmental arrest in embryos. Using a high-fidelity DdCBE (Hifi-DdCBE), we successfully generate mice carrying the pathogenic MT-ND4 G11778A mutation, the most common LHON variant. These mice exhibit hallmark phenotypes, including retinal ganglion cell loss and impaired visual function. Intravitreal delivery of adeno-associated virus encoding TALE-linked deaminases (TALEDs) restores both phenotype and genotype in these mice. Furthermore, optimized TALEDs corrects the ND4 mutation with minimal off-target effects in LHON patient-derived cells, highlighting the potential of mitochondrial base editing as a therapeutic strategy for mtDNA-associated diseases.
Similar content being viewed by others


Introduction
Mitochondria produce ATP and supply energy required for cellular metabolism. They contain circular, double-stranded DNA encoding 13 proteins and 22 tRNAs and 2 rRNAs, essential for cellular respiration via the mitochondrial oxidative phosphorylation system (OXPHOS)1.
Dysfunctional mitochondria promote severe defects in several tissues, especially in neural and metabolic organs where mitochondrial activities are particularly active and crucial2. These disorders are categorized as mitochondrial diseases, which have an incidence of 1 in 50003. Most mitochondrial diseases are caused by point mutations in the mitochondrial DNA (mtDNA)4.
Leber hereditary optic neuropathy (LHON) is a major mitochondrial disease, notably prevalent in America and Europe. LHON, which accounts for nearly 15% of all mitochondrial genetic diseases5, is caused by mutations in mitochondrial complex I subunits. Three mtDNA point mutations, m.G11778A in MT-ND4, m.T14484C in MT-ND6, and m.G3460A in MT-ND1, are responsible for over 90% of LHON cases. Particularly, the MT-ND4 mutation (m.G11778A) is the most common, with a prevalence of ~69%. In most patients, the pathogenic mutation is present in a homoplasmic state and symptoms in patients appear in young adulthood, with around 80% of patients being male. Furthermore, some individuals exhibit incomplete penetrance, whereby the absence of disease symptoms despite carrying the mutation may, in part, result from a lack of environmental factors such as smoking and alcohol consumption, which are known to contribute to the pathogenic mechanism5,6,7. LHON primarily causes a gradual loss of central and color vision due to retinal ganglion cell (RGC) degeneration. This progressive vision loss gradually deteriorates over months, often resulting in a visual acuity of 20/200 or less, making the patient legally blind8. Despite the severity of the symptoms, only a limited number of pharmaceutical treatments are currently available. Idebenone, a synthetic analog of coenzyme Q10, is currently the only approved treatment for LHON9. However, Idebenone is a compound that helps improve mitochondrial function; thus, it only temporarily relieves symptoms and is ineffective for most patients10. Furthermore, the high cost of Idebenone limits access for patients to sustained treatment. Hence, gene therapy may offer a promising and effective treatment for LHON. Gene therapy offers lasting therapeutic benefits from just one dose, positioning it as an effective approach for genetic disorders11. For instance, Luxturna, a gene therapy delivering RPE65 cDNA to patients with an inherited retinal disorder (IRD), demonstrated significant clinical efficacy12. However, a similar genetic approach delivering MT-ND4 cDNA to LHON patients showed no significant improvements13. Recently, Casgevy, the first therapy for genetic disorders using the CRISPR/Cas9 system, provided effective treatment in patients with sickle cell disease (SCD)14. Likewise, gene editing-based therapy may effectively treat mitochondrial genetic disorders. However, applying the CRISPR/Cas9 system to mitochondrial genetic disorders poses challenges, particularly in delivering guide RNA effectively into the mitochondria15,16. To address this limitation, we design a gene therapy tool specifically for mitochondrial genetic disorders using an mtDNA-targeted base editor17.
The mtDNA targeting base editors, including the DddA-derived cytosine base editors (DdCBEs)18 and transcription activator-like effector (TALE)-linked deaminases (TALEDs)19, enable efficient C(G) to T(A) and A(T) to G(C) conversions, respectively, on the mtDNA. However, canonical DdCBEs are limited to conversion only in the 5’–TC–3’ motif, and TALEDs induce broad RNA off-target effects. Therefore, Mok et al. successfully engineered DdCBE via DddA6/11 to enable C(G) to T(A) conversion in non-TC motifs20, and Cho et al. significantly reduced RNA off-target effects using engineered TadA8e (TALED-V28R)21. Using these advanced methods, we aim to generate MT-ND4 mutant mice mimicking human LHON mutations via DddA11. Additionally, we seek to perform mitochondrial-targeted gene editing-based gene therapy using TALED-V28R. However, DddA11 induces widespread off-target effects in the mitochondrial genome, leading to developmental arrest in mouse embryos.
In this study, we utilize high-fidelity DdCBEs (Hifi-DdCBEs), which reduce off-target effects on the mitochondrial genome while enabling effective on-target editing22. Using this tool, we successfully generate MT-ND4 mutant mice mimicking the LHON mutation. In addition, we perform an adeno-associated virus (AAV)-mediated mitochondrial gene editing-based therapy via an intravitreal injection of TALED-V28R to rescue the ND4 mutation in RGCs.
Results
DddA11 targets the mtDNA, inducing widespread off-target effects in the mitochondrial genome and causing developmental arrest in mouse embryos
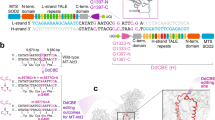
First, we designed a DddA11 construct targeting the MT-ND4 site in the mouse mitochondrial genome. The desired editing site was the m.G11185 locus, which mimics the human pathogenic m.G11778 site. Mutation levels induced by DddA11 were affected by the DddA toxin split site; we investigated editing efficiencies using two plasmid constructs encoding DddA11 with different split sites of the DddA toxin-(L1397N + R1397C) and (L1397C + R1397N)-in the mouse fibroblast (NIH3T3) and mouse melanoma cell lines (B16F10) (Supplementary Fig. 1 a, b). We confirmed increased on-target editing efficiencies in both cell lines after using the m.G11185A and DddA11 (L1397C + R1397N) combination. Thus, the DddA11 construct containing the L1397C + R1397N-encoded mRNAs were microinjected into 1-cell stage embryos (Fig. 1a). We observed a successful on-target editing efficiency of 38–75% in blastocysts; however, the editing efficiencies in pups from the DddA11-injected embryo revealed no difference from the buffer-injected blastocysts (Fig. 1b). Furthermore, the DddA11 genotyping results of the injected blastocysts indicated that DddA11 promotes remarkable C(G) to T(A) conversion; however, the on-target editing was not precise and induced a high frequency of undesired editing (m.G11182A and m.C11188T, which encodes S339N and T341I, hereinafter referred to as S339N and T341I) (Fig. 1c). To test whether these phenomena were unique to the MT-ND4 site, we conducted a similar analysis at another LHON pathogenic site in MT-ND1 (m. G2904, mimicking the human m. G3460 mutation)23. We screened out 10 DddA11 combinations with different TALE sequences in the NIH3T3 cell line (Supplementary Fig. 2a, b). The genotyping results for the MT-ND1 site also revealed the same phenomenon (Supplementary Fig. 2c, d). Thus, we assumed that the C(G) to T(A) conversion induced by DddA11 causes a wide range of off-target effects, prompting us to perform whole mitochondrial genome sequencing. In correlation with our assumption, a wide range of off-target effects in the mitochondrial genome were observed in the whole mitochondrial genome sequencing results following DddA11 injection into the blastocysts (Fig. 1d). Therefore, we infer that DddA11 induces numerous off-target mutations in the mitochondrial genome and leads to blastocyst developmental arrest, making it unsuitable for creating disease models in mice.

a Schematic illustration of DddA11 in mouse MT-ND4 gene. Base editing window is annotated with black, and the TALE binding sequence with gray. Desired editing locus was shown in red (m.G11185A). b Editing efficiency of m.G11185A in buffer injected blastocysts (n = 5), DddA11 injected blastocysts (n = 20), and DddA11 injected pups (n = 35). The exact p-values were 1.33861E-14 in buffer injected blastocysts and DddA11 injected blastocysts, 0.419 in buffer injected blastocysts and DddA11 injected pups. (ns not significant, ***p < 0.001 using Student’s two-tailed t-test). c MT-ND4 gene mutation alleles induced by DddA11 in blastocysts. Yellow bars indicate the frequencies of m.G11185A without undesired editing. Targeted editing loci were shown in red (m.G11185A), and undesired editing loci were in blue. d Mitochondrial genome-wide off-target editing in buffer injected blastocysts and DddA11 injected blastocysts. On-target editing efficiencies were shown in blue (m.G11185A), and off-targets were in black.
Hifi-DdCBEs enable efficient C(G) to T(A) conversion to produce mutant mice
After confirming the off-target effects of DddA11 and its cytotoxicity in mouse embryos, we employed Hifi-DdCBEs incorporating the T1391A variant of DddA11 to avoid embryonic arrest and minimize widespread mitochondrial off-target effects (Fig. 2a). To investigate the toxicity of DddA11 and the safety of Hifi-DdCBEs, we measured cell viability in B16F10 cells for comparison. The results revealed no difference in cell viability between untreated and Hifi-DdCBE-treated cells, whereas DddA11 exhibited significant cellular toxicity (Supplementary Fig. 2e). After confirming the safety of Hifi-DdCBEs, we successfully applied them to produce MT-ND4 mutant mice (Fig. 2b and Supplementary Table 1). The on-target (m.G11185A) editing efficiencies were most prevalent in edited alleles in blastocysts (Fig. 2c) and pups (Fig. 2d). Compared to DddA11, Hifi-DdCBEs achieved effective on-target editing with reduced undesired editing (S339N and T341I) in blastocysts and pups (Fig. 2e). Furthermore, Hifi-DdCBEs promoted minimal off-target effects across the whole mitochondrial genome compared to DddA11 (Fig. 2f and Supplementary Fig. 3). The proportions of widespread off-target cytosine to total cytosine in the Hifi-DdCBE-injected blastocysts and pups were significantly lower compared to those in DddA11-injected blastocysts (Fig. 2g). In addition, we confirmed no genetic mosaicism in several organs of mutant mice through genotyping (Supplementary Fig. 4a) and that these induced mutations were maintained after 35 weeks (Supplementary Fig. 4b). Moreover, mutant loads in mutant mice were transmitted to the offspring (Supplementary Fig. 4c), regardless of the mother’s age (Supplementary Fig. 4d). These results suggest that Hifi-DdCBEs represent an effective method for creating mtDNA mutant mice by reducing genome-wide mitochondrial off-target effects and avoiding developmental arrest.

a Schematic illustration of Hifi-DdCBE in mouse MT-ND4 gene. Base editing window is annotated with black, and the TALE binding sequence with gray. Desired editing locus was shown in red (m.G11185A). b Editing efficiency of m.G11185A in buffer injected blastocysts (n = 5), Hifi-DdCBE injected blastocysts (n = 15), and Hifi-DdCBE injected pups (n = 8). The exact p-values were 7.5902E-13 in buffer injected blastocysts and Hifi-DdCBE injected blastocysts, and 9.87332E-11 in Hifi-DdCBE injected pups. (***p < 0.001 using Student’s two-tailed t-test). Bars indicate median efficiency in each groups. c MT-ND4 gene mutation alleles induced by Hifi-DdCBE in blastocysts. Yellow bars indicate the frequencies of m.G11185A without undesired editing. Desired editing locus was shown in red (m.G11185A), and undesired editing loci (m.G11182A and m.C11188T) were in blue. d MT-ND4 gene mutation alleles induced by Hifi-DdCBE in pups. Yellow bars indicate the frequencies of m.G11185A without undesired editing. Desired editing locus was shown in red (m.G11185A), and undesired editing loci (m.G11182A and m.C11188T) were in blue. e Proportion of MT-ND4 gene mutation alleles induced in DddA11 injected blastocysts (n = 8), Hifi-DdCBE injected blastocysts (n = 8), and Hifi-DdCBE injected pups (n = 5). Alleles are distributed into 4 parts: WT (white), m.G11185A only (sky blue), m.G11185A with m.G11182A and m.C11188T (navy), and only m.G11182A and m.C11188T editing frequencies (gray). f Mitochondrial genome-wide off-target editing in buffer injected blastocyst, Hifi-DdCBE injected blastocysts, and Hifi-DdCBE injected pups. Only G11185A frequencies shown in blue, G11185A with m.G11182A and m.C11188T in red, and off-targets were in black. g Relative ratio of mitochondrial genome-wide off-target activities occurring outside of editing range in buffer injected blastocysts (n = 2), DddA11 injected blastocysts (n = 6), Hifi-DdCBE injected blastocysts (n = 5), and Hifi-DdCBE injected pups (n = 3). The exact p-values were 0.0017 in DddA11 injected blastocyst and Hifi-DdCBE injected blastocyst, 0.0007 in DddA11 injected blastocyst and Hifi-DdCBE injected pup. Error bars indicate standard error of the mean (s.e.m.) for biologically independent samples. (**p < 0.01, ***p < 0.001 using Student’s two-tailed t-test).
MT-ND4 mutant mice exhibit RGC degeneration and RGC-specific genotypes
Based on patient case reports, LHON disease does not indicate other tissue defects. We assumed that mutant mice reflected normal conditions across various tissues and biochemistry. To evaluate potential abnormalities in other tissues, we conducted histological and serum analyses. These assessments revealed that mutant mice did not exhibit any notable abnormalities in other tissues compared to WT (Supplementary Fig. 5a, b). The major cause for loss of vision in LHON patients derives from the degeneration of RGCs. Thus, we examined the histology of the retina to investigate the degeneration of RGCs in MT-ND4 mutant mice. We also intravitreally injected rotenone (a mitochondrial complex 1 inhibitor known to induce an LHON-like phenotype) into wild-type mice as a positive control. In line with previous reports24,25, rotenone treatment reduced retinal thickness and RGC populations. Additionally, hematoxylin and eosin (H&E) staining results indicated that MT-ND4 mutant mice with a heteroplasmy level of m.G11185A exceeding 20% which determined from toe-derived DNA presented a reduced retinal thickness compared to WT and dimethyl sulfoxide (DMSO)-treated control. (Fig. 3a, Supplementary Fig. 6a). Quantitative values also indicated that MT-ND4 mutant mice with over 20% m.G11185A heteroplasmy levels exhibited reduced total retinal thickness, ganglion cell layer (GCL) thickness, and RGC populations (Fig. 3b–d).

a H&E-stained retinal sections of WT, DMSO-treated, Rotenone-treated, and MT-ND4 mutant mice with varying m.G11185A heteroplasmy (<10%, 10–20%, 20–30%, 50–60%, >70%). Enlarged views (×4, ×10, ×40) highlight layer alterations (GCL ganglion cell layer, IPL Inner plexiform layer, INL inner nuclear layer, OPL outer plexiform layer, ONL outer nuclear layer, IS/OS inner segment/outer segment). b Quantifications of total retinal thickness, Rotenone (p = 0.002) vs DMSO, <10% (p = 0.93),10–20% (p = 0.9), 20–30% (p = 0.002), 50–60% (p = 0.002), >70% (p = 0.0004) vs WT. c Quantifications of GCL thickness: Rotenone (p = 0.0002) vs DMSO, <10% (p = 0.9) 10–20% (p = 0.5), 20–30% (p = 0.0002), 50–60% (p = 0.00002), >70% (p = 0.00004) vs WT. d Quantifications of GCL cell counts: Rotenone (p = 0.00001) vs DMSO, <10% (p = 0.1),10–20% (p = 0.4), 20–30% (p = 0.000001), 50–60% (p = 0.00016), >70% (p = 0.0000001) vs WT. Error bars, s.e.m. (b-d: <10%, 10-20%, 20-30%, >70% n = 6; DMSO, Rotenone, 50–60% n = 4). e OCT images at ±10° from optic nerve head show retinal thinning in mutants. f Total retinal thickness, p = 0.006. g inner retinal layer, p = 0.004. h RNFL + GCL, p = 0.01. (f-h: WT n = 4, mutant n = 6). i Immunohistochemistry staining of RBPMS in enlarged eyeball sections. j Quantification: 20–30% (p = 0.03), 60–90% (p = 0.01) vs WT. (WT n = 6, mutant 20–30% n = 3, 60–90% n = 5). k Whole-mount Brn3a immunofluorescence of RGCs (green) and DAPI (blue). l Quantifications of Brn3a+ cells: 20–30% (p = 0.002), 60–90% (p = 0.011). m Quantifications of DAPI+ cells: 20–30% (p = 0.02), 60–90% (p = 0.3); n Quantifications of Brn3a + /DAPI ratio: 20–30% (p = 0.005), 60–90% (p = 0.0005). (l-n: WT n = 6, mutants n = 3). o Editing efficiencies in m.G11185A locus from MT-ND4 mutant mice toe and RGC. Error bars indicate s.e.m. Statistical analyses: ANOVA with Tukey’s HSD (b–d) or Student’s two-tailed t-test (f–n). Significance threshold: p*<0.05, **p < 0.01, ***p < 0.001.
Next, we performed optical coherence tomography (OCT), a noninvasive method, to measure retinal thickness (Fig. 3e). Furthermore, MT-ND4 mutant mice with over 20% m.G11185A heteroplasmy levels revealed reduced total retinal thickness, inner retinal layer thickness (retinal nerve fiber layer [RNFL] + GCL + inner nuclear layer), and RNFL + GCL thickness (Fig. 3f–h). Next, we investigated a potential mechanism for retinal degeneration caused by the MT-ND4 mutation. Thus, we conducted immunohistochemistry (IHC) analysis using an RBPMS antibody, a marker specific to ganglion cells; mutant mice exhibited reduced diaminobenzidine expression, indicating deficient RGCs (Fig. 3i, j). Furthermore, when immunofluorescence was performed on a whole-mount retina using Brn3a, a ganglion cell-specific marker, mutant mice exhibited a reduction in the number of Brn3a-positive RGCs compared to WT control (Fig. 3k, l). Additionally, the reduced number of 4’,6-diamidino-2-phenylindole (DAPI)-positive cells in mutant mice indicated an overall decrease in RGCs, including Brn3a-positive RGCs (Fig. 3m, n). Taken together, our findings indicate that mutant mice exhibit LHON-like retinal phenotypes when harboring over 20% m.G11185A mutation (Supplementary Fig. 6b). Since the major cause of LHON is RGCs degeneration, we conducted IHC using a cleaved caspase-3 antibody to assess the apoptosis levels in RGCs. However, no apoptotic effects were observed in RGCs of mutant mice (Supplementary Fig. 7a–c). Following the results, we assumed that the degeneration of RGC was caused by the caspase-3 independent pathway. Thus, we conducted IHC using the LC3B antibody to assess autophagy-related cell death in RGCs. Compared to WT control, mutant mice exhibited significantly increased LC3B-positive cells. Therefore, we presumed that autophagy mediated the observed RGC death in mutant mice (Supplementary Fig. 7d–h). Additionally, there were several reports that RGC dysfunction leads to an elevated neuroinflammatory response. Interestingly, mutant mice also exhibited higher neuroinflammatory responses in retina (Supplementary Fig. 7i–k). In conclusion, RGC degeneration in ND4 mutant mice appears to be mediated by autophagy, which may also influence neuroinflammatory activity in the retinal phenotypes at relatively low heteroplasmy levels (over 20%). Despite these findings, we sought to clarify why RGC defects appeared even at relatively low heteroplasmy levels. Although similar heteroplasmy levels were observed across various tissues in mutant mice (Supplementary Fig. 4a), we performed genotypic analysis on isolated RGCs to explore underlying tissue-specific differences. Surprisingly, each isolated RGC harbored higher heteroplasmy levels compared to other tissues (Fig. 3o and Supplementary Fig. 8a, b). This suggests that a higher level of m.G11185A heteroplasmy in RGCs, even without homoplasmy, is sufficient to drive the LHON-like phenotype. Since the S339N and T341I editing induced by the editing tool remains a point of concern, we conducted a histological analysis of mutant mice harboring low m.G11185A heteroplasmy but high S339N and T341I heteroplasmy levels. In this case, mutant mice harboring high S339N and T341I heteroplasmy levels exhibited no difference in retinal thickness and RGC numbers compared to WT control, based on histological analysis (Supplementary Fig. 9a–e). Furthermore, we analyzed oxygen consumption rate and ATP concentration to assess mitochondrial function in mutant mouse embryonic fibroblasts (MEFs), which harbor higher S339N and T341I heteroplasmy levels than the m.G11185A locus. Mutant MEF cell lines exhibited no difference in mitochondrial function compared to WT MEF cells (Supplementary Fig. 9f–h). Thus, the MT-ND4 mutant mouse phenotype was expressed by those with the m.G11185A mutation rather than undesired mutations.
sTALED-V28R enables the restoration of the MT-ND4 mutation locus and promotes therapeutic effects on mitochondrial function in vitro
After confirming the histological defects in mutant mice, we sought to restore the phenotypes using sTALED-V28R, the mitochondrial adenine base editors that correct the mutation. Thus, we evaluated the gene editing efficiency of eight sTALED-V28R candidates through lipofection in MEF cells (Supplementary Fig. 10a, b). Our results demonstrated that several candidates achieved on-target mutation base recovery approaching 10% (Supplementary Fig. 10c). Subsequently, we constructed plasmid vectors for AAV packing the sTALED-V28R candidates with the hSyn promoter for improving the delivery efficiency to RGCs and assessed gene editing efficiency in MEF cells (Supplementary Fig. 11a, b). Two pAAV-sTALED-V28R candidates (L1N + R1C and L2C + R1N) achieved 7.3% and 8.4%, respectively. Although the (L2C + R1N) candidates achieved higher on-target base recovery, it also induced undesired m.A11190G editing within the editing window of sTALED-V28R (Supplementary Fig. 11c). We selected the sTALED-V28R (L1N + R1C) encoding option as the final AAV product combination based on the recovery ratios of the on-target mutation (Fig. 4a). To confirm the gene editing efficiency of AAV, we treated MEF cells with AAV at a multiplicity of infection of 100,000 and evaluated the editing efficiency after 7 days. The results indicated an on-target mutation recovery ratio of approximately 8% (Fig. 4b). Overall, these results suggest that AAV-mediated mitochondrial gene editing via sTALED-V28R achieves higher target specificity than plasmid-based approaches.

a Illustration of selected sTALED-V28R-AAV for MT-ND4 mutant mice mitochondrial gene therapy. TALE sequence shown in navy box and desired locus shown in red (m.G11185A), and undesired edits in MT-ND4 mutant MEF cell line were shown in blue (S339N and T341I). b A heatmap showing base recovery in MT-ND4 mutant MEF cell line #6 after treating sTALED-V28R-AAV via infection. The on-target (m.G11185A) locus is represented with an arrow. Data were shown as means from n = 3 biologically independent samples. c ATP concentrations in WT, MT-ND4 mutant #MEF 6, and MT-ND4 mutant #MEF 6 sTALED-V28R AAV-treated cell lines. The exact p-values were 0.04 on MT-ND4 mutant MEF #6 compared with its sTALED-V28R AAV-treated counterpart, 0.0002 on WT compared with MT-ND4 mutant #MEF 6, and 0.0005 on WT compared with sTALED-V28R AAV-treated MT-ND4 mutant #MEF 6. Error bars indicate standard error of the mean (s.e.m.) for independent samples (n = 3). (*p < 0.05, ***p < 0.001 using Student’s two-tailed t-test). d Oxygen consumption rates of WT, MT-ND4 mutant #6 and MT-ND4 mutant #6 sTALED-V28R AAV- treated MEF cell lines. The error bars indicate the s.e.m. for replicates (n = 4).
Next, we investigated whether the mtDNA mutation could impair ATP production and whether AAV treatment could rescue any potential defect. We found an approximately 30% reduction in ATP production in mutant MEF cells compared to WT MEF cells. However, ATP levels in AAV-treated cells were elevated compared to mutant MEF cells (Fig. 4c). Furthermore, AAV-treated mutant MEF cells exhibited an improved oxygen consumption rate compared to mutant MEF cells (Fig. 4d). These results demonstrate the potential of AAV expressing sTALED-V28R as a gene therapeutic modality.
AAV-mediated mitochondrial gene therapy demonstrates therapeutic efficacy in the histological characteristics of MT-ND4 mutant mice
Building on the possibility of AAV-mediated mitochondrial gene therapy in vitro, we investigated the therapeutic effect of AAV-packaged sTALED-V28R in vivo. We manufactured AAV with serotype 2 encoding sTALED-V28R under the control of the hSyn promoter, which is known to have high-expression specificity in RGCs25,26, and delivered the AAV into both eyes of 2-week-old mutant mice through intravitreal injection, a time point prior to RGC maturation aimed to prevent the cell death (Fig. 5a). Eight weeks after injection, at postnatal week 10, we evaluated the therapeutic effects to assess phenotypic improvement. First, we conducted OCT analysis to investigate whether AAV-mediated mitochondrial gene therapy led to phenotypic improvement in mutant mice harboring over 20% heteroplasmy of the m.G11185A mutation (Fig. 5b and Supplementary Fig. 12a). Mutant mice injected with phosphate-buffered saline (PBS) in both eyes served as the negative control group. The retinas of AAV-injected mutant mice exhibited remarkable improvements compared to those of PBS-injected controls. Furthermore, the retinal thickness in AAV-treated mutant mice was comparable to that of WT mice (Fig. 5c–e). To evaluate the therapeutic effects of the AAV-mediated gene therapy on RGCs, we performed immunofluorescence analysis using Brn3a. In line with the OCT results, the number of RGCs in the AAV-injected mutant mice increased by approximately 1.34-fold compared to the PBS-injected control, indicating that the RGC dysfunction observed in the LHON mutant mice was restored by AAV-mediated gene editing (Fig. 5f–i and Supplementary Fig. 12b). Furthermore, as shown previously (in Fig. 3o), mutant mice exhibited elevated heteroplasmy levels at the m.G11185 locus, especially within RGCs, as shown in Fig. 3o and Supplementary Fig. 8b. To investigate whether the observed phenotypic recovery resulted from genotypic restoration, we isolated RGCs from mutant mice treated with either PBS or AAV and conducted genotypic analyses. Although mutant mice exhibited elevated heteroplasmy levels in RGCs compared to other tissues (Fig. 3o and Supplementary Fig. 8b), AAV administration reduced these levels to those comparable to the toe, and in some cases, even lower than in the toe or other tissues (Fig. 5j and Supplementary Fig. 13a), indicating that the m.G11185A mutation in RGCs was successfully corrected by AAV treatment. To more clearly assess the base recovery efficiency mediated by AAV, we administered PBS into the left eye and AAV into the right eye of each mutant mouse, followed by a genotypic analysis of RGCs isolated from both eyes. In all treated mice, heteroplasmy levels of the m.G11185A mutation in RGCs from the AAV-injected (right) eye were reduced compared to those from the PBS-injected (left) eye (Fig. 5k and Supplementary Fig. 13b). Despite variations among individuals, correction frequencies ranged from a minimum of 18.65% to a maximum of 64.55% compared to pre-treatment levels (Fig. 5l). The most significant potential risk associated with gene editing therapies is off-target effects. Therefore, we analyzed the off-target effects of our AAV-based gene therapy and found that there were minimal off-target effects compared to wild-type controls in both nuclear and mtDNA (Supplementary Fig. 14a, b). Following these results, our data suggest that AAV-mediated mitochondrial DNA editing can serve as a promising treatment approach for patients with mitochondrial diseases.

a Schematic illustrations of mitochondrial gene therapy using sTALED-V28R AAVs. Left AAVs in red, right in blue. sTALED-V28R AAVs were intravitreally injected at 2 weeks of age; phenotypic assessments were performed at 10 weeks. Created in BioRender. Kim, K. (2025) https://BioRender.com/70u65f8. b OCT images of retina in WT, PBS-injected, and sTALED-V28R AAVs-injected MT-ND4 mutant mice. Scale bar: 200 μm. c Retinal thickness quantification in WT (n = 3), PBS-injected, and sTALED-V28R AAVs-injected (n = 5) MT-ND4 mutant mice. p = 0.011 (PBS vs AAV), p = 0.6 (AAV vs WT). d Inner retinal layer thickness quantification. p = 0.011 (PBS vs AAV), p = 0.8 (AAV vs WT). e RNFL + GCL thickness quantification. p = 0.007 (PBS vs AAV), p = 0.2 (AAV vs WT). f Brn3a immunofluorescence staining in whole-mount retinas of WT, PBS-injected, and AAV-injected MT-ND4 mutant mice. DAPI (blue) stains nuclei; Brn3a (green) marks RGCs. g Quantification of Brn3a-positive cells. p = 0.001 (PBS vs AAV), p = 0.81 (AAV vs WT). h Quantification of DAPI-positive cells. p = 0.04 (PBS vs AAV), p = 0.02 (AAV vs WT). i Brn3a(+) nuclei (%) quantification. p = 0.003 (PBS vs AAV), p = 0.79 (AAV vs WT). j Editing efficiencies at m.G11185A locus in tissue and RGCs from MT-ND4 mutant mice. Both eyes received AAV; RGC numbers used in genotyping indicated above plots. k Editing efficiencies at m.G11185A locus: left RGCs with PBS (red), right RGCs with AAV (cyan). l Correction frequencies at m.G11185A locus in AAV-injected RGCs vs PBS-injected RGCs. Bars represent median from 6 mice. Error bars = s.e.m. Statistical analysis: two-tailed Student’s t-test (c–e, g–i). Significance: *p < 0.05, **p < 0.01, ***p < 0.001.
AAV-mediated mitochondrial gene therapy exhibits improved visual function in MT-ND4 mutant mice
Following the observed improvements in histological characteristics, we investigated whether genotypic and histological recovery would translate into functional visual improvements. First, we conducted optokinetic nystagmus (OKN) and electroretinogram (ERG) tests to investigate whether RGC degeneration led to functional vision deficiency. Mutant mice aged 17 weeks harboring over 20% m.G11185A heteroplasmy (Supplementary Fig. 15a). Unexpectedly, mutant mice revealed no difference in visual acuity in the OKN test compared to WT mice (Supplementary Fig. 15b). Furthermore, no difference was observed in the ERG test between mutant mice and WT mice for either the a-wave, which represents the activity of photoreceptors within the outer retina, or the b-wave, which indicates the activity of bipolar cells and measures the transmission of signals from photoreceptors to the inner retinal layers (Supplementary Fig. 15c, d). Furthermore, there was no decrease in the photopic negative response (PhNR), a measure of RGC function, in mutant mice compared to WT mice (Supplementary Fig. 15e, f). However, since most LHON patients typically exhibit vision loss at 20–30 years of age, it is unlikely that any defects in visual function would be observed in 17-week-old mice. To further assess the therapeutic effects of AAV, we repeated OKN and ERG tests using 26-week-old mutant mice harboring over 20% m.G11185A heteroplasmy. Mutant mice were divided into two groups: bilateral PBS injections and bilateral AAV injections, with WT mice serving as controls (Fig. 6a). While PBS-treated mutants exhibited reduced visual acuity, AAV-treated mice demonstrated a marked improvement in visual performance relative to the PBS group (Fig. 6b, c). In ERG analysis (Fig. 6d), there were no significant differences in a-wave and b-wave amplitudes among the groups (Fig. 6e, f). However, cone-mediated PhNR, a marker of RGC function, was markedly reduced in PBS-injected mutant mice compared to WT (Fig. 6g). Strikingly, PhNR amplitude was restored in the AAV-injected group to levels comparable to WT (Fig. 6g, h). In summary, these findings show that mutant mice have age-related visual impairment akin to LHON, and AAV-mediated mitochondrial gene therapy can restore both genotypic and functional visual deficits.

a m.G11185A editing efficiency in 26-week-old mice with visual phenotypes, with independent-samples for each group (WT, n = 11, PBS injected ND4 mutant, n = 16, AAV injected ND4 mutant, n = 5). b Schematic illustration of the optokinetic nystagmus (OKN). Created in BioRender. Kim, K. (2025) https://BioRender.com/vhbzqbr. c Visual acuity measured in WT (n = 11), PBS-injected (n = 16), and sTALED-V28R AAVs (n = 5) injected MT-ND4 mutant mice. The exact p-values were 0.011 with PBS-injected and sTALED-V28R AAVs injected, 0.19 on sTALED-V28R AAVs injected MT-ND4 mutant mice compared with WT mice. Error bars indicate standard error of the mean (s.e.m.) for biologically independent mice. (n.s. not significant, using Student’s two-tailed t-test). d Schematic illustration of electroretinography (ERG). Created in BioRender. Kim, K. (2025) https://BioRender.com/lpzv9ih. e a-wave amplitude measured in WT (n = 11), PBS-injected (n = 16), and sTALED-V28R AAVs (n = 5) injected MT-ND4 mutant mice, showing no significant difference between the three groups. Error bars indicate standard error of the mean (s.e.m.) for biologically independent mice. (n.s. not significant, using Student’s two-tailed t-test). f b-wave amplitude measured in WT (n = 11), PBS-injected (n = 16), and sTALED-V28R AAVs (n = 5) injected MT-ND4 mutant mice, showing no significant difference between the three groups. Error bars indicate standard error of the mean (s.e.m.) for biologically independent mice. (n.s. not significant, using Student’s two-tailed t-test). g Photopic negative response (PhNR) measured in WT (n = 11), PBS-injected (n = 16), and sTALED-V28R AAVs (n = 5) injected MT-ND4 mutant mice. The exact p-values were 0.025 with PBS-injected and sTALED-V28R AAVs injected, 0.64 on sTALED-V28R AAVs injected MT-ND4 mutant mice compared with WT mice. Error bars indicate standard error of the mean (s.e.m.) for biologically independent mice. (n.s. not significant, *p < 0.05 using Student’s two-tailed t-test). h Recorded electronic waves used in ERG analysis of WT, PBS-injected, and sTALED-V28R AAVs injected MT-ND4 mutant mice. Individual mouse waves are shown in gray, and the average wave for each group is shown as a bold line.
Effective recovery of the pathogenic LHON mutation in patient-derived cells
After confirming that sTALED-V28R-mediated gene recovery can rescue both the phenotype and genotype of LHON in mice, we aimed to develop TALEDs to correct the corresponding mutation (m.G11778A) in human cells derived from LHON patients. Therefore, we first immortalized cells from urine-derived primary cells donated by an LHON patient with the homoplasmic m.G11778A mutation and transfected 210 combinations of TALED-encoding plasmids, designed to target the mitochondrial target site, into patient-derived cells (Fig. 7a, b). Several TALED pairs were active, correcting the mutation with low efficiencies of up to 4.0%. We chose the most active TALED pair to improve the editing efficiency by replacing the mitochondrial targeting sequence (MTS) with other MTS sequences (Fig. 7c). Among a total of 11 MTSs tested, the MTS derived from the human mitochondrial SDHD gene was the most effective, achieving a targeted A-to-G conversion with a 14% editing frequency. To further enhance the editing activity of the TALED pair, we replaced the split DddA moiety with its orthologs, or DddA11, an engineered DddA variant, and found the TALED pair with the WC03 homolog from Ruminococcus sp. AF17-6 split at S257 and SDHD MTS (termed enhanced split TALED [esTALED] for simplicity) was the most efficient, inducing targeted A-to-G conversion with an editing frequency of 35% ± 1% (Fig. 7d and Supplementary Fig. 16). Under optimal transfection conditions, the m.A11778G targeted esTALED achieved base recovery with an editing frequency of 63%, demonstrating a >10-fold improvement compared to the original esTALED (Fig. 7e). However, this adenine base editor was not specific, inducing unwanted edits at several neighboring positions with high frequencies of up to 62%. To minimize unwanted editing, we incorporated the TadA8e V28R or R111S variant21 in esTALED (shown as esTALED-V28R or -R111S in Fig. 7e-g). Furthermore, we replaced TALE moieties in esTALED to shift the editing window (Supplementary Fig. 17a, b). esTALEDs with TadA8e variants significantly reduced undesired edits while inducing the on-target edit with high frequencies ranging from 29 to 37%. In particular, the most selective base editor, esTALED-V28R-L2 + R2, induced on-target edits 3.4-fold more efficiently than cumulative undesired edits, whereas the esTALED with the wild-type TadA8e deaminase induced on-target edits much less frequently than cumulative undesired edits (Fig. 7f).

a Top: Schematic diagram showing the process of improving and developing sTALED. Bottom: Architecture of sTALED pairs designed to target the m.G11778A in human mitochondrial ND4 gene. Double strand DNA sequence is near the MT-ND4 m.G11778A mutation (yellow) that includes the TALE binding sequence (green). MTS: mitochondrial targeting sequence (orange), Taq: 3X HA or 3X FLAG (black), TALE repeats (chartreuse), DddA N-terminal or C-terminal (cyan), TadA8e adenine deaminase (Red). b–f Comparison of correcting pathogenic mutation induced by plasmid encoding the original sTALED and engineered sTALEDs in urine-derived cells from m.G11778A LHON patient. Untreated sample was used as negative control. Data are shown as means from n = 3 biologically independent samples. b Plots indicate the on-target editing efficiency induced by combinations of 210 with different TALE binding locations each of sTALED-8e. c Bar graphs illustrate the on-target A-to-G editing efficiency by the original sTALED (chartreuse) and engineered MTS-sTALEDs. The hsSDHD MTS-sTALED (orange) shows the best on target editing efficiency. Error bars indicate standard error of the mean (s.e.m.) for biologically independent sample (n = 3). d Bar graphs illustrating the on-target A-to-G editing efficiency by hsSDHD MTS-sTALED (orange) and applying various DddA variants or homologs to hsSDHD MTS-sTALED. The SDHD MTS-WC03 DddA homolog-sTALED (red bar) shows the best on target editing efficiency. Error bars indicate standard error of the mean (s.e.m.) for biologically independent sample (n = 3). e Heatmap showing target A-to-G conversions induced by esSTAED-8e-L2 + R2, esSTAED-V28R-L2 + R2 and esSTAED-R111S-L2 + R2. f Plots indicate the on-target editing efficiency and cumulative undesired editing ratio (On target editing efficiency/Sum of editing efficiency in editing window - On target editing efficiency) induced by combinations of 4 with different TALE binding locations each of esTALED-8e (white plots), esTALED-V28R TadA8e variant (cyan plots), and esTALED-R111S TadA8e variant (red plots). Untreated sample was used as negative control (gray plot). g Allele analysis showing target A-to-G conversions induced by esSTAED-V28R-L2 + R2 (Cyan plot in Fig. 6f). Pathogenicity prediction by AlphaMissense and pathogenicity score is divided into likely benign (0–0.34 score), uncertain (0.34–0.564 score), and likely pathogenic (0.564-1 score)27. Error bars indicate standard error of the mean (s.e.m.) for biologically independent sample (n = 3).
We further examined the edited alleles induced by the esTALED-V28R in immortalized cells with the homoplasmic mutation (Fig. 7g). This adenine base editor achieved the desired single A-to-G conversion without undesired edits at an editing frequency of 22.5% (allele-2). In contrast, the top four undesired edits were induced with low frequencies that ranged from 0.7 to 1.7%. The most prevalent undesired edits at nucleotide position 11780 were detected with a low frequency of 1.7%. We also used AlphaMissense27 to assess the potential pathogenicity of undesired edits. The pathogenicity score for the Ile-to-Val variation was low (0.122), indicating it is likely benign (0.000–0.340). In contrast, the His-to-Arg mutation from the LHON mutation, m.A11778G received a high score (0.862), placing it in the likely pathogenic range (0.564–1.000). The other alleles with a undesired edit at position 11774 had a pathogenicity score of 0.385, indicating a Ser-to-Gly variation in the MT-ND4 gene, which falls in the uncertain range (0.340–0.564). However, these alleles were induced with low frequencies, ranging from 0.71 to 0.97%. Furthermore, restoration of mutation leads to elevating mitochondrial protein complex I activity (Supplementary Fig. 18). Taken together, these results demonstrate that our enhanced sTALED-V28R catalyzes the desired single A-to-G conversion, while minimizing undesired edits, in human LHON patient-derived cells with the homoplasmic mutation, providing a basis for its potential application in future gene therapy.
Discussion
In this study, we utilized Hifi-DdCBE, an improved editing tool with significantly reduced off-target effects on the whole mitochondrial genome, to generate mutant mice with the MT-ND4 mutation, which is the most prevalent LHON mutation found in human patients28. The establishment of a mouse model harboring the MT-ND4 mutation enables in vivo histological analyses and quantitative phenotypic assessments. In addition, our model enables detailed in vivo analysis of disease mechanisms and provides a robust preclinical platform for technically or ethically unfeasible experiments in humans, such as retinal tissue staining and next-generation sequencing of isolated ganglion cells. However, one important consideration in the design and execution of this study was that our mutant mice harbored undesired mutations mediated by Hifi-DdCBEs. Undesired mutations were m.G11182A and m.C11188T, which induce S339N, T341I amino acid substitutions in the ND4 gene. We predicted the pathogenic score via Mutpred229, a computational tool for predicting protein structures and assessing the likelihood of disease-causing effects from genetic mutations. The results revealed that the pathogenicity score of the S339N mutation was 0.462 in both mice and humans, suggesting it is likely benign. Although the T341I mutation in the mouse ND4 gene yields a relatively high MutPred score of 0.67, it is unlikely to significantly impact protein function. This is because threonine is the native residue at position 341 in the mouse ND4 gene, whereas isoleucine is the wild-type residue at the corresponding position in the human ND4 gene. This suggests functional conservation between the two species. Furthermore, no functional defects resulting from undesired editing were observed (Supplementary Fig. 9). These results indicate that the observed defects in the mutant mice were solely attributable to the m.G11185A mutation. Nevertheless, precise introduction of only pathogenic mutations is essential for developing reliable gene-edited animal models. Recently, Zhang et al. demonstrated single-nucleotide editing of the same target site (m.G11185A) in mouse cell lines using mitochondrial base editors (mitoBEs)30. Despite the relatively low editing efficiency, a clean, single editing pattern was observed. Based on these findings, we believe that future studies applying mitoBEs to mouse embryos can be useful in validating whether high on-target efficiency can be achieved without undesired editing in vivo. These insights emphasize the potential and limitations of current mitochondrial base editing tools.
Nevertheless, in our model, mutant mice showed various phenotypes associated with LHON, including reduced total retinal thickness, GCL thickness, and decreased RGC populations. Histological analysis revealed degeneration of ganglion cells mediated by autophagy (Fig. 3 and Supplementary Fig. 7). These findings may serve as indirect evidence for elucidating the pathogenic mechanism of LHON. Additionally, the markedly elevated heteroplasmy levels in RGCs, despite low levels in other organs (Fig. 3o and Supplementary Fig 8b), may explain why phenotypic expression can occur even in the absence of homoplasmy in the LHON patients. However, this raises the possibility that mice may be more sensitive to the G11185A mutation than humans, suggesting species-specific differences in threshold effects. Therefore, further studies are warranted to determine whether heteroplasmy alone is sufficient to elicit disease manifestation and clarify the threshold mechanisms across species. In phenotype assessments, significant differences were observed in the visual acuity of the OKN test and cone system PhNR sections in the ERG results between mutant and WT mice (Fig. 6). PhNR is an index for measuring the function of the inner retinal layers, especially for RGCs31. Given that the cone system mediates color vision, the early presentation of color blindness in LHON patients aligns with our findings of cone system dysfunction in mutant mice32,33. This suggests that the phenotypic results observed in mutant mice reflect the early stages of LHON, as the differences are primarily localized to the cone system34. Several cohort reports have shown that patients with LHON possess cone system defects35,36. Based on this hypothesis, our gene editing method could be a promising tool to rescue vision loss. First, we confirmed AAV-mediated gene therapy inducing remarkable improvements in mitochondrial activity in mutant MEF cells. Next, intravitreally injected AAV into 2-week-old mutant mice to prevent LHON phenotypes, suggesting that mtDNA editing can be a protective treatment before disease onset in potential patients harboring the m.G11185A mutation. In correlation with our hypothesis, genotypes of isolated RGCs from AAV-injected mutant mice exhibited significantly decreased heteroplasmy levels at the m.G11185A locus, and this genotypic rescue was accompanied by phenotypic improvements, resulting in the recovery of visual function. While AAV-mediated gene editing therapy carries potential risks, such as prolonged genome editing from sustained expression and potential immune responses, our results demonstrate its therapeutic potential. This approach could correct pathogenic mutation, potentially enabling recovery of compressed RGCs and improving vision. In conclusion, our mtDNA-targeted gene therapy demonstrates potential as a therapeutic strategy for LHON.
As a proof-of-principle, we also presented esTALEDs to correct the homoplasmic m.G11778A mutation in LHON patient-derived cells. By optimizing MTS and incorporating the WC03 homolog from Ruminococcus sp., we achieved over a 10-fold improvement in editing efficiency. Furthermore, we incorporated TadA8e variants (V28R and R111S) in esTALEDs to minimize unwanted edits. These findings underscore the potential of TALEDs as an efficient tool for mitochondrial gene therapy.
Methods
Ethics statement
This study was approved by the Institutional Animal Care and Use Committee of the Korea University (approval number: KOREA-2024-0019). All procedures followed the guidelines of the Association for Research in Vision and Ophthalmology (ARVO) Statement for the Use of Animals in Ophthalmic and Vision Research. The human subjects research in this study was approved by the Institutional Review Board of Seoul National University Bundang Hospital (No. IRB-B-2211-794-301), and written informed consent was obtained from all subjects. Source: human urine, growth properties: adherent. Donor: LHON m.G11778A patient (Sex: M, Age: 19). We obtained written informed consent from the LHON patient himself.
Mice
We used C57BL/6N mice, which carry the rd8 mutation in the Crb1 gene. Since rd8 affects the outer retina, whereas LHON primarily involves the ganglion cell layer and retinal nerve fiber layer, we distinguished LHON-specific phenotypes from background effects. ND4 mutant mice generated on the C57BL/6N background exhibited RGC abnormalities consistent with LHON. To confirm that these findings were not strain-specific, we also generated m.G11185A mutant mice on the C57BL/6J background and observed the same pathological features (Supplementary Fig. 19). C57BL/6N mice were purchased from Taconic. The 6-week-old female C57BL/6N mice were superovulated by intraperitoneally injecting CARD hyperOva (Cosmo bio) and chorionic gonadotropin human (Sigma-Aldrich) over a 48 h interval. The superovulated female mice were mated with male C57BL/6N mice and sacrificed by obtain 1-cell zygotes from oviducts next day. Zygotes were washed in M2 medium (Sigma-Aldrich) with hyaluronidase (Sigma-Aldrich) and then, washed in M2 medium to remove hyaluronidase. After microinjection, Injected embryos were culture in KSOM medium (Sigma-Aldrich) at 37 °C in humidified atmosphere containing 5% CO2 for next day. And embryos were transferred into the pseudopregnant ICR female mice at two cell stage. In histological analysis, 10 weeks old male mice were used because mitochondrial genetic disease mainly occurred in male, and functional analysis were executed in 17 weeks and 26 weeks old in both male and female mice. Female ICR strain mice for surrogate mothers were purchased from Taconic. The mice were raised in a specific pathogen-free facility in Korea University and Seoul National University with a 12 h dark-light cycle, constant temperature (20–26 °C), and humidity (40–60%).
Cell lines
B16F10 and NIH3T3 cells were purchased from American Type Culture Collection (ATCC) (CRL-1658, CRL-6475). B16F10 cells were cultured in Dulbecco’s Modified Eagle Medium (Welgene) with supplemented 10% fetal bovine serum (FBS) (Welgene). NIH3T3 cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) with 10% bovine calf serum (BCS) (Welgene). Mouse Embryonic Fibroblast (MEF) cell lines were generated with MT-ND4 mutant mice, and cultured in DMEM with 10% FBS, 1% Penicillin (Welgene), 1M L-Glutamine (Welgene). The human subjects research in this study was approved by the Institutional Review Board of Seoul National University Bundang Hospital (No. IRB-B-2211-794-301), and written informed consent was obtained from all subjects. Human Urine-derived cells (UDC) were isolated using the protocol previously described37. These primary UDCs were infected with Lenti-SV40T (puro) lentivirus (abm) and selected with puromycin to obtain immortalized UDCs. The provided protocol was followed in detail. Both primary and immortalized UDCs were cultured on plates coated with 0.1% gelatin (Welgene) using REGM culture medium (Renal epithelial cell growth medium bullet kit) (Lonza). Cells were incubated at 37 °C with 5% CO2 and passed when approaching 80% confluency.
Plasmid construction
We used DddA11, Hifi-DdCBE, sTALED-V28R -encoding plasmids from a previous study21,22. In brief, plasmids encoding DddA11s, Hifi-DdCBEs, sTALED-V28Rs targeting MT-ND1, MT-ND4 locus were constructed with inserting customized TALE array sequence. To removing the stuffer sequences for inserting TALE array, each plasmid was digested with BsaI(Cat#R3733L, New England BioLabs) and Esp3I (Cat#R0734S, New England BioLabs). Digested stuffers and TALE arrays were assembled with golden gate methods via T4 ligase (Cat#M0202S, New England BioLabs). The plasmids encoding sTALEDs were constructed to insert custom-designed TALE array sequences encoding specific target sites. The architectures of the sTALED plasmids were as follows: CMV promoter-various MTS-3X HA or not-TALE N-terminal domain-stuffer-TALE C-terminal domain-DddA homologs half, CMV promoter-various MTS-3X FLAG (or not)-TALE N-terminal domain-stuffer-TALE C-terminal domain-DddA homolog half-TadA8e or TadA8e variants. To utilize various MTS and DddA homologs, human codon-optimized DNA fragments were synthesized (IDT), and master vectors capable of inserting TALE array sequences were generated using the Gibson assembly method (NEBuilder HiFi DNA assembly master mix). TALE array sequences targeting the mitochondrial DNA m.G11778A site in the master vectors were produced using six TALE array module vectors and the Golden Gate assembly method38. The resulting plasmid constructs were chemically transformed into E. coli DH5α (enzynomics), and plasmids from selected antibiotic-resistant colonies were subjected to Sanger sequencing to analyze the identity of the constructs.
MEF cell generation
Pregnant mice on embryonic day 13.5 (E13.5) were euthanized by cervical dislocation. The abdominal cavity was opened, and the uterine horns were carefully excised and placed in a sterile disposable Petri dish. Each embryonic sac was opened to release the embryos, which were then transferred individually into the 6-well plate. The embryos were washed thoroughly with D-PBS (Welgene), and removed visceral tissues. The embryos, now free of visceral tissue, were transferred to fresh wells in a new 6-well plate. Two milliliters of Trypsin-EDTA (Welgene) were added to each well, and the embryos were minced into smaller fragments. The plate was incubated at 37 °C for 30 min to allow enzymatic dissociation. After incubation, 3 ml of MEF medium (DMEM containing 10% FBS, 1% penicillin-streptomycin, and 1% L-glutamine) was added to each well. The mixture was pipetted up and down multiple times to dissociate the tissue into single cells. The resulting cell suspension was transferred to a 100 mm culture dish containing 7 ml of MEF medium and cultured at 37 °C in a humidified incubator with 5% CO2.
Cell transfection
For the transfection, B16F10 and NIH3T3 cells were seeded onto 24-well plates (SPL) with density of 1 x 104 cells. Cells were transfected with 500 ng/µl left, right DddA11, Hifi-DdCBE, sTALED-V28R, using 1.5 µl of Lipofectamine 3000 (Invitrogen) after 24 h. For MEF cell transfections, cells were seeded onto 24-well plates (SPL) with density of 1 x 104 cells, and were transfected with 500 ng/µl left, right sTALED-V28R and pAAV-sTALED-V28R using 3.5 µl of Lipofectamine 3000 after 24 h. For UDC transfections, the electroporation method (NEON, Thermo Fisher, Lonza nucleofector X unit, Lonza) was used. Specifically, the sTALED plasmids confirmed by sanger sequencing were extracted using a midi-prep kit (Qiagen), and 1 µg each sTALED plasmid DNA (total 2 µg) was transfected by applying an electric shock. For NEON, (1350 V, 30 ms, 1 pulse) to 1 x 104 cells and CM-137 program with P3 buffer used in nucleofector X unit to 1 x 106 cells. After 6 days, the transfected cells were harvested and lysed by incubating at 55 °C for 1 h, followed by 95 °C for 10 min, in 100 µL of cell lysis buffer containing 50 mM Tris-HCl, pH 8.0 (Sigma Aldrich) 1 mM EDTA (Sigma Aldrich), and 0.005% sodium dodecyl sulfate (Sigma Aldrich) supplemented with 5 µL of Proteinase K (Qiagen).
mRNA preparation
mRNA templates were prepared using PCR via Q5 polymerase (NEB) with the following primers: F: 5′-CATCAATGGGCGTGGATAG-3′ and R: 5′-GACACCTACTCAGACAATGC-3′. In vitro transcription (IVT) conducted using mMESSAGE mMACHINE T7 Ultra kit (Invitrogen) and purified via a MEGAclear kit (Invitrogen) following the manufacturer’s instructions.
Microinjection
For microinjection, a 50 μl mixture containing left DddA11 or Hifi-DdCBE and right DddA11 or Hifi-DdCBE mRNA (25 ng/μl, each), filled up to volume with injection buffer (0.25 mM EDTA, 10 mM Tris; pH 7.4 in DEPC-treated water), was injected into 1-cell zygotes cytoplasm using a Nikon ECLIPSE Ti micromanipulator and FemtoJet 4i microinjector (Eppendorf).
Genomic DNA extraction for targeted deep sequencing
All the assay methods used in genotyping were identical. Transfected cells, blastocyst-stage embryos and tissues from mice were incubated in lysis buffer (25 mM sodium hydroxide [NaOH], 0.2 mM EDTA; pH 10) at 95 °C for 20 min, after which the pH was adjusted to 7.4 via the addition of 4-(2-hydroxyethyl)−1-piperazineethanesulfonic acid [HEPES (Sigma-Aldrich), free acids without pH adjustment at a final concentration of 50 mM].
Targeted deep sequencing
For targeted deep sequencing, nested first and second PCR were conducted to generate high-throughput sequencing libraries, and final index sequences were merged in third PCR using Q5 DNA polymerase (NEB). Primers used in this study were listed in Supplementary Table 2. Libraries were subjected to paired-end read sequencing via Miseq platform (Illumina). Sequencing results were analyzed via the CRISPR RGEN tool and using source code (https://github.com/ ibs-cge2/prime_editor_analysis, created by BotBot Inc.), as previously described39.
Whole mitochondrial genome sequencing
For whole mitochondrial genome sequencing, genomic DNA was extracted using a DNeasy Blood & Tissue kit (QIAGEN). mtDNA was amplified with two fragments for overlap whole mitochondrial genome by PCR using PrimeSTAR GXL polymerase (TAKARA). Amplified mtDNAs were subjected to tagmentation via DNA prep kit (Illumina) following the manufacturer’s protocol. Library was subjected to paired-end sequencing via the MiniSeq platform (Illumina).
Oxygen consumption rate
Oxygen consumption rates were measured using the Seahorse XFe96 Analyzer (Agilent) according to the manufacturer’s instructions. Cells were suspended at a concentration of 1 × 106 cells/mL, and 100 µL of the suspension was seeded into Seahorse XF96 cell culture microplates (Agilent) 16 h prior to measurement. The analysis was conducted in Seahorse XF DMEM medium (pH 7.4) supplemented with 25 mM glucose and 1 mM sodium pyruvate (Agilent). The XF Cell Mito Stress Test protocol was used, involving sequential injections of 1.5 µM oligomycin, 1 µM FCCP, and a mixture of 0.5 µM rotenone and antimycin A.
Measurement of ATP concentration
ATP concentrations in the brain and liver were determined with an ATP Assay Kit (Abcam) according to the manufacturer’s protocol. MEF cells (1 x 106) were used in the assay. Cells were lysed in ATP buffer. After lysis, the samples were centrifuged at 13,000 × g for 5 min at 4 °C to remove insoluble material. They were then loaded into a 96-well plate (SPL) and diluted in ATP assay buffer at the recommended proportions following the manufacturer’s protocol. The plate was incubated at room temperature for 30 min in the dark. The optical density (OD) was then measured at 570 nm with a microplate reader.
Cell viability assay
Cell viabilities were assessed using 0.4% Trypan Blue staining (Gibco). Cells were suspended with centrifuge and washed by PBS. After washing, resuspended cells were mixed with 0.4% trypan blue and incubate for 5 min. Incubated cells were counted with hematocytometer.
Histological analysis
Preparation and immunofluorescence of retinal whole mount
Isolated whole retinae were fixed in 4% paraformaldehyde at 4 °C for 20 min. After fixation, samples were washed 3 × 10 min at 4 °C in PBS, with cold methanol added dropwise. Then, samples were washed 3 × 10 min at 4 °C in PBST [0.01% Triton X-100(Sigma-Aldrich) in PBS] and then were blocked with superblock (Thermo scientific) overnight at 4 °C. Samples were then incubated in light-blocking brown 1.5 ml tube with anti-brn3a primary antibody (Santa Cruz) diluted 1:200 in superblock for 3 day at 4 °C. After washed 3 × 10 min at 4 °C in PBST, samples were incubated with Alexa Fluor 488 goat anti-mouse secondary antibody (Jackson) diluted 1:500 in PBST for 2 h at 4 °C. Samples washed 3 × 5 min at room temperatures in PBST and were coverslipped with DPX mountant (Sigma). Images were acquired using an LSM 800 confocal microscope (Zeiss).
Preparation and H&E stain of paraffin sections
The eyes from ND4 mutant and wild-type mice were fixed in Davidson’s fixation solution at room temperature for 6 h, while other tissues were fixed in 10% neutral formalin at room temperature for 24 h. After rinsing with distilled water, all samples were embedded in paraffin. Eye samples sectioned at a thickness of 3.5 μm, and other tissues were sectioned at 4 μm. The sectioned samples were stained with H&E and coverslipped with DPX mountant (Sigma).
Immunohistochemistry staining of eyeball paraffin sections
The sections were immersed in a 0.3% H2O2 (Sigma-Aldrich) solution, incubated 15 min at room temperature. The antigen retrieval involved microwave heating with 0.01 M citrate buffer 3 × 15 min. After washing the samples with PBS thrice, they were blocked using the VECTASTAIN® Elite® ABC-HRP kit (vector), and incubated with primary antibodies [RBPMS (Abcam, 1:500), Cleaved Caspase-3 (Cell signaling, 1:1000), LC3B (Abcam, 1:2000), GFAP (Abcam, 1:500)] overnight at 4 °C. Subsequently, the sections were washed with PBS and then incubated with the secondary antibody included in the ABC kit according to the manufacturer’s instructions. Detection was performed using the DAB, and the background was stained using hematoxylin.
Optical coherence tomography (OCT)
Spectral-domain optical coherence tomography (SD-OCT) images were captured over a 55° range using a Spectralis OCT system (Heidelberg Engineering GmbH, Heidelberg, Germany). A total diopter range of 100–120 D was applied to obtain OCT images of the mouse retinae. At postoperative week 8, mice were anesthetized with an intramuscular injection of ketamine (100 mg/kg; Yuhan) and xylazine (10 mg/kg; Bayer AG). To induce mydriasis, a drop of Tropherin (Hanmi Pharm. Co., Ltd.) was applied to the cornea. The thicknesses of the total retinal layer (TRL), inner retinal layer (IRL), and ganglion cell layer with the nerve fiber layer (GCL + RNFL) were measured at eccentricities of ±10° along both the temporal-nasal and inferior-superior axes. The eccentricity of 0° was defined as the center of the optic disc. TRL was defined as the region extending from the inner limiting membrane to the outer border of the retinal pigment epithelium (RPE), while IRL was defined as the region spanning from the inner limiting membrane to the outer border of the inner nuclear layer (INL).
Electroretinogram
ERG recordings were performed using Micron Ganzfeld ERG (Phoenix Research Labs, Pleasanton CA, USA). Mice were dark-adapted overnight at least 12 h before the experiment for scotopic testing (rod cell response). After anesthesia the pupils were dilated as previously described. Once the pupil was adequately dilated we applied hypromellose 2.5% (Goniovisc®) and inserted the electrodes. ERG was recorded with Ganzfeld ERG according to the standard protocol provided with the manual instruments. Scotopic ERGs were obtained in response to increasing flash intensities ranging from −1.7 to 1.9 log cd·s/m2. Photopic ERGs were obtained in response to increasing flash intensities ranging from −0.5 to 4.1 log cd·s/m2. Ten responses to light stimulation were averaged. The a-wave (as a measure of photoreceptor function), b-wave (as a measure of bipolar cell function), amplitude and implicit times of rod and cone responses were determined. Amplitude and implicit time of the different waves were measured from rod-mediated responses (scotopic b-wave) to light flashes of 1.9 log cd·s/m2. Cone-mediated responses (photopic b-wave) from light flashes of 4.1 log cd·s/m2 were recorded. The photopic negative response (PhNR) was also analyzed from the same waveforms evoked by 4.1 log cd·s/m2 stimuli. The PhNR was defined as the most negative deflection following the b-wave under photopic conditions. To minimize noise and enhance measurement reliability a post-b-wave analysis window was selected and within this segment the 10 data points with the lowest amplitude values were identified and averaged to represent the PhNR amplitude.
Optokinetic nystagmus
Spatial thresholds for opto-kinetic tracking of sine-wave gratings were measured weekly, using a virtual optokinetic system (OptoMotry, Cerebral Mechanics, Medicine Hat, Alberta, Canada), as previously described. One to three vertical sine-wave gratings moving at 12°/s, drifting either to the left or right, were projected on four surrounding monitors, while an unstrained mouse stood on an elevated platform in the center of an arena. A video camera was placed on the ceiling of the device, and transmitted the image to the connected computer. Clockwise movement drove the tracking through the left eye, while counterclockwise motion drove it through the right eye. The experimenter judged whether the rat or mouse made or did not make slow tracking movements with its head and body, to follow the drifting grating. Major repositioning of the head and grooming movements were ignored, and the trial was restarted if the presence or absence of tracking was not clear. The maximum spatial frequency capable of driving the head tracking was determined.
AAV production
To construct an AAV production plasmid, sTALED-V28R was cloned into AAV vector backbone (pAAV [Exp]-CMVEGFPWPRE, vector builder). All plasmid construction was done using Gibson assembly. The AAV ITR backbone and the GOI, sTALED-V28R, were amplified using superFi (Thermo Fisher Scientific). The PCR products were purified using Expin™ Combo GP (Cat#112-102, Gene ALL) and fragments were assembled using NEBuilder HiFi DNA assembly master mix according to the manufacturer’s protocol. The purification process involved iodixanol gradient ultracentrifugation, which was conducted at the KIST Virus Facility. The production titers, determined by qPCR, were 2.45 × 1013 GC/ml (genome copies/ml) for AAV-sTALED-V28R-Left and 9.66 × 1012 GC/ml (genome copies/ml) for AAV-sTALED-Right.
Intravitreal injection
2-week-old ND4 mutant mice were anesthetized with isoflurane, and mydriatic agent (tropherine, HanAll Biopharma) was applied to the eyes for pupil dilation, followed by topical anesthesia using a local anesthetic (paracaine, HanAll Biopharma). A small incision was made in the limbus using an insulin syringe. The microliter syringe fitted with a 331/2 gauge needle was inserted at a 30˚ angle posterior to the lens, and 2 μl of the AAV-sTALED mixture (AAV-sTALED-Left: 2.45 × 1013 GC/ml, AAV-sTALED-Right: 9.66 × 1012 GC/ml) or PBS was injected. 8 weeks after the injection, mice were used for analysis. For the positive control, 8-week-old wild-type mice were injected with 2 μl of 15 mM rotenone diluted in DMSO using a microliter syringe, following the same procedure. Mice were analyzed 2 weeks after the injection.
Serum biochemical analysis
serum samples were obtained from 30 weeks wild type and ND4 mutant mice. Whole blood was collected via cardiac puncture. The collected blood was centrifuged at 6000 x g for 20 min to separate the serum.
Biochemical analyses of the serum were performed using a chemistry analyzer (FUJI DRI-CHEM NX500; Fujifilm) according to the manufacturer’s instructions. The levels of creatinine, inorganic phosphate, glucose, lactate dehydrogenase, creatine phosphokinase, aspartate aminotransferase (AST), and alanine aminotransferase (ALT) were measured using the FUJI DRI-CHEM SLIDE reagents CRE-P III (Cat No. 15809475), IP-P (Cat No. 15809580), GLU-P III (Cat No. 15809528), LDH-P III (Cat No. 15809607), CPK-P III (Cat No. 15809463), GOT/AST-P III (Cat No. 15809542), and GPT/ALT-P III (Cat No. 15809554), respectively.
Isolation of single retinal ganglion cell by flow cytometry
Retinal ganglion cells were isolated by flow cytometry, following a previously published protocol (Chintalapudi, S. R. et al.)40, with minor modifications. The whole retinas were dissociated by gently pressing through a sterile 70-μm nylon cell strainer. The strainer was subsequently rinsed with additional PBS (1% FBS), and the cell suspension was centrifuged at 200 × g for 7 min at room temperature. After discarding the supernatant, cells were resuspended in 400 μL of PBS (1% FBS). Cells were incubated on ice for 30 min with a primary antibody cocktail diluted 1:50 in PBS (1% FBS), consisting of anti-mouse CD15-PE (BioLegend), anti-mouse CD48-PE-Cy7 (BioLegend), anti-mouse CD57 (Sigma), and anti-mouse CD90.2-AF700 (BioLegend). After immunolabeling, cells were washed twice with 5 mL of PBS (1% FBS) by centrifugation at 200 × g for 7 min at room temperature. Cells were then incubated with goat anti-mouse secondary antibody (BioLegend) diluted 1:250 in PBS (1% FBS) for 30 min on ice, followed by two additional washes under the same conditions. The final cell pellet was resuspended in 500 μL of PBS (1% FBS), and fluorescence-activated cell sorting (FACS) was performed using a BD FACSAria™ III cell sorter (BD Biosciences). CD90.2+ CD48− CD15− CD57− cells were sorted into individual wells of a 96-well plate.
Complex I activity assay
Mitochondria were isolated from immortalized human UDC G11778A (L1-41) cells (approximately 1 × 107 cells) in the three experimental groups (untreated control and plasmid-transfected, V28R) using the Thermo Scientific Mitochondria Isolation Kit for Cultured Cells (Cat# 89874) according to the manufacturer’s protocol.
Complex I (NADH:ubiquinone oxidoreductase) activity was measured using the Abcam Complex I Activity Assay Kit (ab287847) following the manufacturer’s instructions. Briefly, isolated mitochondria were incubated in the provided assay buffer with NADH and decylubiquinone as substrates. Reactions were initiated by adding NADH and were performed in the presence and absence of the supplied Complex I inhibitor (rotenone) to determine specific activity. The reaction mixture (100 µL per well) was incubated at room temperature, and absorbance at 600 nm (reflecting reduction of the Complex I dye) was recorded in kinetic mode at 30 s intervals for 5 min using a Synergy H1 microplate reader (BioTek). A standard curve of oxidized dye was used to convert absorbance change to enzyme activity units. Protein concentrations were used to normalize Complex I activity.
Statistics and reproducibility
All statistical analyses were presented as error bars indicating the standard error of the mean (s.e.m.), and p-values were calculated using Student’s two-tailed t-test. In Fig. 3b–d, we used ANOVA analysis, following statements. We used One-Way ANOVA to analyze statistical difference among experimental groups, WT, DMSO-treated, Rotenone treated, ND4 mutant (<10%), ND4 mutant (10–20%), ND4 mutant (20–30%), ND4 mutant (50–60%), and ND4 mutant (>70 %). Fig. 3b (Total retinal thickness): The degree of freedom is 41. The value of F is 9.770, which reaches significance with a p-value of 1.297E-06 (p*** < 0.001). Fig. 3c (GCL thickness): The degree of freedom is 41. The value of F is 16.446, which reaches significance with a p-value of 3.033E-09 (p*** < 0.001). Fig. 3d (GCL thickness): The degree of freedom is 41. The value of F is 21.151, which reaches significance with a p-value of 1.152E-10 (p*** < 0.001).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data supporting the results are available in the main text or supplementary materials, and the raw data for all experiments were shown in Source data file, and the high-throughput sequencing data from this study have been deposited in the NCBI Sequence Read Archive (SRA) database under the accession code: (PRJNA1334167) [https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1334167]. Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request. (hjlee102@korea.ac.kr). Source data are provided with this paper.
Code availability
Base editing frequencies and indel frequencies from targeted deep sequencing data were calculated with source code (https://github.com/ibs-cge2/prime_editor_analysis) DOI: 10.5281/zenodo.7726909, and is publicly available as of the date of publication.
References
Willis, E. J. The powerhouse of the cell. Ultrastruct. Pathol. 16, iii–vi (1992).
Ng, Y. S. et al. Mitochondrial disease in adults: recent advances and future promise. Lancet Neurol. 20, 573–584 (2021).
Ng, Y. S. & Turnbull, D. M. Mitochondrial disease: genetics and management. J. Neurol. 263, 179–191 (2016).
Hong, S., Kim, S., Kim, K., and Lee, H. Clinical approaches for mitochondrial diseases. Cells 12. https://doi.org/10.3390/cells12202494 (2023).
Yu-Wai-Man, P. & Chinnery, P. F. Leber hereditary optic neuropathy - therapeutic challenges and early promise. Taiwan J. Ophthalmol. 1, 12–15 (2011).
Kirches, E. LHON: mitochondrial mutations and more. Curr. Genom. 12, 44–54 (2011).
Shemesh, A., Sood, G., Blair, K., & Margolin, E. Leber Hereditary Optic Neuropathy (LHON) (StatPearls, 2024).
Newman, N. J., Carelli, V., Taiel, M. & Yu-Wai-Man, P. Visual outcomes in leber hereditary optic neuropathy patients with the m.11778G>A (MTND4) mitochondrial DNA mutation. J. Neuroophthalmol. 40, 547–557 (2020).
Balta, G., Cristache, G., Barac, A. D., Anton, N., & Barac, I. R. Leber’s hereditary optic neuropathy (LHON): clinical experience and outcomes after long-term idebenone treatment. Life 13. https://doi.org/10.3390/life13102000.
Aleo, S. J. et al. Genetic variants affecting NQO1 protein levels impact the efficacy of idebenone treatment in Leber hereditary optic neuropathy. Cell Rep. Med. 5, 101383 (2024).
Goncalves, G. A. R. & Paiva, R. M. A. Gene therapy: advances, challenges and perspectives. Einstein 15, 369–375 (2017).
Prado, D. A., Acosta-Acero, M. & Maldonado, R. S. Gene therapy beyond luxturna: a new horizon of the treatment for inherited retinal disease. Curr. Opin. Ophthalmol. 31, 147–154 (2020).
Battista, M. et al. Gene therapy for Leber hereditary optic neuropathy. Expert Opin. Biol. Ther. 24, 521–528 (2024).
Singh, A. et al. Revolutionary breakthrough: FDA approves CASGEVY, the first CRISPR/Cas9 gene therapy for sickle cell disease. Ann. Med. Surg. 86, 4555–4559 (2024).
Phan, H. T. L., Lee, H. & Kim, K. Trends and prospects in mitochondrial genome editing. Exp. Mol. Med. 55, 871–878 (2023).
Lim, K. Mitochondrial genome editing: strategies, challenges, and applications. BMB Rep. 57, 19–29 (2024).
Kim, J. S. & Chen, J. Base editing of organellar DNA with programmable deaminases. Nat. Rev. Mol. Cell Biol. 25, 34–45 (2024).
Mok, B. Y. et al. A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature 583, 631–637 (2020).
Cho, S. I. et al. Targeted A-to-G base editing in human mitochondrial DNA with programmable deaminases. Cell 185, 1764–1776 e1712 (2022).
Mok, B. Y. et al. CRISPR-free base editors with enhanced activity and expanded targeting scope in mitochondrial and nuclear DNA. Nat. Biotechnol. 40, 1378–1387 (2022).
Cho, S. I. et al. Engineering TALE-linked deaminases to facilitate precision adenine base editing in mitochondrial DNA. Cell 187, 95–109 e126 (2024).
Lee, S., Lee, H., Baek, G. & Kim, J. S. Precision mitochondrial DNA editing with high-fidelity DddA-derived base editors. Nat. Biotechnol. 41, 378–386 (2023).
Kaplanova, V. et al. Segregation pattern and biochemical effect of the G3460A mtDNA mutation in 27 members of LHON family. J. Neurol. Sci. 223, 149–155 (2004).
Mansergh, F. C., Chadderton, N., Kenna, P. F., Gobbo, O. L. & Farrar, G. J. Cell therapy using retinal progenitor cells shows therapeutic effect in a chemically-induced rotenone mouse model of Leber hereditary optic neuropathy. Eur. J. Hum. Genet. 22, 1314–1320 (2014).
Zhang, X., Jones, D. & Gonzalez-Lima, F. Mouse model of optic neuropathy caused by mitochondrial complex I dysfunction. Neurosci. Lett. 326, 97–100 (2002).
Nieuwenhuis, B. et al. Improving adeno-associated viral (AAV) vector-mediated transgene expression in retinal ganglion cells: comparison of five promoters. Gene Ther. 30, 503–519 (2023).
Cheng, J. et al. Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science 381, eadg7492 (2023).
McFarland, R. et al. Homoplasmy, heteroplasmy, and mitochondrial dystonia. Neurology 69, 911–916 (2007).
Pejaver, V. et al. Inferring the molecular and phenotypic impact of amino acid variants with MutPred2. Nat. Commun. 11, 5918 (2020).
Zhang, X. et al. Precise modelling of mitochondrial diseases using optimized mitoBEs. Nature 639, 735–745 (2025).
Prencipe, M., Perossini, T., Brancoli, G. & Perossini, M. The photopic negative response (PhNR): measurement approaches and utility in glaucoma. Int. Ophthalmol. 40, 3565–3576 (2020).
Jolly, J. K. et al. Structural and functional characteristics of color vision changes in choroideremia. Front. Neurosci. 15, 729807 (2021).
Ventura, D. F. et al. Male prevalence of acquired color vision defects in asymptomatic carriers of Leber’s hereditary optic neuropathy. Investig. Ophthalmol. Vis. Sci. 48, 2362–2370 (2007).
Tanimoto, N., Sothilingam, V. & Seeliger, M. W. Functional phenotyping of mouse models with ERG. Methods Mol. Biol. 935, 69–78 (2013).
Majander, A. et al. The pattern of retinal ganglion cell dysfunction in Leber hereditary optic neuropathy. Mitochondrion 36, 138–149 (2017).
Esmaeil, A., Ali, A. & Behbehani, R. Leber’s hereditary optic neuropathy: update on current diagnosis and treatment. Front. Ophthalmol. 2, 1077395 (2022).
Zhou, T. et al. Generation of human induced pluripotent stem cells from urine samples. Nat. Protoc. 7, 2080–2089 (2012).
Kim, Y. et al. A library of TAL effector nucleases spanning the human genome. Nat. Biotechnol. 31, 251–258 (2013).
Lee, J. et al. Prime editing with genuine Cas9 nickases minimizes unwanted indels. Nat. Commun. 14, 1786 (2023).
Chintalapudi, S. R., et al Isolation of primary murine retinal ganglion cells (RGCs) by flow cytometry. J. Vis. Exp., e55785, https://doi.org/10.3791/55785 (2017).
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grants funded by the Korea government (MSIT) (RS-2024-00334994, RS-2023-00261905, RS-2024-00441068, RS-2023-00220894, RS-2024-00408822, RS-2023-00260462, RS-2025-02303431 and RS-2024-00400118) and Ministry of SMEs and Startups (RS-2023-00272313) and the Korea Institute for Advancement of Technology grants funded by the Korea government (MSIT) (P0028364). Graphical abstract was generated with Biorender. We thank Drs. Jeong-Min Hwang and Hee Kyung Yang for help with LHON patient’s cells. We also thank Dr. Kim Ungsoo Samuel from the Department of Ophthalmology, Chung-Ang University Gwangmyeong Hospital.
Author information
Authors and Affiliations
Contributions
H.L. supervised the research. K.K., J.-S.K., and H.L. conceived the research. S.K., J.K., S.C., S.J., C.J.L., S.L., K.Y.S., J.K.S., K.K., J.-S.K. and H.L. design the study. S.K., J.K., S.C., S.J., C.J.L., K.K., J.-S.K. and H.L. performed and analyzed the main experiments. S.H., J.B., Y.O., S.J., S.P.K., H-S.S., J.H.Y., J.P., S.R., L.K.K., S-Y.L. S.B.L., S.H.C, S-J.P., C.G.C., M.C. and J.P performed the experiments. S.K., J.K., S.C., S.J., C.J.L., K.K., J.-S.K. and H.L. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
J.-S.K. is a co-founder of and holds stock in ToolGen, Inc., Edgene, Inc., and GreenGene Inc. J.-S.K., J.K., and H.L. have filed patent applications related to this work (application number: WO2022060185A1, PCT/KR2021/012872, Korea Patent 10-2024-0119585). The other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Carlos Moraes and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kim, S., Kim, J., Cha, S. et al. In vivo mitochondrial base editing restores genotype and visual function in a mouse model of LHON. Nat Commun 16, 10923 (2025). https://doi.org/10.1038/s41467-025-66600-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-66600-3
