
Abstract
The coordination between epithelial progenitors and their mesenchymal niche is critical for organogenesis and repair, yet the mechanisms governing their competitive interactions remain unclear. Here, we reveal a paradigm of tissue-scale fitness sensing in the lung, where mesenchymal Yap levels antagonize epithelial Yap levels to dictate epithelial stem cell fate. We show that reduced fitness in alveolar fibroblasts (AF1s) via Yap/Taz or Myc deletion leads to their apoptotic elimination and a collapse of the alveolar stem cell niche. This niche collapse triggers a pathological competitive response from the epithelium, which undergoes aberrant bronchiolization that phenocopies human pulmonary fibrosis. Mechanistically, we uncovered a molecular switch that controls mesenchymal fate. During development and fibrosis resolution, Snail1/2 sequesters Yap/Taz to drive an adipogenic program, generating niche-supportive AF1s. Conversely, Yap/Taz-TEAD-Myc binding instructs a myogenic, pro-fibrotic program. Our findings demonstrate that inter-tissue cell competition, governed by a Snail/Yap rheostat, orchestrates lung architecture and provides a framework for targeting the mesenchymal niche to treat fibrotic disease.
Similar content being viewed by others

Introduction
The human lung’s intricate structure optimizes gas exchange through its extensive branching network of airways, culminating in 700 million alveoli1. Spanning an impressive 70 square meters, this network forms during the branching morphogenesis phase of development, where mesenchymal cells encircle the epithelial monolayer2. This crosstalk between epithelial and mesenchymal cells drives branching and differentiation processes. Interactions between these cell populations, along with their matrix, regulate the branching program and transition to the alveolar differentiation phase3,4,5. This epithelial-mesenchymal crosstalk remains essential during adult homeostasis and repair following injury2,6,7.
Early tissue recombination studies revealed that the mesenchyme plays a crucial role in instructing the epithelium, imparting its regional identity8. This instructive function becomes particularly evident in the injured adult lung. Following injury, developmental programs are reactivated to repair tissue damage and restore the organ to homeostasis. This paradigm applies to the lung, involving various signaling pathways such as Fgf, Hippo, and Wnt, which are either upregulated or downregulated in response to lung injury9,10,11. These signals form the foundation for communication and crosstalk between different cell types, both within the epithelium and between epithelial and mesenchymal cells.
In the adult lung, cartilage, airway smooth muscle, alveolar fibroblast 1 (AF1), and pericytes establish various mesenchymal stem cell niches. These niches are essential for maintaining and/or activating adult epithelial stem cells during homeostasis or initiating repair after injury6,9,10,12,13,14. Any disruptions in these interactions during lung development, homeostasis, or repair after injury can lead to the development of pulmonary diseases such as bronchopulmonary dysplasia (BPD), emphysema, and pulmonary fibrosis.
Fibrotic diseases, a major contributor to global morbidity and mortality, remain poorly understood. These diseases can affect various organs, ultimately leading to organ failure15. Fibrosis is believed to account for up to 45% of global deaths, with chronic lower respiratory fibrosis being a leading cause of death in the United States15,16. Idiopathic pulmonary fibrosis (IPF), a prevalent form of interstitial lung disease (ILD), results in alveolar remodeling and progressive loss of pulmonary function, respiratory failure, and death within five years of diagnosis17,18. IPF pathogenesis involves fibrotic remodeling, inflammation, and the loss of lung architecture19. Despite the underlying causes being elusive, genetic and experimental evidence supports the notion that chronic alveolar epithelial injury and inadequate repair of the respiratory epithelium are intrinsic to IPF disease pathogenesis. Histologically, respiratory epithelial cells in the lung parenchyma express atypical proximal airway epithelial and indeterminate cell type markers20,21, including goblet and basal cell (BC) markers, which are typically restricted to conducting airways. Fibrotic lesions and honeycomb structures replace alveoli, which are normally lined by alveolar type 1 (AT1) and AT2 cells. The bronchiolization process that leads to honeycomb structures is driven by intra-epithelial cell competition, where distal airway stem cells with high Yap/Myc levels become supercompetitors and replace the alveolar epithelium with bronchial epithelium22. Some abnormal airway/alveolar epithelial cells attempting to aid in regeneration persist in Krt8+ transitional states, which express genes associated with pro-fibrotic phenotypes23,24,25,26.
In this work, we reveal a paradigm of tissue-scale fitness sensing in the lung, where mesenchymal Yap levels antagonize epithelial Yap levels to dictate epithelial stem cell fate. Mechanistically, we uncovered a molecular switch that controls mesenchymal fate. During development and fibrosis resolution, Snail1/2 sequesters Yap/Taz to drive an adipogenic program, generating niche-supportive AF1s. Conversely, Yap/Taz-TEAD-Myc binding instructs a myogenic, pro-fibrotic program. Our findings demonstrate that inter-tissue cell competition orchestrates lung architecture and provides a framework for targeting the mesenchymal niche to treat fibrotic disease.
Results
Yap/Taz bind to Snail1/2 to drive alveolar fibroblast 1 differentiation during lung development
The pulmonary mesenchyme includes multiple distinct cell lineages with various functions in lung development and in the pathogenesis and progression of debilitating respiratory conditions like idiopathic pulmonary fibrosis (IPF)27,28. Lung mesenchymal lineages are increasingly recognized as highly heterogeneous, including alveolar fibroblast 1 (AF1), adventitial fibroblasts or alveolar fibroblasts 2 (AF2s), peribronchial fibroblasts, airway and vascular smooth muscle cells, pericytes, mesothelial cells. The AF1 lineage which forms the niche for AT2 stem cells in the adult lung arises around E16.5 during lung development. During lung fibrosis, AF1s take on multiple molecular states with distinct roles in facilitating responses to fibrotic lung injury giving rise to injury-induced fibrotic, inflammatory and stress activated fibroblasts populations which then either undergo apoptosis or revert back to AF1s during fibrosis resolution29,30,31. Yap/Taz signaling32,33 mediates the acquisition of some of these molecular states. However, little is still known about how these different lineages arise during lung development and are regulated during lung fibrosis and resolution34.
The role of Hippo signaling during mesenchymal lineage commitment during lung development remains largely unknown. To investigate this, we genetically inactivated Yap/Taz (Yap1f/f;Wwtr1f/f) or the Hippo kinases Mst1/2 (Stk4f/f;Stk3f/f) from the undifferentiated lung mesenchyme using Tbx4rtTa;TetCre starting from E9.5. Subsequently, we performed combined single nuclear RNAseq and ATACseq on E15.5 and E18.5 lungs. For each sample, we sequenced between 5200 and 8200 cells.
To our surprise, we found that inactivating Yap1/Wwtr1 in the lung mesenchyme from E9.5 onwards impairs AF1 differentiation (marked by Scube2), which was confirmed by immunostaining for Plin2, a well-known marker for AF1s (Fig. 1A–F, Supplementary Fig. S1, Fig. 2A). Conversely, inactivating Stk3/4 in the lung mesenchyme from E9.5 onwards accelerates AF1 differentiation (Fig. 1A–F, Supplementary Fig. S1). Neither inactivation of Yap1/Wwtr1 or Stk3/4 using Tbx4rtTa;TetCre starting from E9.5 affected endothelial differentiation (Supplementary Fig. S4B), though E18.5 Tbx4rtTa;TetCre;Yap1f/f;Wwtr1f/f lungs featured increased subpleural vascularization (Supplementary Fig. S4A). These findings suggest that mesenchymal Yap/Taz signaling plays a pivotal role in driving AF1 lineage commitment. This finding is intriguing because upon bleomycin injury, increased mesenchymal Yap/Taz-Tead signaling is believed to promote AF1 to fibrotic fibroblast differentiation32,33.

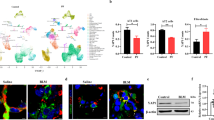
A Graph-based clustering and cell-type or sample annotation of integrated datasets from E15 and E18 ctrl and mutant lungs based on enriched gene expression and chromatin accessibility profiles by using ArchR. AF1ss (AF1 population in Snai1/2 inactivated mesenchyme), AF1yt (AF1 population in Yap/Taz inactivated mesenchyme). Subsetting and clustering of B mesenchyme and C epithelium. Graph-based clustering and cell-type annotation on individual samples of D mesenchyme and E epithelium. F Tead motif accessibility and representative MAGIC imputed gene expression for main mesenchymal and epithelial clusters. G Heatmap of differential transcription factor motif accessibility showing which regulatory factors are predicted to most active in each cell type. AF1ss (AF1 population in Snai1/2 inactivated mesenchyme) contains enrichment for myogenic differentiation and Tead transcription factors (green), AF1 is enriched for adipogenic factors (yellow), AF1yt (AF1 population in Yap/Taz inactivated mesenchyme) lacks Tead binding activity.

A Immunostaining for Plin2 and Acta2 on E15.5 control (n = 7), Tbx4rtTa;TetCre;Yap1f/f;Wwtr1f/f (n = 3; p = 0.02), Tbx4rtTa;TetCre;Snai1/2f/f (n = 3, p = 0.005), and Tbx4rtTa;TetCre;Stk3/4f/f (n = 2, p = 0.04) lungs. Scale bar 50 µm. Quantification of Plin2 expression from images represented. B Co-immunostaining for Rage and Sftpc on E18.5 control, Tbx4rtTa;TetCre;Yap1f/f;Wwtr1f/f, Tbx4rtTa;TetCre;Snai1/2f/f and Tbx4rtTa;TetCre;Stk3/4f/f lungs doxycycline induced from E9.5. Scale bar 50 µm. Nanostring nCounter RNA profiling on control (n = 19) and Tbx4rtTa;TetCre;Yap1f/f;Wwtr1f/f (n = 21) lungs at E18.5 (Sox2 p = <0.000001, Hopx p = <0.000001, Ager p = <0.000001, Sftpc p = 0.000012). Data are graphed on a Log2 scale. C Co-immunostaining for Yap and E-cadherin on E15.5 control (n = 6), Tbx4rtTa;TetCre;Yap1f/f;Wwtr1f/f (n = 3) and Tbx4rtTa;Tet-YapS112A (n = 5) lungs doxycycline induced from E9.5. Scale bar 50 µm. Quantification of epithelial (Tbx4rtTa;TetCre;Yap1f/f;Wwtr1f/f p = 0.0001 and Tbx4rtTa;Tet-YapS112A p = 0.0006) and mesenchymal (Tbx4rtTa;TetCre;Yap1f/f;Wwtr1f/f p = 0.0029 and Tbx4rtTa;Tet-YapS112A p = 0.04) Yap intensity on represented images. D Co-immunostaining for Yap and Acta2 on E15.5 control (n = 5) and Tbx4rtTa;Tet-YapS112A (n = 5) lungs doxycycline induced from E9.5. Scale bar 100 µm. Quantification of average pixel intensity of Acta2 staining on whole lung represented images (p = 0.02). Data are presented as mean values +/− SEM. Two tailed unpaired T-test was used to determine significance. F test was used to determine equal variances and unreported F values indicate equal variance. *p < 0.05, **p < 0.01, ****p < 0.0001.
We were intrigued to explore whether Yap/Taz signaling in AF1 differentiation is partially mediated by a different transcriptional cofactor besides Teads. Snail1 and Snail2, which are highly expressed in AF1s and undifferentiated distal mesenchyme (Supplementary Fig. S2A–E), have been demonstrated to bind Yap and Taz in bone marrow-derived mesenchymal stem cells (MSCs) to regulate their self-renewal and differentiation35,36. In these MSCs, Snail1 and Snail2 form binary complexes with Yap/Taz that not only control Yap/Taz protein levels but also regulate the expression of specific Yap/Taz-target genes35,36. To investigate a potential role for Snail1/2 in regulating mesenchymal lineage commitment during lung development, we added E15.5 and E18.5 Tbx4rtTa;TetCre;Snai1/2f/f lung samples, induced from E9.5, to our combined single nuclear RNAseq and ATACseq analysis.
We discovered that mesenchymal Snail1/2 binds to Yap/Taz to orchestrate an adipogenic differentiation program towards AF1. This is evident from reduced chromatin accessibility of CEBP binding motifs in lung AF1s from Tbx4rtTa;TetCre;Snai1/2f/f and Tbx4rtTa;TetCre;Yap1f/f;Wwtr1f/f mice (Fig. 1G, Supplementary Fig. S2F). Notably, the absence of Snail1/2 increases chromatin accessibility of Tead binding motifs, suggesting that Snail1/2 inhibits Yap/Taz-mediated Tead signaling (Fig. 1G, F, Supplementary Fig. S2F). A closer examination of Snail1/2 knockout mesenchyme compared to control mesenchyme reveals that the loss of Snail1/2 promotes mesenchymal differentiation along a myogenic differentiation program towards a myofibroblast lineage. This lineage exhibits increased chromatin accessibility of Stat1, Stat3, and Stat437,38,39,40 as well as myogenic regulator factors including MyoD, Myf5, and MyoG41,42,43 binding motifs (Fig. 1G, Supplementary Fig. S2F). This suggests that the Snail/Yap rheostat governs a fundamental fate decision between adipogenic and contractile mesenchymal lineages. The specific outcome—differentiation towards myofibroblasts versus airway smooth muscle—is likely dependent on additional developmental cues and the specific mode of Yap/TEAD activation.
Spectra44 (supervised pathway deconvolution of interpretable gene programs) analysis also demonstrates decreased activation of adipogenic or fat cell differentiation pathways and increased activation of myofibroblast differentiation pathways in AF1s upon Snai1/2 inactivation (Supplementary Fig. S3I, Supplementary Data 1). Inactivation of Snai1/2 using Tbx4rtTa;TetCre starting from E9.5 did not appear to drastically affect endothelial cell differentiation (Supplementary Fig. S4).
Mesenchymal Yap antagonizes epithelial Yap to coordinate fate transitions across tissue compartments
We discovered that accelerated AF1 differentiation in the mesenchymal Stk3/4 knockout lungs is accompanied by accelerated alveolar epithelial differentiation (Fig. 1E, Supplementary Fig S3A–H). Conversely, impaired AF1 differentiation in the mesenchymal Yap/Taz knockout lungs hinders the transition from a branching program to an alveolar differentiation program3,4,5 (Fig. 2B, Supplementary Fig. S3A–H), which occurs around E16.5 in parallel with AF1 differentiation. Notably, increased epithelial Yap signaling during lung development also impairs this transition45, implying that mesenchymal Yap levels may antagonize epithelial Yap levels. Indeed, we observe increased Yap levels in the epithelium of Tbx4rtTa;TetCre;Yap1f/f;Wwtr1f/f lungs but decreased epithelial Yap levels in lungs in which we overexpress a dominant, active version of the Hippo transcriptional effector Yap1S112A in the lung mesenchyme (Fig. 2C). This overexpression also activates a robust myogenic differentiation program, leading to increased smooth muscle cell differentiation (Fig. 2D). Together, our findings indicate that lung mesenchymal Yap levels antagonize epithelial Yap levels during lung development to coordinate airway versus alveolar epithelial differentiation.
The transition from a branching program to an alveolar differentiation program3,4,5 during lung development bears a striking resemblance to the binary decision made by distal airway stem cells in response to bleomycin injury. This decision involves either generating more airway epithelium, leading to bronchiolization, or alveolar epithelium. Notably, bronchiolization, a hallmark of pulmonary fibrosis, is mediated by an increase in airway epithelial Yap/Myc signaling22. Since mesenchymal Yap levels antagonize epithelial Yap levels during lung development, thereby coordinating airway versus alveolar epithelial differentiation, we hypothesized that mesenchymal Yap levels could also antagonize bronchiolization in pulmonary fibrosis. To investigate this, we induced bleomycin injury in adult lungs in which we inactivated Yap/Taz in AF1 cells using three distinct Cre lines: Plin2CreERT2, Tbx4rtTa;TetCre, and Scube2CreERT2, while simultaneously lineage tagging them with Rosa26-mTmG. Surprisingly, all three mouse models exhibited a striking increase in bronchiolization 6 weeks after bleomycin injury. This was characterized by an increase in aberrant basaloid (Krt8) and/or basal cells (Krt5) and increased pulmonary fibrosis based on hydroxyproline content compared to control lungs (Fig. 3A–K, Supplementary Fig. S5A–K). We found that inactivating Yap/Taz profoundly affects the fitness and survival of AF1s in the injured environment. Lineage tracing revealed a dramatic reduction in GFP-positive AF1s in Plin2CreERT2;Yapf/f;Wwtrf/f;mTmG lungs post-injury (Fig. 3E), and TUNEL staining confirmed these cells were undergoing apoptosis (Fig. 3L, M). As AF1s form the essential niche for AT2 stem cells, this loss of the mesenchymal support structure was accompanied by AT2 cell loss. We propose that this collapse of the alveolar niche triggers a pathological regenerative response from distal airway stem cells, which, in the absence of proper mesenchymal cues, fill the damaged parenchyma through bronchiolization rather than functional alveolar repair (Fig. 3L, M). Furthermore, since AF1s form the niche for AT2 cells, we observed that this loss of AF1s also leads to AT2 cell death. In contrast, the opposite phenotype was observed in Plin2CreERT2;Stk3/4f/f;mTmG lungs, where we inactivated the Hippo kinases Mst1/2 in AF1 cells. These lungs exhibited decreased bronchiolization 6 weeks after bleomycin injury and no AF1 loss (Fig. 3D, H, J, Supplementary Fig. S5A–K). We therefore propose that bronchiolization in pulmonary fibrosis also involves an epithelial-mesenchymal cell competition based on Yap levels between distal airway stem cells and AF1/myofibroblasts. Note that in the absence of injury Plin2CreERT2;Yap1f/f;Wwtr1f/f;mTmG and Plin2CreERT2;Stk3/4f/f;mTmG lungs appeared normal 9 weeks after tamoxifen induction (Supplementary Fig. S6).

A Mice were placed on tamoxifen chow for 3 weeks, after a 3 week washout, lungs were injured with bleomycin and dissected 6 weeks later. Created in BioRender. Warren, R. (2025) https://BioRender.com/buq1mhz. B–D Immunostaining for GFP and Krt5 on B control (n = 7), C Plin2CreERT2;Yap1f/f;Wwtr1f/f;mTmG (n = 12) lungs 6 weeks after bleomycin injury. Scale bar 500 µm. D Nanostring nCounter RNA analysis for Krt5 on control (n = 42), Plin2CreERT2;Yap1f/f;Wwtr1f/f;mTmG (n = 13, p < 0.0001) and Plin2CreERT2;Stk3/4f/f (n = 11, p < 0.0001). E Image quantification of GFP in images represented in (B, C) (p = 0.004). Immunostaining for Krt8 in F control (n = 4), G Plin2CreERT2;Yap1f/f;Wwtr1f/f (n = 3, p = 0.03), H Plin2CreERT2;Stk3/4f/f (n = 6, p = 0.04), and I Scube2CreERT2;Yap1f/f;Wwtr1f/f (n = 5, p = 0.04) lungs 6 weeks after bleomycin injury. Scale bar 500 µm. J Image quantification of Krt8 in images represented in (F–I). K Hydroxyproline analysis on bleomycin injured Scube2CreERT2;mTmG controls (n = 43) and Scube2CreERT2;Yap1f/f;Wwtr1f/f;mTmG (n = 30, p = 0.04). L, M TUNEL staining and quantification on bleomycin injured control Plin2CreERT2;mTmG (n = 3) and Plin2CreERT2;Yap1f/f;Wwtr1f/f;mTmG (n = 3, p = 0.001) lungs. Data are presented as mean values +/−SEM. Two tailed unpaired T-test was used to determine significance. F test was used to determine equal variances and unreported F values indicate equal variance. *p < 0.05, **p < 0.01, ****p < 0.0001.
Next, we performed single-cell RNAseq on non-injured control and bleomycin-injured control lungs, as well as bleomycin injured Plin2CreERT2;Yap1f/f;Wwtr1f/f lungs. Notably, only Plin2CreERT2;Yap1f/f;Wwtr1f/f lungs exhibited a prominent aberrant basaloid cell population which based on trajectory inference analysis appears to be derived from club cells (Supplementary Fig. S5A–K). This population displayed increased expression of Krt5, Krt17, Yap1, and Gdf15 (Fig. 4A,B, Supplementary Fig. S5A–K) and increased inferred Myc transcriptional activity (Fig. 4C). Spectra analysis also suggests increased Myc target gene expression in the aberrant basaloid cells that appear in Plin2CreERT2;Yap1f/f;Wwtr1f/f lungs after bleomycin injury (Supplementary Fig. S7). Deeper Spectra analysis indicates that this aberrant basaloid population is highly proliferative and metabolically active, likely undergoing rapid division and growth. The combination of active pathways such as Glycolysis, Pyruvate Metabolism, Citric Acid (TCA) Cycle, Oxidative Phosphorylation, Ribosome Biogenesis, Amino Acid Metabolism, Fatty Acid and Ketone Metabolism, Nucleotide Synthesis, Myc target gene activation, mTORC1/PI3K-AKT Signaling,G1/S Transition, DNA Synthesis and Repair, G2/M Transition and the quality control p53 pathway etc., suggests a state often seen in cancer cells, activated stem cells, or rapidly dividing progenitor cells (Supplementary Fig. S7, Supplementary Data 2). A significant number of the active pathways are dedicated to energy production and the creation of molecular building blocks. Taken together, the transcriptional and pathway analyses reveal that the aberrant basaloid population has undergone a profound metabolic reprogramming. They have adopted a hyper-proliferative, cancer-like metabolic state, providing a clear molecular engine for their ‘supercompetitor’ phenotype and their ability to drive pathological tissue remodeling.

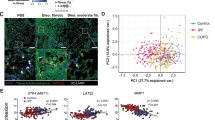
A Clusters from scRNA-seq on non injured control (NI), bleomycin injured control, and bleomycin injured Plin2CreERT2;Yap1f/f;Wwtr1f/f. Aberrant basaloid cell cluster is circled. B Feature plots showing normalized expression of canonical marker genes across the clusters. C Myc inferred transcription factor activity using dorothea.
Surprisingly, at 6 weeks post bleomycin injury, we found no difference between the AF1 populations from control versus Plin2CreERT2;Yap1f/f;Wwtr1f/f injured lungs. This suggests that increased pulmonary fibrosis is a result of the rise in aberrant basaloid cells. The lack of significant transcriptional differences in AF1 populations from control versus Plin2CreERT2;Yap1f/f;Wwtr1f/f injured lungs at 6 weeks post injury could be because most mutant AF1s have already been outcompeted at this time point.
To compare the aberrant basaloid populations observed in Plin2CreERT2;Yap1f/f;Wwtr1f/f lungs after bleomycin injury with those observed in human IPF we integrated our mouse data sets with a human data set26 containing aberrant basaloid cells and found that both cell populations cluster together indicative of their similarity (Fig. 5). Together these data illustrate that this aberrant basaloid population bears striking resemblance to that observed in IPF22,23,24,25,26,46.

A Clusters from scRNA-seq on control not injured (NI), bleomycin injured control, bleomycin injured Plin2CreERT2;Yap1f/f;Wwtr1f/f and human IPF. B Gene expression of marker genes.
Alveolar fibroblast 1 fitness levels are determined by their Myc levels
Yap and Myc work together to control genes essential for cell proliferation. When Myc is activated, it binds extensively to its genomic targets, which are largely pre-associated with TEAD47. At these sites, Yap recruitment appears to depend on Myc and is necessary for complete transcriptional activation. This partnership between Yap and Myc is considered vital for initiating the cell cycle and supporting organ growth47. Since we have recently demonstrated that Myc and Yap cooperate in the lung epithelium to drive cell competition22 we wondered whether inactivation of Myc in AF1s prior to bleomycin injury would also render them vulnerable to becoming outcompeted by bronchial epithelium (Fig. 6A). Indeed, we observe increased bronchiolization based on Krt8 and Krt5 expression (Fig. 6B, D, E) and increased pulmonary fibrosis based on hydroxyproline content (Fig. 6C) in Plin2CreERT2;Mycf/f;mTmG mice 6 weeks after bleomycin injury.

A Mice were placed on tamoxifen chow for 3 weeks, after a 3 week washout, lungs were injured with bleomycin and dissected 6 weeks later. Created in BioRender. Warren, R. (2025) https://BioRender.com/buq1mhz. B Immunostaining for Krt8 (p = 0.03) and Krt5 (p = 0.005) on control (n = 6) and Plin2CreERT2;Mycf/f;mTmG (n = 11) lungs 6 weeks after bleomycin injury. Scale bar 200 µm. C Hydroxyproline analysis on bleomycin injured Plin2CreERT2;mTmG (n = 23) and Plin2CreERT2;Mycf/f;mTmG (n = 34) lungs (p = 0.009). D Image quantification of Krt8 and Krt5 in images represented in (B). E qPCR analysis on RNA from bleomycin injured Cre- controls (n = 19) and Plin2CreERT2;Mycf/f;mTmG (n = 22) lungs for Krt5 (p = 0.03) and Trp63 (p = 0.03) genes. Data are presented as mean values +/− SEM. Two tailed unpaired T-test was used to determine significance. F test was used to determine equal variances and unreported F values indicate equal variance. *p < 0.05, **p < 0.01.
Myofibroblast to alveolar fibroblast 1 dedifferentiation requires a Yap/Taz/Snail1/2 transcriptional program
Finally, we wondered if Snail1 and Snail2 are also crucial for the dedifferentiation of myofibroblasts into AF1s during the resolution of bleomycin-induced pulmonary fibrosis. To investigate this, we inactivated Snai1 and Snai2 in myofibroblasts using Acta2CreERT2;Snai1/2f/f,mTmG mice, starting 2 weeks after bleomycin injury and harvesting the lungs 4 weeks later (Fig. 7A). Single-cell RNA sequencing analysis of these lungs and lineage tracing of myofibroblasts revealed that inactivating Snai1/2 from myofibroblasts after bleomycin injury hinders their dedifferentiation into AF1s and/or impairs their clearance through apoptosis. This is demonstrated by an increase in lineage labeled AF1s/Myofibroblasts (Fig. 7E, F) and a cluster of myofibroblast-like cells (AF1-SS) with elevated expression of fibrosis-related genes (myofibroblast) population, including Mt1, Mt2, and Thbs148 in our single cell analysis (Fig. 7B, C). Note that the Snai1f/f mice we used only have exon3, which is predicted to encode the two C-terminal zinc finger domains as well as the polyadenylation sequence, of the Snai1 gene flanked by LoxP sites49 and therefore a truncated Snai mRNA including exon1 and 2 is still being produced that is detected by 2 of the 3 probes of the 10X flex platform for single cell analysis. Since Snail1 is known to negatively regulate its own expression50, we therefore see an increase in the expression of this truncated Snai1 mRNA in our AF1ss population.

A Mice were injured with bleomycin and 2 weeks after injury, they were placed on tamoxifen chow until dissection at 6 weeks after injury. Created in BioRender. Warren, R. (2025) https://BioRender.com/xo6zwap. B Mesenchymal cell clusters from scRNAseq on non injured control and bleomycin injured control and Acta2CreERT2;Snai1/2f/f lungs tamoxifen induced 2 weeks after bleomycin injury. C Fibrosis promoting pathway activities are upregulated in AF1-SS (AF1 population in Snai1/2 inactivated mesenchyme). D Hydroxyproline analysis of soluble collagen on control (n = 30) and Acta2CreERT2;Snai1/2f/f (n = 7, p = 0.0001). E Co-immunostaining for GFP and Acta2 on bleomycin injured control (n = 3) and Acta2CreERT2;Snai1/2f/f (n = 7, p = 0.02) lungs tamoxifen induced 2 weeks after bleomycin injury. Scale bar 100 µm. F Quantification of GFP in images represented in (E). Data are presented as mean values +/− SEM. Two tailed unpaired T-test was used to determine significance. F test was used to determine equal variances and unreported F values indicate equal variance. *p < 0.05, ***p < 0.001.
Spectra analysis of the AF1-SS population revealed a cell state locked in a pro-fibrotic, pro-inflammatory loop (Fig. S8, Supplementary Data 3). These cells showed sustained activation of myofibroblast differentiation and collagen synthesis pathways, alongside robust signatures of cellular stress (Unfolded Protein Response), inflammation (TNF-α and IL6-JAK-STAT3 signaling), and hypoxia. This molecular signature is indicative of a stalled pathological state that actively prevents the resolution of fibrosis. Consequently, we observed increased pulmonary fibrosis 6 weeks after bleomycin injury, as measured by hydroxyproline content (Fig. 7D).
To further characterize the AF1-SS population we integrated our datasets with those recently published by Tsukui et al.30,48,51 and found that AF1-SS population clusters together with inflammatory fibroblasts in these datasets (Supplementary Figs. S9, S10).
Discussion
The mechanical properties of the lung, such as tension and stiffness, are tightly regulated to maintain organ form and function. These bulk features originate from the mechanical properties of individual cells, which are determined by actomyosin-dependent contractility and intercellular adhesion. Dynamic regulation of these processes allows cells to exert forces on their neighbors, and during regeneration, this machinery serves to sense the growth rates of neighboring cells, ensuring uniform tissue growth. This study uncovers a Snail/Yap rheostat that functions as a core molecular switch controlling the fitness of mesenchymal cells. We demonstrate that this rheostat governs a process of inter-tissue cell competition, providing a new mechanistic framework for how the lung coordinates its architecture during development and repair.
Hippo/Yap signaling serves as a molecular sensor for cell density and a key regulator of tissue force balance. While Yap activity varies between individual cells, the collective pattern of its localization reflects the tissue’s overall mechanical state, making Yap signaling a potent determinant of cellular fitness. Our data suggest that inactivating Yap/Taz in alveolar fibroblast 1 cells (AF1s) reduces their fitness and disrupts the lung’s overall mechanical properties. This perturbation allows distal airway stem cells to increase their own Yap levels and initiate a pathological bronchiolization of the lung parenchyma. We recently observed a similar response when inactivating Yap/Taz in AT2 stem cells, and these findings collectively reveal that different lung cell populations engage in a competitive process to remodel the lung, aligning with the classical cell competition model52,53.
A central finding of this work is that the pathological bronchiolization seen in our injury models is not driven by a primary increase in epithelial aggression, but rather by the primary collapse of the mesenchymal niche. Our data support a clear causal sequence: genetic perturbations that reduce the fitness of AF1s, such as the deletion of Yap/Taz or Myc, lead to their elimination via apoptosis. The loss of these essential niche-supporting cells leads to the secondary death of the alveolar type 2 (AT2) stem cells they maintain. This destruction of the niche creates an epithelial void and generates a powerful, but misdirected, regenerative signal. In the absence of the proper instructive cues from the now-absent AF1s, distal airway stem cells mount a default pathological response, filling the damaged parenchyma with aberrant airway-like structures instead of functional alveoli. This “niche collapse” model provides an alternative possibility of our understanding of fibrotic remodeling, shifting the focus from the “winner” epithelium to the “failing” mesenchyme, such as observed with aging54, as the possible initiating pathological event.
We further identify tissue-scale mechanical cooperation as a crucial factor in orchestrating organ formation and regeneration. This cooperation involves mesenchymal and epithelial cell populations sensing the Yap-defined fitness levels in their neighbors. We find that during both lung development and repair after bleomycin injury, mesenchymal Yap levels and fitness antagonize epithelial Yap levels and stemness. Our data therefore suggest that tissues strive to restore tissue-wide homeostatic Yap levels as quickly as possible following injury. However, attempting to maintain tissue-level function at all costs may lead to aberrant remodeling when there is a great disturbance in the force, such as when millions of cells die off because of a catastrophic injury.
Our analysis of the aberrant basaloid cells that emerge in this context provides a stunning molecular explanation for their “supercompetitor” status. Our pathway analysis of this population reveals a profound metabolic reprogramming, characterized by the coordinated upregulation of glycolysis, the TCA cycle, oxidative phosphorylation, nucleotide synthesis, and Myc target gene activation. This is the classic signature of a hyper-proliferative state, analogous to the Warburg effect in cancer cells or the bioenergetics of rapidly dividing stem cells. This metabolic rewiring provides the functional engine for the “winner” phenotype, allowing these cells to generate the biomass and energy required to rapidly proliferate and overwhelm the compromised alveolar region, connecting the mechanisms of pathological lung remodeling to the broader fields of cancer metabolism and stem cell bioenergetics.
Furthermore, our experiments inactivating Snai1/2 during the resolution phase of fibrosis provide a molecular basis for the persistence of a non-resolving, pathological state. The resulting AF1-SS cell population is trapped in a vicious cycle, defined by the simultaneous activation of pro-fibrotic (collagen synthesis), pro-inflammatory (TNF-α and IL6-JAK-STAT3 signaling), and cellular stress (Unfolded Protein Response) pathways. This molecular signature is indicative of a stalled state that actively prevents the resolution of fibrosis. This finding has direct therapeutic implications, as it suggests that strategies aimed at pharmacologically promoting the Snail1/2-high/Yap-low state in myofibroblasts could represent a different class of pro-resolution therapies for treating pulmonary fibrosis.
In conclusion, our work demonstrates that the lung maintains its form and function through a system of tissue-scale cooperation, where epithelial and mesenchymal populations sense each other’s fitness levels via the Hippo/Yap pathway. These findings provide new insights into how organ size is regulated and how regeneration is coordinated across different tissue compartments. Moving forward, future studies aiming to better characterize this contractile heterogeneity and explore the tissue-level mechanisms that coordinate force balance will be important to find a cure for pulmonary fibrosis.
Methods
Experimental model and subject details
Both male and female mice were used in all experiments. Mice were bred in a pathogen-free environment on a standard laboratory setting of a 12 h light, 12 h dark exposure to mimic the day-night cycle. Room temperature was kept between 21 °C and 24 °C. Stk3/4f/f (RRID:IMSR_JAX:017635), Yap1f/f;Wwtr1f/f55, Tet-Yap1S112A-H2BGFP (RRID:IMSR_JAX:031279), Plin2CreERT2 (kind gift from Dr. Ahlbrecht56), Acta2CreERT2 (kind gift from Dr. Chambon57), Scube2CreERT2 (kind gift from Dr. Sheppard30), Rosa26-mTmG (RRID:IMSR_JAX:007676), Rosa26-Confetti (RRID:IMSR_ JAX:017492), Snai1f/f (kind gift from Dr. Weiss49), Snai2f/f (RRID:IMSR_JAX:037485), TetCre (RRID:IMSR_JAX:006234), Tbx4rtTA (kind gift from Dr. Shi58).
Summary information of genetic mouse models and experiments can be found in Supplementary Table 1. Bleomycin mice were intratracheally instilled with 50 µL bleomycin (0.8–2 U/kg body weight optimized for each strain, batch of bleomycin, and gender) as adults, 8–12 weeks old. For tamoxifen induction, Acta2CreERT2, Plin2CreERT2, Scube2CreERT2 were placed on tamoxifen chow (rodent diet with 400 mg/kg tamoxifen citrate; Harlan Teklad TD.130860). Plin2CreERT2 were placed on tamoxifen chow for 10 days at 2–3 months of age and were intratracheally instilled with bleomycin 3 weeks after being returned to normal rodent chow. Scube2CreERT2 mice were placed on tamoxifen chow at 2 months old for 3 weeks with an additional intraperitoneal tamoxifen injection (0.20 mg/g body weight, Millipore Sigma) on the last day of tamoxifen citrate feed, and after 3 weeks on normal laboratory chow, they were injured with bleomycin. Acta2CreERT2 received bleomycin at 2–3 months of age and were put on tamoxifen chow 3 weeks later for 3 weeks until euthanasia. Tbx4rtTA;TetCre mice were put on doxycycline containing chow (rodent diet with 625 mg/kg doxycycline; Harlan Teklad TD.09761) at weaning and remained on doxycycline chow until euthanasia and were intratracheally instilled with bleomycin at 2–3 months of age on doxycycline. All injured mice were euthanized 6 weeks post bleomycin injury.
Dams of embryonic mice were put on doxycycline at embryonic day 9.5 (E9.5), and embryos were dissected at E13.5, E15.5, and E18.5. All experiments were approved by the Mayo Clinic Institutional Animal Care and Use Committee.
Immunohistochemistry and fluorescence
All staining was done on paraffin sections of formalin-fixed lungs. Immunofluorescent staining was performed with the following primary antibodies: chicken anti-GFP (1:500; GFP-1020; RRID:AB_10000240; Aves Labs Inc.), rabbit anti-Keratin 5 (1:200; clone EP1601Y; MA5-14473; RRID:AB_10979451; Thermo Fisher Scientific), rabbit anti-SFTPC (1:200; WRAB-9337; RRID:AB_2335890; Seven hills bioreagents), goat anti-RAGE (1:500; AF1145; RRID:AB_354628; R&D Systems), rat anti-Keratin 8 (1:100; TROMA-I; RRID:AB_531826; Developmental Studies Hybridoma Bank), mouse anti-alpha actin (smooth muscle actin (SMA), Acta2; 1:500; Clone 1A4; sc-32251; RRID:AB_262054; Santa Cruz Biotechnology Inc.), rabbit anti-Yap (1:100; 4912; RRID:AB_2218911; Cell Signaling), rabbit anti-PLIN2 (1:200, ab52356, RRID:AB_2223599, Abcam), mouse anti-Cadherin (E Monoclonal) (1:200, Clone 36, 610181, RRID:AB_397580, BD Biosciences), rabbit anti-Snail2 (1:100, Clone C19G7, 9585, RRID:AB_2239535). TUNEL assays were performed on paraffin sections of formalin-fixed lungs using the In-Situ Cell Detection Kit, TMR Red (12156792910, Roche).
After deparaffinization, slides were rehydrated through a series of decreasing ethanol concentrations, antigen unmasked by either microwaving in citrate-based antigen unmasking solution (Vector Labs, H-3300) or by incubating sections with proteinase K (7.5 μg/ml) (Invitrogen, 25530-049) for 7 min at 37 °C. Tissue sections were then washed in TBS with 0.1% Tween-20 and blocked with 3% Bovine Serum Albumin (BSA), 0.4% Triton X-100 in TBS for 30 min at room temperature followed by overnight incubation of primary antibodies diluted in 3% BSA, 0.1% Triton X-100 in TBS. The next day, slides were washed in TBS with 0.1% Tween-20 and incubated with secondary antibodies diluted in 3% BSA, 0.1% Triton X-100 in TBS for 3 h at room temperature. All fluorescent staining was performed with appropriate secondary antibodies from Jackson Immunoresearch. Slides were mounted using Vectashield (Vector Labs, H-1000).
Microscopy and imaging
Tissue was imaged using a micrometer slide calibrated Zeiss LSM800 Laser scanning confocal microscope using ZEN imaging software or Leica Stellaris 5 confocal microscope with LASX imaging software. Lungs were imaged using tiled stitched 20× images covering the entire cross-section of the left or lower right lung lobe from ≥6 different lungs. Representative images were chosen. Images were processed and analyzed using Zen blue (Zeiss), LASX (Leica), ImageJ (NIH) and Adobe Photoshop 2024 (Adobe) software. The total area of Krt5, Krt8 or GFP was determined using ImageJ (NIH) and analyzed based on the total lung area. Image quantification and analysis was performed in a double blinded fashion. Each quantification was ≥3 different mouse lungs.
Quantitative real-time PCR
Total mRNA was extracted from lung accessory lobes stored in RNALater (Invitrogen, AM7021) and using Total RNA Kit I (Omega Biotek, R6834-02) according to the manufacturer’s instructions. RNA concentration was determined by spectrophotometry. cDNA was generated using Maxima™ First Strand cDNA Synthesis (Fisher Scientific, FERK1642) according to the manufacturer’s instructions. Gene expression was analyzed by quantitative RT-PCR using Taqman Gene Expression Assays (Applied Biosystems, 4369016) directed against the mouse targets β-glucuronidase (Mm00446953_m1), Krt5 (Mm01305291_g1), Trp63 (Mm00495788_m1). Quantitative real-time PCR was performed using a StepOne Plus system (Applied Biosystems). Data were presented as 2−ΔΔCt with β-glucuronidase as the internal sample control normalized to control group. Each experiment was repeated with samples obtained from ≥3 different lung preparations.
Nanostring
RNA was isolated from lung accessory lobes as described above. 100 ng of RNA was hybridized with a custom RNA probe panel designed by NanoString (NanoString Technologies; DL_1206_C9662) for 16 h according to manufacturer’s instructions. The RNA-probe hybridization was loaded on a NanoString cartridge and processed in a NanoString nCounter. Data was analyzed with Rosalind.bio (Rosalind, Inc) and Log2 Fold Changes were calculated and graphed. Each experiment was repeated with samples obtained from ≥3 different lung preparations.
Hydroxyproline assay
The right lobes were flash frozen in dry ice at the time of harvest and stored at −80 °C. For acid hydrolysis, the lobes were baked in a 70 °C oven without lids for 2 days until completely dry. The weights of dry lobes were measured and 500 µl of 6 N HCl were added to each sample. The lungs were then hydrolyzed in an 85 °C oven for 2 days with occasional vortexing. The hydrolysates were cooled at room temperature and centrifuged at maximum speed for 10 min. The supernatants then were transferred to fresh 1.5 mL tubes and centrifuged at maximum speed for 10 min. Each sample or standard was diluted with citrate-acetate buffer (5% citric acid, 1.2% glacial acetic acid, 7.24% sodium acetate, and 3.4% sodium hydroxide) in a 96-well plate. Chloramine-T solution (1.4% chloramine-T, 10% N-propanol, and 80% citrate-acetate buffer) was added, and the mixture was incubated for 20 min at room temperature. Then, Ehrlich’s solution (1.27 M p-dimethylaminobenzaldehyde, 70% N-propanol, 20% perchloric acid) was added to each sample and the samples were incubated at 65 °C for 20 min. Absorbance was measured at 550 nm. Standard curves were generated for each experiment using reagent hydroxyproline (Sigma H-1637) as a standard. The amount (µg) of hydroxyproline were calculated by comparison to the standard curve.
10× genomics single cell multiome ATAC+ gene expression sequencing
Embryonic day E15.5 or E18.5 lungs were collected and flash-frozen in a dry ice/ethanol slurry. Frozen tissues were homogenized with a dounce homogenizer and nuclei were isolated according to 10× Genomics Nuclei Isolation from Embryonic Mouse Brain for Single Cell Multiome ATAC+ Gene Expression Sequencing. Nuclei viability was assessed, and total nuclei were counted. ATAC and gene expression libraries were then constructed from the nuclei according to the 10× Genomics Chromium Next GEM Single Cell Multiome ATAC+ Gene Expression User Guide. Sequencing was performed on a NextSeq500. Alignment was performed using the ‘Cellranger count’ function provided in 10× Genomics single-cell gene expression software. For secondary analysis we used the R-package ArchR59 to apply standard quality control metrics and unsupervised clustering for the generation of initial UMAP projections and integrate multiomics data.
10× genomics single cell fixed RNA profiling (flex) analysis
Cells from 2 50 μm formalin fixed paraffin embedded (FFPE) tissue sections were deparaffinized and manually dissociated with a pellet pestle using 1.0 mg/mL Liberase TH in RPMI according to manufacturer’s directions (10× Genomics).Cells were counted using Countess II FL Automated Cell Counter (Thermo Fisher Scientific) and up to 10,000 cells per sample were hybridized with mouse WTA Probes (10× Genomics; PN-2000703-2000718) for 17 h according to manufacturer’s instructions. Post-hybridization cells were counted and pooled with equal number of cells per sample for a target recovery of 128,000 cells. GEM generation and final library preparations and sequencing were completed by the Mayo Genomics Research Core according to manufacturer’s instructions on an Illumina NextSeq. Fastq files were analyzed with Cell Ranger Pipeline using 10× Genomics Cloud Analysis. For secondary analysis we used Scanpy60 to apply standard quality control metrics and unsupervised clustering for the generation of initial UMAP projections and integrate data using Harmonypy61 or Scanorama62, trajectory inference was determined using Monocle 363, whereas (supervised pathway deconvolution of interpretable gene programs) analysis was performed using Spectra44. Data from the current study (CTRL NI, CTRL d42 Bleo, Acta2CreERT2;Snai1/2f/f d42 Bleo) was integrated with multiple published lung fibrosis datasets (GSE218666, GSE173878, GSE235212, GSE218665, GSE210341, GSE132771, GSE276546, GSE229523)30,48,51.
In situ for Snai1 and Snai2
Whole-mount in situ hybridization was performed as previously described64 with antisense probes against full length Snai1 or Snai2.
Statistics and reproducibility
All results are expressed as mean values ± SEM. The ‘n’ represents biological replicates and can be found in the figure legends. The significance of differences between 2 sample means was determined by two-tailed unpaired t-test (assuming unequal or equal variances as determined by the F-test of equality of variances). All datasets followed a normal distribution and P values less than 0.05 were considered statistically significant. The number of samples to be used was based on the number of experimental paradigms multiplied by the number in each group that is necessary to yield statistically significant results (based on power analysis, to reject the null hypothesis with 80% power (type I error = 0.05).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
10x Genomics Multiome and Flex RNA profiling generated in this study have been deposited in Zenodo Repository (10.5281/zenodo.17468266). Previously published datasets use in this study include: GSE218666, GSE173878, GSE235212, GSE218665, GSE210341, GSE132771, GSE276546, GSE229523. Further information and requests for resources, data, and reagents should be directed to and will be fulfilled by the Lead Contact, Stijn De Langhe: delanghe.stijn@mayo.edu.
References
Gebb, S. A. & Jones, P. L. Hypoxia and lung branching morphogenesis. Adv. Exp. Med Biol. 543, 117–125 (2003).
Ochoa-Espinosa, A. & Affolter, M. Branching morphogenesis: from cells to organs and back. Cold Spring Harb. Perspect. Biol. 4, https://doi.org/10.1101/cshperspect.a008243 (2012).
Chang, D. R. et al. Lung epithelial branching program antagonizes alveolar differentiation. Proc. Natl Acad. Sci. USA 110, 18042–18051 (2013).
Alanis, D. M., Chang, D. R., Akiyama, H., Krasnow, M. A. & Chen, J. Two nested developmental waves demarcate a compartment boundary in the mouse lung. Nat. Commun. 5, 3923 (2014).
Volckaert, T. et al. Localized Fgf10 expression is not required for lung branching morphogenesis but prevents differentiation of epithelial progenitors. Development 140, 3731–3742 (2013).
Volckaert, T. & De Langhe, S. Lung epithelial stem cells and their niches: Fgf10 takes center stage. Fibrogenes. Tissue Repair 7, 8 (2014).
Hines, E. A. & Sun, X. Tissue crosstalk in lung development. J. Cell Biochem. 115, 1469–1477 (2014).
Shannon, J. M., Nielsen, L. D., Gebb, S. A. & Randell, S. H. Mesenchyme specifies epithelial differentiation in reciprocal recombinants of embryonic lung and trachea. Dev. Dyn. 212, 482–494 (1998).
Volckaert, T. et al. Fgf10-Hippo Epithelial-Mesenchymal Crosstalk Maintains and Recruits Lung Basal Stem Cells. Dev. Cell 43, 48–59.e45 (2017).
Volckaert, T. et al. Parabronchial smooth muscle constitutes an airway epithelial stem cell niche in the mouse lung after injury. J. Clin. Invest. 121, 4409–4419 (2011).
Lyu, H. et al. Niche-mediated repair of airways is directed in an occupant-dependent manner. Cell Rep. 41, 111863 (2022).
Nabhan, A. N., Brownfield, D. G., Harbury, P. B., Krasnow, M. A. & Desai, T. J. Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science 359, 1118–1123 (2018).
Peng, T. et al. Hedgehog actively maintains adult lung quiescence and regulates repair and regeneration. Nature 526, 578–582 (2015).
Lee, J. H. et al. Anatomically and Functionally Distinct Lung Mesenchymal Populations Marked by Lgr5 and Lgr6. Cell 170, 1149–1163.e1112 (2017).
Mannino, D. M., Brown, C. & Giovino, G. A. Obstructive lung disease deaths in the United States from 1979 through 1993. An analysis using multiple-cause mortality data. Am. J. Respir. Crit. Care Med. 156, 814–818 (1997).
Obi, J., Mehari, A. & Gillum, R. Mortality related to chronic obstructive pulmonary disease and co-morbidities in the United States, a multiple causes of death analysis. COPD 15, 200–205 (2018).
King, T. E. Jr., Pardo, A. & Selman, M. Idiopathic pulmonary fibrosis. Lancet 378, 1949–1961 (2011).
Steele, M. P. & Schwartz, D. A. Molecular mechanisms in progressive idiopathic pulmonary fibrosis. Annu. Rev. Med. 64, 265–276 (2013).
Barkauskas, C. E. & Noble, P. W. Cellular mechanisms of tissue fibrosis. 7. New insights into the cellular mechanisms of pulmonary fibrosis. Am. J. Physiol. Cell Physiol. 306, C987–C996 (2014).
Plantier, L. et al. Ectopic respiratory epithelial cell differentiation in bronchiolised distal airspaces in idiopathic pulmonary fibrosis. Thorax 66, 651–657 (2011).
Seibold, M. A. et al. The idiopathic pulmonary fibrosis honeycomb cyst contains a mucocilary pseudostratified epithelium. PLoS One 8, e58658 (2013).
Warren, R. et al. Cell competition drives bronchiolization and pulmonary fibrosis. Nat. Commun. 15, 10624 (2024).
Strunz, M. et al. Alveolar regeneration through a Krt8+ transitional stem cell state that persists in human lung fibrosis. Nat. Commun. 11, 3559 (2020).
Kobayashi, Y. et al. Persistence of a regeneration-associated, transitional alveolar epithelial cell state in pulmonary fibrosis. Nat. Cell Biol. 22, 934–946 (2020).
Adams, T. S. et al. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci. Adv. 6, eaba1983 (2020).
Habermann, A. C. et al. Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci. Adv. 6, eaba1972 (2020).
McCulley, D., Wienhold, M. & Sun, X. The pulmonary mesenchyme directs lung development. Curr. Opin. Genet Dev. 32, 98–105 (2015).
Rock, J. R. et al. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc. Natl Acad. Sci. USA 108, E1475–E1483 (2011).
El Agha, E. et al. Two-way conversion between lipogenic and myogenic fibroblastic phenotypes marks the progression and resolution of lung fibrosis. Cell Stem Cell 20, 261–273.e263 (2017).
Tsukui, T., Wolters, P. J. & Sheppard, D. Alveolar fibroblast lineage orchestrates lung inflammation and fibrosis. Nature 631, 627–634 (2024).
Horowitz, J. C. & Thannickal, V. J. Mechanisms for the resolution of organ fibrosis. Physiology 34, 43–55 (2019).
He, X. et al. Myofibroblast YAP/TAZ activation is a key step in organ fibrogenesis. JCI Insight 7, https://doi.org/10.1172/jci.insight.146243 (2022).
Liu, F. et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 308, L344–L357 (2015).
Morrisey, E. E. & Hogan, B. L. Preparing for the first breath: genetic and cellular mechanisms in lung development. Dev. Cell 18, 8–23 (2010).
Tang, Y. & Weiss, S. J. Snail/Slug-YAP/TAZ complexes cooperatively regulate mesenchymal stem cell function and bone formation. Cell Cycle 16, 399–405 (2017).
Tang, Y., Feinberg, T., Keller, E. T., Li, X. Y. & Weiss, S. J. Snail/Slug binding interactions with YAP/TAZ control skeletal stem cell self-renewal and differentiation. Nat. Cell Biol. 18, 917–929 (2016).
Jang, Y. N. & Baik, E. J. JAK-STAT pathway and myogenic differentiation. JAKSTAT 2, e23282 (2013).
Wang, K., Wang, C., Xiao, F., Wang, H. & Wu, Z. JAK2/STAT2/STAT3 are required for myogenic differentiation. J. Biol. Chem. 283, 34029–34036 (2008).
Wang, F. et al. Noncanonical JAK1/STAT3 interactions with TGF-beta modulate myofibroblast transdifferentiation and fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 323, L698–L714 (2022).
Oh, R. S. et al. RNAi screening identifies a mechanosensitive ROCK-JAK2-STAT3 network central to myofibroblast activation. J. Cell Sci. 131, https://doi.org/10.1242/jcs.209932 (2018).
Hecker, L., Jagirdar, R., Jin, T. & Thannickal, V. J. Reversible differentiation of myofibroblasts by MyoD. Exp. Cell Res. 317, 1914–1921 (2011).
Sanders, Y. Y., Kumbla, P. & Hagood, J. S. Enhanced myofibroblastic differentiation and survival in Thy-1(-) lung fibroblasts. Am. J. Respir. Cell Mol. Biol. 36, 226–235 (2007).
Tapscott, S. J. The circuitry of a master switch: Myod and the regulation of skeletal muscle gene transcription. Development 132, 2685–2695 (2005).
Kunes, R. Z., Walle, T., Land, M., Nawy, T. & Pe’er, D. Supervised discovery of interpretable gene programs from single-cell data. Nat. Biotechnol. 42, 1084–1095 (2024).
Volckaert, T. et al. Hippo signaling promotes lung epithelial lineage commitment by curbing Fgf10 and beta-catenin signaling. Development 146, https://doi.org/10.1242/dev.166454 (2019).
Zhang, Y. et al. GDF15 is an epithelial-derived biomarker of idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 317, L510–L521 (2019).
Croci, O. et al. Transcriptional integration of mitogenic and mechanical signals by Myc and YAP. Genes Dev. 31, 2017–2022 (2017).
Konkimalla, A. et al. Transitional cell states sculpt tissue topology during lung regeneration. Cell Stem Cell 30, 1486–1502.e1489 (2023).
Rowe, R. G. et al. Mesenchymal cells reactivate Snail1 expression to drive three-dimensional invasion programs. J. Cell Biol. 184, 399–408 (2009).
Peiro, S. et al. Snail1 transcriptional repressor binds to its own promoter and controls its expression. Nucleic Acids Res. 34, 2077–2084 (2006).
Fang, Y. et al. RUNX2 promotes fibrosis via an alveolar-to-pathological fibroblast transition. Nature 640, 221–230 (2025).
Morata, G. & Ripoll, P. Minutes: mutants of drosophila autonomously affecting cell division rate. Dev. Biol. 42, 211–221 (1975).
Simpson, P. & Morata, G. Differential mitotic rates and patterns of growth in compartments in the Drosophila wing. Dev. Biol. 85, 299–308 (1981).
Chanda, D. et al. Mesenchymal stromal cell aging impairs the self-organizing capacity of lung alveolar epithelial stem cells. Elife 10, https://doi.org/10.7554/eLife.68049 (2021).
Xin, M. et al. Hippo pathway effector Yap promotes cardiac regeneration. Proc. Natl Acad. Sci. USA 110, 13839–13844 (2013).
Ntokou, A. et al. A novel mouse Cre-driver line targeting Perilipin 2-expressing cells in the neonatal lung. Genesis 55, https://doi.org/10.1002/dvg.23080 (2017).
Wendling, O., Bornert, J. M., Chambon, P. & Metzger, D. Efficient temporally-controlled targeted mutagenesis in smooth muscle cells of the adult mouse. Genesis 47, 14–18 (2009).
Zhang, W. et al. Spatial-temporal targeting of lung-specific mesenchyme by a Tbx4 enhancer. BMC Biol. 11, 111 (2013).
Granja, J. M. et al. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat. Genet. 53, 403–411 (2021).
Wolf, F. A., Angerer, P. & Theis, F. J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019).
Hie, B. L., Kim, S., Rando, T. A., Bryson, B. & Berger, B. Scanorama: integrating large and diverse single-cell transcriptomic datasets. Nat. Protoc. 19, 2283–2297 (2024).
Trapnell, C. et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 32, 381–386 (2014).
De Langhe, S. P. et al. Dickkopf-1 (DKK1) reveals that fibronectin is a major target of Wnt signaling in branching morphogenesis of the mouse embryonic lung. Dev. Biol. 277, 316–331 (2005).
Acknowledgements
This study was supported by NIH R01 HL146461 (S.D.L.), HL132156 (S.D.L.), NIH R35 HL161169 (S.D.L.), NIH T32 HL105355 (R.W.) and Mayo Clinic Brewer Family Career Development Award (R.W.).
Author information
Authors and Affiliations
Contributions
K.K., R.W., and J.K. designed and performed experiments, interpreted the data and prepared the manuscript. T.N. performed experiments. S.D.L. conceived, designed and supervised the study, analyzed and interpreted the data and prepared the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Klinkhammer, K., Warren, R., Knopp, J. et al. Epithelial-mesenchymal cell competition coordinates fate transitions across tissue compartments during lung development and fibrosis. Nat Commun 16, 10956 (2025). https://doi.org/10.1038/s41467-025-66690-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-66690-z
